

Abstract
Aims
When activated, Na+/H+ exchanger-1 (NHE1) produces some of the largest ionic fluxes in the heart. NHE1-dependent H+ extrusion and Na+ entry strongly modulate cardiac physiology through the direct effects of pH on proteins and by influencing intracellular Ca2+ handling. To attain an appropriate level of activation, cardiac NHE1 must respond to myocyte-derived cues. Among physiologically important cues is nitric oxide (NO), which regulates a myriad of cardiac functions, but its actions on NHE1 are unclear.
Methods and results
NHE1 activity was measured using pH-sensitive cSNARF1 fluorescence after acid-loading adult ventricular myocytes by an ammonium prepulse solution manoeuvre. NO signalling was manipulated by knockout of its major constitutive synthase nNOS, adenoviral nNOS gene delivery, nNOS inhibition, and application of NO-donors. NHE1 flux was found to be activated by low [NO], but inhibited at high [NO]. These responses involved cGMP-dependent signalling, rather than S-nitros(yl)ation. Stronger cGMP signals, that can inhibit phosphodiesterase enzymes, allowed [cAMP] to rise, as demonstrated by a FRET-based sensor. Inferring from the actions of membrane-permeant analogues, cGMP was determined to activate NHE1, whereas cAMP was inhibitory, which explains the biphasic regulation by NO. Activation of NHE1-dependent Na+ influx by low [NO] also increased the frequency of spontaneous Ca2+ waves, whereas high [NO] suppressed these aberrant forms of Ca2+ signalling.
Conclusions
Physiological levels of NO stimulation increase NHE1 activity, which boosts pH control during acid-disturbances and results in Na+-driven cellular Ca2+ loading. These responses are positively inotropic but also increase the likelihood of aberrant Ca2+ signals, and hence arrhythmia. Stronger NO signals inhibit NHE1, leading to a reversal of the aforementioned effects, ostensibly as a potential cardioprotective intervention to curtail NHE1 overdrive.
Keywords: SLC9A1, NO, Cyclic nucleotides, Heart, pH
Graphical Abstract
Graphical Abstract.
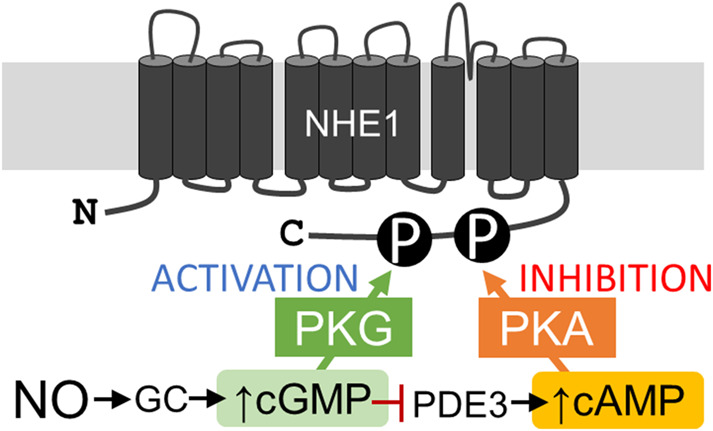
1. Introduction
Sarcolemmal Na+/H+ exchanger-1 (NHE1) is the most powerful regulator of intracellular pH (pHi) in the heart, capable of correcting acid–base disturbances within minutes.1–3 When fully activated, NHE1 can generate acid-extrusion fluxes of the order of tens of mM/min, coupled to a matching influx of Na+ ions.1–3 Such high acid-extrusion fluxes are conducive for cardiomyocyte pHi homeostasis, particularly in periods of elevated metabolic acid production, driven by a heightened demand for cardiac work. The importance of maintaining a favourable pHi is perhaps best exemplified by the exquisite pH-sensitivity of contraction, which halves in strength when pHi falls by just 0.2 units.3–6
Activated NHE1 can become the largest route of Na+ entry into myocytes, meaningfully challenging the corrective capacity of the Na+/K+ ATPase pump.7 An NHE1-evoked rise in intracellular [Na+] reduces the driving force for Ca2+ extrusion by Na+/Ca2+ exchange (NCX), leading to an increase in the cellular content of Ca2+ ions.5,8,9 This effect can be positively inotropic,8,10,11 but beyond a critical level of Ca2+ over-load, it may trigger aberrant Ca2+ signals, such as Ca2+ waves, and hence arrhythmias.12,13 Thus, the cardiomyocyte must carefully balance the need for correcting pHi disturbances against the knock-on effects on Ca2+ signalling. This balancing act is exercised by appropriately controlling NHE1 activity.
A physiological trigger for NHE1 activity is intracellular acidity, but the extent to which this evokes a corrective flux depends largely on the post-translational state of the protein.3,14 Various extrinsic factors, including hormones and neurotransmitters, exercise control over NHE1 through kinase-operated cascades.3,14 However, in order for cardiac NHE1 to be responsive to the state of the heart, its regulation must involve auto- or paracrine factors that act locally. Among these factors is nitric oxide (NO), which provides important regulatory cues for a myriad of cardiac pathways.15–20 However, there have been conflicting reports of an inhibitory21 and activatory effect22 of NO on NHE1. Critically, it is unclear how physiologically-relevant levels of NO stimulation affect NHE1. Earlier measurements of [NO] in cardiac tissue indicated levels in the 10–1000 nM range,23–25 but more recent analyses have narrowed this to the high picomolar/low-nanomolar range.26 Consequently, experimental manoeuvres that had used excessive concentrations of NO donor-substances may not reflect normal physiology.26
The major source of NO in the heart is the neuronal-type synthase nNOS,19 and its influence on Ca2+-handling proteins is well-documented.15,20 NO may affect target-proteins by S-nitros(yl)ation16,18 or through guanylyl cyclase (GC)-dependent cascades.17,27 The latter mechanism canonically triggers cGMP signals, which may secondarily evoke a rise in [cAMP] in cardiomyocytes.28–30 Cyclic nucleotides may regulate the fluxes carried by NHE1 by activating kinases that phosphorylate the C-terminus. Previous studies31–36 have linked transport activity with two functionally prominent residues, Ser648 and Ser703, predicted to be consensus sites for PKA and PKG14. Whereas Ser648 phosphorylation inhibits NHE1 activity,31 phosphorylation at Ser703 has the opposite effect.36 It is thus unclear how such an apparently ambiguous design would be compatible with finely-controlled oversight by NO.
Here, we studied the effects of NO on cardiac NHE1 activity, interrogated by means of direct functional assays performed on isolated adult ventricular myocytes. Given that NHE1 is electroneutral, its activity cannot be inferred from measurements of membrane currents; instead, the most appropriate readout is the recovery of pHi, after accounting for H+ buffering.1 Such measurements are able to resolve inhibitory or activatory effects on transporter fluxes, and they relate more closely to cardiac physiology. Using manipulations that include nNOS knockout, adenoviral nNOS gene delivery, nNOS inhibition, and pharmacological NO titrations, we demonstrate that low levels of NO activate NHE1, whereas higher doses switch to a net inhibitory effect. These dynamic effects were not dependent upon S-nitros(yl)ation16,37 but, instead, involved a careful balancing of activatory cGMP and inhibitory cAMP signals. We show that PKG and PKA can phosphorylate the activatory Ser703 at the C-terminus of NHE1 in vitro, but Ser648, an inhibitory residue, is more strongly phosphorylated by PKA. Finally, we demonstrate how the biphasic modulation of NHE1 by NO affects Ca2+ handling, using the example of spontaneous Ca2+ waves.
2. Methods
2.1 Adult ventricular myocyte isolation
Ventricular myocytes were isolated from hearts by enzymatic digestion and mechanical dispersion. This study used myocytes from wild-type male Sprague-Dawley rats (300–325 g), or 4- to 5-month-old nNOS−/− knockout mice and their wild-type littermates, bred under license (PPL 30/3374). nNOS knockout was achieved by targeted disruption of exon 2 of nNOS.38 Rats were sacrificed by stunning followed by cervical dislocation, and mice were sacrificed by cervical dislocation and exsanguination in accordance with UK Home Office regulations (Schedule I of A(SP)A 1986), approved by national and University ethics committees. Hearts were rapidly removed and rinsed in isolation solution (in mM): 120 NaCl, 4 KCl, 1.2 MgCl2, 11 glucose, 10 HEPES, 2 NaH2PO4, 2.5 pyruvate pH 7.4 at 37°C; supplemented with heparin sodium (5 units/L), and then mounted on a Langendorff perfusion system. Once cleared of blood, the heart was perfused with collagenase type II (0.9 mg/mL, Lorne Laboratories, UK) and protease type XIV (0.09 mg/mL, Sigma, UK) for 10 min, cut into pieces, triturated with a Pasteur pipette and dissociated cells filtered through a 250-µm nylon mesh.
2.2 Neonatal ventricular myocyte isolation and culture
Neonatal rat ventricular myocytes were isolated from 1- to 2-day-old Sprague-Dawley rats. Animals were euthanized by cervical dislocation and exsanguination according to Schedule 1 of the Animals (Scientific Procedures) Acts 1986. Experiments were approved by Oxford University ethical review boards and conform to the guidelines from Directive 2010/63/EU. Cells were isolated from ventricular tissue of excised hearts by enzymatic digestion and plated onto fibronectin-coated Ibidi µ-slides (Ibidi, Germany) then cultured as described previously.39
2.3 MDA-MB-468 cells
MDA-MB-468 cells, obtained from ATCC, were cultured in DMEM with 10% FCS in an atmosphere of 5% CO2. Cells were plated on Ibidi µ-slides (Ibidi, Germany), grown to confluency, and used for superfusion experiments.
2.4 Viral transduction of myocytes
Adult ventricular myocytes were infected with adenovirus containing the gene for nNOS under a CMV promoter,40,41 the FRET-based cAMP sensor H187,42 or the appropriate sham controls. Myocytes were cultured overnight on µ-slides (Ibidi, Germany) in myocyte culture medium (MEM supplemented with 9 mM NaHCO3, 1% L-glutamine, 1% penicillin/streptomycin), 0.5 µM cytochalasin D to preserve cell shape,43 and adenovirus (H187 final titer 5 × 109 viral particles/mL, nNOS final titer: 107 viral particles/mL). After overnight culture, the virus-containing medium was replaced with myocyte culture medium supplemented with 2.5% FBS.
2.5 NO donors and scavengers
Experiments used either sodium nitroprusside (SNP) or 3-ethyl-3-(ethylaminoethyl)-1-hydroxy-2-oxo-1-triazene (NOC12), which are short and long half-life NO donors, respectively. To clamp [NO] to physiological levels, the NO scavenger CPTIO (2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide) was combined with NOC12; additionally, 300 µM urate to scavenge NO2 and ONOO−.44
2.6 Solutions and superfusion
Solutions were delivered at 37°C to a custom-made Perspex superfusion chamber with a coverslip glass bottom (for experiments on adult myocytes) or an Ibidi µ-slide (for experiments on cultured cells). Normal Tyrode contained (in mM) 135 NaCl, 4.5 KCl, 1 CaCl2, 1 MgCl2, 11 glucose, 20 Hepes at pH 7.4. In ammonium-containing solutions, NaCl was iso-osmotically replaced with NH4Cl. In acetate-containing solutions, NaCl was iso-osmotically replaced with NaAcetate.
2.7 Dual microperfusion
The boundary between microstreams was visualized by including 10 mM sucrose into one of the two microstreams. Flows were adjusted to produce a sharp boundary, which was positioned across the middle of a myocyte.45,46
2.8 Fluorescence imaging
To image cAMP levels, the FRET-sensor H187 was excited at 405 nm and fluorescence was measured at 480 ± 10 nm and 530 ± 10 nm in xy-mode on a Zeiss LSM700 confocal system. To image NO levels, DAR-4M was AM-loaded into cells (10 µM DAR-4M AM, for 10 min), and its fluorescence was excited at 514 nm and emission detected at 580 ± 20 nm on a Leica TCS NT system.47 To image Ca2+ waves, Fluo3 was AM-loaded into myocytes (5 µM Fluo3, for 10 min), and its fluorescence was excited at 488 nm and detected >520 nm on a Leica TCS NT confocal system in xt mode. To measure pHi, myocytes were AM-loaded with cSNARF1 (10 µM, for 10 min), and fluorescence was excited at 530 nm and detected simultaneously at 580 ± 10 nm and 640 ± 10 nm on an inverted Olympus microscope with an Orca 05G CCD Camera (Hamamatsu, Japan) and Optosplit (Cairn Research, UK).
2.9 Expression of NHE1 C-terminus (His182)
The C-terminus of NHE1 was expressed in a bacterial (BL21-SI) system, as described previously.48 Bacteria were cultured in LBON media (bacto tryptone 10 g/L, bacto yeast extract 5 g/L, AMP 100 mg/L) at 30°C. After reaching an optical density of 0.6 (600 nm), expression was induced by adding 300 mM NaCl and incubation for 4 h. His182 was purified as described previously.48
See Supplementary material online for Kinase reactions, Western blotting, Phos-tag gel analysis, S-nitros(yl)ation (iodoTMT) assay, organo-mercury enrichment, antibodies, and mass spectrometry.
2.10 Statistics
Differences tested by two-way ANOVA at 5% significance, followed by post hoc test (Fisher’s LSD). Data are reported as means ± SEM, and repeats as ‘number of cells/number of animals’. Flux data are normally distributed continuous variables. For non-normally distributed data comparing two groups, statistical analysis was done using a Mann–Whitney U-test.
3. Results
3.1 PKA and PKG produce distinct patterns of NHE1 phosphorylation
NO signalling in the myocyte can activate PKG via the canonical pathway involving cGMP17,27 and, under certain conditions, PKA through a rise in [cAMP].28–30 To study how NHE1 responds biochemically to these kinases, the C-terminus (wherein most modulatory influences converge) of the human protein was expressed in a bacterial system.48 This fragment, thenceforth referred to as His182, was reacted in vitro with PKA or PKG, and phosphorylation was analysed by Phos-tag gel blotting (Figure 1A). Both PKA and PKG were able to phosphorylate His182, but the mobility shifts were distinct, indicating that the kinases act differentially on NHE1. Two important residues linked directly to regulating NHE1 transport activity,31,36 and predicted substrates for PKA and PKG,14 are Ser648 and Ser703. To test if these residues are phosphorylated, kinase reactions were subject to analysis by mass spectrometry (Figure 1B). This revealed that His182 was phosphorylated on multiple serine residues, including Ser648 and Ser703. Consistent with the Phos-tag analysis, mass spectrometry revealed a differential pattern of His182 phosphorylation by PKA and PKG (Figure 1C). Western blotting using antibodies raised against phosphorylated serine confirmed phosphorylation of His182 by both PKA and PKG, albeit with distinct bands (Figure 1D).
Figure 1.
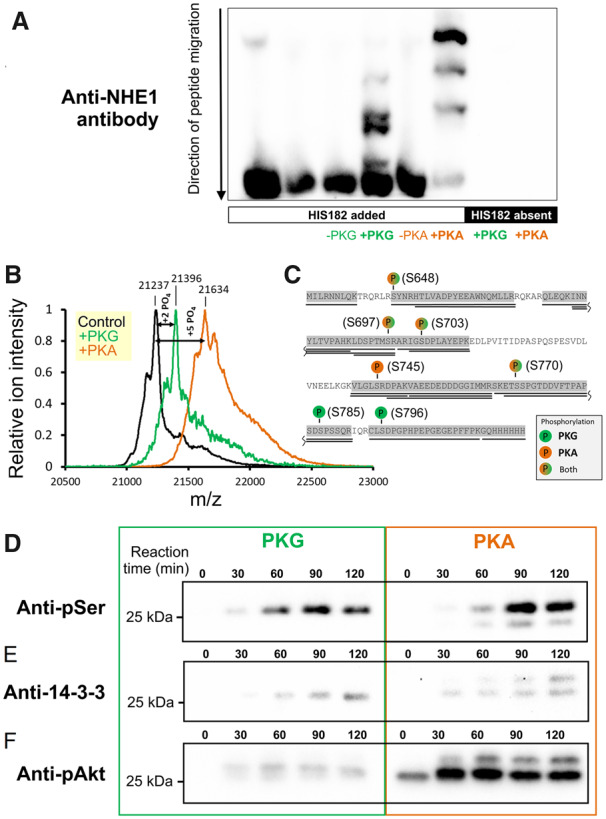
Phosphorylation of the NHE1 C-terminus by PKA and PKG. (A) Various combinations of kinase (PKA or PKG; 8 ng/µL) and protein (His182; 20 ng/µL) were reacted in vitro and run on a Phos-tag gel to seek evidence for differences in phosphorylation pattern. (B) His182 (100 ng/µL) was reacted in vitro with PKA (100 ng/µL) or PKG (50 ng/µL) and analysed by LC-MS/MS mass spectrometry for phosphorylation sites. Results indicated a mean phosphorylation stoichiometry of five and two phosphate groups introduced by PKA and PKG, respectively. (C) Residues identified as being phosphorylated by PKA and PKG. (D) Time course of reaction between His182 (20 ng/µL; plus 500 µM ATP) and either PKG (8 ng/µL; left) or PKA (8 ng/µL; right). Western blots were performed using an antibody raised against phosphorylated serine residues. (E) Western blot performed using antibody against the phosphorylated 14-3-3 binding motif (detecting Ser703 in NHE1). (F) Western blot performed using antibody against phosphorylated Akt substrate (Ser648 in NHE1). Note: blots for PKA- and PKG-reacted His182 were prepared with the same amount of substrate and developed simultaneously with identical exposure time. See Supplementary material online, Figure S1 for loading controls.
To investigate how the phosphorylation status of Ser648 and Ser703 responds to PKA and PKG, antibodies recognizing specific phosphoserine motifs were used for western blotting (see Supplementary material online, Figure S1 for loading controls). An antibody raised against the 14-3-3 binding motif detected Ser703 phosphorylation by PKA and PKG, albeit with a distinct immunoreactivity pattern (Figure 1E). In contrast, an antibody raised against the Akt phosphorylation consensus sequence, i.e. Ser648 on NHE1, showed substantially greater immunoreactivity towards PKA-treated His182, and only weak signal with PKG (Figure 1F). These findings indicate that both PKA and PKG can phosphorylate Ser703, which underpins NHE1 activation,36 but that PKA is considerably more selective for Ser648, a residue responsible for NHE1 inhibition.31 To investigate how these kinases are recruited physiologically by NO to regulate cardiac NHE1, the next experiments measured transport activity in ventricular myocytes subjected to pharmacological or genetic interventions that target PKG- and PKA-mediated cascades.
3.2 Basal NO production activates cardiac NHE1
nNOS knockout mice were used to investigate the effect of basal NO production on pHi control by NHE1 (see Supplementary material online, Figure S2 for confirmation of nNOS gene ablation). Resting pHi was measured in enzymatically isolated cardiomyocytes loaded with the pH reporter dye, cSNARF1, and superfused in physiological CO2/-buffered solution. The frequency distribution of resting [H+]i in nNOS−/− myocytes was, compared to cells from wild-type littermates, broader and extended into the acidic range. Thus, myocytes lacking basal NO production are less able to defend a favourable (mildly alkaline) pHi (Figure 2A). Intrinsic buffering capacity, attributable largely to proteins, was no different between the two experimental groups (Supplementary material online, Figure S3; measured by a step-wise NH4Cl removal method49) To measure NHE1 activity, the transporter was activated by lowering pHi, attained by means of an ammonium prepulse solution manoeuvre.1 For these experiments, myocytes were superfused with Hepes-buffered solution to inactivate -dependent transport. Under these conditions, recovery from an acidic pHi is mediated primarily by NHE1, as demonstrated by its sensitivity to dimethylamiloride (DMA; 30 µM), an NHE1 inhibitor (Figure 2B). Given that NHE1 is exquisitely sensitive to [H+], data were presented as pHi-flux curves, and comparisons are made at matching levels of pHi. NHE1 flux, calculated as the product of buffering capacity and the rate of pHi change during the recovery phase, was slower in nNOS−/− myocytes compared to wild-type cells (Figure 2C), demonstrating that basal NO production has an activatory effect on NHE1. There was no effect of the nNOS knockout on the DMA-insensitive component of flux.
Figure 2.

nNOS-derived NO activates NHE1 activity. (A) Histogram of resting [H+]i in ventricular myocytes isolated from nNOS knockout mice (N = 267 from five mice) or their wild-type littermates (N = 244 from five mice). (B) Time course of ammonium prepulse (mean ± SEM; N = 64 from five nNOS knockout mice and N = 54 from five wild-type littermates). Hepes-buffered superfusates, 37°C. Solution manoeuvre acid-loaded myocytes to interrogate acid-extrusion fluxes during recovery phase (blocked by 30 µM dimethylamiloride, DMA). (C) Flux calculated from the rate of pHi change and buffering capacity (Supplementary material online, Figure S2). NHE1-dependent fluxes were slower in nNOS knockout mice (P < 0.0001, two-way ANOVA). (D) NHE1 flux measured in wild-type rat ventricular myocytes. Hepes-buffered superfusates, 37°C. Treatment with selective nNOS inhibitor AAAN (5 µM; N = 16 from five rats) reduced activity relative to control (N = 18 from five rats) (P < 0.0001, two-way ANOVA). (E) Ammonium prepulse performed on adenovirally infected rat ventricular myocytes cultured overnight. Hepes-buffered superfusates, 37°C. Infected cells were identified by eGFP fluorescence (inset). (F) NHE1 activity was accelerated (P < 0.0001, two-way ANOVA) in myocytes infected with nNOS gene (N = 16 cells from four independent infections) compared to sham infected cells (N = 28 from five independent infection).
Genetic ablation of nNOS may have exerted its activatory effect through a reversible post-translational effect driven by NO, or through a slower up-regulation of NHE1 protein. These two possible mechanisms were tested by measuring the acute response of NHE1 flux to nNOS inhibition with the compound AAAN.50 Wild-type rat myocytes treated with 5 µM AAAN had a left-shifted NHE1-pHi curve (Figure 2D), indicating that basal nNOS activity is exerting a reversible, post-translational effect on NHE1.
Further testing of the effect of nNOS activity on NHE1 used rat myocytes infected with an adenovirus encoding for an nNOS-eGFP fusion protein. Cells incubated overnight with virus produced a good yield of construct expression, as determined by eGFP fluorescence (Figure 2E, inset). Compared to cells incubated overnight with virus containing only the eGFP gene (i.e. sham infection), adenoviral nNOS gene delivery hastened pHi recovery to a more alkaline resting pHi, and increased NHE1 flux (Figure 2F). Overall, these findings demonstrate that basal nNOS-derived NO exerts an activatory influence on NHE1.
3.3 Exogenously delivered NO exerts a dose-dependent effect on NHE1
The actions of NO on NHE1 were further studied using an exogenous source of NO delivered by superfusion. SNP has widely been used as a short half-life NO donor, producing substantial NO release peaking near 600 µM within tens of minutes of addition to superfusates (Supplementary material online, Figure S4). Recovery from an acid-load (Hepes-buffered superfusates) was first measured under control conditions, and repeated in the presence of SNP (1 µM). Exposing the myocytes to a high concentration of NO resulted in a slowing of pHi recovery and a more acidic resting pHi (Figure 3A). This attenuation of NHE1 flux was not due to an NO-independent, time-dependent attrition, because a consecutive pair of control measurements produced a similar recovery (Supplementary material online, Figure S5). The inhibitory effect of SNP was absent in cells pre-treated with 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ; 6 µM), an irreversible inhibitor of GC, indicating the involvement of cGMP signals (Figure 3B). These data suggest that a high concentration of NO released from SNP inhibits NHE1 flux, in apparent contradiction to the activatory effect of basal NO production by nNOS described earlier (see Figure 2). SNP (1 µM) had no effect on Na+-HCO3− cotransport (NBC), the other major acid-extruder in myocytes, measured in CO2/HCO3− buffered superfusates containing DMA to block NHE1 (Figure 3C).
Figure 3.

NO delivered from donors produces a concentration-dependent biphasic effect on NHE1 activity. (A) Wild-type rat myocytes. Hepes-buffered superfusates, 37°C. The short half-life NO donor, sodium nitroprusside (SNP), was added to solutions 4–6 min before activating NHE1. With 1 µM SNP, NO is expected to peak at 600 nM during pHi recovery (control: N = 35 cells from five animals; SNP: N = 31 cells from five animals). (B) Experiments repeated on myocytes that had been pre-treated with 6 µM ODQ (1H-[1,2,4]Oxadiazolo[4,3-a]quinoxalin-1-one), an inhibitor of guanylyl cyclase (GC; ODQ: N = 24 cells from four animals; ODQ+SNP: N = 26 cells from four animals). (C) CO2/-buffered superfusates, 37°C; 30 µM dimethylamiloride (DMA) included to block NHE isoforms. pHi recovery mediated by dependent transporters is unaffected by NO (control: N = 15 cells from three animals; SNP: N = 15 cells from three animals). (D) Wild-type rat myocytes. Hepes-buffered superfusates, 37°C. The long half-life NO donor NOC12 was added to solutions 10 min before activating NHE1. At a concentration of 5 µM, NOC12 is expected to maintain NO above 300 nM for the duration of pHi recovery. (E) NHE1 fluxes. Control (N = 21 from five animals), effect of NOC12 (N = 18 from five animals), and effect of NOC12 on ODQ-pretreated cells (N = 26 from five animals). Significant difference between control and NOC12 (P < 0.0001), but not between control and NOC12+ODQ (P = 0.06). (F) Lower concentrations of NO (<30 nM) were produced by mixing donor NOC12 (300 µM) with scavenger CPTIO (100 µM). (G) NHE1 fluxes. Control (N = 21 from five animals), effect of NOC12/CPTIO (N = 19 from five animals), and effect of NOC12/CPTIO on ODQ-pretreated cells (N = 10 from four animals). Significant difference between control and NOC12 (P < 0.0001), but not between control and NOC12/CPTIO+ODQ (P = 0.08).
The actions of NO on NHE1 were investigated further using NOC12, a donor with longer half-life, thus releasing a steadier flow of NO. Kinetic modelling using published stability constants indicates that myocytes superfused with 5 µM NOC12 are exposed to >300 nM NO (Supplementary material online, Figure S4). At this concentration, NO exerted an inhibitory effect on the rate of pHi recovery and acidified steady-state pHi (Figure 3D), akin to the effect of SNP. The inhibitory effect of 5 µM NOC12, which could be described in terms of a left-shift in the NHE1-pHi curve by ∼0.1 units, was absent in myocytes pretreated with ODQ, confirming that the exogenously released NO was signalling via cGMP (Figure 3E). Although many previous studies have used micromolar doses of donors to interrogate NO signalling, the concentration of NO attained may be considered supra-physiological.26 To measure the effects of lower NO concentrations, NOC12 was combined with a scavenger, CPTIO, to titrate [NO] down to the low-nM range.44 A kinetic model determined that combining 300 µM NOC12 with 100 µM CPTIO (plus 300 µM urate to scavenge the by-product NO2) could maintain [NO]<30 nM27,44 (Supplementary material online, Figure S4). At this lower [NO], pHi recovery was faster and progressed to a more alkaline resting pHi (Figure 3F) indicating NHE1 activation (Figure 3G), which is now consistent with the effect of nNOS-derived NO. This effect, observed as a right-shift in the NHE1-pHi curve, was absent in myocytes pretreated with ODQ (Figure 3G). Thus, high and low concentrations of NO produce opposite effects on NHE1 activity, yet both involve cGMP signals.
3.4 Biphasic modulation of NHE1 by NO is a special property of the adult cardiomyocyte
To determine if the biphasic modulation of NHE1 by NO is a more general property of NHE1-expressing cells, measurements were performed on cultured neonatal rat ventricular myocytes and on a human breast cancer cell line MDA-MB-468, both of which produce NHE1 fluxes that are comparable in magnitude to those in adult ventricular myocytes. However, the biphasic effect of NO on NHE1 was not observed in neonatal myocytes (Supplementary material online, Figure S6A and B) or MDA-MB-468 cells (Supplementary material online, Figure S6C and D). These findings argue that NO signals modulate NHE1 in a highly context-sensitive manner, involving elements that are present in adult cardiac myocytes, but not necessarily in other cell types. Consequently, expression systems and in vitro-based assays are not appropriate for interrogating the mechanisms of cardiac NO-NHE1 interplay; instead, the relevant cascades must be studied in primary adult myocytes.
3.5 NO signals can be transmitted onto remote NHE1 targets and do not involve S-nitros(yl)ation
One possible mechanism through which NO may be regulating NHE1 is S-nitrosylation or S-nitrosation. Indeed, many protein targets have been proposed to undergo this post-translational modification,16,18,37 although the physiological significance of some of these observations has been questioned.51 To test whether sarcolemmal NHE1 is substrate for direct NO reactions, immunoblotting using the so-called TMT-method52 was performed on membrane fractions of cardiac tissue treated with either high or low [NO], delivered by Langendorff-perfusion with 5 µM NOC12 or 300 µM NOC12 plus 100 µM CPTIO, respectively (Supplementary material online, Figure S7A). There was no evidence of any increase in nitros(yl)ation at the band corresponding to NHE1 in response to low or high [NO] (Figure 4A), arguing against S-nitros(yl)ation. As a positive control, an increase in nitros(yl)ation was observed in whole-cell lysates subject to treatment with S-nitrosoglutathione (Supplementary material online, Figure S7B). The lack of NHE1 S-nitros(yl)ation by high levels of [NO] was confirmed using the complementary technique of organo-mercury resin-assisted capture that is considered a more specific test for S-nitros(yl)ated residues (SNO)53 (Figure 4B). Blots were quantified as the ratio of SNO signal to input (NHE1), and this analysis revealed no significant difference between control and NOC12-treated hearts (Figure 4C). As confirmation of the method’s resolving power to detect changes in protein nitros(yl)ation, lysates pretreated with DTT or GSNO produced measurable changes on blots (Supplementary material online, Figure S8).
Figure 4.

NO regulates NHE1 activity via a diffusible messenger and not via S-nitros(yl)ation. (A) Hearts were Langendorff-perfused with normal Tyrode, or Tyrode with high (5 µM NOC12) or low (300 µM NOC12 + 100 µM CPTIO) [NO]. S-nitros(yl)ation assay was performed on membrane fractions of heart lysates. NHE1 staining visible as bands near 90 kDa. Re-probed for TMT as readout of protein S-nitros(yl)ation: no increase in nitros(yl)ation was detectable in the presence of NO donors. (B) Direct NHE1 S-nitros(yl)ation (SNO) was evaluated by an independent method that uses organo-mercury enrichment. (C) No difference in the extent of SNO-enriched NHE1 was observed between control and high NO-treated hearts. N = 7 per group; not significant (P = 0.9); Mann–Witney U-test. (D) Dual microperfusion of isolated rat myocyte. Left microstream produced a NO microdomain from slow-releasing NOC12 (5 µM); NHE1 was inactivated locally by dimethylamiloride (30 µM). Right microstream contained CPTIO to scavenge any spill-over of NO from the left microstream. (E) Measurement of NO microdomain using fluorescent dye DAR-4M, showing gradient of [NO] along length of cell (N = 9 cells from three animals). (F) NHE1 activity in myocyte determined from the pHi recovery following a uniformly-applied ammonium prepulse. Note the magnitude of flux is lower because NHE1 activity in half of the myocyte is blocked with DMA (N = 14 treated with NOC12; N = 12 control experiments from three animals). High [NO] delivered by NOC12 remotely activated NHE1 (P < 0.0001, two-way ANOVA). (E) Remote signalling between NO and NHE1 was absent in cells pretreated with the GC inhibitor ODQ (5 µM), indicating a role for the diffusible messenger cGMP.
The lack of evidence for a direct S-nitros(yl)ation of NHE1 suggests the involvement of cGMP signals and is consistent with the ODQ-sensitivity of NO actions on NHE1 (Figure 3E and G). If the diffusible messenger cGMP were responsible for the coupling between NO and NHE1, then a spatially-confined source of NO would be expected to influence the activity of more remote NHE1 targets. However, with increasing distance from NO-activated GC, the concentration of cGMP will decay as a result of degradation by phosphodiesterases (PDEs). Since NO exerts a biphasic effect on NHE1, spatially-decaying [cGMP] may flip from being an inhibitory signal to becoming activatory. To test for remote signalling by NO, a dual microperfusion system was used to deliver two sharply separated microstreams at a perpendicular angle to a myocyte. One microstream contained NOC12 to release NO, whilst the other contained CPTIO to scavenge any NO spill-over (Figure 4D). The fluorescent dye DAR-4M reported a rise in [NO] that was spatially restricted to the NOC12-exposed half of the cell (Figure 4E). Thus, using dual microperfusion, it is possible to deliver NO to one side of a myocyte, and observe any remote actions on NHE1 at the opposite end.
To trigger NHE1 activity, myocytes were first acid-loaded, by means of ammonium prepulse, and then dually microperfused. One half of the myocyte was exposed to NOC12 at a dose (5 µM) that would inhibit NHE1, if applied uniformly to a cell (Figure 3E). To investigate how this localized source of NO influences NHE1 remotely, the NOC12-containing microstream also included DMA to inactivate NHE1 locally; consequently, any pHi recovery produced by the cell must be attributable to NHE1 activity at the other half of the myocyte. Measurements of NHE1 flux showed a stimulatory effect of NOC12 when the donor was delivered at a distance from NHE1 (Figure 4F). This remote NO-NHE1 coupling was absent in cells pre-treated with ODQ, indicating the involvement of diffusible cGMP signals (Figure 4G). Thus, high [NO], which would locally inhibit NHE1, produces an activatory effect on more remote NHE1 protein. This flipping of polarity can be explained by a [cGMP]-dependence of action: low [cGMP] is activatory, whilst high [cGMP] is inhibitory.
3.6 Crosstalk between cGMP and cAMP produces biphasic modulation of NHE1 by NO
Based on the results of in vitro studies using His182 (Figure 1), the pattern of NHE1 phosphorylation produced by PKG is distinct to that evoked by PKA. Thus, it is possible to explain biphasic NO regulation of cardiac NHE1 by dose-dependent recruitment of kinases. PKG was found to be more potent at phosphorylating Ser703 (linked to activation; Figure 1E) than Ser648 (linked to inhibition; Figure 1F), in comparison to PKA which acted strongly on Ser648.54 Notwithstanding this in vitro evidence, it is not intuitive to explain how a NO signal, acting through its canonical second messenger cGMP, could orchestrate NHE1 activation at low concentrations, but inhibition with stronger stimulations. A plausible mechanism by which a cGMP signal could switch its polarity of action may involve the recruitment of cAMP beyond a threshold [cGMP]. Indeed, previous studies have shown that cGMP-evoking stimuli can produce a secondary rise in [cAMP], arising from the inhibitory effect of cGMP on the cAMP-degradating enzyme phosphodiesterase-3 (PDE3).55
To determine if the cGMP signal evoked with 5 µM NOC12 is sufficient to produce a detectable rise in [cAMP], myocytes were first infected with virus containing the gene for H187, a FRET-based cAMP probe. Superfusion with 5 µM NOC12 produced a measurable rise in [cAMP], reported as a response that was equivalent to the effect of low-dose (∼1 nM) isoprenaline (Iso), a beta-agonist (Figure 5A). Thus, a sufficiently large NO signal can evoke a cAMP response in adult myocytes (Figure 5B).
Figure 5.

Biphasic regulation of NHE1 by NO is achieved by balancing cGMP-cAMP crosstalk. (A) Rat myocyte infected with H187, a FRET-based sensor of cAMP (inset). NOC12 (5 µM) produced a significant increase in signal. As a positive control, F/I (forskolin and IBMX) raises cAMP to levels that saturate the sensor. Isoprenaline (ISO) is a cAMP-mobilizing β-agonist. NO release from NOC12 produces a rise in [cAMP] equivalent to the effect of ∼1 nM Isoprenaline (Iso). (B) Proposed mechanism through which cGMP increases [cAMP]. (C) Effect of 0.5 nM Iso on NHE1 flux (N = 10 Iso-treated cells from three animals; N = 19 controls from three animals), showing significant reduction in flux (P < 0.0001; two-way ANOVA). (D) Effect of 20 µM 8Br-cAMP on NHE1 flux (N = 15 treated cells from four animals; N = 15 control cells from four animals), showing significant reduction in flux (P < 0.0001; two-way ANOVA). (E) Effect of 5 µM NOC12 when adenylyl cyclase activity is inactivated with membrane-permeable inhibitor dideoxyadenosine (ddA; N = 40 myocytes from five animals). NHE1 flux was significantly greater if cAMP production was ablated (P < 0.0001; two-way ANOVA). (F) Effect of two different membrane-permeable cGMP analogues, 8pCPT-cGMP and 8Br-cGMP, on NHE1 flux vs. control (N = 18, 21, 18 cells from four animals). 8pCPT-cGMP (which does not mobilise cAMP) increased NHE1 activity (P < 0.0001; two-way ANOVA), whereas 8Br-cGMP (which evokes a secondary cAMP signal) decreased NHE1 activity (P < 0.0001; two-way ANOVA).
The effects of cAMP on NHE1 were tested in wild-type rat myocytes stimulated with either 0.5 nM Iso or 20 µM 8Br-cAMP, a membrane-permeable cAMP derivative. Engaging cAMP-dependent signalling with these agents produced an acute inhibition of NHE1 (Figure 5C and D), consistent with previous studies showing a PKA-dependent phosphorylation of the inhibitory site at Ser648.3 Thus, a cAMP signal emerging from a NO-evoked rise in [cGMP] may explain the net inhibitory effect of high [NO] on NHE1. To test this, myocytes were exposed to 5 µM NOC12 to activate cGMP signalling, but one group of cells was pretreated with 50 µM dideoxyadenosine (ddA), a membrane-permeable inhibitor of adenylyl cyclase, which is expected to prevent the secondary [cAMP] rise. NHE1 activity was significantly higher in myocytes that were blocked from evoking a cAMP signal, representing the state of NHE1 modulation in response to cGMP alone (Figure 5E).
The coupling between NO-NHE1 was further interrogated by introducing membrane-permeable cGMP derivatives in the absence of an NO donor. The analogue 8Br-cGMP activates canonical cGMP targets, such as PKG, and also inhibits PDE3. In contrast, 8pCPTcGMP, unlike endogenous cGMP, is not broken down by PDEs and also does not inhibit PDE3.56 When applied to myocytes, 20 µM 8Br-cGMP and 10 µM 8pCPTcGMP produced opposite effects on NHE1 flux (Figure 5F). The activatory effect of 8pCPTcGMP, which is unable to evoke secondary cAMP signals, was consistent with the actions of low [NO] (Figure 3G), whereas the effect of 8Br-cGMP is more akin to that of high [NO] (Figure 3E). Thus, the biphasic effect of NO on NHE1 is a result of a [NO]-dependent rise in cGMP, which per se is activatory on NHE1, but switches to becoming inhibitory when a sufficient degree of PDE3 inhibition allows [cAMP] to build-up. This mechanism exploits the distinct patterns of NHE1 phosphorylation produced by PKG and PKA (Figure 1).
3.7 NO control of NHE1 activity produces a biphasic effect on Ca2+ wave frequency
The triggering of NHE1 activity at low pHi evokes a substantial influx of Na+ ions, in the range of several mM/min, which can Ca2+-overload the cardiomyocyte through a well-documented mechanism involving NCX (Figure 6A). Beyond a threshold of Ca2+ loading, myocytes will produce spontaneous Ca2+ release from the sarcoplasmic reticulum (SR), detected as cytoplasmic Ca2+ waves. Modulation of Na+-influx via NHE1 by NO signals is therefore expected to influence the frequency of spontaneous Ca2+ waves.
Figure 6.

Biphasic regulation of NHE1 by NO affects the propensity of aberrant forms of Ca2+ signalling evoked by mild acidosis. (A) Schematic model of mechanism linking mild acidosis (attained with 30 mM acetate) to sarcoplasmic reticulum (SR) overload. (B) Rat myocytes (loaded with pH-sensitive cSNARF1) under superfusion with Hepes-buffered normal Tyrode with [Ca2+] raised to 5 mM to produce Ca2+ overload and increase the incidence of waves. After a 6 min period in control solution (NT, normal Tyrode), cells were exposed to 30 mM acetate (Ac) which acidifies the myocyte and activates NHE1 (13 cells from two animals). (C) Predicted Na+ influx on NHE1, based on pH-sensitivity of flux determined in Figure 2-2, mapped onto the pHi time course. (D) Rat myocyte loaded with Ca2+ dye Fluo3, imaged in line scan mode. Exemplar line scans are shown for a cell in control conditions, and after 6 min of superfusion with 30 mM acetate. Line scan measurements repeated on cells stimulated with either high or low [NO] released from 5 µM NOC12 (producing [NO]>300 nM) or with 300 µM NOC12 plus 100 µM CPTIO (producing [NO]<30 nM), respectively. Results presented as Ca2+ frequency normalized to baseline level (N = 22 for control, 13 for high [NO], 13 for low [NO]; from five animals). Low [NO] increased Ca2+ wave frequency (P < 0.0001; two-way ANOVA), whereas high [NO] decreased Ca2+ wave frequency (P < 0.0001; two-way ANOVA).
To trigger NHE1 activity, the cytoplasm of myocytes was uniformly acidified by exposure to 30 mM acetate, which enters and acidifies the cytoplasm and triggers the activation of NHE1 (Figure 6B). As NHE1 attempts to extrude the acid-load, Na+ ions enter the myocyte (Figure 6C). Ca2+ release events were recorded in Fluo3-loaded myocytes, superfused with solutions containing 5 mM Ca2+ (to raise the likelihood of spontaneous release). The frequency of Ca2+ waves was calculated from the interval between events detected by imaging in linescan mode (Figure 6D). In the absence of NO donors, Ca2+ wave frequency increased by a factor of two within 5 minutes of acidification (Figure 6D). The protocol was repeated in the presence of either low [NO] (300 µM NOC12 plus 100 µM CPTIO) to stimulate NHE1 or high [NO] (5 µM NOC12) to inhibit NHE1. It is well-established that NO can affect Ca2+ waves through various NHE1-independent mechanisms (e.g. on RyR2 channels),37,57,58 and to correct for these actions, the response of Ca2+ wave frequency to acetate was normalized to baseline frequency. In the presence of high [NO], the acetate-evoked rise in Ca2+ wave frequency was attenuated (to only ∼150% of resting levels), consistent with an inhibitory effect on NHE1-dependent Na+-influx (Figure 6D). Thus, inhibition of NHE1 with high doses of NO is anti-arrhythmogenic, similar to the pharmacological effect of NHE1 inhibitors such as cariporide.59 In contrast, low [NO] resulted in a more robust acetate-evoked rise in Ca2+ wave frequency (to ∼300% of resting levels), explained by the larger Na+ influx through activated NHE1 (Figure 6D).
4. Discussion
We provide the first report of a physiologically relevant molecule exercising a biphasic effect on cardiac NHE1, the heart’s most powerful pHi regulator and major Na+ entry pathway. NHE1 activity has a broad remit of actions on cardiac physiology because its activity influences pHi homeostasis and Ca2+ signalling, and thence a myriad of downstream effects. Although resting pHi in myocytes is normally kept near 7.2, deviations can occur pathologically, such as during ischaemia, or physiologically in response to intra- or extracellular signals.54,60–62 Controlled shifts in pHi are implemented physiologically by changing transmembrane H+ ion traffic carried by transporters, such as NHE1. Unlike other regulators of NHE1, NO does not operate through a surface receptor, and thus the coupling to its downstream enzyme (GC) has less ‘gain’ than in the case of G protein-coupled cascades. Instead, the physiological outcomes of NO signalling are fine-tuned by regulating the pattern of gas release. Other examples of dose-dependent actions of NO on the heart have been reported for chronotropy,63,64 but these are the ensemble of effects on various proteins, and not necessarily a convergence onto a single identifiable target.
The regulatory scope of NO signalling on NHE1 can be quantified from the dynamic range of its effect, calculated as the ratio of the highest to the lowest activation state. Based on results such as those in Figure 3, the dynamic range for the NO effect on NHE1 is a factor of two, offering good leverage on cardiac physiology. At the lower end of this activity range, NHE1 is still able to maintain a low intracellular [H+] that is conducive for cellular physiology, but this reduction in homeostatic prowess would permit some degree of pHi fluctuations, serving as bona fide H+-signals within the so-called permissive range.1 When maximally activated, cardiac NHE1 can produce an unprecedented magnitude of H+ extrusion to defend against acid-challenges and maintain an alkaline pHi.
Intriguingly, the biphasic effect of NO on NHE1 was not observed in neonatal rat ventricular myocytes or in a breast cancer cell line (Supplementary material online, Figure S6). This finding indicates that the biochemical details of the NO-NHE1 cascade cannot be resolved in expression systems (e.g. with the use of NHE1 mutants) or with cell-free approaches. Here, we were able to obtain mechanistic insights by studying the responses of primary adult myocytes to pharmacological or genetic manipulations of the putative NO-NHE1 cascade (Figures 2–6), and interpreting these findings in terms of how PKA and PKG differentially phosphorylate the C-terminus of NHE1, as determined using in vitro assays (Figure 1).
The modulation of NHE1 activity by NO is not exercised by S-nitros(yl)ation, tested biochemically by two independent methods (Figure 4A–C), but through the enzyme GC instead (Figures 3 and 4) Canonically, this pathway signals through the diffusible messenger cGMP, but in the heart, adequate cGMP-inhibition of PDE3 can allow a secondary rise in [cAMP], which we confirmed with a FRET-based sensor of cAMP (Figure 5A). Results of in vitro studies (Figure1A–C) show that serine residues of the regulatory C-terminus of NHE1 are substrates for cAMP- and cGMP-dependent kinases. Using antibodies against specific phosphorylation sites, we show that PKA and PKG have distinct selectivity for serine residues. Specifically, PKG showed a preference for phosphorylating Ser703, a residue linked to NHE1 activation. PKA, in contrast, was able to produce a strong phosphorylation of Ser648. Our functional data explain how NO signals are able to use these kinases to orchestrate NHE1 activation and inhibition at low and high [NO], respectively, by recruiting cGMP in a dose-dependent manner, and triggering cAMP only with the higher concentration (Figure 7). Using pharmacological inhibition and knockdown, we show that basal NO production by nNOS was activatory on NHE1. Adenoviral delivery of nNOS, which preferentially targets the sarcolemma,65 or a carefully titrated low (<30 nM) dose of NO (NOC12+CPTIO) produced a further stimulatory effect (Figure 7A).44 In the latter experiments, NO concentrations attained inside myocytes are likely to be attenuated further by chemical reactions involving proteins such as myoglobin (∼200µM myoglobin will scavenge NO with a rate constant of 22 µM/s66). We therefore argue that the low [NO] concentration delivered by mixing NO donor and scavenger is within the physiological range.26 NHE1 activation by this level of NO signalling could be explained by PKG phosphorylation of Ser703, without affecting Ser648 (Figure 1E). Exposure to higher [NO] (>300 nM) released from donors (SNP or NOC12 alone) switched the polarity of NO signalling to net inhibition because of the secondary recruitment of cAMP signalling (Figure 3). Activation of PKA is expected to increase Ser648 phosphorylation, an inhibitory modification (Figure 1F). Indeed, a previous report showing NHE1 inhibition by NO released from a high dose of SNP21 is likely to have involved cAMP signals (Figure 5B). Thus, the crossover in the polarity of the NO-NHE1 cascade occurred around 100 nM, postulated to be above the normal physiological range of NO signalling.26 As a test of our model, a cGMP-analogue that cannot inhibit PDE3 (and hence has no effect on [cAMP]) was found to activate NHE1 (Figure 5F), whereas stimuli that evoked cAMP signals but without a change in cGMP, caused NHE1 inhibition (Figure 5C and D). Furthermore, it was possible to reverse the effect of a nominally inhibitory NO signal (i.e. domain of high [NO]) by imposing a diffusion distance between the source of NO and NHE1, along which the cGMP signal decays and becomes inadequate to raise [cAMP] (Figure 4).
Figure 7.
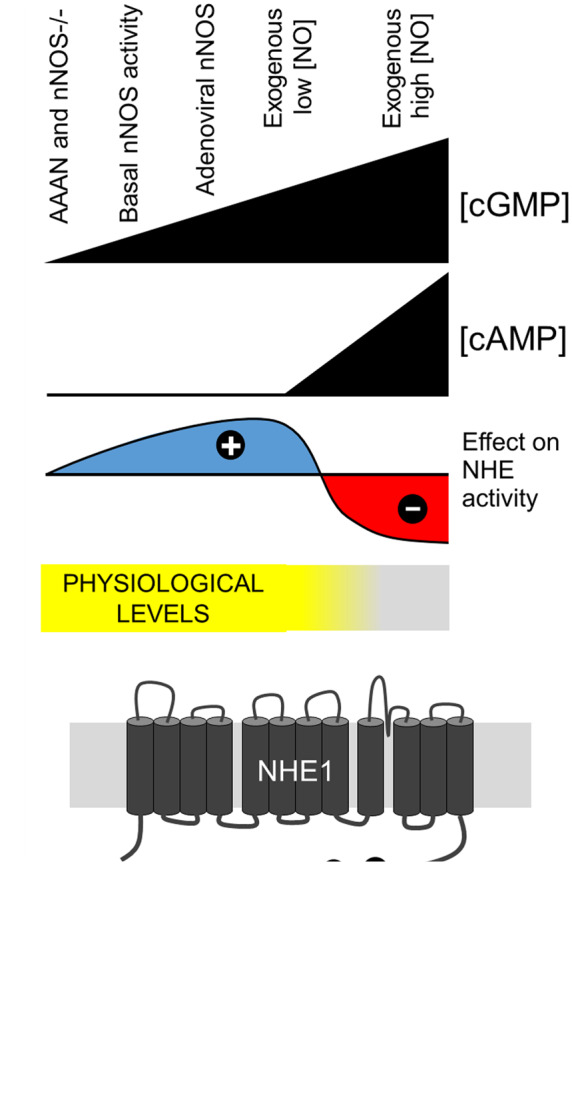
Model of cardiac NHE1 regulation by NO signalling. (A) The various experimental manoeuvres performed in this study are ranked by strength of NO signal. Higher NO stimulations will evoke greater cGMP production. At higher [cGMP], inhibition of PDE3 permits a steady rise in [cAMP]. The balance between cGMP and cAMP signals sets the level of NHE1 activity. Based on literature values for [NO] measured in vivo, NO-evoked NHE1 activation is expected to be within physiological signalling, whereas NO-evoked NHE1 inhibition would occur over the pathophysiological range of elevated [NO]. (B) Schematic of the signalling pathways, evoked by NO, that regulate NHE1 activity by means of a differential activation of PKG-and PKA-dependent cascades.
Given that the actions of NO on NHE1 are highly context-dependent, as shown by their manifestation in adult but not neonatal myocytes (Figure 3, Supplementary material online, Figure S6), it is plausible that pathologic remodelling of myocytes may alter NO-NHE1 coupling further. Patterns of myocardial NO signalling can undergo changes in heart failure,67–69 hypertension,70 and ischaemia/reperfusion injury68,71,72 as a result of altered NOS isoform expression67,68,70,71 and subcellular distribution67,68 or abnormal intracellular chemistry (e.g. NO scavengers73 and NOS co-factors19) In failing hearts, for instance, raised nNOS activity at the sarcolemma67,68 may produce a more intensive release of NO near NHE1, potentially resulting in inhibition.
The tightening of pHi control by NO-activated NHE1 would improve the responsiveness of the myocyte’s pHi-regulatory apparatus to acidic disturbances, such as in periods of increased metabolic demand or failure of perfusion. However, the benefit of this augmentation must be weighed against the additional energetic burden on the Na+/K+ ATP pump required to restore the inward transmembrane [Na+] gradient. The NHE1-driven increase in intracellular [Na+] will also affect Ca2+ signalling by increasing SR Ca2+ loading. At best, this would exert a positive inotropic effect, but more sustained overloading will eventually precipitate an arrhythmia as a result of spontaneous Ca2+ release. Considering the injurious nature of the latter phenomenon, there is a biological niche for agents that protect against NHE1 over-drive. High volumes of NO release would produce such an effect, as shown by the inhibitory effect on Ca2+ wave frequency (Figure 6D). We speculate that basal-to-low levels of NO serve to increase heart contractility by stabilizing pHi at a more alkaline level, and by increasing cellular Ca2+ loading through increased Na+ entry. However, excessive activation of NHE1 can be acutely deleterious to the heart, as demonstrated by ischaemia–reperfusion injury74 when cells become Ca2+ overloaded11 and arrhythmic.75 Chronically overactive NHE1 has been linked to maladaptive hypertrophy, a risk factor in heart failure.76,77 Thus, enhanced NO signalling may serve as a natural NHE1 inhibitor, producing more quiescent Ca2+ signalling by curtailing Na+ influx and allowing the cell to undergo a mild acidification.
Modulation of NHE1 by NO signalling may be relevant to the interpretation of clinical trials, such as the GUARDIAN trial (cariporide)78 because the benefit of targeting NHE1 will depend on the extent to which the protein is functionally active. Patients and animal models with HFpEF (heart failure with preserved ejection fraction) have greatly increased nitrosative stress69 which may be equivalent to levels of NO that inhibit NHE1. In these states, pharmacological inhibition of NHE1 would yield a reduced therapeutic effect, and drugs such as cariporide may not be as effective as anticipated. Compared to HFpEF, myocardial ischaemia is postulated72 to involve more modest increases in NO levels. If such a nitrosative stress activates NHE1, a higher dose of cariporide would be required to adequately suppress Na+ influx. This may explain why the sole beneficial outcomes noted in the GUARDIAN trial were for the highest doses (120 mg) of cariporide.78 However, without information about the state of NO signalling, it is not possible to perform a meta-analysis of patients stratified by endogenous NHE1 activity.
In summary, the biphasic effect of NO provides unprecedented flexibility in controlling NHE1 locally in the heart, and with it, the capacity to modulate cardiac Ca2+ signalling and other pH-sensitive processes.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Supplementary Material
Acknowledgements
We thank Dr Konstantinos Lefkimmiatis for providing the virus for expressing H187, Dr My-Nhan Nguyen for providing the nNOS western blot (Supplementary material online, Figure S2), Johanna Michl for help with phostag blots, Ryan Lam for assistance in western blotting, Andrew Snabaitis for helpful discussions on NHE1 biochemistry, Susann Bruche for help in in vitro experiments, and Ms Chela Nunez-Alonso for technical help in isolating adult and neonatal myocytes. Phenylmercury resin was kindly provided by Professor Harry Ischiropoulos and Dr Paschalis Doulias (University of Pennsylvania, USA).
Conflict of interest: none declared.
Funding
This work was supported by the British Heart Foundation (PG/12/2/29324 to P.S., RG/15/9/31534 to P.S., and CH/12/3/29609 to B.C.).
Translational perspective
Na+/H+ exchanger-1 (NHE1) regulates intracellular [H+] and [Na+], but its over-activation can drive ionic imbalances that affect cardiac contractility and rhythm. Pharmacological control of NHE1 (e.g. with cariporide) has been proposed as cardioprotective in conditions such as ischaemia/reperfusion injury, but trials (e.g. GUARDIAN) failed to demonstrate overall clinical benefit. A confounding factor in these analyses is NHE1 modulation by endogenous factors. We demonstrate that nitric oxide (NO) signalling fine-tunes NHE1, producing stimulation at low levels, turning into inhibition as the signal grows stronger. Thus, evaluations of the therapeutic efficacy of NHE1-blocking drugs should consider NO signalling, a pathway known to undergo changes in disease.
References
- 1. Leem CH, Lagadic-Gossmann D, Vaughan-Jones RD.. Characterization of intracellular pH regulation in the guinea-pig ventricular myocyte. J Physiol 1999;517(Pt 1):159–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yamamoto T, Swietach P, Rossini A, Loh SH, Vaughan-Jones RD, Spitzer KW.. Functional diversity of electrogenic Na+-HCO3- cotransport in ventricular myocytes from rat, rabbit and guinea pig. J Physiol 2005;562:455–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vaughan-Jones RD, Spitzer KW, Swietach P.. Intracellular pH regulation in heart. J Mol Cell Cardiol 2009;46:318–331. [DOI] [PubMed] [Google Scholar]
- 4. Allen DG, Orchard CH.. The effects of changes of pH on intracellular calcium transients in mammalian cardiac muscle. J Physiol 1983;335:555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bountra C, Vaughan-Jones RD.. Effect of intracellular and extracellular pH on contraction in isolated, mammalian cardiac muscle. J Physiol 1989;418:163–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Orchard CH, Kentish JC.. Effects of changes of pH on the contractile function of cardiac muscle. Am J Physiol 1990;258:C967–C981. [DOI] [PubMed] [Google Scholar]
- 7. Garciarena CD, Youm JB, Swietach P, Vaughan-Jones RD. H+- activated Na+ influx in the ventricular myocyte couples Ca2+-signalling to intracellular pH. J Mol Cell Cardiol 2013;61:51–59. [DOI] [PubMed] [Google Scholar]
- 8. Harrison SM, Frampton JE, McCall E, Boyett MR, Orchard CH.. Contraction and intracellular Ca2+, Na+, and H+ during acidosis in rat ventricular myocytes. Am J Physiol 1992;262:C348–C357. [DOI] [PubMed] [Google Scholar]
- 9. Ford KL, Moorhouse EL, Bortolozzi M, Richards MA, Swietach P, Vaughan-Jones RD.. Regional acidosis locally inhibits but remotely stimulates Ca2+ waves in ventricular myocytes. Cardiovasc Res 2017;113:984–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Swietach P, Spitzer KW, Vaughan-Jones RD.. Na+ ions as spatial intracellular messengers for co-ordinating Ca2+ signals during pH heterogeneity in cardiomyocytes. Cardiovasc Res 2015;105:171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Choi HS, Trafford AW, Orchard CH, Eisner DA.. The effect of acidosis on systolic Ca2+ and sarcoplasmic reticulum calcium content in isolated rat ventricular myocytes. J Physiol 2000;529(Pt 3):661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lederer WJ, Tsien RW.. Transient inward current underlying arrhythmogenic effects of cardiotonic steroids in Purkinje fibres. J Physiol 1976;263:73–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Capogrossi MC, Houser SR, Bahinski A, Lakatta EG.. Synchronous occurrence of spontaneous localized calcium release from the sarcoplasmic reticulum generates action potentials in rat cardiac ventricular myocytes at normal resting membrane potential. Circ Res 1987;61:498–503. [DOI] [PubMed] [Google Scholar]
- 14. Hendus-Altenburger R, Kragelund BB, Pedersen SF.. Structural dynamics and regulation of the mammalian SLC9A family of Na+/H+ exchangers. Curr Top Membr 2014;73:69–148. [DOI] [PubMed] [Google Scholar]
- 15. Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, Hobai IA, Lemmon CA, Burnett AL, O'Rourke B, Rodriguez ER, Huang PL, Lima JA, Berkowitz DE, Hare JM.. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature 2002;416:337–339. [DOI] [PubMed] [Google Scholar]
- 16. Lima B, Forrester MT, Hess DT, Stamler JS.. S-nitrosylation in cardiovascular signaling. Circ Res 2010;106:633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Massion PB, Feron O, Dessy C, Balligand JL.. Nitric oxide and cardiac function: ten years after, and continuing. Circ Res 2003;93:388–398. [DOI] [PubMed] [Google Scholar]
- 18. Murphy E, Kohr M, Menazza S, Nguyen T, Evangelista A, Sun J, Steenbergen C.. Signaling by S-nitrosylation in the heart. J Mol Cell Cardiol 2014;73:18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sears CE, Ashley EA, Casadei B.. Nitric oxide control of cardiac function: is neuronal nitric oxide synthase a key component? Philos Trans R Soc Lond B Biol Sci 2004;359:1021–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sears CE, Bryant SM, Ashley EA, Lygate CA, Rakovic S, Wallis HL, Neubauer S, Terrar DA, Casadei B.. Cardiac neuronal nitric oxide synthase isoform regulates myocardial contraction and calcium handling. Circ Res 2003;92:e52–e59. [DOI] [PubMed] [Google Scholar]
- 21. Ito N, Bartunek J, Spitzer KW, Lorell BH.. Effects of the nitric oxide donor sodium nitroprusside on intracellular pH and contraction in hypertrophied myocytes. Circulation 1997;95:2303–2311. [DOI] [PubMed] [Google Scholar]
- 22. Pravdic D, Vladic N, Cavar I, Bosnjak ZJ.. Effect of nitric oxide donors S-nitroso-N-acetyl-DL-penicillamine, spermine NONOate and propylamine propylamine NONOate on intracellular pH in cardiomyocytes. Clin Exp Pharmacol Physiol 2012;39:772–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Balbatun A, Louka FR, Malinski T.. Dynamics of nitric oxide release in the cardiovascular system. Acta Biochim Pol 2003;50:61–68. [PubMed] [Google Scholar]
- 24. Neishi Y, Mochizuki S, Miyasaka T, Kawamoto T, Kume T, Sukmawan R, Tsukiji M, Ogasawara Y, Kajiya F, Akasaka T, Yoshida K, Goto M.. Evaluation of bioavailability of nitric oxide in coronary circulation by direct measurement of plasma nitric oxide concentration. Proc Natl Acad Sci USA 2005;102:11456–11461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fujita S, Roerig DL, Bosnjak ZJ, Stowe DF.. Effects of vasodilators and perfusion pressure on coronary flow and simultaneous release of nitric oxide from guinea pig isolated hearts. Cardiovasc Res 1998;38:655–667. [DOI] [PubMed] [Google Scholar]
- 26. Hall CN, Garthwaite J.. What is the real physiological NO concentration in vivo? Nitric Oxide 2009;21:92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Batchelor AM, Bartus K, Reynell C, Constantinou S, Halvey EJ, Held KF, Dostmann WR, Vernon J, Garthwaite J.. Exquisite sensitivity to subsecond, picomolar nitric oxide transients conferred on cells by guanylyl cyclase-coupled receptors. Proc Natl Acad Sci USA 2010;107:22060–22065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zaccolo M, Movsesian MA.. cAMP and cGMP signaling cross-talk: role of phosphodiesterases and implications for cardiac pathophysiology. Circ Res 2007;100:1569–1578. [DOI] [PubMed] [Google Scholar]
- 29. Meier S, Andressen KW, Aronsen JM, Sjaastad I, Hougen K, Skomedal T, Osnes JB, Qvigstad E, Levy FO, Moltzau LR.. PDE3 inhibition by C-type natriuretic peptide-induced cGMP enhances cAMP-mediated signaling in both non-failing and failing hearts. Eur J Pharmacol 2017;812:174–183. [DOI] [PubMed] [Google Scholar]
- 30. Zhao CY, Greenstein JL, Winslow RL.. Roles of phosphodiesterases in the regulation of the cardiac cyclic nucleotide cross-talk signaling network. J Mol Cell Cardiol 2016;91:215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Snabaitis AK, Cuello F, Avkiran M.. Protein kinase B/Akt phosphorylates and inhibits the cardiac Na+/H+ exchanger NHE1. Circ Res 2008;103:881–890. [DOI] [PubMed] [Google Scholar]
- 32. Cuello F, Snabaitis AK, Cohen MS, Taunton J, Avkiran M.. Evidence for direct regulation of myocardial Na+/H+ exchanger isoform 1 phosphorylation and activity by 90-kDa ribosomal S6 kinase (RSK): effects of the novel and specific RSK inhibitor fmk on responses to alpha1-adrenergic stimulation. Mol Pharmacol 2007;71:799–806. [DOI] [PubMed] [Google Scholar]
- 33. Snabaitis AK, D'Mello R, Dashnyam S, Avkiran M.. A novel role for protein phosphatase 2A in receptor-mediated regulation of the cardiac sarcolemmal Na+/H+ exchanger NHE1. J Biol Chem 2006;281:20252–20262. [DOI] [PubMed] [Google Scholar]
- 34. Lehoux S, Abe J, Florian JA, Berk BC.. 14-3-3 Binding to Na+/H+ exchanger isoform-1 is associated with serum-dependent activation of Na+/H+ exchange. J Biol Chem 2001;276:15794–15800. [DOI] [PubMed] [Google Scholar]
- 35. Pedersen SF, Counillon L.. The SLC9A-C mammalian Na+/H+ exchanger family: molecules, mechanisms, and physiology. Physiol Rev 2019;99:2015–2113. [DOI] [PubMed] [Google Scholar]
- 36. Takahashi E, Abe J, Gallis B, Aebersold R, Spring DJ, Krebs EG, Berk BC.. p90(RSK) is a serum-stimulated Na+/H+ exchanger isoform-1 kinase. Regulatory phosphorylation of serine 703 of Na+/H+ exchanger isoform-1. J Biol Chem 1999;274:20206–20214. [DOI] [PubMed] [Google Scholar]
- 37. Gonzalez DR, Beigi F, Treuer AV, Hare JM.. Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc Natl Acad Sci USA 2007;104:20612–20617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huang PL, Dawson TM, Bredt DS, Snyder SH, Fishman MC.. Targeted disruption of the neuronal nitric oxide synthase gene. Cell 1993;75:1273–1286. [DOI] [PubMed] [Google Scholar]
- 39. Zoccarato A, Surdo NC, Aronsen JM, Fields LA, Mancuso L, Dodoni G, Stangherlin A, Livie C, Jiang H, Sin YY, Gesellchen F, Terrin A, Baillie GS, Nicklin SA, Graham D, Szabo-Fresnais N, Krall J, Vandeput F, Movsesian M, Furlan L, Corsetti V, Hamilton G, Lefkimmiatis K, Sjaastad I, Zaccolo M.. Cardiac hypertrophy is inhibited by a local pool of cAMP regulated by phosphodiesterase 2. Circ Res 2015;117:707–719. [DOI] [PubMed] [Google Scholar]
- 40. Channon KM, Qian H, Neplioueva V, Blazing MA, Olmez E, Shetty GA, Youngblood SA, Pawloski J, McMahon T, Stamler JS, George SE.. In vivo gene transfer of nitric oxide synthase enhances vasomotor function in carotid arteries from normal and cholesterol-Fed rabbits. Circulation 1998;98:1905–1911. [DOI] [PubMed] [Google Scholar]
- 41. Heaton DA, Lei M, Li D, Golding S, Dawson TA, Mohan RM, Paterson DJ.. Remodeling of the cardiac pacemaker L-type calcium current and its beta-adrenergic responsiveness in hypertension after neuronal NO synthase gene transfer. Hypertension 2006;48:443–452. [DOI] [PubMed] [Google Scholar]
- 42. Richards M, Lomas O, Jalink K, Ford KL, Vaughan-Jones RD, Lefkimmiatis K, Swietach P.. Intracellular tortuosity underlies slow cAMP diffusion in adult ventricular myocytes. Cardiovasc Res 2016;110:395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tian Q, Pahlavan S, Oleinikow K, Jung J, Ruppenthal S, Scholz A, Schumann C, Kraegeloh A, Oberhofer M, Lipp P, Kaestner L.. Functional and morphological preservation of adult ventricular myocytes in culture by sub-micromolar cytochalasin D supplement. J Mol Cell Cardiol 2012;52:113–124. [DOI] [PubMed] [Google Scholar]
- 44. Griffiths C, Wykes V, Bellamy TC, Garthwaite J.. A new and simple method for delivering clamped nitric oxide concentrations in the physiological range: application to activation of guanylyl cyclase-coupled nitric oxide receptors. Mol Pharmacol 2003;64:1349–1356. [DOI] [PubMed] [Google Scholar]
- 45. Swietach P, Youm JB, Saegusa N, Leem CH, Spitzer KW, Vaughan-Jones RD.. Coupled Ca2+/H+ transport by cytoplasmic buffers regulates local Ca2+ and H+ ion signaling. Proc Natl Acad Sci USA 2013;110:E2064–E2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Swietach P, Leem CH, Spitzer KW, Vaughan-Jones RD.. Experimental generation and computational modeling of intracellular pH gradients in cardiac myocytes. Biophys J 2005;88:3018–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Steinert JR, Kopp-Scheinpflug C, Baker C, Challiss RA, Mistry R, Haustein MD, Griffin SJ, Tong H, Graham BP, Forsythe ID.. Nitric oxide is a volume transmitter regulating postsynaptic excitability at a glutamatergic synapse. Neuron 2008;60:642–656. [DOI] [PubMed] [Google Scholar]
- 48. Li X, Liu Y, Kay CM, Muller-Esterl W, Fliegel L.. The Na+/H+ exchanger cytoplasmic tail: structure, function, and interactions with tescalcin. Biochemistry 2003;42:7448–7456. [DOI] [PubMed] [Google Scholar]
- 49. Zaniboni M, Swietach P, Rossini A, Yamamoto T, Spitzer KW, Vaughan-Jones RD.. Intracellular proton mobility and buffering power in cardiac ventricular myocytes from rat, rabbit, and guinea pig. Am J Physiol Heart Circ Physiol 2003;285:H1236–H1246. [DOI] [PubMed] [Google Scholar]
- 50. Wang L, Henrich M, Buckler KJ, McMenamin M, Mee CJ, Sattelle DB, Paterson DJ.. Neuronal nitric oxide synthase gene transfer decreases [Ca2+]i in cardiac sympathetic neurons. J Mol Cell Cardiol 2007;43:717–725. [DOI] [PubMed] [Google Scholar]
- 51. Wolhuter K, Whitwell HJ, Switzer CH, Burgoyne JR, Timms JF, Eaton P.. Evidence against stable protein S-nitrosylation as a widespread mechanism of post-translational regulation. Mol Cell 2018;69:438–450.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Qu Z, Meng F, Bomgarden RD, Viner RI, Li J, Rogers JC, Cheng J, Greenlief CM, Cui J, Lubahn DB, Sun GY, Gu Z.. Proteomic quantification and site-mapping of S-nitrosylated proteins using isobaric iodoTMT reagents. J Proteome Res 2014;13:3200–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Doulias PT, Raju K, Greene JL, Tenopoulou M, Ischiropoulos H.. Mass spectrometry-based identification of S-nitrosocysteine in vivo using organic mercury assisted enrichment. Methods 2013;62:165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lagadic-Gossmann D, Vaughan-Jones RD.. Coupling of dual acid extrusion in the guinea-pig isolated ventricular myocyte to alpha 1- and beta-adrenoceptors. J Physiol 1993;464:49–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wen JF, Cui X, Jin JY, Kim SM, Kim SZ, Kim SH, Lee HS, Cho KW.. High and low gain switches for regulation of cAMP efflux concentration: distinct roles for particulate GC- and soluble GC-cGMP-PDE3 signaling in rabbit atria. Circ Res 2004;94:936–943. [DOI] [PubMed] [Google Scholar]
- 56. Geiger J, Nolte C, Butt E, Sage SO, Walter U.. Role of cGMP and cGMP-dependent protein kinase in nitrovasodilator inhibition of agonist-evoked calcium elevation in human platelets. Proc Natl Acad Sci USA 1992;89:1031–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Curran J, Tang L, Roof SR, Velmurugan S, Millard A, Shonts S, Wang H, Santiago D, Ahmad U, Perryman M, Bers DM, Mohler PJ, Ziolo MT, Shannon TR.. Nitric oxide-dependent activation of CaMKII increases diastolic sarcoplasmic reticulum calcium release in cardiac myocytes in response to adrenergic stimulation. PLoS One 2014;9:e87495.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Niggli E, Ullrich ND, Gutierrez D, Kyrychenko S, Polakova E, Shirokova N.. Posttranslational modifications of cardiac ryanodine receptors: Ca(2+) signaling and EC-coupling. Biochim Biophys Acta 2013;1833:866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Russ U, Balser C, Scholz W, Albus U, Lang HJ, Weichert A, Scholkens BA, Gogelein H.. Effects of the Na+/H+-exchange inhibitor Hoe 642 on intracellular pH, calcium and sodium in isolated rat ventricular myocytes. Pflugers Arch 1996;433:26–34. [DOI] [PubMed] [Google Scholar]
- 60. Puceat M, Clement-Chomienne O, Terzic A, Vassort G.. Alpha 1-adrenoceptor and purinoceptor agonists modulate Na-H antiport in single cardiac cells. Am J Physiol 1993;264:H310–H319. [DOI] [PubMed] [Google Scholar]
- 61. Matsui H, Barry WH, Livsey C, Spitzer KW.. Angiotensin II stimulates sodium-hydrogen exchange in adult rabbit ventricular myocytes. Cardiovasc Res 1995;29:215–221. [PubMed] [Google Scholar]
- 62. Gunasegaram S, Haworth RS, Hearse DJ, Avkiran M.. Regulation of sarcolemmal Na(+)/H(+) exchanger activity by angiotensin II in adult rat ventricular myocytes: opposing actions via AT(1) versus AT(2) receptors. Circ Res 1999;85:919–930. [DOI] [PubMed] [Google Scholar]
- 63. Wyeth RP, Temma K, Seifen E, Kennedy RH.. Negative inotropic actions of nitric oxide require high doses in rat cardiac muscle. Pflügers Arch 1996;432:678–684. [DOI] [PubMed] [Google Scholar]
- 64. Mohan P, Sys SU, Brutsaert DL.. Positive inotropic effect of nitric oxide in myocardium. Int J Cardiol 1995;50:233–237. [DOI] [PubMed] [Google Scholar]
- 65. Carnicer R, Suffredini S, Liu X, Reilly S, Simon JN, Surdo NC, Zhang YH, Lygate CA, Channon KM, Casadei B.. The subcellular localisation of neuronal nitric oxide synthase determines the downstream effects of NO on myocardial function. Cardiovasc Res 2017;113:321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Flogel U, Merx MW, Godecke A, Decking UK, Schrader J.. Myoglobin: a scavenger of bioactive NO. Proc Natl Acad Sci USA 2001;98:735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Damy T, Ratajczak P, Shah AM, Camors E, Marty I, Hasenfuss G, Marotte F, Samuel JL, Heymes C.. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet 2004;363:1365–1367. [DOI] [PubMed] [Google Scholar]
- 68. Bendall JK, Damy T, Ratajczak P, Loyer X, Monceau V, Marty I, Milliez P, Robidel E, Marotte F, Samuel JL, Heymes C.. Role of myocardial neuronal nitric oxide synthase-derived nitric oxide in beta-adrenergic hyporesponsiveness after myocardial infarction-induced heart failure in rat. Circulation 2004;110:2368–2375. [DOI] [PubMed] [Google Scholar]
- 69. Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, Luo X, Jiang N, May HI, Wang ZV, Hill TM, Mammen PPA, Huang J, Lee DI, Hahn VS, Sharma K, Kass DA, Lavandero S, Gillette TG, Hill JA.. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019;568:351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Piech A, Dessy C, Havaux X, Feron O, Balligand JL.. Differential regulation of nitric oxide synthases and their allosteric regulators in heart and vessels of hypertensive rats. Cardiovasc Res 2003;57:456–467. [DOI] [PubMed] [Google Scholar]
- 71. Damy T, Ratajczak P, Robidel E, Bendall JK, Oliviero P, Boczkowski J, Ebrahimian T, Marotte F, Samuel JL, Heymes C.. Up-regulation of cardiac nitric oxide synthase 1-derived nitric oxide after myocardial infarction in senescent rats. Faseb J 2003;17:1934–1936. [DOI] [PubMed] [Google Scholar]
- 72. Simon JN, Duglan D, Casadei B, Carnicer R.. Nitric oxide synthase regulation of cardiac excitation-contraction coupling in health and disease. J Mol Cell Cardiol 2014;73:80–91. [DOI] [PubMed] [Google Scholar]
- 73. Vaziri ND, Ni Z, Oveisi F, Trnavsky-Hobbs DL.. Effect of antioxidant therapy on blood pressure and NO synthase expression in hypertensive rats. Hypertension 2000;36:957–964. [DOI] [PubMed] [Google Scholar]
- 74. Karmazyn M. Amiloride enhances postischemic ventricular recovery: possible role of Na+-H+ exchange. Am J Physiol 1988;255:H608–H615. [DOI] [PubMed] [Google Scholar]
- 75. Orchard CH, Cingolani HE.. Acidosis and arrhythmias in cardiac muscle. Cardiovasc Res 1994;28:1312–1319. [DOI] [PubMed] [Google Scholar]
- 76. Karmazyn M, Kilic A, Javadov S.. The role of NHE-1 in myocardial hypertrophy and remodelling. J Mol Cell Cardiol 2008;44:647–653. [DOI] [PubMed] [Google Scholar]
- 77. Cingolani HE, Ennis IL.. Sodium-hydrogen exchanger, cardiac overload, and myocardial hypertrophy. Circulation 2007;115:1090–1100. [DOI] [PubMed] [Google Scholar]
- 78. Chaitman BR. A review of the GUARDIAN trial results: clinical implications and the significance of elevated perioperative CK-MB on 6-month survival. J Card Surg 2003;18(Suppl. 1):13–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.