

Abstract
The upstream stimulatory factor 2 (USF2) is a transcription factor implicated in several cellular processes and among them, tumor development seems to stand out. However, the data with respect to the role of USF2 in tumor development are conflicting suggesting that it acts either as tumor promoter or suppressor. Here we show that absence of USF2 promotes proliferation and migration. Thereby, we reveal a previously unknown function of USF2 in mitochondrial homeostasis. Mechanistically, we demonstrate that deficiency of USF2 promotes survival by inducing mitophagy in a ROS-sensitive manner by activating both ERK1/2 and AKT.
Altogether, this study supports USF2′s function as tumor suppressor and highlights its novel role for mitochondrial function and energy homeostasis thereby linking USF2 to conditions such as insulin resistance, type-2 diabetes mellitus, and the metabolic syndrome.
Keywords: Upstream stimulatory factor 2 (USF2), Proliferation, Migration, Compromised mitochondria, Mitophagy
1. Introduction
The upstream stimulatory factors (USFs), USF1 and USF2, were first identified by their ability to bind the major late adenovirus promoter and were isolated from Hela cell nuclear extracts [1]. In mammals, USFs are encoded by two distinct ubiquitously expressed genes [2] and additionally the alternative splicing of USF2 pre-mRNA gives rise to USF2a and USF2b [3]. All USF proteins belong to the basic helix-loop-helix leucine zipper transcription factor family and act as either homodimer or heterodimer [4] by binding to E-boxes of the DNA-core sequence (5′-CANNTG-3′) in their target genes [4]. The USFs appear to play crucial roles as either transcriptional activators or transcriptional repressors of various genes involved in the stress and immune response, energy metabolism, circadian rhythm as well as cell growth and development [[5], [6], [7]].
From the USFs, USF2 seems to have a more dominant role than USF1. While USF1 knockout mice appear to be normal [8,9], about 50% of the mice deficient for USF2 die shortly after birth and surviving littermates remain smaller than wild-type littermates throughout their entire lifetime [8,9]. Mice deficient in both USF1 and USF2 die during early embryogenesis [8,9]. Thus, these findings suggest that especially USF2 may be important for promoting cellular growth. However, these findings are contrasted by studies showing that USF2 can act as growth suppressor in non-tumorigenic cells as well as in several tumor cells [[10], [11], [12], [13]]. Another layer of complexity comes from findings showing that USF2 may also act as a tumor promoter [14]. Hence, a complete view about USF2's action at the cellular level has not been reached. To overcome this knowledge gap and to avoid the complexity of an organ/tissue interplay as well as the varying mutational background of cancer cells we used mouse embryonic fibroblasts (MEFs) to study the effect of USF2 loss-of-function. We found that MEFs lacking USF2 have a proliferative advantage despite having compromised mitochondria which are mainly cleared through mitophagy in a redox-sensitive manner.
2. Results
2.1. Lack of USF2 affects the size of the nuclear area
To better understand the role of USF2, we generated USF2 deficient MEFs (ΔUSF2) by using the CRISPR-Cas9 technology (Fig. 1A). To investigate whether the USF2 knockout affects cell morphology we performed crystal violet staining. Both control (scrambled) and ΔUSF2 cell lines showed little or no difference in cell shape and size being either rounded or less commonly elongated (Fig. 1B). However, the ΔUSF2 MEFs appeared to display a reduced size of the nuclear area. To quantify the difference, we performed high-content image screens with the Operetta system and found a reduction in the nuclear area size of the ΔUSF2 cells when compared to control cells whereas nuclear roundness was not affected by the lack of USF2. The reduction in nuclear size in the ΔUSF2 cells could be rescued upon re-expression of USF2 in those cells (ΔUSF2+USF2) (Supplementary Fig. 1A). Thus, lack of USF2 reduces nuclear size which could be indicative for an altered proliferative programme.
Fig. 1.

Lack of USF2 in cells affects morphology, proliferation and migration. (A) Representative Western blot analysis of USF2 protein levels in control (scrambled), USF2-deficient (ΔUSF2) and ΔUSF2 MEFs re-expressing a V5-tagged USF2 (ΔUSF2+USF2). (B) Light microscopy images of control, ΔUSF2 and ΔUSF2+USF2 MEFs stained with crystal violet. (C) Real-time proliferation rate of control, ΔUSF2 and ΔUSF2+USF2-MEFs. *significant difference, p ≤ 0.05. (D) Cell cycle distribution of control and ΔUSF2 cells. Histograms display cells in the G1 (blue fraction), S (yellow), and G2/M (green) phase of the cell cycle. (E) Quantification of cell cycle distribution. *significant difference control vs ΔUSF2 cells, p ≤ 0.05. (F, G) Apoptosis in control and ΔUSF2 MEFs was assessed by Annexin-V/P staining, measured by flow cytometry. (H) Real-time cell wound closure analysis of control, ΔUSF2 and ΔUSF2+USF2 MEFs; *significant difference, p ≤ 0.05. (I) Representative images of wound closure of control, ΔUSF2 and ΔUSF2+USF2 MEFs at 0 h, and 16 h after wound introduction. Scale bar 20 μm. (J) Migration was assessed by crystal violet staining and quantified by measuring the absorbance of crystal violet at 595 nm. The OD of ΔUSF2 cells was set to 100%. *significant difference control vs ΔUSF2 cells, **ΔUSF2 cells vs ΔUSF2+USF2 cells, p ≤ 0.05. (K) Representative images of the transwell migration assay. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
2.2. Lack of USF2 promotes cell proliferation
Next, we investigated whether deficiency of USF2 affects cell proliferation and apoptosis. First, we assessed proliferation by using live-cell analysis (IncuCyte®), cell count and bromodesoxyuridine (BrdU) incorporation. The data show that loss of USF2 promoted cell proliferation as indicated by the real-time cell measurements (Fig. 1C). These effects were USF2-specific since rescue of ΔUSF2 cells by reintroduction of USF2 antagonized the proliferative advantage (Fig. 1C). The view that lack of USF2 promotes proliferation was further supported by the higher cell number, and the increased BrdU incorporation into newly synthesized DNA of USF2-deficient cells (Supplementary Fig. 1B and C). Indeed, when assessing the distribution of cells within the cell cycle, we found that more ΔUSF2 than control cells were in the S-phase which is in line with the data from BrdU assays (Fig. 1D and E). By contrast, the analysis of apoptosis by flow cytometry with propidium iodide (PI) and annexin staining revealed no difference between control and ΔUSF2 cells (Fig. 1F and G) indicating that lack of USF2 promotes proliferation rather than inhibition of apoptosis.
2.3. Lack of USF2 promotes cell migration and invasion
As a next step, we analyzed whether the absence of USF2 has also an impact on cell migration and invasion. The data revealed that, like in the proliferation assays, loss of USF2 increased migration in a scratch wound healing assay (Fig. 1H and I). In addition, in transwell migration assays we found that the capacity of ΔUSF2 cells to migrate through a membrane was higher than that of the control cells (Fig. 1J and K). Again, these responses were specific to USF2 as the enhanced migration and invasion of ΔUSF2 cells could be counteracted upon re-expression of USF2. Together, these data show that loss of USF2 increases cellular migration and invasion.
2.4. Lack of USF2 affects mitochondrial morphology and function
As USF2 lacking cells show an increased proliferation, which requires energy, we next investigated whether USF2 deficiency affects mitochondria. To do this, we first carried out electron microscopy. The results showed that several mitochondria of ΔUSF2 cells were enlarged and showed a highly reduced number of cristae and an electrolucent matrix. In addition, large structures with vesicles, lamellar bodies and electron dense structures (lysophagosomes) were present (Fig. 2A). By contrast, mitochondria in the control cells were of normal size and displayed a typical tubular network with normal electron density and cristae structure (Fig. 2A). Next, we checked whether the lack of USF2 also affected mitochondrial function. To this end, we used TMRE staining and high-content fluorescent screening to determine the mitochondrial membrane potential. We found that the TMRE fluorescence was reduced in ΔUSF2 cells indicating a decreased mitochondrial membrane potential (Fig. 2B and C). In addition, we quantified mitochondrial number, area, length, and width and found that they all were affected in the ΔUSF2 cells (Supplementary Fig. 2A–E). The morphological changes were strictly dependent on USF2 as the mitochondria of rescued ΔUSF2 cells appeared normal (Fig. 2A, Supplementary Fig. 2A–E). Together, these data indicate that the knockout of USF2 affects mitochondrial morphology and function.
Fig. 2.

Lack of USF2 alters mitochondrial morphology and function. (A) Control, ΔUSF2 and ΔUSF2+USF2 MEFs cells were subjected to electron microscopy. Representative images of a cross-section showing the mitochondria (arrows), are presented. Original magnification, 6300 ×. Scale bar 1 μm. (B) Control, ΔUSF2 and ΔUSF2+USF2 cells were stained with TMRE and analyzed with the Operetta high-content imaging system. *significant difference control vs ΔUSF2 cells, **ΔUSF2 cells vs ΔUSF2+USF2 cells, p ≤ 0.05. (C) Representative fluorescent images of TMRE stained cells. (D) The oxygen consumption rate (OCR) of control and ΔUSF2 cells was measured under basal conditions and after the sequential addition of 1 μM oligomycin, 1 μM FCCP, and 0.5 μM rotenone/antimycin to allow calculation of basal respiration, ATP production, proton leak, and spare respiratory capacity (SRC). *significant difference, p ≤ 0.05. (E) Loss of USF2 increases ROS levels. Control, ΔUSF2 and ΔUSF2+USF2 MEFs were stained with CellROX® and the fluorescence intensity was analyzed and quantified with the Operetta high-content imaging system. *significant difference control vs ΔUSF2 cells, **ΔUSF2 cells vs ΔUSF2+USF2 cells, p ≤ 0.05. (F) Representative images of cells displaying CellROX® fluorescence.
2.5. Loss of USF2 reduces ATP production in mitochondria
Next, we used ΔUSF2 and control MEFs with the Seahorse XFP Analyzer and measured the mitochondrial oxygen consumption rate (OCR) under basal conditions and after a mitochondrial stress test in real-time. The results reveal that, under both basal and stressed conditions, the mitochondria of ΔUSF2 cells display a reduced OCR and ECAR in comparison to control cells (Fig. 2D, Supplementary Fig. 2). In addition, ΔUSF2 cells produce less ATP, and have reduced proton leak as well as spare respiratory capacity after the mitochondrial stress test when compared to control cells (Fig. 2D). Together, these data show that loss of USF2 negatively affects mitochondrial oxidative phosphorylation and thus mitochondrial ATP production.
2.6. Lack of USF2 increases cellular ROS levels
As compromised mitochondria may give rise to an increased formation of reactive oxygen species (ROS), we next measured cellular ROS levels. To do this, ΔUSF2, control and ΔUSF2+USF2 rescued MEFs were stained with CellROX® reagent and quantified with the Operetta high-content imaging system. As expected, we observed an increase in ROS levels in ΔUSF2 cells and these levels decreased again upon rescue of USF2 (Fig. 2E and F). Together, these data show that the lack of USF2 increases cellular ROS levels.
2.7. The lack of USF2 promotes autophagy
As the lack of USF2 compromises mitochondria which need to be cleared by autophagy involving the lysosomes, we next investigated whether USF2 affects the lysosomal compartment. Therefore, we stained control, ΔUSF2 and ΔUSF2+USF2 rescued MEFs with LysoTracker Red and quantified fluorescence intensity, lysosomal area and number of lysosomes with the Operetta high-content imaging system. We found that ΔUSF2 cells showed an increase in both LysoTracker fluorescence intensity and lysosomal area (Fig. 3A and B) but no change in lysosome number per cell (Supplementary Fig. 3) suggesting that loss of USF2 could promote autophagy by involving the lysosomes.
Fig. 3.
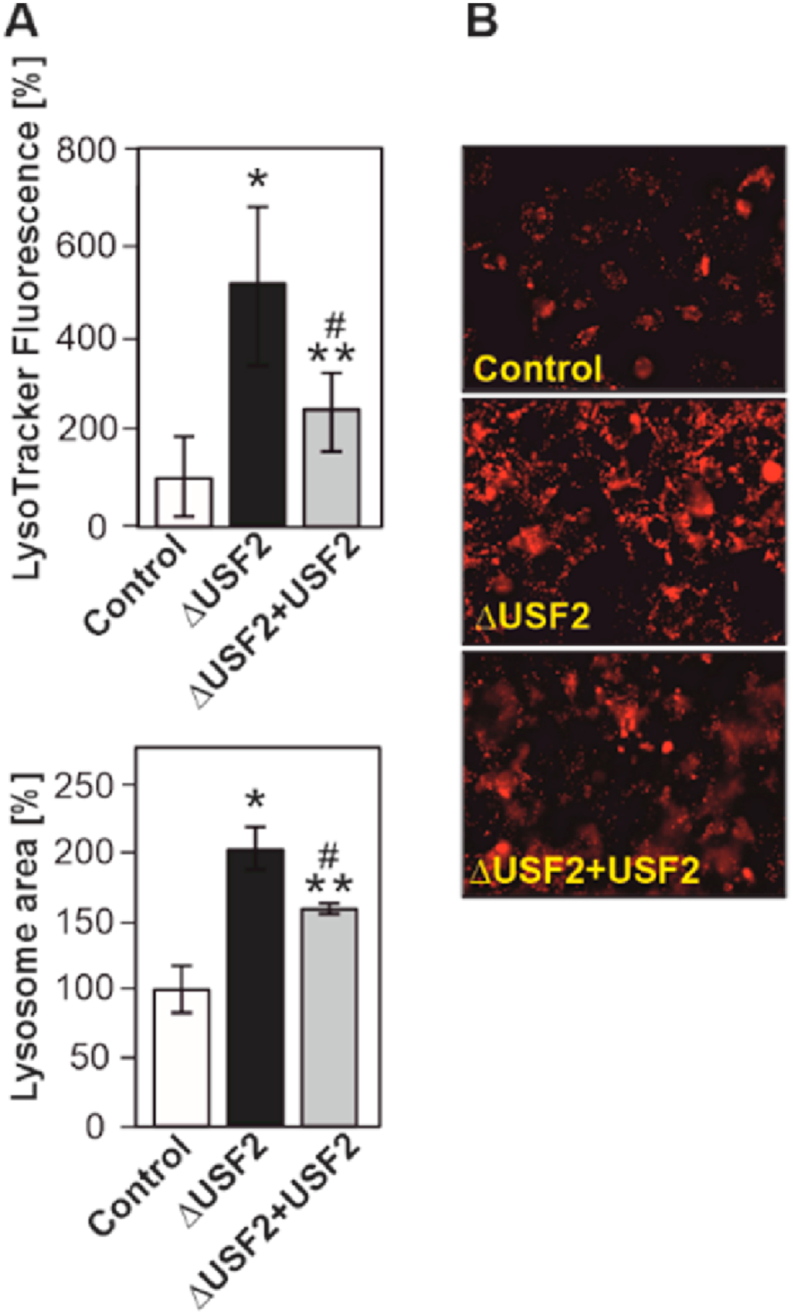
Lack of USF2 alters lysosomes. (A) Control and ΔUSF2 cells were stained with LysoTracker red and quantified with the Operetta high-content imaging system. *significant difference control vs. ΔUSF2 cells, **ΔUSF2 cells vs ΔUSF2+USF2 cells and # control vs ΔUSF2+USF2 cells, p ≤ 0.05. (B) Representative fluorescent images. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
We next tested the expression of several proteins involved in autophagy such as microtubule light chain (LC3), and autophagy related 5 (ATG5), as well as the mitochondrial protein succinate dehydrogenase (SDHB). In line with an activated autophagic process we found that the expression of LC3II, and ATG5 was enhanced in ΔUSF2 cells whereas the expression of SDHB was decreased when compared to control cells. Rescue of USF2-deficient cells with USF2 restored the expression of these proteins nearly to levels seen in the control MEFs (Fig. 4A and B).
Fig. 4.

Lack of USF2 promotes autophagy and activates ERK1/2 and AKT (A, B) Western blot analyses and densitometric quantification of the protein levels of autophagy-related LC3, ATG5 as well as SDHB, USF2, and α-tubulin in control, ΔUSF2, and ΔUSF2+USF2 cells. The protein levels of the corresponding proteins in control cells were set to 100%. *significant difference control vs. ΔUSF2 and **ΔUSF2 cells vs ΔUSF2+USF2 cells, p ≤ 0.05. (C, D) Representative fluorescence images from control, ΔUSF2, and ΔUSF2+USF2 cells in the presence and absence of chloroquine (Clq; 50 μM for 6 h) probed with an antibody against LC3, stained with DAPI and quantified with the Operetta high-content imaging system. The LC3 levels in control cells were set to 100%. *significant difference control vs. ΔUSF2, ** Control chloroquine treated vs ΔUSF2 chloroquine treated cells, and # untreated vs chloroquine treated p ≤ 0.05. (E, F) Western blot analyses and densitometric quantification of the LC3II and p62 protein levels from control, ΔUSF2, and ΔUSF2+USF2 cells in the presence and absence of chloroquine (Clq; 50 μM for 6 h). The LC3II and p62 levels in control cells were set to 100%. *significant difference control vs. ΔUSF2, **Control chloroquine treated vs ΔUSF2 chloroquine treated cells, and # untreated vs chloroquine treated p ≤ 0.05. (G, H) Western blot analyses and densitometric quantification of the protein levels of pERK1/2, ERK1/2, pAKT, AKT, AMPKα, AMPKβ, USF2, V5-tag, and α-tubulin in control, ΔUSF2 and ΔUSF2+USF2 cells. The protein levels of the corresponding phospho (p) protein levels in the control cells were quantified and normalized to their total levels and the corresponding levels in control cells were set to 100%. *significant difference control vs. ΔUSF2, **ΔUSF2 cells vs ΔUSF2+USF2 cells, and # control vs ΔUSF2+USF2 cells, p ≤ 0.05.
To further prove that USF2 affects autophagy, we performed more thorough autophagy experiments [15]. To this end, we first performed immunofluorescent analyses and detected the levels of LC3 in the absence and presence of chloroquine that blocks lysosomal degradation of autophagosomal content. We found that LC3 was forming punctate green dot structures indicating autophagosomes which were higher in number in ΔUSF2 cells. In agreement with an enhanced autophagy in ΔUSF2 cells, the LC3 positive dot number further increased in the presence of chloroquine. By contrast, rescue of ΔUSF2 cells reduced the number of LC3 positive dots to values seen in the control cells under basal conditions as well as in the presence of chloroquine (Fig. 4C and D). We further corroborated these data by measuring the abundance of lipidated autophagosomal LC3II together with the levels of p62 (an LC3-binding protein degraded by autophagy) in the absence and presence of chloroquine by Western blot. We found that ΔUSF2 cells had an increased basal level of LC3II when compared to control cells, and a concurrent decrease in p62. Inhibition of lysosomal degradation by chloroquine caused accumulation of LC3II and p62. The increase in p62 in chloroquine treated cells compared to untreated cells reflects the amount of LC3II and p62 that would have been degraded by autophagy. As the increment is higher in ΔUSF2 cells than in control cells this indicates enhanced autophagic flux in ΔUSF2 cells. Again, rescue of ΔUSF2 cells with USF2 restored the expression of both LC3II and p62 (Fig. 4E and F). Together, these data indicate that lack of USF2 promotes autophagy.
2.8. Loss of USF2 affects ERK1/2 and AKT signaling pathways
The ERK1/2, AMPK, and protein kinase B (AKT) pathways are crucial for controlling proliferation, energy status and survival of cells, respectively. Therefore, we examined the effect of USF2 loss on ERK1/2, AMPKα/β and AKT phosphorylation using Western blot analyses. We found that lack of USF2 increased ERK1/2, and AKT phosphorylation whereas phosphorylation of both AMPKα and β was decreased when compared to control cells (Fig. 4G and H). Rescue of the USF2-deficient cells with USF2 reversed these effects. Together, these data suggest that lack of USF2 promotes proliferation, and survival likely via ERK1/2 and AKT and does not cause a lack of energy despite damaged mitochondria.
2.9. ROS are crucial in the activation of mitophagy by ERK1/2 and AKT
As mitochondria are crucial for the maintenance of ROS homeostasis, we argued whether the induced formation of ROS in ΔUSF2 cells may contribute to mitophagy via ERK1/2 and AKT activation. To address this, we treated cells with the antioxidant N-acetylcysteine (NAC) and measured mitochondrial membrane potential and ERK1/2 and AKT phosphorylation. We found that addition of NAC counteracted the enhanced ROS formation and antagonized the decrease in mitochondrial membrane potential (Fig. 5A) and reduced the enhanced phospho-ERK1/2 and phospho-AKT levels (Fig. 5B and C). In addition, NAC restored SDHB levels indicating a reduced degradation of mitochondria (Fig. 5B and C). Together, these data indicate that ROS generated in the absence of USF2 are important messengers which contribute to ERK1/2 and AKT activation as well as to mitophagy.
Fig. 5.

Lack of USF2 promotes autophagy and activates ERK1/2 and AKT in a redox-sensitive manner. (A) Mitochondrial transmembrane potential in control and ΔUSF2 cells treated with N-acetylcysteine (NAC). Cells were stained with TMRE and analyzed with the Operetta high-content imaging system. *significant difference control vs. ΔUSF2 and **ΔUSF2 cells vs ΔUSF2 cells + NAC, p ≤ 0.05. (B, C) Western blot analyses and densitometric quantification of the protein levels of pERK1/2, ERK1/2, pAKT, AKT and autophagy-related LC3, ATG5 as well as SDHB in control and ΔUSF2 cells treated with NAC. (C) The protein levels of the corresponding phospho (p) ERK and (p) AKT protein levels were quantified and normalized to their respective total levels and the resulting levels in the control cells were set to 100%. *significant difference control vs. ΔUSF2, **ΔUSF2 cells vs ΔUSF2 + NAC cells and # control vs control + NAC, p ≤ 0.05.
2.10. USF2 is crucial for the expression of genes encoding mitochondrial proteins
As USF2 is acting as a nuclear transcription factor we next asked whether USF2 would directly control the transcription of genes encoding mitochondrial proteins. To do this, we first performed gene ontology (GO) enrichment analyses for cellular components by querying the PANTHER classification system (http://www.pantherdb.org) with USF2 ChIP-seq data from the ENCODE Transcription Factor Targets dataset. The analyses revealed that 4088 genes (~32%) from the 12829 potential USF2 target genes of the ENCODE database cluster with cellular organelles (GO:0043226) (Fig. 6A). The first level analysis again showed that 4030 from the 4088 genes (~98,6%) clustered with intracellular organelles (GO:0043229) (Figs. 6B) and 3536 genes clustered with intracellular membrane-bounded organelles (GO:0043231). A further level 2 analysis indicated that from the received 1323 component hits 403 genes (~30,4%) clustered with mitochondria (Fig. 6C). We next included the entries from MitoCarta 2.0 into our analyses and used those entries to check how many genes obtained from the second level Panther analysis and the ENCODE ChIP-seq data are part of the MitoCarta group. We found that 1011 genes from MitoCarta and 394 from the Panther list were among the USF2 target genes of the ENCODE dataset and 297 genes were common in all three datasets (Supplementary. Fig. 4).
Fig. 6.

USF2 is crucial for the expression of genes encoding mitochondrial proteins. (A, B, C) Gene ontology (GO) analyses. 12829 genes from the USF2 ENCODE dataset were queried in the PANTHER classification system for statistical overexpression in the GO cellular component complete test. All results shown are valid for an overall FDR <0.05 adjusted P value calculated via the Benjamini–Hochberg procedure. (D, E) RT-qPCR analyses. Data represent log 2 values of fold changes in ΔUSF2 cells relative to control and normalized to the levels of β-Actin, 18S rRNA and Hprt mRNA. *significant difference, p ≤ 0.05.
Although the above analyses suggest that genes with relevance to mitochondria could be directly regulated by USF2, they do not indicate whether this would result in a transcriptional activation or inhibition. Therefore, we next tested and validated some of those GO results and examined the mRNA levels of genes with importance for mitochondrial function and metabolism in ΔUSF2 and control cells. We found that, in accordance with the autophagy data, the mRNA level of Pink1 was upregulated in the ΔUSF2 cells (Fig. 6D). In addition, Mfn2 (mitofusin 2) mRNA was enhanced whereas the mRNA levels of microsomal glutathione S-transferase 1 (Mgst1), frataxin (Fxn), ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F (Atp5j), and acyl-Coenzyme A dehydrogenase, very long chain (Acadvl) were reduced (Fig. 6D). An interesting pattern could be detected in the expression of the pyruvate dehydrogenase kinase (PDK) isozymes. While Pdk1 and Pdk3 mRNA levels did not display a significant difference, the mRNA levels of Pdk2, the most abundant PDK isoform, were downregulated while those of Pdk4 were strongly upregulated (Fig. 6E). Altogether, these data indicate that USF2 is crucially involved in a transcriptional program that contributes to mitochondrial homeostasis.
3. Discussion
In this study, we explored whether deficiency of the transcription factor USF2 plays an important role for the regulation of cell growth and migration. Our novel findings show that MEFs deficient of USF2 gain an overall advantage in proliferation and migration. Furthermore, we identify USF2 of being crucial to maintain ROS levels as well as mitochondrial morphology and function since lack of USF2 was found to result in abnormal mitochondria which appeared to be cleared by mitophagy. Collectively, these results establish USF2 as being crucial for the control of growth processes and mitochondrial homeostasis.
Earlier results revealing that overexpression of USF2 could decrease the expression of the tumor marker protein PAI-1 [10,11] in hepatocytes, or counteracts the activity of cMYC in the S-phase of the cell cycle in rat embryonic fibroblasts [9] already suggested that USF2 could act as a growth suppressor. Our present data are in agreement with those earlier findings as well as reports showing that deep deletions of USF2 are present in about 3.15% of prostate cancer patients [16] where they associate with a decreased survival. Further support comes from other investigations showing that partial or complete lack of USF2 activity in breast cancer cell lines such as MCF-7, MDA-MB-231 promotes growth [17]. Surprisingly, a recent study using MCF-7 and MDB-MA-231 breast cancer cell lines reported that USF2 overexpression promotes tumor cell growth [14]. While the reasons for the latter discrepancy are unknown, another study from Saos-2 osteosarcoma cells revealed that lack of USF2 function causes increased cell growth in agreement with the present study [18]. Thereby, it appeared that overall USF2 activity seemed to require the presence of a coactivator that has yet to be identified. This coactivator was supposed to associate with the USR, an essential domain within the USF2 protein, in order to allow binding of USF2 to promoters without an initiator element [18].
Cell proliferation, migration and a lack of apoptosis are linked with activation of the ERK1/2/MAPK and protein kinase B (AKT) signaling pathways. Indeed, we found that lack of USF2 enhanced ERK1/2 phosphorylation and increased the number of cells in the S-phase of the cell cycle which is in line with earlier findings, demonstrating that ERK1/2 activity promotes entry of cells from G1 into the S phase [19]. Further, lack of USF2 increased the AKT phosphorylation status. Since AKT is involved in many cellular processes the current data would hint that lack of USF2 could promote survival by attenuating apoptosis or augmenting selective autophagy such as mitophagy. Since our data show that apoptosis was not affected in the USF2-deficient cells, the involvement of AKT appears to be related to the recruitment of LC3II that, as shown here and in an earlier report [20,21], promote recycling of dysfunctional mitochondria.
In that respect, our data also reveal a previously unknown function of USF2 in mitochondrial homeostasis which is underlined by our functional data showing that ΔUSF2 cells had mitochondria with a decreased mitochondrial membrane potential, and an impaired ATP production that were visible as being enlarged with a reduced amount of cristae, and an electrolucent matrix. These findings suggest that USF2 is a transcription factor crucially regulating nuclear expression of mitochondrial proteins. Indeed, our GO analyses support that view, and about 297 genes from the USF2 target genes found in ENCODE were also found in Panther and MitoCarta clustering with mitochondria. The qRT-PCR validation then showed that the mRNA levels of Pink1, Mfn2, Mgst1, Fxn, Atp5j, Acadvl and Pdk2 as well as Pdk4 were differentially regulated upon absence of USF2. Thereby, the induction of Pink1, and Mfn2 go well in line with the enhanced mitophagy [22] whereas the reduced Mgst1 and Fxn mRNA levels provide links for reduced glutathione conjugation of electrophiles and reduced assembly of iron and sulfur clusters [23], respectively. Consequently, these expression patterns fit with the enhanced ROS levels and the uncoupling of mitochondria from other organelles such as the endoplasmic reticulum [24]. In connection with the impaired mitochondrial function of the USF2 lacking cells is also the reduction in Atp5j, and Acadvl expression which goes in line with a reduced mitochondrial ATP synthesis and beta-oxidation capacity [25], respectively.
The current observations are also supported by studies in which depolarization of mitochondria occurred when CDK5, a positive regulator of USF2 [26], was knocked out [27]. Further, knockout of CDK5 in MDA MB-231 and MCF-7 cells enhanced ROS formation via mitochondria [27] in line with our present results showing an increased ROS formation in USF2-deficient cells. The increase in ROS levels could affect redox signaling and regulate ERK1/2 and AKT activity which all have been shown to be involved in the induction of proliferation, survival and mitophagy [28,29].
In support of the latter are also our data showing denser lysophagosomes in the electron microscopy pictures of the USF2 lacking cells which could indicate that those cells dispose dysfunctional mitochondria via lysosomes. Indeed, we found an increase in LysoTracker staining in ΔUSF2 cells which together with the autophagy markers LC3II, ATG5 and p62 are indicative for mitophagy and autophagy. Further, the low phospho-AMPKα/β levels are in line with findings from mice showing that loss of AMPK decreases mitochondrial content and function [[30], [31], [32], [33], [34]].
Although the reasons for induced ROS formation by lack of USF2 may be multiple, ROS are generally implicated to be messengers contributing to ERK1/2 and AKT activation [35]. Thus, it could be reasoned that the loss of USF2 causes mitochondrial stress leading to increased cellular ROS levels which induce mitophagy via ERK1/2 and AKT. The involvement of ROS was then confirmed by the experiments using NAC as antioxidant. Addition of NAC not only counteracted the enhanced ROS formation, phospho-ERK1/2 and phospho-AKT levels but also antagonized the decrease in mitochondrial membrane potential as well as the degradation of mitochondria as indicated by the restored SDHB levels.
Together, our study extends the knowledge about the role of the transcription factor USF2 in health and disease. On the one hand, it appears that USF2 is crucial in regulating cellular growth which is of utmost importance in physiological situations such as regeneration and wound healing or pathological processes such as tumor proliferation and metastasis. On the other hand, and with respect to the major role of USF2 for mitochondrial function and energy homeostasis, it is plausible that alterations in the presence or function of USF2 are very important in conditions such as insulin resistance, type-2 diabetes mellitus, and the metabolic syndrome.
4. Materials and methods
4.1. Materials
All biochemicals and enzymes were of analytical grade and obtained from commercial suppliers.
4.2. Plasmid constructs
The pLenti6/V5-DEST™ vector (Invitrogen, Life Technologies, Finland) has been used for the generation of lentivirus particles expressing USF2 proteins.
4.3. Cell culture
Mouse embryonic fibroblasts (MEFs) were isolated as previously described [36]. Following isolation, cells were cultured in Dulbeccos modified Eagles medium (DMEM, Sigma-Aldrich, Helsinki, Finland), supplemented with 1% non-essential amino acids (Sigma-Aldrich, Helsinki, Finland), 10% fetal bovine serum (Sigma-Aldrich, Helsinki, Finland), and 0.5% ciprofloxacin (MP Biomedicals, Illkrich, France) at 37 °C in an atmosphere of 5% CO2, 79% N2, 16% O2, and 97% humidity and spontaneously immortalized as described [36,37].
4.4. Generation of USF2 knockout MEFs by CRISPR-Cas9-mediated genome editing
A 20-bp guide sequence targeting the fifth exon of mouse USF2 (USF2, ENSMUST00000058860.13) was designed online using Zhang's laboratory web resource (www.genome-engineering.org); a non-targeting, scrambled sequence (OriGene) was used as a negative control. gRNA-encoding oligonucleotides (Sigma-Aldrich) were cloned into the vector SpCas9(BB)-2A-GFP (PX458, Addgene plasmid ID 48138) using standard procedures as described [38]. The generation of the USF2 control and knockout cells via CRISPR-Cas9-mediated non-homologous end-joining (NHEJ) DNA repair and the screening of the clonal cell lines was performed as already described [26,39]. All primer sequences are listed in Table S1. For analyses, a pool of four individual clones was used.
4.5. Overexpression of USF2 in cells via lentiviral infection
Polybrene was added (1 μg/mL) to cells prior lentiviral infection. Infected cells were subjected to selection with 0.5 μg/mL blasticidin for 2–3 weeks before harvesting and analyses. The overexpression of USF2 was verified by Western blot analysis.
4.6. Protein isolation, Western blot, and immunofluorescence
Whole cell extracts from MEFs were prepared as previously described [26]. The protein concentration was measured using the Bradford method [40].
Western blot analyses were performed using standard protocols with antibodies against LC3B (#2775S; 1:1000; Cell Signaling), ATG5 (sc-133158; 1:1000; Santa Cruz Biotechnology), SDHB (sc:13315; 1:1000; Santa Cruz Biotechnology), p62/SQSTM1 (#5114; 1:1000; Cell Signaling), pERK1/2 (#9101; 1:1000; Cell Signaling), total ERK1/2 (#9107; 1:1000; Cell Signaling), pAMPKα/β and total AMPKα/β (AMPK and ACC Antibody Sampler Kit #9957; 1:1000; Cell Signaling), USF-2 (N-18; 1:500; Santa Cruz Biotechnology) as well as monoclonal antibodies against V5-tag (#R960-25; 1:5000; Invitrogen), and α-tubulin (clone B-5-1-2; #T5168, 1:10000; Sigma-Aldrich). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit and goat anti-mouse IgGs (both 1:5000; Bio-Rad) were used as secondary antibodies.
The enhanced chemiluminescence system (ECL, GE Healthcare) and X-ray films were used for detection of proteins. The protein quantifications were performed with Fiji software (http://imagej.net/Ops).
Immunofluorescence was carried out as described [26,39] with an anti- LC3B (#2775S; 1:1000; Cell Signaling) at room temperature for 1 h. Subsequently, cells were washed in PBS three times for 10 min before incubation with FITC-conjugated goat anti-rabbit IgG secondary Ab (1:400; #A16143 Thermo-Fisher). Thereafter, cells were washed, counterstained with DAPI (1 μg/mL) and quantified with the Operetta software “Harmony” (PerkinElmer).
4.7. RNA preparation and quantitative real-time PCR analyses
Isolation of total RNA was performed using the Qiagen RNeasy® Mini Kit (Qiagen, Helsinki, Finland) following the manufacturer's instructions. One μg of total RNA was used for cDNA synthesis with the iScript™ cDNA synthesis Kit (Bio-Rad, Helsinki, Finland). Quantitative real-time PCR was performed in an Applied Biosystems 7500 Real-Time PCR System (Life Technologies, Helsinki, Finland) by using an Applied Biosystem Power SYBR® green PCR master mix (Life Technologies). Primers used are listed in Table S1.
4.8. Proliferation and cell migration assays
Proliferation assays were performed by counting viable cells using a hemocytometer and by real-time quantitative live-cell proliferation analysis using IncuCyte® ZOOM system (Essen BioScience) as already described [26,39]. BrdU incorporation was measured also as described [26,39]. The scratch wound assays were performed by using IncuCyte® ZOOM system (Essen BioScience) and the cell migration assays were conducted in vitro in transwell chambers (Becton Dickinson) without additional migratory stimulants as already described [26,39].
4.9. High-throughput analysis of mitochondria and lysosyme structures
The cells grown on 96-well plates were stained with either 200 nM tetramethylrhodamine (ab113852, TMRE) (Abcam, Cambridge, UK), 75 nM LysoTracker™ (#L12492, Invitrogen, Helsinki, Finland) or 5 μM CellROX® (#C10422, Invitrogen, Helsinki, Finland) for 30 min. Photos of the cells were then taken with the Operetta high-content imaging system (PerkinElmer) by exciting the fluorophores with the respective wavelengths (TMRE Ex/Em = 520–550/560-630 nm, LysoTracker™ Ex/Em = 560–580/560-630 nm and CellROX® Ex/Em = 620–640/650-760 nm). Image segmentation and quantification were performed using the Operetta software “Harmony” (PerkinElmer).
4.10. Transmission electron microscopy
After fixation, cells were immersed in 2% agarose, post-fixed in 1% osmium tetroxide, dehydrated in acetone, and embedded in Epon LX 112 (Ladd Research Industries). Thin sections were cut with a Leica Ultracut UCT ultramicrotome, stained in uranyl acetate and lead citrate, and examined in a Tecnai G2 Spirit transmission electron microscope (FEI Europe). Images were captured by using a Quemesa CCD camera (Olympus Soft Imaging Solutions GmbH).
4.11. Analysis of cell cycle and cell death
Cell apoptosis and cell cycle were measured in synchronized cells by Annexin-V-FLOUS and propidium iodide staining kits (Roche/Sigma-Aldrich, Helsinki, Finland) according to the manufacturer ‘s protocol. Synchronization was achieved by a combination of contact inhibition and serum deprivation. In brief, cells were grown to confluency, thereafter they were incubated without medium change for further 24 hrs. Release into the cell cycle was achieved by subculture and incubation with fresh serum-containing medium as above. The cells were then analyzed for cell cycle parameters and apoptosis using flow cytometry (BD FACSAria™ III cell sorter).
4.12. Seahorse XFp analysis
The mitochondrial function of the cells was determined through real-time measurement of oxygen consumption rate (OCR) using the XFp Extracellular Flux Analyzer (Seahorse, Agilent Technologies, Santa Clara, CA, USA) and Seahorse XFp Cell Mito Stress Tests according to the manufacturer's instructions. The cells were seeded onto XFp cell culture miniplates in triplicates 7 h in advance and mitochondrial stress test was conducted in Seahorse XF base medium supplemented with 10 mM glucose, 1 mM sodium pyruvate and 2 mM l-glutamine in response to oligomycin A (1 μM), FCCP (1 μM) and rotenone/antimycin A (0,5 μM). Assays were analyzed using the Seahorse XF Report Generator software (Wave, Agilent).
5. Data analyses and statistics
All experiments were performed at least three times and representative data are shown. If not otherwise stated data are means ± SD of at least three independent experiments. Statistical analyses were done using two-tailed Student's t-test and statistical differences of p ≤ 0,05 were considered as significant.
GO analyses were performed by querying the PANTHER classification system (http://www.pantherdb.org) [41] with USF2 ChIP-seq data from the ENCODE Transcription Factor Targets dataset (ENCODE Transcription Factor Targets https://www.encodeproject.org/publications/a70b36a7-e15e-45df-be62-7ac43940c16d/). MitoCarta genes were downloaded from Mitocarta 2.0 (https://www.broadinstitute.org/files/shared/metabolism/mitocarta/human.mitocarta2.0.html) [42] and used as above.
Declaration of competing interest
The authors declare that there is no conflict of interest-
Acknowledgments
This work was supported by the Academy of Finland SA296027, the Jane and Aatos Erkko Foundation, the Finnish Cancer Foundation, the Sigrid Juselius Foundation, the University of Oulu, and Biocenter Oulu. The authors are grateful to Virpi Glumoff for FACS sorting with CRISPR-Cas9 constructs, to Aki Manninen and Jaana Träskelin for the assistance in the generation of lentiviral particles (Biocenter Oulu) as well as to the expertise of the Biocenter Oulu Electron Microscopy core facility. Biocenter Oulu is a member of Biocenter Finland.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101750.
Contributor Information
Thomas Kietzmann, Email: Thomas.Kietzmann@oulu.fi.
Elitsa Y. Dimova, Email: Elitsa.Dimova@oulu.fi.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Sawadogo M., Roeder R.G. Interaction of a gene-specific transcription factor with the adenovirus major late promoter upstream of the TATA box region. Cell. 1985;43:165–175. doi: 10.1016/0092-8674(85)90021-2. [DOI] [PubMed] [Google Scholar]
- 2.Sirito M., Lin Q., Maity T. Ubiquitous expression of the 43-and 44-kDa forms of transcription factor USF in mammalian cells. Nucleic Acids Res. 1994;22:427–433. doi: 10.1093/nar/22.3.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yan S., Sloane B.F. Isolation of a novel USF2 isoform: repressor of cathepsin B expression. Gene. 2004;337:199–206. doi: 10.1016/j.gene.2004.05.005. 199-206. [DOI] [PubMed] [Google Scholar]
- 4.Viollet B., Lefrançois-Martinez A.M., Henrion A. Immunochemical characterization and transacting properties of upstream stimulatory factor isoforms. J. Biol. Chem. 1996;271:1405–1415. doi: 10.1074/jbc.271.3.1405. [DOI] [PubMed] [Google Scholar]
- 5.Galibert M.D., Boucontet L., Goding C.R. Recognition of the E-C4 element from the C4 complement gene promoter by the upstream stimulatory factor-1 transcription factor. J. Immunol. 1997;159:6176–6183. [PubMed] [Google Scholar]
- 6.Corre S., Galibert M. USF as a key regulatory element of gene expression. M-S (Med. Sci.) 2006;22:62–67. doi: 10.1051/medsci/200622162. [DOI] [PubMed] [Google Scholar]
- 7.Horbach T., Götz C., Kietzmann T. Protein kinases as switches for the function of upstream stimulatory factors: implications for tissue injury and cancer. Front Pharmacology. 2015;6:3. doi: 10.3389/fphar.2015.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vallet V.S., Casado M., Henrion A.A. Differential roles of upstream stimulatory factors 1 and 2 in the transcriptional response of liver genes to glucose. J. Biol. Chem. 1998;273:20175–20179. doi: 10.1074/jbc.273.32.20175. [DOI] [PubMed] [Google Scholar]
- 9.Sirito M., Lin Q., Deng J.M. Overlapping roles and asymmetrical cross-regulation of the USF proteins in mice. Proc. Natl. Acad. Sci. U.S.A. 1998;95:3758–3763. doi: 10.1073/pnas.95.7.3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Samoylenko A., Roth U., Jungermann K. The upstream stimulatory factor-2a inhibits plasminogen activator inhibitor-1 gene expression by binding to a promoter element adjacent to the hypoxia-inducible factor-1 binding site. Blood. 2001;97:2657–2666. doi: 10.1182/blood.v97.9.2657. [DOI] [PubMed] [Google Scholar]
- 11.Dimova E.Y., Kietzmann T. Cell type-dependent regulation of the hypoxia-responsive plasminogen activator inhibitor-1 gene by upstream stimulatory factor-2. J. Biol. Chem. 2006;281:2999–3005. doi: 10.1074/jbc.M512078200. [DOI] [PubMed] [Google Scholar]
- 12.Nupponen N.N., Kakkola L., Koivisto P. Genetic alterations in hormone-refractory recurrent prostate carcinomas. Am. J. Pathol. 1998;153:141–148. doi: 10.1016/S0002-9440(10)65554-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen N., Szentirmay M.N., Pawar S.A. Tumor-suppression function of transcription factor USF2 in prostate carcinogenesis. Oncogene. 2006;25:579–587. doi: 10.1038/sj.onc.1209079. [DOI] [PubMed] [Google Scholar]
- 14.Tan Y., Chen Y., Du M. USF2 inhibits the transcriptional activity of Smurf1 and Smurf2 to promote breast cancer tumorigenesis. Cell Signals. 2019;53:49–58. doi: 10.1016/j.cellsig.2018.09.013. [DOI] [PubMed] [Google Scholar]
- 15.Klionsky D.J., Abdalla F.C., Abeliovich H. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abida W., Cyrta J., Heller G. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 2019;116:11428–11436. doi: 10.1073/pnas.1902651116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ismail P.M., Lu T., Sawadogo M. Loss of USF transcriptional activity in breast cancer cell lines. Oncogene. 1999;18:5582–5591. doi: 10.1038/sj.onc.1202932. [DOI] [PubMed] [Google Scholar]
- 18.Qyang Y., Luo X., Lu T. Cell-type-dependent activity of the ubiquitous transcription factor USF in cellular proliferation and transcriptional activation. Mol. Cell Biol. 1999;19:1508–1517. doi: 10.1128/mcb.19.2.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sebolt-Leopold J.S., Dudley D.T., Herrera R. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat. Med. 1999;5:810–816. doi: 10.1038/10533. [DOI] [PubMed] [Google Scholar]
- 20.Ryan B.J., Hoek S., Fon E.A. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem.Sci. 2015;40:200–210. doi: 10.1016/j.tibs.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 21.Rüb C., Wilkening A., Voos W. Mitochondrial quality control by the pink1/parkin system. Cell Tissue Res. 2017;367:111–123. doi: 10.1007/s00441-016-2485-8. [DOI] [PubMed] [Google Scholar]
- 22.Rojo M., Legros F., Chateau D. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J. Cell Sci. 2002;115:1663–1674. doi: 10.1242/jcs.115.8.1663. [DOI] [PubMed] [Google Scholar]
- 23.Cavadini P., O’Neill H.A., Benada O. Assembly and iron-binding properties of human frataxin, the protein deficient in Friedreich ataxia. Hum. Mol. Genet. 2002;11:217–227. doi: 10.1093/hmg/11.3.217. [DOI] [PubMed] [Google Scholar]
- 24.Santel A., Fuller M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001;114:867–874. doi: 10.1242/jcs.114.5.867. [DOI] [PubMed] [Google Scholar]
- 25.Seminotti B., Leipnitz G., Karunanidhi A. Mitochondrial energetics is impaired in very long-chain acyl-CoA dehydrogenase deficiency and can be rescued by treatment with mitochondria-targeted electron scavengers. Hum. Mol. Genet. 2019;28:928–941. doi: 10.1093/hmg/ddy403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chi T.F., Horbach T., Götz C. Cyclin-dependent kinase 5 (CDK5)-Mediated phosphorylation of upstream stimulatory factor 2 (USF2) contributes to carcinogenesis. Cancers. 2019;11 doi: 10.3390/cancers11040523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Navaneethakrishnan S., Rosales J.L., Lee K. Loss of Cdk5 in breast cancer cells promotes ROS-mediated cell death through dysregulation of the mitochondrial permeability transition pore. Oncogene. 2018;37:1788–1804. doi: 10.1038/s41388-017-0103-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gibson S.B. A matter of balance between life and death: targeting reactive oxygen species (ROS)-induced autophagy for cancer therapy. Autophagy. 2010;6:835–837. doi: 10.4161/auto.6.7.13335. [DOI] [PubMed] [Google Scholar]
- 29.Petry A., Djordjevic T., Weitnauer M. NOX2 and NOX4 mediate proliferative response in endothelial cells. Antioxid Redox Signal. 2006;8:1473–1484. doi: 10.1089/ars.2006.8.1473. [DOI] [PubMed] [Google Scholar]
- 30.O’Neill H.M., Maarbjerg S.J., Crane J.D. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc. Natl. Acad. Sci. U.S.A. 2011;108:16092–16097. doi: 10.1073/pnas.1105062108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanner C.B., Madsen S.R., Hallowell D.M. Mitochondrial and performance adaptations to exercise training in mice lacking skeletal muscle LKB1. Am. J. Physiol. Endocrinology and Metabolism. 2013;305:1018. doi: 10.1152/ajpendo.00227.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeppesen J., Maarbjerg S.J., Jordy A.B. LKB1 regulates lipid oxidation during exercise independently of AMPK. Diabetes. 2013;62:1490–1499. doi: 10.2337/db12-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lantier L., Fentz J., Mounier R. AMPK controls exercise endurance, mitochondrial oxidative capacity, and skeletal muscle integrity. FASEB J. 2014;28:3211–3224. doi: 10.1096/fj.14-250449. [DOI] [PubMed] [Google Scholar]
- 34.Mottillo E.P., Desjardins E.M., Crane J.D. Lack of adipocyte AMPK exacerbates insulin resistance and hepatic steatosis through Brown and beige adipose tissue function. Cell Metabol. 2016;24:118–129. doi: 10.1016/j.cmet.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J., Wang X., Vikash V. 2016. ROS and ROS-Mediated Cellular Signaling Oxidative Medicine and Cellular Longevity; p. 4350965. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu J. Preparation, culture, and immortalization of mouse embryonic fibroblasts. Curr.Protoc.Mol.Biol. Chapter. 2005;28 doi: 10.1002/0471142727.mb2801s70. Unit 28.1. [DOI] [PubMed] [Google Scholar]
- 37.Parrinello S., Samper E., Krtolica A. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat. Cell Biol. 2003;5:741–747. doi: 10.1038/ncb1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ran F.A., Hsu P.D., Wright J. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dimova E.Y., Jakupovic M., Kubaichuk K. The circadian clock protein CRY1 is a negative regulator of HIF-1 alpha. iScience. 2019;13:284–304. doi: 10.1016/j.isci.2019.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 41.Mi H., Muruganujan A., Ebert D. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019;47:D419–D426. doi: 10.1093/nar/gky1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Calvo S.E., Clauser K.R., Mootha V.K. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44:D1251–D1257. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.