

Abstract
Co-occurrence of tau and α-synuclein pathologies in a subset of Alzheimer’s disease patients has led to the idea that mixed pathologies may play a unique characteristic role in the Alzheimer’s disease neurodegenerative cascade. To understand the aetiology of such mixed pathologies, we investigated cross-seeding by human recombinant tau and human recombinant α-synuclein fibrillar species in a mouse model of tauopathy (Line PS19) or synucleinopathy (Line M20). Unilateral hippocampal injection of tau fibrils or α-synuclein fibrils, and to a lesser extent tau + α-synuclein copolymer fibrils prepared from co-incubating individual recombinant monomers, induced robust phosphorylated tau pathology in PS19 mice relative to control mice. Though the tau + α-synuclein copolymer fibrils did not modulate induction of pathologies at the site of injection, examination of the whole brain showed that these copolymers exacerbated neuroanatomic transmission of seeded tau pathology compared to tau fibril-injected mice. Only α-synuclein fibrils, but not tau alone or tau + α-synuclein copolymers, triggered modest levels of endogenous phosphorylated α-synuclein pathology. Overall, data from the PS19 mice suggest that human α-synuclein fibrils can efficiently cross-seed human tau and have a modest priming effect on mouse α-synuclein, and the presence of tau fibrils does not exacerbate the priming process. In M20 mice, unilateral hippocampal injection of α-synuclein fibrils or tau fibrils induced robust bilateral phosphorylated α-synuclein pathology, while tau + α-synuclein copolymer injection resulted in restricted phosphorylated α-synuclein pathology predominantly in the ipsilateral cortex. This suggests that human tau fibrils can also induce human α-synuclein pathogenesis, and the presence of combinatorial seeds is not synergistic. None of these aggregates induced phosphorylated tau pathology in M20 mice, showing that mouse tau cannot be primed efficiently by human tau fibrils or human α-synuclein fibrils. Neuropathological analysis of the whole brain of M20 mice showed that tau + α-synuclein copolymer-injected mice had lower abundance of bilaterally transmitted α-synuclein pathologies relative to α-synuclein fibril-injected mice. Thus, the tau + α-synuclein copolymer fibrils show robust transmission properties preferentially in rodent model of tauopathies but not in synucleinopathy, probably signifying an enhanced cooperative relationship between tau and α-synuclein in the tau seeding process. Together, our data highlight the unique cross-seeding properties of tau and αSyn in neurodegenerative proteinopathies.
Keywords: mixed pathology, cross-seeding, copolymer, transmission, neuroanatomic connectivity
Using transgenic mouse models of tauopathy and synucleinopathy, Williams et al. show that recombinant tau and α-synuclein can cross-seed each other and copolymers of tau and α-synuclein fibrils preferentially potentiate CNS transmission of induced tau pathology.
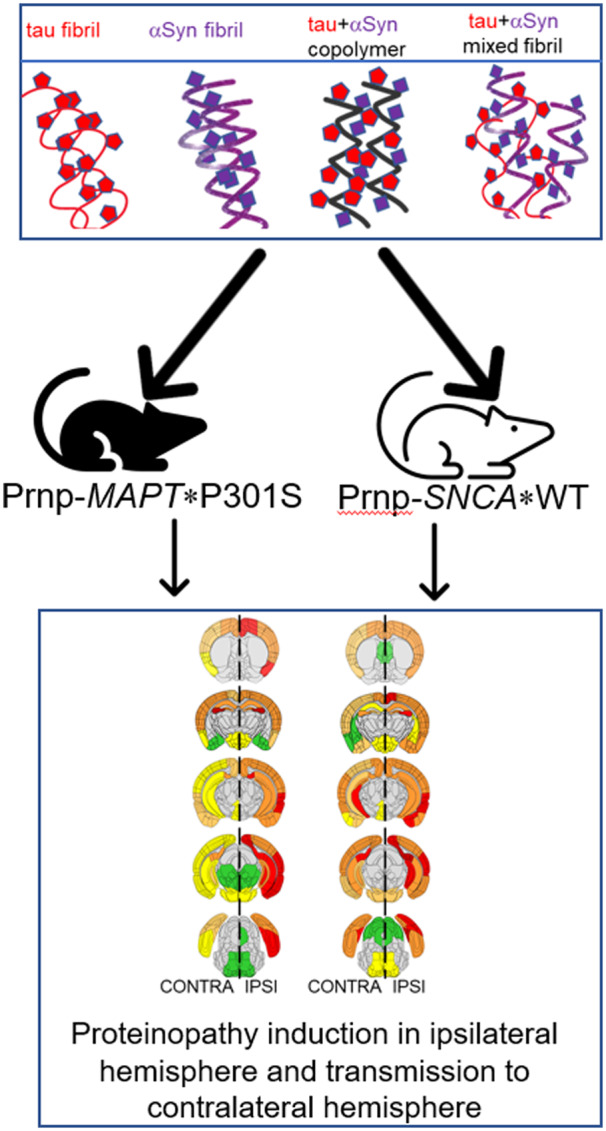
Graphical Abstract
Graphical Abstract.
Introduction
The term ‘mixed pathologies’ has come to denote the concomitant presence of various neuropathologic lesions in clinically diagnosed Alzheimer’s disease patients (James and Bennett, 2019). For example, in several community-based cohorts such as ROSMAP, MRC CFAS, CC75C, Vantaa, VITA and 90+ study, there is evidence of robust α-synuclein (αSyn) pathology in the brains of Alzheimer’s disease patients (Rahimi and Kovacs, 2014). Specifically, a subset of these Alzheimer’s disease patients display neocortical Lewy bodies (LBs) which are primarily composed of αSyn, in addition to classical Alzheimer-type amyloid β (Aβ) plaques and tau-containing neurofibrillary tangles (NFTs). In the ROSMAP cohort, the prevalence of the co-occurrence of αSyn is ∼20% whereas in the MRC CFAS and Vantaa studies, the frequency increases to ∼39% (Rahimi and Kovacs, 2014). Interestingly the Contursi kindred, the first reported family with a rare A53T αSyn mutation resulting in Parkinson’s disease, exhibits not only the expected αSyn pathology but additionally, robust levels of neuritic and perikaryal inclusions of tau protein (Duda et al., 2002). In general, the prevalence of mixed pathologies is higher in older patients (Schneider et al., 2007) and minority background patients (Barnes et al., 2015). This poses unique challenges to neuropsychiatric rehabilitation for these patients as they may have a different trajectory of disease progression and severity compared to patients with predominantly classical homogenous pathologies. It is also possible that in patients with mixed pathologies, disease-modifying therapy targeting only one protein may have little to no effect on disease progression. Taken together, such neuropathological heterogeneity can pose serious challenges in terms of biomarker evaluation, disease prognosis, clinical severity and treatment.
The aetiology of mixed pathologies in Alzheimer’s disease type dementias is unknown. One explanation is that Alzheimer’s disease-typical pathologies (Aβ and tau) and LB disorder (LBD)-typical pathologies (αSyn) might interact with each other and facilitate pathogenesis (Spires-Jones et al., 2017). Though cross-seeding between tau and Aβ has been well-documented in mouse models (Götz et al., 2001; Bolmont et al., 2007; He et al., 2018), whether a similar synergistic relationship exists between tau and αSyn is poorly understood. So far, molecular and cellular evidence of a direct interaction between tau and αSyn has been provided mainly by three studies (Jensen et al., 1999; Giasson et al., 2003; Guo et al., 2013). Using affinity chromatography and direct binding assays in vitro, the C-terminal domain of αSyn was shown to bind to the microtubule-binding repeat regions of recombinant tau protein inducing its phosphorylation at Ser262/356 (Jensen et al., 1999). The first in vivo demonstration of a possible pathogenic interaction between tau and αSyn came from the human A53T mutant αSyn transgenic mice which displays tau spheroid pathology in addition to LB-type αSyn inclusions (Giasson et al., 2003). Furthermore, a more recent report showed that αSyn fibrils can cross-seed tau pathology in the human P301S tau transgenic PS19 mouse model of tauopathy (Guo et al., 2013). This last report demonstrated that aggregated αSyn derived from patient brains can adopt specific conformations that can trigger tau misfolding in PS19 mice (Guo et al., 2013).
Studying how heterogeneous cross-seeds influence the templated seeding and subsequent intra-CNS transmission of proteinopathy can potentially enable us to understand the aetiology of mixed pathologies in Alzheimer’s disease type dementias. In this study, we specifically wanted to address the question whether co-fibrillized polymers of tau and αSyn possess unique cross-seeding properties compared to individual fibrils themselves in vivo. To determine the relative potency and characteristic strain-like properties of tau fibrils, αSyn fibrils, tau + αSyn copolymer seeds or a mixture of pre-aggregated tau and αSyn fibrils, we injected these unilaterally in the hippocampus of transgenic mouse models of tauopathy (Line PS19) or synucleinopathy (Line M20). We found that while the copolymers do not lead to exacerbated pathological burden compared to cohorts injected with tau or αSyn fibrils alone in both lines of mice, the tau + αSyn copolymer resulted in more widespread transmission of the induced pathology in PS19 mice, indicating a cooperative cross-seeding phenomenon between tau and αSyn in tauopathies.
Materials and methods
Mice and study design
Mouse husbandry and experimental procedures were performed in accordance with the protocols and policies approved by the Institutional Animal Care and Use Committee at the University of Florida. All mice were maintained under a 12 hour light/dark cycle and had access to water and food ad libitum. The mice were maintained under specific pathogen-free conditions and all surgeries were done under anaesthesia using sterile conditions by a trained surgeon. No adverse events (such as inability to recover from anaesthesia or sudden death) were noted following surgery. The PS19 mice were initially obtained from Jackson Labs and maintained in house on a B6/C3H background, developing age-progressive tauopathy and hindlimb paralysis at 9–12 months of age (Yoshiyama et al., 2007). Line M20 mice express human WT αSyn and do not develop any intrinsic phenotypes or αSyn pathology in their lifetime (Giasson et al., 2002). Two separate groups of mice (PS19 and M20) were set up to be injected with either tau fibrils, αSyn fibrils, tau + αSyn copolymers or tau + αSyn fibril mixture at 2 months of age in the right hippocampi. Mouse cohorts are tabulated in Supplementary Table 1. The groups were then aged for 3 months post-surgery and analysed for neuropathology. Euthanasia was performed with intracardiac perfusion of phosphate buffered saline (PBS) containing heparin and the brains were fixed in 10% normal buffered formalin. The fixed brains were then sliced coronally at indicated bregma locations and processed for paraffin embedding. All antibodies used in this study are tabulated in Supplementary Table 2.
The human patient slides were obtained from University of Florida Neuromedicine Human Brain Tissue Bank following institutional regulations and complete de-identification. The DLB patients were both females, aged 78 and 81 years.
In vitro aggregation of αSyn and K18 proteins and Thioflavin T assay
Recombinant N-terminal truncated human αSyn (residues 21–140) and the K18 human tau fragment (residues Q244-E372 of 2N/4R human tau) with an added methionine residue at the N-terminus were cloned in the pRK172 plasmid as described previously (Giasson et al., 2003). In previous in vitro studies, the 21–140 αSyn fibrils were able to induce pathology similar to full-length αSyn fibrils (Waxman and Giasson, 2010). Heparin-induced K18 tau aggregates have been shown to assemble into frank paired helical fragments (PHFs) capable of seeding human tau (Shammas et al., 2015).
αSyn and tau proteins were expressed in Escherichia coli BL21 (DE3) and purified utilizing chromatography as previously described (Giasson et al., 2003). All recombinant proteins were diluted in sterile PBS, pH 7.4 (Invitrogen) and concentrations were determined using bicinchoninic acid assay (Pierce, Waltham, MA) with bovine serum albumin as the standard protein. To prepare copolymers of αSyn and K18 tau, the purified proteins were diluted separately in sterile PBS, mixed together in a 1:1 ratio and assembled into amyloid fibrils with continuous shaking at 1050 rpm at 37°C for 5 days. Three separate fibrillization conditions were utilized which were 4 mg/ml αSyn alone to generate αSyn fibrils, 4 mg/ml K18 tau with 50 µM heparin to generate tau fibrils (Chakrabarty et al., 2015; Sorrentino et al., 2017) or 2 mg/ml αSyn mixed with 2 mg/ml K18 tau to generate tau + αSyn copolymer fibrils (Giasson et al., 2003). Amyloid formation for each species was assessed by thioflavin T fluorometry as described previously (Crystal et al., 2003). To generate the kinetic curve, 75 or 150 µM solution of each monomer diluted in PBS was shaken for 5 days at 1050 rpm. Aliquots were retrieved at indicated time points, mixed with 25 μM thioflavin T in PBS buffer and fluorescence intensities were monitored immediately at 480 nm with excitation at 450 nm using a Hitachi F-4500 fluorescence spectrophotometer.
For cell culture experiments, fibrils were further diluted to 2 mg/ml and fragmented using bath sonication for 1 h prior to seeding experiments as described earlier (Chakrabarty et al., 2015). For generating mixed fibrils, pre-aggregated K18 tau and pre-aggregated αSyn fibrils (fibrillized separately), generated as described above, were individually sonicated and were mixed in a 1:1 ratio immediately prior to use to generate the mixed tau + αSyn fibril preparation. Henceforth, the terms fibrils and aggregates have been used interchangeably. For in vivo injection, all fibrillar species were used at 1 mg/ml concentration in a volume of 2 µl.
Electron microscopy and slot blot analysis of fibrils
To investigate the presence of tau and αSyn in fibrillar species, a modified protocol for immuno-labelling followed by electron microscopy (EM) imaging was used (Giasson et al., 2003). For fibrils prepared from αSyn alone, K18 tau alone, or both copolymerized together, fibrils underwent centrifugation at 100, 000 g for 30 min and then the fibril containing pellets were suspended in microscopy grade water. Centrifugation was repeated twice to remove salts, after which fibrils were adsorbed onto 300 mesh carbon-coated copper grids (Electron Microscopy Sciences, Hatfield, PA). Grids were blocked with 2% bovine serum albumin in PBS for 10 min and sequentially incubated with primary antibodies diluted in 2% bovine serum albumin in PBS: affinity purified anti-rabbit 3026 antibody (20 µg/ml, specific for total human 0N/3R tau; Strang et al., 2017) and anti-mouse 94-3A10 antibody (20 µg/ml, specific for αSyn residues 130–140; Dhillon et al., 2017) were used. Following repeated washes with PBS, the grids were sequentially incubated with anti-mouse and anti-rabbit IgG secondary antibodies conjugated to 6 nm or 10 nm colloidal gold, respectively (Electron Microscopy Sciences, Hatfield, PA). Fibrils underwent negative staining with 1% uranyl acetate and were visualized with a Hitachi 7600 transmission electron microscope. Images were processed using Adobe Photoshop.
For slot blot analysis, different amounts of proteinaceous aggregates were added in duplicate (in a total volume of 100 µl) into each well of a slot blot apparatus (Minifold II, Schleicher & Schuell) and transferred onto 0.25 µm nitrocellulose membrane (Biorad) by vacuum aspiration. The filter blots were dried overnight, rehydrated in PBS, blocked in 0.5% casein and incubated overnight at 4°C in primary antibodies (tau-specific 81A11 antibody or αSyn-specific 94-3A10 antibody) diluted in block. The blots were then incubated in highly cross-adsorbed IRDye secondary antibodies (1:20, 000; Licor Biosciences) at room temperature for an hour and then images scanned using the Odyssey scanner.
HEK293T transfection, biochemical fractionation and immunoblotting
HEK293T cells were transfected using a modified calcium phosphate protocol (Chakrabarty et al., 2015). The cDNAs encoding full-length human WT αSyn and human P301L mutant 0N/4R tau were previously cloned in the mammalian expression vector pcDNA3.1(+) (Waxman and Giasson, 2011). For each of three replicate wells per condition, 1.5 μg of pcDNA3.1 vector expressing P301L tau or WT αSyn was used for transfection. One hour after transfection, either αSyn fibrils (1 µM), K18 tau fibrils (1 µM), or copolymerized tau + αSyn fibrils (containing either 0.5 or 1 µM of each protein) were added to the indicated final concentrations, with the concentrations being based on that of the monomeric subunits. Cells were lysed for biochemical fractionation 64 h post-transfection. Transfected cells were washed in PBS, lysed in 200 μl/well detergent extraction buffer (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 20 mM NaF) containing protease inhibitors and sedimented at 100, 000 g for 30 min at 4°C. SDS containing sample buffer was added for the detergent-soluble and detergent-insoluble fractions and boiled for 10 min. The detergent-insoluble fraction was probe sonicated for 1 min and boiled for 10 min before separating on SDS-PAGE (10% for tau transfected cells and 15% for αSyn transfected cells). Antibody 4110 (specific for residues 2–15 in human αSyn) was used to detect total αSyn (Sorrentino et al., 2018) and antibody 3026 (specific for recombinant 0N/3R human tau) was used to detect total tau (Strang et al., 2017).
Hippocampal stereotactic injections
Mice were aged to 2 months and unilaterally injected in the right hippocampus (coordinates from Bregma: A/P −2.2, L −1.6, D/V −1.2) with either tau, αSyn, tau + αSyn copolymerized fibrils or tau + αSyn mixed fibrils. Fibrils (2 µl of 1 mg/ml) were injected at a rate of 0.2 µl per minute. Control mice were injected with sterile PBS in the hippocampus or left uninjected. All the mice were allocated randomly to each experimental cohort. Mice were analysed after 3 months. The mouse numbers for the PS19 injection group were: Control, n = 5, tau fibrils, n = 6, αSyn fibrils, n = 5, tau + αSyn copolymers, n = 7 and tau + αSyn mixed fibrils, n = 5. All mice were female in the PS19 cohort. We decided to use only one sex to limit the inter-animal variability in this group. Additionally, because it was recently determined that prion-related tau seeding activity is not affected by sex differences in these mice especially at younger ages, we used mice of one sex (Woerman et al., 2018). The mouse numbers for the M20 injection group were: Control, n = 4, tau fibrils, n = 4, αSyn fibrils, n = 4 and tau + αSyn copolymers, n = 4 and tau + αSyn mixed fibrils, n = 5 (Supplementary Table 1). Mice were balanced for sex in M20 cohort.
Immunohistochemistry and Immunofluorescence
Immunohistochemical analysis was performed with the following antibodies: 7F2 (1:1000; B. Giasson; Strang et al., 2017), PHF1 (1:1000; Peter Davies), CP13 (1:1000; Peter Davies); MC1 (1:1000; Peter Davies), 3026 (1:500; B. Giasson; Strang et al., 2017), 81 A (1:3000; B. Giasson; Rutherford et al., 2016), 5G9 (1:500; B. Giasson; Sorrentino et al., 2018), EP1536Y (1:1000; Abcam); GFAP (1:1000; Dako), Iba-1 (1:1000; Wako), p62 (1:1000; Proteintech) and synaptophysin (1:500; Thermofisher) (Supplementary Table 2). For all antibodies, except Iba1 which required citrate buffer pH 6.0 (Target Retrieval Solution, Dako), antigen retrieval was performed by steaming in water for 25 min. The slides were incubated in 3% hydrogen peroxide (Fisher Scientific) for 20 min to block endogenous peroxidase activity and then washed three times in PBS for 5 min each. Next, slides were blocked in 2% FBS (Hyclone, GE) for 45 min before the appropriate primary antibody was diluted in block solution and incubated on the slides overnight at 4°C. The following day, slides were washed three times with PBS for 5 min each. Then the appropriate secondary antibody (ImmPRESS Polymer Reagent, Vector Labs) was applied for 30 min at room temperature. Following PBS washes, colour was developed using 3,3ʹ-diaminobenzidine (Vector® DAB, Vector Labs) and slides were counterstained with haematoxylin (Sigma, MO). Next, brain sections were dehydrated in a series of ethanol, cleared in xylene, mounted in Cytoseal-60 media (Fisher Scientific) and coverslipped. For immunofluorescence, additional steps included using Autofluorescence Eliminator Reagent (Millipore) following antigen retrieval step, using Alexa Fluor labelled (488 and 594 nm) secondary antibodies (Invitrogen) and cover-slipping using DAPI containing Fluoromount G (Southern Biotech).
Gallyas silver impregnation protocol
We used a modified protocol derived from Kuninaka et al. (2015). Briefly, sections were rehydrated in water and placed in 5% periodic acid for 5 min. Following incubation in alkaline silver iodide solution for 1 min, sections were washed in 0.5% acetic acid for 10 min. The slides were then incubated in fresh developer solution for 5 min. After development, slides were washed in 0.5% acetic acid for 3 min and then water for 5 min. Next, sections were placed in 0.1% gold chloride for 5 min, then rinsed in water before being incubated in 1% sodium thiosulphate solution for 5 min and washed in tap water. Counterstaining was done according to manufacturer’s instructions. Finally, sections were dehydrated, cleared and mounted.
Analysis of histochemical and immunofluorescence images
The initial slides were stained with haematoxylin and eosin, their location identified using Paxinos and Franklin’s mouse brain atlas and further slides were prepared according to desired bregma locations. The images of antibody stained slides were captured using Aperio Scanscope XT image scanner (Leica Biosystems, Vista, CA, USA). We generated semi-quantitative heat maps to assess spatial distribution of immunoreactivity using 4–6 individual samples corresponding to the indicated area of interest. Semi-quantitative analysis was performed on 7F2, 81A, MC1, p62, GFAP and Iba-1 positive neuropathology that was scored by two blinded individuals on a scale from 0 to 3 (0: no pathology; 1: low pathology; 2: medium pathology; 3: high pathology) at the assigned coronal CNS level (bregma −2.03 was designated as injection level). After the separate brain regions were scored for each individual mouse, the average values were imported into Microsoft Excel where an adapted heat map programme from Vertex42 (see https://www.vertex42.com/blog/help/excel-help/dynamicgeographic-heat-map-in-excel.html, 9 September 2019 accessed) was used to generate pathology distribution maps. The coronal mouse brain illustrations were adapted from the Allen Mouse Brain Atlas at the indicated Bregma (see http://mouse.brain-map.org/, 10 September 2019 accessed). The schematic for the connectivity map was derived from data found in the Allen Brain Atlas: Mouse Brain Connectivity Atlas (Oh et al., 2014).
Fluorescently labelled sections were visualized on an Olympus BX60 microscope with a colour camera. Total fluorescence intensity was measured using ImageJ (NIH) by subtracting the mean fluorescence of background readings normalized to the area from the integrated density.
Statistics
Immunoblot results and neuropathology scores were analysed using one-way ANOVA. The immunohistochemical data were assessed by two blinded observers individually and then collated for data analysis. All data were assembled using Adobe Photoshop Elements.
Availability of data and material
All data generated during this study are included in this published article [and its Supplementary information files].
Results
Recombinant tau + αSyn copolymers potentiate tau seeding in cell culture
Overlapping patterns of intracellular inclusions composed of tau and αSyn are frequently observed in the neocortex of DLB patients (Supplementary Fig. 1A and B). Co-immunofluorescence studies showed that such tau and αSyn pathologies co-localize in individual cells in this region (Supplementary Fig. 1C–E). Based on post-mortem studies, it has been thought that such Alzheimer’s disease-type tau pathologies co-occurring with αSyn lead to worse prognosis in these patients (Howlett et al., 2015; Irwin et al., 2017; Ferman et al., 2018). In a previous in vitro study, recombinant αSyn was shown to facilitate aggregation of recombinant tau in the absence of heparin leading to the formation of copolymerized fibrils containing both tau and αSyn (Giasson et al., 2003). This was suggestive of a synergistic association between tau and αSyn, as observed in pathological inclusions in DLB patients or Alzheimer’s disease patients with mixed pathologies. Here, using a systematic set of in vitro and in vivo experiments, we specifically wanted to examine the relative seeding properties of recombinant tau or αSyn aggregates relative to in vitro generated tau + αSyn copolymers in an effort to understand the synergistic cooperation between tau and αSyn leading to the formation of mixed pathologies in neurodegenerative dementias.
We generated recombinant human WT K18 tau fibrils and recombinant human WT αSyn fibrils as described previously (Giasson et al., 2003). Recombinant human K18 tau monomers and recombinant human αSyn monomers were co-incubated in equimolar quantities to generate the copolymerized fibrils, henceforth referred to as tau + αSyn copolymers (Giasson et al., 2003). While αSyn and tau + αSyn copolymers could be generated spontaneously by shaking, K18 tau fibrillization required the presence of heparin. We established that these tau + αSyn copolymers indeed contained both tau and αSyn by a slot blotting assay using antibodies against total tau and total αSyn (Fig. 1A and B). Assuming that antibody-mediated tau and αSyn detection limits are equivalent and that both tau and αSyn epitopes are equally available for antibody binding, αSyn is overrepresented by a factor of ∼3 in the copolymers relative to tau at the lowest level of copolymer bound to the membrane. Though the relative composition of individual protein filaments cannot be quantitatively assessed, this method shows that both tau and αSyn in these copolymers are not cryptic and remain available for epitope recognition (Fig. 1A and B). Analysis of fibril growth rate using thioflavin T revealed that the tau + αSyn copolymers closely followed the aggregation kinetics of K18 tau, while the αSyn monomers were slower to reach saturation (Supplementary Fig. 2A). We then analysed the ultrastructure of the human WT K18 tau fibrils, human WT αSyn fibrils and tau + αSyn copolymers using immuno-EM following immunolabelling with antibodies specific for total tau or total αSyn (Fig. 1C). Immunogold labelling showed that recombinant K18 tau and αSyn fibrils appeared to have unique representation as exemplified in the immuno-EM micrographs (Fig. 1C). Furthermore, the tau + αSyn copolymers contain both tau and αSyn, as indicated by immunolabelling with both the tau-specific antibody and αSyn-specific antibody (Fig. 1C; arrowheads, tau; arrows, αSyn).
Figure 1.

Tau and αSyn copolymerized fibrils potentiate tau seeding in cell culture. (A–C) Characterization of human K18 tau fibril, human αSyn fibril and tau+αSyn copolymer preparations used in this study by slot blotting and immuno-EM. Selected amounts (µg) of aggregated proteins, as indicated on the left panel, were transferred onto two identical nitrocellulose membranes and were probed with an antibody raised against total tau (A) or an antibody raised against total αSyn (B). The control row consists of two consecutive slots coated with buffer and a third slot (to the far right) coated with 2.5 µg of bovine serum albumin protein (A, B). The fibrillar preparations were examined using immuno-EM following simultaneous incubation of the aggregates with 10 nm gold particle conjugated total tau antibody (indicated by arrowheads) and 6 nm gold particle conjugated αSyn antibody (indicated by arrows) (C). The tau+αSyn copolymers display immunoreactivity to both tau and αSyn specific antibodies. Scale Bar, 50 nm. To ascertain the seeding activity of different aggregates, HEK293T cells were transfected with plasmids expressing human P301L tau (D, E) or human WT αSyn (F, G) and treated with αSyn fibril, K18 tau fibril or tau+αSyn copolymer as indicated. Two different doses of tau+αSyn copolymers were used in both experiments. Cells were biochemically fractionated into Triton-soluble and Triton-insoluble fractions. Western blots depicting Triton-soluble and Triton-insoluble levels of tau (D) or αSyn (F) in seeded HEK293T cells. The molecular weights are designated in the right side of the western blots and type of exogenous seeds added depicted on the left. Quantification of Triton-insoluble tau levels normalized to Triton-soluble tau levels of the corresponding sample was done using ImageJ (E, G). Three experimental replicates are shown here. Full-length blots are presented in Supplementary Fig. 2B–C. One-way ANOVA with Tukey’s post hoc analysis. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We next investigated the relative efficiencies of these fibrils in inducing intracellular seeding of tau or αSyn in in vitro assays. HEK293T cells were transfected with either full-length human P301L tau or full-length human WT αSyn and treated with either 1 µM of K18 tau fibrils, 1 µM αSyn fibrils, 1 µM of tau + αSyn copolymers or 2 µM of tau + αSyn copolymers. We generated two different copolymer preparations containing increasing amounts of tau and αSyn monomers (1:1 ratio) in order to assess whether the in vitro seeding activity was dose-dependent. Subcellular fractionation of cells using buffers containing detergent was used to evaluate the extent of intracellular seeded proteinopathy, with the Triton X-100 insoluble fraction indicating the seeded protein content and the supernatant indicating levels of soluble cellular protein. Since we used N-terminally truncated αSyn (lacking 1–20 residues) to generate the αSyn aggregates which otherwise does not alter its aggregation properties in vitro or in vivo (Waxman and Giasson, 2010), we were able to use an antibody specific to these N-terminal residues to establish that the intracellular seeded αSyn originated from full-length αSyn protein expressing from the HEK293T cells. The copolymers were also generated using this truncated αSyn monomer. For detecting intracellular seeded tau, we used an antibody that would react with residues within the K18 fragment of the exogenous aggregates as well as the overexpressing tau protein. In HEK293T cells overexpressing human P301L tau, we found that application of exogenous K18 tau fibrils induced insoluble tau compared to cells that were not treated with any fibrils (Fig. 1D and E; P < 0.05 compared to αSyn aggregate treated cells). Exogenous αSyn fibrils also induced modest levels of tau seeding under similar conditions (Fig. 1D and E). Interestingly, application of tau + αSyn copolymers dramatically enhanced tau seeding compared to cells treated with tau fibrils or αSyn fibrils alone (Fig. 1D and E; P < 0.0001 compared to tau or αSyn aggregate treated cells). In fact, we observed that 2 µM of copolymers was superior to the lower dose of 1 µM, suggesting that this process is dose-dependent (Fig. 1D and E; P < 0.01).
We next tested the seeding activity of these different fibrils preparations on HEK293T cells expressing human WT αSyn. We found that αSyn fibrils, but not K18 tau fibrils, robustly seeded insoluble αSyn in these cells (Fig. 1F and G; P < 0.001). In these cells, tau + αSyn copolymer did not boost αSyn seeding relative to αSyn fibrils—in fact, while the higher concentration of copolymer fibrils (2 µM) led to seeding activity comparable to αSyn fibrils alone, the lower concentration of copolymer fibrils (1 µM) appeared to suppress accumulation of seeded insoluble αSyn relative to αSyn fibril (Fig. 1F and G; P < 0.05). Given that our slot blot as well as EM data showed availability of αSyn epitopes within the copolymers, these data are suggestive of unique differences in copolymer-mediated seeding depending on the cellular substrate. Taken together, our data suggest that while the tau + αSyn copolymer seeds showed a synergistic effect on tau seeding, they showed little to negative effect on αSyn seeding process in this cellular seeding model.
Tau + αSyn copolymers induce ptau pathology in PS19 mouse model of tauopathy
To test the relative seeding activities of tau fibrils, αSyn fibrils or tau + αSyn copolymers, we injected these aggregates into the right dorsal hippocampus of 2-month-old female PS19 mice. The PS19 line is a widely used mouse model of tauopathy that develops NFTs, neuronal loss and brain atrophy by ∼9 months of age (Yoshiyama et al., 2007). Additionally, at younger ages, these mice are amenable to experimental paradigms that test propagation of seeded tau following intracerebral injection of recombinant tau fibrils or human brain-derived lysates (Iba et al., 2013; Guo et al., 2016; Narasimhan et al., 2017; Woerman et al., 2017). Though PS19 mice show inter-animal variability, it was recently determined that prion-related tau seeding activity is not affected by sex differences in these mice, especially at younger ages (Woerman et al., 2018). Therefore, we used only female mice in this study to limit the complexity of the study design. Following unilateral injection of various proteinaceous aggregates, we analysed the mice at 5 months of age for induction of phosphorylated tau (ptau) using ptau specific 7F2 antibody (specific for pT205 residue) and PHF1 antibody (specific for pS396/S404 residues). 7F2 and PHF1 immunoreactivity at the site of injection was used to impute a semi-quantitative heatmap based on blinded assessment of immunoreactivity intensity on a scale of 0–3 (Supplementary Fig. 3A). Both the 7F2 and PHF1 antibodies recognize phosphorylated residues on tau outside of the K18 filaments (residues 244–372) used as exogenous seeds, therefore establishing that the ptau pathology is generated by the tau present in the transgenic animal. We found that PS19 mice injected with tau or αSyn fibrils exhibited robust bilateral intracellular and neuropil 7F2 immunoreactivity relative to PBS injected or naïve mice that displayed negligible ptau immunoreactivity around the site of injection at bregma −2.0 (Fig. 2A and Supplementary Fig. 4A–D). The ptau was spread throughout the hippocampus, cortex and hypothalamus while the caudate putamen and thalamus were spared in the tau fibril or αSyn fibril-injected cohort. Similar to the tau fibril-injected mice, the tau + αSyn copolymer aggregate injected mice also showed robust ptau pathology (Fig. 2A). Both the pattern of spread (Fig. 2A) as well the neuropathology score in the ipsilateral and contralateral hemispheres (Supplementary Fig. 4A–D, P < 0.0001 compared to control mice) were equivalent in the three PS19 cohorts injected with the tau fibril, αSyn filbril and tau + αSyn copolymers. Examination of the PHF1 epitope similarly showed that the tau fibril-injected and αSyn fibril-injected cohorts accumulated somatodendritic PHF1-reactive inclusions in both the ipsilateral and contralateral hippocampus and cortex, indicating development of pathologic tau while control mice exhibited predominantly diffuse neuropil PHF1 immunoreactivity (Supplementary Fig. 5A). Relative to K18 tau injected cohort, the tau + αSyn copolymer-injected mice had lower burden of intracellular PHF1 immunoreactivity in the cortex (Supplementary Fig. 5A). Overall, this indicated that though tau + αSyn copolymers induced robust ptau pathology, they did not exacerbate tauopathy relative to the tau fibril-injected mice at the site of injection.
Figure 2.

Induction of tauopathy in PS19 mice seeded with different fibrillar species. Representative images from the ipsilateral (‘IPSI’) and contralateral (‘CONTRA’) hippocampus showing pathology in PS19 mice. Tau, αSyn or tau+αSyn copolymerized fibrils were injected into the right hippocampi of 2-month-old PS19 mice. Five-month-old mice were analysed for ptau (A), pretangle MC1-immunoreactive tau (B) and p62 (C). Age-matched PS19 mice (uninjected naive) were used as controls. Semi-quantitative analyses performed on a scale of 0 (no pathology) to 3 (high pathology) for different epitopes tested were colour-coded onto heat maps and depicted under each panel. The key to heat map scores is provided at the bottom of the figure. The ipsilateral side is shown on the right and contralateral side is depicted on the left of the heatmaps. The analysis was done at bregma −2.0 (area of injection). The insets indicate magnified area of interest indicated by a small box within the main panel. Scale Bar, Inset, 50 µm; main panel, 200 µm. n = 5–7 female mice/group.
Next, we used the conformation-sensitive anti tau antibody MC1, the stress-inducible autophagy marker protein p62 and Gallyas silver staining to examine whether the ptau inclusions in these mice were similar to NFT-type aggregated tau. We found that both tau and αSyn fibril-injected PS19 mice showed MC1 immunostaining in the ipsi- and contralateral brain hemispheres (Fig. 2B). The major brain areas in these mice with MC1 immunoreactivity were hippocampus, motor cortex, somatosensory cortex and auditory cortex. However, in the tau + αSyn copolymer-injected mice, we observed very limited MC1 immunoreactivity localized in the hippocampus and cortical amygdala area of the ipsilateral hemisphere (Fig. 2B). Quantitative assessment of the neuropathology score revealed that the copolymer-injected mice showed lower MC1 immunoreactivity relative to the tau injected (P < 0.0001 in ipsilateral cortex and hippocampus; P < 0.001 in contralateral and hippocampus) and αSyn fibril-injected cohort (P < 0.0001 in ipsilateral cortex and hippocampus; P < 0.01 in contralateral hippocampus) (Supplementary Fig. 4E–H).
The autophagy associated adaptor protein, p62, is generally associated with formation of neuropathological inclusions (Deng et al., 2017). Consistent with this, we found that p62 is upregulated in the ipsilateral hippocampus and auditory cortex of tau fibril and αSyn fibril-injected mice (Fig. 2C and Supplementary Fig. 4I and J, P < 0.01 in ipsilateral hippocampus and P < 0.0001 in ipsilateral cortex). In the contralateral hippocampus, only K18 tau showed robust p62 immunoreactivity (Supplementary Fig. 4K, P < 0.0001 relative to control). In the contralateral cortex, neither tau aggregates nor tau + αSyn copolymer-injected mice displayed detectable levels of p62 (Supplementary Fig. 4L). Interestingly, the αSyn aggregate injected mice had sparse p62 immunoreactivity in the contralateral cortex (Supplementary Fig. 4I–L).
We performed Gallyas silver staining on these mice brains and observed low amounts of silver-positive inclusion pathology distributed across the hippocampus and cortex of K18 fibril and αSyn fibril-injected mice (Supplementary Fig. 5B), indicating that the MC1 and p62-immunopositive cells indeed contained frank NFTs. The tau + αSyn copolymer-injected mice or control mice did not display any detectable Gallyas positive structures (Supplementary Fig. 5B).
Next, we examined whether any of these fibrillar species induced pathological inclusions of αSyn in the PS19 mice. Consistent with our earlier studies on wild-type mice (Sorrentino et al., 2017), we observed that injection of αSyn fibrils led to accumulation of modest levels of 81A-immunopositive (antibody reactive to pSer129 αSyn) intracellular inclusions in the hippocampus and cortex, suggesting that the human αSyn fibrils are capable of seeding endogenous mouse αSyn (Supplementary Fig. 5C). However, neither tau K18 fibrils nor tau + αSyn copolymers were able to induce detectable levels of intracellular αSyn inclusions (Supplementary Fig. 5C).
We wanted to examine the relative contents of intracellular inclusions in the PS19 mice seeded with tau fibrils, αSyn fibrils or tau + αSyn copolymerized fibrils. Therefore, we performed co-immunofluorescence studies to co-localize tau and αSyn immunoreactivity in the hippocampus and cortex of these mice using a human tau antibody (3026 antibody specific for total human tau; Strang et al., 2017) and mouse αSyn antibody (5G9 antibody specific for mouse Syn; Sorrentino et al., 2018). In all the cohorts of the fibril-injected mice, we observed both human tau (arrows, Supplementary Fig. 6) and mouse αSyn (arrowheads, Supplementary Fig. 6) in individual cells. We observed rare instances of human tau and mouse αSyn protein co-localizing in the same cells in the cortex or hippocampus of tau fibril-injected mice (Supplementary Fig. 6A, cortex) but could not detect co-localized signal in the tau + αSyn copolymer-injected mice. This suggests that the subsequent transmission of the cross-seeds may be independent of an additive pathologic cascade or that the copolymer-induced biologically active seeds generated in vivo may not be detectable by these antibodies and may require antibodies recognizing specialized conformers.
We also tested the levels of neuroinflammation in these PS19 mice using astrocyte-specific GFAP antibody and microglia-specific Iba-1 antibody. We observed increased levels of bilateral astrocytosis in the hippocampus, retrosplenial area, fimbria and internal capsule in both the tau fibril and αSyn fibril-injected mice relative to control PS19 mice (Fig. 3A). In both the ipsilateral and contralateral hippocampus, tau fibril, αSyn fibril and tau + αSyn copolymer-injected mice showed increased astrocytosis (Supplementary Fig. 4M and O; P < 0.0001 compared to control), though the GFAP burden was lower in tau + αSyn copolymer-injected mice (Supplementary Fig. 4M and O; P < 0.05 in ipsilateral hemisphere and P < 0.01 in contralateral hemisphere compared to tau fibril and αSyn fibril-injected mice). In the ipsilateral cortex, both tau fibril (P < 0.001) and αSyn fibril (P < 0.01) injected mice showed GFAP immunoreactivity relative to control mice (Fig. 3A, Supplementary Fig. 4N). The tau + αSyn copolymer-injected mice had lower burden of GFAP immunostaining that was predominantly localized to internal capsule and ipsilateral auditory cortex (Fig. 3A andSupplementary Fig. 4N, P < 0.01 compared to tau and P < 0.05 compared to αSyn). None of the cohorts showed measurable GFAP immunostaining in the contralateral cortex (Fig 3A;Supplementary Fig. 4P). Iba-1 reactivity, indicative of microgliosis was observed in the ipsilateral hippocampus, retrosplenial area and cortex of the tau fibril and αSyn fibril-injected mice relative to control PS19 mice (Fig. 3B). Microgliosis in the tau + αSyn copolymer-injected mice was also observed in similar areas as these two cohorts, albeit to a lesser extent (Fig. 3B andSupplementary Fig. 4Q, P < 0.001 relative to K18 tau mice and αSyn in ipsilateral hippocampus). Only the ipsilateral hippocampus of the fibril-injected mice (but not ipsilateral cortex or contralateral cortex and contralateral hippocampus) showed significantly increased Iba-1 immunostaining relative to control mice (Supplementary Fig. 4Q–T).
Figure 3.

Induction of gliosis in PS19 mouse model of tauopathy seeded with different fibrillar species. Representative images from the ipsilateral (‘IPSI’) and contralateral (‘CONTRA’) hippocampus showing gliosis in PS19 mice. Tau, αSyn or tau+αSyn copolymerized fibrils were injected into the right hippocampi of 2-month-old PS19 mice. Age-matched PS19 mice (PBS injected or uninjected naive) were used as controls. Five-month-old mice were analysed for GFAP-immunoreactive astrogliosis (A) and Iba-1-immunoreactive microgliosis (B). Semi-quantitative analyses performed on a scale of 0 (no pathology) to 3 (high pathology) for different epitopes tested were colour-coded onto heat maps and depicted under each panel. The key to heat map scores is provided at the bottom of the figure. The ipsilateral side is shown on the right and contralateral side is depicted on the left on the heat map. The analysis was done at bregma −2.0 (area of injection). The insets indicate magnified area of interest indicated by the small box within the main panel. Scale Bar, Inset, 50 µm (A); Inset, 25 µm (B); main panel, 200 µm. n = 5–7 female mice/group.
We were curious whether a simple mixture of pre-aggregated tau fibrils and pre-aggregated αSyn fibrils would have differential seeding properties relative to the copolymerized fibrils in PS19 mice. These mixed fibrils were generated by mixing pre-existing individual fibrils (1 µM each) immediately before brain injection as opposed to the copolymers that were generated from incubating individual monomers. To establish the seeding properties of such mixed aggregates containing both tau and αSyn, we delivered a freshly prepared mixture of pre-aggregated tau and αSyn unilaterally into the dorsal hippocampus of PS19 mice. Surprisingly, we observed that relative to the tau + αSyn copolymer-injected mice, the tau + αSyn mixed aggregate injected mice had lower burden of ptau at the site of injection in the ipsilateral hemisphere (Supplementary Fig. 7A–C, P < 0.05 in ipsilateral hippocampus relative to tau and P < 0.001 in ipsilateral cortex relative to tau, αSyn and tau + αSyn copolymer-injected mice). In particular, we observed that these mixed fibril-induced tauopathy was transmitted less robustly to the contralateral hippocampus, cortex and amygdala relative to tau + αSyn copolymer seeded mice (Supplementary Fig. 7A, D and E, P < 0.001 in contralateral hippocampus and P < 0.0001 in contralateral cortex relative to tau, αSyn and tau + αSyn copolymer-injected mice).
Since the presence of mixed pathologies can have detrimental effects on clinical phenotype and disease progression in humans [reviewed in Spires-Jones et al. (2017)], we were curious whether any of these seeded mice showed abnormalities in synaptic homeostasis. We immunostained for synaptophysin, a pre-synaptic protein, that is a gold standard in evaluating Alzheimer’s disease-type synaptic abnormalities (de Wilde et al., 2016) in the ipsilateral cortex and hippocampus of our mouse cohorts (Supplementary Fig. 8A). We did not find any significant changes in synaptophysin density in the ipsilateral cortex (Supplementary Fig. 8B). In the ipsilateral hippocampus, we observed reduced trends (non-significant, P > 0.05) of synaptophysin density in the K18 tau and tau + αSyn mixed fibril-injected mice compared to control mice (Supplementary Fig. 8C). Interestingly, we observed increased synaptic density in αSyn fibril and tau + αSyn copolymer-injected mice relative to K18 tau mice (Supplementary Fig. 8C, P < 0.01). The tau + αSyn copolymer-injected mice had higher synaptophysin density compared to control mice (P < 0.05) and tau + αSyn mixed fibril-injected mice (P < 0.01) (Supplementary Fig. 8C). Future experiments will need to be done to understand how the induction and transmission of cross-seeded proteinopathy alter behavioural outcomes on a functional and cellular level.
Tau + αSyn copolymers reinforce the extent of pathology transmission in PS19 mouse model of tauopathy
The semi-quantitative heatmap analysis of PS19 mice showed that injection of tau + αSyn copolymer fibrils resulted in comparatively lower levels of ptau pathology burden at the level of injection compared to tau fibril-injected mice (Fig. 2A andSupplementary Fig. 5A). We next wanted to compare the extent of transmission of the human tau cross-seeded by the tau + αSyn copolymer seeds or the K18 tau seeds alone in neuroanatomically connected parts of the brain. To study the CNS distribution pattern of ptau neuropathology along the anterio-posterior axis, we dissected the whole brains of tau + αSyn copolymer-injected mice and tau fibril-injected mice at five different bregma locations and stained with 7F2 antibody (Fig. 4A). We observed robust ptau pathology widely distributed throughout the brain in both the K18 tau fibril and tau + αSyn copolymer-injected mice (Fig. 4A; representative images of brain areas, Supplementary Fig 9A). In tau + αSyn copolymer-injected mice, there was more efficient contralateral propagation along the anterio-posterior axis, compared to tau fibril-injected mice which appeared to accumulate higher levels of ptau pathology in and around the site of injection (Fig. 4A; Supplementary Fig 9A). In particular, we noticed comparatively higher ptau burden in ectorhinal cortex, perirhinal cortex, entorhinal cortex, pyramidal layer, hypothalamus pons, inferior colliculus and periaqueductal grey areas in the contralateral hemisphere of tau + αSyn copolymer-injected mice compared to tau fibril-injected mice. Based on known anterograde and retrograde neuroanatomic connectivity to the dorsal hippocampus (Oh et al., 2014), which was the site of the injection, we mapped out the proximal brain areas that are neuroanatomically connected to this specific region (Fig. 4B and C). While we observed that both the injected cohorts had equivalent spread of ptau in major brain areas connected via anterograde connections, we found that the ptau pathology was more widely distributed along retrogradely connected pathways in the tau + αSyn copolymer-injected mice brains compared to tau fibril-injected mice (areas with ptau pathology marked in red font, Fig. 4B and C; representative images of brain areas, Supplementary Fig. 9A). This suggests that tau + αSyn copolymer seeded tauopathy is more efficiently and selectively propagated along the neuroanatomic connections compared to K18 tau fibrils themselves.
Figure 4.
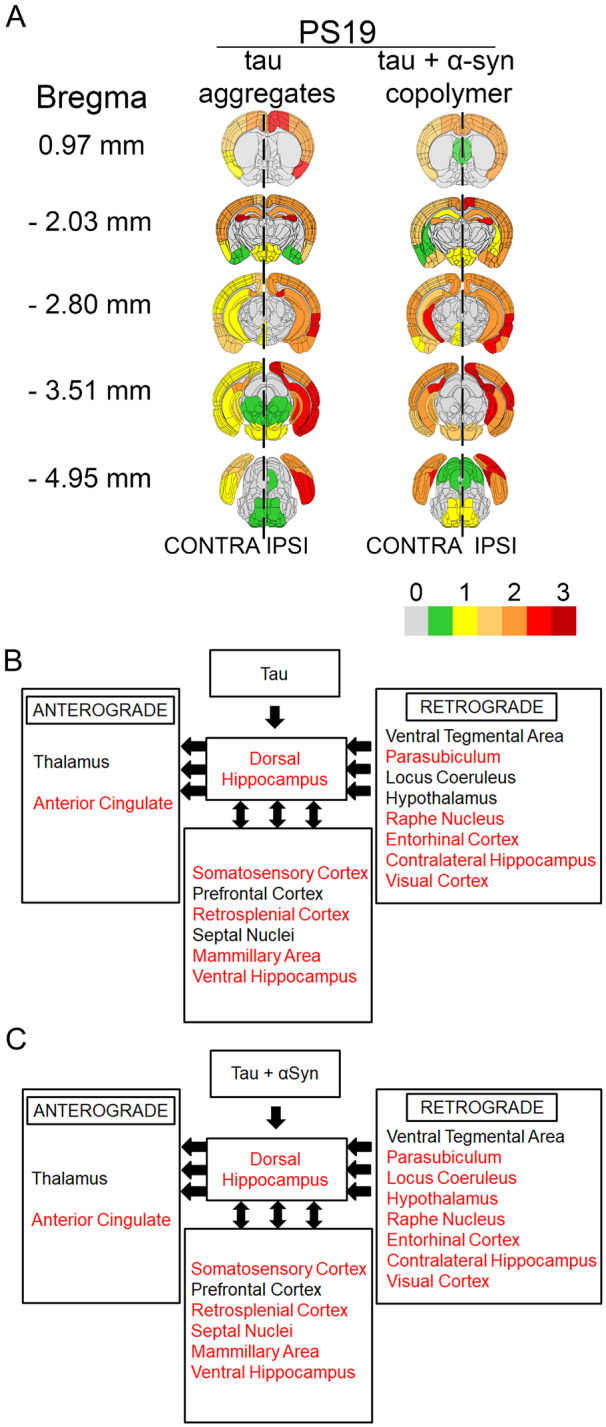
Tau + αSyn copolymers accelerate the transmission of tau pathology in PS19 mice. Semi-quantitative analyses were performed by assigning pathology severity in seeded PS19 mice on a scale of 0 (no pathology) to 3 (high pathology) corresponding to 7F2 immunoreactive ptau pathology which was then colour-coded onto heat maps (A). The key to heat map scores is provided at the bottom of the figure. The ipsilateral (‘IPSI’) side is shown on the right and contralateral (‘CONTRA’) side is depicted on the left with a dotted line demarcating the central axis. Five coronal planes are shown from top to bottom: bregma 0.97, −2.03, −2.80, −3.51 and −4.95 mm. Scores were averaged between mice for each group. Diagrams show different brain regions neuroanatomically connected to the dorsal hippocampus via anterograde pathways or retrograde pathways (left-pointing arrows) or both pathways (double-headed arrows) (B, C). Brain regions showing 7F2 immunoreactive ptau pathology is indicated. Red text indicates brain regions showing ptau pathology and black text indicates regions showing no detectable ptau pathology. (B) The relative neuroanatomic distribution of ptau pathology in tau fibril-injected mice and (C) shows the same for tau+αSyn copolymer-injected mice. For graphical representation of the neuropathology scores, see Supplementary Fig. 4A–D; for representative images of stained brain areas, see Supplementary Fig. 9A. n = 4 female mice/group.
Tau + αSyn copolymers show restricted induction of pathology in the M20 mouse model of synucleinopathy
We next investigated the in vivo seeding efficiency of tau, αSyn, tau + αSyn copolymers or tau + αSyn mixed fibrils in a mouse model of synucleinopathy. For this experiment, we selected the M20 mice that overexpress WT human αSyn but do not intrinsically develop αSyn inclusion pathology or an overt phenotype in their lifetime (Giasson et al., 2002). We have previously shown that intracerebral injection of human αSyn fibrils leads to induction and widespread CNS transmission of pathological αSyn inclusions (Sacino et al., 2014; Sorrentino et al., 2017), allowing this model to be used to study prion-like synuclein seeding and transmission aetiologies. Because this model does not intrinsically develop any αSyn pathology, it is very convenient in terms of assessing the transmission of seeded pathology along neuroanatomic connectivity.
To investigate the relative potencies of tau fibrils, αSyn fibrils, tau + αSyn copolymers or tau + αSyn mixed fibrils in inducing inclusion pathology in synucleinopathies, we injected these fibrils into the right dorsal hippocampus of 2-month-old M20 mice and aged them to 5 months. The burden of pathological αSyn in the brains of these mice was assessed using an antibody raised against pSer129 αSyn (clone 81A; Rutherford et al., 2016), an epitope that has been used widely in the field as a sensitive marker of abnormally aggregated αSyn. Consistent with our previous observations (Sorrentino et al., 2017), we did not observe any αSyn pathology in M20 mice injected with PBS (Fig. 5A). M20 mice injected with αSyn fibrils developed robust 81A immunopositive intracellular αSyn pathology in ipsilateral hippocampus and ipsilateral and contralateral cortices (Fig. 5A andSupplementary Fig. 10A–D, P < 0.001 in ipsilateral hippocampus and P < 0.0001 in ipsilateral and contralateral cortex compared to control mice). The tau fibril-injected mice also developed similar patterns of intracellular αSyn pathology in the cortex (Fig. 5A andSupplementary Fig. 10B and D, P < 0.0001 in ipsilateral and contralateral cortex), though there were no detectable inclusions in the hippocampus (Supplementary Fig. 10A and C). Since human K18 tau does not seed mouse αSyn (Supplementary Fig. 5C, tau aggregate panel), this suggests that human K18 tau aggregates can actually prime human αSyn, suggesting some form of cooperativity between the human proteins. On the other hand, the tau + αSyn copolymer-injected mice developed intracellular αSyn pathology predominantly only in the ipsilateral cortex (Supplementary Fig. 10B, P < 0.0001 relative to control mice) and less robustly in the contralateral cortex while sparing the retrospenial cortex and amygdala (Supplementary Fig. 10D, P < 0.01 relative to αSyn mice). No pathology was detected in the hippocampus of tau + αSyn copolymer-injected mice (Fig. 5A andSupplementary Fig. 10A, P < 0.001 in ipsilateral hippocampus relative to αSyn mice).
Figure 5.

Induction of synucleinopathy and gliosis in M20 mice seeded with different fibrillar species. Representative images from the ipsilateral (‘IPSI’) and contralateral (‘CONTRA’) hippocampus or cortex showing pathology in M20 mice. Tau, αSyn or tau+αSyn copolymers were injected into the right hippocampi of 2-month-old M20 mice. Age-matched mice (PBS injected or naive) were used as controls. Five-month-old mice were analysed for pSer129 αSyn immunoreactivity (A, 81A), astrogliosis (B, GFAP) and microgliosis (C, Iba-1). Semi-quantitative analyses performed on a scale of 0 (no pathology) to 3 (high pathology) for different epitopes tested were colour-coded onto heat maps and depicted under each panel. The key to heat map scores is provided at the bottom of the figure. The ipsilateral side is shown on the right and contralateral side is depicted on the left on the heat map. The analysis was done at bregma −2.0 (area of injection). The insets indicate magnified area of interest indicated by the small box within the main panel. Inset, 25 µm (A, C); Inset, 50 µm (B); main panel, 200 µm. n = 4 mice/group
We next tested the induction of astrogliosis in fibril-injected M20 mice using the GFAP antibody and Iba-1 antibody (Fig. 5B and C and Supplementary Fig. 10E–L). It was interesting to note that while astrocytosis was affected in all the areas examined, microgliosis was altered predominantly in the hippocampus. GFAP immunoreactivity was increased in the hippocampus, thalamus and striatum of both hemispheres of αSyn-injected mice (Fig. 5B andSupplementary Fig. 10E–H, P < 0.05 in ipsilateral hippocampus, P < 0.01 in ipsilateral cortex, P < 0.001 in contralateral cortex compared to control). The K18 tau-injected mice did not show notable changes in GFAP immunostaining in the contralateral or ipsilateral hemispheres (Fig. 5B andSupplementary Fig. 10E–H). The tau + αSyn copolymer-injected mice showed modest changes in the GFAP immunohistochemical burden in the ipsilateral hippocampus (P < 0.05 compared to control; Supplementary Fig. 10E) while showing reduced GFAP in ipsilateral cortex and contralateral cortex compared to αSyn mice (Supplementary Fig. 10F and H; P < 0.01).
Microgliosis levels were increased in the ipsilateral hippocampus, striatum and thalamus of αSyn fibrils, tau fibril and tau + αSyn copolymer-injected mice (Fig. 5C andSupplementary Fig. 10I–L). Compared to αSyn fibril-injected mice, tau fibril-injected mice showed lower Iba-1 reactivity in the ipsilateral and contralateral hippocampus (Supplementary Fig. 10I, K; P < 0.0001) but displayed modest increases in the thalamus and striatum areas (Fig. 5C). The tau + αSyn copolymer-injected mice showed increased Iba-1 reactivity in the cortex compared to control mice but this was comparatively lower than that induced by αSyn fibril alone (Supplementary Fig. 10I and K). None of the fibrillar species altered microgliosis in ipsilateral or contralateral cortices to a significant degree (Fig. 5C andSupplementary Fig. 10J and L).
Although human tau fibrils induced αSyn inclusions in M20 mice (Fig. 5A), these did not induce ptau inclusions as examined by 7F2 antibody in the cortex or hippocampus adjoining the area of injection (Supplementary Fig. 11A). Indeed, none of tau fibril, αSyn fibril or tau + αSyn copolymer-injected mice showed any 7F2 immunoreactivity (Supplementary Fig. 11A). Similarly, we noticed that injection of all three fibrillar species did not induce intracellular tau phosphorylation at the PHF1 epitope in the ipsilateral and contralateral hemispheres (Supplementary Fig. 11B). None of these mice developed MC1 pathology, which typically represents misfolded tau regarded as a late feature in tauopathies (Supplementary Fig. 11C). These mice also did not develop intracellular accumulation of the autophagy regulator protein, p62 (Supplementary Fig. 11D).
We next examined the efficacy of a mixed preparation of tau fibrils and αSyn fibrils on induction of neuropathology in M20 mice. The intracellular αSyn pathology burden in the ipsilateral hemisphere of mice injected with this 1:1 mixture of pre-aggregated tau fibril and αSyn fibril resulted in robust 81A immunopositive inclusions in the ipsilateral and contralateral cortex and hippocampus (Supplementary Fig. 12). Interestingly, the αSyn pathology was equally distributed in the ipsilateral and contralateral hemispheres of the tau + αSyn mixed fibril-injected mice in contrast to the tau + αSyn copolymer injected cohort that shows restricted spread to the contralateral hemisphere (Supplementary Fig. 12A, D and E, P < 0.01 in hippocampus and P < 0.001 in contralateral cortex relative to tau + αSyn copolymer-injected mice). Indeed, the relative prevalence of αSyn pathology in tau + αSyn mixed fibril-injected mice was equivalent to mice injected with αSyn fibrils (Supplementary Fig. 12B–E), suggesting that the mixed fibrillar species had higher propensity of transmission to the contralateral hemisphere relative to the tau + αSyn copolymer-injected mice.
Tau + αSyn copolymers show reduced transmission of pathology in M20 mouse model of synucleinopathy compared to αSyn fibrils
In order to understand whether tau + αSyn copolymers had a modifying effect on transmission of seeded αSyn pathology relative to αSyn fibril alone, we assessed the pathological burden and distribution of αSyn pathology across the whole brains of M20 mice (Fig. 6). Our semi-quantitative analysis of 81A immunostaining at different bregma locations showed differential patterns of distribution of pSer129 αSyn pathology in M20 mice seeded with αSyn versus the mice seeded with the tau + αSyn copolymers. Overall, we observed that the tau + αSyn copolymers were not efficient seeding substrates for transmission to the contralateral hemisphere, indicating that there was no apparent additive effect on αSyn pathology induced with the tau + αSyn copolymers compared to αSyn fibrils alone (Fig. 6A, representative images of brain areas, Supplementary Fig. 9B). In particular, several brain areas that displayed robust pSer129 αSyn pathology in the αSyn-injected mice (mamillary area, ventral tegmental area, locus ceruleus, raphe nucleus and visual cortex) were not affected in the tau + αSyn copolymer-injected mice (Fig. 6A and Supplementary Fig. 9B). Using neuroanatomic connectivity patterns derived from the Allen Brain Atlas (Oh et al., 2014), we further examined the antero-posterior distribution of pSer129-αSyn pathology in brain regions connected anterogradely and retrogradely to the dorsal hippocampus, the site of these fibril injections (Fig 6B and C). In the tau + αSyn copolymer injected M20 mice, we observed that there were fewer retrogradely connected brain areas that developed pSer129-αSyn pathology when compared to mice seeded with αSyn alone (areas with αSyn pathology indicated by red font, Fig 6B and C; representative areas of the brain, Supplementary Fig. 9B). Overall, in the M20 mice, the tau + αSyn copolymers were relatively inefficient in both inducing hippocampal pathology and transmission of seeded αSyn pathology along neuroanatomic connections.
Figure 6.
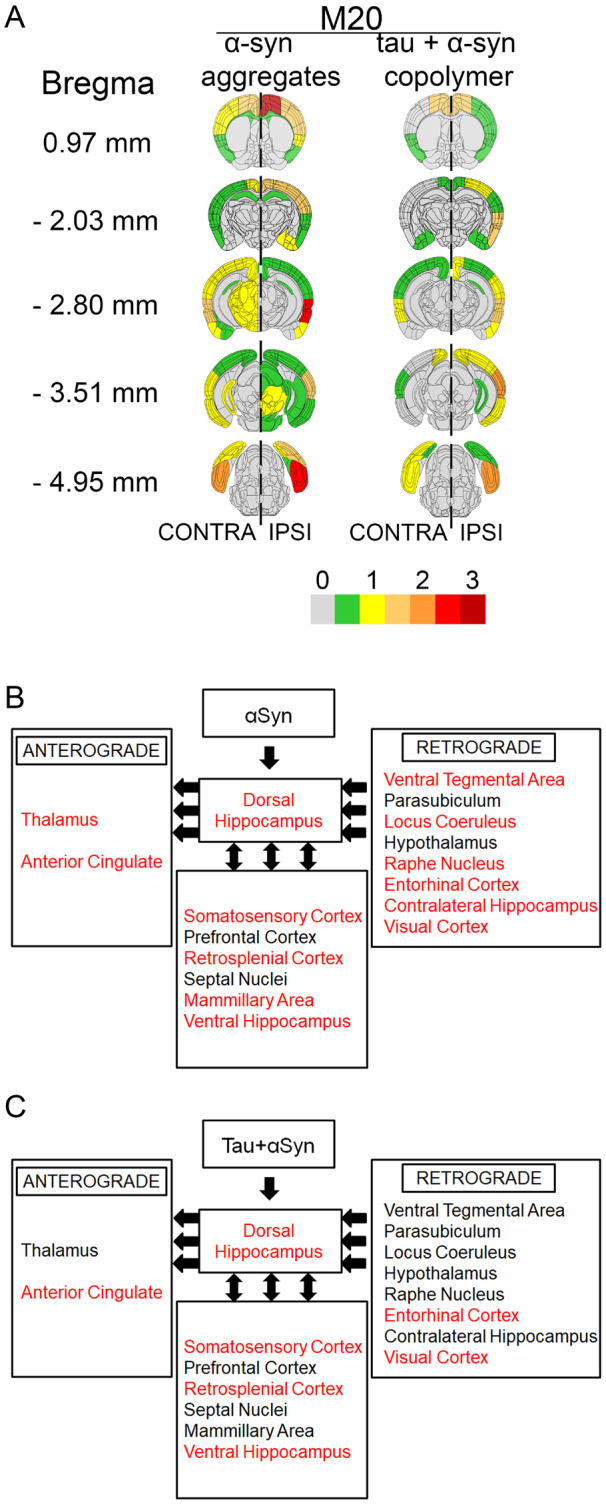
Seeding with tau + αSyn copolymers lead to reduced distribution of αSyn pathology compared to αSyn fibrils injected M20 mice. Semi-quantitative analyses of pathological burden in M20 mice was done by assigning pSer129 αSyn pathology (81A immunoreactivity) intensity on a scale of 0 (no pathology) to 3 (high pathology) which was then colour-coded onto heat maps (A). The key to heat map scores is provided at the bottom of the figure. The ipsilateral (‘IPSI’) side is shown on the right and contralateral (‘CONTRA’) side is depicted on the left with a dotted line demarcating the central axis. Five coronal planes are shown from top to bottom: bregma 0.97, −2.03, −2.80, −3.51 and −4.95 mm. Scores were averaged between mice for each group. The ipsilateral side is shown on the right and contralateral side is depicted on the left. (B, C) Diagrams show different brain regions neuroanatomically connected to the dorsal hippocampus via anterograde or retrograde pathways (left-pointed arrows) or both pathways (double-headed arrows). Brain regions showing pSer129 αSyn pathology are indicated. Red text indicates brain regions showing pαSyn pathology and black text indicates regions showing no detectable pαSyn pathology. (B) The relative neuroanatomic distribution of pαSyn pathology in αSyn fibril-injected mice and (C) shows the same for tau+αSyn copolymer-injected mice. For graphical representation of the neuropathology scores, see Supplementary Fig. 10A–D; for representative images of stained brain areas, see Supplementary Fig. 9B. n = 4 mice/group.
Discussion
In Alzheimer’s disease and LBD patients, presence of mixed pathologies of tau and αSyn is associated with higher risk of dementia (Schneider et al., 2007) and shorter survival (Irwin et al., 2017). Several studies have shown that tau and αSyn are present as co-aggregates in patient brains (Arima et al., 2000; Ishizawa et al., 2003; Colom-Cadena et al., 2013), thus raising the possibility that these co-occurring pathological lesions may result from specific molecular interactions between these different proteins. Though there is evidence that tau and αSyn can indeed co-aggregate in vitro (Jensen et al., 1999; Giasson et al., 2003; Guo et al., 2013), the molecular properties of such cross-seeds remain obscure. Given that prion-type templated seeding and transmission is one of the major pathways utilized by both tau and αSyn to achieve CNS-wide spread, we wanted to examine whether copolymerized tau + αSyn fibrillar species displayed unique seeding properties relative to tau fibrils or αSyn fibrils in cross-seeding tau or αSyn in vivo.
Towards this goal, we delivered K18 tau fibrils or WT αSyn fibrils alone, or as preformed copolymerized fibrils or as a mixture of pre-aggregated fibrils and assessed whether there is a synergistic interaction between these proteins leading to the development of mixed pathologies in PS19 mice or M20 mice. In PS19 mice, we observed that (i) tau and tau + αSyn copolymer fibrils induce widespread ptau pathology, though the copolymers do not potentiate the induction of mis-folded tau at the site of injection; (ii) tau + αSyn copolymers led to more widespread transmission of tau pathology in neuroanatomically connected brain regions compared to tau fibrils alone; (iii) tau + αSyn mixed aggregates were inefficient seeding substrates compared to tau + αSyn copolymers; (iv) αSyn fibrils can also induce robust tauopathy, indicating cooperativity of interaction and (v) in spite of efficient heterotypic cross-seeding, we rarely observed co-localized intracellular inclusions containing both tau and αSyn. In M20 mice, we observed that (i) αSyn fibrils were relatively more efficient in induction and transmission of αSyn pathologies compared to tau + αSyn copolymers; (ii) mixed preparation of pre-aggregated tau and αSyn resulted in better induction of αSyn pathology at the site of injection as well as transmission to the contralateral side relative to the copolymer fibrils; (iii) K18 tau aggregates could induce αSyn pathologies, showing cooperativity and (iv) human K18 tau fibrils or tau + αSyn copolymers were unable to prime endogenous mouse tau. Overall, we found that both human tau and human αSyn can cross-seed each other and that tau + αSyn copolymers are better cross-seeding substrates in mouse models of tauopathies relative to synucleinopathies.
Recombinant tau protein requires exogenous factors such as heparin or polyanions to polymerize in vitro (Gamblin et al., 2003), while αSyn can self-polymerize. Interestingly, αSyn can itself induce tau polymerization in vitro and in cell culture in the absence of any other factors (Giasson et al., 2003; Kotzbauer et al., 2004; Waxman and Giasson, 2011; this study), suggesting some form of cooperativity. Indeed, specific conformers of recombinant αSyn can cross-seed human tau in vivo (Guo et al., 2013), consistent with our data. Importantly, we show that while human K18 tau and αSyn can cross-seed each other, these cross-seeds have differential effects on neuroanatomic spread of tauopathy or synucleinopathy. Whether these observations are related to the morphological, structural or biochemical properties of these cross-seeds remains to be investigated. Several groups have hypothesized that such cross-seeded fibrils may form different conformational strains with unique properties that correspond with phenotypic diversity and clinical severity in Alzheimer’s disease and related dementias (Sanders et al., 2014; Fitzpatrick et al., 2017; Falcon et al., 2018). Given this, our observation that tau + αSyn copolymers specifically enhance tauopathy spread but not synucleinopathy may be consistent with the idea that cross-seeds may form different strains that may determine phenotypic diversity and clinical severity in the ADRD spectrum.
Tau and αSyn pathologies co-exist in a spectrum of dementias, such as in Alzheimer’s disease, Parkinson’s disease and DLB (Spires-Jones et al., 2017), though different brain regions are selectively vulnerable to these individual pathologies. For example, in Alzheimer’s disease, the earliest NFTs are frequently observed in the entorhinal cortex whereas in LBDs, LBs are found in the neocortex, and in Parkinson’s disease, the LBs initiate in the substantia nigra. However, pure proteinopathies are now increasingly being recognized as rarer than instances of patients or even elderly healthy controls bearing multiple co-occurring pathologies of different mis-folded proteins (Kovacs et al., 2013; Dugger et al., 2014; White et al., 2016; Abner et al., 2017; Irwin et al., 2017; Robinson et al., 2018). Several studies have also shown that clinical severity is a measure of the burden of co-pathologies in Alzheimer’s disease and LB spectrum disorders. There is evidence that Alzheimer’s disease patients who have LB pathology undergo a more aggressive course of disease compared to those who have a more pure presentation with primarily Aβ plaques and NFTs (Coulthard and Love, 2018). Likewise in LBDs, presence of mixed pathologies was generally associated with worsened clinical phenotype and reduced survival (Irwin et al., 2017). Overall, though the pathologic link between co-occurring pathologies to disease severity in Alzheimer’s disease and LBDs is established, the aetiology of mixed pathologies in primary tauopathies still remain fairly uncertain barring a few notable studies (Robinson et al., 2018; Tan et al., 2018). By providing an experimental platform to compare the relative efficiencies of tau and αSyn cross-seeds in mouse models, we believe that our study has laid the foundation for testing the relevance of mixed or cross-seeds in differentially triggering neuropathologies and their effect in learning and memory impairment as a measure of clinical variability observed in patients.
Overall, our in vivo and in vitro data show that tau + αSyn copolymers were more efficient in seeding tauopathy but not synucleinopathy. However, there were some findings in mouse models that were not strictly recapitulated in cell culture studies. For example, αSyn fibrils could induce robust pathological ptau and pαSyn pathologies in PS19 mice, suggesting that αSyn fibrils can directly cross-seed tau, consistent with an earlier study (Guo et al., 2013). But, the seeding effect of αSyn fibrils on tau expressing cells in culture was not so dramatic. Strikingly, we also found that tau fibrils can induce αSyn pathology in the M20 model, though we did not observe this in cell culture. These somewhat disparate findings can be explained based on the fact that cell cultures do not fully recapitulate the anatomic connectivity, functional complexity and selective vulnerability of an intact nervous system.
A limitation of our study is that we characterized pathological distribution, not progression, as we only analysed tissue from one time point following the delivery of fibrillar seeds at a young age. To correctly delineate how cross-seeding alters disease severity and clinical presentation, multiple timepoints would be necessary to map out the initial priming, induction and subsequent transmission of pathologies across neuroanatomically connected brain regions. Additionally, we could not detect the occurrence of intracellular mixed pathologies in the seeded mouse models, at least in the mouse models that we examined. This may be related to either such species being transient or our current antibody repertoire being unable to detect the conformation of biologically active seeds. Using antibodies or other sensitive methods that can detect oligomeric species of tau or αSyn or other transient seed-competent tau or αSyn species will be informative in determining the nature of cross-seeds that result in altered transmission across neuroanatomic pathways. Future studies would also investigate whether our present observations using exogenous seeds derived from truncated recombinant proteins (with K18 tau or 21–140 αSyn, as used in this study) can recapitulate the functional and clinical outcomes of seeding-competent cross-seeds derived from human brains or with cross-seeds prepared from full-length recombinant proteins. For example, though K18 tau forms the core amyloid structure of PHFs and has been used widely by many groups including ours (Shammas et al., 2015), heparin-induced PHFs comprised of shorter tau sequences adopt structures that are different from those found in human patients (Zhang et al., 2019). Further experimentation using human brain-derived materials from primary or secondary tauopathies and LB disorders can illuminate the biophysical and functional complexity of cross-seeds and their role in modulating disease progression and clinical outcomes in neurodegenerative proteinopathies.
Supplementary material
Supplementary material is available at Brain Communications online.
Funding
We acknowledge support from Alzheimer’s Association AARG-16-443352 (P.C.), McKnight Brain Institute (P.C.), 1 FLORIDA Alzheimer’s Disease Center, National Institutes on Aging F30 AG063446 (Z.S.) and National Institutes on Aging T32 AG061892 (T.W.).
Competing interests
The authors report no competing interests.
Supplementary Material
Glossary
- αSyn =
α-synuclein
- Aβ =
amyloid β
- APOE =
Apolipoprotein E
- CC75C =
Cambridge city over-75s cohort
- EM =
electron microscopy
- LB =
Lewy body
- LBD =
Lewy body disorder
- MRC CFAS =
Medical Research Council Cognitive Function and Aging Study
- NFT =
neurofibrillary tangle
- pαSyn =
αSyn phosphorylated at Ser129
- ptau =
phosphorylated tau
- PBS =
Phosphate buffered saline
- ROSMAP =
Religious Orders Study and Memory and Aging Project
- VITA =
Vienna Trans-Danube Aging
- WT =
wild type
References
- Abner EL, Kryscio RJ, Schmitt FA, Fardo DW, Moga DC, Ighodaro ET, et al. Outcomes after diagnosis of mild cognitive impairment in a large autopsy series. Ann Neurol 2017; 81: 549–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arima K, Mizutani T, Alim MA, Tonozuka-Uehara H, Izumiyama Y, Hirai S, et al. NACP/alpha-synuclein and tau constitute two distinctive subsets of filaments in the same neuronal inclusions in brains from a family of parkinsonism and dementia with Lewy bodies: double-immunolabeling fluorescence and electron microscopic studies. Acta Neuropathol 2000; 100: 115–21. [DOI] [PubMed] [Google Scholar]
- Barnes LL, Leurgans S, Aggarwal NT, Shah RC, Arvanitakis Z, James BD, et al. Mixed pathology is more likely in black than white decedents with Alzheimer dementia. Neurology 2015; 85: 528–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolmont T Clavaguera F Meyer-Luehmann M Herzig MC Radde R Staufenbiel M, et al. Induction of tau pathology by intracerebral infusion of amyloid-beta-containing brain extract and by amyloid-beta deposition in APP x Tau transgenic mice. Am J Pathol 2007; 171: 2012–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty P, Hudson Iii VJ, Sacino AN, Brooks MMT, D’Alton S, Lewis J, et al. Inefficient induction and spread of seeded tau pathology in P301L mouse model of tauopathy suggests inherent physiological barriers to transmission. Acta Neuropathol 2015; 130: 303–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colom-Cadena M, Gelpi E, Charif S, Belbin O, Blesa R, Martí MJ, et al. Confluence of alpha-synuclein, tau, and beta-amyloid pathologies in dementia with Lewy bodies. J Neuropathol Exp Neurol 2013; 72: 1203–12. [DOI] [PubMed] [Google Scholar]
- Coulthard EJ, Love S. A broader view of dementia: multiple co-pathologies are the norm. Brain 2018; 141: 1894–7. [DOI] [PubMed] [Google Scholar]
- Crystal AS, Giasson BI, Crowe A, Kung M-P, Zhuang Z-P, Trojanowski JQ, et al. A comparison of amyloid fibrillogenesis using the novel fluorescent compound K114. J Neurochem 2003; 86: 1359–68. [DOI] [PubMed] [Google Scholar]
- de Wilde MC, Overk CR, Sijben JW, Masliah E. Meta-analysis of synaptic pathology in Alzheimer's disease reveals selective molecular vesicular machinery vulnerability. Alzheimer's Dement 2016; 12: 633–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Purtell K, Lachance V, Wold MS, Chen S, Yue Z. Autophagy receptors and neurodegenerative diseases. Trends Cell Biol 2017; 27: 491–504. [DOI] [PubMed] [Google Scholar]
- Dhillon J-KS, Riffe C, Moore BD, Ran Y, Chakrabarty P, Golde TE, et al. A novel panel of alpha-synuclein antibodies reveal distinctive staining profiles in synucleinopathies. PLoS One 2017; 12: e0184731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Mabon ME, Miller DC, Golbe LI, Lee VM-Y, et al. Concurrence of alpha-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathol 2002; 104: 7–11. [DOI] [PubMed] [Google Scholar]
- Dugger BN, Adler CH, Shill HA, Caviness J, Jacobson S, Driver-Dunckley E, et al. Concomitant pathologies among a spectrum of Parkinsonian disorders. Parkinsonism Relat Disord 2014; 20: 525–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon B, Zhang W, Schweighauser M, Murzin AG, Vidal R, Garringer HJ, et al. Tau filaments from multiple cases of sporadic and inherited Alzheimer's disease adopt a common fold. Acta Neuropathol 2018; 136: 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferman TJ, Aoki N, Crook JE, Murray ME, Graff-Radford NR, van Gerpen JA, et al. The limbic and neocortical contribution of alpha-synuclein, tau, and amyloid beta to disease duration in dementia with Lewy bodies. Alzheimer's Dement 2018; 14: 330–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, et al. Cryo-EM structures of tau filaments from Alzheimer's disease. Nature 2017; 547: 185–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamblin TC, Berry RW, Binder LI. Modeling tau polymerization in vitro: a review and synthesis. Biochemistry 2003; 42: 15009–17. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 2002; 34: 521–33. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, et al. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 2003; 300: 636–40. [DOI] [PubMed] [Google Scholar]
- Götz J, Chen F, Van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 2001; 293: 1491–5. [DOI] [PubMed] [Google Scholar]
- Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, et al. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell 2013; 154: 103–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Narasimhan S, Changolkar L, He Z, Stieber A, Zhang B, et al. Unique pathological tau conformers from Alzheimer's brains transmit tau pathology in nontransgenic mice. J Exp Med 2016; 213: 2635–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z Guo JL Mcbride JD Narasimhan S Kim H Changolkar L, et al. Amyloid-β plaques enhance Alzheimer's brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med 2018; 24: 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett DR, Whitfield D, Johnson M, Attems J, O'Brien JT, Aarsland D, et al. Regional multiple pathology scores are associated with cognitive decline in Lewy body dementias. Brain Pathol 2015; 25: 401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's-like tauopathy. J Neurosci 2013; 33: 1024–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DJ, Grossman M, Weintraub D, Hurtig HI, Duda JE, Xie SX, et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol 2017; 16: 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizawa T, Mattila P, Davies P, Wang D, Dickson DW. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol 2003; 62: 389–97. [DOI] [PubMed] [Google Scholar]
- James BD, Bennett DA. Causes and patterns of dementia: an update in the era of redefining Alzheimer's disease. Annu Rev Public Health 2019; 40: 65–84. [DOI] [PubMed] [Google Scholar]
- Jensen PH, Hager H, Nielsen MS, Hojrup P, Gliemann J, Jakes R. Alpha-synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J Biol Chem 1999; 274: 25481–9. [DOI] [PubMed] [Google Scholar]
- Kotzbauer PT, Giasson BI, Kravitz AV, Golbe LI, Mark MH, et al. Fibrillization of alpha-synuclein and tau in familial Parkinson's disease caused by the A53T alpha-synuclein mutation. Exp Neurol 2004; 187: 279–88. [DOI] [PubMed] [Google Scholar]
- Kovacs GG, Milenkovic I, Wöhrer A, Höftberger R, Gelpi E, Haberler C, et al. Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol 2013; 126: 365–84. [DOI] [PubMed] [Google Scholar]
- Kuninaka N, Kawaguchi M, Ogawa M, Sato A, Arima K, Murayama S, et al. Simplification of the modified Gallyas method. Neuropathology 2015; 35: 10–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan S, Guo JL, Changolkar L, Stieber A, McBride JD, Silva LV, et al. Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J Neurosci 2017; 37: 11406–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SW, Harris JA, Ng L, Winslow B, Cain N, Mihalas S, et al. A mesoscale connectome of the mouse brain. Nature 2014; 508: 207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimi J, Kovacs GG. Prevalence of mixed pathologies in the aging brain. Alzheimers Res Ther 2014; 6: 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 2018; 141: 2181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford NJ, Brooks M, Giasson BI. Novel antibodies to phosphorylated alpha-synuclein serine 129 and NFL serine 473 demonstrate the close molecular homology of these epitopes. Acta Neuropathol Commun 2016; 4: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacino AN, Brooks M, McKinney AB, Thomas MA, Shaw G, Golde TE, et al. Brain injection of alpha-synuclein induces multiple proteinopathies, gliosis, and a neuronal injury marker. J Neurosci 2014; 34: 12368–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014; 82: 1271–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007; 69: 2197–204. [DOI] [PubMed] [Google Scholar]
- Shammas SL, Garcia GA, Kumar S, Kjaergaard M, Horrocks MH, Shivji N, et al. A mechanistic model of tau amyloid aggregation based on direct observation of oligomers. Nat Commun 2015; 6: 7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino ZA, Brooks MMT, Hudson V, 3rd, Rutherford NJ, Golde TE, Giasson BI, et al. Intrastriatal injection of alpha-synuclein can lead to widespread synucleinopathy independent of neuroanatomic connectivity. Mol Neurodegen 2017; 12: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino ZA, Vijayaraghavan N, Gorion K-M, Riffe CJ, Strang KH, Caldwell J, et al. Physiological C-terminal truncation of alpha-synuclein potentiates the prion-like formation of pathological inclusions. J Biol Chem 2018; 293: 18914–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones TL, Attems J, Thal DR. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol 2017; 134: 187–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strang KH, Goodwin MS, Riffe C, Moore BD, Chakrabarty P, Levites Y, et al. Generation and characterization of new monoclonal antibodies targeting the PHF1 and AT8 epitopes on human tau. Acta Neuropathol Commun 2017; 5: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan RH, Yang Y, Halliday GM. Multiple neuronal pathologies are common in young patients with pathologically proven frontotemporal lobar degeneration. Neuropathol Appl Neurobiol 2018; 44: 522–32. [DOI] [PubMed] [Google Scholar]
- Waxman EA, Giasson BI. A novel, high-efficiency cellular model of fibrillar alpha-synuclein inclusions and the examination of mutations that inhibit amyloid formation. J Neurochem 2010; 113: 374–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, Giasson BI. Induction of intracellular tau aggregation is promoted by alpha-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci 2011; 31: 7604–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White LR, Edland SD, Hemmy LS, Montine KS, Zarow C, Sonnen JA, et al. Neuropathologic comorbidity and cognitive impairment in the Nun and Honolulu-Asia Aging Studies. Neurology 2016; 86: 1000–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woerman AL, Kazmi SA, Patel S, Freyman Y, Oehler A, Aoyagi A, et al. MSA prions exhibit remarkable stability and resistance to inactivation. Acta Neuropathol 2018; 135: 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woerman AL, Patel S, Kazmi SA, Oehler A, Freyman Y, Espiritu L, et al. Kinetics of human mutant tau prion formation in the brains of 2 transgenic mouse lines. JAMA Neurol 2017; 74: 1464–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang S-M, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007; 53: 337–51. [DOI] [PubMed] [Google Scholar]
- Zhang W, Falcon B, Murzin AG, Fan J, Crowther RA, Goedert M, et al. Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer's and Pick's diseases. Elife 2019; 8: e43584. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated during this study are included in this published article [and its Supplementary information files].