

ABSTRACT
Mammalian genome structure is closely linked to function. At the scale of kilobases to megabases, CTCF and cohesin organize the genome into chromatin loops. Mechanistically, cohesin is proposed to extrude chromatin loops bidirectionally until it encounters occupied CTCF DNA-binding sites. Curiously, loops form predominantly between CTCF binding sites in a convergent orientation. How CTCF interacts with and blocks cohesin extrusion in an orientation-specific manner has remained a mechanistic mystery. Here, we review recent papers that have shed light on these processes and suggest a multi-step interaction between CTCF and cohesin. This interaction may first involve a pausing step, where CTCF halts cohesin extrusion, followed by a stabilization step of the CTCF-cohesin complex, resulting in a chromatin loop. Finally, we discuss our own recent studies on an internal RNA-Binding Region (RBRi) in CTCF to elucidate its role in regulating CTCF clustering, target search mechanisms and chromatin loop formation and future challenges.
KEYWORDS: CTCF, Cohesin, TADs, Loop Extrusion, Convergent Rule, Binding Polarity, RNA-Binding Region, PDS5, NIPBL
Introduction
Mammalian genomes face the dual challenge of safely packaging and storing around two meters of DNA inside the nucleus, while retaining access to several processes including transcription, replication and DNA repair. Accordingly, genome structure is intimately linked to genome function and mammalian genomes are organized at multiple scales. At the chromosomal scale, chromosomes occupy discrete territories [1] and at the small scale, 147 base pairs of DNA are wrapped around histone octamers into nucleosomes [2]. In this review, we will focus on the intermediate scale of mammalian interphase genome organization (kilobases to megabases), where genomes appear to be organized by two major mechanisms [3].
First, a poorly understood compartmentalization mechanism organizes the genome into two major A- and B-compartments [4,5]. A-compartments largely correspond to gene-rich and transcriptionally active euchromatic regions, that replicate earlier in S-phase and tend to associate with nuclear speckles [6,7]. In contrast, B-compartments largely correspond to gene-poor condensed heterochromatin that is largely transcriptionally inactive, tends to replicate late in S-phase, and is often associated with the nuclear lamina and the nucleolus [8–10]. A/B compartments can be further divided into several subcompartments [11–14]. While the molecular mechanisms of compartmentalization remain poorly understood, preferential A-A and B-B interactions can largely explain the segregation of chromosomes into compartments. In fact, polymers of distinct A/B segments, known as block copolymers, naturally undergo microphase separation [3,12,15]. As such, compartmentalization leads to global segregation. Loci in an A compartment on a given chromosome are more likely to interact with other A compartment loci both on the same chromosome and on other chromosomes. By causing both preferential intra- and inter-chromosomal interactions, A/B compartmentalization is visible as a ‘plaid’ or ‘checkerboard’ pattern in Hi-C contact maps [4,5] (Figure 1).
Figure 1.
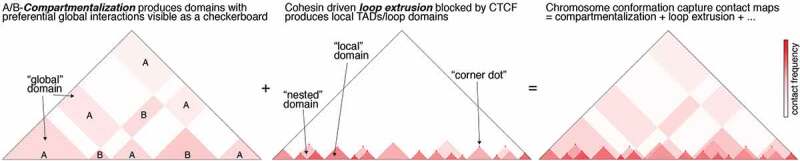
A simplified illustration of how contact map features are shaped by A/B-compartmentalization and loop extrusion. Highly simplified sketches of hypothetical contact maps produced by chromosome conformation capture methods such as Hi-C. Left: Hypothetical contact map produced by A/B-compartmentalization. Compartmentalization generates both local and global domains. Middle: Hypothetical contact map produced by loop extrusion. Loop extrusion generates strictly local maps demarcated by strong convergent CTCF binding sites, and sometimes forms nested domains. Right: Real contact maps are affected by both A/B-compartmentalization and CTCF/cohesin-mediated loop extrusion – as well as a number of other processes especially at the fine-scale [129,130] – and are therefore the sum of all of these processes. This makes interpreting and classifying ‘domains’ in Hi-C contact maps highly challenging [9,30].
Second, increasing evidence suggests that a loop extrusion mechanism organizes genomes into local domains known as Topologically Associating Domains (TADs) or Loop Domains [16–19]. During interphase, the multi subunit cohesin complex [20] is hypothesized to extrude DNA bi-directionally at a speed of ~0.5–1.0 kb/second [16–19,21–23], until it is blocked by a boundary. The insulator protein, CCCTC-Binding Factor (CTCF), appears to be the primary boundary factor in mammals, at least at the level of TADs and loops visible in Hi-C [24]. This model is supported by the observations that loss of cohesin [25–28] eliminates essentially all TADs and loop domains as measured by Hi-C, whereas loss of CTCF affects many TADs and loop domains [28,29], though the effect of CTCF loss on TADs is markedly weaker than cohesin loss. Unlike A/B-compartments, TADs are local domains, albeit often nested local domains (Figure 1). We note that nomenclature and domain classification remain a challenge in the field: although TADs appear to be formed by loop extrusion, not all TADs are anchored by visible loops (‘corner dots’) and not all loops give rise to TADs. Furthermore, since TADs and compartment domains can appear similar in contact maps, distinguishing these is also a challenge [9,30].
Notably, CTCF binding sites are asymmetric and CTCF-mediated loops are largely bridged by convergent CTCF DNA binding sites [11,31–33]. Moreover, inversion of a single CTCF binding site can be sufficient to disrupt a loop and TAD, and rearrange the 3D folding of hundreds of kilobases of DNA [18,31,32,34], though not all inversions had this effect [31,32]. What this means is that a loop-extruding cohesin complex with a diameter of ~50 nm [20] is somehow able to distinguish whether it approaches a comparatively tiny DNA-bound CTCF protein (~3-5 nm) from the N-terminal or C-terminal side [35]. Though how this works remains a mechanistic mystery, a series of very recent papers have shed new light on this process [28,36–42].
In this Extra View article, we will focus on how CTCF and cohesin interact to regulate genome folding into TADs and loops, and place our own recent studies in the context of these new findings [36,43]. For other important aspects of 3D genome organization including the role of TADs in regulating gene expression and other functions, how to interpret Hi-C contacts maps, nomenclature, distinguishing TAD and compartment domains (Figure 1), the wealth of available technologies for probing nuclear organization, and other exciting areas, we refer the reader to a number of excellent recent reviews [9,30,44–46].
Cohesin
Cohesin belongs to the Structural Maintenance of Chromosomes (SMC) family of protein complexes. SMC complexes appear to organize chromosomal DNA topology in all living organisms from bacteria to eukaryotes. SMC complexes are ancient and their evolution likely preceded histones [20]. Here, we will focus on mammalian cohesin, which contains two proteins, SMC1 and SMC3, that dimerize at the hinge (Figure 2a). Antiparallel coiled coils (~50-nm) connect the hinge to ATPase head domains, which bind the N- and C-terminus of a kleisin subunit, RAD21, thereby forming a ring [47]. RAD21 can be bound by regulatory proteins such as HAWKs (HEAT repeat containing proteins Associated With Kleisins) including STAG1/STAG2 (SA1/SA2), PDS5A/PDS5B, and NIPBL [40,48–50]. For simplicity, herein we will refer to STAG1 or STAG2 as STAG and PDS5A or PDS5B as PDS5, though it is important to note that STAG1- and STAG2-cohesin play somewhat different roles [38,51–53]. The STAG protein is essentially always associated with cohesin unlike NIPBL and PDS5. NIPBL regulates cohesin loading on DNA [54–56] and is also required for loop extrusion in vitro [21,23]. PDS5, together with WAPL, regulates cohesin release from DNA [57–59]. Cohesin can switch between a single compartment state (S-K) and a two-compartment state (S and K) in an ATP-dependent manner (Figure 2a; see also [20,60–62] for a more comprehensive discussion). Despite great progress, many mechanistic and structural ambiguities remain and it is important to note that although cohesin-mediated DNA loop extrusion has now been observed in vitro [21–23], it remains to be demonstrated in vivo. Moreover, though models have been proposed [20,54,61,63,64], the molecular mechanism of cohesin extrusion remains unclear as does whether monomeric or dimeric cohesin extrudes loops [21,23,65]. Although cohesin can topologically enclose DNA inside its ring, loop extrusion seems to involve non- or pseudo-topological DNA engagement [21,23].
Figure 2.
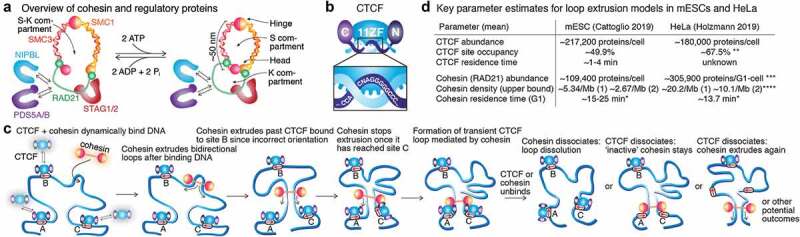
Overview of cohesin, CTCF, and loop extrusion. (a) Overview of mammalian cohesin and some of its regulatory proteins. (b) Overview of CTCF with N-terminal, 11 Zinc Fingers, and C-terminal domains. (c) Simplified sketch of cohesin-mediated loop extrusion and the convergent CTCF rule. (d) Summary of key parameters constraining loop extrusion models in mouse embryonic stem cells (mESCs) [65] and human HeLa cells [83], with mESC residence times taken from [70]. * These are cohesin G1 residence times (both STAG1 and STAG2), but after these studies were published it was found that STAG2-cohesin binds DNA substantially more dynamically than STAG1-cohesin [38], suggesting that putative loop extruding G1 cohesins have at least two residence times. ** Estimated from [83] (~180,000 and ~120,000 CTCF proteins and sites per HeLa cell) with added assumption that 45% of CTCF proteins are bound to cognate sites (~45%, i.e. mean of mESC and U2OS in [70]). *** 305,900 is the mean of the LC-MS and FCS estimates reported in [83]. **** Cohesin density is estimated from ~159,437 dynamically bound (~13.7 min residence time) cohesin proteins (SCC1-mEGFP) in G1 and the reported HeLa genome sizes 7.9 Gb, both taken from [83]. It is important to note that these are genomic averages: e.g. CTCF residence time is for an average site (some sites will have slower and faster binding), cohesin density may not be uniform throughout the genome, and since the two in vitro cohesin loop extrusion papers disagreed on whether cohesin is monomeric [23] or dimeric [21], densities for both monomeric [1] and dimeric [2] are shown.
CTCF
CTCF is an 11-Zinc Finger (ZF) DNA-Binding protein that is conserved across most animals, but absent from plants, C. elegans and yeast [66,67]. Mammalian CTCF has unstructured N- and C-terminal domains flanking the 11-ZF DNA-binding domain [68,69] (Figure 2b). Depending on the antibody used and the bioinformatic threshold, CTCF binds ~40,000–90,000 sites in mammalian genomes, of which ~30-60% are cell-type specific and with around half in intergenic regions and the other half at promoters, in introns or exons [39,66,70–72]. Consistent with CTCF regulating cohesin positioning on chromatin, but not cohesin loading onto chromatin, >90% of all cohesin ChIP-Seq peaks co-localize with CTCF [39,70,73–75], but CTCF depletion does not affect the amount of cohesin on chromatin, only its location [42,74,76].
Loop extrusion
In the simplest formulation of the loop extrusion model, cohesin loads randomly on chromatin and begins extruding loops bidirectionally (Figure 2c). CTCF binds cognate sites and ‘passively’ waits for a cohesin complex to arrive. CTCF will block the extruding cohesin complex if, and only if, the CTCF site on DNA is in a convergent orientation such that cohesin first encounters its N-terminal domain (thus site ‘B’ is skipped in Figure 2c). We refer to cohesin’s preference for occupied CTCF binding sites in the convergent orientation as the convergent rule [11,18,31–34]. Since both CTCF and cohesin bind DNA dynamically [70], there is a significant probability that either CTCF or cohesin dissociates from DNA before they encounter each other. But if cohesin reaches two convergent and occupied CTCF binding sites, a CTCF loop is stabilized for an unknown duration and may appear as a ‘corner dot’ in a Hi-C map (Figure 1). Though the lifetime of such CTCF loops remain unknown, we have previously argued that they are likely dynamic [24,70]. Regardless, the loop may dissociate when CTCF or cohesin dissociates from DNA. If CTCF dissociates first, it is not known if cohesin remains or continues to extrude (Figure 2c). Polymer simulations of the simple loop extrusion model with just cohesin and CTCF generate contact maps similar to experimental maps at the level of TADs and loop domains [17,18,38,44,77]. This is a simplified picture of loop extrusion and it is important to note that loop extrusion inside the cell is likely not this simple: the transcriptional machinery, for example, is likely to also serve as a partial boundary to cohesin-mediated loop extrusion [76,78–80].
Key parameters for the simple loop extrusion model are: CTCF residence time, probability that a CTCF binding site is occupied, cohesin residence time, mean density of loop extruding cohesins on chromatin (upper bound estimated from G1 phase), and extrusion speed. Though the speed of extrusion in live cells remains unknown, various direct and indirect estimates have been reported: ~22.5 kb/min (cohesin in HCT116 cells [26]); ~6-12 kb/min (condensin II in chicken cells [81]); ~54 kb/min (bacterial SMC complex [82]), and ~30-60 kb/min on naked DNA in vitro [21,23]. The speed of cohesin extrusion is likely to differ between different local genomic regions and cell types. The other parameters have recently been estimated in mouse embryonic stem cells (mESC) [65] and human HeLa cells [83] (Figure 2d). One key insight is that CTCF forms a ‘permeable boundary’: all but the strongest CTCF binding sites are only occupied some of the time (on average, ~50% in mESC; ~68% in HeLa) [65,83]. Thus, an extruding cohesin can skip a convergent CTCF binding site a significant fraction of the time. This can explain why different loops may form at different cells, or in the same cell at different points in time [65,70,83], as well as the formation of nested loops and domains in Hi-C maps [30]. Whether cohesin exists and functions as a monomer, dimer, or oligomer remains debated [62,65,84,85] as does whether monomeric or dimeric cohesin extrudes loops [21,23]. Practically, this means that upper bound estimates of the density of loop extruding cohesin vary by a factor of two: ~2.67 to 5.34 per Mb in mESCs [65] and ~10-20 per Mb in HeLa [83] (Figure 2d). Thus, quantitative constraints on the loop extrusion model are beginning to emerge and obtaining such data for more cell types will further help reveal if and how loop extrusion parameters are regulated in a cell-type specific manner to regulate genome structure and function.
One-step vs. multi-step CTCF-cohesin interactions to explain the convergent rule
The fact that cohesin would first encounter the N-terminus of CTCF when encountering a convergent CTCF site was originally used as a mechanistic explanation for the convergent rule. Given that cohesin extrudes loops very rapidly, this would require a near-instantaneous and near-deterministic interaction between cohesin and the CTCF N-terminus. However, if this interaction was a rapid and efficient ‘lock and key’ type interaction, it is not immediately clear why cohesin when encountering an incorrectly oriented CTCF binding site could not just extrude past the CTCF C-terminus to stably halt at the N-terminus (Figure 3a). Though it is conceivable that such a binding interface might be ‘directionally sensitive’ and only be properly presented from one orientation (Figure 3b).
Figure 3.
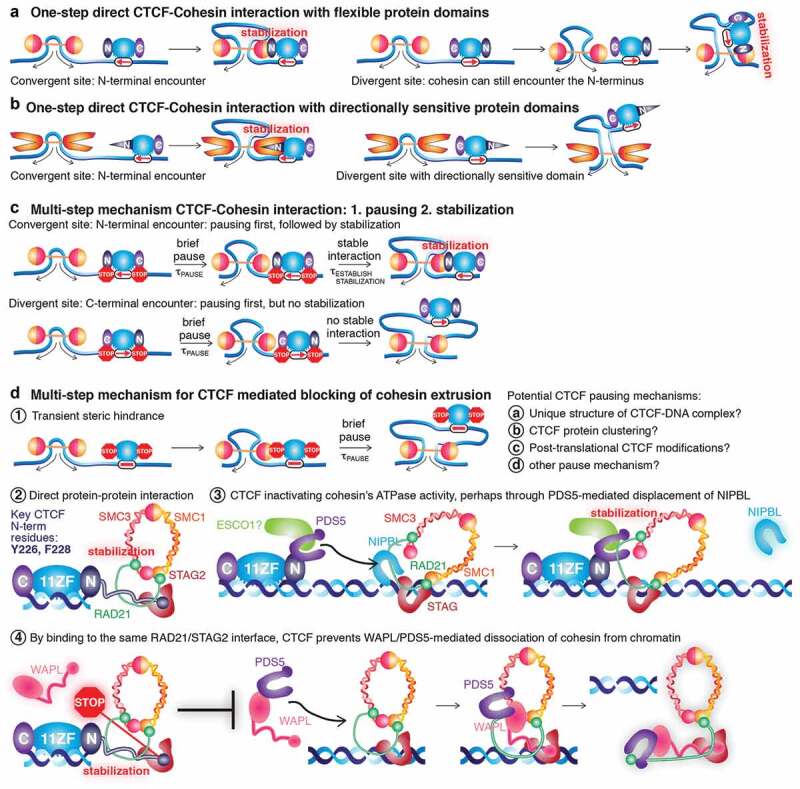
One-step vs. Multi-step CTCF-cohesin interaction mechanisms. (a) One-step CTCF-cohesin mechanism. If 1-step mechanism, it is not clear why cohesin could not extrude past the C-terminal domain of CTCF to interact with the N-terminal domain on the other side. (b) One-step CTCF-cohesin mechanism with directionally sensitive domains. For a one-step mechanism to work, the N-terminal CTCF domain and cohesin would both have to exhibit a directional sensitivity as illustrated. (c) Multi-step CTCF-cohesin mechanism. For an N-terminal encounter, pausing is eventually followed by stabilization. For a C-terminal encounter, pausing is not followed by stabilization, so cohesin eventually extrudes past or dissociates. (d) Instead of a one-step mechanism, a multi-step mechanism would involve transient pausing of cohesin next to CTCF (1), followed by stabilization of cohesin only from the N-terminal side of CTCF, through either direct protein-protein interaction (2), CTCF ‘turning OFF’ the cohesin motor (ATPase) perhaps mediated via PDS5A/B and/or ESCO1 (3), or through CTCF preventing WAPL-mediated release of cohesin from chromatin by CTCF binding to the same RAD21/STAG2 interface as WAPL does (4). It is important to note both that these mechanisms are not mutually exclusive, and that many other mechanisms could contribute.
Instead, recent studies [28,36–42] have proposed and substantiated a multi-step mechanism that can better explain the convergent rule involving pausing and stabilization [39–42] (Figure 3c). Along the lines of what is proposed in [42], cohesin extrusion is near-deterministically paused by CTCF, either dependent on or independent of CTCF binding site orientation. Cohesin pauses for a period, τPAUSE, after which it will eventually extrude past CTCF. Next, assume that the N-terminus of CTCF can directly or indirectly stabilize cohesin, but that establishing stabilization is moderately slow though faster than the pause period (τESTABLISH STABILIZATION < τPAUSE) and also that the C-terminus cannot stabilize cohesin. Now if cohesin faces the C-terminal side of CTCF, cohesin will pause for duration, τPAUSE, but since cohesin cannot be stabilized, cohesin will eventually extrude past or dissociate from DNA. Since cohesin extrudes quickly and since establishing stabilization is slow, it is extremely unlikely that stabilization will occur once cohesin extrudes over the N-terminus from a C-terminal encounter. In contrast, when cohesin encounters the N-terminal side first, it will pause and during the pause period τPAUSE, it is very likely that stabilization is established before cohesin can escape since τESTABLISH STABILIZATION < τPAUSE. Once stabilized, a loop will be stabilized with duration τLOOP.
Such a multi-step mechanism would be able to explain the convergent rule. Conceptually, this means the convergent rule can be divided into at least two components: 1) the mechanism of pausing and 2) the mechanism of stabilization. Cohesin stabilization would likely be essential for forming sufficiently stable CTCF-loops to be visible as ‘corner peaks’ in Hi-C maps, whereas both pausing and stabilization would contribute to TAD insulation. Therefore, interfering with the stabilization mechanism would be expected to affect ‘loops’ more strongly in Hi-C maps than TADs, and this was indeed observed [40]. We therefore next consider potential mechanisms of pausing and stabilization.
Mechanism of CTCF-mediated pausing of cohesin extrusion
At the most simplified level, given the size differences between CTCF (~3-5 nm) and cohesin (~50 nm), it is not immediately clear how CTCF could near-deterministically halt and pause an extruding cohesin (Figure 3d, 1). However, CTCF is an unusual DNA-binding protein and binds DNA for minutes [70,86,87] instead of seconds as seen for conventional transcription factors [88,89], and may have a unique ability to halt cohesin [90]. Here we consider a number of non-mutually exclusive possibilities.
First, CTCF is uniquely able to position ~20 nucleosomes around its DNA-binding sites [91–95], suggesting a mechanism whereby CTCF binding generates a unique chromatin microenvironment that could potentially serve as a steric hindrance pause signal. Consistently, CTCF has been reported to form a highly unusual DNA structure where it binds [96]. Thus, CTCF may simply pause cohesin through the unique 3D conformation of the CTCF-DNA complex [39], perhaps in a CTCF orientation independent manner. Though analyses of CTCF paralog CTCFL are informative here: CTCFL has a nearly identical 11ZF domain and motif preference to CTCF. While CTCFL cannot rescue CTCF-mediated genome organization [41], a CTCF-CTCFL chimera with the N-terminus and ZF1 + 2 from CTCF and ZF3-11 and C-terminus from CTCFL was able to largely rescue cohesin ChIP-Seq binding [39], though loop-resolution Hi-C analyses of this chimera were not performed. Beyond CTCF-DNA interactions, this roadblock model may also include CTCF-RNA interactions [36,37,39].
Second, we previously showed that CTCF forms clusters [70], whereby several to tens of CTCF proteins come together. Similarly, CTCF forms larger foci in senescent cells [97]. Recently, we showed that CTCF clustering is mediated partially through an internal RNA-Binding Region (RBRi) in CTCF, and consistent with CTCF clustering playing a role in genome organization, around one-third of all CTCF loops are lost in ΔRBRi-CTCF mESCs [36]. Mechanistically, a bulky cluster of CTCF may more efficiently pause an extruding cohesin – perhaps in a binding site orientation independent manner – which may explain why impaired CTCF clustering might also impair cohesin pausing, thereby leading to loop disruption. Thus, the current data is also consistent with CTCF clustering contributing to pausing cohesin.
Third, CTCF is subject to a number of post-translational modifications [66], some of which are quite large such as SUMOylation [98] and poly-ADP-ribosylation [99–101]. Early reports suggested that poly-ADP-ribosylation contributes to the insulation function of CTCF [99,100], and mutation of the 11 amino acids in the N-terminus reported to be poly-ADP-ribosylated as well as treatment with a PARP inhibitor, moderately decreased CTCF’s ability to stabilize cohesin at its binding sites as measured by ChIP-Seq [39]. These results are consistent with poly-ADP-ribosylation playing some role, albeit not an absolutely required one, in CTCF’s ability to pause cohesin extrusion.
It remains highly unclear exactly how CTCF might pause an extruding cohesin, and there may well be many other potential mechanisms such as DNA supercoiling and the chirality of the double helix [102]. It is also important to note that the three mechanisms discussed above – nucleosome positioning/chromatin structure, CTCF clustering, and poly-ADP-ribosylation – are not mutually exclusive but may instead be synergistic (Figure 3d, 1). As such, more work is urgently needed.
Mechanism of CTCF-mediated stabilization of cohesin
Originally, a small region C-terminal to CTCF’s 11 Zinc Finger domain was reported to be necessary and sufficient for binding the STAG2 subunit of cohesin and this was assumed to be the only direct interaction between CTCF and the cohesin complex [103]. However, subsequent studies found that ΔRBRi-CTCF, whose deletion encompasses this region, co-immunoprecipitated cohesin equally well as wild-type CTCF [36,41,104], indicating that this region is not required for the biochemical CTCF-cohesin interaction, though it does contribute to loop formation [36]. Instead, despite different experimental systems and different read-outs, the recent papers all reached remarkable agreement that it is the CTCF-N-terminus as well as the first two Zinc Fingers that play key roles in the CTCF-cohesin interaction, though notably, neither region is autonomously sufficient [39–42,105]. Notably, placing the CTCF N-terminus on the C-terminal side does not fully recapitulate function [41,42]. Here, we discuss three mechanistic models, that are not mutually exclusive, for the interaction between CTCF and cohesin: direct protein-protein interaction, CTCF inactivating cohesin’s ATPase function, and CTCF stabilizing cohesin on DNA by antagonizing WAPL-mediated cohesin release (Figure 3d).
First, the simplest model invokes direct protein-protein interaction between CTCF and cohesin (Figure 3d, 2). This model is supported by the observed CTCF-cohesin co-immunoprecipitation [36,40,41,70,104,106] and Li et al. recently elucidated the structural basis for this interaction [40] by solving the crystal structure of a short CTCF peptide (N-terminal amino acids 222–231) bound to the RAD21-STAG2 interface. Thus, the CTCF N-terminus binds both RAD21 and STAG2, albeit relatively weakly (~0.6 μM) [40]. Thus, it is clear that a direct CTCF-cohesin protein-protein interaction is at play in vitro [40]. However, since it is relatively weak (~0.6 μM), it will be important to study in the future if there are additional interactions and how this interaction functions in vivo.
Second, CTCF could regulate cohesin’s ATPase activity directly or indirectly through cohesin regulatory proteins such as PDS5 or ESCO1. If CTCF could turn OFF ‘extrusion’, this might ‘lock’ cohesin in place and thereby stabilize a CTCF-cohesin loop (Figure 3d, 3). This model is suggested by studies by Wutz et al. [28,38]. WAPL binds PDS5 and releases cohesin from chromatin [58,59]. Depletion of WAPL greatly increases cohesin’s residence time on chromatin [58], leading to its reorganization into structures called ‘vermicelli’ and compaction of DNA [107]. Thus, in Hi-C maps, WAPL depleted cells exhibit many more loops or ‘corner peaks’ [28,108]. PDS5 depletion also increases cohesin’s residence time in G1 and leads to even more pronounced vermicelli than depleting WAPL alone [28]. Therefore, PDS5-depleted Hi-C maps ought to resemble WAPL-depleted Hi-C maps, but surprisingly, PDS5-depleted Hi-C maps show fewer loops, more closely resemble CTCF-depleted Hi-C maps, and strongly violate the convergent rule [28]. Specifically, if the CTCF binding site orientation played no role, 25% of CTCF loops would be expected to be convergent instead of the observed 65–92% [11,31,32,109]. But upon PDS5 depletion, only 30.9% of loops are bridged by convergent CTCF sites, implicating the PDS5 proteins in mediating the convergent rule [28]. Since NIPBL stimulates cohesin’s ATPase activity [110,111], is required for vermicelli formation [108], binds to the same cohesin interface as PDS5, and therefore competes with PDS5 for cohesin binding [49], Wutz et al. proposed that PDS5 could inactivate cohesin’s ATPase activity in a CTCF-dependent manner [28]. Consistently, NIPBL is required for cohesin extrusion in vitro [21,23] and is sub-stoichiometric compared to cohesin [111]. Thus, if following CTCF-mediated pausing of cohesin extrusion, the CTCF N-terminus and PDS5 could displace NIPBL or otherwise inactivate cohesin’s ATPase activity, this would mechanistically explain the convergent rule and stabilization of cohesin by CTCF. In this model, the role of PDS5 may depend on whether or not it partners with CTCF (turn off NIPBL) or WAPL (unload cohesin from DNA). For this model to explain bidirectional extrusion where cohesin can stop independently on the left and on the right, a dimeric form of cohesin associating with two NIPBL proteins which can be independently dissociated by CTCF/PDS5 would likely be required [21,62,65,85]. Finally, we note that direct support for PDS5’s ability to inhibit extrusion comes from in vitro single-molecule studies where WAPL-PDS5 were found to inhibit cohesin translocation on DNA [112].
Third, CTCF could stabilize cohesin on DNA by counteracting cohesin’s release from DNA by WAPL-PDS5 [58,59] (Figure 3d, 4). Cohesin exists in a dynamic equilibrium of binding and dissociation from chromatin and ~40% of cohesins are specifically associated with chromatin in G1 in mESCs [70]. This population may represent loop extruding cohesins. Intriguingly, Li et al. found that CTCF and WAPL bind the same RAD21-STAG2 interface of cohesin, with CTCF binding more strongly than WAPL [40]. Li et al. therefore proposed a model where CTCF stabilizes cohesin and loops by counteracting WAPL-mediating cohesin release from DNA. Consistently, CTCF depletion [38] and mutation of key RAD21-STAG2 interacting amino acids in CTCF (Y226A, F228A) [40] decreased cohesin’s residence time on DNA as measured by iFRAP. Relatedly, Wutz et al. recently found that STAG1-cohesin exhibits more SMC3 acetylation and a more stable residence time on DNA compared to STAG2-cohesin, which is less acetylated and binds DNA more dynamically [38]. Whereas ESCO2 acetylates cohesin only during S-Phase [113], ESCO1 acetylates cohesin throughout the cell cycle including during G1 [113], and ESCO1 is recruited to cohesin by PDS5 [114]. Thus, a related mechanism through which CTCF could stabilize cohesin would be CTCF-PDS5 mediated recruitment of ESCO1, which then acetylates and stabilizes cohesin at convergent CTCF sites [38]. Consistent with this model, RNAi-mediated depletion of ESCO1 and CTCF both decreased cohesin’s residence time [38] and SMC3 acetylation [38,76]. Consistent with CTCF and ESCO1 protecting cohesin from WAPL, co-depleting CTCF and WAPL results in the same cohesin residence time as WAPL depletion alone [38]. Taken together, the work of Li et al. and Wutz et al. suggest that CTCF stabilizes cohesin on DNA by counteracting WAPL-mediated release of cohesin from chromatin [38,40]. Potential mechanisms, which may be synergistic, include CTCF binding to the same RAD21-STAG2 interface as WAPL and thereby outcompeting WAPL [40] as well as CTCF-PDS5 mediated recruitment of ESCO1 to acetylate cohesin and thereby stabilize cohesin [38].
While recent work has greatly increased our knowledge of the direct and indirect ways CTCF and cohesin might form a stabilized loop maintenance complex and clarified the relevant domains and amino acids required, it is important to stress that we still do not understand the molecular mechanism. In this regard it is important to note that the three mechanisms discussed here – direct protein-protein interaction, CTCF inactivating cohesin’s ATPase activity and CTCF preventing WAPL-mediated release of cohesin from chromatin – are not mutually exclusive. Rather, they may be synergistic.
Role of an internal RNA-binding region (RBRi) in CTCF in regulating CTCF clustering, target search, and chromatin looping
As an Extra View article to our recent two papers on the functions of an internal RNA-Binding Region (RBRi) in CTCF [36,43], we next discuss these and place them in the context of the proposed multi-step mechanism outlined above (Figure 3). Our studies were motivated by our previous observation that CTCF forms small clusters in mouse and human cells [70]. To understand the mechanism, we took a biochemical approach and generated an mESC line where the two endogenous alleles carry distinct epitope tags (3xFLAG-Halo-CTCF and V5-SNAPf-CTCF). FLAG- and V5-tagged CTCF co-IP’ed consistent with prior studies [99,104,115], but CTCF co-IP was insensitive to DNase but sensitive to RNase A treatment [36,104]. This suggests that CTCF self-associates in a manner that is directly or indirectly mediated by RNA [36,104]. Consistently, treatment with RNase reduces CTCF binding to chromatin [37,116]. The C-terminal Domain of CTCF was originally reported to be required for RNA binding and designated as CTCF’s RNA-Binding Region (RBR) [104,117] and deletion of amino acids 576–614 just C-terminal to the zinc finger domain was reported to drastically decrease RNA binding [104]. We refer to this internal 576–614 region as the RBRi and it largely corresponds to mouse CTCF exon 10, which we homozygously deleted in an mESC line where CTCF has been homozygously Halo-tagged to generate Halo-ΔRBRi-CTCF (Halo-CTCFΔ576-611) [36]. Notably, although the RBRi is physiologically important (the growth rate of ΔRBRi-CTCF mESCs is ~2-fold slower than wt-CTCF mESCs), ΔRBRi-CTCF only showed modestly reduced and not abolished RNA-binding in vivo and in vitro [36]. Consistently, subsequent studies found that ZF1 and ZF10 also contribute to CTCF RNA-Binding and likely bind RNA more strongly than the RBRi [37,118]. It is important to note that whether CTCF-RNA interactions in vivo play more of a structural scaffold role or a direct interaction role remains unclear [119]. Regardless, using super-resolution PALM imaging we found that CTCF clustering is significantly reduced in ΔRBRi-CTCF mESCs, suggesting that CTCF clustering is partially mediated by the RBRi. Given the recent interest in Liquid-Liquid Phase Separation (LLPS) [120], it is worth noting that although CTCF exhibits many features associated with LLPS (Protein-RNA interactions, clustering, intrinsically disordered regions [68]), CTCF clustering is most likely not due to LLPS, at least not in mESCs and human U2OS cells. A key LLPS prediction is that protein over-expression above a critical concentration would cause all additional protein to enter the new phase [120–122]. However, even upon extremely high CTCF over-expression, CTCF remains relatively homogenously distributed in the nucleus [70], ruling out CTCF LLPS. Instead, it is worth noting that many TADs are demarcated by clustered CTCF binding sites [123].
In a parallel project, we had discovered that CTCF exhibits highly anomalous diffusion inside the nucleus [43]. CTCF exists in a DNA bound state (~49% bound to cognate sites and ~19% nonspecifically interacting with chromatin in mESCs) as well as a ‘freely’ diffusing state (~32%) [70]. We found that ‘freely diffusing’ CTCF exhibited anisotropic diffusion, such that following a step in one direction, CTCF is substantially more likely to return backwards. We developed a computational pipeline to analyze CTCF Single-Particle Tracking (SPT) data [43] and found that CTCF’s tendency to ‘return backwards’ primarily manifests itself at ~200 nm displacements. We explored a number of anomalous diffusion models [124,125], and found that the only model that can quantitatively explain our data is a model where CTCF gets transiently trapped in small ~200 nm zones inside the nucleus (typically for ms to tens of ms). We call this model Anisotropic Diffusion through transient Trapping in Zones (ADTZ) [43]. Our SPT data and theory suggested the existence of zones that trap CTCF and we therefore hypothesized that the zones would correspond to CTCF clusters. This is because clustering is due to (in)direct CTCF self-association, and self-association can also explain trapping. If this model is correct, 1) ΔRBRi-CTCF should exhibit strongly reduced anisotropic diffusion at ~200 nm scales and 2) wt-CTCF should exhibit anisotropic displacements predominantly in the vicinity of CTCF clusters. Both predictions were experimentally confirmed [43]. Thus, CTCF exhibits a novel mode of nuclear diffusion (ADTZ), which is likely mediated by CTCF clustering and the RBRi. Functionally, we found that the residence time for binding to cognate DNA sites is unaffected in ΔRBRi-CTCF, but that the specifically bound fraction is reduced. This means that it takes ΔRBRi-CTCF ~2.5-fold longer to find a cognate CTCF DNA-binding site compared to wt-CTCF [43]. Thus, CTCF exhibits a novel ‘facilitated’ or ‘guided’ nuclear target search mechanism where RBRi-mediated interactions accelerate the cognate DNA target search, without affecting the strength of cognate DNA binding itself [43].
Given the roles of the RBRi in mediating CTCF clustering and target search and its physiological importance [36,43], we performed high-resolution Micro-C as well as ChIP-Seq and RNA-Seq to assess 3D genome organization in ΔRBRi-CTCF mESCs [36]. In the context of the multi-step CTCF-cohesin mechanism (Figure 3b), it is interesting that ΔRBRi-CTCF mESCs exhibit only moderate TAD and insulation defects, but that loops/corner peaks were strongly affected: of 14,372 loops, 57% (8,189 loops) and 39% (5,490 loops) were weakened by 1.5- and 2-fold, respectively. Thus, CTCF loops fall into at least two distinct categories, RBRi-independent and RBRi-dependent (Figure 4a-b). And we proposed a model where CTCF clustering might facilitate blocking or pausing of cohesin-mediated extrusion (Figure 4c).
Figure 4.
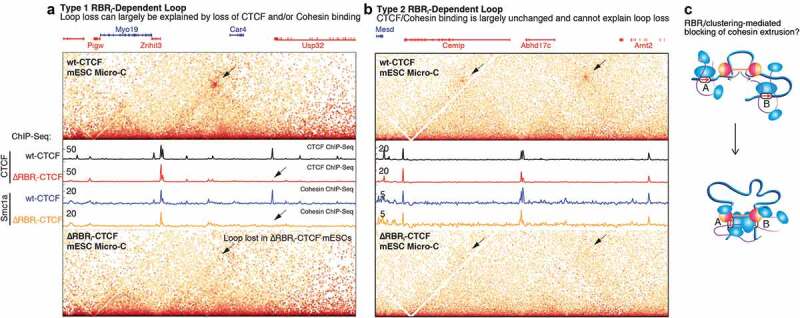
Distinct classes of chromatin loops. (a-b) Micro-C maps and CTCF and Cohesin (Smc1a) ChIP-Seq shown for wt-CTCF mESCs and ΔRBRi-CTCF mESCs, illustrating Type 1 RBRi-dependent loops that can be explained by loss of CTCF/cohesin binding (a) and Type 2 RBRi-dependent loops that cannot be explained by loss of CTCF/cohesin binding (b). (c) Sketch of a role for CTCF clustering in blocking cohesin extrusion. Figures 4a-c are partially reproduced and edited from [36] with permission.
Surprisingly, we found that RBRi-dependent loops can be further subdivided into two classes or types (Figure 4a,b). The loop in Figure 4a is an example of type 1 RBRi-dependent loops, anchored by two CTCF/Cohesin binding sites, at least one of which is lost upon RBRi deletion. The left site is RBRi-independent: CTCF binds equally well with and without the RBRi and cohesin is similarly recruited equally well with and without the RBRi. In contrast, CTCF and cohesin binding at the right-hand CTCF binding site is fully lost after RBRi-deletion in Figure 4a. The right site is thus RBRi-dependent. Since one CTCF-anchor is lost, this straightforwardly explains why the loop is RBRi-dependent and lost. Thus, type 1 losses are RBRi-dependent because at least one anchor is RBRi-dependent [36].
However, we were surprised to find a second category, type 2 RBRi-dependent loops (Figure 4b). Despite being bridged by CTCF/Cohesin anchors, these loops are lost without strong or obvious changes to CTCF/Cohesin binding, as measured by ChIP-Seq (Figure 4b) [36]. Notably, deletion of the ZF1 and ZF10 RBRs produced two similar types of loops: 1) loops lost that could be explained by loss of CTCF binding and 2) loops lost without obvious changes to CTCF binding as measured by ChIP-Seq [37]. Taken together, this suggests that while CTCF/Cohesin binding is necessary for loop formation, it is not sufficient. This means that ChIP-Seq is insufficient to read out the function of CTCF/cohesin mutants. Mechanistically, this suggests that CTCF and cohesin can form a ‘loop-incompetent’ complex, that is nevertheless sufficiently stabilized to generate a clear ChIP-Seq signal. This may explain why not all CTCF sites form loops and TADs. How ‘loop-competent’ and ‘loop-incompetent’ CTCF/Cohesin complexes differ mechanistically is an important question for future studies.
Experimental considerations for studying CTCF-cohesin interactions
Although recent papers have greatly increased our knowledge of how CTCF and cohesin interact, form chromatin loops, and insulate [28,36–42], the seemingly multi-step (Figure 3b-c) nature of the interaction has led to several surprising findings that should inform future studies and which we discuss here. Although CTCF/Cohesin DNA binding (typically measured with ChIP-Seq), TAD insulation and loop/corner peak formation are clearly related, it was found that they can be decoupled to a surprising extent [28,36–42]. First, loops/corner peaks can be disrupted without obvious changes to CTCF/cohesin binding as measured by ChIP-Seq [36,37] (Figure 4b). Second, loops/corner peaks can be near-completely disrupted with only moderate effects on TADs and insulation scores [36,37,40]. This point was clearly demonstrated by Li et al. in their Hi-C maps of mutant CTCF (Y226A, F228A HAP1 cells (Figure 5) [40]. Loops were almost entirely lost (from 2,756 loops to just 98 in mutant cells), yet, although TADs and insulation were clearly weakened, the effect was much more modest. Thus, loop-resolution Hi-C or Micro-C is necessary to distinguish effects on TADs and loops [36,37,40]. Similarly, since some CTCF mutants affect only a subset of loops [36,37], genome-wide analyses are necessary. In conclusion therefore, CTCF and Cohesin ChIP-Seq combined with loop-resolution Hi-C or Micro-C is required to fully ascertain the function of CTCF/Cohesin mutants, which unfortunately makes these experiments rather expensive.
Figure 5.
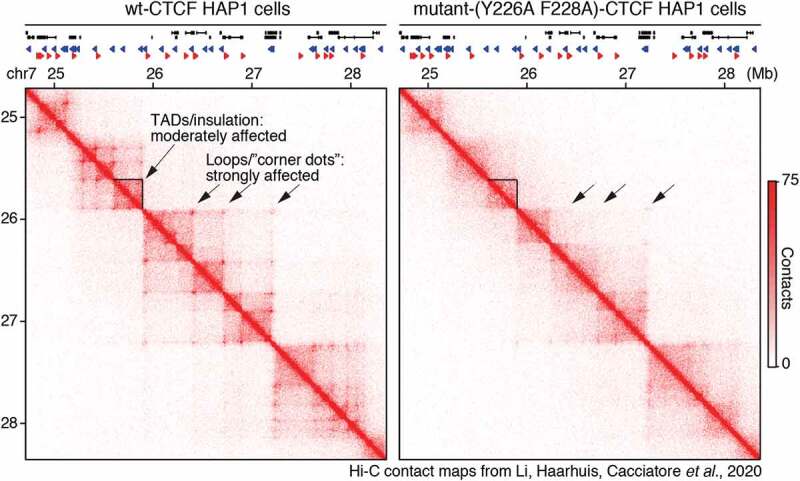
CTCF-mediated loops can be disrupted with only modest effects on TADs/insulation. (a-b) Hi-C contact matrices at 10 kb resolution of the HOXA locus in wt-CTCF and Y226A/F228A-CTCF HAP1 cells from [40].
Figures 5 is partially reproduced and edited from [40] with permission.
Moreover, since CTCF-null mice are embryonic lethal [126,127], studying CTCF mutants is experimentally challenging. One approach is generating endogenous mutants using genome-editing [36,40,43]. Advantages of this approach include: 1) it is clean – there is no endogenous protein left and 2) effects on physiology (e.g. growth rate, differentiation) are easily assessed; 3) gene expression is driven by endogenous regulatory elements. However, disadvantages include: 1) cannot study lethal mutations; 2) cannot distinguish primary from secondary effects; 3) has a low throughput. Thus, an alternative approach is to express wt- or mutant-CTCF in an inducible degron line (e.g. AID-tagged CTCF [28,29]) [37,41,42] or alternatively in a CTCF-mutant background [39] (though this is challenging to generalize and leads to co-expression of two versions of CTCF). The inducible complementation approach [37,41,42] is more general and 1) allows studying both lethal and non-lethal mutants; 2) allows some distinction between direct and secondary effects; 3) allows higher throughput. However, disadvantages include: 1) some AID-clones tend to have significant residual wt-protein; 2) it can difficult to achieve physiological expression. For the second point, it is important to consider both the mean and variance of the expression level distribution. E.g. in the inducible complementation system [41,42], the mean CTCF transgene expression was ~10-fold lower than endogenous CTCF level (~217,200 [65] vs. ~21,720 CTCF proteins per mESC). But the variance of transgene expression was also much greater (seemingly ~10-fold by FACS [41]). Greatly altered expression levels impairs our ability to distinguish effects of mutants from altered expression level. Therefore, since the inducible complementation approach is more generally applicable, an important future step will be to develop a next-generation version where the mean and variance of transgene expression matches those of endogenous CTCF. Ensuring endogenous expression levels will likely be especially important for cohesin, since overexpression of just one sub-unit causes the overexpressed subunit to primarily exist as monomers instead of being incorporated into cohesin complexes [70] and improper dosage of cohesin sub-units or regulatory proteins cause a range of diseases known as cohesinopathies [128].
Conclusion and outlook
The 3D genome field is moving at an incredible pace and the last year alone has seen a number of important breakthroughs including: 1) the direct observation of cohesin extrusion in vitro for the first time [21–23], which strongly substantiates the loop extrusion model; 2) great new insights suggesting a multi-step CTCF-cohesin interaction mechanism to explain the convergent rule [28,36–42]; 3) the first structural insights into the CTCF-cohesin interaction [40]; 4) the development and application to mammalian cells of a chromosome conformation capture technique, Micro-C [36,129,130], which is capable of capturing enhancer-promoter interactions relevant to gene regulation that are otherwise largely invisible in Hi-C [131]. We end by highlighting a subset of the key challenges that lie ahead.
First, although the recent studies discussed here have shed great light on the interaction between CTCF and cohesin, suggested a multi-step mechanism for inhibition of loop extrusion, and revealed some of the necessary proteins and domains [28,36–42], we still do not understand the molecular mechanism. Furthermore, although referred to as the ‘convergent rule’, not all convergent CTCF sites form corner dots in Hi-C maps, and not all corner dots are bridged by convergent CTCF sites [11,31,32,109]. The development of in vitro [21,23] and cell-free extract [22] systems for loop extrusion now makes it possible to study the role of regulatory proteins (e.g. CTCF, WAPL, ESCO1, PDS5, nucleosomes, etc.) and mutants to probe the molecular mechanism of loop extrusion and its inhibition. Such approaches are likely to be particularly informative over the next few years.
Second, our mechanistic understanding is severely hampered by the current lack of structural insights. Full structural elucidation of full-length CTCF, cohesin and cohesin regulatory proteins at each step of the loop extrusion, pausing and stabilization process would yield profound insights.
Third, translation of these insights from in vitro to in vivo will be crucial, and the development and application of super-resolution live-cell imaging approaches will be necessary here.
Fourth, although TADs and loops were initially reported to be cell-type invariant [11,132], it has now become clear that cell differentiation is associated with widespread changes to TAD and loop organization [133–140]. Consistently, mutating RBRs in CTCF only disrupts a subset of loops [36,37] and CTCF binding can be regulated [141,142]. Understanding how and to which extent cell-type specific regulation of CTCF/cohesin-mediated chromatin looping takes place and is instructive for gene regulation is another important aspect to clarify in the future.
Fifth and along those lines, the roles of CTCF, cohesin and TADs in regulating gene expression remain controversial [143] and, although the experimental systems differ, the studies of CTCF mutants discussed here reported deregulation of several hundreds to a couple of thousand genes [36,37,39–41]. Distinguishing the roles of CTCF, cohesin and TADs in maintaining gene expression from establishing/inducing gene expression may be informative here [144,145].
Sixth, Micro-C has recently revealed the existence of ‘micro domains’ at the scale of kb to tens of kb, which are frequently anchored by proteins other than CTCF/cohesin including the transcriptional machinery [129,130]. Understanding the structure, regulation and function of these ‘micro domains’ will be important.
In summary, recent studies now suggest a multi-step mechanism for the interaction of CTCF and cohesin to explain aspects of the convergent rule [28,36–42], though we are likely still years away from understanding the molecular mechanisms. The 3D genome field shows no sign of slowing down and promises to be a particularly exciting field to continue to disentangle over the coming years.
Acknowledgments
The CTCF-cohesin interaction models were inspired by discussions at the MIT Chromosome Club between the Mirny, Vos and Hansen labs. I am grateful to Seychelle Vos, Claudia Cattoglio, Tsung-Han Stanley Hsieh, James Rhodes, Michele Gabriele, Viraat Goel, and Jin Harvey Yang for insightful comments on drafts. I would like to thank Iain Finley Davidson for discussions regarding the Holzmann 2019 calculations in Figure 2d and thank Benjamin Rowland, Daniel Panne, Elzo de Wit, and Kyle Muir for kindly providing the Hi-C map file used in Figure 5. This work was supported by the National Institutes of Health under Grant R00GM130896.
Funding Statement
This work was supported by the National Institutes of Health under Grant R00GM130896;National Institute of General Medical Sciences [R00GM130896].
Extra view
Hansen AS, Hsieh THS, Cattoglio C, Pustova I, Saldana-Meyer R, Reinberg D, Darzacq X, Tjian R. Distinct Classes of Chromatin Loops Revealed by Deletion of an RNA-Binding Region in CTCF. Molecular Cell, 2019, doi:10.1016/j.molcel.2019.07.039
And also
Hansen AS; Amitai A; Cattoglio C; Tjian R; Darzacq X. Guided nuclear exploration increases CTCF target search efficiency. Nature Chemical Biology, 2019, doi:10.1038/s41589-019-0422-3
Disclosure statement
I declare that no competing financial interests exist.
References
- [1].Cremer T, Cremer C.. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet. 2001;2(4):292–301. [DOI] [PubMed] [Google Scholar]
- [2].Luger K, Mäder AW, Richmond RK, et al. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389(6648):251–260. [DOI] [PubMed] [Google Scholar]
- [3].Nuebler J, Fudenberg G, Imakaev M, et al. Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proceed Nat Acad Sci. 2018;115(29):E6697–E6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lieberman-Aiden E, van Berkum NL, Williams L, et al. Science. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hildebrand EM, Dekker J. Mechanisms and functions of chromosome compartmentalization. Trends Biochem Sci. 2020;45(5):385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen Y, Zhang Y, Wang Y, et al. Mapping 3D genome organization relative to nuclear compartments using TSA-Seq as a cytological ruler. J Cell Biol. 2018;217(11):4025–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chen Y, Belmont AS. Genome organization around nuclear speckles. Curr Opin Genet Dev. 2019;55:91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Guelen L, Pagie L, Brasset E, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453(7197):948–951. [DOI] [PubMed] [Google Scholar]
- [9].Rowley MJ, Corces VG. Organizational principles of 3D genome architecture. Nat Rev Genet. 2018;19(12):789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Quinodoz SA, Ollikainen N, Tabak B, et al. Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell. 2018;174(744–757):e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rao SSP, Huntley MH, Durand NC, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159(7):1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Falk M, Feodorova Y, Naumova N, et al. Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature. 2019;570(7761):395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sati S, Bonev B, Szabo Q, et al. 4D Genome Rewiring during Oncogene-Induced and Replicative Senescence. Mol Cell. 2020;78(3):522–538.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rosencrance CD, Ammouri HN, Yu Q, et al. Chromatin hyperacetylation impacts chromosome folding by forming a nuclear subcompartment. Mol Cell. 2020;78(112–126):e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Erdel F, Rippe K. Formation of chromatin subcompartments by phase separation. Biophys J. 2018;114(10):2262–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Alipour E, Marko JF. Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Res. 2012;40(22):11202–11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fudenberg G, Imakaev M, Lu C, et al. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016;15(9):2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sanborn AL, Rao SSP, Huang SC, et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A. 2015;112(47):E6456-E6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Riggs AD. DNA methylation and late replication probably aid cell memory, and type I DNA reeling could aid chromosome folding and enhancer function. Philos. Trans. R. Soc. London. B, Biol. Sci. 1990;326:285–297. [DOI] [PubMed] [Google Scholar]
- [20].Yatskevich S, Rhodes J, Nasmyth K. Organization of Chromosomal DNA by SMC Complexes. Annu. Rev. Genet. 2019;53(1):445–482. [DOI] [PubMed] [Google Scholar]
- [21].Kim Y, Shi Z, Zhang H, et al. Human cohesin compacts DNA by loop extrusion. Science. 2019;366(6471):1345–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Golfier S, Quail T, Kimura H, et al. Cohesin and condensin extrude DNA loops in a cell-cycle dependent manner. Elife. 2020;9:e53885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Davidson IF, Bauer B, Goetz D, et al. DNA loop extrusion by human cohesin. Science. 2019;366(6471):1338–1345. [DOI] [PubMed] [Google Scholar]
- [24].Hansen AS, Cattoglio C, Darzacq X, et al. Recent evidence that TADs and chromatin loops are dynamic structures. Nucleus. 2018;9(1):20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Schwarzer W, Abdennur N, Goloborodko A, et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature. 2017;551(7678):51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rao SSP, Huang SC, Glenn St Hilaire B, et al. Cohesin loss eliminates all loop domains. Cell. 2017;171(2):305–320.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gassler J, Brandão HB, Imakaev M, et al. A mechanism of cohesin-dependent loop extrusion organizes zygotic genome architecture. Embo J. 2017;36(24):3600–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wutz G, Várnai C, Nagasaka K, et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. Embo J. 2017;36(24):3573–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nora EP, Goloborodko A, Valton AL, et al. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell. 2017;169(5):930–944.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Beagan JA, Phillips-Cremins JE. On the existence and functionality of topologically associating domains. Nat Genet. 2020;52(1):8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].de Wit E, Vos ESM, Holwerda SJB, et al. Determines Chromatin Looping. Mol Cell. 2015;60(4):676–684. [DOI] [PubMed] [Google Scholar]
- [32].Guo Y, Xu Q, Canzio D, et al. CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell. 2015;162(4):900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Vietri Rudan M, Barrington C, Henderson S, et al. Comparative Hi-c reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep. 2015;10(8):1297–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].van Bemmel JG, Galupa R, Gard C, et al. The bipartite TAD organization of the X-inactivation center ensures opposing developmental regulation of Tsix and Xist. Nat Genet. 2019;51(6):1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nagy G, Czipa E, Steiner L, et al. Motif oriented high-resolution analysis of ChIP-seq data reveals the topological order of CTCF and cohesin proteins on DNA. BMC Genomics. 2016;17(1):637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hansen AS, Hsieh T-HS, Cattoglio C, et al. Distinct classes of chromatin loops revealed by deletion of an RNA-binding region in CTCF. Mol Cell. 2019;76(3):395–411.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Saldaña-Meyer R, Rodriguez-Hernaez J, Escobar T, et al. Essential for CTCF-mediated genome organization. Mol Cell. 2019;76(3):412–422.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wutz G, Ladurner R, St Hilaire BG, et al. ESCO1 and CTCF enable formation of long chromatin loops by protecting cohesinSTAG1 from WAPL. Elife. 2020;9:e52091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pugacheva EM, Kubo N, Loukinov D, et al. CTCF mediates chromatin looping via N-terminal domain-dependent cohesin retention. Proc Natl Acad Sci. 2020; 117(4):2020–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Li Y, Haarhuis JHI, Sedeño Cacciatore Á, et al. The structural basis for cohesin–CTCF-anchored loops. Nature. 2020;578(7795):472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nishana M, Rodriguez-Hernaez J, Ha C, et al. Defining the relative and combined contribution of CTCF and CTCFL to genomic regulation. Genome Biol. 2020;21:1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nora EP, Caccianini L, Fudenberg G, et al. Molecular basis of CTCF binding polarity in genome folding. bioRxiv. 2019. https://www.biorxiv.org/content/10.1101/2019.12.13.876177v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hansen AS, Amitai A, Cattoglio C, et al. Guided nuclear exploration increases CTCF target search efficiency. Nat Chem Biol. 2019. DOI: 10.1038/s41589-019-0422-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fudenberg G, Abdennur N, Imakaev M, et al. Emerging evidence of chromosome folding by loop extrusion. Cold Spring Harb Symp Quant Biol. 2017;82:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Dekker J, Mirny L. The 3D genome as moderator of chromosomal communication. Cell. 2016;164(6):1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kempfer R, Pombo A. Methods for mapping 3D chromosome architecture. Nat Rev Genet. 2019. DOI: 10.1038/s41576-019-0195-2. [DOI] [PubMed] [Google Scholar]
- [47].Gruber S, Haering CH, Nasmyth K. Chromosomal Cohesin Forms a Ring. Cell. 2003;112(6):765–777. [DOI] [PubMed] [Google Scholar]
- [48].Ouyang Z, Yu H. Releasing the cohesin ring: A rigid scaffold model for opening the DNA exit gate by Pds5 and Wapl. BioEssays. 2017;39(4):1600207. [DOI] [PubMed] [Google Scholar]
- [49].Kikuchi S, Borek DM, Otwinowski Z, et al. Crystal structure of the cohesin loader Scc2 and insight into cohesinopathy. Proceed Nat Acad Sci. 2016;113(44):12444–12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wells JN, Gligoris TG, Nasmyth KA, et al. Evolution of condensin and cohesin complexes driven by replacement of Kite by Hawk proteins. Curr Biol. 2017;27(1):R17–R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Casa V, Moronta Gines M, Gade Gusmao E, et al. Redundant and specific roles of cohesin STAG subunits in chromatin looping and transcriptional control. Genome Res. 2020;30(4):515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Remeseiro S, Cuadrado A, Gómez-López G, et al. A unique role of cohesin-SA1 in gene regulation and development. Embo J. 2012;31(9):2090–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kojic A, Cuadrado A, De Koninck M, et al. Distinct roles of cohesin-SA1 and cohesin-SA2 in 3D chromosome organization. Nat Struct Mol Biol. 2018;25(6):496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Higashi TL, Eickhoff P, Simoes JS, et al. A Structure-Based Mechanism for DNA Entry into the Cohesin Ring. BioRxiv. 2020. https://www.biorxiv.org/content/10.1101/2020.04.21.052944v12020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Shi Z, Gao H, Bai X, et al. Cryo-EM structure of the human cohesin-NIPBL-DNA complex. Science. 2020;eabb0981. DOI: 10.1126/science.abb0981 [DOI] [PubMed] [Google Scholar]
- [56].Ciosk R, Shirayama M, Shevchenko A, et al. Cohesin’s binding to chromosomes depends on a separate complex consisting of Scc2 and Scc4 proteins. Mol Cell. 2000;5(2):243–254. [DOI] [PubMed] [Google Scholar]
- [57].Gandhi R, Gillespie PJ, Hirano T. Human wapl is a cohesin-binding protein that promotes sister-chromatid resolution in mitotic prophase. Curr Biol. 2006;16(24):2406–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kueng S, Hegemann B, Peters BH, et al. Wapl controls the dynamic association of cohesin with chromatin. Cell. 2006;127(5):955–967. [DOI] [PubMed] [Google Scholar]
- [59].Ouyang Z, Zheng G, Tomchick DR, et al. Structural basis and IP6 requirement for Pds5-dependent cohesin dynamics. Mol Cell. 2016;62(2):248–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chapard C, Jones R, van Oepen T, et al. Sister DNA entrapment between juxtaposed smc heads and kleisin of the cohesin complex. Mol Cell. 2019;75(224–237):e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Hassler M, Shaltiel IA, Haering CH. Towards a unified model of SMC complex function. Curr Biol. 2018;28(21):R1266–R1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Skibbens RV. Condensins and cohesins – one of these things is not like the other! J Cell Sci. 2019;132(3):jcs220491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Nichols MH, Corces VG. A tethered-inchworm model of SMC DNA translocation. Nat Struct Mol Biol. 2018;25(10):906–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Marko JF, De Los Rios P, Barducci A, et al. DNA-segment-capture model for loop extrusion by structural maintenance of chromosome (SMC) protein complexes. Nucleic Acids Res. 2019;47(13):6956–6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Cattoglio C, Pustova I, Walther N, et al. Determining cellular CTCF and cohesin abundances to constrain 3D genome models. Elife. 2019;8:e40164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ong C-T, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet. 2014;15(4):234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Lobanenkov VV, Nicolas RH, Adler VV, et al. A novel sequence-specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5′-flanking sequence of the chicken c-myc gene. Oncogene. 1990;5(12):1743–1753. [PubMed] [Google Scholar]
- [68].Martinez SR, Miranda JJL. CTCF terminal segments are unstructured. Protein Sci. 2010;19(5):1110–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bonchuk A, Kamalyan S, Mariasina S, et al. N-terminal domain of the architectural protein CTCF has similar structural organization and ability to self-association in bilaterian organisms. Sci Rep. 2020;10(1):2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hansen AS, Pustova I, Cattoglio C, et al. CTCF and cohesin regulate chromatin loop stability with distinct dynamics. Elife. 2017;6:e25776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kim TH, Abdullaev ZK, Smith AD, et al. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128(6):1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Nakahashi H, Kwon K-RK, Resch W, et al. A genome-wide map of CTCF multivalency redefines the CTCF code. Cell Rep. 2013;3(5):1678–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Rubio ED, Reiss DJ, Welcsh PL, et al. CTCF physically links cohesin to chromatin. Proceed Nat Acad Sci. 2018;105(24):8309–8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Wendt KS, Yoshida K, Itoh T, et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451(7180):796–801. [DOI] [PubMed] [Google Scholar]
- [75].Parelho V, Hadjur S, Spivakov M, et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. 2008;132(3):422–433. [DOI] [PubMed] [Google Scholar]
- [76].Busslinger GA, Stocsits RR, van der Lelij P, et al. Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl. Nature. 2017;544(7651):503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Banigan EJ, van den Berg AA, Brandão HB, et al. Chromosome organization by one-sided and two-sided loop extrusion. Elife. 2020;9: DOI: 10.7554/elife.53558.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Brandão HB, Paul P, van den Berg AA, et al. RNA polymerases as moving barriers to condensin loop extrusion. Proceedings of the National Academy of Sciences. 2019;116(41):20489–20499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Heinz S, Texari L, Hayes MGB, et al. Transcription elongation can affect genome 3D structure. Cell. 2018;174(1522–1536):e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Olan I, Parry AJ, Schoenfelder S, et al. Transcription-driven cohesin repositioning rewires chromatin loops in cellular senescence. bioRxiv. 2019;823–831. https://www.biorxiv.org/content/10.1101/823831v1.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Gibcus JH, Samejima K, Goloborodko A, et al. A pathway for mitotic chromosome formation. Science. 2018;359(6376):eaao6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Wang X, Brandão HB, Le TBK, et al. Bacillus subtilis SMC complexes juxtapose chromosome arms as they travel from origin to terminus. Science. 2017;355(6324):524–527.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Holzmann J, Politi AZ, Nagasaka K, et al. Absolute quantification of cohesin, CTCF and their regulators in human cells. Elife. 2019;8:e46269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Nasmyth K. Cohesin: a catenase with separate entry and exit gates? Nat Cell Bio. 2011;13(10):1170–1177. [DOI] [PubMed] [Google Scholar]
- [85].Zhang N, Kuznetsov SG, Sharan SK, et al. A handcuff model for the cohesin complex. J Cell Biol. 2008;183(6):1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Agarwal H, Reisser M, Wortmann C, et al. Direct observation of cell-cycle-dependent interactions between CTCF and Chromatin. Biophys J. 2017;112(10):2051–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Kieffer-Kwon K-R, Nimura K, Rao SSP, et al. Myc regulates chromatin decompaction and nuclear architecture during B cell activation. Mol Cell. 2017;67(566–578):e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Teves SS, An L, Hansen AS, et al. A dynamic mode of mitotic bookmarking by transcription factors. Elife. 2016;5:e22280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Mazza D, Abernathy A, Golob N, et al. A benchmark for chromatin binding measurements in live cells. Nucleic Acids Res. 2012;40(15):e119–e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Davidson IF, Goetz D, Zaczek MP, et al. Rapid movement and transcriptional re-localization of human cohesin on DNA. Embo J. 2016;35(24):2671–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Clarkson CT, Deeks EA, Samarista R, et al. CTCF-dependent chromatin boundaries formed by asymmetric nucleosome arrays with decreased linker length. Nucleic Acids Res. 2019;47(21):11181–11196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Fu Y, Sinha M, Peterson CL, et al. The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLOS Genet. 2008;4(7):e1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Teif VB, Beshnova DA, Vainshtein Y, et al. Nucleosome repositioning links DNA (de)methylation and differential CTCF binding during stem cell development. Genome Res. 2014;24(8):1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Kelly TK, Liu Y, Lay FD, et al. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012;22(12):2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Owens N, Papadopoulou T, Festuccia N, et al. CTCF confers local nucleosome resiliency after DNA replication and during mitosis. Elife. 2019;8:e47898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].MacPherson MJ, Sadowski PD. The CTCF insulator protein forms an unusual DNA structure. BMC Mol. Biol. 2010;11(1):101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Zirkel A, Nikolic M, Sofiadis K, et al. HMGB2 loss upon senescence entry disrupts genomic organization and induces CTCF clustering across cell types. Mol Cell. 2018;70(730–744):e6. [DOI] [PubMed] [Google Scholar]
- [98].MacPherson MJ, Beatty LG, Zhou W, et al. The CTCF insulator protein is posttranslationally modified by SUMO. Mol Cell Biol. 2009;29(3):714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Yu W, Ginjala V, Pant V, et al. Poly(ADP-ribosyl)ation regulates CTCF-dependent chromatin insulation. Nat Genet. 2004;36(10):1105–1110. [DOI] [PubMed] [Google Scholar]
- [100].Farrar D, Rai S, Chernukhin I, et al. Mutational analysis of the poly (ADP-ribosyl) ation sites of the transcription factor CTCF provides an insight into the mechanism of its regulation by poly (ADP-ribosyl) ation. Mol Cell Biol. 2010;30(5):1199–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Zhang Y, Wang J, Ding M, et al. Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat Methods. 2013;10(10):981–984. [DOI] [PubMed] [Google Scholar]
- [102].Racko D, Benedetti F, Dorier J, et al. Are TADs supercoiled? Nucleic Acids Res. 2018;47(2):521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Xiao T, Wallace J, Felsenfeld G. Specific sites in the C terminus of CTCF interact with the SA2 subunit of the cohesin complex and are required for cohesin-dependent insulation activity. Mol Cell Biol. 2011;31(11):2174–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Saldaña-Meyer R, González-Buendía E, Guerrero G, et al. CTCF regulates the human p53 gene through direct interaction with its natural antisense transcript, Wrap53. Genes Dev. 2014;28(7):723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Li J, Huang K, Hu G, et al. An alternative CTCF isoform antagonizes canonical CTCF occupancy and changes chromatin architecture to promote apoptosis. Nat Commun. 2019;10(1):1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Justice M, Carico ZM, Stefan HC, et al. A WIZ/Cohesin/CTCF complex anchors DNA loops to define gene expression and cell identity. Cell Rep. 2020;31(2):107503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Tedeschi A, Wutz G, Huet S, et al. Wapl is an essential regulator of chromatin structure and chromosome segregation. Nature. 2013;501(7468):564–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Haarhuis JHI, van der Weide RH, Blomen VA, et al. The Cohesin Release Factor WAPL Restricts Chromatin Loop Extension. Cell. 2017;169(693–707):e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Tang Z, Luo OJ, Li X, et al. CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell. 2015;163(7):1611–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Murayama Y, Uhlmann F. Biochemical reconstitution of topological DNA binding by the cohesin ring. Nature. 2014;505(7483):367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Rhodes J, Mazza D, Nasmyth K, et al. Scc2/Nipbl hops between chromosomal cohesin rings after loading. Elife. 2017;6:e30000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Kanke M, Tahara E, Huis in’t Veld PJ, et al. Cohesin acetylation and Wapl-Pds5 oppositely regulate translocation of cohesin along DNA. Embo J. 2016;35(24):2686–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Alomer RM, da Silva EML, Chen J, et al. Esco1 and Esco2 regulate distinct cohesin functions during cell cycle progression. Proceed Nat Acad Sci. 2017;114(37):9906–9911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Minamino M, Ishibashi M, Nakato R, et al. Esco1 acetylates cohesin via a mechanism different from that of esco2. Curr Biol. 2015;25(13):1694–1706. [DOI] [PubMed] [Google Scholar]
- [115].Yusufzai TM, Tagami H, Nakatani Y, et al. CTCF tethers an insulator to subnuclear sites, suggesting shared insulator mechanisms across species. Mol Cell. 2004;13(2):291–298. [DOI] [PubMed] [Google Scholar]
- [116].Thakur J, Fang H, Llagas T, et al. Architectural RNA is required for heterochromatin organization. bioRxiv. 2019;784835. https://www.biorxiv.org/content/10.1101/784835v1. [Google Scholar]
- [117].Kung JT, Kesner B, An JY, et al. Locus-specific targeting to the X chromosome revealed by the RNA interactome of CTCF. Mol Cell. 2015;57(2):361–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].He C, Sidoli S, Warneford-Thomson R, et al. High-resolution mapping of RNA-binding regions in the nuclear proteome of embryonic stem cells. Mol Cell. 2016;64(2):416–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Barutcu AR, Blencowe BJ, Rinn JL. Differential contribution of steady-state RNA and active transcription in chromatin organization. EMBO Rep. 2019;20(10):e48068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].McSwiggen DT, Mir M, Darzacq X, et al. Evaluating phase separation in live cells: diagnosis, caveats, and functional consequences. Genes Dev. 2019;33(23–24):1619–1634. available at: http://genesdev.cshlp.org/content/early/2019/10/08/gad.331520.119.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Banani SF, Lee HO, Hyman AA, et al. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017;18(5):285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Shin Y, Brangwynne CP. Liquid phase condensation in cell physiology and disease. Science. 2017;357(6357):eaaf4382. [DOI] [PubMed] [Google Scholar]
- [123].Kentepozidou E, Aitken SJ, Feig C, et al. Clustered CTCF binding is an evolutionary mechanism to maintain topologically associating domains. Genome Biol. 2020;21:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Amitai A. Chromatin configuration affects the dynamics and distribution of a transiently interacting protein. Biophys J. 2018;114(4):766–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Metzler R, Jeon J-H, Cherstvy AG, et al. Anomalous diffusion models and their properties: non-stationarity, non-ergodicity, and ageing at the centenary of single particle tracking. Phys Chem Chem Phys. 2014;16:24128–24164. [DOI] [PubMed] [Google Scholar]
- [126].Heath H, de Almeida CR, Sleutels F, et al. CTCF regulates cell cycle progression of αβ T cells in the thymus. Embo J. 2008;27(21):2839–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Fedoriw AM, Stein P, Svoboda P, et al. Transgenic RNAi reveals essential function for CTCF in H19 gene imprinting. Science. 2004;303(5655):238–240. [DOI] [PubMed] [Google Scholar]
- [128].Banerji R, Skibbens RV, Iovine MK. How many roads lead to cohesinopathies? Dev Dyn. 2017;246(11):881–888. [DOI] [PubMed] [Google Scholar]
- [129].Hsieh T-HS, Cattoglio C, Slobodyanyuk E, et al. Resolving the 3D landscape of transcription-linked mammalian chromatin folding. Mol Cell. 2020;78(3):539–553.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Krietenstein N, Abraham S, Venev SV, et al. Ultrastructural details of mammalian chromosome architecture. Mol Cell. 2020;78(3):554–565.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Gasperini M, Hill AJ, McFaline-Figueroa JL, et al. Framework for mapping gene regulation via cellular genetic screens. Cell. 2019;176(377–390):e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Dixon JR, Selvaraj S, Yue F, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485(7398):376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Bonev B, Mendelson Cohen N, Szabo Q, et al. Multiscale 3D genome rewiring during mouse neural development. Cell. 2017;171(557–572):e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Pękowska A, Klaus B, Xiang W, et al. Gain of CTCF-anchored chromatin loops marks the exit from naive pluripotency. Cell Syst. 2018;7(482–495):e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Beagan JA, Duong MT, Titus KR, et al. YY1 and CTCF orchestrate a 3D chromatin looping switch during early neural lineage commitment. Genome Res. 2017;27(7):1139–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Stadhouders R, Vidal E, Serra F, et al. Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat Genet. 2018;50(2):238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Di Giammartino DC, Kloetgen A, Polyzos A, et al. KLF4 is involved in the organization and regulation of pluripotency-associated three-dimensional enhancer networks. Nat Cell Bio. 2019;21(10):1179–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Phillips-Cremins JE, Sauria MEG, Sanyal A, et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153(6):1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Barrington C, Georgopoulou D, Pezic D, et al. Enhancer accessibility and CTCF occupancy underlie asymmetric TAD architecture and cell type specific genome topology. Nat Commun. 2019;10(1):2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Niu L, Shen W, Shi Z, et al. Systematic Chromatin Architecture Analysis in Xenopus tropicalis Reveals Conserved Three-Dimensional Folding Principles of Vertebrate Genomes. bioRxiv. 2019. https://www.biorxiv.org/content/10.1101/2020.04.02.021378v1. [Google Scholar]
- [141].Kaaij LJT, Mohn F, van der Weide RH, et al. The ChAHP complex counteracts chromatin looping at CTCF sites that emerged from SINE expansions in mouse. Cell. 2019;178(1437–1451):e14. [DOI] [PubMed] [Google Scholar]
- [142].Barisic D, Stadler MB, Iurlaro M, et al. SWI/SNF selectively mediate binding of distinct transcription factors. Nature. 2019;569(7754):136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Mir M, Bickmore W, Furlong EEM, et al. Chromatin topology, condensates and gene regulation: shifting paradigms or just a phase? Development. 2019;146(19):dev182766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Cuartero S, Weiss FD, Dharmalingam G, et al. Control of inducible gene expression links cohesin to hematopoietic progenitor self-renewal and differentiation. Nat Immunol. 2018;19(9):932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Ibrahim DM, Mundlos S. The role of 3D chromatin domains in gene regulation: a multi-facetted view on genome organization. Curr Opin Genet Dev. 2020;61:1–8. [DOI] [PubMed] [Google Scholar]