

Abstract
In recent years, engineering of various human tissues in microphysiologically relevant platforms, known as organs-on-chips (OOCs), has been explored to establish in vitro tissue models that recapitulate the microenvironments found in native organs and tissues. However, most of these models have overlooked the important roles of immune cells in maintaining tissue homeostasis under physiological conditions and in modulating the tissue microenvironments during pathophysiology. Significantly, gradual progress is being made in the development of more sophisticated microphysiologically relevant human-based OOC models that allow the studies of the key biophysiological aspects of specific tissues or organs, interactions between cells (parenchymal, vascular, and immune cells) and their extracellular matrix molecules, effects of native tissue architectures (geometry, dynamic flow or mechanical forces) on tissue functions, as well as unravelling the mechanism underlying tissue-specific diseases and drug testing. In this Progress Report, we discuss the different components of the immune system, as well as immune OOC platforms and immunocompetent OOC approaches that have simulated one or more components of the immune system. We also outline the challenges to recreate a fully functional tissue system in vitro with a focus on the incorporation of the immune system.
Keywords: organ-on-a-chip, immunocompetent, microenvironment, extracellular matrix, immune cell
Graphical Abstract
This Progress Report discusses the different components of the immune system, as well as immune organ-on-a-chip (OOC) platforms and immunocompetent OOC approaches that have simulated one or more components of the immune system. The challenges to recreate fully functional tissue systems in vitro are also outlined with a focus on the incorporation of the immune system.

1. Introduction
Organs are composed of two or more tissue types that are orderly organized and work together in a unique three-dimensional (3D) structure. Each tissue, in turn, is a group of similar cells, held together by the cell-cell junctions and extracellular matrix (ECM), that work together for a specific function.[1] Living cells and tissues experience spatiotemporal dynamics in biochemical gradients and biomechanical forces (e.g., fluid shear stresses, cyclic strains, compressions) in their respective microenvironments, which are crucial for the survival, growth, and functions of these cells and tissues.[2] Each tissue possesses an ECM with a unique topological, biophysical, and biochemical composition, i.e., ECM is tissue-specific, and is markedly heterogeneous.[1] The ECM is a non-cellular 3D macromolecular network composed of collagens, proteoglycans, glycosaminoglycans, fibronectin, elastin, laminins, and glycoproteins.[3–6] ECM components bind with each other and also with cell surface adhesion receptors known as integrins, forming a complex network that surrounds cells within a tissue.[4,7] Cell surface adhesion receptors activate signal transduction from the ECM to the cells, which is crucial for the regulation of cellular functions, such as survival, growth, migration, and differentiation, and thus maintaining normal tissue homeostasis.[8] In addition, ECM is highly dynamic in nature undergoing continuous remodeling, a process mediated by ECM-degrading enzymes, such as metalloproteinases, during normal and pathological conditions.[8,9]
Immune cells, which produce cytokines, hormones, neuropeptides, and other soluble molecules, mediate both pro-inflammatory and anti-inflammatory responses and are distributed throughout the body.[10] Immune cells are well-organized, positioned, and equipped that they constantly surveil the body system to detect invading pathogens or foreign materials.[10] Upon threat recognition, the immune system coordinates an inflammatory signaling cascade that eliminates destroyed cells and pathogens as well as contributes towards restoration of tissue homeostasis.[11] In fact, the immune system comprises of the lymphoid organs, such as the bone marrow, spleen, and thymus, and the lymphatic system including lymph nodes and lymphatic vasculature.[12] In addition, leukocytes (white blood cells) and lymphocytes (B and T lymphocytes) play important roles within the immune system.[13] While most of the immune cells originate in the bone marrow (a source of lymphocyte stem cells) and circulate to peripheral tissues through the blood and the lymph,[14] some immune cells such as microglia, resident macrophages in the central nervous system (CNS)[15] and Kupffer cells, resident macrophages in liver,[16] originate from the progenitor cells during early development in the embryonic yolk sac. While B cells mature in the bone marrow, the thymus serves as the site of T cell maturation. Lymph nodes act as the sites for lymphocyte activation and proliferation as well as the sites of antigen presentation between antigen-presenting cells (APCs) and lymphocytes. Similarly, the spleen is another site for lymphocyte activation and proliferation, and the site of interactions between blood-borne antigens and lymphocytes.[14,17]
The immune system and its mechanism of actions, including innate and adaptive responses, immunological memory, and their relationship with diseases have been extensively investigated in the context of infections and injuries.[18] Although the structure and immunological functions of primary and secondary lymphoid organs have been studied extensively, the immune responses in nonlymphoid tissues or organs such as liver, lung, kidney, muscle, reproductive organs, and gastrointestinal tract, as well as tumors, are less-studied. Recent reports provide evidences that, diverse types of both innate and adaptive immune cells are present in nonlymphoid tissues (tissue-resident immune cells) that interact with the surrounding environments, directly by cell-cell interactions or indirectly through cytokines and interleukins (ILs). For example, liver harbors abundant CD8+ T cells, invariant natural killer T (NKT) cells, mucosal associated invariant T (MAIT) cells, and γδT cells, in addition to the resident macrophages known as Kupffer cells.[19] Similarly, kidney possesses NKT cells, MAIT cells, and γδ T cells.[20] CNS consists of macrophages-like cells known as microglia along with monocytes, dendritic cells, natural killer (NK) cells, T cells, and B cells.[21] Similarly, immune cells residing in the adipose tissue include eosinophils, anti-inflammatory M2 macrophages, and type 2 innate lymphoid cells.[22] These immune cells sense signals from their tissue-specific environment and play pivotal roles in the initiation, propagation, and regulation of complex physiological processes,[10] such as neuron functions in the central nervous system and peripheral tissues,[23,24] regulation of adipose tissues and metabolic homeostasis[25,26], repair of the muscular tissues[27,28], as well as in pregnancy,[29] bone homeostasis[30], and senescence,[31] demonstrating that the tissue-specific immunity is critical for tissue homeostasis, regeneration, and repair as well as immunological surveillance against pathogens[32] and tumors[33] (Figure 1). Therefore, understanding the tissue-specific immune system and its interactions within the tissues and with other physiological systems are key to providing in-depth knowledge of physiology of the tissues and organs, to diagnose the pathological conditions, and to improve therapeutic strategies in a number of diseases including but not limited to, inflammatory, infectious, and autoimmune diseases, wound healing, and cancers.
Figure 1. Contributions of the immune system in the regulation of complex physiological processes of the different organ systems.
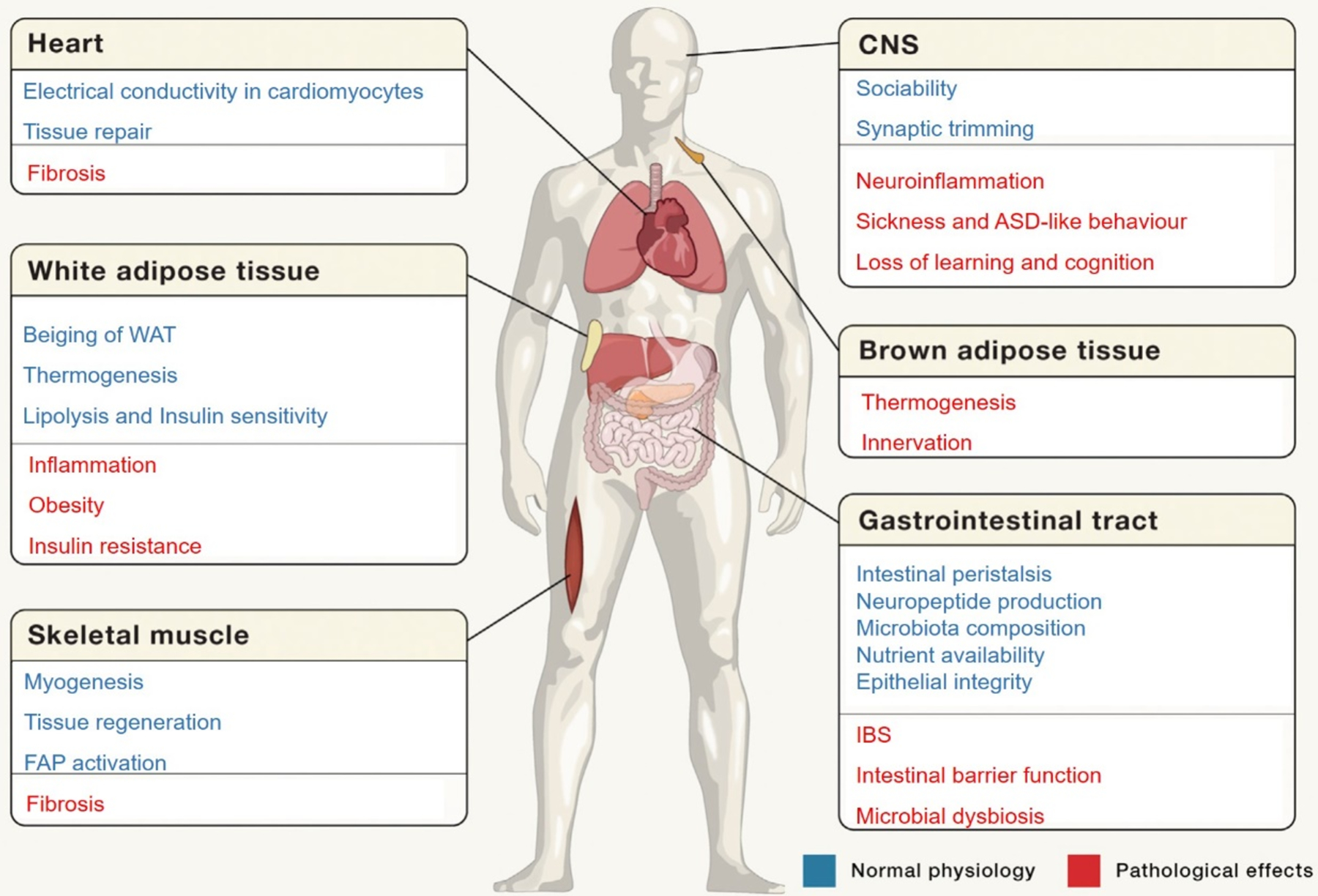
For example, the immune cells play crucial role in the normal functioning of the CNS, heart conductivity and heart tissue repair, white/brown adipose tissues, and muscle tissue homeostasis. Alteration of the immune system results in diseases, such as fibrosis, obesity, and neurological disorders including ASD and IBS, among others. Reproduced with permission.[10] Copyright 2018, Elsevier. ASD: autism spectrum disorder, CNS: central nervous system, FAP: fibroblast activation protein, IBS: irritable bowel syndrome, WAT: white adipose tissue.
Despite considerable technological progress, existing animal models and conventional cell culture models have limitations. The use of animal models is costly, time-consuming, and most importantly they do not completely recapitulate the human physiology and pathology. In vitro two-dimensional (2D), static cell cultures grown as flat monolayers poorly reproduce in vivo tissue structures and dynamic responses due to the lack of cellular and tissue architectures under normal and diseased conditions. Organ-on-a-chip (OOC) models have emerged as alternatives to conventional 2D cell culture systems and animal models. OOC models utilize microfabrication and microfluidic technologies to create micrometer-sized cell culture platforms combined with continuous flow systems mimicking the physiologically relevant conditions and functionalities of tissues and organs.[34–37] Most OOC models with microscale fluidic channels are fabricated with the polydimethylsiloxane (PDMS) elastomer using soft lithography.[38] These OOC models enable to incorporate different cell types, including cell lines or primary cells, as well as stem and progenitor cells, in combination with precise applications of other factors found in vivo, such as ECM, cellular interactions, physiologically relevant fluid flow and physical forces including biomechanical stresses and strains, as well as electrical stimulation, to achieve intended changes in local physiological conditions.
Although several unique in vitro OOC platforms, including those of the liver, heart, kidney, and lung, etc., have demonstrated different levels of human physiological functions and responses to drug exposures,[39,40] they still do not reflect the full complexity of human physiology, such as the immune responsiveness. In addition, while there is a limited amount of in vitro models of tissues that are an integral part of the human immune system such as the spleen, lymph node, and thymus, in vitro models for inflammation processes as well as autoimmune diseases are still rare. In this Progress Report, we discuss immune OOC models of tissues that are an integral part of the human immune system and other relevant immunocompetent OOC models that have incorporated immune functions such as inflammation, as well as outline the challenges to recreate fully functional tissue system in vitro.
2. Immune organ-on-a-chip
The organs of the immune system are categorized into primary and secondary lymphoid organs. Primary lymphoid organs include the bone marrow and the thymus, where B and T lymphocytes are generated, whereas secondary lymphoid organs include the lymph nodes, spleen, and mucosa-associated lymphoid tissues including Peyer’s patches, where immune cells are activated and propagated for antigen-specific immunity.[41] Each type of the lymphoid organs is characterized by diversified cell types, dynamic ECMs, and constant migration of immune cells. Recent advances towards engineering immune tissues-on-chips have improved the relevance of in vitro models and allowed better understanding of the immune system, including development of immune cells as well as their recruitment, selection, and activation.[42] These in vitro immune OOC models can also provide the platforms for the development of immunotherapies against infections, cancers, and autoimmunity, among others.[43] However, the structural complexity of the immune tissues as well as spatiotemporally dynamic cell-cell and cell-ECM interactions complicate and thus limit their successful engineering in vitro.[44]
Nevertheless, continuous efforts have been made in the development of microfluidic immune tissue-on-a-chip models. An inflammatory microfluidic chip was developed to mimic the binding of activated Jurkat T cells to cell adhesion molecules expressed by vascular endothelial cells.[45] Different ratios of intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin were immobilized on the surfaces of the microfluidic channels and allowed for binding of migrating Jurkat T cells. In addition, immunosuppressive drugs, tacrolimus and cyclosporine A, were applied that inhibited T cell interactions under the physiological conditions (Figure 2A). Another study reported a microfluidic platform that replicated the migration of neutrophils and monocytes to an inflammation site that allowed the real-time measurement of leukocyte chemotaxis in the presence of soluble mediators.[46] In addition, it was found that lipoxin A4 and resolvin D1 inhibited the trafficking of neutrophils and monocytes towards leukotriene B4 as well as reduction of elastase release by neutrophils and monocytes in the presence of Lipoxin A4. Similarly, migration of T-cells and dendritic cells (DCs) from one chamber to another having bacteria was monitored in a microfluidic infection-on-a-chip device to study their immune responses towards infections.[47] Migration of T-cells (MF2.2D9 cells) or DCs (isolated from bone marrow of C57BL/6 mice) loaded at the migratory compartment towards the chemoattractant, chemokine (C-C motif) ligand (CCL)19-loaded activator channel was demonstrated. Furthermore, a microfluidic device was used to study the chemotactic behavior of DC cells in the presence of gradients of CCL21 and CCL19 and showed that the immobilized CCL21 and CCL19 gradients played an important role in DC chemotaxis.[48] Likewise, a microfluidic device was designed for trapping and pairing of lymphocytes, using a passive hydrodynamics-based cell-loading protocol.[49] This study demonstrated the activation of CD8+ T cells with different T-cell receptor (TCR) affinities when paired with APCs (CD40-activated B cells). It also suggested that the differences in TCR affinities influenced the Ca2+ mobilization and the production of cytokines by CD8+ T cells. The secretions of interferon (IFN)-γ by T cells was similar for both low- and high-TCR affinities, whereas the interleukin (IL)-2 production was significantly higher for the high-TCR affinity (Figure 2B). Each of these microfluidic immune OOCs used one or few components or cells of the immune system has provided limited information on the immune system. Thus, better microfluidic chip platforms that emulate the complex microenvironments and components of immune tissues are required in the future.
Figure 2. Immune OOC models.
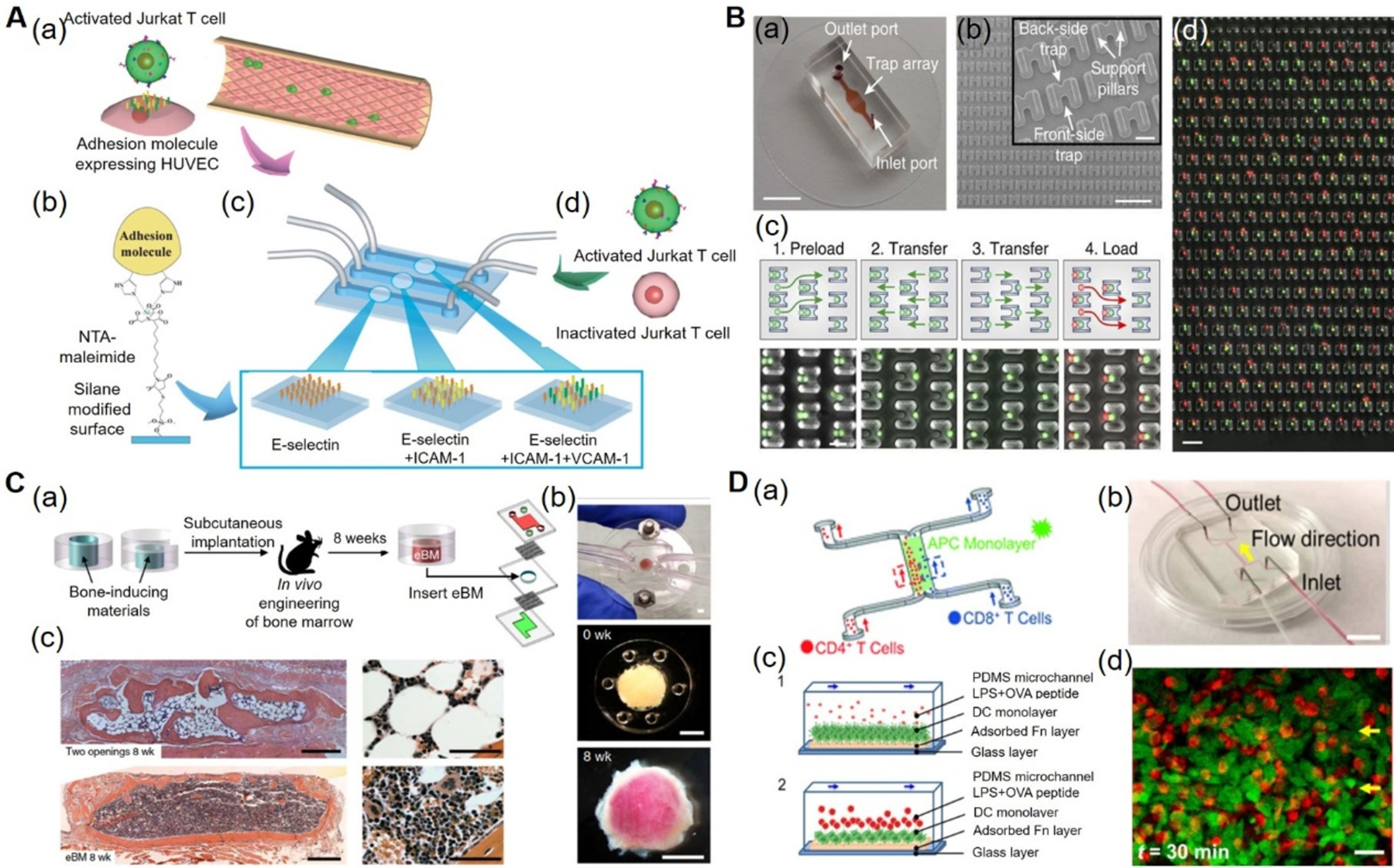
A) Inflammatory-mimetic microfluidic chip. (a) Schematic of the Inflammatory mimetic microfluidic chip. Activated Jurkat T-cells and HUVECs. (b) Adhesion molecules such as E-selectin, ICAM-1, and VCAM-1 were immobilized in the fluidic channel by treating with MPTMS and NTA-maleimide. (c) Schematics of vascular endothelia formed with adhesion molecule-expressing HUVECs. (d) Infusion of activated or drug-treated Jurkat T-cells into the channels to study the T-cell interactions. Reproduced with permission.[45] Copyright 2014, Royal Society of Chemistry. B) Microfluidic device for immune cell pairing. (a) Photograph of microfluidic cell-pairing device. (b) Scanning electron micrographs of the cell trap. (c) Cell-loading and pairing protocol. (d) Phase-contrast and fluorescence images showing pairing of primary mouse lymphocytes (red) and DCs (green) in the traps. Reproduced under the Creative Commons Attribution 4.0.[49] International Public License for open access articles. C) Bone marrow-on-a-chip. (a) Schematics of a bone marrow-on-a-chip in which eBM within a PDMS device formed in vivo was later explanted and cultured in a microfluidic system. (b) Bone marrow-on-a-chip microdevice used to culture the eBM in vitro (top), the PDMS device containing the matrix and bone-inducing factors before implantation (middle), and the white cylindrical bone with pink marrow visible within the eBM explanted after 8 weeks (bottom). (c) Hematoxylin and eosin staining of the eBM formed in the PDMS device with two openings (top) and one opening (bottom) at 8 weeks of implantation. Reproduced with permission.[63] Copyright 2014, Springer Nature. D) Immune cell-cell interaction-on-a-chip. (a) Schematics of the immune cell-cell interaction-on-chip. (b) The PDMS chip consisting of one main flow channel for cells with two inlets and two outlets. (c) Schematics showing the dynamic interactions between DCs and T-cells, in the presence of LPS and OVA peptides (1) and loading of T-cells and their interactions with DCs (2). (d) Confocal images showing the T-cell and DC interactions within 10 mins and 30 mins. Reproduced with permission.[78] Copyright 2016, Royal Society of Chemistry. DCs: dendritic cells, eBM: engineered bone marrow, HUVECs: human umbilical vein endothelial cells, ICAM-1: intercellular adhesion molecule-1, LPS: lipopolysaccharides, MPTMS: (3-mercaptopropyl)trimethoxysilane, NTA-maleimide: nitrilotriacetic acid-maleimide, OVA: ovalbumin, PDMS: polydimethylsiloxane, OOC: organ-on-a-chip, VCAM-1: vascular cell adhesion molecule-1.
2.1. Bone marrow-on-a-chip
Bone marrow represents the origin of immune cells and is a complex environment where mesenchymal stem cells, immune cells, osteoblasts, osteoclasts, adipose tissues, and interactions among each other.[50] In the bone marrow, immune cells originated from the hematopoietic lineage are present near the bone surfaces, forming stem cell niches and microenvironments.[51,52] The endothelial cells and pericytes of the vasculature found within the bone marrow form sinusoidal, leaky vessels, enabling the migration of cells between the bone marrow and the circulatory system.[53] Similarly, adipocytes present in the bone marrow are important players in health and in disease progression.[54] Moreover, during bone cancer progression or metastases to the bone from other parts of the body via blood vessels, the cancer cells receive abundant growth factors and obtain opportunities to interact with mesenchymal and hematopoietic progenitor cells at different stages of differentiation.[55,56] Most importantly, the bone marrow provides favorable microenvironments such as hypoxia and chemokine gradients for survival of the cancer cells during neoplastic processes, increasing the survival and colonization of metastatic cancer cells in this new environment.[57–59]
Although the different cells and their relationships in the bone marrow are understood to varying degrees, the interactions of various components of the bone marrow and the progression of various diseases including malignant neoplasms are not fully understood. Development of in vitro 3D tissue models of healthy as well as primary malignancies of the bone marrow provides great insights into its normal physiology and disease status, as well as paves the way towards improved therapeutics by developing of effective targeted bone marrow drugs.[60] To mimic the dynamics of the bone marrow microenvironment and to study the interactions between osteoblasts and cells in the bone marrow, a 3D microfluidic ossified tissue culture system was developed. It comprised of an eight‐chambered microfluidic culture device seeded with a human fetal osteoblast cell line (hFOB 1.19) and mononuclear cells from the bone marrows of multiple myeloma patients.[61] This platform maintained essential components of the bone marrow microenvironment within a perfused dynamic environment, including the endosteal surface and its ECM, stromal cells, growth factors, and cytokines from the patients’ plasma. It was demonstrated that the 3D microfluidic ossified tissue platform enabled the propagation of primary human multiple myeloma cells in vitro by culturing mononuclear cells isolated from the patient bone marrow biopsies.
In another study, a bone marrow‐on‐a‐chip was prepared using a hydroxyapatite-coated zirconium oxide scaffold comprised of human mesenchymal stromal cells and cord blood-derived multipotent hematopoietic stem and progenitor cells (HSPCs).[62] This device enabled HSPCs culture for 28 days and HSPCs were found to remain in their primitive state (CD34+CD38− ). In addition, these HSPCs had the potential of differentiating into their various progenies (granulocytes, erythrocytes, macrophages, megakaryocytes). Another bone marrow-on-a-chip was developed to study toxicity of radiation exposure.[63] A PDMS chip with a central cylindrical channel (1-mm high × 4-mm diameter) having open ends was microfabricated. The cavity was filled with a collagen type I hydrogel supplemented with the osteogenic demineralized bone powder and bone morphogenetic proteins (BMP2 and BMP4), which was then implanted subcutaneously in the mouse. The implant was harvested after 8 weeks and cultured in vitro in a similarly shaped microfluidic culture system (Figure 2C). Histological results showed that the explant was composed mostly of adipocytes and a low level of hematopoietic cells. Thus, to reduce the adipocytes in the implants, the PDMS device was modified by sealing the top end of the central cavity, followed by the subcutaneous implantation. The modified implant generated a cylindrical engineered bone marrow consisting of a hematopoietic cell composition similar to that of the native bone marrow. This engineered bone marrow was used to study the cell behaviors under γ-radiation exposure. It was found that the proportion of hematopoietic stem cells, hematopoietic progenitors, lymphoid cells, and myeloid cells were significantly reduced, depending on the dose of radiation.
Another bone marrow-on-a-chip model was developed to study the effect of a chemotherapeutic drug, cytarabine, on acute lymphoblastic leukemia.[64] The PDMS microfluidic platform consisted of four microfluidic channels, with one inlet and one outlet. Human bone marrow stromal cells (BMSCs), leukemic cells (SUP-B15), and osteoblasts were encapsulated in a collagen hydrogel and seeded in the channels. The cells in the microfluidic chip were treated with cytarabine and compared for its effect in 2D and 3D cultures under static and dynamic conditions. Under both dynamic and static conditions, coordinated cell-cell interactions among the three cell types were observed in 3D cultures while in the 2D static cultures, interactions among the cells were absent. Further, it was shown that leukemic cells were less sensitive towards the cytarabine in 3D cultures as compared to the 2D cultures. These results demonstrated the importance of the surrounding microenvironment in controlling cellular behaviors.
Recently, a vascularized bone-marrow-on-a-chip was reported that comprised of a top hematopoietic channel filled with the BMSCs and CD34+ cells-encapsulated fibrin gel, a bottom vascular channel lined with human umbilical vein endothelial cells (HUVECs) , and a porous PDMS membrane that separated the top and bottom channels.[65] The medium, supplemented with progenitor cell-supporting cytokines, including thrombopoietin, stem cell factor, and Fms-related tyrosine kinase 3 (Flt3) ligand, as well as granulocyte colony stimulating factor (G-CSF) and erythropoietin that induce myeloerythroid proliferation and differentiation, was perfused through vascular channel. Within 2 weeks of the culture period, the cell microenvironment consisting of multiple hematopoietic lineages was formed, similar to that found in the native bone marrow. Flow cytometric analysis suggested that the cells within the bone marrow chip were mostly of neutrophil and erythroid lineages. The bone marrow hematopoiesis was further assessed by labeling cells with 5-ethynyl-2’-deoxyuridine (EdU). It was found that the EdU+ cells within the neutrophil lineage were mostly an immature CD16lo population, while the EdU+ cells within the erythroid lineage were largely an immature CD71+CD235− phenotype. The hematotoxicity of 5-fluorouracil was predicted by infusing the drug through the vascular channel for 2 days and compared that with treated static suspension and gel cultures. Moreover, this bone marrow chip was used to reproduce a hematopoietic disease, the Shwachman-Diamond syndrome (SDS), by culturing CD34+ cells from SDS patients with normal BMSCs and endothelial cells. The bone marrow chip was found to display hematopoietic as well as cell-specific defects in both the neutrophil and erythroid lineages. This vascularized bone marrow chip represented a clinically relevant human hematopoiesis model that recapitulated important features of the bone marrow pathophysiology. Bone marrow abnormalities commonly occur due to drug toxicity, radiation exposure, as well as various immune and genetic disorders. The current in vitro bone marrow-on-chip models recapitulated some aspects of the bone marrow, related diseases, and drug toxicities and perhaps, improved bone marrow models with complex structural and functional properties would expand their applications towards understanding the bone marrow and the development of drugs.
2.2. Lymph node-on-a-chip
Lymph nodes are small bean-shaped glands distributed throughout the body and are connected by the lymphatic vessels.[66] The lymph nodes are critical for protective responses to pathogens and a range of other harmful stimuli from damaged or dying cells as well as abnormally behaving cells such as those infected and cancerous, for maintaining matured immune cells, and for initiating the antigen-specific adaptive immune responses.[67–69] Structurally, the lymph node consists of the outer cortex, the middle paracortex, and the inner medulla. Clusters of B cells mostly reside in the cortex region whereas T cells and DCs are present in the paracortex region. The medulla region possesses blood vessels, medullary sinuses (lymphatic channels), and medullary cords (aggregates of lymphoid tissues containing plasma cells, macrophages, and B cells).[70] The APCs recognize pathogens, abnormal cells, and foreign particles, process them and then present antigens to naive T cells via major histocompatibility complex molecules, together with co-stimulatory molecules and cytokines.[13,71,72] Once APCs present antigens to T cells, these T cells proliferate and differentiate into either helper T cells (CD4+ T cells) or cytotoxic T cells (CD8+ T cells), and memory cells. On the other hand, B cells present antigens to T-helper cells, around the area between the follicle and paracortex. After receiving signals from helper T cells, B cells proliferate and generate antigen-specific antibodies.[73]
The lymph nodes along with other components of the lymphatic system are involved in fluid homeostasis, immune cell trafficking, as well as initiation and modulation of the immune responses.[13] Structural and functional abnormalities of the components of the lymphatic system can contribute to various diseases, including immune disorders, cancer, and lymphedema, among others.[74–77] Despite advances towards understanding the structures and functions of the lymphatic system, promising tools and technologies are needed to study its role in pathophysiology of various diseases as well as to develop immunotherapeutics. To this end, a microfluidic chip device of the paracortical region of the lymph node was developed to investigate the dynamic interactions between DCs and T cells under shear stress.[78] The device consisted of one main channel with two inlets and two outlets, in which DCs were maintained while CD8+ T cells and CD4+ T cells were introduced into the channel by continuous flows (Figure 2D). The DCs were first activated with lipopolysaccharide (LPS) and then incubated with the ovalbumin (OVA) peptides, OVAII or OVAI, for presenting antigens to CD4+ or CD8+ T cells, respectively. It was found that the attachment of antigen-specific CD8+ T cells and CD4+ T cells on DCs depended on the amount of shear stresses. Antigen-specific T cells were found to attach to DCs at low shear stresses (0.01–0.15 dyne cm−2), which were released at a higher stress (12 dyne cm−2).
Similarly, to mimic the migration of DCs and antigen presentation to T cells in the lymph node, a microfabricated system was developed.[79] This device consisted of top DC and bottom T-cell compartments. The DCs were subjected to a chemokine CCL19 gradient, and migration of these DCs towards the T cell compartment was monitored. It was found that in response to the chemokine gradient, the DCs migrated and activated the T cells, as assessed by the increased calcium level in the T cells. In another study, a bioreactor was developed that consisted of two cell culture compartments separated by a double-layered oxygenating membrane.[80,81] This membrane-based 3D bioreactor mimicked the architecture of the lymph node. DCs were immobilized within macroporous agarose sheets inside the bioreactor and peripheral blood mononuclear cells (PBMCs) were perfused with culture medium containing the hepatitis A (HavrixTM) vaccine. The results showed that HavrixTM induced an early proinflammatory response that decreased rapidly within the first days of culture. However, subsequent re-stimulation of immune cells with antigen-loaded DCs or only antigens induced the production of tumor necrosis factor-alpha (TNF-α) and T-helper 2 cell-promoting cytokines (IL-6 and IL-10). The addition of dexamethasone suppressed the production of cytokines by the immune cells. In addition to the cellular immunity as monitored by cytokines released by immune cells, humoral immunity was analyzed by assessing the activation of B cells and formation of plasma cells as well as the secretion of antibodies, immunoglobulin M (IgM) and immunoglobulin G (IgG).
Interestingly, a lymph node slice-on-a-chip was designed, in which a slice of a murine lymph node was placed on a device to study the effect of local chemical stimuli on the lymph node tissue.[82] The PDMS device comprised of three layers, the top layer with a perfusion chamber for tissue slice culture, the middle layer with ten stimulus ports, and the bottom layer with ten parallel microchannels. The lymph node slices within the chips were treated with fluorescein isothiocyanate (FITC)-dextran (40 and 70 kDa) and monitored for distribution, accumulation, and removal of these molecules over time. The FITC-dextran molecules were cleared within 30–80 s from the slices, based on their molecular weights. Similarly, glucose-conjugated bovine serum albumin was tested, and it was found that their retention was higher in the B-cell zone as compared to the T-cell zone. Although the fabrication of a native lymph node in vitro is difficult, primarily due to its complex architectural and structural organization, the current lymph node-on-chip devices were able to recreate some of the functions of the lymph node such as responses to chemokine gradients, migration of immune cells, and antigen presentation, among others. Since the major functions of the lymph node is to filter foreign particles, pathogens, and cancer cells, an improved lymph node-on-chip device should reflect its ability to simulate the immunological responses with respect to foreign particles, pathogens, and cancer cells.
2.3. Lymphatic vessel-on-chip
Lymphatic vessels are thin-walled valved structures that facilitate the drainage of lymph (a collection of interstitial tissue fluid, proteins, and inflammatory cells) from the peripheral blood vessels and its return to the venous circulation, thus maintaining the fluid homeostasis.[83,84] Moreover, they are the important part of the immune system that serve as conduits for delivering antigen-loaded DCs, T cells, and neutrophils to draining lymph nodes during immune surveillance and infection, and also facilitate the absorption of lipids from the digestive tract.[13,85–87] Since lymphatic vessels are an important part of the lymphatic system that plays a vital role in immune modulation, in regulation of lipid absorption and inflammation, as well as in interstitial fluid homeostasis, understanding the detailed mechanisms of the lymphatic system is significant to fight against various infections and diseases and to develop therapeutic interventions.
To this end, a microcirculation-on-a-chip was developed that possessed both blood and lymphatic vessels.[88] The PDMS device consisted of partially overlapped top and bottom channels that were separated by a porous membrane. Normal human dermal microvascular endothelial cells (HMVEC-d) and normal human lymphatic microvascular endothelial cells (HMVEC-dLy) were cultured on opposite sides of the porous poly(ethylene terephthalate) (PET) membrane, thus forming the upper vascular endothelial layer and the lower lymphatic endothelial layer. Permeation of the high- and low-molecular weight molecules from the blood vascular channel to the lymphatic channel was assessed using tetramethylrhodamine isothiocyanate (TRITC)-dextran (formula weight (Fw) = 40,000) and Lucifer Yellow (Fw = 457) molecules, respectively. During the 1-h permeability test, only a small amount of Lucifer Yellow was leaked through the vascular endothelial layer whereas there was no TRITC-dextran leakage, demonstrating the formation of a confluent layer of endothelium with tight junctions. The formation of cell-cell tight junctions was also as shown by claudin-5 and vascular endothelial (VE)-cadherin expressions. Furthermore, the addition of an inflammation-promoting substance, histamine, resulted in alteration of expressions and localizations of tight junction proteins, and thus increasing the permeability of in the endothelial layer.
Similarly, a microscale lymphatic vessel (μLYMPH) system was developed to study the biology of lymphatic vessels in the tumor microenvironment (TME).[89] The device was fabricated by placing a PDMS rod across a gel chamber, which was then covered with collagen type I gel. After polymerization of the gel, the PDMS rod was removed to form a hollow lumen where primary human lymphatic endothelial cells or HUVECs were seeded. The diffusion assay, using 10-kDa and 70-kDa Texas Red-dextran, showed the leaky lymphatic vessels with higher permeability (1.38×10−5 ± 0.29×10−5 cm s−1) when compared to that of blood vessels (0.68×10−5 ± 0.13×10−5 cm s−1). In the μLYMPH system, the vascular endothelial layer was found to produce a comparatively higher level of vascular endothelial growth factor-C (VEGF-C), hepatocyte growth factor (HGF), endoglin, placental growth factor, and IL-8 , whereas the lymphatic endothelial layer produced higher levels of follistatin, endothelin-1, and G-CSF. Moreover, this platform was used to study the crosstalk between lymphatic vessels and the TME by culturing breast cancer-associated fibroblasts (CAFs) in the collagen hydrogel. In the presence of CAFs, secretions of G-CSF and HGF were increased by 8-fold and 15-fold, respectively, whereas the productions of IL-6 and IL-8 were increased by 20-fold and 15-fold, respectively. On the other hand, secretions of VEGF-C, endoglin, and endothelin-1 were decreased by at least 10-fold. The barrier function of the lymphatic vessels was significantly disrupted due to the IL-6 secreted by CAFs. The function of the vessels was completely restored when the IL-6/IL-6R signaling pathway was blocked with an IL-6-neutralizing antibody, demonstrating the potential of the device for screening therapeutic targets.
Although the lymphatic vasculature has been less-studied than the blood vessels, recently it has become an attractive area of research. The above-mentioned lymphatic vessel-on-chip models are significant tools to study lymphatic vessels and their functions. Nevertheless, they can be further modified to simulate structurally and physiologically similar lymphatic vessels by incorporating more relevant cell types and biological components. Considering the immune relevance, lymphatic vessel-on-a-chip platforms should facilitate the study of the migration of immune cells and transport of antigens through the lymphatic system during inflammation or other diseased conditions as well as their impact on the immune system.
2.4. Spleen-on-a-chip
Spleen is the largest secondary lymphoid organ and is an important part of the immune system.[90] Spleen serves as the main site for filtration of bloodborne pathogens, metabolism of iron, and homeostasis of erythrocytes.[91,92] On the basis of structures and functions, it is divided into the red pulp zone and the white pulp zone, separated by an interface termed the perifollicular zone.[90] The red pulp consists of macrophages that are specialized for phagocytosis of pathogens as well as damaged or aged red blood cells (RBCs) and also plays an important role in iron recycling from aging RBCs. The perifollicular zone contains both macrophages and DCs. Macrophages engulf pathogens whereas DCs recognize foreign antigens and present them to the lymphocytes in the white pulp zone. The white pulp is the main immunological site of the spleen, which is similar to a lymph node in structure and function. It is comprised of T-cell and B-cell regions, responsible for antigen-specific immunity.[93]
A microfluidic splenon-on-a-chip was developed to mimic the hydrodynamic properties of the splenon, the basic functional unit of the spleen and its filtering functions.[94] The chip consisted of a fast-flow channel, a slow-flow channel, and microconstrictions. The closed fast-flow channel represented the microcirculation in the spleen where blood flows directly to the splenic venules bypassing the spleen’s filtration process, while the open slow-flow channel having a pillar matrix represented the microcirculation where blood passes through the filtration beds of the red pulp that facilitate the phagocytosis of unhealthy RBCs by macrophages. The microconstrictions at the end of the slow-flow channels represented the inter-endothelial slits (IES). Results demonstrated that aging RBCs had less deformability than fresh RBCs. In addition, the filtration ability of device was evaluated using healthy and malaria-infected RBCs. Infected reticulocytes (immature red blood cells) were observed to be more deformable as compared to noninfected reticulocytes when peripheral blood of mice infected with the Plasmodium yoelii 17X-GFP strain was passed through the IES.
In another study, an extracorporeal microfluidic device having two adjacent fluidic channels was reported.[95] One channel represented the vascular channel with blood flow and the other represented spleen’s venous sinusoid with saline under slow flow. The blood and the sinusoid channels were interconnected through a series of open rectangular slits. This device was used to filter magnetically opsonized pathogens and endotoxins from the blood of patients with sepsis. Mannose-binding lectin (MBL) was genetically engineered, in which carbohydrate recognition domains were retained whereas collagen-helix regions were removed. The carbohydrate recognition site and MBL were conjugated with IgG1 Fc, followed by biotinylation of Fc-containing MBL (FcMBL) by the conjugation of alanine-lysine-threonine sequence. Magnetic nanobeads were added to the contaminated blood, which was then flown through a spiral mixer and incubation loop to enhance the binding of nanobeads with pathogens before entering the vascular channel and the isotonic saline was perfused through the sinusoid channel. It was observed that, the system was able to remove ~90% of live Staphylococcus aureus and Escherichia coli (E. coli) from the contaminated blood within 1 h. Further, the engineered magnetic nanobeads exhibited significant binding to Candida albicans, Saccharomyces cerevisiae, Klebsiella pneumoniae, and Staphylococcus epidermidis.
Despite its important roles in the immune system, iron metabolism, and erythrocyte homeostasis, very few attempts have been made to engineer the spleen in vitro. Considering its critical role in filtration and to better understand it functions in immunology, it is essential to recreate the in vitro spleen models with high structural and physicochemical precisions.
3. Other relevant immunocompetent OOC models
3.1. Skin-on-a-chip
The skin, the largest organ of the body, represents an active immune organ and the first line of defense to protect against pathogens.[96] The skin is comprised of three major layers, the epidermis, the dermis, and the hypodermis.[97] The major immune-like cells present in the epidermis layer are keratinocytes that express Toll-like receptors for detecting pathogens and specialized DCs, known as the Langerhans cells.[98,99] The immune cells present in the dermis layer are DCs, T cells, and mast cells.[98] Upon recognition of invading pathogens, keratinocytes in the epidermal layer secrete antimicrobial peptides, proinflammatory cytokines, and chemokines that activate the immune cells in the dermal layer, initiating an immune response.[100,101]
Several skin-on-a-chip models have been established. A simplistic skin-on-a-chip model was developed, which simulated an epidermal layer of the skin.[102] Neonatal human epidermal keratinocytes (HEK cells) were patterned on collagen patches within microfluidic channels and it was found that the HEK cells maintained 93–99% viability under continuous perfusion of medium for 72 h. In another study, a skin-on-a-chip model was fabricated that possessed the outer epidermal and inner dermal equivalents.[103] The microfluidic device consisted of four poly(methyl methacrylate) (PMMA) layers, two microporous track-etched polycarbonate membranes placed between second and third PMMA layers, and a circular polytetrafluoroethylene (PTFE) membranes placed between the third and fourth PMMA layers. The human primary foreskin-derived dermal fibroblasts encapsulated in a fibrin-based matrix were used to fabricate the dermal layer, whereas N/TERT-1 keratinocytes were cultured on the top of the dermal layer to form the epidermal layer (Figure 3A). The medium was perfused through the top epidermal layer and below the porous membrane to maintain air-liquid and liquid-liquid interfaces, respectively. The study demonstrated that differentiation and morphogenesis of the epidermal layer were improved under continuous perfusion and controlled microenvironment. The barrier function was also enhanced, as shown by immunohistochemical analysis of different markers and trans-epidermal electrical resistance (TEER) measurements using Ag/AgCl electrodes, as well as permeability test conducted with caffeine.
Figure 3. Immunocompetent OOC models.
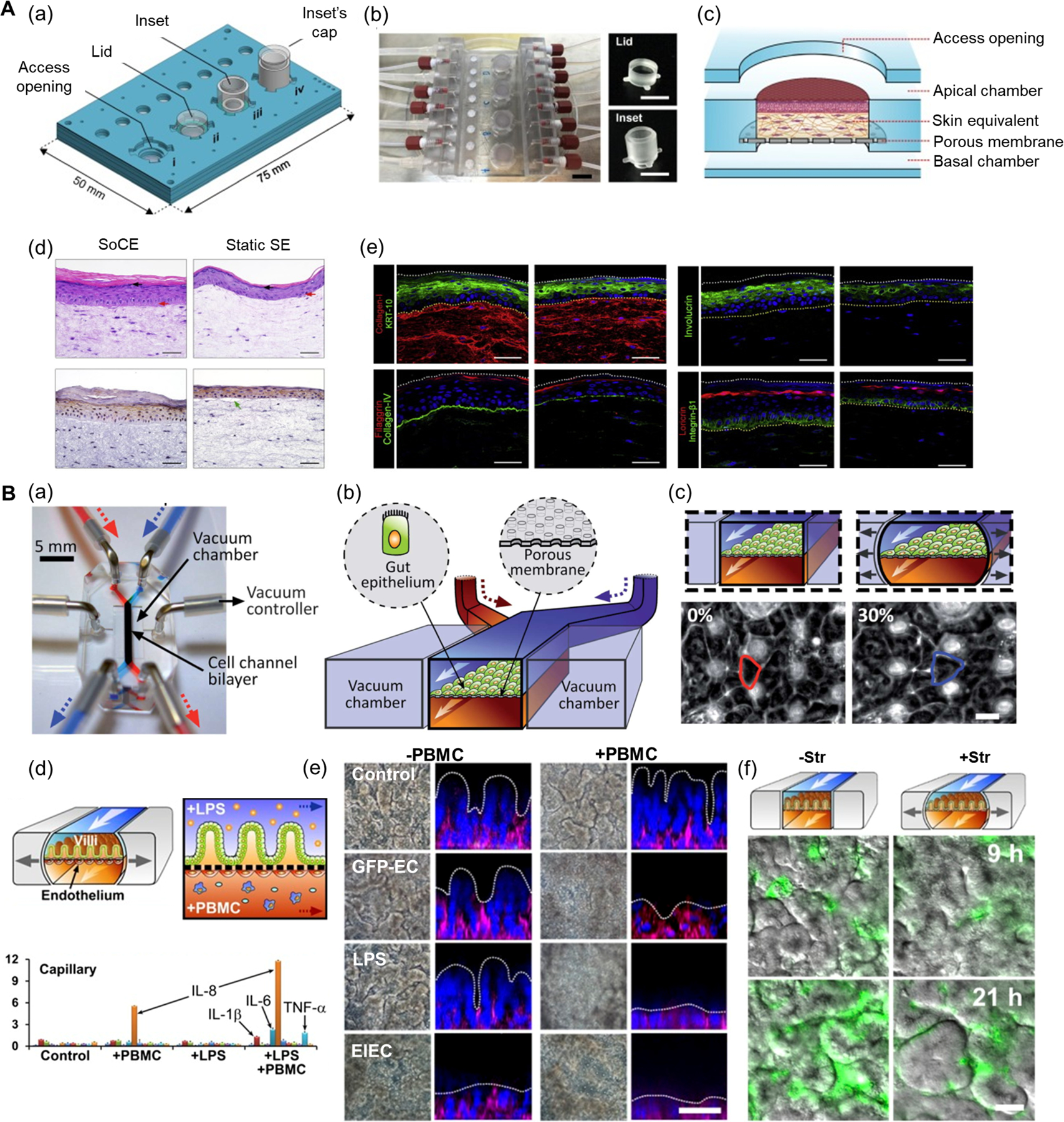
A) Skin-on-a-chip model. (a) Schematics of the four-chamber skin-on-a-chip device with each unit having different operational configurations. (b) Photographs of the skin-on-a-chip device with interchangeable lid and inset. (c) Schematics of the skin-on-a-chip for human fibroblasts and keratinocytes coculture. (d) Histological images of the epidermal layer from skin-on-a-chip compared to skin equivalents grown in static culture inserts. (e) Fluorescent images showing the expressions of keratinocyte differentiation markers KRT-10 (green), filaggrin (red), involucrin (green), and loricrin (red); constituents of basement membrane zone collagen type IV (green), integrin-β1 (green), and dermal matrix protein collagen type I (red). Reproduced under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0.[103] International Public License for open access articles. B) Human gut-on-a-chip. (a) Photograph of the gut-on-a-chip device. (b) Schematics of the gut-on-a-chip device with epithelial cell layer grown on the porous membrane and vacuum chambers on the sides. (c) Schematics (top) and phase-contrast images (bottom) of the intestinal monolayer within the gut-on-a-chip with or without mechanical strain (30%). Reproduced with permission.[124] Copyright 2012, Royal Society of Chemistry. (d) The schematics showing the interface between the intestinal villus epithelium and vascular endothelium (top) and secretions of proinflammatory cytokines after costimulation with LPS and PBMCs (bottom). (e) Phase-contrast images and fluorescence confocal micrographs of intestinal villus damage in response to GFP-EC, LPS, and EIEC in the absence or the presence of PBMCs. (f) Overlaid fluorescence and DIC microscopic views showing growth of GFP-EC on the intestinal villi in the absence (-Str) or presence (+Str) of cyclic stretching motions (10% in cell strain, 0.15 Hz in frequency) for 9 h and 21 h. Reproduced under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0.[125] International Public License for open access articles. DIC: differential interference contrast, EIEC: enteroinvasive Escherichia coli, GFP-EC: GFP-labeled Escherichia coli, LPS: lipopolysaccharides, Str: cyclic stretching motion.
Similarly, vascularized skin-on-a-chip models have been developed. For examples, a perfusable skin-on-a-chip device was reported that possessed an epidermal layer, a dermal layer, and a vascular channel.[104] The normal human dermal fibroblasts-loaded collagen was filled in the chamber across which nylon wires were strung. The hollow channels were formed by removing these nylon wires, which were then were seeded with HUVECs. Next, normal human epidermal keratinocytes seeded into a square-shaped silicone rubber barrier were placed over the dermal layer and cultured under continuous perfusion. Hematoxylin and eosin staining, and immunostaining of the skin equivalent showed the expressions of type IV collagen, cytokeratin (CK)10, and CK15 in the epidermal layer. Similarly, immunostaining of cluster of differentiation 31 (CD31) and zonula occludens-1 (ZO-1) revealed the formation of endothelial layers in the channels. Moreover, permeability of the skin tissue model was tested by diffusing isosorbide dinitrate and caffeine in medium through the skin tissue and it was found that the amount of isosorbide dinitrate in the diffused medium was 3–4-fold higher as compared to caffeine.
Likewise, a vascularized skin-on-a-chip device was constructed to recreate skin inflammation and edema.[105] The chip consisted of three PDMS layers and the porous PET membranes placed in between the PDMS layers. The adjacent PET membrane and PDMS layers were bonded by the 3-aminopropyltriethoxysilane-based bonding method. The porous PET membranes allowed the diffusion of nutrients, cytokines, and drugs from one layer to another of the device. The human keratinocyte cells (HaCaTs) and HS27 fibroblasts were first seeded onto the upper and the middle layer, respectively. After overnight incubation, the chip was turned over, and then HS27 fibroblasts were seeded onto the middle layer whereas HUVECs on the bottom layer. The HaCaTs and HS27 fibroblasts formed the epidermal and dermal layers of the skin, respectively, while HUVECs formed the vascular layer. TNF-α-induced skin inflammation was further analyzed by measuring the expression of proinflammatory cytokines, IL-1β, IL-6, and IL-8. Inhibition of TNF-α-induced inflammation was studied by treating the damaged skin with dextran. When dextran was administrated to the inflamed skin, the expressions of IL-1β, IL-6, and IL-8 were decreased, depending on the dose of dextran used.
The skin-on-a-chip model that has incorporated immune cells other than keratinocytes was developed by Ramadan and Ting.[106] The device consisted of two chambers whereby HaCaT cells were seeded in the upper chamber forming the epidermis layer and U937 human leukemic monocyte lymphoma cells were seeded after 1 week to form a layer of DCs. These layers were separated by a porous PET membrane that facilitated intercommunications between the HaCaT cells and the DCs. The continuous perfusion of culture medium significantly improved the formation of tight junctions as evidenced by TEER measurement. To investigate the response of HaCaT cells to other external stimuli, the HaCaT cell layer was treated with LPS, neutral substances such as cobalt sulfate, and glycerol, as well as skin allergens such as nickel sulfate and 2, 4-dinitrochlorobenzene (DNCB). It was observed that TEER values significantly decreased when incubated with nickel sulfate and DNCB, whereas no significant change was seen with LPS, cobalt sulfate, or glycerol, suggesting that nickel sulfate and DNCB disturbed the tight HaCaT cell layer. Further, when the HaCaT cell layer was exposed to a continuous UV irradiation for 24 h, TEER value was decreased. In addition, significant increases in IL-6 and IL-1β levels were observed in the U937 cell monoculture as compared to those of the HaCaT monoculture. Similarly, the expressions of cytokines were found to increase in HaCaT/U937 coculture when treated with LPS, suggesting the prevention of LPS diffusion through the keratinocyte barrier formed in the model.
The skin is a complex and dynamic organ that functions as physical, chemical, and microbiological barriers to external stimuli and pathogens. In addition, the skin contains various types of immune cells that are crucial for host defense and tissue homeostasis.[107] Even though the current skin-on-a-chip models incorporate some of the structural and functional properties of the skin, most of them are still very simple and lack essential cellular and structural components of the skin such as the outermost stratum corneum, vasculature, skin appendages including sweat glands and hair follicles, and importantly, the interplay between the key immune and immune-like cells of the epidermal and dermal layers. These in vitro systems do not yet emulate the complexity of the native skin tissue.
3.2. Gut-on-a-chip
The gastrointestinal (GI) tract forms an important part of the immune system. Since the exogenous microorganisms including bacteria, viruses, and fungi, can enter the gut along with the food taken, the inner lining of the GI tract constantly encounters foreign pathogens. Besides, the GI tract is densely populated with symbiotic microorganisms, forming a symbiotic microbial community termed the gut microbiota.[108–110] The mucosal immune system of the GI tract includes the intestinal epithelial barrier, the lamina propria, and the gut-associated lymphoid tissue (GALT).[111] Further, Peyer’s patches, mesenteric lymph nodes, and isolated lymphoid follicles form the GALT and represent the lymphatic organ of the GI tract.[111] Although the primary function of the intestinal epithelium is the absorption of nutrients, it contains specialized lineages of cells, including microfold cells (M cells), enterocytes, goblet cells, Paneth cells, and endocrine cells, that play important roles in the GI tract immunity.[112,113] The lamina propria, which is located below the epithelium, possess most of the immune cells in the intestine, and thus is key for preventing invasion by pathogens.[114,115] The Peyer’s patches are found throughout the ileum and are the major inductive sites for mucosal immune responses. Each Peyer’s patch contains a subepithelial dome (SED) region just beneath the follicle-associated epithelium (FAE) and are characterized by the presence of M cells. These consist of large B-cell areas and smaller T-cell regions within a subepithelial dome.[116] In addition to B cells and T cells, DCs and macrophages are also present in the Peyer’s patches.[117] The M-cells are specialized intestinal epithelial cells present in the FAE that transport antigens and microbes to DCs in the GALT located within the SED. Besides, studies have shown that the gut microbiota also regulate the mucosal immune system directly or by producing metabolites such as short-chain fatty acids and adenosine triphosphate.[118,119] The gut microenvironment, including the gut microbial population and their metabolites, gets altered by diet, drugs, stress, and infections.[120–123] As such, the mucosal immunity must constantly adapt in response to the changes in the gut environment. Intestinal epithelial cells become stimulated by such alterations and interact with the immune cells present in the lamina propria and GALT to modulate mucosal immune responses. Therefore, the GI tract represents a critical part of the immune system that needs to be studied in detail to better understand the complex functioning of the gut microbiota and various aspects of the gut mucosal immunity.
A human gut-on-a-chip was developed to study the effect of peristalsis and shear stress on the epithelial layer of an intestine.[124] The device consisted of two channels separated by a PDMS porous membrane coated with the hydrogel mix of collagen type I and Matrigel. Caco-2 human epithelial colorectal adenocarcinoma cells were seeded on the top surface of the hydrogel-coated membrane and cultured under perfusion of medium through both microchannels to recreate the shear stress due to fluid flow in the intestinal lumen (Figure 3B a–c). Mechanical deformation of the epithelial cell layer was generated by applying cyclic suction to the vacuum chambers present on either side of the microfluidic channel, causing regular stretching and relaxation of the hydrogel-coated porous membrane, thus recreating the peristaltic motions of the human intestine. It was observed that planar columnar epithelium layer of Caco-2 cells, under continuous medium flow and cyclic strains for longer times, spontaneously formed undulations and folds, similar to the native morphology of the normal intestinal villi. In addition, the Caco-2 cell layer, with or without strain, exhibited 3- to 4-fold higher peak TEER levels as compared to that in static transwell culture. Furthermore, the ability of the intestinal epithelial cell layer to sustain bacterial growth was assessed by co-culturing Lactobacillus rhamnosus GG (L. hamnosus GG) on the surface of the Caco-2 cell layer for 7 days under continuous flows and cyclic strains, and it was found that the monolayer remained intact with also improved barrier function.
The gut-on-a-chip device with a similar setup was used to recreate a disease model of small intestinal bacterial overgrowth and inflammation.[125] Eight different probiotics, and a pathogenic E. coli were cocultured with the Caco-2 epithelial cell layer and the contributions of several factors (inflammatory cytokines, LPS, PBMCs, and mechanical forces) were analyzed, either individually or in combination, on bacterial overgrowth, inflammation, and subsequent villus injury in the intestine (Figure 3B d–f). The study revealed that the simultaneous administration of PBMCs and LPS induced the secretion of proinflammatory cytokines by epithelial cells such as IL-8, IL-6, IL-1β, and TNF-α that acted together to damage the intestinal barrier and induce villus injury. In addition, gene microarray analysis of the mechanically active gut-on-a-chip in the presence or absence of living commensal microbes suggested distinct gene expression profiles compared to those of the cells cultured in a static transwell format, and the phenotype of Caco2 cells was changed significantly when commensal gut microbes were co-cultured for 72 h.
Likewise, a microfluidic human-microbial crosstalk-on-a-chip model was developed consisting of three co-laminar channels, a perfusion chamber, a human cell chamber, and a microbial cell chamber.[126] The polycarbonate porous membrane was sandwiched between the human cell and microbial cell chambers. Furthermore, the microfluidic chip was integrated with oxygen sensors that allowed real-time monitoring of oxygen dissolved in the perfusion and microbial chambers. The Caco-2 cells were seeded on the collagen-coated microporous membrane and formation of the epithelial monolayer was evaluated by TEER measurement and immunostaining of the tight junction protein, occludin. To demonstrate the ability of the device to maintain the culture of anaerobic microbes, commensal L. rhamnosus GG was cultured under anaerobic conditions. The growth of the bacterial strain was supported while retaining the tight junctions of the Caco2 cell monolayer. Moreover, non-cancerous colonic cells (CCD-18Co) were cultured in the human microchamber and primary CD4+ T cells in the perfusion microchamber in the absence or presence of L. rhamnosus GG over 48 h, for which no marked difference in CCD-18Co cell viability was observed.
Alterations in the composition of normal gut microbes are closely associated with multiple diseases and most of the current gut-on-a-chip models have focused on the host-microbe interactions. These in vitro microfluidic platforms have allowed the co-culture of human intestinal cells and microbial cells under controlled environments and would facilitate the investigations of host-microbiome interactions to better-understand the relationship between the gut microbiome and the development of the diseases. Nevertheless, gut-on-a-chip models should also be explored to incorporate the mucosal immune system for better recapitulation of the gut-microbe interactions, and to investigate the relationship of the mucosal immune system with other organ systems of the body for applications such as drug screening and delivery as well as pharmacokinetic studies of therapeutics.
3.3. Tumor-on-a-chip
Tumor tissues are composed of various cell types including cancer cells, stromal cells such as CAFs,[127] immune cells, vasculature, lymphatic vessels, and ECM components such as collagen responses.[128,129] In addition, the tumor stromal tissues represent the dynamic source of a variety of cytokines and growth factors that influence tumor progression and drug.[130] The interactions between the cancer cells and the stroma affect different aspects of tumor behaviors such as tumorigenesis, tumor invasion, metastasis, angiogenesis, and resistance to therapeutic agents.[127,129] Biomolecules produced by cancerous or stromal cells mediate the dynamics of solid tumors. For example, E-cadherin acts as a suppressor of cancer invasiveness.[131] Furthermore, the TME contains cellular components of both innate and adaptive immune systems and plays crucial roles in the development of a tumor (Figure 4A).[132] For example, M2 macrophages accelerate tumor progression.[133–135] Tumor-on-a-chip systems represent promising in vitro platforms capable of recapitulating important biochemical, physiological, and mechanical aspects of the TME, but typically with much lower variability compared to the native in vivo tumors.[136–138] Nevertheless, these tumor-on-a-chip systems facilitate a better understanding of cancer pathophysiology leading to improved preclinical assessments of anticancer drug efficacies.
Figure 4. Immune microenvironments in tumors and in vitro tumor-on-a-chip models.
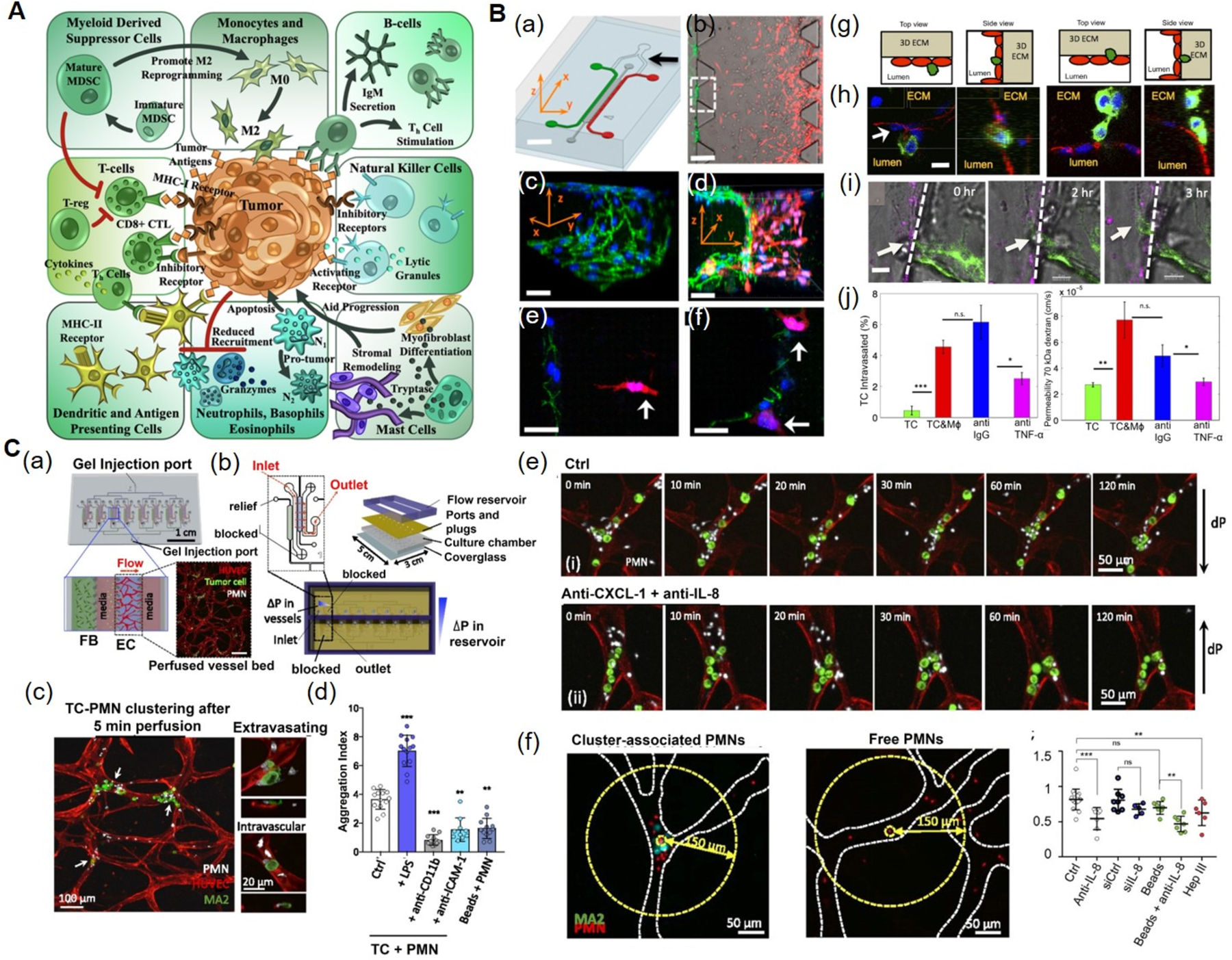
A) The TME comprises of tumor cells, as well as cellular components of both the innate and adaptive immune systems. Reproduced under the Creative Commons Attribution License 4.0.[132] International License for open access articles. B) Microfluidic tumor-vascular interface model. (a) Endothelial channel (green), tumor channel (red), and 3D ECM (dark gray). b) Phase-contrast image showing the invasion of fibrosarcoma cells (HT1080, red) through the ECM (gray) towards the endothelium (MVECs, green). (c) VE-cadherin and nuclei staining showing the endothelial monolayer. (d) 3D rendering of a confocal image showing the invasion of tumor cells and adhesion to the endothelium. (e) Invasion of HT1080 cells (white arrow) towards the endothelium. (f) HT1080 cells attached to the endothelial monolayer. (g) Schematics showing the endothelial layer, the tumor cells, and the 3D ECM within the device. (h) Confocal images showing intravasation of a MDA-MB-231 cell (green) across the MVEC layer (stained red for VE-cadherin). (i) Time lapse confocal images showing the intravasaton of a MDA-MB-231 cell (white arrow) across a HUVEC monolayer (magenta) in the presence of RAW264.7. (j) Intravasation of MDA-MB-231 cells and endothelial permeability of 70 kDa dextrans across a HUVEC monolayer in the presence of macrophages. Blocking of macrophage-secreted TNF-α resulted in a significant reduction in both intravasation and permeability compared to the IgG antibody control. Reproduced under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0.[139] International Public License for open access articles. C) Multiplexed microvasculature device for dynamic inflamed neutrophil and the tumor cell interactions. (a) Schematics of device consisting of eight independent hydrogel regions where microvascular networks are formed. Inset depicts a confocal image of one perfused vascular network. (b) Schematics and photographs showing different components of the device. (c) PMN and TC clusters in microvessels, highlighting extravasating and nonextravasated A375-MA2 cells in TC-PMN clusters. (d) Degree of TC-PMN aggregate formation during intravascular arrest. (e) The dispersion of cluster-associated PMNs from TCs over 2 h, in the absence and presence of anti-CXCL-1+anti-IL-8. (f) Cluster-associated and free PMNs arrested intraluminally, and fraction of PMNs remaining in individual clusters over 6 h in the presence of IL-8 and/or CXCL-1 neutralizing antibodies, or when tumor cells are replaced with 15 μm polystyrene beads. Reproduced under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0.[141] International Public License for open access articles. CXCL-1: chemokine (C-X-C motif) ligand-1, 3D: three-dimensional, HUVECs: human umbilical vein endothelial cells, IL-8: interleukin-8, IgG: immunoglobulin G, MVECs: human microvascular endothelial cells, PMN: LPS-stimulated neutrophils, TC: tumor cell, TME: tumor microenvironment, TNF-α: tumor necrosis factor-alpha.
A 3D microfluidic model of the tumor-vascular interface was developed to study cancer cell invasion into the circulatory system.[139] The device consisted of two side channels and one central chamber (Figure 4B). Breast carcinoma cells (MDA-MB-231 overexpressing green fluorescent protein (GFP)-tagged MenaINV) or fibrosarcoma cells (HT1080) embedded in collagen were seeded on one side channel, whereas human microvascular endothelial cells (MVECs) or HUVECs were seeded into another side channel. These tumor and endothelial side channels were interconnected through a central 3D ECM channel filled with the collagen hydrogel (Figure 4B a–f). Murine macrophages (RAW264.7 cells) were incorporated inside the 3D ECM to study the role of macrophages in inducing carcinoma intravasation. It was found that macrophages induced tumor cell intravasation as indicated by the migration of tumor cells from the tumor channel towards the apical surface inside the vascular channel. The percentages of tumor cells that had intravasated were similar for MVECs and HUVECs (Figure 4B g–j). This study provided a microfluidic-based approach that allowed real-time visualization and quantification of the tumor cell invasion and intravasation in the presence of macrophages as well as interactions between the tumor cells and the endothelial layer.
Similarly, a 3D microfluidic model was fabricated to study the extravasation of breast cancer cells in a vascularized bone-mimicking (BMi) microenvironment using primary human BMSCs, osteoblast-differentiated BMSCs (OBs), and GFP-HUVECs encapsulated in a fibrin gel creating a perfusable microvascular network concentrically wrapped with mural cells.[140] GFP-HUVECs, BMSCs, and OBs embedded in a fibrin gel were seeded in the central channel whereby GFP-HUVECs formed the vasculature, and BMSCs and OBs formed a BMi microenvironment. The formation of the vascularized BMi was confirmed by immunostaining of endothelial markers (VE-cadherin and ZO-1) and bone markers (osteocalcin and alkaline phosphatase). To study the cancer extravasation, breast cancer cells were introduced from the side medium channels and it was demonstrated the extravasation of the cancer cells into the BMi microenvironment whereby breast cancer cells were flown into, adhered to, and metastasized through the vascular networks. The tumor cell extravasation rate was found to be higher for the BMi microenvironment when compared to the muscle microenvironment formed using myoblasts (C2C12 cells), and control microenvironment without any cells. In addition, it was shown that when the A3 adenosine receptors on the breast cancer cells were blocked with adenosine, the extravasation rate of the cancer cells was decreased significantly whereas the permeability of the vasculature was increased as compared to the BMi microenvironment.
A multiplexed PDMS microvasculature device was developed to study the dynamic interactions between the inflamed neutrophils and the tumor cells.[141] The device consisted of a single injection port and eight independent vasculature regions where microvascular networks were formed with HUVECs encapsulated in a fibrin hydrogel (Figure 4C a, b). When LPS-stimulated neutrophils, derived from human blood, and A375-MA2 melanoma cells were perfused together through the microvascular bed, tumor cells-neutrophils aggregates were formed as a result of both physical trapping in narrow capillaries and adhesion between neutrophils and endothelial ICAM-1(Figure 4C c–f). Further, these heterotypic aggregates were found to migrate continuously in a confined manner, driven by neutrophil-secreted IL-8 and tumor cell-secreted chemokine (C-X-C motif) ligand-1 (CXCL-1), leading to the disruption of endothelial barrier and increased extravasation of tumor cells.
The current tumor-on-a-chip models represent simplistic tumor systems that are suited for drug testing and cancer and/or immune cell motility studies. The more complex in vitro models should be engineered considering the dynamic interactions between cancer cells, the tumor stromal microenvironment, and the immune system. The in vitro recapitulation of the complex native TME and dynamics is challenging but remains an aspiration for developing more sophisticated and physiologically relevant models that are critical for understanding physiopathology of tumors, anticancer drug screening, and personalized cancer therapy.
3.4. Cancer immunotherapy-on-a-chip
In recent years, cancer immunotherapy has become a potential approach that harnesses patient’s immune system to recognize and selectively ablate cancer cells.[142–144] For cancer immunotherapy, a variety of strategies such as modulation of the immune system, administration of the exogenous cytokines (IL-2 and IFN-α), engineered T cells (chimeric antigen receptor-modified T cells (CAR-T) and TCR-T cells), and immune checkpoint blockade with antibodies targeting the immune check point inhibitors (programmed cell death protein 1 (PD-1)/PD-1 ligand (PD-L1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)), have been used to activate the immune system and to fight cancer.[145–150] Other immunotherapies include cell-based immunotherapy whereby naïve or engineered immune cells, such as cytotoxic CD8+ T cells or natural killer cells (NK cells), are administrated into the bloodstream to destroy the tumor cells by secreting TNF-α, perforins, or apoptosis-inducing proteins such as TNF-related apoptosis-inducing ligand (TRAIL) and granzymes.[151–157]
To this end, different microfluidic systems have been developed to facilitate the investigation of various aspects of cancer immunotherapy such as immune cell migration, modification of TCRs, evaluation of NK cell cytotoxicity, T cell infiltration, and immune checkpoint blockade, among others. A simple microfluidic chip was developed to study the time-dependent migration and cytotoxicity of TCR-engineered T cells.[158] The device consisted of a central tumor channel in which hepatocellular carcinoma cells that expressed both GFP and hepatitis B virus (HBV) envelope protein HBsAg (HepG2-Env cells), were seeded in the collagen type I hydrogel, whereas TCR-engineered T cells that expressed TCR recognizing HLA-A0201/HBV Env183–191 epitope complexes were added to the medium channel, allowing the interactions between the liver cancer cells and T cells in the 3D space. It demonstrated the ability of TCR-T cells to migrate and ablate the tumor cells under different oxygen levels (2% or 20%) and in the presence of cytokines, interferon gamma (IFN-γ) and TNF-α. It was also shown that the TCR-T cells migrated towards the central hydrogel region only in the presence of the HepG2-Env cells, suggesting that migration of the TCR-T cells was induced by the chemokines produced by the cancer cells, as revealed by the cytokine/chemokine profiling assay (Regulated on Activation, Normal T Cell Expressed and Secreted (RANTES), fractalkine, IFN-γ, CXCL-8, VEGF, CXCL-10, macrophage inflammatory protein (MIP)-1α, and MIP-1β). This study demonstrated the tumor-specific T cell migration and tumor cell killing, as well as the significance of oxygen and the effects of inflammatory cytokines for desired activity of the T cells.
In another study, a similar microfluidic device consisting of a middle cancer cell channel and two side fluidic channels, was developed to study the effect of monocytes on HBV-specific TCR-T cell’s efficacy to kill tumor cells as well as the role of PD-L1/PD-1 signaling.[159] Human primary monocytes and hepatocellular carcinoma cells expressing pre-S1 coding region of HBV and GFP (HepG2-preS1-GFP) embedded in a collagen gel were introduced together in the central channel, whereas HBV-specific TCR-T cells were flown through one fluidic channel, mimicking the intrahepatic TME. It was shown that both retrovirally transduced (Tdx) and mRNA-electroporated HBV-specific TCR-T cells ablated the HepG2-preS1-GFP cells. However, the expression of PD-1 and the cytotoxic efficacy of HBV-specific TCR-T cells were not altered by the presence of monocytes. In contrast, the monocytes could suppress only Tdx HBV-specific TCR-T cell cytotoxicity via PD-L1/PD-1.
Recently, the microphysiological 3D lung cancer and breast cancer models have been created to investigate the antitumor activity of receptor tyrosine kinase-like orphan receptor 1 (ROR1)-specific CAR-T cells, in which the decellularized submucosa and mucosa of porcine jejunum was used as the ECM scaffold for culturing A549 non-small cell lung cancer cells and MDA-MB-231 cells under static or dynamic culture conditions.[160] It was demonstrated that the ROR1-CAR T cells rapidly infiltrated and migrated through the 3D tumor tissues and exhibited a potent antitumor effect, as shown by the secretion of caspase-cleaved keratin 18, a tumor cell apoptotic marker. Similarly, a 3D microfluidic human ovarian cancer model was developed by embedding human epidermal growth factor receptor 2 (HER2)+ ovarian cancer cells (SKOV3) in a micropatterned gelatin methacryloyl (GelMA) hydrogel and micromilled hypoxia device to establish an oxygen gradient across micropatterned cancer cells.[161] It consisted of perfusion channels for the delivery of HER2-targeting 4D5 scFv CAR-T cells and to study the infiltration as well as the cytotoxicity of CAR-T cells under heterogeneous oxygen concentration conditions within the tumor. The enhanced cytotoxic activity of CAR-T cells was observed under the physiologically relevant oxygen level, whereas immunosuppression was increased under the hypoxic condition as shown by increased PD-L1 expression by the tumor cells under hypoxia culture. However, during the given time period, the inhibition of expression of PD-L1 did not enhance the cytotoxicity of CAR-T cells.
In an another study, a higher-throughput microfluidic plastic array culture (CACI-IMPACT) device was designed to study the cytotoxic efficacy of lymphocytes to ablate cancer cells in 3D microenvironments.[162] Rail-based microstructures, comprised of the low-rail (cancer cell) zone, the high-rail (immune cell) zone, and the reservoir wall, were placed in microwells and integrated to form a 2×6 rectangular arrays of the microwells. HeLa cells encapsulated within the collagen hydrogel were patterned in a low-rail zone, whereas NK-92 cell suspension was loaded in the high-rail zone and allowed to migrate towards the HeLa cells in the low-rail zone (Figure 5A a, b). The migration and cytotoxicity of NK-92 cells against HeLa cells within the collagen matrix was monitored for 24 h by live imaging and compared to those in 2D culture. It was found that the migration of NK-92 cells was markedly decreased in the presence of 3D collagen ECM, resulting in reduced cytotoxicity as compared to 2D culture (Figure 5A c, d).
Figure 5. Immunotherapy-on-a-chip models.
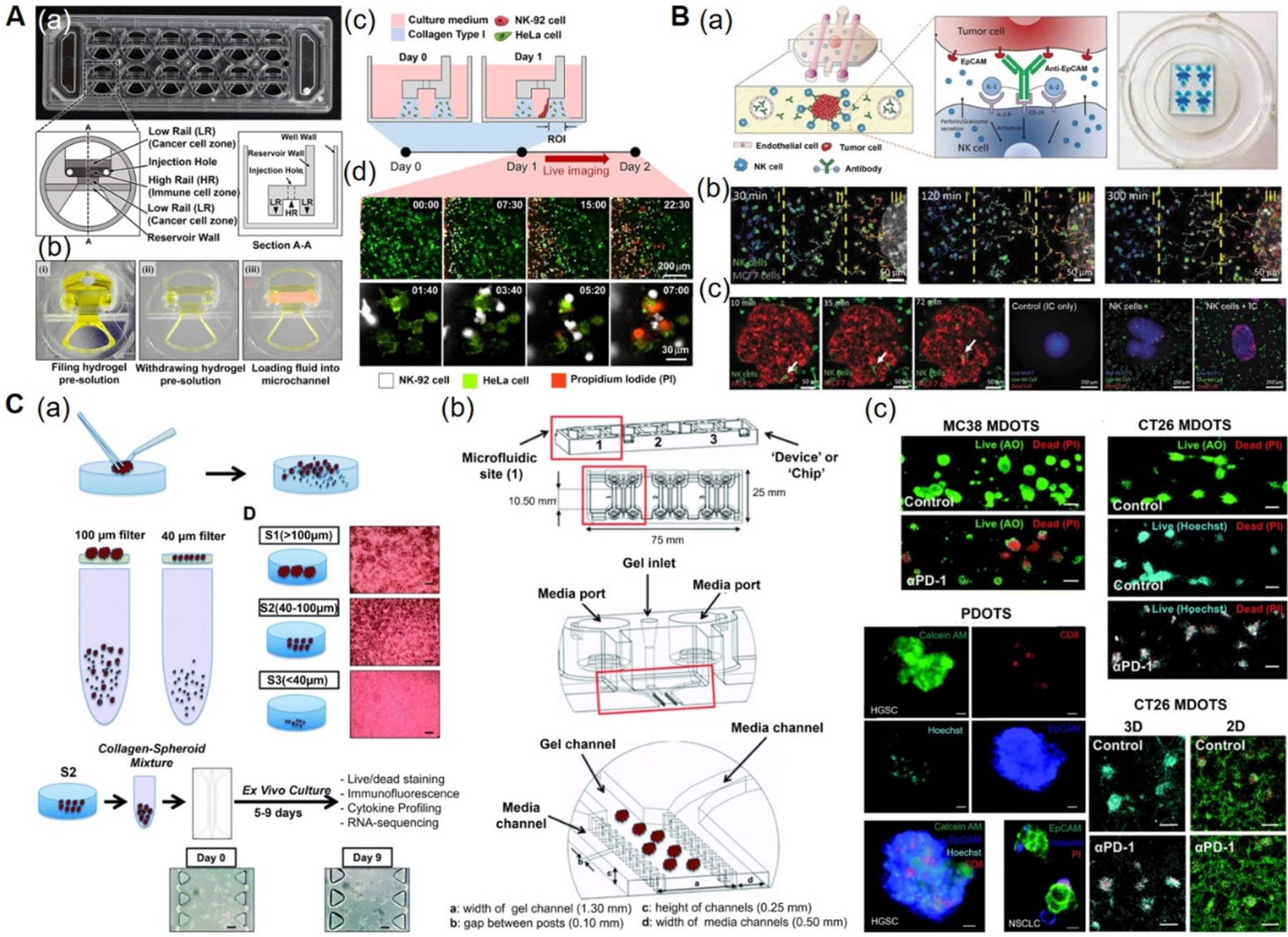
A) CACI-IMPACT platform for 3D cytotoxicity assay. (a) Photograph and schematics of microfluidic device consisting of rail-based microstructures integrated in a 2 × 6 rectangular array of wells. (b) Photographs showing the compartments within the device. Each well consists of two LRs for hydrogel patterning and one HR forming a channel for fluid flow after hydrogel crosslinking. (c) Schematics showing the loading of HeLa cells embedded in collagen into LRs at Day 0 and NK-92 cells into a microchannel after 24 h. The device was tilted to 90° for 20 min to accumulate NK-92 cells on one side of the collagen gel. (d) Time-lapse images showing the migration of NK-92 cells towards the collagen matrix containing HeLa cells. Time is indicated in hour:minute in the top right corner of each image. Reproduced under the Creative Commons Attribution 4.0.[162] International Public License for open access articles. B) Microfluidic device to study ADCC in NK cells. (a) Schematics (left) and photographs (right) of the device consisting of central chamber with a tumor spheroid in collagen hydrogel and two flanking lateral lumens with endothelial cells mimicking blood vessels. NK cells and/or antibodies were embedded in the hydrogel or perfused through the lateral lumens. (b) Time-lapse images showing directional migration of NK-92 cells (labeled in green) towards MCF7 spheroid (labeled in red). The field of view was divided in three regions: distal (I), central (II), and proximal (III) to analyze NK cell migration. (c) Time-lapse images showing the migration of NK-92 cells within the tumor sphere using an ameboid movement (white arrow), followed by the images showing the anti-EpCAM immunocytokine, IC65-induced cell cytotoxicity, when used in combination with NK-92.CD16V cells. Reproduced with permission.[45,163] Copyright 2018, Taylor & Francis. C) 3D microfluidic device for MDOTS/PDOTS culture. (a) Schematics showing the preparation and culture of MDOTS/PDOTS (S2 fraction) from murine or patient-derived tumor specimens. (b) Schematics showing the different components of the microfluidic device. Each device comprised of center gel region and side media channels separated by posts, gel loading port and media ports. (c) Fluorescence images showing the live/dead analysis of MDOTS in the presence of anti-PD-1 antibody or isotype control IgG, in 3D microfluidic and 2D cultures. Reproduced under the Creative Commons Attribution-NonCommercial 3.0[166] Unported Licence for open access articles. ADCC: antibody-dependent cell cytotoxicity, AO: acridine orange, CACI-IMPACT: cytotoxicity assay for cancer immunotherapy-injection molded plastic array culture platform, 2D: two-dimensional, 3D: three-dimensional, EpCAM: epithelial cell adhesion molecule, HR: high rail, IC: immunocytokine, IgG: immunoglobulin G, LR: low rail, MDOTS: murine-derived multicellular organotypic tumor spheroids, NK: natural killer, PDOTS: patient-derived multicellular organotypic tumor spheroids, PD-1:programmed cell death protein 1, PI: propidium iodide.
Moreover, a 3D breast cancer model was developed to study NK cell immunotherapies and antibody-dependent cell cytotoxicity (ADCC), in which breast cancer cell (MCF-7) spheroids were encapsulated in a collagen hydrogel, individually or in combination with NK-92 or the CD16-positive NK-92, whereas two lateral lumens were seeded with HUVECs (Figure 5B a).[163] The anti-epithelial cell adhesion molecule (EpCAM) antibody was introduced through the endothelialized channel or directly incorporated in the hydrogel to understand the dynamics of antibodies supplied and their effect on NK cytotoxicity. The results indicated that antibodies present in the TME were taken up by the tumor cells, whereas the penetration capacity of the antibodies into the spheroids was hindered by endothelial tight junctions. Further, NK-92 cells displayed chemotactic migration and penetration into the tumor spheroids within a few hours, as demonstrated by the secretions of different chemokines as well as the expressions of chemokine receptors (CXCL-12-CXCR4, CCL-28-CCR3, CXCL-8-CXCR1/CXCR2, and CXCL-5-CXCR2). ADCC-induced cytotoxicity was studied by adding immunocytokine (IL-2-conjugated anti-EpCAM antibody) to the CD16-positive NK-92 cells. The addition of the IL-2-conjugated anti-EpCAM antibody significantly enhanced cytotoxicity of NK cells in 2D, while the cytotoxic effect of NK cells was limited to the surface of the spheroids (Figure 5B b, c). The solid tumor immunotherapies have been facing challenges because in the solid tumors, transport of antibodies and/or infiltration of immune cells from the vasculature and into a dense mass of tumor is required. The simple model of solid tumor immunotherapy presented in this study represents an important tool that would improve the understanding of antibody-mediated immunotherapy of solid tumors.
Also, a 3D microfluidic device was developed to assess the migration of IFN-DCs towards SW620 colorectal cancer cells, and their interactions.[164] The PDMS device consisted of central immune-chamber connected to two side tumor-chambers by narrow microgrooves. The behavior of IFN-DCs towards SW620 cells was monitored in the presence or absence of the combined antitumor drug (an immunomodulator IFN-α and an epigenetic drug romidepsin). It was found that IFN-DCs migrated towards drug-treated tumor-chamber rather than the untreated one and that the drug induced apoptosis of SW620 cells, which were phagocytosed by the IFN-DCs. It was further shown that drug-treated SW620 cells expressed enhanced levels of chemokines such as CCL2, CCL4, CXCL-1, CXCL-9, and CXCL-12, and chemokine receptors such as CCR1, CCR5, CCR7, CXCR3, and CXCR4, among others.
More interestingly, the murine-derived multicellular organotypic tumor spheroids (MDOTS) or patient-derived multicellular organotypic tumor spheroids (PDOTS) embedded in collagen hydrogels were cultured in a microfluidic device to achieve the complexity and heterogeneity of relevant cell types in the TME.[165,166] The MDOTS and PDOTS were derived from murine and patient-derived tumor specimens, respectively. The S2 fraction of either MDOTS or PDOTS was resuspended in collagen and cultured in the center gel region of the microfluidic device (Figure 5C a, b). Treatment of murine colon adenocarcinoma cell line-derived MDOTS (MC38 MDOTS or CT26 MDOTS) with anti-PD-1 antibody resulted in CD8+ T cell-mediated tumor cell killing (Figure 5C c). Further, immune cell profiling of MDOTS and PDOTS showed the heterogeneity in the immune cell population, comprising CD4+ T cells, CD8+ T cells, B cells, monocytes, and DCs. T cells were found to express PD-1, TIM-3 (T-cell immunoglobulin and mucin-domain containing-protein 3 and CTLA-4) whereas DCs, tumor-associated macrophages, and myeloid-derived suppressor cells were found to express PD-L1/PD-L2. The impact of TANK binding kinase 1 (TBK1)/inhibitor of κB kinase ε (IKKε) was evaluated in MDOTS and it was found that innate immune signaling kinases, TBK1 and IKKε, not only promoted autocrine/paracrine cytokine signaling but also induced T cell activation. Further, addition of TBK1/IKKε inhibitor to PD-1 blockade increased killing of CT26 MDOTS while decreasing the levels of CCL-4, CCL-3, and IL-1β. In addition, it induced the production of cytokine G-CSF, involved in activated innate immune responses. The responses to PD-1 and PD-1/CTLA-4 blockade were also evaluated in PDOTS, where the upregulations of CCL19 and CXCL-13 were observed in majority of the samples along with the induction of cytokines, IFN-γ, TNF-α, and IL-2. Besides, a microfluidic cyclic olefin copolymer device, known as EVIDENT (Ex Vivo Immuno-oncology Dynamic Environment for Tumor biopsies), was developed that could accommodate twelve tumor biopsy fragments and allowed to interact with tumor-infiltrating lymphocytes (TILs) in a dynamic microenvironment.[167] The device consisted of an array of posts in the center for tumor fragment culture while medium carrying TILs was flowed around the tumor fragments. This system was used to study the effect of immune checkpoint inhibitors (ICIs) using MC38 colon tumor and human non-small cell lung cancer biopsies. The device further enabled real-time imaging of the interactions between TILs and tumor cells, TILs-mediated tumor cell killing, and studying the effect of various ICIs.
The recent progress on immunotherapy via engineered CAR-T cells, checkpoint inhibitors and cancer vaccines has revolutionized the treatment of various cancer types. However, while exciting successes have been achieved in immunotherapy of nonsolid tumors (e.g., leukemia), it is still limited in the case of solid tumors. Immunotherapy-on-a-chip models incorporating immune-TME hold great promise as novel approaches to investigate different immunotherapies and their consequences, as well as to understand the mechanisms of actions of immunotherapy. These in vitro approaches would also help to screen the toxicities and efficacy of current cancer immunotherapeutic drugs.
4. Concluding remarks and perspectives
The understanding of the immune system has changed gradually over the last decade and it is becoming clearer that the immune system has critical physiological roles that extends far beyond its conventional role in defense mechanism against infections and diseases. Although considerable progress has been made towards understanding the role of the immune system in homeostatic regulation, inflammation, and tumor progression, more thorough studies are still needed to gain in-depth knowledge on the contributions of the immune system in the regulation of other complex physiological processes including those in the central nervous system, cardiovascular system, and metabolisms.
The OOC approach offers the opportunities to reproduce the structures and functions of immune system organs or tissues in vitro. Development of physiologically and pathophysiologically relevant immune OOCs and/or immunocompetent OOCs would provide powerful tools to study normal and pathophysiology of the immune system and its interactions with other organs, as well as for pre-clinical drug testing for diseases including inflammatory diseases, autoimmune diseases, and cancers. Further, as the main route of immune cell trafficking and the site of immune responses, incorporation of microvasculature is also crucial while modeling the dynamic tissue microenvironments and pathological processes. In fact, in order to perform their functions in immune surveillance and immune response regulation, cells of the immune system must repeatedly move in and out of the tissue microenvironments and the circulation, including both vascular and lymphatic systems.[168,169] OCC platforms have been explored to recapitulate immune cell trafficking and inflammatory responses under normal and diseased conditions such as study of adhesion and migration of neutrophils across the endothelium,[170] effect of endothelial wall shear stress and inflammatory cytokines (e.g., TNF-α) on adhesion of immune cells (neutrophils or monocytes) to the endothelium,[171–175] adhesion and transmigration of monocytes across the endothelium in the presence of low- or high-density lipoproteins[176] and titanium microbead implants as the foreign bodies,[177] the influence of high dietary salt on inflammatory monocytic cell adhesion,[178] and adhesion and transmigratory behaviors of neutrophils in the presence of chemoattractant IL-8.[179] However, most of the immune OOCs, immunocompetent OOCs, and circulatory devices developed so far are only prototype models of the real organs or tissues (Table 1). There are several challenges of studying the immune system using the OOC technology, with the main reason being the structural and functional complexity of the immune system both in immune organs and within the other tissues or organs. Most of the OOC devices could capture the simplified innate immune components including macrophages and/or neutrophils, or functional units of the immune organs such as the bone marrow and the lymph node with limited functionality, rather than the key steps of the immune repertoire within the adaptive immune system. Despite the challenges such as appropriate scaling of the organs, vascularization of tissues and inclusion of different components of immune system among others, studies are underway to retain the greater complexity in engineering of microfluidic OOCs in vitro to provide important insights into the complicated immunological systems and their relationships with different organs of the body.
Table 1.
Summary of immune and immunocompetent organ-on-a-chip systems cited in this review.
| Models | Cell types | ECM hydrogels | Key findings | Refs. |
|---|---|---|---|---|
| BM-on-a-chip | ||||
| hFOB 1.19 human fetal osteoblasts and mononuclear cells from bone marrow | Endosteal surfacesand its ECM from patients | Ossified tissue formation | [61] | |
| BMSCs and HSPCs | — | Differentiation of HSPCs into various progenies (granulocytes, erythrocytes, macrophages, megakaryocytes) | [62] | |
| Hematopoietic cell composition from in vivo implantation | Collagen | Effect of γ-radiation exposure on BM | [63] | |
| BMSCs, OBs, and SUP-B15 leukemic cells | Collagen | Chemotherapeutic effect cytarabine on acute lymphoblastic Leukemia. | [64] | |
| Human CD34+ cells, BMSCs, and HUVECs | Fibrin | BM hematopoiesis and hematotoxicity of 5-FU | [65] | |
| LN-on-a-chip | ||||
| DCs, CD8+T cells and CD4+T cells | — | Antigen presentation to CD8+ T-cells and CD4+ T-cells by DCs | [78] | |
| DCs and T cells | — | Migration of DCs and presentation of the antigens to T-cells | [79] | |
| DCs and PBMCs | Agarose | Induction of proinflammatory response | [80,81] | |
| Spleen-on-a-chip | ||||
| Fresh and malaria-infected RBCs | — | Deformation of infected RBCs and noninfected red blood cells | [94] | |
| LV-on-chip | ||||
| HMVEC-d and HMVEC-dLy | — | Permeation of the high- and low-MW molecules from the vascular channel to the LV | [88] | |
| LECs, HUVECs, CAFs | Collagen | Permeability of LV and crosstalk between LV and breast cancer cells | [89] | |
| Skin-on-chip | ||||
| HEK human epidermal keratinocytes | Collagen | Simulation of an epidermal layer of the skin | [102] | |
| HDFs and N/TERT-1 keratinocytes | Fibrin | Differentiation and morphogenesis of the epidermal layer | [103] | |
| NHDFs and HUVECs | Collagen | Percutaneous absorption of isosorbide dinitrate and caffeine | [104] | |
| HaCaT human keratinocytes, HS27 human foreskin fibroblasts, and HUVECs | — | TNF-α-induced skin Inflammation | [105] | |
| HaCaT human keratinocytes, U937 human leukemic monocyte lymphoma cell line, and DCs | — | Response of HaCaT cells to skin allergens (nickel sulfate and 2, 4 dinitrochlorobenzene) | [106] | |
| Gut-on-a-chip | ||||
| Caco-2 human colon epithelial cells | Collagen and Matrigel | Mechanical deformation of the epithelial layer under cyclic strain | [124] | |
| Caco-2 human colon epithelial cells and PBMCs | — | Induction of intestinal bacterial overgrowth and inflammation by LPS and PBMCs | [125] | |
| Caco-2 human colon epithelial cells, CCD-18Co non-cancerous colonic cells, and L. rhamnosus GG | Collagen | Co-culture of human cells and microbes | [126] | |
| Tumor-on-chip | ||||
| MDA-MB-231 cells, HT1080 fibrosarcoma cells, MVECs, HUVECs and RAW264.7 murine macrophages | Collagen | Tumor cell invasion and intravasation in the presence of macrophages, and interactions between tumor cells and an endothelial layer | [139] | |
| BMSCs, GFP-HUVECs, OBs and MDA-MB-231 cells | Fibrin | Extravasation of breast cancer cells within a vascularized bone-mimicking microenvironment | [140] | |
| Cancer immunotherapy-on-chip | ||||
| HepG2-Env hepatocellular carcinoma cells and TCR-T cells | Collagen | Tumor-cell-specific T-cell migration and cancer cell killing | [158] | |
| Monocytes from PBMCs, HepG2-preS1-GFP and HBV-TCR-T cells | Collagen | Efficacy of HBV-TCR-T cells to kill HepG2-preS1-GFP cells | [159] | |
| ROR1-CAR-T cells, A549 non-small cell lung cancer cells, MDA-MB-231 breast carcinoma cells | Decellularized mucosa of small intestine | Migration and antitumor activity of ROR1-CAR-T cells | [160] | |
| SKOV3 ovarian cancer cells | GelMA hydrogel | Infiltration and antitumor activity of CAR-T cells under heterogeneous oxygen concentration conditions | [161] | |
| HeLa cervical cancer cells and NK-92 human natural killer cells | Collagen | Infiltration, migration and cytotoxicity of NK-92 cells | [162] | |
| MCF-7 breast cancer cells, NK-92 human natural killer cells CD16+ NK-92 human natural killer cells and HUVECs | Collagen | Penetration capacity of anti-EpCAM antibody into the MCF-7 spheroids | [163] | |
| SW620 colorectal cancer cells and IFN-DCs | — | Migration of IFN-DCs towards SW620cells in the presence or absence of an IFN-α and an epigenetic drug romidepsin | [164] | |
| MC38 and CT26 murine colon adenocarcinoma cells-derived MDOTS, or PDOTS | Collagen | Immune cell profiling of MDOTS/PDOTS and the effect of PD-1 or PD-1/CTLA-4 blockade on MDOTS/PDOTS | [165,166] | |
BM: Bone marrow, BMSCs: human bone marrow stromal cells, CAFs: cancer-associated fibroblasts, DCs: dendritic cells, ECM: extracellular matrix, 5-FU: 5-fluorouracil, GFP-HUVECs: green fluorescent protein-HUVECs, HMVEC-d cells: human dermal microvascular endothelial cells, HUVECs: human umbilical vein endothelial cells, HMVEC-dLy cells: human lymphatic microvascular endothelial cells, HSPCs: cord blood-derived multipotent hematopoietic stem and progenitor cells, HDFs: human primary foreskin-derived dermal fibroblasts, HBV-TCR-T cells: hepatitis B virus-specific TCR-T cells, HepG2-preS1-GFP cells: hepatocellular carcinoma cells expressing pre-S1 coding region of HBV and GFP, IFN-DCs: interferon-DCs, LN: Lymph node, LV: Lymphatic vessel, LECs: human lymphatic endothelial cells, MW: molecular weight, MVECs: human microvascular endothelial cells, MDOTS: multicellular organotypic tumor spheroids, NHDFs: normal human dermal fibroblasts, OBs: osteoblast-differentiated from BMSCs, PBMCs: peripheral blood mononuclear cells, PDOTS: patient-derived multicellular organotypic tumor spheroids, RBCs: red blood cells, ROR1-CAR-T cells: receptor tyrosine kinase-like orphan receptor 1-specific chimeric antigen receptor-modified T cells, TCR-T cells: T-cell receptor engineered T-cells
Acknowledgments
This work was supported by funding from the National Institutes of Health (K99CA201603, R00CA201603, R21EB025270, R21EB026175, R01EB028143, R03EB027984), the Brigham Research Institute, and the American Fund for Alternatives to Animal Research (AFAAR).
References
- [1].Frantz C, Stewart KM, Weaver VM, J Cell Sci. 2010, 123, 4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Barthes J, Özçelik H, Hindié M, Ndreu-Halili A, Hasan A, Vrana NE, Biomed Res Int. 2014, 921905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Johnson A, Lewis J, Morgan D, Raff M, Roberts K, Walter P, Garland Press, 2015. [Google Scholar]
- [4].Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK, Adv Drug Deliv Rev. 2016, 97, 4. [DOI] [PubMed] [Google Scholar]
- [5].Lewin B, Cassimeris L, Lingappa V, Plopper G, Cells. 2007.
- [6].Neal CA, Nelson-Brantley JG, Detamore MS, Staecker H, Mellott AJ, J Vis Exp. 2018, e56523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kusindarta DL, Wihadmadyatami H, Tissue Eng. 2018, 65. [Google Scholar]
- [8].Bonnans C, Chou J, Werb Z, Nat. Rev. Mol. Cell Biol 2014, 15, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cox TR, Erler JT, Dis Model Mech. 2011, 4, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rankin LC, Artis D, Cell. 2018, 173, 554. [DOI] [PubMed] [Google Scholar]
- [11].Medzhitov R, Nature 2008, 454, 428. [DOI] [PubMed] [Google Scholar]
- [12].Janeway CA Jr, Travers P, Walport M, Shlomchik MJ, in Immunobiology: The Immune System in Health and Disease. 5th edition, Garland Science, 2001. [Google Scholar]
- [13].Randolph GJ, Ivanov S, Zinselmeyer BH, Scallan JP, Annu Rev Immunol. 2017, 35, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P, in Molecular Biology of the Cell. 4th edition, Garland Science, 2002. [Google Scholar]
- [15].Ginhoux F, Lim S, Hoeffel G, Low D, Huber T, Front. Cell. Neurosci 2013, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Soysa DR, Crispe IN, J Immunol. 2016, 196, 126.18. [Google Scholar]
- [17].Janeway CA Jr, Travers P, Walport M, Shlomchik MJ, in Immunobiology: The Immune System in Health and Disease. 5th edition, Garland Science, 2001. [Google Scholar]
- [18].Harvill ET, mBio. 2013, 4, e00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang Y, Zhang C, Front. Immunol 2019, 10, 1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Turner J-E, Becker M, Mittrücker H-W, Panzer U, J. Am. Soc. Nephrol 2018, 29, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Korin B, Ben-Shaanan TL, Schiller M, Dubovik T, Azulay-Debby H, Boshnak NT, Koren T, Rolls A, Nat. Neurosci 2017, 20, 1300. [DOI] [PubMed] [Google Scholar]
- [22].Chung K-J, Nati M, Chavakis T, Chatzigeorgiou A, Rev Endocr Metab Disord. 2018, 19, 283. [DOI] [PubMed] [Google Scholar]
- [23].Nayak D, Roth TL, McGavern DB, Annu. Rev. Immunol 2014, 32, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nimmerjahn A, Kirchhoff F, Helmchen F, Science 2005, 308, 1314. [DOI] [PubMed] [Google Scholar]
- [25].Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, Thome JJ, Farber DL, Lutfy K, Seale P, Nature 2015, 519, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, Mathis D, Nature 2012, 486, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C, Cell 2013, 155, 1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tidball JG, Nat. Rev. Immunol 2017, 17, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Alecsandru D, García-Velasco JA, Curr Opin Obstet Gynecol. 2015, 27, 231. [DOI] [PubMed] [Google Scholar]
- [30].Takayanagi H, Nat. Rev. Immunol 2007, 7, 292. [DOI] [PubMed] [Google Scholar]
- [31].López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G, Cell 2013, 153, 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fan X, Rudensky AY, Cell 2016, 164, 1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ribatti D, Oncotarget 2017, 8, 7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Huh D, Torisawa Y.-s., Hamilton GA, Kim HJ, Ingber DE, Lab Chip 2012, 12, 2156. [DOI] [PubMed] [Google Scholar]
- [35].Zhang B, Radisic M, Lab Chip 2017, 17, 2395. [DOI] [PubMed] [Google Scholar]
- [36].Caplin JD, Granados NG, James MR, Montazami R, Hashemi N, Adv. Healthc. Mater 2015, 4, 1426. [DOI] [PubMed] [Google Scholar]
- [37].Ronaldson-Bouchard K, Vunjak-Novakovic G, Cell Stem Cell 2018, 22, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bhatia SN, Ingber DE, Nat. Biotechnol 2014, 32, 760. [DOI] [PubMed] [Google Scholar]
- [39].Shanti A, Teo J, Stefanini C, Pharmaceutics. 2018, 10, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Polini A, Loretta L, Barra A, Zhang YS, Calabi F, Gigli G, Drug Discov. Today 2019, 24, 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Brodin P, Davis MM, Nat. Rev. Immunol 2017, 17, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shah SB, Singh A, Curr Opin Hematol. 2017, 24, 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bucktrout SL, Bluestone JA, Ramsdell F, Genome Med. 2018, 10, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Nowarski R, Jackson R, Flavell RA, Cell 2017, 168, 362. [DOI] [PubMed] [Google Scholar]
- [45].Kim SK, Moon WK, Park JY, Jung H, Analyst. 2012, 137, 4062. [DOI] [PubMed] [Google Scholar]
- [46].Jones CN, Dalli J, Dimisko L, Wong E, Serhan CN, Irimia D, Proc Natl Acad Sci USA 2012, 109, 20560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gopalakrishnan N, Hannam R, Casoni GP, Barriet D, Ribe JM, Haug M, Halaas Ø, Lab Chip 2015, 15, 1481. [DOI] [PubMed] [Google Scholar]
- [48].Haessler U, Pisano M, Wu M, Swartz MA, Proc Natl Acad Sci USA 2011, 108, 5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dura B, Dougan SK, Barisa M, Hoehl MM, Lo CT, Ploegh HL, Voldman J, Nat. Commun 2015, 6, 5940. [DOI] [PubMed] [Google Scholar]
- [50].Zhao E, Xu H, Wang L, Kryczek I, Wu K, Hu Y, Wang G, Zou W, Cell Mol Immunol. 2012, 9, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Yin T, Li L, J Clin Invest 2006, 116, 1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Weilbaecher KN, Guise TA, McCauley LK, Nat. Rev. Cancer 2011, 11, 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Morrison SJ, Scadden DT, Nature 2014, 505, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Rosen CJ, Ackert-Bicknell C, Rodriguez JP, Pino AM, Crit. Rev. Eukaryot. Gene Expr 2009, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Xu W.-t., Bian Z.-y., Fan Q.-m., Li G, Tang T.-t., Cancer Lett. 2009, 281, 32. [DOI] [PubMed] [Google Scholar]
- [56].Tsukamoto S, Honoki K, Fujii H, Tohma Y, Kido A, Mori T, Tsujiuchi T, Tanaka Y, Int J Oncol 2012, 40, 163. [DOI] [PubMed] [Google Scholar]
- [57].Makhoul I, Montgomery CO, Gaddy D, Suva LJ, Nat. Rev. Endocrinol 2016, 12, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Dunn LK, Mohammad KS, Fournier PG, McKenna CR, Davis HW, Niewolna M, Peng XH, Chirgwin JM, Guise TA, PloS one 2009, 4, e6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Sasaki S, Baba T, Nishimura T, Hayakawa Y, Hashimoto S.-i., Gotoh N, Mukaida N, Cancer Lett. 2016, 378, 23. [DOI] [PubMed] [Google Scholar]
- [60].Zhou BO, Yu H, Yue R, Zhao Z, Rios JJ, Naveiras O, Morrison SJ, Nat. Cell Biol 2017, 19, 891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zhang W, Lee WY, Siegel DS, Tolias P, Zilberberg J, Tissue Eng Part C Methods 2014, 20, 663. [DOI] [PubMed] [Google Scholar]
- [62].Sieber S, Wirth L, Cavak N, Koenigsmark M, Marx U, Lauster R, Rosowski M, J Tissue Eng Regen Med. 2018, 12, 479. [DOI] [PubMed] [Google Scholar]
- [63].Torisawa Y.-s., Spina CS, Mammoto T, Mammoto A, Weaver JC, Tat T, Collins JJ, Ingber DE, Nat. Methods 2014, 11, 663. [DOI] [PubMed] [Google Scholar]
- [64].Bruce A, Evans R, Mezan R, Shi L, Moses BS, Martin KH, Gibson LF, Yang Y, PLoS One 2015, 10, e0140506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Chou DB, Frismantas V, Milton Y, David R, Pop-Damkov P, Ferguson D, MacDonald A, Bölükbaşı ÖV, Joyce CE, Teixeira LSM, Nat. Biomed. Eng 2020, 1. [Google Scholar]
- [66].Buettner M, Bode U, Clin. Exp. Immunol 2012, 169, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Itano AA, Jenkins MK, Nat Immunol 2003, 4, 733. [DOI] [PubMed] [Google Scholar]
- [68].Choi I, Lee S, Hong Y-K, Cold Spring Harb Perspect Med. 2012, 2, a006445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Irvine DJ, Stachowiak AN, Hori Y, “Lymphoid tissue engineering: invoking lymphoid tissue neogenesis in immunotherapy and models of immunity”, presented at Semin Immunol, 2008. [DOI] [PubMed] [Google Scholar]
- [70].Willard-Mack CL, Toxicol. Pathol 2006, 34, 409. [DOI] [PubMed] [Google Scholar]
- [71].Bousso P, Nat. Rev. Immunol 2008, 8, 675. [DOI] [PubMed] [Google Scholar]
- [72].Qi H, Kastenmüller W, Germain RN, Annu Rev Cell Dev Biol 2014, 30, 141. [DOI] [PubMed] [Google Scholar]
- [73].Pape KA, Catron DM, Itano AA, Jenkins MK, Immunity 2007, 26, 491. [DOI] [PubMed] [Google Scholar]
- [74].Padera TP, Meijer EF, Munn LL, Annu. Rev. Biomed. Eng 2016, 18, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Liao S, Padera TP, Lymphat Res Biol. 2013, 11, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kataru RP, Baik JE, Park HJ, Wiser I, Rehal S, Shin JY, Mehrara BJ, Front Immunol. 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Shimony A, Tidhar D, Ann Plast Surg 2008, 60, 228. [DOI] [PubMed] [Google Scholar]
- [78].Moura Rosa P, Gopalakrishnan N, Ibrahim H, Haug M, Halaas Ø, Lab Chip 2016, 16, 3728. [DOI] [PubMed] [Google Scholar]
- [79].Mitra B, Jindal R, Lee S, Xu Dong D, Li L, Sharma N, Maguire T, Schloss R, Yarmush ML, RSC Adv 2013, 3, 16002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Giese C, Lubitz A, Demmler CD, Reuschel J, Bergner K, Marx U, J. Biotechnol 2010, 148, 38. [DOI] [PubMed] [Google Scholar]
- [81].Giese C, Demmler CD, Ammer R, Hartmann S, Lubitz A, Miller L, Müller R, Marx U, Artif Organs. 2006, 30, 803. [DOI] [PubMed] [Google Scholar]
- [82].Ross AE, Belanger MC, Woodroof JF, Pompano RR, Analyst 2017, 142, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Swartz MA, Hubbell JA, Reddy ST, “Lymphatic drainage function and its immunological implications: from dendritic cell homing to vaccine design”, presented at Semin Immunol. , 2008. [DOI] [PubMed] [Google Scholar]
- [84].Jackson DG, J Clin Cell Immunol. 2014, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Randolph GJ, Angeli V, Swartz MA, Nat. Rev. Immunol 2005, 5, 617. [DOI] [PubMed] [Google Scholar]
- [86].Teijeira A, Russo E, Halin C, “Taking the lymphatic route: dendritic cell migration to draining lymph nodes”, presented at Semin Immunopathol, 2014. [DOI] [PubMed] [Google Scholar]
- [87].Hunter MC, Teijeira A, Halin C, Front Immunol. 2016, 7, 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Sato M, Sasaki N, Ato M, Hirakawa S, Sato K, Sato K, PloS one 2015, 10, e0137301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Gong MM, Lugo-Cintron KM, White BR, Kerr SC, Harari PM, Beebe DJ, Biomaterials. 2019, 214, 119225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Lewis SM, Williams A, Eisenbarth SC, Sci Immunol. 2019, 4, eaau6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Timens W, Immunol. 1991, 142, 316. [DOI] [PubMed] [Google Scholar]
- [92].Bronte V, Pittet MJ, Immunity. 2013, 39, 806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Mebius RE, Kraal G, Nat. Rev. Immunol 2005, 5, 606. [DOI] [PubMed] [Google Scholar]
- [94].Rigat-Brugarolas L, Elizalde-Torrent A, Bernabeu M, De Niz M, Martin-Jaular L, Fernandez-Becerra C, Homs-Corbera A, Samitier J, Del Portillo H, Lab Chip 2014, 14, 1715. [DOI] [PubMed] [Google Scholar]
- [95].Kang JH, Super M, Yung CW, Cooper RM, Domansky K, Graveline AR, Mammoto T, Berthet JB, Tobin H, Cartwright MJ, Nat. Med 2014, 20, 1211. [DOI] [PubMed] [Google Scholar]
- [96].Salmon J, Armstrong C, Ansel J, West J Emerg Med 1994, 160, 146. [PMC free article] [PubMed] [Google Scholar]
- [97].Yousef H, Alhajj M, Sharma S, StatPearls, 2019.
- [98].Matejuk A, Arch Immunol Ther Exp 2018, 66, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Baker B, Ovigne JM, Powles A, Corcoran S, Fry L, Br J Dermatol 2003, 148, 670. [DOI] [PubMed] [Google Scholar]
- [100].Schauber J, Gallo RL, J Allergy Clin Immunol 2008, 122, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Diamond G, Beckloff N, Weinberg A, Kisich KO, Curr Pharm Des. 2009, 15, 2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].O’Neill AT, Monteiro-Riviere NA, Walker GM, Cytotechnology. 2008, 56, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Sriram G, Alberti M, Dancik Y, Wu B, Wu R, Feng Z, Ramasamy S, Bigliardi PL, Bigliardi-Qi M, Wang Z, Mater. Today 2018, 21, 326. [Google Scholar]
- [104].Mori N, Morimoto Y, Takeuchi S, Biomaterials. 2017, 116, 48. [DOI] [PubMed] [Google Scholar]
- [105].Wufuer M, Lee G, Hur W, Jeon B, Kim BJ, Choi TH, Lee S, Sci. Rep 2016, 6, 37471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Ramadan Q, Ting FCW, Lab Chip 2016, 16, 1899. [DOI] [PubMed] [Google Scholar]
- [107].Kabashima K, Honda T, Ginhoux F, Egawa G, Nat. Rev. Immunol 2019, 19, 19. [DOI] [PubMed] [Google Scholar]
- [108].Eloe-Fadrosh EA, Rasko DA, Annu Rev Med. 2013, 64, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Mahler GJ, Esch MB, Glahn RP, Shuler ML, Biotechnol Bioeng. 2009, 104, 193. [DOI] [PubMed] [Google Scholar]
- [110].Okumura R, Takeda K, Exp Mol Med. 2017, 49, e338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Ahluwalia B, Magnusson MK, Öhman L, Scand J Gastroenterol. 2017, 52, 1185. [DOI] [PubMed] [Google Scholar]
- [112].Peterson LW, Artis D, Nat. Rev. Immunol 2014, 14, 141. [DOI] [PubMed] [Google Scholar]
- [113].Scaldaferri F, Pizzoferrato M, Gerardi V, Lopetuso L, Gasbarrini A, J Clin Gastroenterol. 2012, 46, S12. [DOI] [PubMed] [Google Scholar]
- [114].McDermott AJ, Huffnagle GB, Immunology. 2014, 142, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Makita S, Kanai T, Nemoto Y, Totsuka T, Okamoto R, Tsuchiya K, Yamamoto M, Kiyono H, Watanabe M, J. Immunol 2007, 178, 4937. [DOI] [PubMed] [Google Scholar]
- [116].Reboldi A, Cyster JG, Immunol Rev. 2016, 271, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Jung C, Hugot J-P, Barreau F, Int J Inflam. 2010, 2010, 823710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, Takahashi M, Fukuda NN, Murakami S, Miyauchi E, Hino S, Atarashi K, Onawa S, Fujimura Y, Lockett T, Clarke JM, Topping DL, Tomita M, Hori S, Ohara O, Morita T, Koseki H, Kikuchi J, Honda K, Hase K, Ohno H, Nature. 2013, 504, 446. [DOI] [PubMed] [Google Scholar]
- [119].Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, Yagita H, Ishii N, Evans R, Honda K, Takeda K, Nature. 2008, 455, 808. [DOI] [PubMed] [Google Scholar]
- [120].Zhang M, Yang X-J, World J Gastroenterol. 2016, 22, 8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Theriot CM, Koenigsknecht MJ, Carlson PE Jr., Hatton GE, Nelson AM, Li B, Huffnagle GB, Z Li J, Young VB, Nat. Commun 2014, 5, 3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Bailey MT, Dowd SE, Parry NMA, Galley JD, Schauer DB, Lyte M, Infect Immun. 2010, 78, 1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Singh P, Teal TK, Marsh TL, Tiedje JM, Mosci R, Jernigan K, Zell A, Newton DW, Salimnia H, Lephart P, Sundin D, Khalife W, Britton RA, Rudrik JT, Manning SD, Microbiome. 2015, 3, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Kim HJ, Huh D, Hamilton G, Ingber DE, Lab Chip. 2012, 12, 2165. [DOI] [PubMed] [Google Scholar]
- [125].Kim HJ, Li H, Collins JJ, Ingber DE, Proc Natl Acad Sci USA. 2016, 113, E7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Shah P, Fritz JV, Glaab E, Desai MS, Greenhalgh K, Frachet A, Niegowska M, Estes M, Jäger C, Seguin-Devaux C, Zenhausern F, Wilmes P, Nat. Commun 2016, 7, 11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Erdogan B, Ao M, White LM, Means AL, Brewer BM, Yang L, Washington MK, Shi C, Franco OE, Weaver AM, J. Cell Biol 2017, 216, 3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Kim J-Y, Fluri DA, Marchan R, Boonen K, Mohanty S, Singh P, Hammad S, Landuyt B, Hengstler JG, Kelm JM, J. Biotechnol 2015, 205, 24. [DOI] [PubMed] [Google Scholar]
- [129].Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, Kleeff J, Friess H, hepatology, Nat. Rev. Gastroenterol. Hepatol 2012, 9, 454. [DOI] [PubMed] [Google Scholar]
- [130].Han B, Qu C, Park K, Konieczny SF, Korc M, Cancer Lett. 2016, 380, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Vleminckx K, Vakaet L Jr, Mareel M, Fiers W, Van Roy F, Cell. 1991, 66, 107. [DOI] [PubMed] [Google Scholar]
- [132].Bregenzer ME, Horst EN, Mehta P, Novak CM, Raghavan S, Snyder CS, Mehta G, PloS one. 2019, 14, e0216564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Biswas SK, Mantovani A, Nat Immunol. 2010, 11, 889. [DOI] [PubMed] [Google Scholar]
- [134].Lan J, Sun L, Xu F, Liu L, Hu F, Song D, Hou Z, Wu W, Luo X, Wang J, Cancer Res. 2019, 79, 146. [DOI] [PubMed] [Google Scholar]
- [135].Bai J, Adriani G, Dang T-M, Tu T-Y, Penny H-XL, Wong S-C, Kamm RD, Thiery J-P, Oncotarget. 2015, 6, 25295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Heylman C, Sobrino A, Shirure VS, Hughes CC, George SC, Exp Biol Med (Maywood) 2014, 239, 1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Mannino RG, Santiago-Miranda AN, Pradhan P, Qiu Y, Mejias JC, Neelapu SS, Roy K, Lam WA, Lab Chip. 2017, 17, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Zhang YS, Zhang Y-N, Zhang W, Drug Discov Today. 2017, 22, 1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB, Kamm RD, Proc Natl Acad Sci USA. 2012, 109, 13515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Jeon JS, Bersini S, Gilardi M, Dubini G, Charest JL, Moretti M, Kamm RD, Proc Natl Acad Sci USA. 2015, 112, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Chen MB, Hajal C, Benjamin DC, Yu C, Azizgolshani H, Hynes RO, Kamm RD, Proc Natl Acad Sci USA. 2018, 115, 7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Kruger S, Ilmer M, Kobold S, Cadilha BL, Endres S, Ormanns S, Schuebbe G, Renz BW, D’Haese JG, Schloesser H, Heinemann V, Subklewe M, Boeck S, Werner J, von Bergwelt-Baildon M, J Exp Clin Cancer Res. 2019, 38, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Im A, Pavletic SZ, oncology J Hematol. Res 2017, 10, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Sahin U, Türeci Ö, Science. 2018, 359, 1355. [DOI] [PubMed] [Google Scholar]
- [145].Sanmamed MF, Chen L, Cell. 2018, 175, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Xia A-L, Xu Y, Lu X-J, J. Med. Genet 2019, 56, 1. [DOI] [PubMed] [Google Scholar]
- [147].Rosenberg SA, J. Immunol 2014, 192, 5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, N Engl J Med 2014, 371, 1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, Yu Z, Yang J, Wang B, Sun H, Nature. 2018, 560, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Zhang Y, Li Y, Cancer Cell Int. 2019, 19, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Guillerey C, Huntington ND, Smyth MJ, Nat Immunol. 2016, 17, 1025. [DOI] [PubMed] [Google Scholar]
- [152].Joyce JA, Fearon DT, Science. 2015, 348, 74. [DOI] [PubMed] [Google Scholar]
- [153].Brehm C, Huenecke S, Esser R, Kloess S, Quaiser A, Betz S, Zimmermann O, Soerensen J, Passweg JR, Klingebiel T, Cancer Immunol Immunother. 2014, 63, 821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Leclerc M, Voilin E, Gros G, Corgnac S, de Montpréville V, Validire P, Bismuth G, Mami-Chouaib F, Nat. Commun 2019, 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Farhood B, Najafi M, Mortezaee K, J. Cell. Physiol 2019, 234, 8509. [DOI] [PubMed] [Google Scholar]
- [156].Stagg J, Smyth M, Drug News Perspect. 2007, 20, 155. [DOI] [PubMed] [Google Scholar]
- [157].Ye B, Stary CM, Gao Q, Wang Q, Zeng Z, Jian Z, Gu L, Xiong X, J. Immunol. Res 2017, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Pavesi A, Tan AT, Koh S, Chia A, Colombo M, Antonecchia E, Miccolis C, Ceccarello E, Adriani G, Raimondi MT, Kamm RD, Bertoletti A, JCI insight. 2017, 2, e89762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Lee SWL, Adriani G, Ceccarello E, Pavesi A, Tan AT, Bertoletti A, Kamm RD, Wong SC, Front. Immunol 2018, 9, 416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Wallstabe L, Göttlich C, Nelke LC, Kühnemundt J, Schwarz T, Nerreter T, Einsele H, Walles H, Dandekar G, Nietzer SL, Hudecek M, JCI insight. 2019, 4, e126345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Ando Y, Siegler EL, Ta HP, Cinay GE, Zhou H, Gorrell KA, Au H, Jarvis BM, Wang P, Shen K, Adv Healthc Mater. 2019, 8, 1900001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Park D, Son K, Hwang Y, Ko J, Lee Y, Doh J, Jeon NL, Front. Immunol 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Ayuso JM, Truttschel R, Gong MM, Humayun M, Virumbrales-Munoz M, Vitek R, Felder M, Gillies SD, Sondel P, Wisinski KB, Patankar M, Beebe DJ, Skala MC, Oncoimmunology. 2018, 8, 1553477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Parlato S, De Ninno A, Molfetta R, Toschi E, Salerno D, Mencattini A, Romagnoli G, Fragale A, Roccazzello L, Buoncervello M, Canini I, Bentivegna E, Falchi M, Bertani FR, Gerardino A, Martinelli E, Natale C, Paolini R, Businaro L, Gabriele L, Sci. Rep 2017, 7, 1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, Bowden M, Deng J, Liu H, Miao D, He MX, Walker W, Zhang G, Tian T, Cheng C, Wei Z, Palakurthi S, Bittinger M, Vitzthum H, Kim JW, Merlino A, Quinn M, Venkataramani C, Kaplan JA, Portell A, Gokhale PC, Phillips B, Smart A, Rotem A, Jones RE, Keogh L, Anguiano M, Stapleton L, Jia Z, Barzily-Rokni M, Cañadas I, Thai TC, Hammond MR, Vlahos R, Wang ES, Zhang H, Li S, Hanna GJ, Huang W, Hoang MP, Piris A, Eliane J-P, Stemmer-Rachamimov AO, Cameron L, Su M-J, Shah P, Izar B, Thakuria M, LeBoeuf NR, Rabinowits G, Gunda V, Parangi S, Cleary JM, Miller BC, Kitajima S, Thummalapalli R, Miao B, Barbie TU, Sivathanu V, Wong J, Richards WG, Bueno R, Yoon CH, Miret J, Herlyn M, Garraway LA, Van Allen EM, Freeman GJ, Kirschmeier PT, Lorch JH, Ott PA, Hodi FS, Flaherty KT, Kamm RD, Boland GM, Wong K-K, Dornan D, Paweletz CP, Barbie DA, Cancer Discov. 2018, 8, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Aref AR, Campisi M, Ivanova E, Portell A, Larios D, Piel BP, Mathur N, Zhou C, Coakley RV, Bartels A, Bowden M, Herbert Z, Hill S, Gilhooley S, Carter J, Cañadas I, Thai TC, Kitajima S, Chiono V, Paweletz CP, Barbie DA, Kamm RD, Jenkins RW, Lab on a Chip 2018, 18, 3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Moore N, Doty D, Zielstorff M, Kariv I, Moy L, Gimbel A, Chevillet J, Lowry N, Santos J, Mott V, Lab Chip. 2018, 18, 1844. [DOI] [PubMed] [Google Scholar]
- [168].Carman CV, Martinelli R, Front Immunol. 2015, 6, 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Jackson DG, Front Immunol. 2019, 10, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Lamberti G, Prabhakarpandian B, Garson C, Smith A, Pant K, Wang B, Kiani MF, Anal Chem. 2014, 86, 8344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Lu H, Koo LY, Wang WM, Lauffenburger DA, Griffith LG, Jensen KF, Anal Chem. 2004, 76, 5257. [DOI] [PubMed] [Google Scholar]
- [172].Cicha I, Beronov K, Ramirez EL, Osterode K, Goppelt-Struebe M, Raaz D, Yilmaz A, Daniel WG, Garlichs CD, Atherosclerosis. 2009, 207, 93. [DOI] [PubMed] [Google Scholar]
- [173].Rouleau L, Copland IB, Tardif JC, Mongrain R, Leask RL, Ann Biomed Eng. 2010, 38, 2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Venugopal Menon N, Tay HM, Pang KT, Dalan R, Wong SC, Wang X, Li KHH, Hou HW, APL Bioeng. 2018, 2, 016103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Chen Z, Tang M, Huang D, Jiang W, Li M, Ji H, Park J, Xu B, Atchison LJ, Truskey GA, Leong KW, Lab Chip. 2018, 18, 2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Robert J, Weber B, Frese L, Emmert MY, Schmidt D, von Eckardstein A, Rohrer L, Hoerstrup SP, PLoS One. 2013, 8, e79821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Sharifi F, Htwe SS, Righi M, Liu H, Pietralunga A, Yesil-Celiktas O, Maharjan S, Cha BH, Shin SR, Dokmeci MR, Vrana NE, Ghaemmaghami AM, Khademhosseini A, Zhang YS, Adv Healthc Mater. 2019, 8, e1801425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Wild J, Soehnlein O, Dietel B, Urschel K, Garlichs CD, Cicha I, Thrombosis and haemostasis 2014, 112, 183. [DOI] [PubMed] [Google Scholar]
- [179].Wu W-H, Punde TH, Shih P-C, Fu C-Y, Wang T-P, Hsu L, Chang H-Y, Liu C-H, Sens Actuators B Chem. 2015, 209, 470. [Google Scholar]