

Abstract
The Hippo-Yap/Taz pathway, originally identified as a central developmental regulator of organ size, has been found perturbed in many types of human tumors, and linked to tumor growth, survival, evasion, metastasis, stemness, and drug resistance. Beside these tumor-cell-intrinsic functions, Hippo signaling also plays important immune regulatory roles. In this review, we will summarize and discuss recent breakthroughs in our understanding of how various components of the Hippo-Yap/Taz pathway influence the tumor immune microenvironment, including their effects on the tumor secretome and immune infiltrates, their roles in regulating crosstalk between tumor cells and T cells, and finally their intrinsic functions in various types of innate and adaptive immune cells. While further research is needed to integrate and reconcile existing findings and to discern the overall effects of Hippo signaling on tumor immunity, it is clear that Hippo signaling functions as a key bridge connecting tumor cells with both the adaptive and innate immune systems. Thus, all future therapeutic development against the Hippo-Yap/Taz pathway should take into account their multi-faceted roles in regulating tumor immunity in addition to their growth-regulatory functions. Given that immune therapies have become the mainstay of cancer treatment, it is also important to pursue how to manipulate Hippo signaling to boost response or overcome resistance to existing immune therapies.
Keywords: Hippo pathway, Mst1, Mst2, Yap, Taz, Innate Immunity, Adaptive Immunity, Cancer Immunity
Immune therapies have shown extraordinary promises in treatment of many types of cancer, although resistance (primary or acquired) remains prevalent1. Existing evidence suggests that to elicit durable and effective immune response to eliminate established tumors, several key steps are required including reversing the immunosuppressive tumor microenvironment (TME), promoting the infiltrations of active antigen-presenting cells (APCs) and effector T cells, inducing immunogenic tumor cell death and increasing antigen presentations, and enhancing the effector functions of tumor-infiltrating CD4+ and CD8+ T cells1,2.
To overcome these barriers to tumor rejection, combination therapies are being explored1,2. However, these efforts have been hampered by a lack of clear understanding of the mechanisms that underlie the complex crosstalk between tumor cells and the immune cells, between immune cells and other stromal cells within the TME, and between subpopulations of the immune cells within the TME and beyond. In this review, we will summarize the emerging roles of the Hippo-Yap/Taz pathway in regulations of the above-mentioned crosstalk and their effects on tumor immunity, discuss recent literature that directly implicates various components of the Hippo-Yap/Taz pathway in immune cell differentiation and activation, and deliberate how therapies that target the Hippo-Yap/Taz pathway could be potentially integrated with existing immune therapies to increase response rate and improve outcome.
Canonical and noncanonical Hippo-Yap/Taz signaling
The canonical Hippo-Yap/Taz pathway, composed of the Mst1/2-Lats1/2 core kinase cascade, is responsible for phosphorylating and inactivating two homologous transcription regulators Yap and Taz (Fig. 1). Specifically, facilitated by scaffolding proteins Merlin, Kibra, Sav1 and Mob1, the Mst1/2 kinases phosphorylate and activate the Lats1/2 kinases, which in turn phosphorylate Yap and Taz at multiple sites3,4. Lats1/2-mediated phosphorylation of Yap and Taz creates binding sites for the highly abundant 14-3-3 proteins, which trap Yap/Taz in the cytoplasm3,4, and also primes Yap/Taz for further phosphorylation by Casein kinase 1δ/ε, creating phospho-degron motifs that recruit the β-TrCP-SCF E3 ubiquitin complex, causing ubiquitination and proteasomal degradation of Yap/Taz5,6.
Figure 1. Schematic representation of the tumor-cell-intrinsic roles of Yap/Taz in regulating tumor immunity.
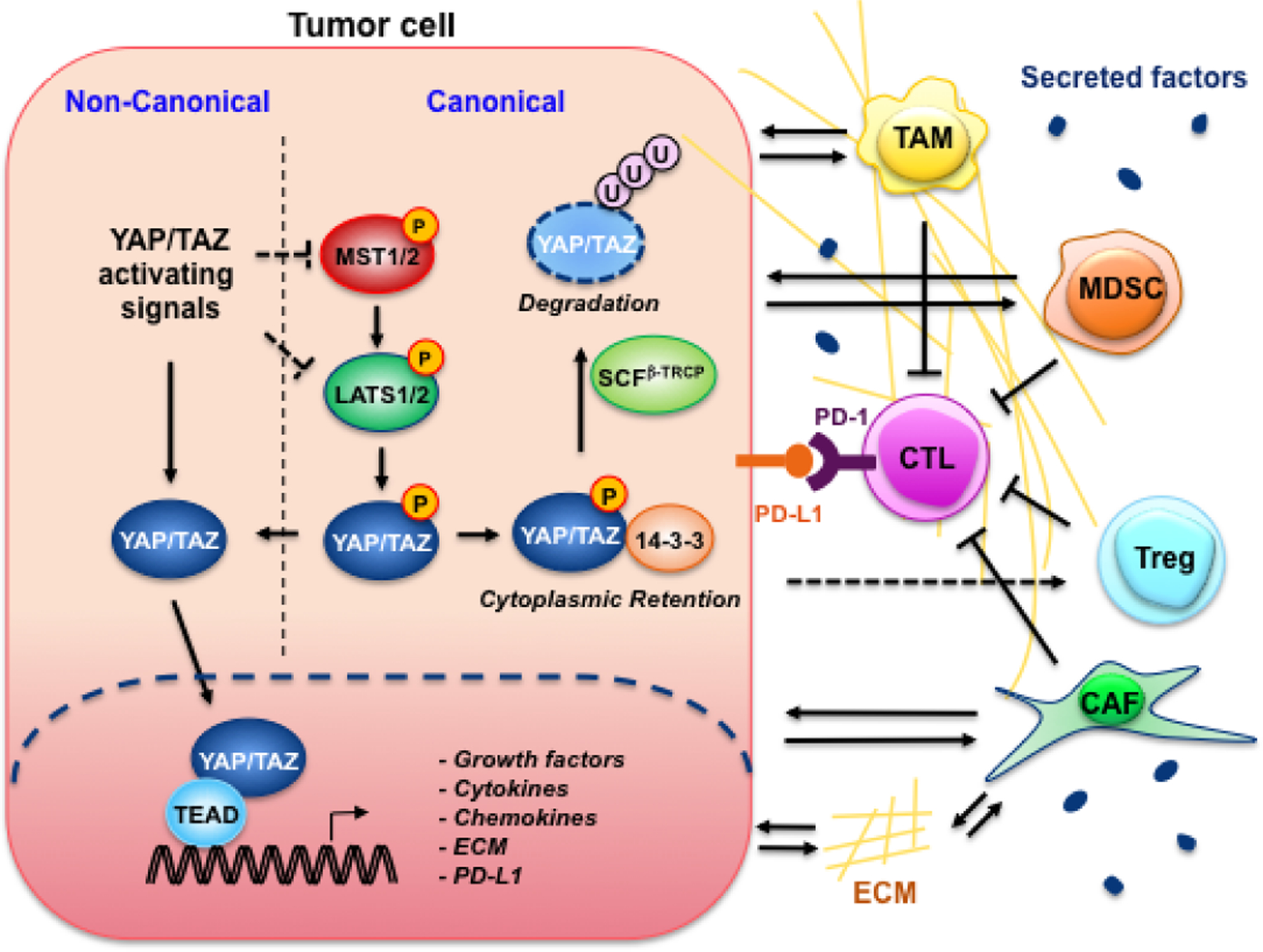
In tumor cell, Yap and/or Taz activated through either suppression of the canonical Hippo pathway or non-canonical activating signals translocate into the nucleus and partner with the Tead family of transcription factors to promote the expression of genes including PD-L1, growth factors, cytokines, chemokines, and extracellular matrix (ECM) proteins12–15,18–22,25. PD-L1 translocate onto the plasma membrane where it binds to PD-1 expressed on the surface of cytotoxic T cells (CTLs), and directly inactivates CTLs. Moreover, tumor cells release Yap/Taz-induced growth factors, cytokines and chemokines, orchestrating an immune suppressive TME that activates and recruits tumor-associated macrophage (TAM), myeloid-derived suppressor cell (MDSC), regulatory T cells (Treg), and cancer-associated fibroblast (CAF), indirectly suppressing CTLs.
In response to various oncogenic signals or through direct genetic mutations, Hippo signaling is often inactivated in tumor cells, resulting in dephosphorylation and nuclear translocation of Yap and Taz7. Once inside the nucleus, Yap/Taz, which possess transactivation domains but no DNA-binding domains, partners with Tead1–4 (containing DNA-binding domains but no transactivation domains) and other transcription factors to promote the expression of genes that are important for tumor cell proliferation and survival7. It should be noted, however, other noncanonical Hippo-independent mechanisms also regulate subcellular localization and/or transcriptional activities of Yap and Taz7,8. Conversely, upstream Mst1/2 and Lats1/2 kinases can phosphorylate and regulate substrates that are outside the Hippo-Yap/Taz pathway9,10.
Efforts are ongoing in developing small molecule inhibitors to target the Hippo-Yap/Taz pathway7. To successfully incorporate Hippo-targeting agents with existing cancer therapies, particularly immune therapies, it is important to consider the immune modulatory effects of this pathway in addition to their growth regulatory roles in tumor cells. In this review, we will discuss the emerging roles of both canonical and noncanonical Hippo signaling in regulations of TME and immune response.
Tumor cell intrinsic roles of Hippo-Yap/Taz signaling in regulating tumor immunity
Tumor cells employ multiple mechanisms to evade immune surveillance, including suppression of antigen production and/or presentation, activation of cell surface immune checkpoint ligands such as PD-L1, and secretion of cytokines that promote the activation and recruitment of cancer associated fibroblasts (CAFs) and immune suppressive myeloid cell populations, orchestrating a TME that is hostile to the recruitment and/or functionalities of effector T cells11. Intriguingly, recent studies have implicated the Hippo pathway effectors Yap and Taz in several aspects of the above-mentioned tumor cell intrinsic mechanisms of immune suppression.
Potential direct transcriptional regulation of PD-L1 by Yap/Taz in tumor cells
The immune checkpoint receptor PD-1 and its ligand PD-L1 function as a dominant immune checkpoint pathway that normally prevents recognition of self-antigens by T cells11. Tumor cells exploit this pathway to evade host immunity by upregulating PD-L1, which interacts with PD-1 receptors expressed on activated T cells causing apoptosis or anergy of those T cells11.
Four groups recently reported that Yap/Taz suppress T-cell-mediated killing of tumor cells by directly transcribing PD-L1 in human melanoma, lung and breast cancer cells (Fig. 1), and two of the reports further showed strong correlations between the levels of nuclear Yap and PD-L1 in primary NSCLC and melanoma samples12–15. Therapeutic antibodies that target PD-L1 or PD-1 have been successfully employed in the clinics to reactivate T cells and cause tumor regression in many types of aggressive cancers, although the majority of patients are upfront resistant or subsequently relapse from anti-PD-1/PD-L1 treatment11. Therefore, it would be interesting in the future to determine how Hippo-Yap/Taz signaling status may influence response to PD-1/PD-L1 therapies.
Yap-mediated expression of tumor secreted factors drive the polarization and recruitment of immune suppressive MDSCs and TAMs
Myeloid-derived suppressor cells (MDSCs) and tumor associated macrophages (TAMs) are heterogeneous populations of immune cells of the myeloid lineage, which not only potently suppress T-cell function within the TME, but also secrete cytokines that promote tumor cell proliferation, survival and metastasis16. High numbers of tumor-infiltrating MDSCs and TAMs often correlate with early local or metastatic relapse and resistance to T-cell checkpoint therapies, leading to poor survival in patients17.
Using a syngeneic mouse model, DePinho and Wang groups demonstrated Yap-dependent recruitment of MDSCs into subcutaneous tumors derived from a murine Pten−/−Smad4−/− prostate tumor cell line18. Mechanistically, the authors showed Yap binds to the promoter and induces the expression of Cxcl5 in prostate tumor cells, which recruits MDSCs via its cognate receptor Cxcr218. Independently, our group showed that Yap drives not only the recruitment but also the polarization of MDSCs cells by coordinately upregulating Il-6, Csf1–3, Tnfα, Il-3, Cxcl1/2, and Ccl2 expression in the genetically engineered p48-Cre;KrasLSL-G12D/+;p53LSL-R172H/+ (KPC) pancreatic cancer model19. We further showed that Yap binds to the promoters and directly promotes the transcription of Il-6 and the Csfs in PDAC cells19. Notably, another group also reported the direct transcriptional regulation of Il-6 by Yap in breast cancer stem cells20.
Beside MDSCs, we found that deletion of Yap from Kras:p53 mutant pancreatic neoplastic epithelial (KPYC) cells induced massive influx of MHCII+ M1-like macrophages within the TME19. Importantly, the downregulation of MDSCs and reprogramming of TAMs in KPYC pancreata were accompanied by activation of CD8+ T cells, indicating that Yap mediates T cell suppression at least in part by inducing the accumulation of MDSCs and TAMs in KPC pancreata19. Consistent with our finding, two other studies showed that activation of Yap in liver epithelial cells leads to early recruitment of TAMs by upregulating Ccl2 and Csf1 expression, which prevents immune clearance of Yap-activated tumor-initiating cells and promote hepatocellular carcinoma development21,22.
Together, these studies reveal that Yap-controlled tumor secreted factors drive the recruitment of immune suppressive myeloid cells across multiple tumor types (Fig. 1).
Yap-mediated activation of CAFs fosters immune suppression
CAFs are heterogeneous populations of fibroblast-like cells activated by tumor-secreted growth factors and cytokines within the TME. Beside secreting extracellular matrix (ECM) proteins that form the tumor stroma and growth factors that stimulate angiogenesis and tumor growth, CAFs have been shown to release a large number of immune suppressive cytokines that promote immune evasion23.
PDAC evokes a highly desmoplastic stromal reaction, which is the consequence of pro-fibrotic activation of CAFs by PDAC cells24. We previously demonstrated that KPYC pancreata showed dramatic reduction in the overall number of CAFs and collagen buildup around early lesions, likely due to downregulation of Ctgf, Cyr61, Cox2, Il1a, Il6, Mmp7 and possibly other tumor-secreted cytokines25. Moreover, multiple studies showed that Yap promotes matrix stiffening and enhances cell tension in fibroblasts and tumor cells by increasing the expression of cytoskeleton regulators26–30. In a feed-forward loop, increased mechanical tension caused by ECM buildup was found to further enhance Yap activities in both tumor epithelial cells and CAFs through Integrin-FAK-SRC and Rap2-Arhgap29-RhoA signaling26,29,31–35. Intriguingly, a recent study showed that treatment of a small molecule FAK inhibitor reduced tumor fibrosis and the recruitment of immunosuppressive cells, and rendered the KPC PDAC model responsive to adoptive T cell therapy and PD-1 antagonists36. Collectively, these findings point to the essential roles for YAP in CAF activation, tumor stroma buildup and mechanotransduction, which also contribute to the establishment of immune suppressive TME (Fig. 1).
Supraphysiological Yap/Taz activation could trigger immune rejection
While overwhelming evidence supports the roles for Yap/Taz in orchestrating tumor immune evasion, excessive Yap/Taz activation was found to cause immune rejection in both autochthonous and syngeneic models36–40. In the mouse liver, overexpression of constitutively active Yap (Yap5SA, in which all five inhibitory Lats1/2-phosphorylation sites were mutated) or deletion of both Lats1 and Lats2 was shown to induce DNA damage and p53-mediated senescence or cell death, triggering T-cells dependent immune clearance21,40. Similar observations were made with syngeneic injections of Lats1/2 KO or Yap5SA-overexpressing melanoma, head and neck squamous cell carcinoma and breast cancer lines, which were attributed to increased secretion of nucleic-acid-rich extracellular vesicles by these cells and activation of TLR signaling39. It remains to be determined, however, whether the observed increase in extracellular nucleic acids were independent of or resultant from elevated DNA damage or cell stress triggered by hyper-activation of Yap/Taz. Notably, the immune clearance triggered by hyper-activation of Yap/Taz was reminiscent of that of oncogenic RAS or BRAF41–44, suggesting that similar to other proto-oncogenes, excessive Yap/Taz activation can trigger p53-mediated senescent and/or apoptotic programs, which in turn elicit immune recognition and clearance.
Direct regulations of the adaptive and innate immune systems by Hippo-Yap/Taz signaling
With the increasing interest in exploiting the Hippo-Yap/Taz pathway as targets for regenerative or cancer therapies8, it is important to understand the physiological roles of Hippo-Yap/Taz signaling in the immune system. In this section, we will summarize recent studies linking both canonical and noncanonical Hippo-Yap/Taz signaling to regulations of the differentiation and functions of various types of immune cells.
Mst1 regulates the development of B- and T- lymphocytes
Mst1 is highly expressed in lymphatic tissues and Mst1 deficiency causes B- and T- lymphopenia in both humans and mouse models45,46. In B-cell development, Mst1 was shown to be required for the transcription of CD19, a glycoprotein that is specifically expressed in B cells and required for B-cell function (Fig. 2)47. In T-cell development, Mst1 phosphorylates and stabilizes transcription factors Foxo1/3, which are important for maintaining the proliferation and survival of naïve T cells48–50. Moreover, Mst1 is required for thymocyte trafficking and antigen recognition within the thymus (Fig. 2)51, and the egress of mature T cells from the thymus to peripheral lymphoid organs52,53. Although the precise mechanisms by which Mst1 promotes T cell migration and antigen recognition remain unclear, it likely involves myosin-mediated trafficking and membrane clustering of integrin LFA-1, which binds to ICAM-1 on antigen-presenting cells (APCs) to initiate the immunological synapse formation51,54–57.
Figure 2. Schematic representation of direct regulatory roles of canonical and noncanonical Hippo-Yap/Taz signaling in immune cells.
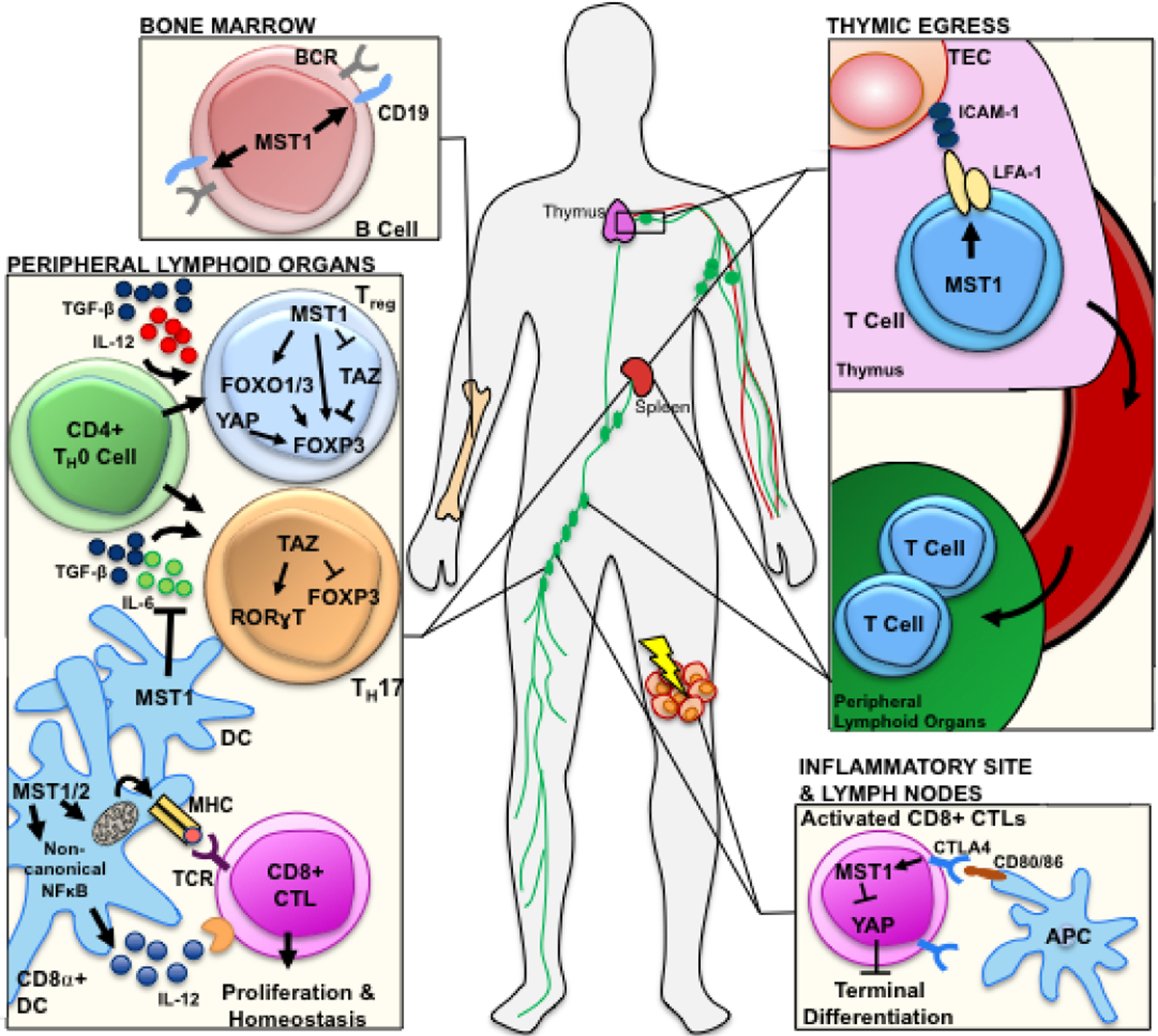
In developing lymphocytes, Mst1 promotes the CD19 expression in B cells and induces T cell thymic migration and egression by promoting LFA-1 membrane clustering47,51,54–57. During CD4+ T cell differentiation, Mst1 promotes the expression of the Treg transcription factor Foxp3 through both Taz-dependent and Taz-independent mechanisms50,59,60. Independent of canonical Hippo signaling, Yap promotes Treg differentiation by increasing Foxp3 expression via activin-TGF-β signaling62. Taz promotes TH17 differentiation by directly bolstering RORγt transcription, while Mst1 expression in dendritic cells (DC) inhibits IL-6 secretion, a cytokine necessary for TH17 differentiation60,61. Mst1/2 maintains mitochondrial respiration and dynamics and noncanonical NF-κB-mediated IL-12 production in CD8α+ DCs, a major type of antigen-presenting cells (APCs) for CD8+ CTLs66. Finally, CTLA4-CD80/86 engagement between CTLs and APCs activates Mst1 in CTLs, resulting inactivation of Yap and terminal differentiation64.
Antagonizing functions of Mst1 and Taz balance Treg/TH17 differentiation
In response to various cytokines, naive CD4+ T cells can differentiate into distinct helper T cell subsets, including TH1, TH2 and TH17 cells, as well as Foxp3+ regulatory T (Treg) cells. In particular, the imbalance between the pro-inflammatory TH17 cells and the immunosuppressive Tregs, whose differentiation is connected by their shared dependence on TGF-β, has emerged as a prominent factor in autoimmunity and cancer immune escape58. The presence of TGF-β drives the conversion of naive T cells into Treg cells through Smad2/3-mediated transcriptional activation of Foxp3, whereas TGF-β in combination with pro-inflammatory cytokines IL-6 or IL-21 causes differentiation into TH17 cells by inducing TH17 lineage-defining transcriptional factor RORγt and IL-1758.
A series of studies showed that Mst1 promotes Treg differentiation, while reducing differentiation towards the TH17 lineage through both cell-autonomous and non-autonomous mechanisms (Fig. 2)50,59–61. Within CD4+ T cells, Mst1 was reported to promote Foxp3 expression and Treg differentiation by phosphorylating and stabilizing Foxo1/3, blocking TCR-stimulated Akt activation, preventing Foxp3 de-acetylation by Sirt1, or suppressing Taz activity (also see below)50,59,60. Beside these cell-autonomous mechanisms, Mst1 was shown to inhibit the secretion of IL-6 in dendritic cells, thereby indirectly suppressing TH17 differentiation61.
Opposite of Mst1, Taz, which is specifically upregulated in TH17 cells, was found to skew T cell differentiation along the TH17 lineage at the expense of Tregs (Fig. 2)60. Mechanistically, Taz directly binds to and bolster the activity of TH17 transcription factor RORγt, while inducing proteasomal degradation of Foxp3 by inhibiting Tip60-mediated acetylation of Foxp360. Importantly, defects in Treg differentiation caused by Mst1 deficiency was rescued by co-deletion of Taz, suggesting that canonical Hippo-Taz signaling likely plays a central role in Treg/TH17 differentiation60.
Yap amplifies TGF-β signaling and maintains immune suppressive function in Tregs
Unlike Mst1 and Taz, T-cell specific knockout of Yap had little effect on T cell lineage commitment in vivo60,62. Nevertheless, Yap was found to be specifically induced in the Treg population, and amplify TGF-β signaling by transcribing Activin/Acvr1c (Fig. 2)62. Genetic or chemical inhibition of Yap dramatically impaired the immune suppressive function of Tregs in vitro and in vivo, and boosted anti-tumor immune response either as a mono-therapy or in combination with tumor vaccine or PD-1 treatment62. These exciting results highlight the promise of Yap as a potential target for overcoming resistance to immune therapy.
Hippo signaling activated by CTLA4-CD80 engagement contributes to terminal differentiation of CD8+ T cells
CTLA4 is a member of the immunoglobulin superfamily that is expressed by activated T cells to prevent over-stimulation of T cells by APCs63. CTLA4 binds to CD80/CD86 (also known as B7–1/2) expressed on APCs with greater affinity than the T cell co-stimulatory molecule CD28, thus blocking CD28-B7 binding and dampening TCR signaling63. Although much is unknown about the roles of Hippo-Yap signaling in regulation of CD8+ T-cell activation, an early study reported that activating the CD8+ T cell with antigen and IL-2 in vitro induces the expression of the core Hippo pathway components, including Yap (Fig. 2)64. Furthermore, the authors showed that engagement of CTLA4 with CD80 triggers the activation of Hippo signaling resulting in phosphorylation and degradation of Yap, which in turn causes downregulation of Eomes and upregulation of Blimp-1 and terminal differentiation of CD8+ T cells64. These findings, while remain to be confirmed in vivo, imply a possible role for the Hippo-Yap signaling in regulation of cytotoxic T cells.
Mst1/2 maintains metabolic homeostasis and IL-12 signaling in CD8α+ dendritic cells
Baf3+CD8α+ dendritic cells are the major APCs for CD8+ T cells in response to viruses, bacteria and tumors65. A recent study found that deletion of Mst1/2, but not Lats1/2 or Yap/Taz, selectively blocks Baf3+CD8α+ DCs from activating CD8+ T cell66. Mechanistically, the authors showed that Mst1/2 maintain mitochondrial function and dynamics on one hand, and promote noncanonical NF-κB signaling and IL-12 production on the other hand, thus coordinating metabolic reprogramming with cytokine signaling (Fig. 2)66. This finding suggests that activation of Mst1/2 could potentially be used to boost tumor immunity by activating Baf3+CD8α+ dendritic cells.
Crosstalk between Mst1/2 (Hippo) and TLR signaling
Toll-like receptors (TLRs) are a major subgroup of membrane bound pattern recognition receptors (PRRs) used by innate immune cells to detect pathogen-associated molecular patterns (PAMPs) commonly shared by microorganisms67. Upon binding by PAMPs, TLRs undergo conformational changes that trigger the binding of adapter protein MyD88 or TRIF67. Recruitment of MyD88 to TLRs initiates NFκB signaling by facilitating the assembly of oligomeric IRAK4-IRAK1/2-TRAF6 complex and TRAF6-mediated formation of K63 poly-ubiquitin chains, which serve as scaffolds for the phosphorylation and activation of the IKK complex by TAK167. Activated IKK complex then phosphorylates IκB, causing its degradation, freeing NFκB to translocate into the nucleus to activate the expression of inflammatory cytokines67. In addition, bacterial engagement of a subset of TLRs leads to phagocytosis and the assembly of TRAF6-ECSIT complexes on the phagosomes, which recruit mitochondria to augment ROS production and bacteria killing67. On the other hand, binding of TRIF to TLRs triggers IRF3 signaling by recruiting E3 ligase TRAF3, IKK-like kinases - TBK1 and IKKε, and IRF3, where intermolecular trans-phosphorylation, facilitated by Lys63-linked ubiquitination, causes IRF3 to dimerize and translocate into the nucleus, where it promotes the expression of type I interferons (IFNs) and other inflammatory cytokines67.
A series of recent studies showed that bacteria-induced TLR signaling activates the Hippo pathway in both Drosophila and mammalian cells68–71. In Drosophila, it was shown that bacteria-induced assembly of a Toll-MyD88-Pelle (homologue of IRAK) complex phosphorylates and inactivates Cka (homolog of Striatin), an essential subunit of the Hippo-inhibitory STRIPAK PP2A complex, resulting in activation of the Hippo kinase (Mst1/2 homologue) and suppression of Yorkie (Yap/Taz homologue) in the Drosophila immune organ - fat bodies (Fig. 3a)68. In mammalian cells, Boro et al. reported that IRAK1/4 directly interact with Mst1/2 and promote Mst1/2 phosphorylation and activation following bacteria infection (Fig. 3b)70, whereas Yuan et al. found that IRF3 activated by bacteria infection directly binds to the promoter of the STK4 (Mst1) gene inducing its expression (Fig. 3b)71.
Figure 3. Crosstalk between Toll-like-receptor (TLR) signaling and the Hippo pathway in Drosophila and mammalian cells.
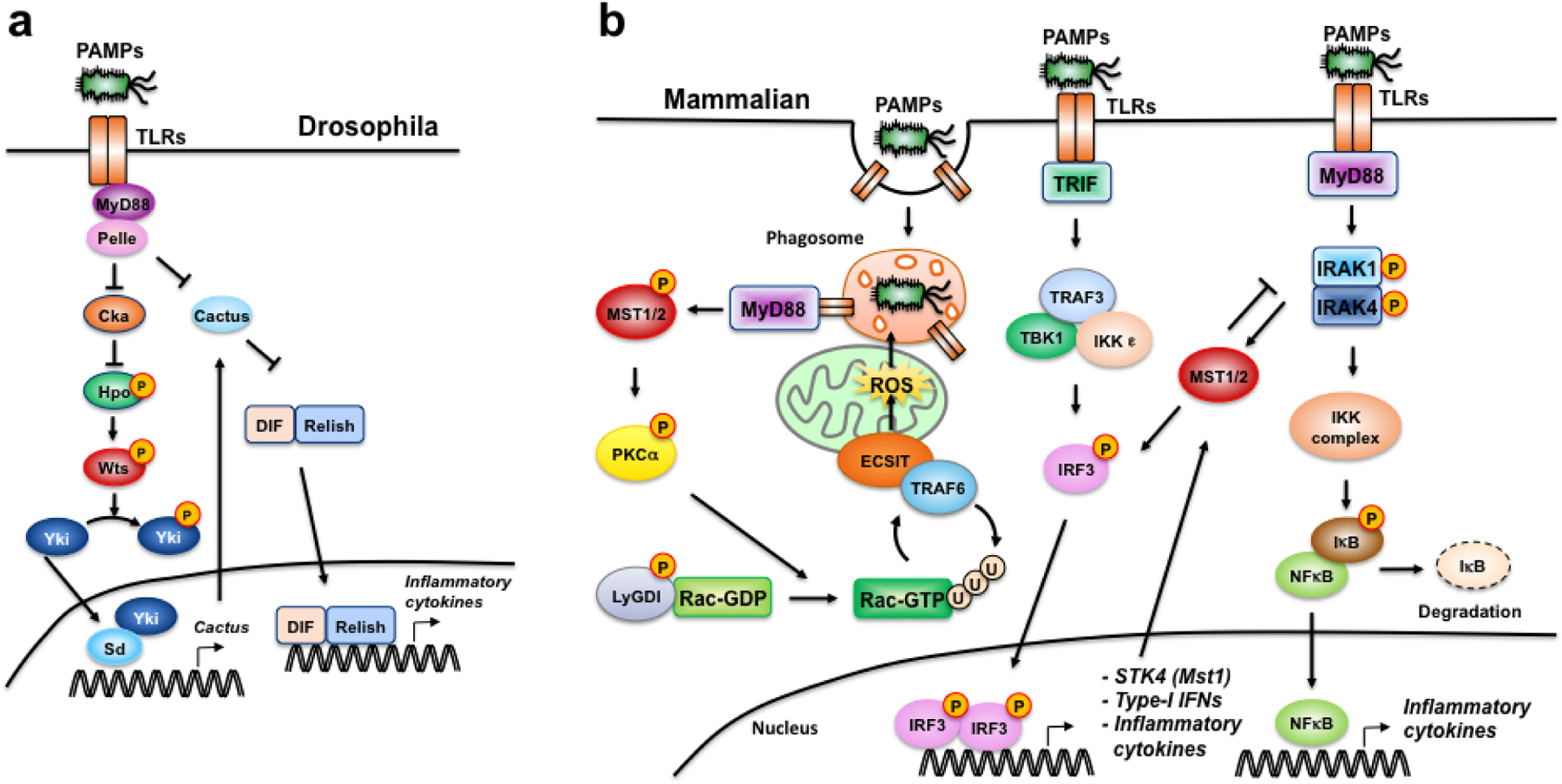
(a) Toll-Hpo signaling crosstalk in Drosophila. Upon PAMP recognition, Toll-MyD88-Pelle (homolog of IRAK) complex inactivates Cka (homolog of Striatin), resulting in Hpo (homolog of Mst1/2) phosphorylation and activation of canonical Hpo signaling68. Hpo-mediated Yki (homolog of Yap/Taz) phosphorylation prevents Yki nuclear translocation and transcription of Cactus (homolog of IκB), boosting NF-κB activation68,73. (b) TLR-Mst1/2 crosstalk in mammalian cells. During bacteria-induced phagocytosis, TLR-MyD88 signaling activates Mst1/2, which phosphorylates PKCα and inactivates LyGDI, allowing Rac1 to become fully-activated to promote TRAF6-ECSIT-mediated mitochondrial recruitment and delivery of ROS to phagosomes69. Downstream of TLR-MyD88, IRAK1/4 directly phosphorylates and activates Mst1, which phosphorylates IRAK1 leading to its degradation and inhibition of NF-κB signaling in a negative feedback loop, while promoting IRF3 activation, which induces the transcription of STK4 (Mst1) in a feed-forward loop70–72.
Upon activation by TLR signaling, Hippo or Mst1/2 kinases have been found to influence all three branches of TLR signaling. Two studies showed that Mst1/2 promote TLR-mediated activation of IRF3 signaling and IFN-β production in response to bacterial infection of murine cells70,72. Li et al. also reported that opposite of its stimulatory effect on IRF3 signaling, Mst1 inhibits TLR4-mediated NF-κB activation by binding to and phosphorylating IRAK1, targeting it for degradation (Fig. 3b)72. However, this finding was contradicted by two Drosophila studies, which showed that Yorkie directly transcribes Cactus (homolog of IkB), and loss of Hippo or activation of Yorkie leads to elevated Cactus mRNA levels and reduced anti-microbial peptides, whereas knockdown of Yorkie reduces Cactus levels while upregulating NF-κB transcription factors, DIF and Relish and other anti-microbial peptides (Fig. 3a)68,73. Finally, a critical role for Mst1/2 in TLR-induced mitochondrial recruitment to phagosomes was recently described (Fig. 3b)69. The authors found that Mst1/2 phosphorylate and activate PKC-α, which in turn phosphorylates and inactivates LyGDI, a cytosolic inhibitor of small GTPase Rac69. Free of LyGDI inhibition, Rac conjugates with GTP and undergoes TRAF6-mediated K63 poly-ubiquitination to become fully activated, allowing the formation of TRAF6-ECSIT complex that mediates mitochondrion-phagosome juxtaposition69.
TLR agonists have been shown great promises as adjuvant therapies in eliciting innate immune response, enhancing antigen presentation and reducing tumor immune tolerance to enhance response to standard immune-, radio- and chemo- therapies67. Thus, it would be important to further dissect the crosstalk between TLR signaling and Mst1/2 and other components of the Hippo pathway in the tumor context and resolve the discrepancies between existing studies.
The roles of Hippo-Yap/Taz signaling in cytosolic nucleic acid sensing
Beside membrane-bound TLRs, cells express a variety of cytosolic viral RNA/DNA sensors, including RIG-I-like receptors for detection of double-stranded RNA, and cGAS, AIM2, IFI16 and RNA polymerase III for detection of cytosolic DNA74. The same viral sensors are also responsible for detection of damage-associated molecular patterns (DAMPs) released by infected or tumor cells75. Once loaded, these sensors engage mitochondrial-associated MAVS, ER-located STING and/or other adaptors, causing them to self-aggregate, which in turn triggers the assembly of the TRAFs/TBK1/IKKε/IRF3 complex, dimerization and activation of IRF3, and the production of type I IFN74,75.
A number of recent studies have identified potential roles for Hippo-Yap/Taz signaling in regulation of cytosolic nucleic acid sensing in the context of viral infection. Contrary to the above-described positive roles in TLR-IRF3 signaling in response to bacteria infections70,72, Mst1 was recently shown to inhibit viral-induced IRF3 activation through both direct inhibitory phosphorylation of IRF3 and blockade of its activator - TBK1 (Fig. 4)76. Two other studies showed that Yap and/or Taz also inhibit IRF3 signaling and type I IFN production, rendering cells more susceptible to viral infection77,78. Zhang et al. found that viral infection cause Yap and Taz to translocate into the cytoplasm where they directly interact with TBK1 and prevent IRF3 activation by blocking TBK1 ubiquitination and subsequent recruitment of adaptors/substrates (Fig. 4)78. In contrast, Wang et al. showed that Yap does not affect the phosphorylation levels of TBK1, IKKε and IRF3, but rather directly binds to IRF3 and inhibits its dimerization and nuclear translocation (Fig. 4)77. Furthermore, Wang et al. demonstrated that viral-activated kinase IKKε phosphorylates and triggers lysosomal degradation of Yap, thereby relieving Yap-mediated inhibition of the cellular antiviral response (Fig. 4)77. Contrary to these two studies, a third study by Jiao et al. found that viral infection triggers IRF3-mediated nuclear translocation of Yap, and IRF3 complexes with Yap and Tead to further augment their transcriptional activities (Fig. 4)79. A notable difference between the three studies was viral exposure time: ~48 hours in Jiao et al. study versus 1 and 8 hours in studies by Zhang et al. and Wang et al., respectively. Further kinetic studies will be necessary to determine whether acute versus chronic viral infections could exert differential effects on the Hippo-Yap/Taz pathway, and vise versa. Given that oncolytic viruses and other activators of cytosolic nucleic acid sensing pathways have been successfully used to boost the efficacies of T-cell based immune therapies in clinical and preclinical studies75,80–83, it would be also important to understand how the statuses of Hippo signaling in tumor cells and tumor-infiltrating APCs influence the response to these combination therapies.
Figure 4. The roles of Mst1, Yap and Taz in cytosolic nucleic acid sensing.
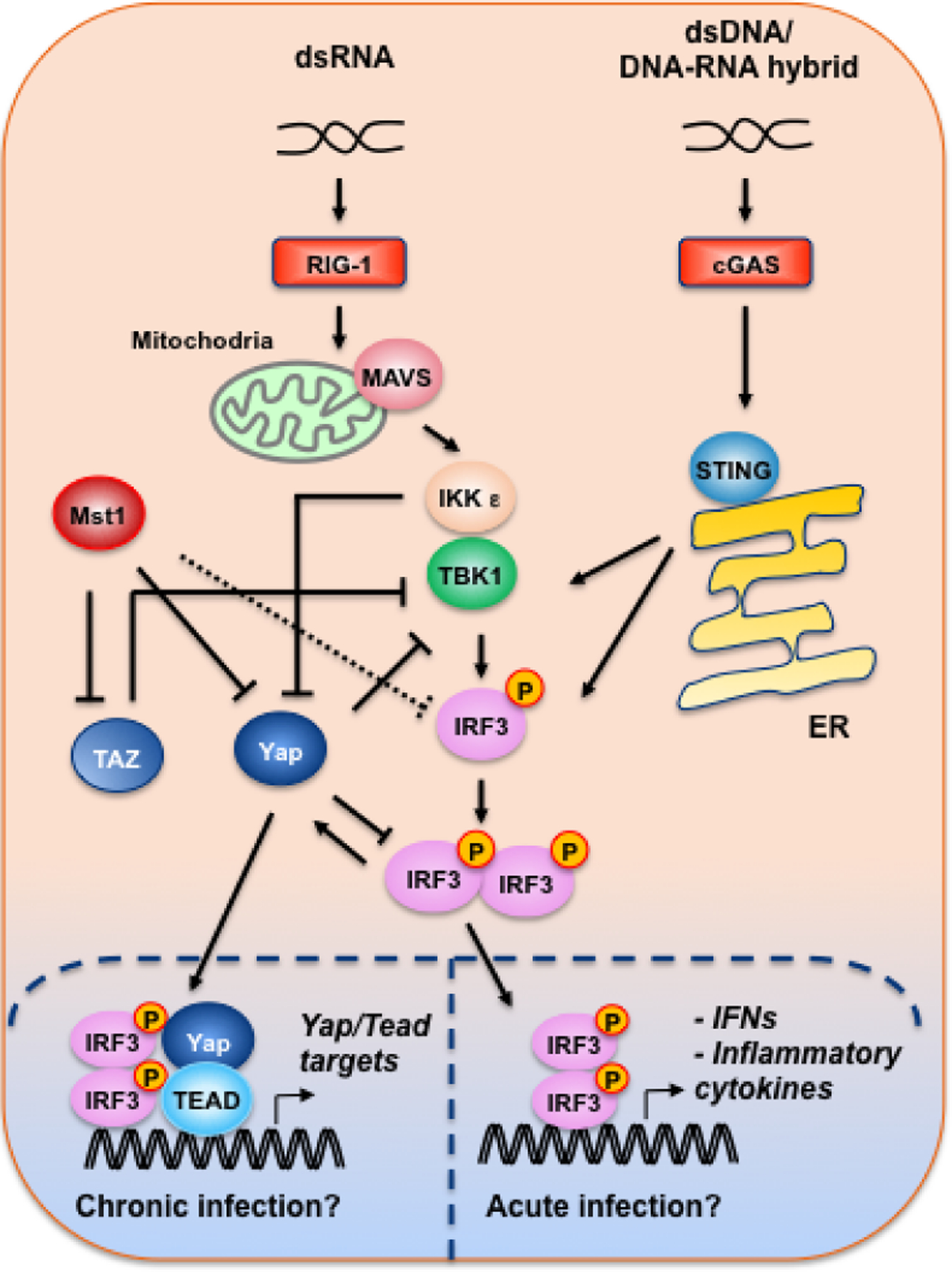
dsRNA and dsDNA or DNA-RNA hybrid activate the RIG-1-MAVS and cGAS-STING cytosolic sensors, respectively, leading to the activation of the TBK1-IKKε complex. In contrast to its reported roles in promoting bacteria-induced TLR-IRF3 signaling70,72, Mst1 was found to directly phosphorylate IRF3, inhibiting IRF3 activity in response to viral infections76. Yap and Taz were shown to directly bind TBK1 and block its interaction with other adaptors/substrates78. Moreover, Yap was reported to directly interfere with IRF3 dimerization, and IKKε relieves Yap inhibition of IRF3 by directly phosphorylating and targeting Yap for lysosomal degradation77. Finally, it was reported that during chronic (>48 hours) viral infection, dimerized IRF3 promotes Yap nuclear translocation and complexes with Yap and Tead in inducing the expression Yap/Tead target genes85.
Implications to Cancer Immune Therapies
Existing studies indicate that within tumor cells, Yap/Taz promote not only the expression of PD-L1, which directly inactivates T cells and promotes immune evasion, but also the production of various immune suppressive cytokines that attract immune suppressive myeloid cells to further suppress T cell functions. Although not yet demonstrated in a tumor setting, Yap/Taz were recently shown to promote Treg recruitment and suppress inflammatory response following heart injury by promoting IFN-γ production in epicardial cells84. Thus, blocking Yap/Taz activities within tumor cells will likely provide the dual benefits of suppressing tumor growth and reversing the immune suppressive TME.
Beyond controlling TME from within tumor cells, Hippo-Yap/Taz signaling also directly regulates the differentiation, homeostasis and functions of various subtypes of immune cells. From the extensive literature on the Mst1/2 kinases in T cell development and functions, the consensus is that Mst1/2 play key roles in directing the migration and egress of naïve T cells from the thymus, and the differentiation of CD4+ T cell towards the Treg lineage. Mst1 is also essential for B cell development by maintaining CD19 expression and BCR signaling. Within the innate immune cell populations, Mst1/2 promote TLR signaling, type I IFN production and phagocytosis in response to bacterial infection, and are specifically required for maintaining metabolic homeostasis and T-cell-priming activities of Baf3+CD8α+ dendritic cells - the key APCs for CD8+ T cell activation. Hence, systematic delivery of drugs that activate Mst1/2 kinases should in theory enhance overall immunity and boost response to existing immune checkpoint therapies. However, the immune-boosting effects of Mst1/2 might be dampened by their roles in promoting Treg differentiation and in inhibiting cytosolic nucleic acid sensing, a prospect that requires further investigation.
Even though the majority of the immune-regulatory activities of Mst1/2 are through noncanonical mechanisms, their canonical downstream effectors Yap/Taz also play key roles in T cell differentiation and innate immune response. During lineage commitment of CD4+ T cells, Taz functions opposite of Mst1/2 in promoting TH17 differentiation at the expense of Tregs, while Yap drives Treg differentiation by transcriptionally amplifying TGF-β signaling, implying decoupling of Yap from the canonical Hippo pathway in Treg cells. Yap/Taz have also been implicated in regulations of IRF3 signaling and type I IFN production in response to viral infections, although their precise effects and mechanisms remain controversial. Interestingly, most of the reported regulatory functions of Yap and Taz in immune cells appear to be independent of their transcriptional activities or at least not through their conventional transcriptional partners Tead1–4. This is intriguing given that the abilities of Yap/Taz to bind to Tead1–4 are critical to their growth-promoting activities in non-immune cells and strategies to block Yap/Taz-Tead interactions are being actively pursued as potential cancer therapies8. Thus, further studies to elucidate the divergence of Hippo-Yap/Taz signaling in immune cells from other cell types and how different approaches of Yap/Taz blockade differentially affect the immune system will be key to determine how to best target this important signaling network to maximize the anti-tumor activities.
ACKNOWLEDGEMENT
Research in Yi lab is currently supported by NIH (R01CA187090), Toulmin Pilot Award, Sher Grant and Cancer Center Support Grant (CA051008). Shannon White is a current recipient of National Science Foundation Graduate Research Fellowship (NSF-GRF#2018265935).
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
REFERENCES
- 1.Zappasodi R, Merghoub T, Wolchok JD. Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer Cell. 2018; 33: 581–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moynihan KD, Irvine DJ. Roles for innate immunity in combination immunotherapies. Cancer Res. 2017; 77: 5215–5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 2007; 21: 2747–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lei Q-Y, Zhang H, Zhao B, Zha Z-Y, Bai F, Pei X-H et al. TAZ Promotes Cell Proliferation and Epithelial-Mesenchymal Transition and Is Inhibited by the Hippo Pathway. Mol Cell Biol 2008; 28: 2426–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu C-Y, Zha Z-Y, Zhou X, Zhang H, Huang W, Zhao D et al. The Hippo Tumor Pathway Promotes TAZ Degradation by Phosphorylating a Phosphodegron and Recruiting the SCF β-TrCP E3 Ligase. J Biol Chem 2010; 285: 37159–37169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao B, Li L, Tumaneng K, Wang C-Y, Guan K-L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF -TRCP. Genes Dev 2010; 24: 72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moon S, Yeon Park S, Woo Park H. Regulation of the Hippo pathway in cancer biology. Cell Mol Life Sci 2018; 75: 2303–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu V, Plouffe SW, Guan K-L. The Hippo pathway in organ development, homeostasis, and regeneration. Curr Opin Cell Biol 2017; 49: 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galan JA, Avruch J. MST1/MST2 Protein Kinases: Regulation and Physiologic Roles. Biochemistry 2016; 55: 5507–5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furth N, Aylon Y. The LATS1 and LATS2 tumor suppressors: beyond the Hippo pathway. Cell Death Differ 2017; 24: 1488–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma,1,* Padmanee Hu-Lieskovan,2 Siwen Wargo 3 Jennifer A. and Ribas2 Antoni. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell Rev 2017; 168 http://ac.els-cdn.com/S009286741730065X/1-s2.0-S009286741730065X-main.pdf?_tid=8c86f758-4a19-11e7-b4b0-00000aab0f6c&acdnat=1496686105_798335971803e859ab3b6cce754a7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim MH, Kim CG, Kim S-K, Shin SJ, Choe EA, Park S-H et al. YAP-induced PD-L1 expression drives immune evasion in BRAFi-resistant melanoma. Cancer Immunol Res 2018; : canimm.0320.2017. [DOI] [PubMed] [Google Scholar]
- 13.Lee BS, Park D Il, Lee DH, Lee JE, Yeo M, Park YH et al. Hippo effector YAP directly regulates the expression of PD-L1 transcripts in EGFR-TKI-resistant lung adenocarcinoma. Biochem Biophys Res Commun 2017; 491: 493–499. [DOI] [PubMed] [Google Scholar]
- 14.Janse van Rensburg HJ, Azad T, Ling M, Hao Y, Snetsinger B, Khanal P et al. The Hippo Pathway Component TAZ Promotes Immune Evasion in Human Cancer through PD-L1. Cancer Res 2018; 78: 1457–1470. [DOI] [PubMed] [Google Scholar]
- 15.Miao J, Hsu P-C, Yang Y-L, Xu Z, Dai Y, Wang Y et al. YAP regulates PD-L1 expression in human NSCLC cells. Oncotarget 2017; 8: 114576–114587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schupp J, Krebs FK, Zimmer N, Trzeciak E, Schuppan D, Tuettenberg A. Targeting myeloid cells in the tumor sustaining microenvironment. Cell. Immunol 2017. doi: 10.1016/j.cellimm.2017.10.013. [DOI] [PubMed] [Google Scholar]
- 17.Sica A, Massarotti M. Myeloid suppressor cells in cancer and autoimmunity. J. Autoimmun 2017; 85: 117–125. [DOI] [PubMed] [Google Scholar]
- 18.Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S et al. Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov 2016; 6: 80–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murakami S, Shahbazian D, Surana R, Zhang W, Chen H, Graham GT et al. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene 2017; 36: 1232–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim T, Yang SJ, Hwang D, Song J, Kim M, Kyum Kim S et al. A basal-like breast cancer-specific role for SRF-IL6 in YAP-induced cancer stemness. Nat Commun 2015; 6. doi: 10.1038/ncomms10186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo X, Zhao Y, Yan H, Yang Y, Shen S, Dai X et al. Single tumor-initiating cells evade immune clearance by recruiting type II macrophages. Genes Dev 2017; 31: 247–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim W, Khan SK, Liu Y, Xu R, Park O, He Y et al. Hepatic Hippo signaling inhibits protumoural microenvironment to suppress hepatocellular carcinoma. Gut. 2017. doi: 10.1136/gutjnl-2017-314061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016; 30: 1002–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.von Ahrens D, Bhagat TD, Nagrath D, Maitra A, Verma A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J Hematol Oncol 2017; 10: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang W, Nandakumar N, Shi Y, Manzano M, Smith A, Graham G et al. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci Signal 2014; 7: ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol 2013; 15: 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med 2016; 22: 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foster CT, Gualdrini F, Treisman R. Mutual dependence of the MRTF–SRF and YAP–TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev 2017; 31: 2361–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu F, Lagares D, Choi KM, Stopfer L, Marinković A, Vrbanac V et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Cell Mol Physiol 2015; 308: L344–L357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nardone G, Oliver-De La Cruz J, Vrbsky J, Martini C, Pribyl J, Skládal P et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nat Commun 2017; 8. doi: 10.1038/ncomms15321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N et al. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013; 154: 1047–1059. [DOI] [PubMed] [Google Scholar]
- 32.Furukawa KT, Yamashita K, Sakurai N, Ohno S. The Epithelial Circumferential Actin Belt Regulates YAP/TAZ through Nucleocytoplasmic Shuttling of Merlin. Cell Rep 2017; 20: 1435–1447. [DOI] [PubMed] [Google Scholar]
- 33.Sansores-Garcia L, Bossuyt W, Wada KI, Yonemura S, Tao C, Sasaki H et al. Modulating F-actin organization induces organ growth by affecting the Hippo pathway. EMBO J 2011; 30: 2325–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elosegui-Artola A, Oria R, Chen Y, Kosmalska A, Pérez-González C, Castro N et al. Mechanical regulation of a molecular clutch defines force transmission and transduction in response to matrix rigidity. Nat Cell Biol 2016; 18: 540–548. [DOI] [PubMed] [Google Scholar]
- 35.Meng Z, Qiu Y, Lin KC, Kumar A, Placone JK, Fang C et al. RAP2 mediates mechanoresponses of the Hippo pathway. Nature 2018; 560: 655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 2016; 22: 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ehmer U, Zmoos AF, Auerbach RK, Vaka D, Butte AJ, Kay MA et al. Organ Size Control Is Dominant over Rb Family Inactivation to Restrict Proliferation InVivo. Cell Rep 2014; 8: 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyamura N, Hata S, Itoh T, Tanaka M, Nishio M, Itoh M et al. YAP determines the cell fate of injured mouse hepatocytes in vivo. Nat Commun 2017; 8. doi: 10.1038/ncomms16017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moroishi T, Hayashi T, Pan W-W, Fujita Y, Holt MV, Qin J et al. The Hippo Pathway Kinases LATS1/2 Suppress Cancer Immunity. Cell 2016; 167: 1525–1539.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee D-H, Park JO, Kim T-S, Kim S-K, Kim T-H, Kim M-C et al. LATS-YAP/TAZ controls lineage specification by regulating TGFβ signaling and Hnf4α expression during liver development. Nat Commun 2016; 7: 11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol 2007; 9: 493–505. [DOI] [PubMed] [Google Scholar]
- 42.Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011; 479: 547–551. [DOI] [PubMed] [Google Scholar]
- 43.Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 2013; 15: 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cisowski J, Sayin VI, Liu M, Karlsson C, Bergo MO. Oncogene-induced senescence underlies the mutual exclusive nature of oncogenic KRAS and BRAF. Oncogene 2016; 35: 1328–1333. [DOI] [PubMed] [Google Scholar]
- 45.Abdollahpour H, Appaswamy G, Kotlarz D, Diestelhorst J, Beier R, Schäffer AA et al. The phenotype of human STK4 deficiency. Blood 2012; 119: 3450–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nehme NT, Schmid JP, Debeurme F, André-Schmutz I, Lim A, Nitschke P et al. MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood 2012; 119: 3458–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bai X, Huang L, Niu L, Zhang Y, Wang J, Sun X et al. Mst1 positively regulates B-cell receptor signaling via CD19 transcriptional levels. Blood Adv 2016; 1: 219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou D, Medoff BD, Chen L, Li L, Zhang X, Praskova M et al. The Nore1B/Mst1 complex restrains antigen receptor-induced proliferation of naïve T cells. Proc Natl Acad Sci U S A 2008; 105: 20321–20326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi J, Oh S, Lee D, Oh HJ, Park JY, Lee SB et al. Mst1-FoxO signaling protects Naive T lymphocytes from cellular oxidative stress in mice. PLoS One 2009; 4: e8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Du X, Shi H, Li J, Dong Y, Liang J, Ye J et al. Mst1/Mst2 regulate development and function of regulatory T cells through modulation of Foxo1/Foxo3 stability in autoimmune disease. J Immunol 2014; 192: 1525–35. [DOI] [PubMed] [Google Scholar]
- 51.Ueda Y, Katagiri K, Tomiyama T, Yasuda K, Habiro K, Katakai T et al. Mst1 regulates integrin-dependent thymocyte trafficking and antigen recognition in the thymus. Nat Commun 2012; 3. doi: 10.1038/ncomms2105. [DOI] [PubMed] [Google Scholar]
- 52.Dong Y, Du X, Ye J, Han M, Xu T, Zhuang Y et al. A Cell-Intrinsic Role for Mst1 in Regulating Thymocyte Egress. J Immunol 2009; 183: 3865–3872. [DOI] [PubMed] [Google Scholar]
- 53.Mou F, Praskova M, Xia F, Van Buren D, Hock H, Avruch J et al. The Mst1 and Mst2 kinases control activation of rho family GTPases and thymic egress of mature thymocytes. J Exp Med 2012; 209: 741–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Katagiri K, Imamura M, Kinashi T. Spatiotemporal regulation of the kinase Mst1 by binding protein RAPL is critical for lymphocyte polarity and adhesion. Nat Immunol 2006; 7: 919–928. [DOI] [PubMed] [Google Scholar]
- 55.Katagiri K, Katakai T, Ebisuno Y, Ueda Y, Okada T, Kinashi T. Mst1 controls lymphocyte trafficking and interstitial motility within lymph nodes. EMBO J 2009; 28: 1319–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu X, Jaeger ER, Wang X, Lagler-Ferrez E, Batalov S, Mathis NL et al. Mst1 directs myosin IIa partitioning of low and higher affinity integrins during T cell migration. PLoS One 2014; 9. doi: 10.1371/journal.pone.0105561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishikimi A, Ishihara S, Ozawa M, Etoh K, Fukuda M, Kinashi T et al. Rab13 acts downstream of the kinase Mst1 to deliver the integrin LFA-1 to the cell surface for lymphocyte trafficking. Sci Signal 2014; 7. doi: 10.1126/scisignal.2005199. [DOI] [PubMed] [Google Scholar]
- 58.Knochelmann HM, Dwyer CJ, Bailey SR, Amaya SM, Elston DM, Mazza-McCrann JM et al. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell Mol Immunol 2018; 15: 458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li J, Du X, Shi H, Deng K, Chi H, Tao W. Mammalian Sterile 20-like Kinase 1 (Mst1) Enhances the Stability of Forkhead Box P3 (Foxp3) and the Function of Regulatory T Cells by Modulating Foxp3 Acetylation. J Biol Chem 2015; 290: 30762–30770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Geng J, Yu S, Zhao H, Sun X, Li X, Wang P et al. The transcriptional coactivator TAZ regulates reciprocal differentiation of TH17 cells and Treg cells. Nat Immunol 2017; 18: 800–812. [DOI] [PubMed] [Google Scholar]
- 61.Li C, Bi Y, Li Y, Yang H, Yu Q, Wang J et al. Dendritic cell MST1 inhibits Th17 differentiation. Nat Commun 2017; 8: 14275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ni X, Tao J, Barbi J, Chen Q, Park BV, Li Z et al. YAP is essential for Treg mediated suppression of anti-tumor immunity. Cancer Discov 2018; : CD-17–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lo B, Abdel-Motal UM. Lessons from CTLA-4 deficiency and checkpoint inhibition. Curr Opin Immunol 2017; 49: 14–19. [DOI] [PubMed] [Google Scholar]
- 64.Thaventhiran JED, Hoffmann A, Magiera L, de la Roche M, Lingel H, Brunner-Weinzierl M et al. Activation of the Hippo pathway by CTLA-4 regulates the expression of Blimp-1 in the CD8+ T cell. Proc Natl Acad Sci 2012; 109: E2223–E2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseño CG, Iwata A et al. Transcriptional Control of Dendritic Cell Development. Annu Rev Immunol 2016; 34: 93–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Du X, Wen J, Wang Y, Karmaus PWF, Khatamian A, Tan H et al. Hippo/Mst signalling couples metabolic state and immune function of CD8α+dendritic cells. Nature 2018; 558: 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cen X, Liu S, Cheng K. The Role of Toll-Like Receptor in Inflammation and Tumor Immunity. Front Pharmacol 2018; 9: 878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu B, Zheng Y, Yin F, Yu J, Silverman N, Pan D. Toll Receptor-Mediated Hippo Signaling Controls Innate Immunity in Drosophila. Cell 2016; 164: 406–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Geng J, Sun X, Wang P, Zhang S, Wang X, Wu H et al. Kinases Mst1 and Mst2 positively regulate phagocytic induction of reactive oxygen species and bactericidal activity. Nat Immunol 2015; 16: 1142–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boro M, Singh V, Balaji KN. Mycobacterium tuberculosis-triggered Hippo pathway orchestrates CXCL1/2 expression to modulate host immune responses. Sci Rep 2016; 6. doi: 10.1038/srep37695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yuan L, Mao Y, Luo W, Wu W, Xu H, Wang XL et al. Palmitic acid dysregulates the Hippo–YAP pathway and inhibits angiogenesis by inducing mitochondrial damage and activating the cytosolic DNA sensor cGAS–STING–IRF3 signaling mechanism. 2017. doi: 10.1074/jbc.M117.804005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li W, Xiao J, Zhou X, Xu M, Hu C, Xu X et al. STK4 regulates TLR pathways and protects against chronic inflammation-related hepatocellular carcinoma. J Clin Invest 2015; 125: 4239–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dubey SK, Tapadia MG. Yorkie Regulates Neurodegeneration Through Canonical Pathway and Innate Immune Response. Mol. Neurobiol 2017; : 1–15. [DOI] [PubMed] [Google Scholar]
- 74.Luecke S, Paludan SR. Molecular requirements for sensing of intracellular microbial nucleic acids by the innate immune system. Cytokine 2017; 98: 4–14. [DOI] [PubMed] [Google Scholar]
- 75.Iurescia S, Fioretti D, Rinaldi M. Targeting Cytosolic Nucleic Acid-Sensing Pathways for Cancer Immunotherapies. Front Immunol 2018; 9: 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meng F, Zhou R, Wu S, Zhang Q, Jin Q, Zhou Y et al. Mst1 shuts off cytosolic antiviral defense through IRF3 phosphorylation. Genes Dev 2016; 30: 1086–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang S, Xie F, Chu F, Zhang Z, Yang B, Dai T et al. YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKK[epsiv]-mediated phosphorylation. Nat Immunol 2017; 18: 733–743. [DOI] [PubMed] [Google Scholar]
- 78.Zhang Q, Meng F, Chen S, Plouffe SW, Wu S, Liu S et al. Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ-mediated TBK1 blockade. Nat Cell Biol 2017; 19: 362–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jiao S, Guan J, Chen M, Wang W, Li C, Wang Y et al. Targeting IRF3 as a YAP agonist therapy against gastric cancer. J Exp Med 2018; 215: 699–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017; 170: 1109–1119.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Samson A, Scott KJ, Taggart D, West EJ, Wilson E, Nuovo GJ et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med 2018; 10: eaam7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P et al. Localized Oncolytic Virotherapy Overcomes Systemic Tumor Resistance to Immune Checkpoint Blockade Immunotherapy. Sci Transl Med 2014; 6: 226ra32–226ra32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu Z, Ravindranathan R, Kalinski P, Guo ZS, Bartlett DL. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat Commun 2017; 8: 14754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ramjee V, Li D, Manderfield LJ, Liu F, Engleka KA, Aghajanian H et al. Epicardial YAP/TAZ orchestrate an immunosuppressive response following myocardial infarction. J Clin Invest 2017; 127: 899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jiao S, Guan J, Chen M, Wang W, Li C, Wang Y et al. Targeting IRF3 as a YAP agonist therapy against gastric cancer. J Exp Med 2018. doi: 10.1084/jem.20171116. [DOI] [PMC free article] [PubMed] [Google Scholar]