

Abstract
Despite the approval of oncolytic virus therapy for advanced melanoma, its intrinsic limitations that include the risk of persistent viral infection and cost-intensive manufacturing motivate the development of analogous approaches that are free from the disadvantages of virus-based therapies. Herein, we report a nanoassembly comprised of multivalent host-guest interactions between polymerized paclitaxel (pPTX) and nitric oxide incorporated polymerized β-cyclodextrin (pCD-pSNO) that through its bioactive components and when used locoregionally recapitulates the therapeutic effects of oncolytic virus. The resultant pPTX/pCD-pSNO exhibits significantly enhanced cytotoxicity, immunogenic cell death, dendritic cell activation and T cell expansion in vitro compared to free agents alone or in combination. In vivo, intratumoral administration of pPTX/pCD-pSNO results in activation and expansion of dendritic cells systemically, but with a corresponding expansion of myeloid-derived suppressor cells and suppression of CD8+ T cell expansion. When combined with antibody targeting cytotoxic T lymphocyte antigen-4 that blunts this molecule’s signaling effects on T cells, intratumoral pPTX/pCD-pSNO treatment elicits potent anticancer effects that significantly prolong animal survival. This formulation thus leverages the chemo- and immunotherapeutic synergies of paclitaxel and nitric oxide and suggests the potential for virus-free nanoformulations to mimic the therapeutic action and benefits of oncolytic viruses.
Keywords: Cancer, Chemotherapy, Immunotherapy, Nitric oxide, Paclitaxel
Graphical Abstract
Synthetic oncolytic virus comprised of self-assembled nanoparticles leverages chemoimmunotherapeutic synergies of co-formulated paclitaxel and nitric oxide for anti-cancer therapy. Locoregional application recapitulates distinctive mechanisms of oncolytic virus, enabling synergistic local chemotherapeutic effects and systemic expansion of dendritic cells that improve anti-cancer immunotherapy effects.
1. Introduction
Immunotherapy has emerged this decade as the most efficacious anti-cancer treatment class for multiple disease types. This is due in large part to both the stunning improvements in patient survival when effective as well as the potential to impart lifelong immunity to protect against disease recurrence. With these advancements, much of the therapeutic benefit conferred by other more established anti-cancer treatments are attributed at least in part to their immunomodulatory effects. For example, paclitaxel (PTX) is one of the most widely used anticancer drugs that not only exerts direct cytotoxic effects to cancer cells,[1] but also augments antitumor immunity by inducing immunogenic cell death (ICD) of tumors,[2] maturing dendritic cells (DCs),[3–5] improving antigen presentation,[5] activating T helper 1 (TH1) cells[6–8] and cytotoxic CD8+ T cells (CTLs),[6,7,9] and inhibiting Foxp3+ regulatory T cells (Tregs).[7,9–11] As such, the potential for immunotherapy to synergize with traditional treatments has been proposed and clinically verified in multiple therapeutic settings.[12,13]
A relatively small but growing class of immunotherapy is based on the application of oncolytic viruses (OVs). Although diverse in application, their use overall focuses on leveraging the direct lytic functions of OVs to eliminate rapidly dividing cancer cells.[3,14–17] Several OVs in clinical use are genetically modified to express granulocyte-macrophage colony-stimulating factor (GM-CSF), including T-VEC, which was approved by the US FDA in 2015 as an immunotherapy via intralesional administration for advanced melanoma. Each of these properties serve to manifest in OV immunotherapy functions, including the generation of neo-antigens by selectively replicating in cancer cells[15,16] and recruitment of antigen-presenting cells to initiate antitumor immune response,[15,16] respectively. As a result, T-VEC exhibits considerably high response rate (~60% in injected lesions and ~40% in uninjected lesions) compared to ~15% and 30-40 % response rate for aCTLA-4 and aPD-1, respectively[17] although mechanisms underlying their therapeutic benefit are still being established.[3,14–17] Overall, it is thought that OV therapy will continue to grow as a cancer treatment class over the next decade, and there are dozens of ongoing clinical trials.[16] However, OVs have multiple intrinsic limitations including their cost-intensive manufacturing process due to the live virus cultures, potential to induce persistent viral infections in normal (non-target) cells, risk of unintended infections in household and medical workers, and potential ecosystem disturbance due to the use of recombinant DNA.[15] In particular, the therapeutic effects of OV include an inevitable, yet poorly controllable negative feedback loop, as an enhanced immune response initiated by OV infection in turn contributes to suppress virus replication.[15] A virus-free strategy to mimic OV is thus highly attractive not only to improve the therapeutic potential, but also to broaden its eligibility across various types of tumor.
To this end, drug delivery systems (DDSs) enhance the therapeutic efficacy and biosafety of a multitude drug classes, by controlling and directing their biodistribution and activity. In the context of cancer chemotherapy this typically focuses on increasing drug cytotoxic effects against tumors and minimizing them against host cells.[18] The small, gaseous nitric oxide (NO) has attracted significant attention in anticancer chemotherapy owing to its chemosensitizing effects via inhibition of drug efflux mechanisms.[19–21] Interestingly, NO participates in various immunological regulatory functions,[22] such as protection of DCs from tumor-induced apoptosis,[23] activation of TH1 cells,[24–27] DC maturation,[25,28] enhancement of DC migration,[29] inhibition of Tregs,[27] and improvement of the survival and differentiation of CTLs.[30] Furthermore, intracellular NO level has been reported to be closely associated with chemotherapeutic drug-induced ICD of cancer cells.[31–33] Despite these favorable characteristics, to the best of our knowledge, the potential of NO in the fields of (chemo)immunotherapy has not been reported.
Herein, we report a PTX and NO co-formulation DDS that when applied locoregionally to a growing melanoma augments anti-tumor drug effects. By advancing our self-assembled PTX nanoparticles (NPs),[34] the multivalent host-guest interactions between polymerized PTX (pPTX) and thiol-containing polymerized cyclodextrin (pCD-pSH) allow for self-assembled NPs (pPTX/pCD-pSH). Thiol groups are further converted to S-nitrosothiol (-SNO), which upon decay results in NO release that has both chemosensitizing and immune modulatory effects.[19–33] In so doing, the NPs (pPTX/pCD-pSNO) can simultaneously deliver NO and PTX that are released within the chemical environments of the intracellular space after cellular uptake. When applied in a locoregional manner, formulation into pCD nanocarriers improves PTX’s anti-cancer effects in vitro and in vivo. The immune modulatory effects of PTX are also increased by NO co-delivery, including increased ICD and DC maturation in vitro, and anti-tumor and immune stimulatory effects in vivo. When combined with antibodies blocking the immune checkpoint regulated by cytotoxic T lymphocyte antigen (CTLA)-4, pPTX/pCD-pSNO resulted in significant improvement of the abscopal effect of treatment. These results support the extension of DDS principles to chemoimmunotherapy to mimic the immunological basis thought to underlie OV therapy as a potentially safer and more controllable virus-free treatment approach for cancer therapy.
2. Results
2.1. Preparation and Characterizations of pPTX/pCD-pSNO
The pPTX and pCD were synthesized by conjugating activated hydroxyl groups of PTX and β-CD individually onto poly(maleic anhydride) as previously reported[34] (Figure S1, S2). Resulting from each reaction were pPTX and pCD containing ~19 PTX and ~12 CD on the individual polymer backbones (Figure 1A). pCD was thiolated by reacting cystamine with carboxyl groups of pCD, followed by reduction of resulting disulfide bonds, rendering pCD-pSH (Figure S2). The feasibility of tert-butyl nitrite-mediated S-nitrosothiol formation on free thiols of pCD-pSH was confirmed by 343 nm (π→ π*) absorbance of S-NO bond (Figure S3). Complexation of pPTX and pCD-SH to form pPTX/pCD-pSH NPs by host-guest interactions between individual polymers was performed in 20% ethanol, followed by dialysis against deionized (D.I.) water and freezing drying. In a subfraction of pPTX/pCD-pSH NPs, thiol groups were converted to S-nitrosothiol to create pPTX/pCD-pSNO NPs (Figure 1A). Compared to NP formed from unthiolated pPTX/pCD, pPTX/pCD-pSH and pPTX/pCD-pSNO NPs were found to have a significantly smaller size (60-70 nm versus 133 nm,[34] respectively) (Figure 1B–D and Table 1). As we previously reported, electrostatic repulsion of negatively charged carboxylic anions of polymeric backbones and attractive inclusion forces of CD and PTX, which have opposing effects, allows the formation of the NPs. Supporting the concept that decreased electrostatic repulsion by the additional conjugation of cysteamine onto the carboxylate anions contributes to the reduced size of pPTX/pCD-pSH and pPTX/pCD-pSNO, reduced negative charges were observed in pPTX/pCD-pSH and pPTX/pCD-pSNO compared to pPTX/pCD (Figure 1E, Table 1). Successful conjugation of S-nitrosothiol groups on pPTX/pCD-pSNO was confirmed by specific vibration peak of NO in Fourier-transform infrared spectroscopy (FT-IR) (Figure 1F) and reduced free thiol levels after S-nitrosylation as measured by Ellman’s assay (Figure 1G). Saville and Griess assays revealed that the total SNO contents are 19.7 nmol in 1 mg of pPTX/pCD-pSNO and the conversion efficacy of SNO from free thiols is 84.6±0.2 % (Figure 1G).
Figure 1.
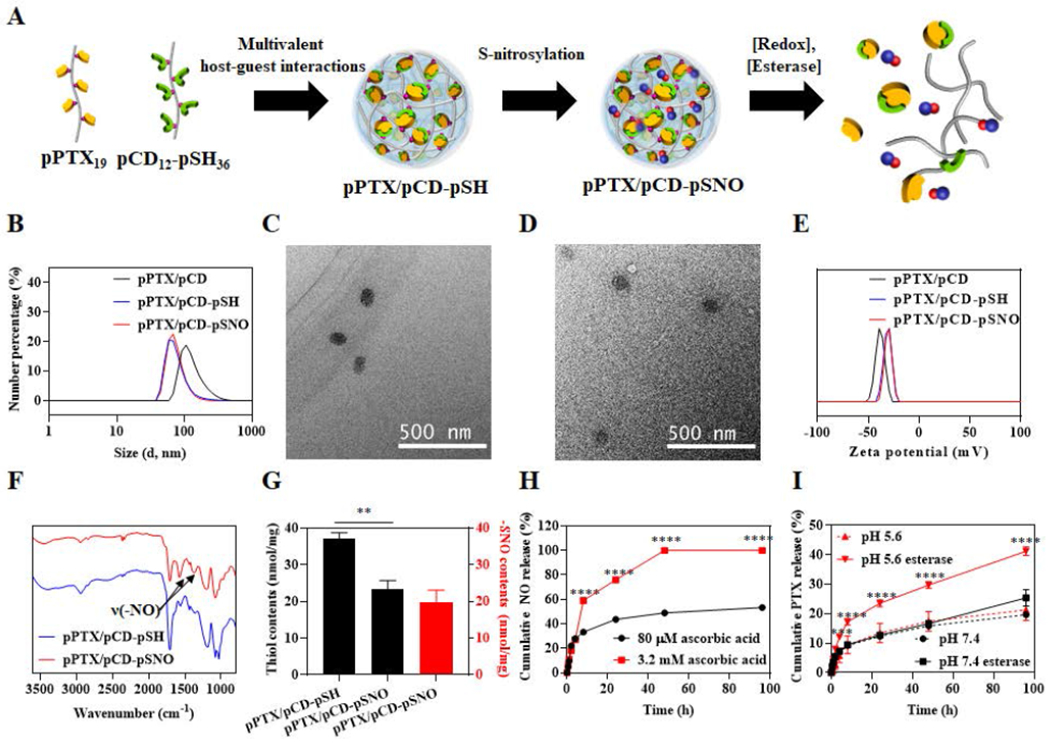
Synthesis and characterizations of pPTX/pCD-pSNO NPs. (A) Schematic of pPTX/pCD-pSNO NP preparation and NO and PTX release mechanisms. (B) Dynamic light scattering (DLS)-based hydrodynamic size distributions of pPTX/pCD, pPTX/pCD-pSH and pPTX/pCD-pSNO NPs. (C) Transmission electron microscopy (TEM) images of pPTX/pCD-pSH NPs. (D) TEM images of pPTX/pCD-pSNO NPs. (E) Zeta potential-based surface charges of pPTX/pCD, pPTX/pCD-pSH and pPTX/pCD-pSNO NPs. (F) FT-IR of pPTX/pCD-pSH and pPTX/pCD-pSNO NPs. (G) Quantification of thiol content of pPTX/pCD-pSH and pPTX/pCD-pSNO NPs by Ellmans’ assay (black box) and quantification of –SNO groups in pPTX/pCD-pSNO NPs by Saville and Griess assays (red box). (H) Cumulative NO release graph under intracellular and extracellular redox conditions. (I) Cumulative PTX release graph at different pH with or without esterase. ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05.
Table 1.
The summarized size and zeta potential of pPTX/pCD, pPTX/pCD-pSH and pPTX/pCD-pSNO NPs.
| Size (d, nm)a) | Size (d, nm)b) | Zeta (mV)b) | |
|---|---|---|---|
| pPTX/pCD | - | 133.1±9.5 | −40.13±0.04 |
| pPTX/pCD-pSH | 60.8±10.9 | 79.9±7.1 | −31.83±0.55 |
| pPTX/pCD-pSNO | 60.5±8.1 | 72.4±4.4 | −30.63±0.32 |
determined by TEM,
determined by DLS.
NO release from pPTX/pCD-pSNO was measured under intracellular (pH 7.4, PBS, 3.2 mM ascorbic acid) and extracellular conditions (pH 7.4, PBS, 80 μM ascorbic acid) (Figure 1H).[35–37] Owing to the redox-responsive behavior of S-nitrosothiol,[38] a burst NO release was observed under intracellular redox conditions. Likewise, PTX release profiles were investigated at the physiological pH and intracellular endolysosomal pH in the presence or absence of esterase (Figure 1I). Interestingly, only esterase at endolysosomal pH led to burst PTX release from pPTX/pCD-pSNO although acidic pH or esterase individually had no effects on the PTX release compared to the control group (pH 7.4 without esterase), which is attributed to the pH-dependent activity of esterases.[39–41] Together, these results demonstrate that S-nitrosothiol and PTX conjugated pPTX/pCD-pSNO NPs exhibit preferential release of NO and PTX under intracellular compared to extracellular conditions.
2.2. Chemotherapeutic Effects and ICD in Cancer Cells in vitro
The chemotherapeutic potential of the pPTX/pCD-pSNO NPs, and specifically the potential for NO to augment the cytotoxic effects of PTX chemotherapy, was investigated by evaluating cytotoxicity in various cell lines commonly used to model taxane-sensitive cancers[42,43] including murine B16F10 melanoma, E0771 mammary carcinoma, and EL4 lymphoma and human LS174T colon carcinoma (Figure 2A–D and Table 2). In all cell lines tested, pPTX/pCD-pSNO NPs exhibited higher cytotoxicity than pPTX/pCD-pSH NPs and free PTX, despite the insignificant cytotoxicity of GSNO up to 1.45 μM (Figure S4 and Table 2), a – SNO concentration comparable to that found in 10 μM pPTX/pCD-pSNO. These results indicate that while NO does not directly exert cytotoxicity, it acts as a sensitizer in PTX-mediated cytotoxicity. In particular, the combination index at IC50 (CI50) was evaluated to be 0.22-0.81 in all four tested cancer cell lines (Table 3), indicative of high synergy between NO and PTX in pPTX/pCD-pSNO NPs. Among various cancer cell lines, B16F10 was selected as a model for in vitro mechanism studies due to the high observed synergism in this model system, as well as the fact that T-VEC was approved to treat melanoma. The synergistic effects of PTX in combination with NO when delivered via pPTX/pCD-pSNO NPs were associated with enhanced late apoptosis and necrosis measured via an Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) assay (Figure 2E, S5).
Figure 2. In vitro effects of pPTX/pCD-pSNO NPs on cancer cells.
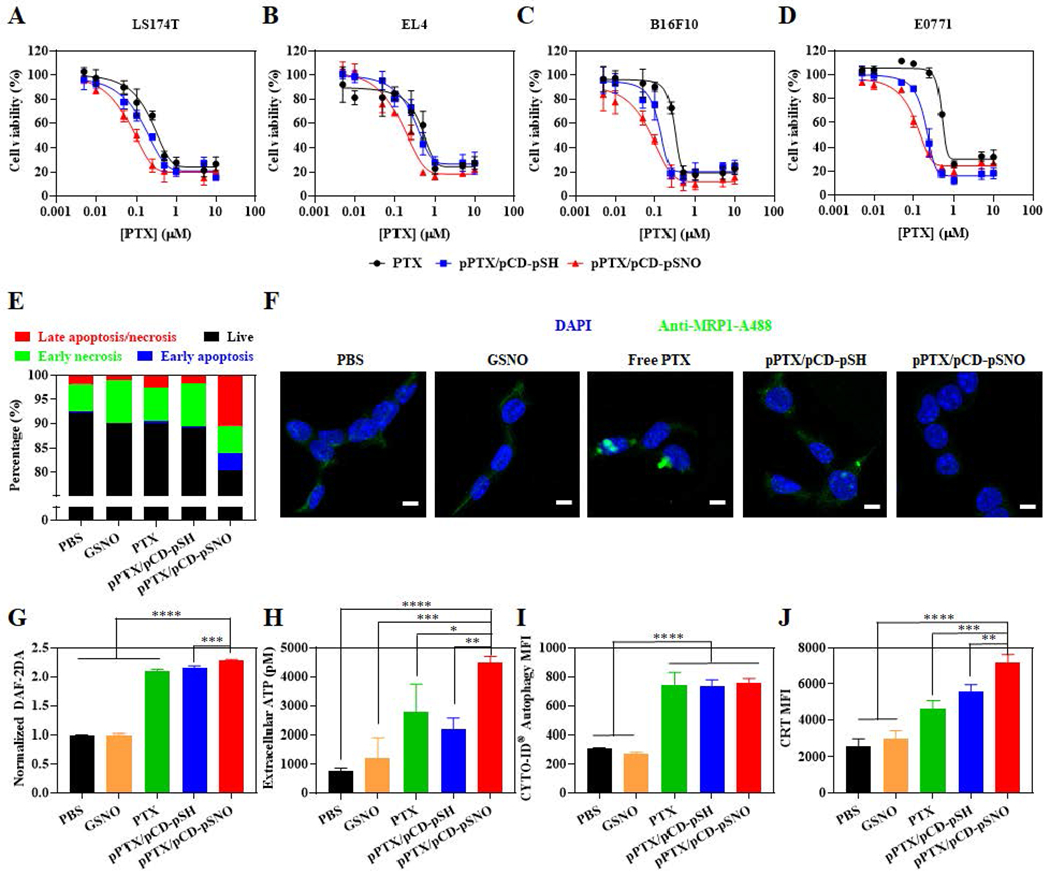
Alamar blue-assisted cytotoxicity testing of free PTX, pPTX/pCD-pSH NPs and pPTX/pCD-pSNO NPs in (A) LS174T, (B) EL4, (C) B16F10, and (D) E0771 cell lines. (E) Quantification of apoptosis and necrosis in B16F10 by Annexin V/PI assay-assisted flow cytometry ([PTX] = 0.05 μM, [-SNO] = 7.2 nM, 1 d incubation). (F) CLSM images of MPR 1 expression (green) in B16F10 ([PTX] = 1 μM, [-SNO] = 144.5 nM, 1 d incubation). Scale bar is 10 μm. (G) Quantification of intracellular NO in B16F10 ([PTX] = 1 μM, [-SNO] = 144.5 nM, 1d incubation). (H) Quantification of extracellular ATP released from B16F10 ([PTX] = 1 μM, [-SNO] = 144.5 nM, 1 d incubation). (I) Quantification of intracellular autophagy in B16F10 ([PTX] = 0.1 μM, [-SNO] = 144.5 nM, 1 d incubation) (J) Quantification of CRT in B16F10 ([PTX] = 0.1 μM, [-SNO] = 144.5 nM, 1 d incubation). ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05.
Table 2.
The summarized IC50 of GSNO, PTX, pPTX/pCD-pSH and pPTX/pCD-pSNO NPs in LS174T, EL4, B16F10 and E0771 cancer cell lines.
| PTX IC50 (μM) | −SNO IC50 (μM) | |||||||
|---|---|---|---|---|---|---|---|---|
| LS174T | EL4 | B16F10 | E0771 | LS174T | EL4 | B16F10 | E0771 | |
| GSNO | - | - | - | - | 56.3±15.8 | 16.5±7.0 | 16.9±4.1 | 20.6±0.6 |
| Free PTX | 0.38±0.10 | 0.68±0.08 | 0.42±0.19 | 0.58±0.11 | - | - | - | - |
| pPTX/pCD-pSH | 0.19±0.04 | 0.46±0.13 | 0.19±0.12 | 0.19±0.03 | - | - | - | - |
| pPTX/pCD-pSNO | 0.11±0.05 | 0.27±0.10 | 0.09±0.01 | 0.15±0.06 | (15.4±7.2) x10−3 | (39.4±13.7) x10−3 | (12.7±0.5) x10−3 | (22.1±8.2) x10−3 |
Table 3.
The summarized combination index of pPTX/pCD-pSNO compared to the mixture of free PTX and free GSNO, or the mixture of pPTX/pCD-pSH NPs and free GSNO.
| combination index at IC50 (CI50) | vs. Free PTX+Free GSNO | vs. PTX/SH-NP+Free GSNO | ||||||
|---|---|---|---|---|---|---|---|---|
| LS174T | EL4 | B16F10 | E0771 | LS174T | EL4 | B16F10 | E0771 | |
| pPTX/pCD-pSNO | 0.28 | 0.40 | 0.22 | 0.26 | 0.57 | 0.60 | 0.47 | 0.81 |
As a major described mechanism of NO-mediated chemosensitization involves the inhibition of drug efflux mediated by P-glycoproteins and multi-drug resistant proteins (MRPs),[20–22] MRP1 expression was investigated after treatment with NPs (Figure 2F). Reduced MRP1 expression was observed in pPTX/pCD-pSNO NPs compared to control groups, demonstrating that pPTX/pCD-pSNO NPs showed synergistic anticancer effects by inhibiting the drug efflux mechanism and enhancing cellular accumulation of PTX. Indeed, the NO-mediated suppression of MRP1 expression resulted in the enhanced uptake of pPTX/pCD-pSNO compared to pPTX/pCD-pSH (Figure S6). The insignificant effects of free GSNO in MRP1 expression may be attributed to the inefficient NO transport mechanism of free S-nitrosothiols.[44,45] The primary mechanism of exogenous small molecular S-nitrosothiols in intracellular NO transport involves the transfer of nitroso groups to L-cysteine to make S-nitroso-L-cysteine that can be easily taken up by amino acid transporter L system (L-AT). Because of these inefficient pathways with multiple steps and cell line-dependent metabolism pathways, GSNO treatment does not always increase the intracellular concentrations of NO.[44,46] Indeed, free GSNO treatment showed same intracellular NO level with the PBS control group, while pPTX/pCD-pSNO NPs significantly increased the intracellular concentrations of NO compared to PBS, free PTX, and pPTX/pCD-pSH NPs (Figure 2G). These results emphasize the pivotal role of NO delivery systems in modulating intracellular NO concentrations. Importantly, free PTX and pPTX/pCD-pSH NPs induced similar MRP1 expression with PBS even though they exhibited higher intracellular NO concentrations than PBS (Figure 2F, G). Our results are correlated with previous reports describing that chemotherapy increases intracellular NO concentrations or upregulates drug efflux mechanisms,[47–51] which suggests that exogenously delivered NO and endogenously generated NO modulate cellular functions via different pathways.[52,53]
The oncolytic virus T-VEC that is approved for the treatment of melanoma via intralesional injection is engineered to induce expression of granulocyte-macrophage colony-stimulating factor (GM-CSF) to recruit antigen presenting cells, including DCs, to boost adaptive immunity during the simultaneous induction of ICD. Adenosine triphosphate (ATP) release and calreticulin (CRT) expression are important ICD markers, which represent “find-me” and “eat-me” signals to DCs, respectively. pPTX/pCD-pSNO NPs showed significantly higher ATP release compared to control groups (Figure 2H). We expected that the higher ATP release of pPTX/pCD-pSNO than control groups is associated with autophagy.[54] However, similar autophagy levels were observed in free PTX, pPTX/pCD-pSH NPs and pPTX/pCD-pSNO NPs, although they are higher than PBS and GSNO (Figure 2I). These results imply that pPTX/pCD-pSNO NPs augment the extracellular ATP release via both PTX-mediated autophagy and unknown mechanisms mediated by NO.[54] In addition, higher CRT expression was detected in pPTX-pCD-pSNO NPs than control groups (Figure 2J). These up-regulated ATP and CRT levels imply that pPTX/pCD-pSNO NPs can recruit DCs into the tumor bed and induce DCs to engulf the dying cancer cells.
In summary, pPTX/pCD-pSNO NPs improve cytotoxic effects of PTX on cancer cells by interfering with drug efflux mechanisms and augment ICD via combined effects of PTX and NO.
2.3. Effects of pPTX/pCD-pSNO NPs on Activation of Dendritic Cells in vitro and in vivo
The direct effects of pPTX/pCD-pSNO on DCs were investigated in vitro in bone marrow-derived DCs (BMDCs). No treatment group induced cytotoxicity to the BMDCs up to 50 μM PTX (Figure S7), consistent with previous reports demonstrating a lack of cytotoxicity by free PTX concentrations from 10-100 μM.[55,56] Compared to PBS, GSNO, free PTX, and free PTX+free GSNO, pPTX/pCD-pSH and pPTX/pCD-pSNO NPs induced higher expression of DC activation markers MHCII+ and CD86+ as well as pro-inflammatory cytokines interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α) at higher than 10 μM PTX, suggesting that the drug delivery system based on multivalent host-guest interactions contributes to improving the adjuvant effects of PTX (Figure 3A–D). In particular, pPTX/pCD-pSNO showed higher expression of DC markers and pro-inflammatory cytokines even including interleukin-12 (IL-12p40) directly associated with CD8+ T cell activation, than pPTX/pCD-pSH at 20-50 μM PTX (Figure 3A–D, S8). These results imply that a high dose of NO is required to enhance the adjuvant effects of PTX.
Figure 3. Effects of pPTX/pCD-pSNO NPs on DCs in vitro and in vivo.
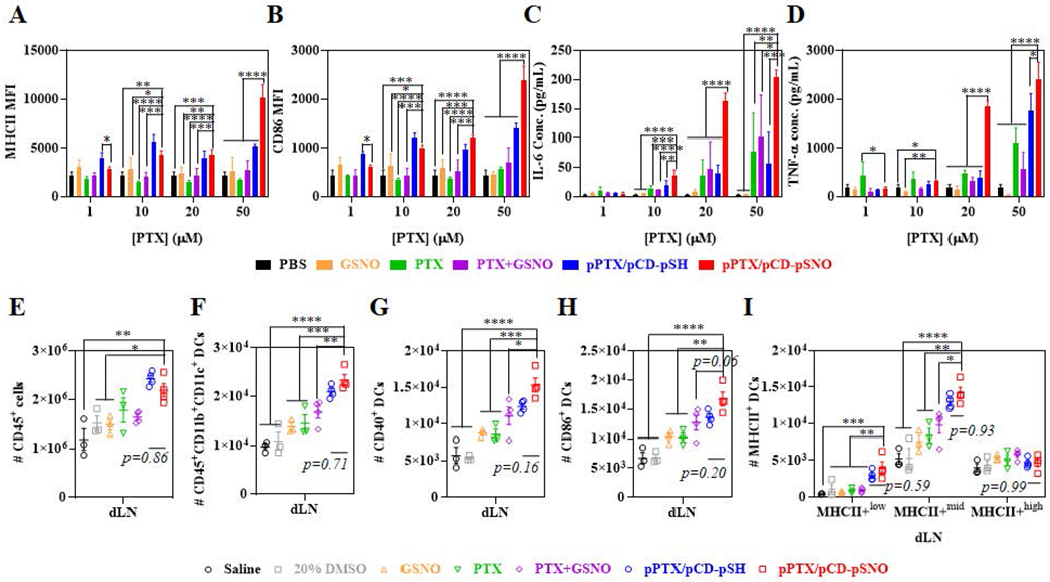
Dose dependent in vitro activation of BMDCs, evaluated by (A) MHCII and (B) CD86. Dose dependent in vitro cytokine production by BMDCs, (C) IL-6 and (D) TNF-α. (A-D) The GSNO, PTX+GSNO and pPTX/pCD-pSNO contains 144.5 nM -SNO groups per 1 μM PTX. (E-I) Number of immune cells in draining lymph nodes (dLNs) 1 d after dorsal injection into tumor-free mouse (C57BL/6J); (E) CD45+ cells, (F) CD45+CD11b+CD11c+ DCs, (G) CD40+ activated CD45+CD11b+CD11c+ DCs, (H) CD86+ CD45+CD11b+CD11c+ DCs, and (I) MHCII+low, MHCII+mid, and MHCII+high CD45+CD11b+CD11c+ DCs. ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05.
In order to investigate the direct adjuvant effects of pPTX/pCD-pSNO in vivo, the levels and activation status of DCs were investigated in the draining lymph nodes (dLNs) one day after its intradermal administration. The expectation was that if bioactive in vivo, pPTX/pCD-pSNO would result in an increase in DC maturation within dLNs, to which nanoformulations drain or are trafficked by migratory cells after skin injection.[57] The number of CD45+ cells in dLN was significantly increased in pPTX/pCD-pSH and pPTX/pCD-pSNO treated animals (Figure 3E), while spleen cellularity was unchanged (Figure S9). pPTX/pCD-pSNO treatment resulted in the highest number of CD45+CD11b+CD11c+ cells (referred to as DCs[58] in all in vivo analyses and results henceforth) in dLN, although the increase compared to pPTX/pCD-pSH was statistically insignificant (Figure 3F). In addition, similar trends were observed in the number of activated (CD40+, CD86+ and MHCII+) DCs in dLNs (Figure 3G–I). In contrast, no significant changes of the DCs were observed in spleen (Figure S10), consistent with the drug formulation effects being restricted to lymphoid tissue-resident DCs within dLN within the experimental timeframe explored here (24 hr). It should be noted that while pPTX/pCD-pSNO led to highest activation of DCs compared to control groups in vivo as well as in vitro, the difference with pPTX/pCD-pSH in vivo was less substantial.
Observations of modulation of DC levels and activation by pPTX/pCD-pSNO would suggest the potential for this system to augment DC-mediated T cell activation. Accordingly, effects of pPTX/pCD-pSNO on DC-mediated CD8+ T cell activation were evaluated by an in vitro mixed lymphocyte reaction (MLR) using mitomycin C-pretreated whole splenocytes as stimulating cells (Figure S11). Treatment with free PTX+free GSNO, pPTX/pCD-pSH, and pPTX/pCD-pSNO showed higher allogeneic CD8+ T cell stimulating capacity than PBS, free PTX, and free GSNO treatment. pPTX/pCD-pSNO also resulted in higher proliferation of CD8+ T cells compared to PBS, GSNO and PTX, while the increase compared to pPTX/pCD-pSH was not statistically significant. While pPTX/pCD-pSNO NPs were observed to increase DC activation compared to pPTX/pCD-pSH, the same trend was not observed in T cell activation, perhaps implying inefficacy of DC-mediated T cell instruction in vitro due to immune checkpoints or other immune cells.
Taken together, pPTX/pCD-pSNO NPs recapitulate key therapeutic features of OV; chemotherapeutic effects mimicking direct lytic effects of OVs, and activation of DCs and ICD induction mirroring that of GM-CSF produced by OV inoculation (Scheme 1).
Scheme 1. Schematic illustration comparing OV and synthetic OV based on pPTX/pCD-pSNO NPs.
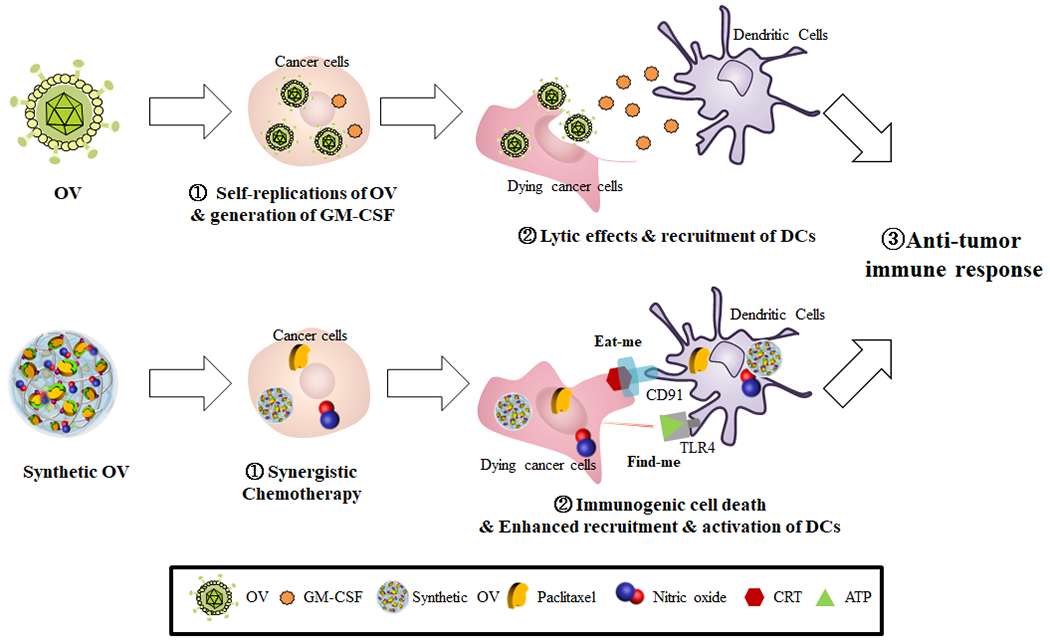
OV self-replicates and generate GM-CSF in the cancer cells, which facilitates lytic effect on cancer cells and recruitments of antigen-presenting cells. Synthetic OV based on pPTX/pCD-pSNO NPs exert synergistic chemotherapy and induce immunogenic cell death on cancer cells, which allow expansion and activation of DCs.
2.4. Therapeutic Potential and Immune Profiles of the pPTX/pCD-pSNO NPs in vivo
A dual tumor model was employed to investigate the antitumor effects and systemic immune response of pPTX/pCD-pSNO. First, biodistribution experiments were conducted to evaluate the potential for NO to augment systemic rather than local availability of PTX. 105 B16F10-OVA cells were implanted in the left dorsal skin as a primary tumor (1° tumor) on day 0 and again in the right dorsal skin as a secondary tumor (2° tumor) on day 3. Alexa Fluor™ 647-labelled pPTX/pCD-pSH and pPTX/pCD-pSNO were injected in the 1° tumor on day 6, and imaged using IVIS (In Vivo Imaging System) spectrum instrument at 0 h, 4 h, and 24 h. After IVIS imaging, the animals were sacrificed to quantify the fluorescence in homogenized tissues (Figure S12). Both pPTX/pCD-pSH and pPTX/pCD-pSNO were detected in the injected tumor (primary tumor, 1° tumor) at high levels, while negligible amounts were observed in the blood, spleen and contralateral tumor (secondary tumor, 2° tumor). Overall, there was no statistical difference in biodistribution profiles between pPTX/pCD-pSH and pPTX/pCD-pSNO.
Next, the local versus abscopal therapeutic potential of pPTX/pCD-pSNO was evaluated in vivo. The dual tumor model was implemented as any effect on the 2° tumor by treatment of the 1° tumor would be indirect, as in governed by any systemic immune response initiated from 1° tumor rather than the direct effects of PTX. Although similar immune effects would presumably manifest within the 1° tumor, they would be superseded by effects of the injected formulations on eliciting direct cytotoxicity, ICD, activation of DCs and other immune cells (Figure 2, 3). Accordingly, after each sample (dose equivalent to 10 mg/kg PTX) was administered to the 1° tumor on day 7, the animals were sacrificed on day 18 for immune phenotyping. Free PTX+free GSNO and pPTX/pCD-pSNO were found to exhibit equivalent anticancer effects as free PTX and pPTX/pCD-pSH in the treated 1° tumor, respectively (Figure 4A, S13), despite synergistic and additional effects of NO in cytotoxicity and ICD in vitro (Figure 2). However, pPTX/pCD-pSH and pPTX/pCD-pSNO showed higher therapeutic effects than free PTX and free PTX+free GSNO in 1° tumor, perhaps due to size-dependent tissue retention improved by NP formulation. Interestingly, free PTX and pPTX/pCD-pSH treatment resulted in larger secondary tumors than saline, free PTX+free GSNO, and pPTX/pCD-pSNO treatments (Figure 4B, S14).
Figure 4. Tumor growth curves and immune cell profiles in various tissues.
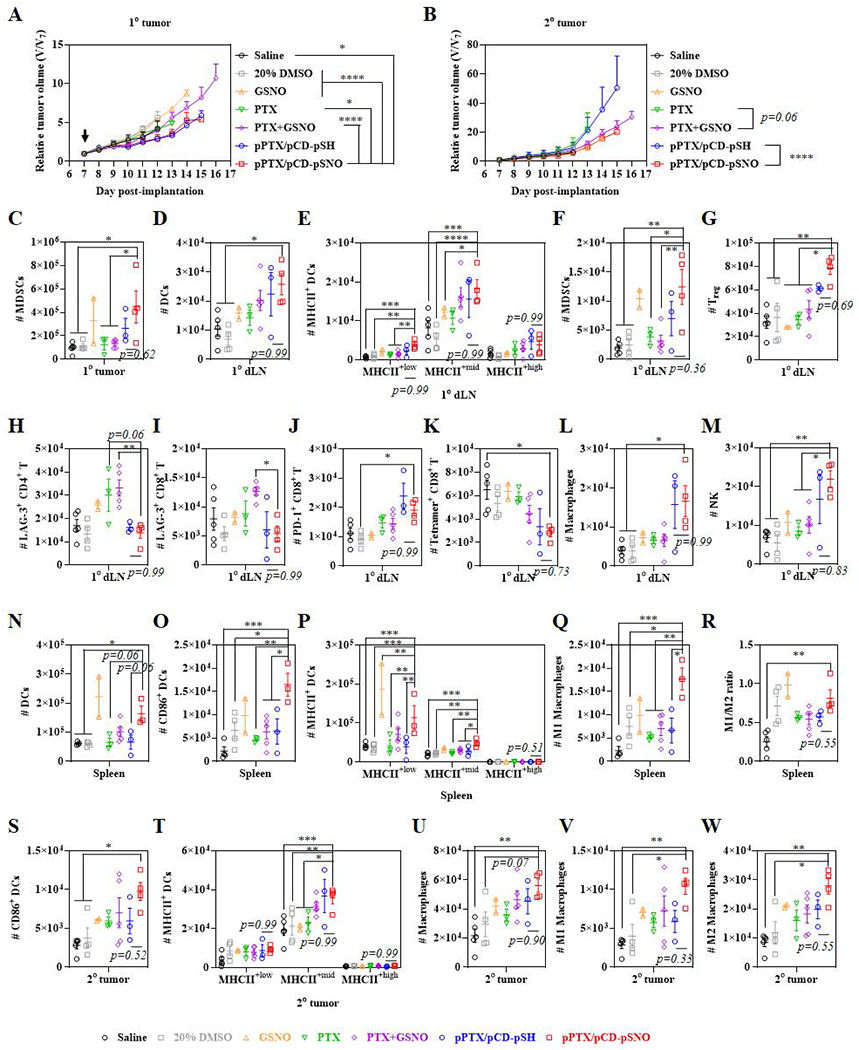
Relative tumor size of (A) 1° tumor and (B) 2° tumor after one time i.t. injection. An arrow indicates injection date. Immune cells were profiled 18 d after inoculations of 1° tumors. Number of (C) CD45+CD11b+CD11c−Gr1+ MDSCs in 1° tumor. Number of immune cells in 1° dLN; (D) CD45+CD11b+CD11c+ DCs, (E) MHCII+low, MHCII+mid, and MHCII+high CD45+CD11b+CD11c+ DCs, (F) CD45+CD11b+CD11c−Gr1+ MDSCs, (G) CD45+CD3+CD4+CD25+Foxp3+ Treg, (H) LAG-3+ CD45+CD3+CD4+ T cells, (I) LAG-3+ CD45+CD3+CD8+ T cells, (J) PD-1+ CD45+CD3+CD8+ T cells, (K) Tetramer+ CD45+CD3+CD8+ T cells, (L) CD45+CD11b+F4/80+ macrophages, and (M) CD45+CD3−NK1.1+ NK cells. Number of immune cells in spleen; (N) CD45+CD11b+CD11c+ DCs, (O) CD86+ CD45+CD11b+CD11c+ DCs, (P) MHCII+low, MHCII+mid, and MHCII+high CD45+CD11b+CD11c+ DCs, (Q) CD45+CD11b+F4/80+CD86+ M1 macrophages, and (R) ratio of M1 to M2 (CD45+CD11b+F4/80+CD206+). Number of immune cells in 2° dLN; (S) CD86+ CD45+CD11b+CD11c+ DCs, (T) MHCII+low, MHCII+mid, and MHCII+high CD45+CD11b+CD11c+ DCs, (U) CD45+CD11b+F4/80+ macrophages, (V) CD45+CD11b+F4/80+CD86+ M1 macrophages, and (W) CD45+CD11b+F4/80+CD206+ M2 macrophages. ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05.
The immunomodulatory effects of treatment were assessed by profiling various immune cells including DCs, T cells, natural killer (NK) cells, myeloid-derived suppressor cells (MDSCs), macrophages and B cells within the 1° and 2° tumors, their dLNs (1° dLN and 2° dLN, respectively) and the spleen (Figure 4C–W, S15–S36). In the 1° tumor, of all cell types considered, only the frequency of MDSCs was found to be changed, with the number of MDSCs increasing in the pPTX/pCD-pSNO group (Figure 4C, S17–S20). The expansion of MDSCs is not surprising, considering that tumor-expressed iNOS recruits MDSCs via VEGF-dependent mechanism.[59] Tetramer+ CD8+ T cells were higher in pPTX/pCD-pSH and pPTX/pCD-pSNO than control groups although the difference is statistically insignificant (Figure S19D), In the 1° dLN, the number of DCs and MHCII+ DCs was significantly expanded in pPTX/pCD-pSH and pPTX/pCD-pSNO groups compared to control groups including saline, DMSO, free PTX and free PTX+free GSNO (Figure 4D, 4E, S21), which is in accordance with the ICD and direct DC activation effects (Figure 2H–J, Figure 3). As was observed in the 1° tumor, the number of MDSCs in the 1° dLN was also augmented in the pPTX/pCD-pSNO group (Figure 4F), which resulted in the significant expansion of Treg (Figure 4G) although the number of CD3+, CD4+ and CD8+ T cells was insignificantly increased (Figure S22, S23). Interestingly, free PTX and free PTX+free GSNO showed higher number of LAG-3+ CD4+ and LAG-3+ CD8+ T cells compared to pPTX/pCD-pSH and pPTX/pCD-pSNO, while pPTX/pCD-pSH and pPTX/pCD-pSNO had similar levels with saline and 20% DMSO control groups (Figure 4H,I). On the other hand, the number of PD-1+ CD8+ T cells was increased in pPTX/pCD-pSH and pPTX/pCD-pSNO compared to control groups (Figure 4J), suggesting more T cells were antigen experienced as a result of treatment. Contrariwise, the number of tetramer+ CD3+ T cells and tetramer+ CD8+ T cells was also significantly decreased in pPTX/pCD-pSH and pPTX/pCD-pSNO (Figure 4K, S22), which can be partially attributed to the MDSCs that inhibit proliferation of antigen-specific CD8+ T cells without affecting the number of CD8+ T cells.[60–62] The number of macrophages was increased, while M1/M2 ratios were maintained amongst treatment groups (Figure 4L, S24C). There were insignificant differences in B cells (Figure S24D), but the number of NK cells were significantly augmented in pPTX/pCD-pSH and pPTX/pCD-pSNO groups (Figure 4M). Overall, the anticancer effects from higher cytotoxicity (Figure 2C), expansion of DCs and NK cells, and restoration of LAG-3+ T cells seem to be compromised by the increase of MDSCs, Treg, and PD-1+ CD8+ T and decrease of tetramer+ CD8+ T cells, which led for pPTX/pCD-pSNO to show marginal enhancement of antitumor effects in 1° tumor compared to the control groups including saline, DMSO, free PTX, free GSNO, and free PTX+free GSNO. In addition, higher cytotoxic effects of pPTX/pCD-pSNO (Figure 2C) than pPTX/pCD-pSH seemed to be offset by slightly higher populations of MDSCs and Treg in pPTX/pCD-pSNO (Figure 4C,F,G), which resulted in the similar 1° tumor size between pPTX/pCD-pSH and pPTX/pCD-pSNO.
The potential for immunity resulting from treatment-associated immunomodulation within the 1° tumor microenvironment to propagate systemically was evaluated (Figure 4N–R, S25–S28). In the spleen, the number of DCs (Figure 4N), CD86+ DCs (Figure 4O), and MHCII+ DCs (Figure 4P) was found to be significantly higher in pPTX/pCD-pSNO than in control groups including saline, DMSO, free PTX, free PTX+free GSNO, and pPTX/pCD-pSH, as contrasted with insignificant higher number of DCs and MCHII+ DCs in pPTX/pCD-pSNO than that in pPTX/pCD-pSH in the 1° dLN. Despite the classical paradigm that DCs generally migrate from peripheral tissues to lymphoid organs, there have been a series of reports demonstrating that a fraction of matured DCs in peripheral tissues enter the bloodstream and relocate into distant lymphoid organs and peripheral tissues.[63–66] Accordingly, the observed DC expansion and differentiation in spleen (Figure 4N–P) despite the negligible accumulations of pPTX/pCD-pSNO (Figure S12) would be partially ascribed to the systemic migration of mature DCs. In particular, the number of M1 macrophages is significantly expanded (Figure 4Q), which resulted in the increase of M1/M2 ratio (Figure 4R), as contrasted with negligible changes of macrophages in 1° tumor and 1° dLN. Except for the DCs and macrophages, there were no changes in the number of T cells, and NK cells, and statistically insignificant increases in MDSCs, and B cells (Figure S26–S28).
Immune profiles of the 2° tumor and its dLN (2° dLN) were next evaluated to determine the effects of systemic immunity induced by treatment localized to the 1° tumor (Figure 4S–W, S29–S36). As was observed in spleen, CD86+ DCs and MHCII+ DCs were expanded in the 2° tumor of pPTX/pCD-pSH and pPTX/pCD-pSNO treated mouse despite the insignificant difference in the number of activated DCs between these groups (Figure 4S,T,S29–S32). These results could be attributed to the relocation of mature DCs from circulation,[63–66] suggesting the ability of the pPTX/pCD-pSNO to induce DC activation systemically, despite its locoregional application. In addition, the number of total macrophages, M1 macrophages and M2 macrophages was augmented in pPTX/pCD-pSNO, which resulted in a constant M1/M2 ratio (Figure 4U–W). Beyond DCs and macrophages in the 2° tumor, there were statistically insignificant expansions in T cells, NK cells, MDSCs, and B cells within both the 2° tumor and 2° dLN (Figure S29–S36). Overall, the average number of CD86+ DCs (Figure 4S), M1 macrophages (Figure 4V), CD40+ DCs (Figure S29C), NK cells (Figure S31F), and B cells in the 2° tumor (Figure S32C), and CD45+ DCs (Figure S33C), MHCII+mid DCs (Figure S33E), CD8+ T/Treg ratio (Figure S35E), and NK cells in the 2° dLN (Figure S35F) is in line with the secondary tumor size that is smaller in free PTX+free GSNO and pPTX/pCD-pSNO treated groups than PTX and pPTX/pCD-pSH treated groups.
In summary, pPTX/pCD-pSNO NPs facilitate the systemic expansion and activation of DCs, but did not lead to a corresponding expansion of differentiated and activated CD8+ T cells, potentially due to the simultaneous expansion of MDSCs.
2.5. Combined pPTX/pCD-pSNO NPs and aCLTA-4 Therapy in vivo
High expression of CD86 by DCs motivated the exploration of the potential therapeutic synergies of immune checkpoint blockade with aCTLA-4 mAb with pPTX/pCD-pSNO treatment, as engagement of DC-expressed CD80/86 with CTLA-4 instead of CD28 on T cells is well-known to inhibit the expansion of T cells.[67,68] Indeed, co-administration of PTX and aCTLA-4 has been reported to lead efficient regression of tumor growth, effects associated with expansion of both activated CD8+ and CD4+ T cells.[12] As such, the therapeutic potential of aCTLA-4 in pPTX/pCD-pSNO treatments was investigated in a dual tumor model (Figure 5). Mice were treated three times with an intraperitoneal (i.p.) injection of aCTLA-4 1 d after pPTX/pCD-pSNO i.t. treatment (Figure 5A). aCTLA-4 itself slightly delayed tumor growth in the 1° tumor despite its negligible effects on 2° tumor size (Figure 5B,C). pPTX/pCD-pSH+aCTLA-4 led to higher anticancer effects on the 1° tumor than saline and saline+aCTLA-4 (Figure 5B). However, pPTX/pCD-pSH treatment led to significantly larger 2° tumor size than saline (p=0.14) and saline+aCTLA4 (p=0.03), implying therapeutic limits of aCTLA-4 in PTX-based chemoimmunotherapy (Figure 5C). Remarkably, pPTX/pCD-pSNO+aCTLA-4 showed the most robust tumor suppression in both 1° and 2° tumors (Figure 5B,C) without any significant change of body weight (Figure 5D). In particular, pPTX/pCD-pSNO+aCTLA-4 led to prolonged survival rate, while aCTLA-4 and pPTX/pCD-pSH+aCTLA-4 exhibited minimal effects on animal survival (Figure 5E). This demonstrates the synergistic benefits of aCTLA-4 in combination with PTX-based therapy to be augmented by NO co-delivery.
Figure 5. In vivo therapeutic effects of aCTLA-4 with pPTX/pCD-pSNO.
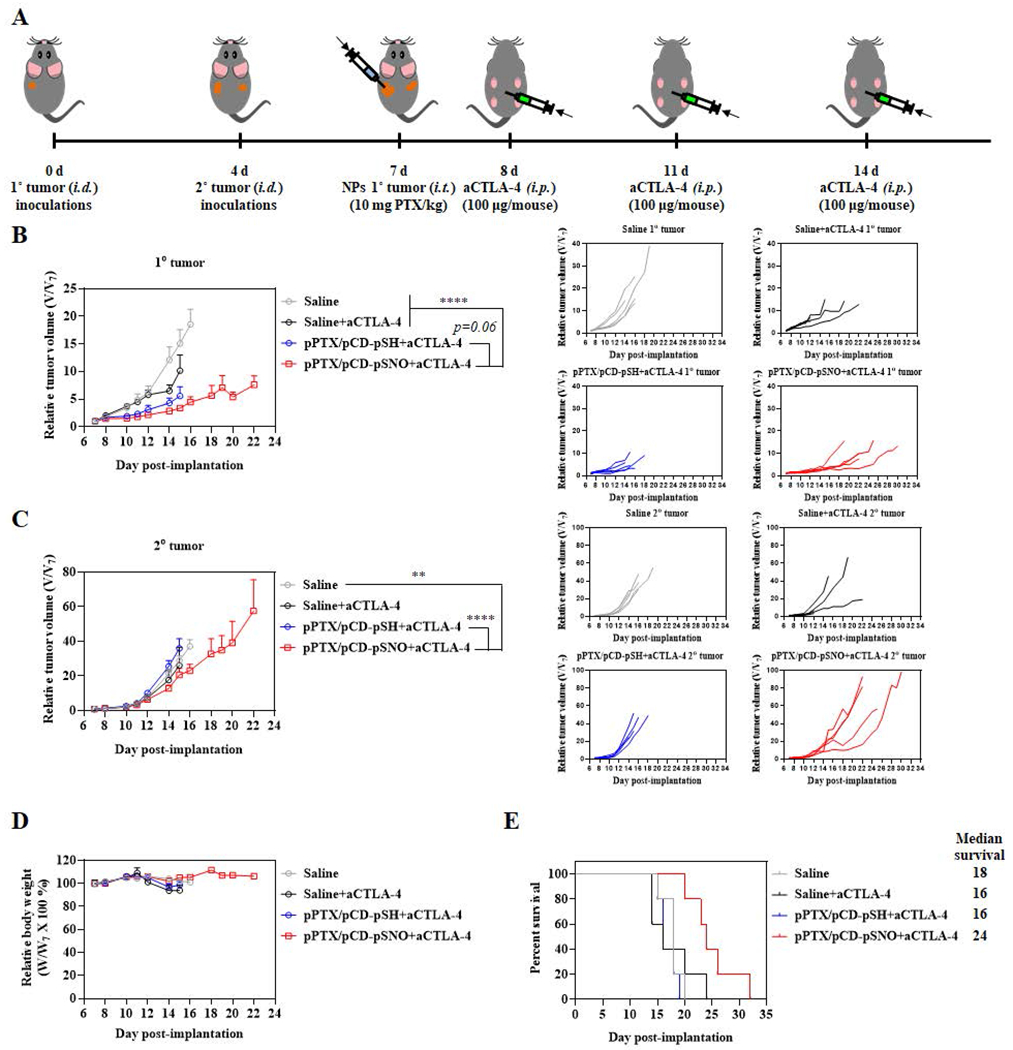
(A) Outline of the disease model and therapy regimen. Relative size of (B) 1° tumor (C) 2° tumors. (D) Relative body weight during therapy. (E) Survival curves during treatment. ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05.
3. Discussion
PTX is one of the most widely used chemotherapeutic drugs in the clinic, with a well-documented ability to induce ICD and activate DCs. However, anti-cancer mechanisms of even this common drug are weakly understood; fragmentary in vitro results have indicated the immune-stimulatory effects of PTX,[1–11,68] however, which are diametric to (pre)clinical therapeutic results.[69–71] Herein, we provide additional experimental insights to understand the action of PTX. As correlated with previous reports, PTX itself led to marginal ICD and slight activation of DCs in vitro (Figure 2H,2J,3A–D,S8). In addition, larger 2° tumor sizes and expansion of splenic Tregs were observed in free PTX-treated mouse compared to saline- and 20% DMSO-treated ones (Figure 4B,S26D). That is, PTX in addition to inducing direct tumor toxicity can be considered as a locoregional DC-stimulator and systemic immune suppressive agent in one.
NO is one of the most versatile endogenous gaseous molecules, which regulates various biological and pathophysiological pathways including cardiovascular homeostasis, neurotransmission, angiogenesis, immune response, and the cell cycle.[19,38] The presence of endogenous NO and its rapid transformation into innocuous ions ameliorate the concerns about side effects associated with the administrations of exogenous drugs and agents, which promote the development of various NO-releasing functional groups (NO-donors) and NO delivery systems.[19,38] NO has not been well explored in (chemo)immunotherapy despite its recent use in improving chemotherapy and continuous evidence in NO’s immune modulatory effects. As with the PTX, the immunological functions of NO have been controversial.[22] In our study, GSNO had negligible effects on ICD and DC activations in vitro (Figure 2H–J,3A–D). However, GSNO slightly matured DCs (Figure 4N,P) and expanded MDSCs (Figure 4F,S32A,S36A) in vivo, which is in line with previous reports.[28,59–62] Nevertheless, there were negligible changes in CD8+ T cells and Treg populations in GSNO-treated mouse in contrast to previous reports describing the NO-induced inhibition of the Tregs in vitro and expansion of CTL in vitro/in vivo.[27,30]
The DDS comprised of multivalent host-guest interactions (pPTX/pCD-pSH) significantly enhanced the effects of PTX in ICD, DC activation and T cell expansion (Figure 2H,2J,3A–D,S11). In addition, incorporation of NO onto pPTX/pCD-pSH to form pPTX/pCD-pSNO further enhanced the formulation’s beneficial effects on ICD and DC activation (Figure 2H,2J,3A–D). However, pPTX/pCD-pSNO showed marginal enhancement of CD8+ T cell expansion compared to pPTX/pCD-pSH in an in vitro MLR assay (Figure S11), despite robust DC activation, implying the potential involvement of other immune cells in NO and PTX-involved T cell instruction. pPTX/pCD-pSH and pPTX/pCD-pSNO also showed very complex in vivo immune modulatory effects. pPTX/pCD-pSNO significantly expanded MDSCs in the 1° tumor and 1° dLN (Figure 4C,F), which may partially contribute to the increase in Tregs in the 1° dLN (Figure 4G). Considering the insignificant effects of PTX on MDSC populations (Figure 4C,F,S28A,S32A,S36A), the expansion of MDSCs of pPTX/pCD-pSNO treated groups seems to be mainly governed by the MDSC-recruiting and expanding effects of NO. Indeed, P. Jayaraman et al. reported that tumor-expressed inducible nitric oxide synthase modulates STAT3 expression and reactive oxygen species production by MDSC as well as expression of VEGF by tumor cells, which recruits and induces MDSCs to foster immunosuppressive environments.[59] Most importantly, pPTX/pCD-pSH and pPTX/pCD-pSNO failed to augment CD8+ T cell populations despite the significant overall expansion of activated DCs in analyzed tissues (Figure 4,S17–S36). pPTX/pCD-pSH and pPTX/pCD-pSNO even decreased populations of tetramer+ CD8+ T cells in 1° dLN (Figure 4K). These results apparently conflict with in vitro MLR results (Figure S11). However, given that mitomycin-C pretreatment inhibits the proliferation of splenocytes including MDSCs, DCs, Tregs, macrophages, effects of MDSCs and Tregs on allogenic CD8+ T cell instructions are limited. Yet all immune cells including MDSCs, Tregs, and macrophages can be directly influenced by NPs in vivo. Likewise, pPTX/pCD-pSNO increased Treg populations in the 1° dLN (Figure 4G). Although NO has been reported to decrease Tregs under artificial in vitro environments comprised of CD4+ T cells and DCs,[27] the expanded MDSC would result in the expansion of Tregs by depletion of arginine, production of IL-10 and TGF-β, and CD40-CD40L interactions.[72] Therefore, the expanded MDSCs measured in vivo seem to result in the different CD8+ T cell proliferation in contrast to results from in vitro MLR assays. In addition to DCs, T cells and MDSCs, pPTX/pCD-pSNO regulated the populations of NK cells and macrophages (Figure 4L,M,Q,R,U,V,W). Finally, combination therapy with aCTLA-4 was explored as pPTX/pCD-pSNO treatment failed to result in CD8+ T cell expansion (Figure 4,S17–S36) despite systemic DC activation (Figure 4). Once this T cell-intrinsic immune checkpoint was blocked, the therapeutic effects by pPTX/pCD-pSNO on both the 1° and 2° tumor were robustly improved, which resulted in significantly prolonged survival (Figure 5). In particular, pPTX/pCD-pSNO with aCTLA-4 showed improved response rate (~60% in injected lesions and ~40% in uninjected lesions) with significantly prolonged survival compared to pPTX/pCD-pSNO (~20% in both injected and uninjected lesions) (Figure S13,S14).
Lymphocyte infiltration into tumors has emerged as one of the strongest indicators of favorable prognosis.[73] As such, therapeutic strategies that augment the immunogenic microenvironment of tumors become of increasing clinical relevance. Nanoformulated DDSs in cancer therapy applications have typically focused on improving selective delivery to tumors, however, their translational impact has been limited save few.[74] However, the concept of locoregional immunomodulation is emerging with immune-modulating scaffolds[58,75] and OV.[3,14–17] Relatively straightforward in superficial tumors such as in skin and breast, intralesional administration can also be implemented for solid tumors within deep tissues using interventional radiology approaches. The potential for DDS to enable substantial improvements in drug retention and targeting as well as enabling controlled, co-delivery of synergistic agents after locoregional administration to improve therapeutic effects is thus feasible in principle in translational settings.
The concept of virus-mediated anticancer therapy emerged due to the advances in molecular biology and genetic engineering in 1990s[14,17] with the OV field taking firm shape only in the early 2000s when T-VEC was first reported.[17,76] T-VEC was developed based on herpes simplex virus type 1 whose ICP34.5 genes were deleted and two copies of GM-CSF inserted.[3,14–17] T-VEC at an early stage showed marginal therapeutic effects and immune response,[76] however, continuous optimization led to its successful clinical translation.[17] In addition, various chemotherapeutic drugs and checkpoint inhibitors have been explored to enhance the therapeutic outcomes of OV.[77–81] Nevertheless, uncertainty of potential genetic interference into host cells is indicated as one of the main problems of OV. Herein, pPTX/pCD-pSNO recapitulated some of the major distinctive anti-tumor mechanisms of OVs (Scheme 1). pPTX/pCD-pSNO killed cancer cells via synergistic chemotherapeutic effects of NO and PTX, analogous to the lytic effects of OVs. The induced ICD also resulted in an expansion of DC populations in vivo, similar to the effects of OV-derived GM-CSF. In particular, the significant overall activation of DCs in treated as well as systemic tissues (Figure 4) demonstrates the ability of locoregional pPTX/pCD-pSNO treatment to result in systemic DC activation. aCTLA-4 combination therapy also contributed to significantly enhanced anticancer effects in primary and distant tumor (Figure 5).
4. Conclusion
In summary, PTX and NO DDSs were developed via multivalent host-guest interactions between polymerized β-CD (pCD) and polymerized PTX (pPTX), followed by the S-nitrosylation of thiol groups in pCD-pSH. The resultant pPTX/pCD-pSNO NPs demonstrate NO and PTX release that is hastened within intracellular chemical microenvironments. After cellular uptake, these complexed NP induce synergistic cytotoxicity and ICD on cancer cells as well as DC activation to expand T cells in vitro. Although locoregional pPTX/pCD-pSNO treatment was able to exert significant expansion and activation of systemic DCs, instruction and expansion of CD8+ T cells was impaired, potentially due to the interference of various enhanced immune cell population including MDSCs cells in vivo. When used in combination with aCTLA-4, pPTX/pCD-pSNO exerted enhanced tumor suppression in both 1° and 2° tumors with prolonged survival. This system thus serves as a guidepost for developing synthetic drug formulations mimicking the functions of OV in cancer immunotherapy.
5. Experimental Section
Materials:
Paclitaxel (PTX) was obtained from LC laboratories. Ethanol and anhydrous N,N-dimethylformamide (DMF) were purchased from VWR. Poly(methyl vinyl ether-alt-Maleic anhydride) (Mn ~80,000), poly(isobutylene-alt-maleic anhydride) (Mw ~6,000), lithium hydride (LiH), β-cyclodextrin (β-CD), dithiothreitol (DTT), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS) cystamine dihydrochloride, tert-butyl nitrite, L-glutathione, sodium nitrite (NaNO2), esterase from porcine liver (e code 3.1.1.1), mercuric chloride (HgCl2), sulfanilamide, N-(1-naphthyl)ethylenediamine dihydrochloride (NED), and collagenase D (Roche) were obtained from Sigma Aldrich. Dialysis tube (3.5 kD) and Float-A-Lyzer G2 (3.5-5KD) were purchased from Spectrumlabs. Dimethyl sulfoxide-d6 was obtained from Cambridge Isotope Laboratories, Inc. Holey carbon grid for TEM (200 mesh, 50 μm) was purchased from Electron Microscopy Sciences. Amicon ultra centrifugal tube (MWCO 3K), Alexa Fluor™ 647 C2 maleimide, alamarblue, annexin V/propidium Iodide (PI) kit, fixable far red dead cell stain dye (ex. 633 or 635 nm), lysotracker™ Red DND-99 (Invitrogen-Molecular Probes), ACK lysis buffer (Lonza), ENLITEN ATP assay kit, mitomycin C (Cayman Chem.), and FoxP3 staining kit (eBioscience) were obtained from Thermo Fisher scientific. Nunc Lab-Tek™ II chamber slide and vectashield mounting medium for confocal microscope was obtained from Nunc Inc. and Vector Laboratories, respectively. Cyto-ID autophagy detection kit was purchased from Enzo Life Sciences. MRP1 antibody (IU2H10) and calreticulin (CRT) antibody (1G6A7) were obtained from Novus Biologicals. Other antibodies, Zombie Aqua™ fixable viability dye, and MojoSort™ Mouse CD8 Naïve T Cell Isolation Kit were purchased from Biolegend. hamster anti-mouse CTLA4 (clone 9H10) was obtained from BioXCell.
Synthesis and Characterizations of pPTX and pCD-pSH Polymers:
pPTX and pCD were synthesized by slight modification of a procedure reported by R. Namgung et al.[34] In brief, 100 mg of PTX and 16 mg of LiH was added in 5 mL anhydrous DMF in dry Ar (g) atmosphere. After 1 d, the resultant activated PTX solution was added dropwise to 480 mg of poly(methyl vinyl ether-alt-Maleic anhydride) (Mn ~80,000) in 19 mL DMF, followed by 1 h stirring. The pPTX was yielded after 2 d dialysis (MWCO 3.5K) against deionized water (DW) and 2 d lyophilization. 2 g of β-CD in 15 mL anhydrous DMF was activated by 16.5 mg of LiH in dry Ar (g) atmosphere for 1 d. The resultant activated β-CD was added dropwise into the 200 mg of poly(isobutylene-alt-maleic anhydride) (Mw ~6,000) in 5 mL anhydrous DMF and stirred for 1 d. After 2 d dialysis (MWCO 3.5K) against DW, the solution was lyophilized to afford the pCD.
In order to synthesize pCD-pSH, 400 mg EDC and 300 mg NHS was added to 500 mg of pCD in 20 mL MES buffer, followed by 4 hr incubation. 470 mg of cystamine dihydrochloride in 10 mL sodium phosphate buffer (pH 7.2) is added dropwise to the EDC/NHS activated pCD solution and 2 mL of 0.1 M NaOH was added to the solution. After 1 d of stirring, 500 mg DTT was added to the solution to reduce disulfide bonds. After 2 h reduction, pCD-pSH was purified by three times centrifugal dialysis with Amicon ultra centrifugal tube (MWCO 3K) and freeze drying.
The conjugation ratio of PTX, CD and cysteamine in polymers was confirmed by 1H nuclear magnetic resonance spectroscopy (1H NMR) with Bruker Advance 300 MHz FT-NMR.
In order to evaluate the S-nitrosothiol formations on pCD-pSH, pCD-pSNO was synthesized. Briefly, 20 mg of pCD-pSH in 4 mL anhydrous ethanol was reacted with 20 μL t-butyl nitrite under dark for 1.5 h. After 1.5 h dialysis (MWCO 3.5K) against DW and 2 d lyophilization, the pCD-pSNO was yielded as a powder. UV-vis spectrum was recorded by Synergy H4 microplate reader (BioTek, Winooski, VT) to demonstrate the formation of S-nitrosothiols.
Preparations of pPTX/pCD-pSNO NPs:
Prior to the preparation of pPTX/pCD-pSNO NPs, pPTX/pCD-pSH NPs were synthesized by dissolving pPTX and pCD-pSH (molar ratio of PTX/CD=1) in 20% (v/v) ethanol to obtain a 0.1% (w/v) solution with 1 d vigorous stirring, 1 d dialysis (MWCO 3.5K) against DW and 2 d lyophilization. In order to prepare pPTX/pCD-pSNO NPs, 15 mg of pPTX/pCD-pSH in 15 mL absolute ethanol was reacted with 15 μL t-butyl nitrite under dark conditions for 1.5 h, dialyzed for 1.5 h (MWCO 3.5K) against DW and freezing-dried for 2 d. The resultant dried pPTX/pCD-pSNO NPs were stored at −20 °C.
Characterization of pPTX/pCD-pSNO NPs:
The size and surface charge of NPs were measured by DLS and Zetasizer Nano ZS (Malvern Instruments, UK). The morphology of the NPs was characterized by high-resolution FEI Tecnai G2 F30 TEM (FEI company, USA). The specific absorbance of S-nitrosothiols in NPs was detected by Synergy H4 microplate reader. FT-IR spectra was recorded by Bruker vortex 70 instrument (Bruker Optik GmbH, Germany). The free thiol contents of NPs were quantified by Ellman’s assay and –SNO contents in the NPs were measured by Saville and Griess assay. In order to investigate the NO release, Float-A-Lyzer G2 (3.5-5KD) dialysis bag containing 1 mL of 1 mg/mL pPTX/pCD-pSNO NPs in 3.2 mM ascorbic acid (pH 7.4, PBS) or 80 μM ascorbic acid (pH 7.4, PBS) was immersed in 50 mL incubation buffer (PBS) at 37 °C under oscillation at 90 r/min. At predetermined time intervals, the dialysis bag was placed into the fresh 50 mL PBS and the withdrawn incubation buffers were used to determine nitrite contents by using Griess assay. For the PTX release test, Float-A-Lyzer G2 (3.5-5KD) dialysis bag containing 1 mL of 1 mg/mL pPTX/pCD-pSNO NPs in PBS (pH 7.4), PBS (pH 5.6), 10 units/mL esterase (pH 7.4, PBS), or 10 units/mL esterase (pH 5.6, PBS) was immersed in 50 mL incubation buffer (PBS) at 37 °C under oscillation at 90 r/min. At predetermined time intervals, the dialysis bag was placed into the fresh 50 mL PBS and the withdrawn incubation buffers were used to determine PTX contents by detecting 240 nm absorbance in Synergy H4 microplate reader. The NO and PTX release results are presented as mean ± standard deviation (n = 3) and statistically analyzed with two-way ANOVA analysis in Prism software (Graphpad). ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05.
In Vitro Cytotoxic Effects and Mechanisms:
Human colorectal cancer LS174T, murine lymphoma EL4, murine melanoma B16F10, or murine breast cancer E0771 cells were seeded onto 96-well plates at a density of 103 cells/well in 100 μL medium (DMEM or RPMI) containing 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin, and incubated at 37 °C CO2 incubator overnight. 2 μL of each sample (PBS, free PTX, Free GSNO, pPTX/pCD-pSH NPs and pPTX/pCD-pSNO NPs) dissolved in DMSO was added to 98 μL media in cell-laden wells and after incubation for 2 d at 37 °C in a CO2 incubator, 5 μL of AlamarBlue® agent was added to each well. After 40 min, the fluorescence (excitation 560 nm, emission 595 nm) was measured by Synergy H4 microplate reader (BioTek, Winooski, VT). The fluorescence of medium without cells and PBS-treated cells represent blank and 100 % cell viability, respectively. In order to investigate the apoptosis and necrosis, PBS, GSNO, free PTX, pPTX/pCD-pSH or pPTX/pCD-pSNO (dose equivalent to 50 nM PTX and/or 7.2 nM –SNO) was added to 2X105 B16F10 cells/well seeded in 6-well plates one day before treatment. After 24 h incubation with treatments, cells were trypsinized, followed by incubation with annexin V-FITC and PI for 15 min. After washing with cold PBS three times, the cells were immediately analyzed by LSR Fortessa flow cytometry (BD Biosciences). For MRP1 detection, PBS, GSNO, free PTX, pPTX/pCD-pSH or pPTX/pCD-pSNO (dose equivalent to 1 μM PTX and/or 144.5 nM –SNO) was added to 4X104 B16F10 cells/well seeded in Nunc Lab-Tek™ II chamber slide 1 d before treatment. After 24 h incubation with treatment, the cells were washed with PBS three times and then fixed with 4% paraformaldehyde for 30 min. After washing with PBS containing 1% BSA, permeabilizing with 0.25% triton X-100 for 15 min, and washing with PBS containing 1% BSA, the cells were stained with 1 μL MRP1-FITC (IU2H10) for 2 h. After washing three times with PBS, cells were stained with DAPI for 5 min. After washing three times, the samples were detected by The Zeiss LSM 700 System consisting of an LSM 700 scan head mounted on a AxioObserver Z1 inverted microscope stage. The cellular uptake of NPs was investigated with Alexa Fluor™ 647 labelled NPs. pPTX/pCD-pSH was labelled with Alexa Fluor™ 647 via maleimide and thiol reactions. Alexa Fluor™ 647-labelled pPTX/pCD-pSNO was also prepared after S-nitrosylation of Alexa Fluor™ 647-labelled pPTX/pCD-pSH. The labelled NPs (dose equivalent to 1 μM PTX and/or 144.5 nM –SNO) were added to 4X104 B16F10 cells/well seeded in Nunc Lab-Tek™ II chamber slide 1d before treatment. After 4 h incubation with treatment of Alexa Fluor™ 647 labelled NPs, the cells were washed with PBS three times and incubated with lysotracker™ Red DND-99 for 30 min. The cells were fixed with 4% paraformaldehyde for 30 min after washing with PBS three times. After washing three times with PBS, cells were stained with DAPI for 5 min. After washing three times, the samples were detected by The Zeiss LSM 700 System consisting of an LSM 700 scan head mounted on a AxioObserver Z1 inverted microscope stage. In order to investigate the intracellular level of NO, PBS, GSNO, free PTX, pPTX/pCD-pSH or pPTX/pCD-pSNO (dose equivalent to 1 μM PTX and/or 144.5 nM –SNO) were treated to 2X105 B16F10 cells/well which was seeded in 6-well plates 1 d before treatment. After 30 min incubation with treatment, 2 μL DAF-2DA was added to the cells. After 24 h incubation, 2 μL live/dead (Far Red Dead Cell Stain Kit for 633 or 635 nm excitation, Thermofisher Scientific) was added to the cells, followed by trypsinization and three washes with cold PBS. After fixing with 4% paraformaldehyde for 30 min and washing with PBS three times, the cells were analyzed by LSR Fortessa flow cytometry (BD Biosciences). For ATP detection, 1X104 B16F10 cells/well were seeded into 96-well plates and incubated overnight at 37 °C 5% CO2. PBS, GSNO, free PTX, pPTX/pCD-pSH or pPTX/pCD-pSNO (dose equivalent to 1 μM PTX and/or 144.5 nM –SNO) were added to the cells and incubated for 1 d. The supernatants were transferred to new 96-well plates, followed by measurement of chemiluminescence in accordance with the manufacturer’s protocols (ENLITEN(R) ATP ASSAY, Promega). In order to investigate the mechanism of ATP release, autophagy was measured by using Autophagy Detection CYTO-ID kit (Enzy lifesciences). Briefly, 2X105 B16F10 cells/well were seeded into 6-well plates, followed by overnight incubation at 37 °C 5% CO2. Cells were treated with PBS, GSNO, free PTX, pPTX/pCD-pSH or pPTX/pCD-pSNO (dose equivalent to 0.1 μM PTX and/or 14.4 nM –SNO). After 1 d incubation, cells were incubated with 1 μL live/dead fixable dye (Far Red Dead Cell Stain Kit for 633 or 635 nm excitation, Thermofisher Scientific) and CYTO-ID agents for 30 min. In order to prevent the effects of starvation and trypsin on autophagy, cells were washed with 5% FBS in PBS twice and harvested by using 1 mM EDTA in 5% FBS containing PBS. Finally, the cells were fixed with 4% paraformaldehyde and then analyzed by LSR Fortessa flow cytometry (BD Biosciences). For CRT detection, 2X105 B16F10 cells/well were seeded into 6-well plates. After overnight incubation at 37 °C 5% CO2, PBS, GSNO, free PTX, pPTX/pCD-pSH or pPTX/pCD-pSNO (dose equivalent to 0.1 μM PTX and/or 14.4 nM –SNO) were treated to the cells. After 1 d incubation, cells were harvested and stained with live/dead (Far Red Dead Cell Stain Kit for 633 or 635 nm excitation, Thermofisher Scientific), fixed with 4% paraformaldehyde, permeabilized with triton X100, and stained with CRT-FITC antibody (clone 1G6A7, NOVUS BIOLOGICALS) for the flow cytometry. All results are presented as mean ± standard deviation (n = 3-4) and statistically analyzed with one-way ANOVA analysis in Prism software (Graphpad). ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05.
In Vitro DC Activation and Cytokine Production:
Animal use was approved by the POSTECH Biotech Center Ethics Committee with the relevant guidelines and regulations. Bone marrow cells were obtained from 6-week-old female C57BL/6 mice by flushing the tibia and femur. Obtained bone marrow was filtered with 40 μm nylon mesh filters and incubated in ACK lysing buffer (Gibco) for 3 min in RT to eliminate RBCs. To generate BMDCs, bone marrow cells were seeded at 2.5X104 cells/well in 96 well plate and cultured in DMEM with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 20 ng/mL GM-CSF (PeproTech), and 0.1% 2-mercaptoethanol (Gibco). Seeded cells were cultured for 5 d by changing half of media with fresh media at every other day. On day 6, BMDCs were treated with PBS, PTX, GSNO, PTX+GSNO, pPTX/pCD-pSH, and pPTX/pCD-pNO in various concentrations (1-50 μM) and incubated for 24 hr at 37 °C. Cell culture supernatant samples were collected to measure secreted IL-6, IL-12p40, and TNF-α by ELISA (Abcam). BMDCs from each well were collected and stained for 30 min at 4 °C with antibodies. The fluorophore list is following; CD11c-PE/Cy7 (clone:N418, Biolegend), MHCII-BV 650 (clone:M5/114.15.2, BD bioscience), CD86-APC (clone:GL1, BD bioscience), and Ghost dye™ violet 510. (Tonbo Biosciences). Fixed BMDCs were analyzed by LSR Fortessa flow cytometry (BD Biosciences). The results are presented as mean ± standard deviation of the mean (n = 4) and statistically analyzed with two-way ANOVA analysis in Prism software (Graphpad). ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05.
In Vitro MLR Assay:
The MLR assay was conducted with the modified methods reported in Yuan et al.[8] Animal experiments proceeded with IACUC approval in a PRL at the Georgia Institute of Technology. Spleens of 8-12 week old C57BL/6 mice were harvested and lymphocytes isolated by grinding through 70 μm strainers with syringe plunger. Red blood cells were removed by ACK lysis buffer (Lonza). C57BL/6 CD8+ T cells were purified from the splenocytes by using MojoSort™ Mouse CD8 Naïve T Cell Isolation Kit (Biolegend). BALB/C splenocytes from 8-12 week old animals were harvested by grinding spleens through 70 μm strainer with syringe plunger and red blood cells were removed by ACK lysis buffer (Lonza). All splenocytes were stored on ice <2 hr prior to use. BALB/C splenocytes at 5X107 cells/mL were treated with 50 μg/mL Mitomycin C for 30 min in 37 °C CO2, after which cells were washed with PBS three times and seeded into 96-well plates at the density of 4X105 cell/well. C57BL/6 CD8+ T cells were seeded at the density of 4X105 cell/well into the 96-well plates that BALB/C splenocytes were seeded. Each sample (PBS, free PTX, Free GSNO, Free PTX+free GSNO, pPTX/pCD-pSH NPs and pPTX/pCD-pSNO NPs) was added to the co-cultures at various doses (matched between treatment groups). After 3 d incubation at 37 °C CO2 incubator, 20 μL of AlamarBlue® agents were added to each well. After 40 min, the fluorescence (excitation 560 nm, emission 595 nm) was measured by Synergy H4 microplate reader (BioTek, Winooski, VT). S.I means stimulation index, which was calculated by follows:
The results are presented as mean ± standard deviation (n = 4) and statistically analyzed with two-way ANOVA analysis in Prism software (Graphpad). ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05.
In Vivo DC Profiles:
Animal experiments were IACUC approved and performed in the PRL at the Georgia Institute of Technology. Saline, 20% DMSO, free GSNO in 20% DMSO, free PTX in 20% DMSO, free PTX+free GSNO in 20% DMSO, pPTX/pCD-pSH in saline or pPTX/pCD-pSNO in saline (dose equivalent to 2 mg/kg PTX) were administrated to C57BL/6 mouse with 8-12 weeks old via dorsal injection. After 24 h, the draining lymph node (dLN), non-draining lymph node (ndLN), and spleen were harvested by sacrificing the mouse. Splenocytes were harvested by grinding the spleens through 70 μm strainer with syringe plunger and red blood cells were removed by ACK lysis buffer (Lonza). Lymphocytes were harvested by incubating each dLN and ndLN in 1 mg/mL collagenase D in 37 °C CO2 incubator for 75 min and grinding the lymph nodes through 70 μm strainer with syringe plunger. All splenocytes and lymphocytes were stored on ice <2 hr prior to use. All cells were stained with the following steps: incubation with 2.4G2 on ice for 5 min, staining with Zombie Aqua™ fixable viability dye at room temperature for 30 min, staining with antibody mixtures on ice for 30 min, and fixation with 2% paraformaldehyde on ice for 15 min. After each step, cells were washed with PBS or FACS buffer. Finally, the cells were analyzed by LSR Fortessa flow cytometry (BD Biosciences). The antibody stain contained CD40-FITC (clone HM40-3), CD45-PerCP (clone 30-F11), CD86-PE (clone GL-1), CD11c-PE/Cy7 (clone N418), MHCII-APC (clone M5/114.15.2), and CD11b-Alexafluor700 (clone M1/70). Results are presented as mean ± standard error of the mean (n =3-4) and statistically analyzed with one-way ANOVA analysis in Prism software (Graphpad). ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05.
NP In Vivo Biodistribution:
Animal experiments were IACUC approved and performed in the PRL at the Georgia Institute of Technology. Alexa Fluor™ 647-labelled pPTX/pCD-pSH and pPTX/pCD-pSNO were prepared as described above. Dual tumor model was established by inoculating 105 B16F10-OVA in 30 μL saline in left dorsal skin on day 0 and in right dorsal skin on day 3, respectively (Figure S12). On day 6, fluorescence-labelled NPs (30 μL) were intratumorally administered into the primary tumor on the left dorsal side. After 24 h, mice were sacrificed and each tissue harvested in 1.4 mm Zirconium bead filled tubes (OPS Diagnostics, Lebanon, NJ) and homogenized with FastPrep-24 (MP Biomedicals, CA, USA) to quantify fluorescence. In order to establish standard curves for fluorescently labels NPs in each tissue, untreated tumor-bearing mice were sacrificed at the time of experimental animal sacrifice. Different concentrations of fluorescence-labelled NPs were added to each tissue homogenate, and the resultant dose-dependent fluorescence was used as a standard curve for each tissue.
Therapeutic Efficacy and Immune Cell Profiles of NPs in Dual Tumor Model:
Animal experiments were IACUC approved and performed in the PRL at the Georgia Institute of Technology. The dual tumor model was established by inoculating 1X105 B16F10-OVA (30 μL) in left dorsal skin on day 0 and in right dorsal skin on day 4, respectively. On day 7, saline, 20% DMSO, free GSNO in 20% DMSO, free PTX in 20% DMSO, free PTX+free GSNO in 20% DMSO, pPTX/pCD-pSH in saline or pPTX/pCD-pSNO in saline (dose equivalent to 10 mg/kg PTX) were administrated to the left tumor (1° tumor). The tumor size was calculated by a prolate ellipsoid formulation (ab2/2, where a is the length and b is the width, respectively). The results are presented as mean ± standard error of the mean (n = 5) and statistically analyzed with two-way ANOVA analysis in Prism software (Graphpad). ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05.
On day 18, 1° tumor, 2° tumor, 1° dLN, 2° dLN and spleen were harvested and cell suspensions generated as described above, save in the case of tumor and LN samples that were treated with 1 mg/mL collagenase D in 37 °C CO2 incubator for 4 h and 75 min, respectively, prior to homogenization and staining. Cells were stained as described above. In the case of SIINFEKL-MHCI--PE tetramer (NIH Tetramer Core Facility, Atlanta, Georgia) staining, incubation was performed on ice prior to staining with the mAb panel. Intracellular staining with FoxP3 staining was performed according to the manufacturer’s protocols after mAb staining. After each step, cells were washed with PBS or FACS buffer. Finally, the cells were analyzed by LSR Fortessa flow cytometry (BD Biosciences). The antibodies and fluorophores used are as follows. Panel 1 includes CD40-FITC (clone HM40-3), CD45-PerCP (clone 30-F11), CD86-PE (clone GL-1), CD11c-PE/Cy7 (clone N418), GR1-APC (clone RB6-BC5), CD11b-Alexafluor700 (clone M1/70), MHCII-APC/Cy7 (clone M5/114.15.2), CD206-BV421 (clone C068C2), B220-BV711 (clone RA3-6B2), F4/80-BV785 (clone BM8) and Zombie Aqua™ fixable dye. Panel 2 includes CD45-PerCP (clone 30-F11), SIINFEKL-MHCI--PE (tetramer, NIH Tetramer Core Facility), CD4-PE/Cy7 (clone GK1.5), LAG-3-APC (clone C9B7W), CD25-Alexafluor700 (clone PC61), CD8-APC/Cy7 (clone 53-6.7), FoxP3-BV421 (clone MC-14), NK1.1-BV605 (clone PK136), CDε3-BV711 (145-2C11), PD-1-BV785 (clone 29F.1A12) and Zombie Aqua™ fixable dye. The results are presented as mean ± standard error of the mean (n =2-5) and statistically analyzed with one-way ANOVA analysis in Prism software (Graphpad). ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05.
Therapeutic Efficacy of NPs with aCTLA-4 in Dual Tumor Model:
Animal experiments were IACUC approved and performed in the PRL at the Georgia Institute of Technology. Dual tumor model was established by implanting 1X105 B16F10-OVA (30 μL) in left dorsal skin on day 0 and right dorsal skin on day 4, respectively. On day 7, saline, 20% DMSO, free GSNO in 20% DMSO, free PTX in 20% DMSO, free PTX+free GSNO in 20% DMSO, pPTX/pCD-pSH in saline or pPTX/pCD-pSNO in saline (dose equivalent to 10 mg/kg PTX) were administrated to the left tumor (1° tumor). On day 8, 11, and 14, 100 μg aCTLA-4 in 30 μL saline were intraperitoneally administered. Tumor size was calculated by a prolate ellipsoid formulation (ab2/2, where a is the length and b is the width, respectively). The results are presented as mean ± standard error of the mean (n = 5) and statistically analyzed with two-way ANOVA analysis.
Supplementary Material
Acknowledgements
Research reported in this publication was supported by R01CA207619 and S10OD016264 from the US National Institutes of Health, a Predoctoral Fellowship from the American Heart Association, and Department of Defense CA150523. We thank the NIH Tetramer Core Facility to supply SIINFEKL-MHCI-PE tetramer. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Jihoon Kim, Parker H. Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, 315 Ferst Dr NW, Atlanta, Georgia 30332; George W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology, 315 Ferst Dr NW, Atlanta, Georgia 30332, USA.
Lauren F. Sestito, Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology, 313 Ferst Dr NW, Atlanta, Georgia 30332, USA and Emory University, 201 Dowman Drive, Atlanta, Georgia 30322, USA
Sooseok Im, School of Interdisciplinary Bioscience and Bioengineering, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea.
Won Jong Kim, School of Interdisciplinary Bioscience and Bioengineering, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea; Department of Chemistry, POSTECH, Pohang 37673, Republic of Korea.
Susan N. Thomas, Parker H. Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, 315 Ferst Dr NW, Atlanta, Georgia 30332; George W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology, 315 Ferst Dr NW, Atlanta, Georgia 30332, USA; Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology, 313 Ferst Dr NW, Atlanta, Georgia 30332, USA and Emory University, 201 Dowman Drive, Atlanta, Georgia 30322, USA; Winship Cancer Institute, Emory University School of Medicine, 1365-C Clifton Road NE, Atlanta, Georgia 30322, USA
References
- [1].Skwarczynski M, Hayashi Y, Kiso Y, J. Med. Chem 2006, 49, 7253. [DOI] [PubMed] [Google Scholar]
- [2].Kepp O, Senovilla L, Kroemer G, Oncotarget 2014, 5, 5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lichty BD, Breitbach CJ, Stojdl DF, Bell JC, Nat. Rev. Cancer 2014, 14, 559. [DOI] [PubMed] [Google Scholar]
- [4].Kaneno R, Shurin GV, Tourkova IL, Shurin MR, J. Trans. Med 2009, 7, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Shurin GV, Tourkova IL, Kaneno R, Shurin MR, J. Immunol 2009, 183, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pfannenstiel LW, Lam SSK, Emens LA, Jaffee EM, Armstrong TD, Cell. Immunol 2010, 263, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Thomas SN, Vokali E, Lund AW, Hubbell JA, Swartz MA, Biomaterials 2014, 35, 814. [DOI] [PubMed] [Google Scholar]
- [8].Yuan L, Wu L, Chen J, Wu Q, Hu S, Vaccine 2010, 28, 4402. [DOI] [PubMed] [Google Scholar]
- [9].Sevko A, Kremer V, Falk C, Umansky L, Shurin MR, Shurin GV, Umansky V, J. Immunotoxicol 2012, 9, 275. [DOI] [PubMed] [Google Scholar]
- [10].Zhang L, Dermawan K, Jin M, Liu R, Zheng H, Xu L, Zhang Y, Cai Y, Chu Y, Xiong S, Clin. Immunol 2008, 129, 219. [DOI] [PubMed] [Google Scholar]
- [11].Zhu Y, Liu N, Xiong SD, Zheng YJ, Chu YW, Scand. J. Immunol 2011, 73, 301. [DOI] [PubMed] [Google Scholar]
- [12].Jure-Kunkel M, Masters G, Girit E, Dito G, Lee F, Hunt JT, Humphrey R, Cancer Immunol. Immunother 2013, 62, 1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yan Y, Kumar AB, Finnes H, Markovic SN, Park S, Dronca RS, Dong H, Front. Immunol 2018, 9, 1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chiocca EA, Nat. Rev. Cancer 2002, 2, 938. [DOI] [PubMed] [Google Scholar]
- [15].Melcher A, Parato K, Rooney CM, Bell JC, Mol. Ther 2011, 19, 1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kaufman HL, Kohlhapp FJ, Zloza A, Nat. Rev. Drug Discov 2015, 14, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rehman H, Silk AW, Kane MP, Kaufman HL, J. Immunother. Cancer 2016, 4, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].El-Sawy HS, Al-Abd AM, Ahmed TA, El-Say KM, Torchilin VP, ACS Nano 2018, 12, 10636. [DOI] [PubMed] [Google Scholar]
- [19].Kim J, Yung BC, Kim WJ, Chen X, J. Control. Release 2017, 263, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chung M-F, Liu H-Y, Lin K-J, Chia W-T, Sung H-W, Angew. Chem. Int. Ed 2015, 54, 9890. [DOI] [PubMed] [Google Scholar]
- [21].Pramanick S, Kim J, Kim J, Saravanakumar G, Park D, Kim WJ, Bioconjugate Chem. 2018, 29, 885. [DOI] [PubMed] [Google Scholar]
- [22].Bogdan C, Trends Immunol. 2015, 36, 161. [DOI] [PubMed] [Google Scholar]
- [23].Perrotta C, Falcone S, Capobianco A, Camporeale A, Sciorati C, De Palma C, Pisconti A, Rovere-Querini P, Bellone M, Manfredi AA, Clementi E, Cancer Res. 2004, 64, 3767. [DOI] [PubMed] [Google Scholar]
- [24].Paolucci C, Burastero SE, Rovere-Querini P, De Palma C, Falcone S, Perrotta C, Capobianco A, Manfredi AA, Clementi E, J. Leuk. Biol 2003, 73, 253. [DOI] [PubMed] [Google Scholar]
- [25].Niedbala W, Wei X-Q, Piedrafita D, Xu D, Liew FY, Eur. J. Immunol 1999, 29, 2498. [DOI] [PubMed] [Google Scholar]
- [26].Niedbala W, Wei X-Q, Campbell C, Thomson D, Komai-Koma M, Liew FY, Proc. Natl. Acad. Sci. USA 2002, 99, 16186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lee S-W, Choi H, Eun S-Y, Fukuyama S, Croft M, J. Immunol 2011, 186, 6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Fernández-Ruiz V, González A, López-Moratalla N, Immunol. Lett 2004, 93, 87. [DOI] [PubMed] [Google Scholar]
- [29].Giordano D, Magaletti DM, Clark EA, Blood 2006, 107, 1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Niedbala W, Cai B, Liu H, Pitman N, Chang L, Liew F-Y, Proc. Natl. Acad. Sci. USA 2007, 104, 15478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].De Boo S, Kopecka J, Brusa D, Gazzano E, Matera L, Ghigo D, Bosia A, Riganti C, Mol. Cancer 2009, 8, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tarr JM, Young PJ, Morse R, Shaw DJ, Haigh R, Petrov PG, Johnson SJ, Winyard PG, Eggleton P, J. Mol. Biol 2010, 401, 799. [DOI] [PubMed] [Google Scholar]
- [33].Kopecka J, Campia I, Brusa D, Doublier S, Matera L, Ghigo D, Bosia A, Riganti C, J. Cell. Mol. Med 2011, 15, 1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Namgung R, Lee YM, Kim J, Jang Y, Lee B-H, Kim I-S, Sokkar P, Rhee YM, Hoffman AS, Kim WJ, Nat. Commun 2014, 5, 3702. [DOI] [PubMed] [Google Scholar]
- [35].May JM, Qu Z-C, Mol. Cell Biochem 2009, 325, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Candelario-Jalil E, Akundi RS, Bhatia HS, Lieb K, Appel K, Muñoz E, Hüll M, Fiebich BL, J. Neuroimmunol 2006, 174, 39. [DOI] [PubMed] [Google Scholar]
- [37].Lee HJ, Kim DE, Park DJ, Choi GH, Yang D-N, Heo JS, Lee SC, Colloids Surf. B Biointerfaces 2016, 146, 1. [DOI] [PubMed] [Google Scholar]
- [38].Kim J, Saravanakumar G, Choi HW, Park D, Kim WJ, J. Mater. Chem. B 2014, 2, 341. [DOI] [PubMed] [Google Scholar]
- [39].Barker DL, Jencks WP, Biochemistry 1969, 8, 3890. [DOI] [PubMed] [Google Scholar]
- [40].Farb D, Jencks WP, Arch. Biochem. Biophys 1980, 203, 227. [DOI] [PubMed] [Google Scholar]
- [41].Lee C-S, Park W, Park S-J, Na K, Biomaterials 2013, 34, 9227. [DOI] [PubMed] [Google Scholar]
- [42].Sofias AM, Dunne M, Storm G, Allen C, Adv. Drug Deliver. Rev 2017, 122, 20. [DOI] [PubMed] [Google Scholar]
- [43].Bernabeu E, Cagel M, Lagomarsino E, Moretton M, Chiappetta DA, Int. J. Pharm 2017, 526, 474. [DOI] [PubMed] [Google Scholar]
- [44].Broniowska KA, Diers AR, Hogg N, Biochim. Biophys. Acta 2013, 1830, 3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhang Y, Hogg N, Proc. Natl. Acad. Sci. USA 2004, 101, 7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Song Q, Tan S, Zhuang X, Guo Y, Zhao Y, Wu T, Ye Q, Si L, Zhang Z, Mol. Pharmaceutics 2014, 11, 4118. [DOI] [PubMed] [Google Scholar]
- [47].Ramanathan B, Jan K-Y, Chen C-H, Hour T-C, Yu H-J, Pu Y-S, Cancer Res. 2005, 65, 8455. [DOI] [PubMed] [Google Scholar]
- [48].Al-alami O, Sammons J, Martin JH, Hassan HT, Leuk. Res. 1998, 22, 939. [DOI] [PubMed] [Google Scholar]
- [49].Su GMI, Davey MW, Davey RA, Int. J. Cancer 1998, 76, 702. [DOI] [PubMed] [Google Scholar]
- [50].Melguizo C, Prados J, Luque R, Ortiz R, Caba O, Álvarez PJ, Gonzalez B, Aranega A, Int. J. Mol. Sci 2012, 13, 16624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wang N-N, Zhao L-J, Wu L-N, He M-F, Qu J-W, Zhao Y-B, Zhao W-Z, Li J-S, Wang J-H, Asian Pac. J. Cancer Prev 2013, 14, 4983. [DOI] [PubMed] [Google Scholar]
- [52].Nakano A, Liu GS, Heusch G, Downey JM, Cohen MV, J. Mol. Cell Cardiol 2000, 32, 1159. [DOI] [PubMed] [Google Scholar]
- [53].Gansauge S, Gansauge F, Nussler AK, Rau B, Poch B, Schoenberg MH, Beger HG, FEBS Lett. 1997, 410, 160. [DOI] [PubMed] [Google Scholar]
- [54].Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, Kepp O, Metivier D, Galluzzi L, Perfettini J-L, Zitvogel L, Kroemer G, Cell Death Differ. 2014, 21, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].John J, Ismail M, Riley C, Askham J, Morgan R, Melcher A, Pandha H, BMC Immunol. 2010, 11, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tanaka H, Matsushima H, Mizumoto N, Takashima A, Cancer Res. 2009, 69, 6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Rohner NA, Thomas SN, ACS Biomater. Sci. Eng 2017, 3, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kim J, Li WA, Choi Y, Lewin SA, Verbeke CS, Dranoff G, Mooney DJ, Nat. Biotech 2015, 33, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Jayaraman P, Parikh F, Lopez-Rivera E, Hailemichael Y, Clark A, Ma G, Cannan D, Ramacher M, Kato M, Overwijk WW, Chen S-H, Umansky VY, Sikora AG, J. Immunol 2012, 188, 5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI, Nat. Med 2007, 13, 828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Nagaraj S, Schrum AG, Cho H-I, Celis E, Gabrilovich DI, J. Immunol 2010, 184, 3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Nagaraj S, Youn J-I, Gabrilovich DI, J. Immunol 2013, 191, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bonasio R, von Andrian UH, Curr. Opin. Immunol 2006, 18, 503. [DOI] [PubMed] [Google Scholar]
- [64].Randolph GJ, Ochando J, Partida-Sanchez S, Annu. Rev. Immunol 2008, 26, 293. [DOI] [PubMed] [Google Scholar]
- [65].Hill S, Edwards AJ, Kimber I, Knight SC, Immunology 1990, 71, 277. [PMC free article] [PubMed] [Google Scholar]
- [66].de Vries IJM, Krooshoop DJEB, Scharenborg NM, Joost Lesterhuis W, Diepstra JHS, Van Muijen GNP, Strijk SP, Ruers TJ, Boerman OC, Oyen WJG, Adema GJ, Punt CJA, Figdor CG, Cancer Res. 2003, 63, 12. [PubMed] [Google Scholar]
- [67].Francis DM, Thomas SN, Adv. Drug Deliver. Rev 2017, 114, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kim J, Manspeaker MP, Thomas SN, Acta Biomater. 2019, 88, 1. [DOI] [PubMed] [Google Scholar]
- [69].Ashraf Javeed. M., Riaz A, Ghafoor A, Afzal S, Mukhtar MM, Eur. J. Pharm. Sci 2009, 38, 283. [DOI] [PubMed] [Google Scholar]
- [70].Lee M, Jeon YJ, Mol. Pharmacol 2001, 59, 248. [DOI] [PubMed] [Google Scholar]
- [71].Chan OTM, Yang L-X, Cancer Immunol. Immunother. 2000, 49, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Gabrilovich DI, Ostrand-Rosenberg S, Bronte V, Nat. Rev. Immunol 2012, 12, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Denkert C, von Minckwitz G, Darb-Esfahani S, Lederer B, Heppner BI, Weber KE, Budczies J, Huober J, Klauschen F, Furlanetto J, Schmitt WD, Blohmer J-U, Karn T, Pfitzner BM, Kümmel S, Engels K, Schneeweiss A, Hartmann A, Noske A, Fasching PA, Jackisch C, van Mackelenbergh M, Sinn P, Schem C, Hanusch C, Untch M, Loibl S, Lancet Oncol. 2018, 19, 40. [DOI] [PubMed] [Google Scholar]
- [74].Lammers T, Kiessling F, Ashford M, Hennink W, Crommelin D, Storm G, Nat. Rev. Mater 2016, 1, 16069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Stephan SB, Taber AM, Jileaeva I, Pegues EP, Sentman CL, Stephan MT, Nat. Biotech 2015, 33, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Liu BL, Robinson M, Han Z-Q, Branston RH, English C, Reay P, McGrath Y, Thomas SK, Thornton M, Bullock P, Love CA, Coffin RS, Gene Ther. 2003, 10, 292. [DOI] [PubMed] [Google Scholar]
- [77].Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, Merghoub T, Wolchok JD, Allison JP, Sci. Transl. Med 2014, 6, 226ra32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, Olszanski AJ, Malvehy J, Cebon J, Fernandez E, Kirkwood JM, Gajewski TF, Chen L, Gorski KS, Anderson AA, Diede SJ, Lassman ME, Gansert J, Hodi FS, Long GV, Cell 2017, 170, 1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].LaRocca CJ, Warner SG, Clin. Trans. Med 2018, 7, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Bommareddy PK, Shettigar M, Kaufman HL, Nat. Rev. Immunol 2018, 18, 498. [DOI] [PubMed] [Google Scholar]
- [81].Garofalo M, Saari H, Somersalo P, Crescenti D, Kuryk L, Aksela L, Capasso C, Madetoja M, Koskinen K, Oksanen T, Mäkitie A, Jalasvuori M, Cerullo V, Ciana P, Yliperttula M, J. Control. Release 2018, 283, 223. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.