

Abstract
Eslicarbazepine acetate (ESL) is a dibenzazepine anticonvulsant approved as adjunctive treatment for partial-onset epileptic seizures. Following first pass hydrolysis of ESL, S-licarbazepine (S-Lic) represents around 95% of circulating active metabolites. S-Lic is the main enantiomer responsible for anticonvulsant activity and this is proposed to be through the blockade of voltage-gated Na+ channels (VGSCs). ESL and S-Lic both have a voltage-dependent inhibitory effect on the Na+ current in N1E-115 neuroblastoma cells expressing neuronal VGSC subtypes including Nav1.1, Nav1.2, Nav1.3, Nav1.6, and Nav1.7. ESL has not been associated with cardiotoxicity in healthy volunteers, although a prolongation of the electrocardiographic PR interval has been observed, suggesting that ESL may also inhibit cardiac Nav1.5 isoform. However, this has not previously been studied. Here, we investigated the electrophysiological effects of ESL and S-Lic on Nav1.5 using whole-cell patch clamp recording. We interrogated two model systems: (1) MDA-MB-231 metastatic breast carcinoma cells, which endogenously express the “neonatal” Nav1.5 splice variant, and (2) HEK-293 cells stably over-expressing the “adult” Nav1.5 splice variant. We show that both ESL and S-Lic inhibit transient and persistent Na+ current, hyperpolarise the voltage-dependence of fast inactivation, and slow the recovery from channel inactivation. These findings highlight, for the first time, the potent inhibitory effects of ESL and S-Lic on the Nav1.5 isoform, suggesting a possible explanation for the prolonged PR interval observed in patients on ESL treatment. Given that numerous cancer cells have also been shown to express Nav1.5, and that VGSCs potentiate invasion and metastasis, this study also paves the way for future investigations into ESL and S-Lic as potential invasion inhibitors.
Keywords: anticonvulsant, cancer, epilepsy, eslicarbazepine acetate, Nav1.5, S-licarbazepine, voltage-gated Na+ channel
Introduction
Eslicarbazepine acetate (ESL) is a member of the dibenzazepine anticonvulsant family of compounds which also includes oxcarbazepine and carbamazepine (Almeida and Soares-da-Silva, 2007). ESL has been approved by the European Medicines Agency and the United States Federal Drug Administration as an adjunctive treatment for partial-onset epileptic seizures (Sperling et al., 2015). ESL is administered orally and rapidly undergoes first pass hydrolysis to two stereoisomeric metabolites, R-licarbazepine and S-licarbazepine (S-Lic; also known as eslicarbazepine; Figures 1A, B) (Almeida et al., 2005; Almeida et al., 2008; Perucca et al., 2011). S-Lic represents around 95% of circulating active metabolites following first pass hydrolysis of ESL and is the enantiomer responsible for anticonvulsant activity (Potschka et al., 2014; Sierra-Paredes et al., 2014). S-Lic also has improved blood brain barrier penetration compared to R-licarbazepine (Alves et al., 2008). Although S-Lic has been shown to inhibit T type Ca2+ channels (Brady et al., 2011), its main activity is likely through blockade of voltage-gated Na+ channels (VGSCs) (Hebeisen et al., 2015). ESL offers several clinical advantages over other older VGSC-inhibiting antiepileptic drugs, e.g. carbamazepine, phenytoin; it has a favourable safety profile (Brown and El-Mallakh, 2010; Hebeisen et al., 2015), reduced induction of hepatic cytochrome P450 enzymes (Galiana et al., 2017), low potential for drug-drug interactions (Falcao et al., 2012; Zaccara et al., 2015), and takes less time to reach a steady state plasma concentration (Bialer and Soares-da-Silva, 2012).
Figure 1.
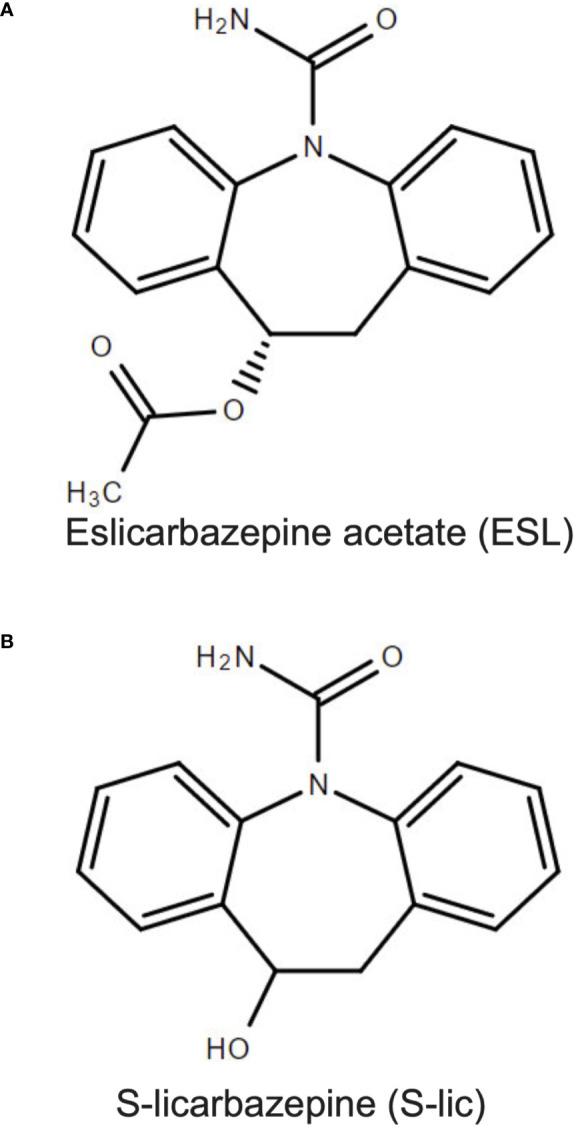
Chemical structures of eslicarbazepine acetate and S-licarbazepine. (A) eslicarbazepine acetate; (9S)-2-carbamoyl-2-azatricyclo[9.4.0.038]pentadeca-1 (15),3,5,7,11,13-hexaen-9-yl acetate. (B) S-licarbazepine; (10R)-10-hydroxy-2-azatricyclo[9.4.0.03,8]pentadeca-1(11),3,5,7,12,14-hexaene-2-carboxamide. Structures were drawn using Chemspider software.
VGSCs are composed of a pore-forming α subunit in association with one or more auxiliary β subunits, the latter modulating channel gating and kinetics in addition to functioning as cell adhesion molecules (Catterall, 2014). There are nine α subunits (Nav1.1-Nav1.9), and four β subunits (β1-4) (Goldin et al., 2000; Brackenbury and Isom, 2011). In postnatal and adult CNS neurons, the predominant α subunits are the tetrodotoxin-sensitive Nav1.1, Nav1.2, and Nav1.6 isoforms (Van Wart and Matthews, 2006) and it is therefore on these that the VGSC-inhibiting activity of ESL and S-Lic has been described. In the murine neuroblastoma N1E-115 cell line, which expresses Nav1.1, Nav1.2, Nav1.3, Nav1.6, and Nav1.7, ESL and S-Lic both have a voltage-dependent inhibitory effect on the Na+ current (Bonifacio et al., 2001; Hebeisen et al., 2015). In this cell model, S-Lic has no effect on the voltage-dependence of fast inactivation, but significantly hyperpolarises the voltage-dependence of slow inactivation (Hebeisen et al., 2015). S-Lic also has a lower affinity for VGSCs in the resting state than carbamazepine or oxcarbazepine, thus potentially improving its therapeutic window over first- and second-generation dibenzazepine compounds (Hebeisen et al., 2015). In acutely isolated murine hippocampal CA1 neurons, which express Nav1.1, Nav1.2 and Nav1.6 (Westenbroek et al., 1989; Yu et al., 2006; Royeck et al., 2008), S-Lic significantly reduces the persistent Na+ current, a very slow-inactivating component ~1% the size of the peak transient Na+ current (Saint, 2008; Doeser et al., 2014). Moreover, in contrast to carbamazepine, this effect is maintained in the absence of β1 (Uebachs et al., 2010; Doeser et al., 2014).
In healthy volunteers, ESL has not been associated with cardiotoxicity and the QT interval remains unchanged on treatment (Vaz-Da-Silva et al., 2012). However, a prolongation of the PR interval has been observed (Vaz-Da-Silva et al., 2012), suggesting that caution should be exercised in patients with cardiac conduction abnormalities (Zaccara et al., 2015). Prolongation of the PR interval suggests that ESL may also inhibit the cardiac Nav1.5 isoform, although this has not previously been studied. Nav1.5 is not only responsible for the initial depolarisation of the cardiac action potential (George, 2005), but is also expressed in breast and colon carcinoma cells, where the persistent Na+ current promotes invasion and metastasis (Roger et al., 2003; Fraser et al., 2005; House et al., 2010; Nelson et al., 2015a). Inhibition of Nav1.5 with phenytoin or ranolazine decreases tumor growth, invasion and metastasis (Yang et al., 2012; Driffort et al., 2014; Nelson et al., 2015b). Thus, it is of interest to specifically understand the effect of ESL on the Nav1.5 isoform.
In the present study we investigated the electrophysiological effects of ESL and S-Lic on Nav1.5 [1] endogenously expressed in the MDA-MB-231 metastatic breast carcinoma cell line, and [2] stably over-expressed in HEK-293 cells. We show that both ESL and S-Lic inhibit transient and persistent Na+ current, hyperpolarise the voltage-dependence of fast inactivation, and slow the recovery from channel inactivation. These findings highlight, for the first time, the potent inhibitory effects of ESL and S-Lic on the Nav1.5 isoform.
Materials and Methods
Pharmacology
ESL (Tokyo Chemical Industry UK Ltd) was dissolved in DMSO to make a stock concentration of 67 mM. S-Lic (Tocris) was dissolved in DMSO to make a stock concentration of 300 mM. Both drugs were diluted to working concentrations of 100–300 µM in extracellular recording solution. The concentration of DMSO in the recording solution was 0.45% for ESL and 0.1% for S-Lic. Equal concentrations of DMSO were used in the control solutions. DMSO (0.45%) had no effect on the Na+ current (Supplementary Figure 1).
Cell Culture
MDA-MB-231 cells and HEK-293 cells stably expressing Nav1.5 (a gift from L. Isom, University of Michigan) were grown in Dulbecco’s modified eagle medium supplemented with 5% FBS and 4 mM L-glutamine (Simon et al., 2020). Molecular identity of the MDA-MB-231 cells was confirmed by short tandem repeat analysis (Masters et al., 2001). Cells were confirmed as mycoplasma-free using the DAPI method (Uphoff et al., 1992). Cells were seeded onto glass coverslips 48 h before electrophysiological recording.
Electrophysiology
Plasma membrane Na+ currents were recorded using the whole-cell patch clamp technique, using methods described previously (Yang et al., 2012; Nelson et al., 2015a). Patch pipettes made of borosilicate glass were pulled using a P-97 pipette puller (Sutter Instrument) and fire-polished to a resistance of 3–5 MΩ when filled with intracellular recording solution. The extracellular recording solution for MDA-MB-231 cells contained (in mM): 144 NaCl, 5.4 KCl, 1 MgCl2, 2.5 CaCl2, 5.6 D-glucose, and 5 HEPES (adjusted to pH 7.2 with NaOH). For the extracellular recording solution for HEK-293 cells expressing Nav1.5, the extracellular [Na+] was reduced to account for the much larger Na+ currents and contained (in mM): 60 NaCl, 84 Choline Cl, 5.4 KCl, 1 MgCl2, 2.5 CaCl2, 5.6 D-glucose, and 5 HEPES (adjusted to pH 7.2 with NaOH). The intracellular recording solution contained (in mM): 5 NaCl, 145 CsCl, 2 MgCl2, 1 CaCl2, 10 HEPES, 11 EGTA, (adjusted to pH 7.4 with CsOH) (Brackenbury and Djamgoz, 2006). Voltage clamp recordings were made at room temperature using a Multiclamp 700B or Axopatch 200B amplifier (Molecular Devices) compensating for series resistance by 40–60%. Currents were digitized using a Digidata interface (Molecular Devices), low pass filtered at 10 kHz, sampled at 50 kHz and analysed using pCLAMP 10.7 software (Molecular Devices). Leak current was subtracted using a P/6 protocol (Armstrong and Bezanilla, 1977). Extracellular recording solution ± drugs was applied to the recording bath at a rate of ~1.5 ml/min using a ValveLink 4-channel gravity perfusion controller (AutoMate Scientific). Each new solution was allowed to equilibrate in the bath for ~4 min following switching prior to recording at steady state.
Voltage Clamp Protocols
Cells were clamped at a holding potential of -120 mV or -80 mV for ≥250 ms, dependent on experiment (detailed in the Figure legends). Five main voltage clamp protocols were used, as follows:
To assess the effect of drug perfusion and wash-out on peak current in real time, a simple one-step protocol was used where cells were held at -120 mV or -80 mV for 250 ms and then depolarised to -10 mV for 50 ms.
To assess the voltage-dependence of activation, cells were held at -120 mV for 250 ms and then depolarised to test potentials in 10 mV steps between -120 mV and +30 mV for 50 ms. The voltage of activation was taken as the most negative voltage which induced a visible transient inward current.
To assess the voltage-dependence of steady-state inactivation, cells were held at -120 mV for 250 ms followed by prepulses for 250 ms in 10 mV steps between -120 mV and +30 mV and a test pulse to -10 mV for 50 ms.
To assess recovery from fast inactivation, cells were held at -120 mV for 250 ms, and then depolarised twice to 0 mV for 25 ms, returning to -120 mV for the following intervals between depolarisations (in ms): 1, 2, 3, 5, 7, 10, 15, 20, 30, 40, 50, 70, 100, 150, 200, 250, 350, 500. In each case, the second current was normalized to the initial current and plotted against the interval time.
Curve Fitting and Data Analysis
To study the voltage-dependence of activation, current-voltage (I-V) relationships were converted to conductance using the following equation:
G = I/(Vm – Vrev), where G is conductance, I is current, Vm is the membrane voltage and Vrev is the reversal potential for Na+ derived from the Nernst equation. Given the different recording solutions used, Vrev for Na+ was +85 mV for MDA-MB-231 cells and +63 mV for HEK-Nav1.5 cells.
The voltage-dependence of conductance and availability were normalized and fitted to a Boltzmann equation:
G = Gmax/[1 + exp ((V1/2 – Vm)/k)], where Gmax is the maximum conductance, V1/2 is the voltage at which the channels are half activated/inactivated, Vm is the membrane voltage and k is the slope factor.
Recovery from inactivation data (It/It=0) were normalized, plotted against recovery time (Δ;t) and fitted to a single exponential function:
τ = A1 + A2 exp (-t/t0), where A1 and A2 are the coefficients of decay of the time constant (τ), t is time and t0 is a time constant describing the time dependence of τ.
The time course of inactivation was fitted to a double exponential function:
I = Af exp (-t/τ f) + As exp (-t/τ s) + C, where Af and As are maximal amplitudes of the slow and fast components of the current, τf and τs are the fast and slow decay time constants and C is the asymptote.
Statistical Analysis
Data are presented as mean and SEM unless stated otherwise. Statistical analysis was performed on the raw (non-normalized) data using GraphPad Prism 8.4.0. Pairwise statistical significance was determined with Student’s paired t-tests. Multiple comparisons were made using ANOVA and Tukey post-hoc tests, unless stated otherwise. Results were considered significant at P < 0.05.
Results
Effect of Eslicarbazepine Acetate and S-Licarbazepine on Transient and Persistent Na+ Current
Several studies have clearly established the inhibition of neuronal VGSCs (Nav1.1, Nav1.2, Nav1.3, Nav1.6, Nav1.7 and Nav1.8) by ESL and its active metabolite S-Lic (Bonifacio et al., 2001; Doeser et al., 2014; Hebeisen et al., 2015; Soares-da-Silva et al., 2015). Given that ESL prolongs the PR interval (Vaz-Da-Silva et al., 2012), potentially via inhibiting the cardiac Nav1.5 isoform, together with the interest in inhibiting Nav1.5 in carcinoma cells to reduce invasion and metastasis (Driffort et al., 2014; Martin et al., 2015; Nelson et al., 2015b; Elajnaf et al., 2018; Djamgoz et al., 2019), it is also relevant to evaluate the electrophysiological effects of ESL and S-Lic on this isoform. We therefore evaluated the effect of both compounds on Nav1.5 current properties using whole-cell patch clamp recording, employing a two-pronged approach: (1) recording Nav1.5 currents endogenously expressed in the MDA-MB-231 breast cancer cell line (Roger et al., 2003; Fraser et al., 2005; Brackenbury et al., 2007), and (2) recording from Nav1.5 stably over-expressed in HEK-293 cells (HEK-Nav1.5) (Patino et al., 2011).
Initially, we evaluated the effect of both compounds on the size of the peak Na+ current in MDA-MB-231 cells. Na+ currents were elicited by depolarising the membrane potential (Vm) to -10 mV from a holding potential (Vh) of -120 or -80 mV. Application of the prodrug ESL (300 μM) reversibly inhibited the transient Na+ current by 49.6 ± 3.2% when the Vh was -120 mV (P < 0.001; n = 13; ANOVA + Tukey test; Figures 2A, D). When Vh was set to -80 mV, ESL (300 μM) reversibly inhibited the transient Na+ current by 79.5 ± 4.5% (P < 0.001; n = 12; ANOVA + Tukey test; Figures 2C, E). We next assessed the effect of ESL in HEK-Nav1.5 cells. Application of ESL (300 μM) inhibited Nav1.5 current by 74.7 ± 4.3% when Vh was -120 mV (P < 0.001; n = 12; Figures 2F, I) and by 90.5 ± 2.8% when Vh was -80 mV (P < 0.001; n = 14; Figures 2H, J). However, the inhibition was only partially reversible (P < 0.001; n = 14; Figures 2F, H–J). Application of ESL at a lower concentration (100 µM) elicited a similar result (Supplementary Figures 2A–J and Supplementary Table 1). Together, these data suggest that ESL preferentially inhibited Nav1.5 in the open or inactivated state, since the current inhibition was greater at more depolarised Vh.
Figure 2.

Effect of eslicarbazepine acetate on Nav1.5 currents. (A) Representative Na+ currents in an MDA-MB-231 cell elicited by a depolarisation from -120 to -10 mV in physiological saline solution (PSS; black), eslicarbazepine acetate (ESL; 300 μM; red) and after washout (grey). Dotted vertical lines define the time period magnified in (B). (B) Representative persistent Na+ currents in an MDA-MB-231 cell elicited by a depolarisation from -120 to -10 mV. (C) Representative Na+ currents in an MDA-MB-231 cell elicited by a depolarisation from -80 to -10 mV. (D) Normalized Na+ currents in MDA-MB-231 cells elicited by a depolarisation from -120 to -10 mV. (E) Normalized Na+ currents in MDA-MB-231 cells elicited by a depolarisation from -80 to -10 mV. (F) Representative Na+ currents in a HEK-Nav1.5 cell elicited by a depolarisation from -120 to -10 mV in PSS (black), ESL (300 μM; red) and after washout (grey). Dotted vertical lines define the time period magnified in (G). (G) Representative persistent Na+ currents in a HEK-Nav1.5 cell elicited by a depolarisation from -120 to -10 mV. (H) Representative Na+ currents in a HEK-Nav1.5 cell elicited by a depolarisation from -80 to -10 mV. (I) Normalized Na+ currents in HEK-Nav1.5 cells elicited by a depolarisation from -120 to -10 mV. (J) Normalized Na+ currents in HEK-Nav1.5 cells elicited by a depolarisation from -80 to -10 mV. Results are mean + SEM. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; one-way ANOVA with Tukey tests (n = 12–14). NS, not significant.
We next tested the effect of the active metabolite S-Lic. S-Lic (300 μM) inhibited the transient Na+ current in MDA-MB-231 cells by 44.4 ± 6.1% when the Vh was -120 mV (P < 0.001; n = 9; ANOVA + Tukey test; Figures 3A, D). When Vh was set to -80 mV, S-Lic (300 µM) inhibited the transient Na+ current by 73.6 ± 4.1% (P < 0.001; n = 10; ANOVA + Tukey test; Figures 3C, E). However, the inhibition caused by S-Lic (300 μM) was only partially reversible (P < 0.05; n = 10; ANOVA + Tukey test; Figures 3A, C–E). In HEK-Nav1.5 cells, S-Lic (300 μM) inhibited Nav1.5 current by 46.4 ± 3.9% when Vh was -120 mV (P < 0.001; n = 13; ANOVA + Tukey test; Figures 3F, I) and by 74.0 ± 4.2% when Vh was -80 mV (P < 0.001; n = 12; ANOVA + Tukey test; Figures 3H, J). Furthermore, the inhibition in HEK-Nav1.5 cells was not reversible over the duration of the experiment. Application of S-Lic at a lower concentration (100 µM) elicited a broadly similar result (Supplementary Figures 3A–J & Supplementary Table 1). Together, these data show that channel inhibition by S-Lic was also more effective at more depolarised Vh. However, unlike ESL, channel blockade by S-Lic persisted after washout, suggesting higher target binding affinity for the active metabolite and/or greater trapping of the active metabolite in the cytoplasm.
Figure 3.

Effect of S-licarbazepine on Nav1.5 currents. (A) Representative Na+ currents in an MDA-MB-231 cell elicited by a depolarisation from -120 to -10 mV in physiological saline solution (PSS; black), S-licarbazepine (S-Lic; 300 μM; red) and after washout (grey). Dotted vertical lines define the time period magnified in (B). (B) Representative persistent Na+ currents in an MDA-MB-231 cell elicited by a depolarisation from -120 to -10 mV. (C) Representative Na+ currents in an MDA-MB-231 cell elicited by a depolarisation from -80 to -10 mV. (D) Normalized Na+ currents in MDA-MB-231 cells elicited by a depolarisation from -120 to -10 mV. (E) Normalized Na+ currents in MDA-MB-231 cells elicited by a depolarisation from -80 to -10 mV. (F) Representative Na+ currents in a HEK-Nav1.5 cell elicited by a depolarisation from -120 to -10 mV in PSS (black), S-Lic (300 μM; red) and after washout (grey). Dotted vertical lines define the time period magnified in (G). (G) Representative persistent Na+ currents in a HEK-Nav1.5 cell elicited by a depolarisation from -120 to -10 mV. (H) Representative Na+ currents in a HEK-Nav1.5 cell elicited by a depolarisation from -80 to -10 mV. (I) Normalized Na+ currents in HEK-Nav1.5 cells elicited by a depolarisation from -120 to -10 mV. (J) Normalized Na+ currents in HEK-Nav1.5 cells elicited by a depolarisation from -80 to -10 mV. Results are mean + SEM. *P ≤ 0.05; ***P ≤ 0.001; one-way ANOVA with Tukey tests (n = 9-13). NS, not significant.
We also assessed the effect of both compounds on the persistent Na+ current measured 20–25 ms after depolarisation to -10 from -120 mV. In MDA-MB-231 cells, ESL (300 μM) inhibited the persistent Na+ current by 77 ± 34% although the reduction was not statistically significant (P = 0.13; n = 12; paired t test; Figure 2B, Table 1). In HEK-Nav1.5 cells, ESL (300 μM) inhibited persistent current by 76 ± 10% (P < 0.01; n = 12; paired t test; Figure 2G, Table 1). S-Lic (300 μM) inhibited the persistent Na+ current in MDA-MB-231 cells by 66 ± 16% (P < 0.05; n = 9; paired t test; Figure 3B, Table 2). In HEK-Nav1.5 cells, S-Lic (300 μM) inhibited persistent current by 35 ± 16% (P < 0.05; n = 11; Figure 3G, Table 2). Application of both compounds at a lower concentration (100 µM) elicited a similar result (Supplementary Table 1). In summary, both ESL and S-Lic also inhibited the persistent Na+ current.
Table 1.
Effect of eslicarbazepine acetate (300 μM) on Na+ current characteristics in MDA-MB-231 and HEK-Nav1.5 cells.1
| A. MDA-MB-231 cells | ||||
|---|---|---|---|---|
| Parameter | Control | ESL | P value | N |
| Vthres (mV) | -45.7 ± 1.7 | -45.0 ± 1.4 | 0.58 | 13 |
| Vpeak (mV) | 3.1 ± 2.1 | -3.9 ± 2.7 | 0.056 | 13 |
| Activation V½ (mV) | -19.3 ± 1.4 | -22.0 ± 1.5 | 0.095 | 12 |
| Activation k (mV) | 10.6 ± 0.7 | 9.3 ± 0.8 | 0.076 | 12 |
| Inactivation V½ (mV) | -80.6 ± 0.7 | -86.7 ± 1.2 | < 0.001 | 13 |
| Inactivation k (mV) | -4.8 ± 0.4 | -7.4 ± 1.7 | 0.139 | 13 |
| Peak current density at -10 mV (pA/pF) | -14.8 ± 3.9 | -8.0 ± 2.5 | < 0.001 | 13 |
| Persistent current density at -10 mV (pA/pF) | -0.15 ± 0.05 | -0.02 ± 0.07 | 0.13 | 12 |
| Tp at -10 mV (ms) | 2.1 ± 0.2 | 1.9 ± 0.2 | < 0.01 | 13 |
| τf at -10 mV (ms) | 1.3 ± 0.1 | 1.3 ± 0.2 | 0.954 | 13 |
| τs at -10 mV) (ms) | 10.0 ± 2.3 | 6.9 ± 2.0 | 0.289 | 13 |
| τr (ms) | 6.0 ± 0.5 | 8.7 ± 0.7 | < 0.05 | 10 |
| B. HEK-Nav1.5 cells | ||||
| Parameter | Control | ESL | P value | N |
| Vthres (mV) | -55.0 ± 1.7 | -54.0 ± 2.2 | 0.758 | 10 |
| Vpeak (mV) | -26.0 ± 2.2 | -24.0 ± 4.3 | 0.591 | 10 |
| Activation V½ (mV) | -39.4 ± 1.3 | -44.2 ± 1.8 | < 0.05 | 10 |
| Activation k (mV) | 5.3 ± 1.3 | 3.8 ± 0.7 | 0.361 | 10 |
| Inactivation V½ (mV) | -78.2 ± 2.5 | -88.3 ± 2.7 | < 0.001 | 10 |
| Inactivation k (mV) | -6.9 ± 0.4 | -9.8 ± 0.7 | < 0.001 | 10 |
| Peak current density at -10 mV (pA/pF) | -154.4 ± 24.0 | -33.1 ± 4.7 | < 0.001 | 12 |
| Persistent current density at -10 mV (pA/pF) | -0.61 ± 0.15 | -0.12 ± 0.05 | < 0.01 | 12 |
| Tp at -10 mV (ms) | 1.4 ± 0.2 | 1.9 ± 0.2 | < 0.001 | 14 |
| τf at -10 mV (ms) | 0.9 ± 0.1 | 1.2 ± 0.1 | < 0.001 | 12 |
| τs at -10 mV (ms) | 6.6 ± 0.8 | 20.8 ± 8.5 | 0.128 | 12 |
| τr (ms) | 4.5 ± 0.4 | 7.1 ± 0.6 | < 0.001 | 10 |
1ESL, eslicarbazepine acetate (300 µM); Vthres, threshold voltage for activation; Vpeak, voltage at which current was maximal; V½, half (in)activation voltage; k, slope factor for (in)activation; Tp, time to peak current; τf, fast time constant of inactivation; τs, slow time constant of inactivation; τr, time constant of recovery from inactivation. The holding potential was -120 mV. Results are mean ± SEM. Statistical comparisons were made with paired t-tests.
Table 2.
Effect of S-licarbazepine (300 μM) on Na+ current characteristics in MDA-MB-231 and HEK-Nav1.5 cells.1
| A. MDA-MB-231 cells | ||||
|---|---|---|---|---|
| Parameter | Control | S-Lic | P value | N |
| Vthres (mV) | -34.4 ± 2.0 | -35.7 ± 2.0 | 0.603 | 7 |
| Vpeak (mV) | 11.43 ± 4.4 | 10.0 ± 4.9 | 0.818 | 7 |
| Activation V½ (mV) | -12.9 ± 1.3 | -13.7 ± 1.4 | 0.371 | 7 |
| Activation k (mV) | 11.0 ± 0.5 | 11.9 ± 0.8 | 0.520 | 7 |
| Inactivation V½ (mV) | -71.8 ± 2.5 | -76.8 ± 2.2 | < 0.05 | 7 |
| Inactivation k (mV) | -6.8 ± 0.9 | -6.0 ± 1.2 | 0.302 | 7 |
| Peak current density at -10 mV (pA/pF) | -12.0 ± 3.1 | -6.9 ± 2.5 | < 0.001 | 9 |
| Persistent current density at -10 mV (pA/pF) | -1.3 ± 0.4 | -0.6 ± 0.2 | < 0.05 | 9 |
| Tp at -10 mV (ms) | 4.5 ± 0.4 | 5.1 ± 0.7 | 0.103 | 9 |
| τf at -10 mV (ms) | 3.8 ± 1.1 | 3.2 ± 0.4 | 0.553 | 7 |
| τs at -10 mV (ms) | 25.7 ± 7.0 | 27.1 ± 12.0 | 0.920 | 7 |
| τr (ms) | 6.8 ± 0.4 | 13.5 ± 1.0 | < 0.01 | 7 |
| B. HEK-Nav1.5 cells | ||||
| Parameter | Control | S-Lic | P value | N |
| Vthres (mV) | -50.0 ± 1.9 | -51.3 ± 3.5 | 0.598 | 9 |
| Vpeak (mV) | -18.0 ± 4.2 | -30.0 ± 5.6 | < 0.001 | 9 |
| Activation V½ (mV) | -32.8 ± 3.1 | -40.5 ± 3.4 | < 0.01 | 9 |
| Activation k (mV) | 5.9 ± 0.9 | 4.5 ± 1.1 | < 0.05 | 9 |
| Inactivation V½ (mV) | -75.9 ± 2.6 | -79.3 ± 4.1 | 0.116 | 9 |
| Inactivation k (mV) | -6.5 ± 0.4 | -8.1 ± 0.5 | < 0.05 | 9 |
| Peak current density at -10 mV (pA/pF) | -140.9 ± 26.8 | -77.2 ± 17.0 | < 0.001 | 13 |
| Persistent current density at -10 mV (pA/pF) | -0.9 ± 0.2 | -0.5 ± 0.2 | < 0.05 | 11 |
| Tp at -10 mV (ms) | 1.8 ± 0.5 | 2.3 ± 0.6 | < 0.01 | 13 |
| τf at -10 mV (ms) | 1.0 ± 0.04 | 1.3 ± 0.06 | < 0.001 | 11 |
| τs at -10 mV (ms) | 6.3 ± 0.5 | 7.3 ± 0.5 | < 0.05 | 11 |
| τr (ms) | 5.7 ± 0.7 | 8.0 ± 1.2 | < 0.01 | 10 |
1S-Lic, S-licarbazepine (300 µM); Vthres, threshold voltage for activation; Vpeak, voltage at which current was maximal; V½, half (in)activation voltage; k, slope factor for (in)activation; Tp, time to peak current; τf, fast time constant of inactivation; τs, slow time constant of inactivation; τr, time constant of recovery from inactivation. The holding potential was -120 mV. Results are mean ± SEM. Statistical comparisons were made with paired t-tests.
Effect of Eslicarbazepine Acetate and S-Licarbazepine on Voltage Dependence of Activation and Inactivation
We next investigated the effect of ESL (300 µM) and S-Lic (300 µM) on the I-V relationship in MDA-MB-231 and HEK-Nav1.5 cells. A Vh of -120 mV was used for subsequent analyses to ensure that the elicited currents were sufficiently large for analysis of kinetics and voltage dependence, particularly for MDA-MB-231 cells, which display smaller peak Na+ currents (Tables 1, 2). Neither ESL nor S-Lic had any effect on the threshold voltage for activation (Figures 4A–D; Tables 1, 2). ESL also had no effect on the voltage at current peak in either cell line (Figures 4A–D; Tables 1, 2). Although S-Lic had no effect on voltage at current peak in MDA-MB-231 cells, it was significantly hyperpolarised in HEK-Nav1.5 cells from -18.0 ± 4.2 to -30.0 ± 5.6 mV (P < 0.001; n = 9; paired t test; Figures 4A–D; Tables 1, 2).
Figure 4.

Effect of eslicarbazepine acetate and S-licarbazepine on the current-voltage relationship. (A) Current-voltage (I–V) plots of Na+ currents in MDA-MB-231 cells in physiological saline solution (PSS; black circles) and in eslicarbazepine acetate (ESL; 300 μM; red squares). (B) (I–V) plots of Na+ currents in HEK-Nav1.5 cells in PSS (black circles) and ESL (300 μM; red squares). (C) I–V plots of Na+ currents in MDA-MB-231 cells in PSS (black circles) and S-licarbazepine (S-Lic; 300 μM; red squares). (D) I–V plots of Na+ currents in HEK-Nav1.5 cells in PSS (black circles) and S-Lic (300 μM; red squares). Currents were elicited using 10 mV depolarising steps from -80 to +30 mV for 30 ms, from a holding potential of -120 mV. Results are mean ± SEM (n = 7–13).
ESL had no significant effect on the half-activation voltage (V½) or slope factor (k) for activation in MDA-MB-231 cells (Figure 5A; Table 1). The activation k in HEK-Nav1.5 cells was also unchanged but the activation V½ was significantly hyperpolarised by ESL from -39.4 ± 1.3 to -44.2 ± 1.8 mV (P < 0.05; n = 10; paired t test; Figure 5B; Table 1). S-Lic also had no significant effect on the activation V½ or k in MDA-MB-231 cells (Figure 5C; Table 2). However, the V½ of activation in HEK-Nav1.5 cells was significantly hyperpolarised from -32.8 ± 3.1 to -40.5 ± 3.4 mV (P < 0.01; n = 9; paired t test; Figure 5D; Table 2) and k changed from 5.9 ± 0.9 to 4.5 ± 1.1 mV (P < 0.05; n = 9; paired t test; Figure 5D; Table 2).
Figure 5.

Effect of eslicarbazepine acetate and S-licarbazepine on activation and steady-state inactivation. (A) Activation and steady-state inactivation in MDA-MB-231 cells in physiological saline solution (PSS; black circles) and in eslicarbazepine acetate (ESL; 300 μM; red squares). (B) Activation and steady-state inactivation in HEK-Nav1.5 cells in PSS (black circles) and ESL (300 μM; red squares). (C) Activation and steady-state inactivation in MDA-MB-231 cells in PSS (black circles) and S-licarbazepine (S-Lic; 300 μM; red squares). (D) Activation and steady-state inactivation in HEK-Nav1.5 cells in PSS (black circles) and S-Lic (300 μM; red squares). For activation, normalized conductance (G/Gmax) was calculated from the current data and plotted as a function of voltage. For steady-state inactivation, normalized current (I/Imax), elicited by 50 ms test pulses at -10 mV following 250 ms conditioning voltage pulses between -120 and +30 mV, applied from a holding potential of -120 mV, was plotted as a function of the prepulse voltage. Results are mean ± SEM (n = 7–13). Activation and inactivation curves are fitted with Boltzmann functions.
As regards steady-state inactivation, in MDA-MB-231 cells, ESL significantly hyperpolarised the inactivation V½ from -80.6 ± 0.7 to -86.7 ± 1.2 mV (P < 0.001; n = 13; paired t test) without affecting inactivation k (Figure 5A; Table 1). ESL also hyperpolarised the inactivation V½ in HEK-Nav1.5 cells from -78.2 ± 2.5 to -88.3 ± 2.7 mV (P < 0.001; n = 10; paired t test), and changed the inactivation k from -6.9 ± 0.4 to -9.8 ± 0.7 mV (P < 0.001; n = 10; paired t test; Figure 5B; Table 1). S-Lic also significantly hyperpolarised the inactivation V½ in MDA-MB-231 cells from -71.8 ± 2.5 to -76.8 ± 2.2 mV (P < 0.05; n = 7; paired t test) without affecting inactivation k (Figure 5C; Table 2). However, the inactivation V½ in HEK-Nav1.5 cells was not significantly altered by S-Lic, although the inactivation k significantly changed from -6.5 ± 0.4 to -8.1 ± 0.5 mV (P < 0.05; n = 9; paired t test; Figure 5D; Table 2). In summary, both ESL and S-Lic affected various aspects of the voltage dependence characteristics of Nav1.5 in MDA-MB-231 and HEK-Nav1.5 cells, predominantly hyperpolarising the voltage dependence of inactivation.
Effect of Eslicarbazepine Acetate and S-Licarbazepine on Activation and Inactivation Kinetics
We next studied the effect of both compounds on kinetics of activation and inactivation. In MDA-MB-231 cells, ESL (300 μM) significantly accelerated the time to peak current (Tp), upon depolarisation from -120 to -10 mV, from 2.1 ± 0.2 to 1.9 ± 0.2 ms (P < 0.01; n = 13; paired t test; Table 1). However, in HEK-Nav1.5 cells, ESL significantly slowed Tp from 1.4 ± 0.2 to 1.5 ± 0.2 ms (P < 0.001; n = 14; paired t test; Table 1). S-Lic (300 μM) had no significant effect on Tp in MDA-MB-231 cells but significantly slowed Tp in HEK-Nav1.5 cells from 1.8 ± 0.5 to 2.3 ± 0.6 ms (P < 0.01; n = 13; paired t test; Table 2).
To study effects on inactivation kinetics, the current decay following depolarisation from -120 to -10 mV was fitted to a double exponential function to derive fast and slow time constants of inactivation (τf and τs). Neither ESL nor S-Lic had any significant effect on τf or τs in MDA-MB-231 cells (Tables 1, 2). However, in HEK-Nav1.5 cells, ESL significantly slowed τf from 0.9 ± 0.1 to 1.2 ± 0.1 ms (P < 0.001; n = 12; paired t test; Table 1) and slowed τs from 6.6 ± 0.8 to 20.8 ± 8.5 ms, although this was not statistically significant. S-Lic significantly slowed τf from 1.0 ± 0.04 to 1.3 ± 0.06 ms (P < 0.001; n = 11; paired t test; Table 2) and τs from 6.3 ± 0.5 to 7.3 ± 0.5 ms (P < 0.05; n = 11; paired t test; Table 2). In summary, both ESL and S-Lic elicited various effects on kinetics in MDA-MB-231 and HEK-Nav1.5 cells, predominantly slowing activation and inactivation.
Effect of Eslicarbazepine Acetate and S-Licarbazepine on Recovery From Fast Inactivation
To investigate the effect of ESL and S-Lic on channel recovery from fast inactivation, we subjected cells to two depolarisations from Vh of -120 to 0 mV, changing the interval between these in which the channels were held at -120 mV to facilitate recovery. Significance was determined by fitting a single exponential curve to the normalized current/time relationship and calculating the time constant (τr). In MDA-MB-231 cells, ESL (300 μM) significantly slowed τr from 6.0 ± 0.5 to 8.7 ± 0.7 ms (P < 0.05; n = 10; paired t test; Figure 6A, Table 1). Similarly, in HEK-Nav1.5 cells, ESL significantly slowed τr from 4.5 ± 0.4 to 7.1 ± 0.6 ms (P < 0.001; n = 10; paired t test; Figure 6B, Table 1). S-Lic (300 μM) also significantly slowed τr in MDA-MB-231 cells from 6.8 ± 0.4 to 13.5 ± 1.0 ms (P < 0.01; n = 7; paired t test; Figure 6C, Table 2). Finally, S-Lic also significantly slowed τr in HEK-Nav1.5 cells from 5.7 ± 0.7 to 8.0 ± 1.2 ms (P < 0.01; n = 10; paired t test; Figure 6D, Table 2). In summary, both ESL and S-Lic slowed recovery from fast inactivation of Nav1.5.
Figure 6.

Effect of eslicarbazepine acetate and S-licarbazepine on recovery from inactivation. (A) Recovery from inactivation in MDA-MB-231 cells in physiological saline solution (PSS; black circles) and in eslicarbazepine acetate (ESL; 300 μM; red squares). (B) Recovery from inactivation in HEK-Nav1.5 cells in PSS (black circles) and ESL (300 μM; red squares). (C) Recovery from inactivation in MDA-MB-231 cells in PSS (black circles) and S-licarbazepine (S-Lic; 300 μM; red squares). (D) Recovery from inactivation in HEK-Nav1.5 cells in PSS (black circles) and S-Lic (300 μM; red squares). The fraction recovered (It/Ic) was determined by a 25 ms pulse to 0 mV (Ic), followed by a recovery pulse to -120 mV for 1–500 ms, and a subsequent 25 ms test pulse to 0 mV (It), applied from a holding potential of -120 mV, and plotted as a function of the recovery interval. Data are fitted with single exponential functions which are statistically different between control and drug treatments in all cases. Results are mean ± SEM (n = 7–10).
Discussion
In this study, we have shown that ESL and its active metabolite S-Lic inhibit the transient and persistent components of Na+ current carried by Nav1.5. We show broadly similar effects in MDA-MB-231 cells, which express endogenous Nav1.5 (Roger et al., 2003; Fraser et al., 2005; Brackenbury et al., 2007), and in HEK-293 cells over-expressing Nav1.5. Notably, both compounds were more effective when Vh was set to -80 mV than at -120 mV, suggestive of depolarised state-dependent binding. In addition, the inhibitory effect of ESL was reversible whereas inhibition by S-Lic was less so. As regards voltage-dependence, both ESL and S-Lic shifted activation and steady-state inactivation curves, to varying extents in the two cell lines, in the direction of more negative voltages. ESL and S-Lic had various effects on activation and inactivation kinetics, generally slowing the rate of inactivation. Finally, recovery from fast inactivation of Nav1.5 was significantly slowed by both ESL and S-Lic.
To our knowledge, this is the first time that the effects of ESL and S-Lic have specifically been tested on the Nav1.5 isoform. A strength of this study is that both the prodrug (ESL) and the active metabolite (S-Lic) were tested using two independent cell lines, one endogenously expressing Nav1.5, the other stably over-expressing Nav1.5. MDA-MB-231 cells also express Nav1.7, although this isoform is estimated to be responsible for only ~9% of the total VGSC current (Fraser et al., 2005; Brackenbury et al., 2007). MDA-MB-231 cells also express endogenous β1, β2, and β4 subunits (Chioni et al., 2009; Nelson et al., 2014; Bon et al., 2016). MDA-MB-231 cells predominantly express the developmentally regulated “neonatal” Nav1.5 splice variant, which differs from the “adult” variant over-expressed in the HEK-Nav1.5 cells by seven amino acids located in the extracellular linker between transmembrane segments 3 and 4 of domain 1 (Fraser et al., 2005; Brackenbury et al., 2007; Djamgoz et al., 2019). Notably, however, there were no consistent differences in effect of either ESL or S-Lic between the MDA-MB-231 and HEK-Nav1.5 cells, suggesting that the neonatal vs. adult splicing event, and/or expression of endogenous β subunits, does not impact on sensitivity of Nav1.5 to these compounds. This finding contrasts another report showing different sensitivity of the neonatal and adult Nav1.5 splice variants to the amide local anaesthetics lidocaine and levobupivacaine (Elajnaf et al., 2018). Our findings suggest that the inhibitory effect of S-Lic on Nav1.5 is less reversible than that of ESL. This may be explained by the differing chemical structures of the two molecules possibly enabling S-Lic to bind the target with higher affinity than ESL. Most VGSC-targeting anticonvulsants, including phenytoin, lamotrigine and carbamazepine, block the pore by binding via aromatic-aromatic interaction to a tyrosine and phenylalanine located in the S6 helix of domain 4 (Lipkind and Fozzard, 2010). However, S-Lic has been proposed to bind to a different site given that it was found to block the pore predominantly during slow inactivation (Hebeisen et al., 2015). Alternatively, the hydroxyl group present on S-Lic (but not ESL) may become deprotonated, potentially trapping it in the cytoplasm.
The findings presented here broadly agree with in vitro concentrations used elsewhere to study effects of ESL and S-Lic on Na+ currents. For example, using a Vh of -80 mV, 300 µM ESL was shown to inhibit peak Na+ current by 50% in N1E-115 neuroblastoma cells expressing Nav1.1, Nav1.2, Nav1.3, Nav1.6, and Nav1.7 (Bonifacio et al., 2001). S-Lic (250 µM) also blocks peak Na+ current by ~50% in the same cell line (Hebeisen et al., 2015). In addition, S-Lic (300 µM) reduces persistent Na+ current by ~25% in acutely isolated murine hippocampal CA1 neurons expressing Nav1.1, Nav1.2, and Nav1.6 (Westenbroek et al., 1989; Yu et al., 2006; Royeck et al., 2008; Doeser et al., 2014). Similar to the present study, ESL was shown to hyperpolarise the voltage-dependence of steady-state inactivation in N1E-115 cells (Bonifacio et al., 2001). On the other hand, similar to our finding in HEK-Nav1.5 cells, S-Lic has no effect on steady-state inactivation in N1E-115 cells (Hebeisen et al., 2015). Again, in agreement with our own findings for Nav1.5, S-Lic slows recovery from inactivation in N1E-115 cells (Hebeisen et al., 2015). These observations suggest that the sensitivity of Nav1.5 to ESL and S-Lic is broadly similar to that reported for neuronal VGSCs. In support of this, Nav1.5 shares the same conserved residues proposed for Nav1.2 to interact with ESL (Figure 7) (Shaikh et al., 2014).
Figure 7.

Clustal alignment of amino acid sequences of Nav1.1-Nav1.9 (SCN1A-SCN11A). ESL was proposed previously (Shaikh et al., 2014) to interact with the highlighted amino acids in Nav1.2. An alignment of Nav1.2 [UniProtKB - Q99250 (SCN2A_HUMAN)] with Nav1.1 [UniProtKB - P35498 (SCN1A_HUMAN)], Nav1.3 [UniProtKB - Q9NY46 (SCN3A_HUMAN)], Nav1.4 [UniProtKB - P35499 (SCN4A_HUMAN)], Nav1.5 [UniProtKB - Q14524 (SCN5A_HUMAN)] Nav1.6 [UniProtKB - Q9UQD0 (SCN8A_HUMAN)], Nav1.7 [UniProtKB - Q15858 (SCN9A_HUMAN)], Nav1.8 [UniProtKB - Q9Y5Y9 (SCN10A_HUMAN)], and Nav1.9 [UniProtKB - Q9UI33 (SCN11A_HUMAN)] shows that the interacting amino acids highlighted in yellow are conserved between Nav1.2 and Nav1.5, along with most other isoforms. Asterisks indicate conserved residues. Colon indicates conservation between groups of strongly similar properties - scoring >0.5 in the Gonnet PAM 250 matrix. Period indicates conservation between groups of weakly similar properties - scoring ≤0.5 in the Gonnet PAM 250 matrix.
Notably, the concentrations used in this study are at or above those achieved in clinical use (e.g. ESL 1,200 mg once daily gives a peak plasma concentration of ~100 µM) (Hebeisen et al., 2015). However, it has been argued that the relatively high concentrations which have been previously tested in vitro are clinically relevant given that S-Lic has a high (50:1) lipid:water partition co-efficient and thus would be expected to reside predominantly in the tissue membrane fraction in vivo (Bialer and Soares-da-Silva, 2012). Our study suggests that a clinically relevant plasma concentration (100 µM) would inhibit peak and persistent Nav1.5 currents. Future work investigating the dose-dependent effects of ESL and S-Lic would be useful to aid clinical judgements.
The data presented here raise several implications for clinicians. The observed inhibition of Nav1.5 is worthy of note when considering cardiac function in patients receiving ESL (Zaccara et al., 2015). Although the QT interval remains unchanged for individuals on ESL treatment, prolongation of the PR interval has been observed (Vaz-Da-Silva et al., 2012). Further work is required to establish whether the basis for this PR prolongation is indeed via Nav1.5 inhibition. In addition, it would be of interest to investigate the efficacy of ESL and S-Lic in the context of heritable arrhythmogenic mutations in SCN5A, as well as the possible involvement of the β subunits (Brackenbury and Isom, 2008; Uebachs et al., 2010; Doeser et al., 2014; Rivaud et al., 2020). The findings presented here are also relevant in the context of Nav1.5 expression in carcinoma cells (Fraser et al., 2014). Given that cancer cells have a relatively depolarised Vm, it is likely that Nav1.5 is mainly in the inactivated state with the persistent Na+ current being functionally predominant (Yang and Brackenbury, 2013; Yang et al., 2020). Increasing evidence suggests that persistent Na+ current carried by Nav1.5 in cancer cells contributes to invasion and several studies have shown that other VGSC inhibitors reduce metastasis in preclinical models (Roger et al., 2003; Fraser et al., 2005; House et al., 2010; Yang et al., 2012; Driffort et al., 2014; Besson et al., 2015; Nelson et al., 2015a; Nelson et al., 2015b). Thus, use-dependent inhibition by ESL would ensure that channels in malignant cells are particularly targeted, raising the possibility that it could be used as an anti-metastatic agent (Martin et al., 2015). This study therefore paves the way for future investigations into ESL and S-Lic as potential invasion inhibitors.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Author Contributions
TL, SC, and WB contributed to the conception and design of the work. TL, LB, and WB contributed to acquisition, analysis, and interpretation of data for the work. TL, SC, and WB contributed to drafting the work and revising it critically for important intellectual content. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Cancer Research UK (A25922) and Breast Cancer Now (2015NovPhD572).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2020.555047/full#supplementary-material
Abbreviations
ESL, eslicarbazepine acetate; HEK-Nav1.5, HEK-293 cells stably expressing Nav1.5; I-V, current-voltage; k, slope factor; PSS, physiological saline solution; S-Lic, S-licarbazepin; Tp, time to peak current; τf, fast time constant of inactivation; τs, slow time constant of inactivation; τr, time constant of recovery from inactivation; VGSC, voltage-gated Na+ channel; Vm, membrane potential; Vh, holding potential; Vpeak, voltage at which current was maximal; Vrev, reversal potential; Vthres, threshold voltage for activation; V1/2, half-activation voltage.
References
- Almeida L., Soares-da-Silva P. (2007). Eslicarbazepine acetate (BIA 2-093). Neurotherapeutics. 4 (1), 88–96. 10.1016/j.nurt.2006.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida L., Falcao A., Maia J., Mazur D., Gellert M., Soares-da-Silva P. (2005). Single-dose and steady-state pharmacokinetics of eslicarbazepine acetate (BIA 2-093) in healthy elderly and young subjects. J. Clin. Pharmacol. 45 (9), 1062–1066. 10.1177/0091270005279364 [DOI] [PubMed] [Google Scholar]
- Almeida L., Minciu I., Nunes T., Butoianu N., Falcão A., Magureanu S.-A., et al. (2008). Pharmacokinetics, Efficacy, and Tolerability of Eslicarbazepine Acetate in Children and Adolescents With Epilepsy. J. Clin. Pharmacol. 48 (8), 966–977. 10.1177/0091270008319706 [DOI] [PubMed] [Google Scholar]
- Alves G., Figueiredo I., Falcao A., Castel-Branco M., Caramona M., Soares-Da-Silva P. (2008). Stereoselective disposition of S- and R-licarbazepine in mice. Chirality. 20 (6), 796–804. 10.1002/chir.20546 [DOI] [PubMed] [Google Scholar]
- Armstrong C. M., Bezanilla F. (1977). Inactivation of the sodium channel. II. Gating Curr. Exp. J. Gen. Physiol. 70 (5), 567–590. 10.1085/jgp.70.5.567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson P., Driffort V., Bon E., Gradek F., Chevalier S., Roger S. (2015). How do voltage-gated sodium channels enhance migration and invasiveness in cancer cells? Biochim. Biophys. Acta 1848 (10 Pt B), 2493–2501. 10.1016/j.bbamem.2015.04.013 [DOI] [PubMed] [Google Scholar]
- Bialer M., Soares-da-Silva P. (2012). Pharmacokinetics and drug interactions of eslicarbazepine acetate. Epilepsia. 53 (6), 935–946. 10.1111/j.1528-1167.2012.03519.x [DOI] [PubMed] [Google Scholar]
- Bon E., Driffort V., Gradek F., Martinez-Caceres C., Anchelin M., Pelegrin P., et al. (2016). SCN4B acts as a metastasis-suppressor gene preventing hyperactivation of cell migration in breast cancer. Nat. Commun. 7, 13648. 10.1038/ncomms13648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacio M. J., Sheridan R. D., Parada A., Cunha R. A., Patmore L., Soares-da-Silva P. (2001). Interaction of the novel anticonvulsant, BIA 2-093, with voltage-gated sodium channels: comparison with carbamazepine. Epilepsia. 42 (5), 600–608. 10.1046/j.1528-1157.2001.43600.x [DOI] [PubMed] [Google Scholar]
- Brackenbury W. J., Djamgoz M. B. (2006). Activity-dependent regulation of voltage-gated Na+ channel expression in Mat-LyLu rat prostate cancer cell line. J. Physiol. 573 (Pt 2), 343–356. 10.1113/jphysiol.2006.106906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackenbury W. J., Isom L. L. (2008). Voltage-gated Na+ channels: potential for beta subunits as therapeutic targets. Expert Opin. Ther. Targets. 12 (9), 1191–1203. 10.1517/14728222.12.9.1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackenbury W. J., Isom L. L. (2011). Na Channel beta Subunits: Overachievers of the Ion Channel Family. Front. Pharmacol. 2:53. 10.3389/fphar.2011.00053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackenbury W. J., Chioni A. M., Diss J. K., Djamgoz M. B. (2007). The neonatal splice variant of Nav1.5 potentiates in vitro metastatic behaviour of MDA-MB-231 human breast cancer cells. Breast Cancer Res. Treat. 101 (2), 149–160. 10.1007/s10549-006-9281-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady K., Hebeisen S., Konrad D., Soares-da-Silva P. (2011). The effects of eslicarbazepine, R-licarbazepine, oxcarbazepine and carbamazepine on ion transmission Cav3.2 channels. Epilepsia. 52, 260. [Google Scholar]
- Brown M. E., El-Mallakh R. S. (2010). Role of eslicarbazepine in the treatment of epilepsy in adult patients with partial-onset seizures. Ther. Clin. Risk Manage. 6, 103–109. 10.2147/tcrm.s6382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W. A. (2014). Structure and function of voltage-gated sodium channels at atomic resolution. Exp. Physiol. 99 (1), 35–51. 10.1113/expphysiol.2013.071969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chioni A. M., Brackenbury W. J., Calhoun J. D., Isom L. L., Djamgoz M. B. (2009). A novel adhesion molecule in human breast cancer cells: voltage-gated Na+ channel beta1 subunit. Int. J. Biochem. Cell Biol. 41 (5), 1216–1227. 10.1016/j.biocel.2008.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djamgoz M. B. A., Fraser S. P., Brackenbury W. J. (2019). In Vivo Evidence for Voltage-Gated Sodium Channel Expression in Carcinomas and Potentiation of Metastasis. Cancers (Basel). 11 (11), 1–20. 10.3390/cancers11111675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doeser A., Soares-da-Silva P., Beck H., Uebachs M. (2014). The effects of eslicarbazepine on persistent Na(+) current and the role of the Na(+) channel beta subunits. Epilepsy Res. 108 (2), 202–211. 10.1016/j.eplepsyres.2013.11.022 [DOI] [PubMed] [Google Scholar]
- Driffort V., Gillet L., Bon E., Marionneau-Lambot S., Oullier T., Joulin V., et al. (2014). Ranolazine inhibits NaV1.5-mediated breast cancer cell invasiveness and lung colonization. Mol. Cancer. 13 (1), 264. 10.1186/1476-4598-13-264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elajnaf T., Baptista-Hon D. T., Hales T. G. (2018). Potent Inactivation-Dependent Inhibition of Adult and Neonatal NaV1.5 Channels by Lidocaine and Levobupivacaine. Anesth Analg. 127 (3), 650–660. 10.1213/ANE.0000000000003597 [DOI] [PubMed] [Google Scholar]
- Falcao A., Fuseau E., Nunes T., Almeida L., Soares-da-Silva P. (2012). Pharmacokinetics, drug interactions and exposure-response relationship of eslicarbazepine acetate in adult patients with partial-onset seizures: population pharmacokinetic and pharmacokinetic/pharmacodynamic analyses. CNS Drugs 26 (1), 79–91. 10.2165/11596290-000000000-00000 [DOI] [PubMed] [Google Scholar]
- Fraser S. P., Diss J. K., Chioni A. M., Mycielska M. E., Pan H., Yamaci R. F., et al. (2005). Voltage-gated sodium channel expression and potentiation of human breast cancer metastasis. Clin. Cancer Res. 11 (15), 5381–5389. 10.1158/1078-0432.CCR-05-0327 [DOI] [PubMed] [Google Scholar]
- Fraser S. P., Ozerlat-Gunduz I., Brackenbury W. J., Fitzgerald E. M., Campbell T. M., Coombes R. C., et al. (2014). Regulation of voltage-gated sodium channel expression in cancer: hormones, growth factors and auto-regulation. Philos. Trans. R. Soc. Lond B. Biol. Sci. 369 (1638), 20130105. 10.1098/rstb.2013.0105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiana G. L., Gauthier A. C., Mattson R. H. (2017). Eslicarbazepine Acetate: A New Improvement on a Classic Drug Family for the Treatment of Partial-Onset Seizures. Drugs R. D. 17 (3), 329–339. 10.1007/s40268-017-0197-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George A. L., Jr. (2005). Inherited disorders of voltage-gated sodium channels. J. Clin. Invest. 115 (8), 1990–1999. 10.1172/JCI25505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin A. L., Barchi R. L., Caldwell J. H., Hofmann F., Howe J. R., Hunter J. C., et al. (2000). Nomenclature of voltage-gated sodium channels. Neuron 28, 365–368. 10.1016/S0896-6273(00)00116-1 [DOI] [PubMed] [Google Scholar]
- Hebeisen S., Pires N., Loureiro A. I., Bonifacio M. J., Palma N., Whyment A., et al. (2015). Eslicarbazepine and the enhancement of slow inactivation of voltage-gated sodium channels: a comparison with carbamazepine, oxcarbazepine and lacosamide. Neuropharmacology 89, 122–135. 10.1016/j.neuropharm.2014.09.008 [DOI] [PubMed] [Google Scholar]
- House C. D., Vaske C. J., Schwartz A. M., Obias V., Frank B., Luu T., et al. (2010). Voltage-gated Na+ channel SCN5A is a key regulator of a gene transcriptional network that controls colon cancer invasion. Cancer Res. 70 (17), 6957–6967. 10.1158/0008-5472.CAN-10-1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkind G. M., Fozzard H. A. (2010). Molecular model of anticonvulsant drug binding to the voltage-gated sodium channel inner pore. Mol. Pharmacol. 78 (4), 631–638. 10.1124/mol.110.064683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin F., Ufodiama C., Watt I., Bland M., Brackenbury W. J. (2015). Therapeutic value of voltage-gated sodium channel inhibitors in breast, colorectal and prostate cancer: a systematic review. Front. Pharmacol. 6, 273. 10.3389/fphar.2015.00273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters J. R., Thomson J. A., Daly-Burns B., Reid Y. A., Dirks W. G., Packer P., et al. (2001). Short tandem repeat profiling provides an international reference standard for human cell lines. Proc. Natl. Acad. Sci. U. S. A. 98 (14), 8012–8017. 10.1073/pnas.121616198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M., Millican-Slater R., Forrest L. C., Brackenbury W. J. (2014). The sodium channel beta1 subunit mediates outgrowth of neurite-like processes on breast cancer cells and promotes tumour growth and metastasis. Int. J. Cancer. 135 (10), 2338–2351. 10.1002/ijc.28890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M., Yang M., Millican-Slater R., Brackenbury W. J. (2015. a). Nav1.5 regulates breast tumor growth and metastatic dissemination in vivo. Oncotarget. 6 (32), 32914–32929. 10.18632/oncotarget.5441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M., Yang M., Dowle A. A., Thomas J. R., Brackenbury W. J. (2015. b). The sodium channel-blocking antiepileptic drug phenytoin inhibits breast tumour growth and metastasis. Mol. Cancer. 14 (1), 13. 10.1186/s12943-014-0277-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patino G. A., Brackenbury W. J., Bao Y. Y., Lopez-Santiago L. F., O’Malley H. A., Chen C. L., et al. (2011). Voltage-Gated Na+ Channel beta 1B: A Secreted Cell Adhesion Molecule Involved in Human Epilepsy. J. Neurosci. 31 (41), 14577–14591. 10.1523/JNEUROSCI.0361-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perucca E., Elger C., Halász P., Falcão A., Almeida L., Soares-da-Silva P. (2011). Pharmacokinetics of eslicarbazepine acetate at steady-state in adults with partial-onset seizures. Epilepsy Res. 96 (1), 132–139. 10.1016/j.eplepsyres.2011.05.013 [DOI] [PubMed] [Google Scholar]
- Potschka H., Soerensen J., Pekcec A., Loureiro A., Soares-da-Silva P. (2014). Effect of eslicarbazepine acetate in the corneal kindling progression and the amygdala kindling model of temporal lobe epilepsy. Epilepsy Res. 108 (2), 212–222. 10.1016/j.eplepsyres.2013.11.017 [DOI] [PubMed] [Google Scholar]
- Rivaud M. R., Delmar M., Remme C. A. (2020). Heritable arrhythmia syndromes associated with abnormal cardiac sodium channel function: ionic and non-ionic mechanisms. Cardiovasc. Res. 116, 1557–1570. 10.1093/cvr/cvaa082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger S., Besson P., Le Guennec J. Y. (2003). Involvement of a novel fast inward sodium current in the invasion capacity of a breast cancer cell line. Biochim. Biophys. Acta 1616 (2), 107–111. 10.1016/j.bbamem.2003.07.001 [DOI] [PubMed] [Google Scholar]
- Royeck M., Horstmann M. T., Remy S., Reitze M., Yaari Y., Beck H. (2008). Role of axonal NaV1.6 sodium channels in action potential initiation of CA1 pyramidal neurons. J. Neurophysiol. 100 (4), 2361–2380. 10.1152/jn.90332.2008 [DOI] [PubMed] [Google Scholar]
- Saint D. A. (2008). The cardiac persistent sodium current: an appealing therapeutic target? Br. J. Pharmacol. 153 (6), 1133–1142. 10.1038/sj.bjp.0707492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh S., Rizvi S. M., Hameed N., Biswas D., Khan M., Shakil S., et al. (2014). Aptiom (eslicarbazepine acetate) as a dual inhibitor of beta-secretase and voltage-gated sodium channel: advancement in Alzheimer’s disease-epilepsy linkage via an enzoinformatics study. CNS Neurol. Disord. Drug Targets 13 (7), 1258–1262. 10.2174/1871527313666140917121600 [DOI] [PubMed] [Google Scholar]
- Sierra-Paredes G., Loureiro A. I., Wright L. C., Sierra-Marcuño G., Soares-da-Silva P. (2014). Effects of eslicarbazepine acetate on acute and chronic latrunculin A-induced seizures and extracellular amino acid levels in the mouse hippocampus. BMC Neurosci. 15 (1), 134. 10.1186/s12868-014-0134-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon A., Yang M., Marrison J. L., James A. D., Hunt M. J., O’Toole P. J., et al. (2020). Metastatic breast cancer cells induce altered microglial morphology and electrical excitability in vivo. J. Neuroinflam. 17 (1), 87. 10.1186/s12974-020-01753-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares-da-Silva P., Pires N., Bonifácio M. J., Loureiro A. I., Palma N., Wright L. C. (2015). Eslicarbazepine acetate for the treatment of focal epilepsy: an update on its proposed mechanisms of action. Pharmacol. Res. Perspect. 3 (2), e00124. 10.1002/prp2.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling M. R., Abou-Khalil B., Harvey J., Rogin J. B., Biraben A., Galimberti C. A., et al. (2015). Eslicarbazepine acetate as adjunctive therapy in patients with uncontrolled partial-onset seizures: Results of a phase III, double-blind, randomized, placebo-controlled trial. Epilepsia 56 (2), 244–253. 10.1111/epi.12894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uebachs M., Opitz T., Royeck M., Dickhof G., Horstmann M. T., Isom L. L., et al. (2010). Efficacy loss of the anticonvulsant carbamazepine in mice lacking sodium channel beta subunits via paradoxical effects on persistent sodium currents. J. Neurosci. 30 (25), 8489–8501. 10.1523/JNEUROSCI.1534-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uphoff C. C., Gignac S. M., Drexler H. G. (1992). Mycoplasma contamination in human leukemia cell lines. I. Comparison of various detection methods. J. Immunol. Methods 149 (1), 43–53. 10.1016/s0022-1759(12)80047-0 [DOI] [PubMed] [Google Scholar]
- Van Wart A., Matthews G. (2006). Impaired firing and cell-specific compensation in neurons lacking nav1.6 sodium channels. J. Neurosci. 26 (27), 7172–7180. 10.1523/JNEUROSCI.1101-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz-Da-Silva M., Nunes T., Almeida L., Gutierrez M. J., Litwin J. S., Soares-Da-Silva P. (2012). Evaluation of Eslicarbazepine acetate on cardiac repolarization in a thorough QT/QTc study. J. Clin. Pharmacol. 52 (2), 222–233. 10.1177/0091270010391789 [DOI] [PubMed] [Google Scholar]
- Westenbroek R. E., Merrick D. K., Catterall W. A. (1989). Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron. 3 (6), 695–704. 10.1016/0896-6273(89)90238-9 [DOI] [PubMed] [Google Scholar]
- Yang M., Brackenbury W. J. (2013). Membrane potential and cancer progression. Front. Physiol. 4, 185. 10.3389/fphys.2013.00185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M., Kozminski D. J., Wold L. A., Modak R., Calhoun J. D., Isom L. L., et al. (2012). Therapeutic potential for phenytoin: targeting Na(v)1.5 sodium channels to reduce migration and invasion in metastatic breast cancer. Breast Cancer Res. Treat. 134 (2), 603–615. 10.1007/s10549-012-2102-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M., James A. D., Suman R., Kasprowicz R., Nelson M., O’Toole P. J., et al. (2020). Voltage-dependent activation of Rac1 by Nav 1.5 channels promotes cell migration. J. Cell Physiol. 235 (4), 3950–3972. 10.1002/jcp.29290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F. H., Mantegazza M., Westenbroek R. E., Robbins C. A., Kalume F., Burton K. A., et al. (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9 (9), 1142–1149. 10.1038/nn1754 [DOI] [PubMed] [Google Scholar]
- Zaccara G., Giovannelli F., Cincotta M., Carelli A., Verrotti A. (2015). Clinical utility of eslicarbazepine: current evidence. Drug Des. Dev. Ther. 9, 781–789. 10.2147/DDDT.S57409 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.