

A multidisciplinary approach yields novel inhibitors and reveals substrate specificity, structure, and mechanisms of CoV-2 PLpro.
Abstract
Viral papain-like cysteine protease (PLpro, NSP3) is essential for SARS-CoV-2 replication and represents a promising target for the development of antiviral drugs. Here, we used a combinatorial substrate library and performed comprehensive activity profiling of SARS-CoV-2 PLpro. On the scaffold of the best hits from positional scanning, we designed optimal fluorogenic substrates and irreversible inhibitors with a high degree of selectivity for SARS PLpro. We determined crystal structures of two of these inhibitors in complex with SARS-CoV-2 PLpro that reveals their inhibitory mechanisms and provides a molecular basis for the observed substrate specificity profiles. Last, we demonstrate that SARS-CoV-2 PLpro harbors deISGylating activity similar to SARSCoV-1 PLpro but its ability to hydrolyze K48-linked Ub chains is diminished, which our sequence and structure analysis provides a basis for. Together, this work has revealed the molecular rules governing PLpro substrate specificity and provides a framework for development of inhibitors with potential therapeutic value or drug repurposing.
INTRODUCTION
The global epidemic of three coronaviruses has emerged in this century so far. In November 2002 in Foshan, China, the first known case of human infected with severe acute respiratory syndrome coronavirus (SARS-CoV) has been reported (1). By July 2003, more than 8000 SARS cases were detected in 27 countries. The main symptoms of SARS-CoV infection were influenza-like and included fever, headache, malaise, shivering, and diarrhea. Only a few cases of infection occurred between December 2003 and January 2004 (2). The implementation of infection control measures has ended the global SARS outbreak. Ten years after the SARS pandemic, a new coronavirus, Middle East respiratory syndrome coronavirus (MERS-CoV) was diagnosed in Saudi Arabia man (3). Due to international travels of infected people, MERS-CoV has spread worldwide. A total of 2502 laboratory-confirmed cases of MERS-CoV infection were reported from September 2012 to the end of December 2019, including 858 associated deaths. In December 2019, a novel coronavirus, SARS-CoV-2, formerly known as the 2019 novel coronavirus (2019-nCoV) was identified in Wuhan, China (4, 5). Current studies indicate that this coronavirus is similar to SARS-CoV. Although these three coronaviruses—SARS-CoV, MERS-CoV, and SARS-CoV-2—are identified as a highly pathogenic into the human population, there is no effective antiviral treatment. Therefore, current studies are focused on rapid development of vaccines and antiviral drugs to prevent and treat coronavirus infection.
One of the attractive antiviral drug targets is the SARS-CoV–encoded cysteine protease—papain-like protease (PLpro) (6). This enzyme recognizes the tetrapeptide LXGG motif found in-between viral proteins nsp1 and nsp2, nsp2 and nsp3, and nsp3 and nsp4 (nsp1/2, nsp2/3, and nsp3/4) (7, 8). The hydrolysis of the peptide bond on the carboxyl side of glycine at the P1 position leads to the release of nsp1, nsp2, and nsp3 proteins, which are essential for viral replication. The in vitro studies have shown that SARS-CoV PLpro harbors two other proteolytic activities, removal of ubiquitin (Ub) and Ub-like (Ubl) protein ISG15 (interferon-induced gene 15) from cellular proteins (9–11). Ubiquitinated and ISGylated substrates are more efficiently hydrolyzed by SARS-CoV PLpro than small substrates containing C-terminal LRGG motif (11, 12). These results indicated a more complex mechanism of substrate recognition than only the interaction of S4-S1 pockets of enzyme with tetrapeptide fragment. Further studies revealed that SARS-CoV PLpro have two distinct Ub binding subsites (SUb1 and SUb2) and recognize Lys48-linked polyUb chains for polyUb chain editing and/or deubiquitination of polyubiquitinated proteins (13–15).
Due to the deubiquitinating and deISGylating activities of SARS-CoV PLpro, this enzyme performs an important role in the innate immune response during viral infection (16, 17). SARS-CoV PLpro is involved in inhibiting the production of cytokines and chemokines that are responsible for the activation of the host innate immune response against viral infection (18–20). For these reasons, this enzyme is an important molecular target in the design of SARS-CoV antiviral drugs. Despite substantial research efforts in the development of SARS-CoV inhibitors, efficacy data of these compounds from clinical trials are missing (21–23). Nevertheless, we hypothesize that information gained over the past years for the SARS-CoV PLpro could be immediately translated into the timely study of SARS-CoV-2 PLpro to accelerate new antivirals development and drug retargeting approaches.
A molecular understanding of CoV-2 PLpro substrate specificity, structure, and mechanism would greatly facilitate development of effective PLpro inhibitors by enabling rational design and research on drug retargeting. In this study, we first performed comprehensive activity profiling of SARS-CoV-2 PLpro using our novel chemical approach, HyCoSuL (Hybrid Combinatorial Substrate Library) (24). The results reveal the molecular rules governing PLpro substrate specificity. Leveraging this information, we next designed and biochemically characterized potent inhibitors (VIR250 and VIR251) harboring high selectivity for SARS-CoV-2 PLpro and the related SARSCoV-1 PLpro versus other proteases. We determined crystal structures of VIR250 and VIR251 in complex with SARS-CoV-2 PLpro, which reveals their inhibitory mechanisms and provides a structural basis for the observed substrate specificity profiles. The unexpected finding that the P4 amino acids of VIR250 and VIR251 occupy opposite sides of the broad S4 pocket of SARS-CoV-2 PLpro and that there are additional regions of this pocket that are unengaged by either inhibitor raise the possibility that our structures will inform future drug discovery efforts. Last, we examined processing of Ub and Ubl protein variants by SARS-CoV-1 and -2 PLpro. These studies revealed that SARS-CoV-2 PLpro harbors deISGylating activities similar to SARSCoV-1 PLpro, but its ability to hydrolyze K48-linked Ub chains is substantially diminished, which our sequence and structural analyses provide a basis for. This finding is important, given the role of Ub and ISG15 conjugation in evasion of the host innate immune response. Together, our data also give a hope for design of a drug that can act as a pan-selective inhibitor against both SARS-CoV PLpro and SARS-CoV-2 PLpro and may have some universal value against emerging coronaviruses in the near future.
RESULTS
Substrate specificity profile
SARS-CoV-2 PLpro recognizes the tetrapeptide LXGG motif found in between viral proteins nsp1 and nsp2, nsp2 and nsp3, and nsp3 and nsp4 (Fig. 1A) (7, 8). Hydrolysis of the peptide bond on the carboxyl side of glycine at the P1 position leads to the release of nsp1, nsp2, and nsp3 proteins, which are essential for viral replication. SARS-CoV-2 PLpro also harbors deubiquitinating and deISGylating activities and recognizes the conserved LRGG motif at the C terminus of these proteins. (Fig. 1A). Our previous studies of SARS-CoV-1 PLpro substrate preferences using a combinatorial substrate library containing only natural amino acids revealed that this protease recognizes LXGG motif at P4-P1 positions with broad substrate specificity at P3 position (25). These results suggest that more detailed mapping of binding pocket architecture should facilitate the design of new, active substrates and optimal peptide sequences for inhibitor development efforts. To achieve this goal, we developed a defined and combinatorial substrate library (HyCoSuL) containing wide variety of nonproteinogenic amino acids (24).
Fig. 1. Activity profiling of SARS-CoV-1 and 2 PLpro protease.
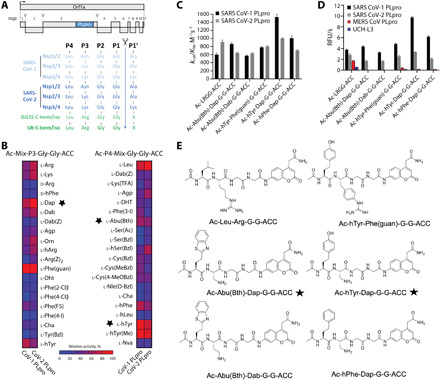
(A) Top: Schematic representation of Orf1a from SARS-hCoV-2, indicating the processed nonstructural proteins (nsps). PLpro cleavage sites are indicated by scissors. Bottom; Sequence of alignment of P4-P1’ amino acids of the indicated PLpro cleavage sites. (B) SARS-CoV-1 PLpro and SARS-CoV 2 PLpro substrate specificity profiles at the P3 and P4 positions presented as heat maps. Asterisk indicates P3 and P4 groups used throughout the study. (C) kcat/KM for tetrapeptide fluorogenic substrates toward SARS-CoV-1 PLpro and SARS-CoV-2 PLpro. (D) The rate of tetrapeptide substrate hydrolysis by DUBs. ([S] = 10 μM; SARS-CoV PLpro concentration, 0.2 μM; SARS-CoV-2 PLpro concentrations, 0.1 μM; MERS-CoV PLpro concentration, 2.5 μM; UCH-L3 concentration, 8.8 μM). (E) Tetrapeptide substrate structures. RFU/s, relative fluorescence unit per second.
Since tetrapeptide fluorogenic substrates are not very efficiently hydrolyzed by enzymes exhibiting deubiquitinating activity, we designed and synthesized the P2 defined library with a general structure of Ac-LRXG-ACC (X: 19 natural and 109 nonproteinogenic amino acids) and a HyCoSuL, where three positions were fixed and one position contains an equimolar mixture of 19 amino acids (Mix) (P3 sublibrary: Ac-Mix-P3-Gly-Gly-ACC, P4 sublibrary: Ac-P4-Mix-Gly-Gly-ACC; P3 and P4—a natural or nonproteinogenic amino acid) (26). By design of libraries with tailored peptide scaffold toward deubiquitinases (DUBs), we could reach the highest possible concentration of individual fluorogenic substrates in each sublibrary during the assay.
P2 library screening revealed that SARS-CoV and SARS-CoV-2 PLpro have very high substrate specificity at this position—only glycine can be accepted (fig. S1). Both proteases exhibit a broad substrate preference at P3 position (Fig. 1B). The S3 pocket of SARS-CoV and SARS-CoV-2 PLpro can tolerate not only positively charged residues like Phe(guan), Dap, Dab, Arg, Lys, Orn, and hArg but also hydrophobic amino acids, such as hTyr, Phe(F5), Cha, Met, Met(O), Met(O)2, d-hPhe (amino acid structures presented in table S1). These enzymes do not recognize acidic residues and most d-amino acids (the exception are d-Arg, d-hPhe, d-Lys, and d-Phg). The S4 pocket of SARS-CoV and SARS-CoV-2 PLpro can accommodate hydrophobic residues only, among natural amino acids, practically only leucine can be tolerated (being the best hit for SARS-CoV-2 PLpro) (Fig. 1B). SARS-CoV PLpro recognized two nonproteinogenic residues better than leucine at P4 position [hTyr and hTyr(Me)]. Other bulky amino acids are also accepted [≥30%, hPhe, Abu(Bth), Phe(3-I), Cys(Bzl), Cys(MeBzl), Cys(4-MeOBzl), hSer(Bzl), and Dht] (Fig. 1B).
Design and kinetic analysis of tetrapeptide fluorogenic substrates
To validate the library-screening data, we designed optimal tetrapeptide fluorogenic substrates to find optimal sequences recognized by SARS-CoV and SARS-CoV-2 PLpro. We analyzed both SARS PLpro substrate specificity profile at the P4-P2 positions and selected the most preferred amino acids [P2: Gly; P3: Dap and Phe(guan); P4: hTyr, hPhe, and Abu(Bth)] (Fig. 1E). Kinetic analysis revealed that some designed substrates were better recognized by SARS-CoV-1 PLpro with Ac-hTyr-Dap-Gly-Gly-ACC being almost 2.5 times more efficiently cleaved than endogenous Ac-LRGG-ACC. In the case of SARS-CoV-2 PLpro, we did not find substantial difference between Ac-LRGG-ACC and all tested substrates (Fig. 1C). It is important to notice that substitution of Arg in P3 position by relatively small Dap did not affect binding to S3 pocket and yields very good substrates (Fig. 1C). Thus, data obtained from combinatorial screening translate very well into individual substrates and demonstrate very high level of similarity between two investigated enzymes.
Next, we wanted to see whether incorporation of nonproteinogenic amino acids in P4 and P3 positions of peptide sequence can result in selective tetrapeptide substrates. We tested the substrates with four enzymes that exhibit deubiquitinating activity—SARS-CoV PLpro, SARS-CoV-2 PLpro, MERS-CoV PLpro, and human DUB UCH-L3. We have found that none of the substrates with nonproteinogenic amino acids in the sequence were substantially recognized at 10 μM either by MERS-CoV PLpro (2.5 μM) or human DUB UCH-L3 (8 μM) (Fig. 1D). In line with previous data, Ac-LRGG-ACC was recognized by all four enzymes (Fig. 1D).
Development of PLpro inhibitors
To further analyze selectivity of peptide sequences with nonproteinogenic amino acids, we converted two substrates [Ac-hTyr-Dap-Gly-Gly-ACC and Ac-Abu(Bth)-Dap-Gly-Gly-ACC] into inhibitors by exchanging the fluorescent tag to a reactive group—vinylmethyl ester (VME). A VME group was selected due to its broad reactivity toward DUBs (inhibitor selectivity is determined by tetrapeptide sequence). The results from kinetic analysis of SARS-CoV PLpro and SARS-CoV-2 PLpro inhibitors reflected those of substrate hydrolysis (Fig. 2, A and B). Ac-hTyr-Dap-Gly-Gly-VME (hereafter referred to as VIR251) was more potent but less selective inhibitor toward these enzymes than Ac-Abu(Bth)-Dap-Gly-Gly-VME (hereafter referred to as VIR250). Both compounds exhibit high selectivity for SARS-PLpro variants and robustly inhibit both SARS-CoV PLpro and SARS-CoV-2 PLpro activities. In contrast, practically no inhibition of human DUB UCH-L3 and only a slight inhibition of MERS-PLpro was observed (Fig. 2, A and B). Furthermore, incubation of HeLa lysates with Ub-VME yields a cross-linking profile that is unaltered by titrations of VIR250 or VIR251 (Fig. 2C). Since a major cross-linking target of Ub-VME is known to be human DUB enzymes, these data suggest that VIR250 and VIR251 do not cross-react with human DUBs. This is an important finding in search for a selective antiviral molecule with minimal cross-reactivity with human DUBs.
Fig. 2. Characterization of nonnatural amino acid-containing inhibitors VIR250 and VIR251 and their crystal structures in complex with SARS-CoV-2 PLpro.
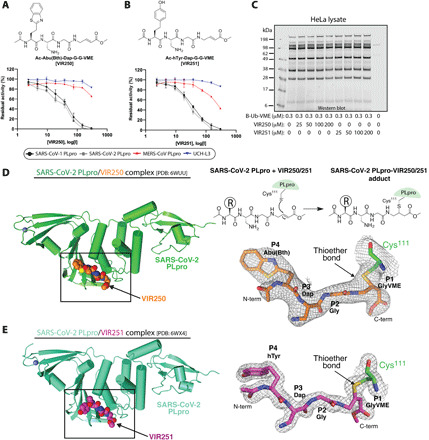
(A and B) DUB inhibition by designed inhibitors ([I] = 2.3–300 μM; SARS-CoV PLpro concentration, 0.3 μM; SARS-CoV-2 PLpro concentration, 0.1 μM; MERS-CoV PLpro concentration, 2.5 μM; UCH-L3 concentration, 8 μM). (C) HeLa lysate selectivity assay. HeLa cell lysate first incubated for 30 min with the indicated inhibitor and next for 30 min with Biotin-Ub-VME followed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot using streptavidin Alexa Fluor 647. (D) Top right: Schematic of the cross-linking of VIR250 and VIR251 to SARS-CoV-2 PLpro. Left: SARS-CoV-2 PLpro is shown as green ribbon representation and VIR250 is shown as spheres with carbon (orange) nitrogen (blue) oxygen (red) and sulfur (yellow). Bottom right: 2FoFc electron density map (1.0 σ) for VIR250 is shown as gray mesh. Carbon atoms of catalytic Cys111 of SARS-CoV-2 PLpro are shown as green sticks. The thioether linkage between Cys111 and VIR250 is indicated with an arrow. (E) SARS-CoV-2 PLpro/VIR251 structure presented as in (E) except with SARS-CoV-2 PLpro and VIR251 carbon atoms colored cyan and magenta, respectively. The electron density map is contoured at 1.5 σ.
Structures of CoV-2 PLpro in complex with VIR250 and VIR251
We next set out to determine crystal structures of SARS-CoV-2 PLpro in complex with VIR250 and VIR251 to gain insights into the molecular mechanism by which these molecules inhibit SARS-CoV-2 PLpro activity as well as the basis for the observed substrate selectivity profile. Catalytic cysteine-111 of CoV-2 PLpro engages in Michael Addition to the β carbon of the vinyl group of the VME warheads of VIR250 and VIR251, resulting in formation of a covalent thioether linkage (Fig. 2D). Large-scale cross-linking reactions yielded CoV-2 PLpro-VIR250 and CoV-2 PLpro-VIR251 complexes of yield and purity sufficient for growth of diffraction quality crystals. The structure of CoV-2 PLpro in complex with VIR250 (Fig. 2D) [Protein Data Bank (PDB): 6WUU] was determined by molecular replacement using the recently determined structure of apo CoV-2 PLpro (PDB: 6W9C) and was resolved to 2.79 Å resolution with R/Rfree values of 0.195/0.230 (table S2). This structure was used as the molecular replacement search model for determination of the structure of CoV-2 PLpro in complex with VIR251 (Fig. 2E) (PDB: 6WX4). The CoV-2 PLpro/VIR251 structure was resolved to 1.65 Å resolution and refined to R/Rfree values of 0.170/0.196 (table S2).
Comparison of apo CoV-2 PLpro to CoV-2 PLpro/VIR250 and CoV-2 PLpro/VIR251 complexes reveal similar overall structures with the exception of the β14-β15 loop that is situated proximal to the active site and undergoes a conformational change in that is likely due to inhibitor binding (fig. S2) (see below). This analysis shows that there are also slight rigid body rotations of the finger and Ubl domains of CoV-2 PLpro that are likely due to crystal packing effects. Analysis of the structures reveal extensive electron density projecting from the catalytic Cys111 side chain of CoV-2 PLpro into which all the atoms of VIR250 and VIR251 could unambiguously be placed (Fig. 2, D and E). Furthermore, the covalent bond between Cys111 and both VIR250 and VIR251 are clear (Fig. 2, D and E). As anticipated, both VIR250 and VIR251 inhibitors occupy the S4-S1 pockets of CoV-2 PLpro in proximity to the active site and adopt similar structures with the exception of the orientation of the P4 substituents, which will be discussed in greater detail below. The P4 position is the only region of chemical divergence between VIR250 and VIR251, with an Abu(Bth) in VIR250 and an h Tyr in VIR251 (Fig. 2, A and B).
Molecular recognition of VIR250 and VIR251
Analysis of the CoV-2 PLpro/VIR250 (PDB: 6WUU) and CoV-2 PLpro/VIR251 (PDB: 6WX4) complexes reveals a similar network of interacting residues with ~560 Å2 from a total of ~775 Å2 solvent accessible area of VIR251 and ~600 Å2 from a total of ~800 Å2 solvent accessible area of VIR250 buried upon complex formation. With the exception of the P4 positions of VIR250 and VIR251, which engage largely in hydrophobic interactions with CoV-2 PLpro, most of the interactions at the P1-P3 positions of both inhibitors are mediated through polar interactions and hydrogen bonds (Fig. 3, A and B). At the P1 position of VIR250, GlyVME is covalently linked via thioether bond to catalytic Cys111 of CoV-2 PLpro and engages in a backbone-backbone hydrogen bond to Gly271 (Fig. 3A). At the P2 VIR250 position, Gly engages in two backbone-backbone hydrogen bonds to Gly163, and van der Waals contacts to Leu163 and Tyr164 of CoV-2 PLpro and P3 Dap of VIR250 participates in a backbone-backbone hydrogen bond with Gly271 (Fig. 3A). The network of backbone-backbone hydrogen bonds participated in at the P3-P1 positions of VIR250 are fully conserved in VIR251 (Fig. 3B). In contrast, while the methylester group from the GlyVME warhead of VIR250 engages in a hydrogen bond with His272 from the catalytic triad of CoV-2 PLpro, the corresponding methylester of VIR251 participates in hydrogen bonds with Trp106 and Asn109 side chains, which are proposed to contribute to oxyanion hole stabilization (Fig. 3, A and B). Trp106 adopts a different conformation and is poorly ordered in the VIR250 complex (Fig. 3A).
Fig. 3. Molecular recognition of VIR250 and VIR251 by SARS-CoV-2 PLpro.
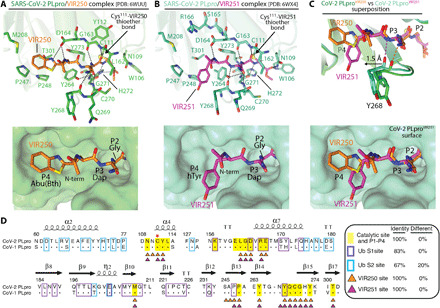
(A) Top: Network of contacts between SARS-CoV-2 PLpro (green) and VIR250 (orange) with involved residues shown as sticks with red oxygen atoms, blue nitrogen atoms, and yellow sulfur atoms. Hydrogen bonds are indicated by dashed lines. Bottom: The VIR250 binding pocket of CoV-2 PLpro is shown as green surface with VIR250 shown as sticks. The P2-P4 positions are labeled. (B) Network of contacts between SARS-CoV-2 PLpro (cyan) and VIR251 (magenta) shown as in (A). (C) Superimposition of the CoV-2 PLpro/VIR250 and CoV-2 PLpro/VIR251 structures presented as in (A) and (B). Only the surface of Cov-2 PLpro from the PLpro/VIR251 structure is shown in the bottom panel. (D) Structure-based sequence alignment CoV-1 and CoV-2 PLpro with secondary structure was shown above sequence. Conserved residues, dots; catalytic cysteine, red star. Residues buried at the CoV-2 PLpro/VIR250 and CoV-2 PLpro/VIR251 interfaces are indicated by orange and magenta triangles below the alignment, respectively. Residues involved in contacts with the S1 Ub residue 73 to 76 (P1-P4), S1 Ub, and S2 Ub based on the SARS-CoV-2 PLpro/K48 diUb structure (PDB: 5E6J) are shaded yellow and boxed purple and cyan, respectively.
There are important differences in how the side chains of the P3 and P4 positions of VIR250 and VIR251 engage CoV-2 PLpro. The side-chain amine of Dap at the P3 position of VIR250 engages in a hydrogen bond with the backbone carbonyl oxygen of Tyr268 and the P4 Abu(Bth) projects toward Met208, Pro247, Pro248, and Thr301 where it engages in a network of van der Waals interactions (Fig. 3A). In contrast, it is the backbone amine of P3 Dap that engages in the hydrogen bond to the carbonyl oxygen of Tyr268, and unexpectedly, hTyr at the P4 position projects toward the opposite side of the S4 pocket compared to Abu(Bth) from VIR250 by extending toward Pro248, Tyr264, and Tyr268 of CoV-2 PLpro and participating in a distinct network of van der Waals interactions (Fig. 3B). This new network of interactions is facilitated by a 1.5-Å shift of the β14-β15 loop (Asn267, Tyr268, and Gln269) toward the hTyr of VIR251 (Fig. 3C), thereby facilitating many novel contacts that would be unable to occur in the absence of this shift. Notably, all of the CoV-2 PLpro residues involved in contacts to both VIR250 and VIR251 are fully conserved in SARS CoV-1 PLpro, and the overall structures of the two SARS PLpro variants are very similar in the catalytic site of the enzyme that likely accounts for the ability of these inhibitors to target both enzymes (Figs. 3C and 5).
Fig. 5. A molecular basis for the observed Ub/Ubl processing profiles of SARS-CoV-1 PLpro and SARS-CoV-2 PLpro.
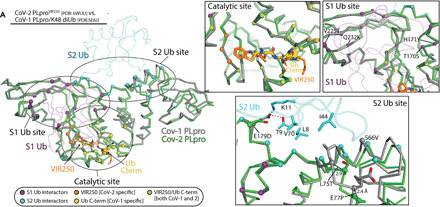
(A) The previously determined CoV-1 PLpro/K48 diUb complex structure (PDB: 5E6J) was superimposed onto our CoV-2 PLpro/VIR250 structure. Selected Cα atoms are shown as spheres to highlight residues involved in the different interfaces in the structures. The Cα atoms of residues contacting the S1 Ub and S2 Ub in the CoV-1 PLpro/K48 diUb complex structure are shown as purple and cyan spheres, respectively. The Cα atoms of conserved residues contacting both VIR250 and the Ub C terminus (LRGG) in the CoV-2 PLpro/VIR250 and CoV-1 PLpro/K48 diUb structures are shown as green spheres. Residues involved specifically in contacts to VIR250 and the Ub C terminus are shown as orange and yellow spheres, respectively. Magnified view of the indicated interfaces are shown to the right and residues that are different between CoV-1 and CoV-2 PLpro are written with respect to CoV-1 sequence.
In terms of how our structures correlate with the observed substrate selectivity profiles described above, P2 dependence on Gly is the result of residues from the β14-β15 and α5-α6 loops of CoV-2 PLpro (notably, Leu162, Tyr264, Cys270, Gly271, and Tyr273) clamping down on top of the P2 position, leaving no room for side-chain atoms at the R position (Fig. 3, A to C). The preference for positive and hydrophobic residues and selection against acidic residues at the P3 position is likely the result of its broader pocket and proximity to the acidic carbonyl oxygens of Tyr268, Gln269, and Leu162, the side chain of Asp164, as well as the hydrophobic side chains of Leu162 and Tyr268 (Fig. 3, A to C). At the P4 position, the strong preference for bulky hydrophobic residues can be explained by the hydrophobic nature of the P4 binding pocket that is largely formed by residues Met208, Pro247, Pro248, Tyr264, and Tyr268 (Fig. 3, A and B). Notably, the very deep and broad nature of the S4 pocket of SARS-CoV-2 PLpro has been exploited by the Abu(Bth) and h Tyr sidechains at P4 of VIR250 and VIR251, which, as noted above, project toward different ends of the S4 pocket and engage in distinct networks of contacts (Fig. 3C). With that said, there remain regions at the deepest parts of this pocket, particularly an acidic patch formed by Asp164, Tyr273, and Thr301 that could potentially be exploited for development of more potent inhibitors.
Processing of Ub and Ubl variants by CoV-1 and CoV-2 PLpro
Studies carried out for SARS-CoV-1 PLpro revealed that this enzyme has a Ub binding domain and efficiently process full Ub fluorogenic substrates (12). We wanted to see whether this is also the case for SARS-CoV-2 PLpro. To that end, we used Ub activity-based probe (ABP) for labeling of both enzymes. In this ABP, biotin was used as detection tag and VME as an irreversible warhead that cross-links to the catalytic cysteines. To test its sensitivity, we performed SDS–polyacrylamide gel electrophoresis (SDS-PAGE) analysis followed by nitrocellulose membrane transfer and visualization with fluorescent streptavidin (Fig. 4A). We observed substantial labeling of both proteases by Biotin-Ub-VME at a concentration twice higher than the enzyme concentration (200 nM); however, SARS-CoV-1 PLpro was more efficiently labeled compared with SARS-CoV-2 PLpro. We next decided to explore the Ub and Ubl protein substrate specificity of SARS-CoV-2 PLpro by testing its reactivity with a panel of Ub/Ubl ABPs. This panel of probes includes a (nonhydrolyzable) K48-linked diUb linked at the C terminus to a peptide with a vinyl amide (VA) warhead (K48 diUb-VA; fig. S3, probe 1), a (nonhydrolyzable) K63-linked diUb linked at the C terminus to a propargyl amide (PA) warhead (K63 diUb-PA; fig. S3, probe 2), mono Ub-PA (fig. S3, probe 3), and murine ISG15 C-terminal domain (CTD)–vinyl pentynyl sulfone (VPS) (ISG15CTD-VPS; fig. S3, probe 4). The results of this analysis indicated a preference of SARS-CoV-2 PLpro for recognition of ISG15 and monoUb and a poor ability to recognize K48-linked and K63-linked diUb (Fig. 4B). As expected, preincubation of SARS-CoV-2 PLpro with VIR251 completely blocked its ability to react with all of the probes (Fig. 4B). The apparent lack of reactivity with the K48-linked diUb probe and the increased processing of ISG15CTD-VPS relative to monoUb-PA was unexpected since the very highly related SARS-CoV-1 PLpro has been demonstrated to robustly process K48-linked polyUb chains and to exhibit a preference for Ub over ISG15 (13, 15).
Fig. 4. Processing of Ub and Ubl variants by SARS-CoV-1 PLpro and SARS-CoV-2 PLpro.
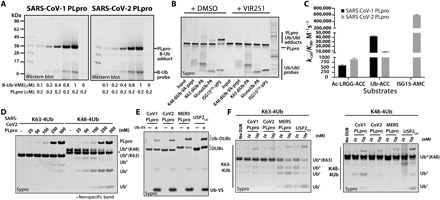
(A) SARS-CoV-1 PLpro and SARS-CoV-2 PLpro labeling by Biotin-Ub-VME. Recombinant enzymes were incubated with the indicated B-Ub-VME concentrations for 45 min at 37°C, analyzed by SDS-PAGE, and subjected to Western blot using fluorescent streptavidin Alexa Fluor 647 conjugate. (B) SARS-CoV-2 PLpro was pretreated with dimethyl sulfoxide (DMSO) or VIR251 for 30 min at 37°C and subsequently incubated with the indicated activity based Ub/Ubl probe at RT for 2 min. This was followed by SDS-PAGE and sypro staining. (C) Kinetic parameters of selected substrates for SARS-CoV PLpro and SARS-CoV-2 PLpro. Asterisk indicates that CoV-1 PLpro/ISG15-AMC pair were not analyzed in this study but kcat/KM values have previously been determined (13, 15). (D) The indicated tetraUb chains were incubated with CoV-2 PLpro for 30 min at 37°C, subjected to SDS-PAGE and sypro staining. (E) Ub-Vinyl Sulfone labeling. Ub-VS was incubated for 30 min at 37°C with the indicated protease, subjected to SDS-PAGE and sypro staining. (F) The indicated tetraUb chains were incubated with the indicated protease and analyzed as in (D).
To more thoroughly examine these differences, we performed a comparison of the kinetics of SARS-CoV-1 and CoV-2 PLpro processing of LRGG-ACC, Ub-ACC, and ISG15-AMC fluorogenic substrates. The results of this experiment show that SARS-CoV-2 PLpro processes Ub-ACC fourfold less efficiently compared to SARS-CoV-1 PLpro and that SARS-CoV-2 PLpro processes ISG15-AMC 60-fold more efficiently than Ub-ACC (Fig. 4C). Furthermore, SARS-CoV-2 PLpro, like SARS-CoV-1 PLpro, more robustly processes K48 tetraUb compared to K63 tetraUb (Fig. 4D), and cross-links to the ABP Ub-VS similarly to SARS-CoV-1 PLpro, MERS PLpro, and the human DUB USP2CD (Fig. 4E). Yet, in side-by-side comparison, SARS-CoV-2 PLpro demonstrates a substantially diminished ability to process K48 tetraUb compared to SARS-CoV-1 PLpro (Fig. 4F). This was an unexpected finding, as we and others have shown before that SARS-CoV-1 PLpro displays a preference for recognition of K48 diUb linkages over ISG15 (13, 15). As expected and shown before, both MERS PLpro and USP2CD efficiently processes both types of Ub chains.
The substantially diminished ability of SARS-CoV-2 PLpro to process K48 polyUb chains compared to SARS-CoV-1 PLpro was unexpected considering the very high overall similarity between the enzymes (83% identity, 9% similarity) (Fig. 3D). To try to reconcile this apparent contradiction, we compared our SARS-CoV-2 PLpro structures with the previously reported structure of SARS-CoV-1 PLpro in complex with K48 diUb (Fig. 5) (PDB: 5E6J) (15). This structure revealed three key interfaces: (i) the catalytic site that accommodates the C terminus of Ub with L73, R74, G75, and G76 constituting the S4-S1 residues; (ii) a binding site for the “S1 Ub,” which is the Ub N-terminal to the cleavage site in the K48 polyUb chain; and (iii) a binding site for the “S2 Ub,” which is N-terminal to the S1 Ub (Fig. 5). Comparative analysis of the catalytic sites of SARS-CoV-1 and 2 PLpro shows a 100% conservation of residues involved in contacts to the S4-S1 positions of S1 Ub, VIR250, and VIR251 (Fig. 3D) and expectedly a very similar structure in this region (Fig. 5). While the Ub S1 site harbors more variability than the catalytic site, the overall amino acid conservation is still very high (83% identity, 17% similarity) (Fig. 3D) and the structures align well in this region (Fig. 5).
In contrast to the catalytic and S1 Ub sites, the S2 Ub site of SARS-CoV-2 PLpro harbors much less conservation at the amino acid level (67% identity, 13% similarity) compared to SARS-CoV-1 PLpro (Fig. 3D), and there are several structural differences at these regions important for molecular recognition of the S2 Ub (Fig. 5). A key interaction surface at this interface is formed by the Ile44 hydrophobic patch (formed by Leu8, Ile44, and Val70), and in the SARS-CoV-1 PLpro/K48 diUb structure, the Ile44 patch of the S2 Ub engages in a network of hydrophobic contacts with Phe70, Leu75, and others (Fig. 5). This residue is changed to a threonine in SARS-CoV-2, which would be unable to engage in a similar network of contacts with S2 Ub as leucine. Furthermore, much of the SARS-CoV-2 structure in proximity to the S2 Ub site adopts a slightly different structure including a ~3-Å translation of Thr75 of SARS-CoV-2 PLpro relative to Leu75 of SARS-CoV-1 PLpro and other notable amino acid changes of S66V and E77P (Fig. 5). Last, Glu179 of SARS-CoV-1 PLpro engages in hydrogen bonds to Thr9 and Lys11 of S2 Ub (Fig. 5). This residue has changed to an aspartate in SARS-CoV-2 PLpro, which is a conservative change, but the shorter aspartate side chain is unable to engage in a similar set of contacts (Fig. 5). Based on our analysis, we posit that the diminished ability of SARS-CoV-2 PLpro to process K48 polyUb is largely due to the aforementioned differences at the S2 Ub binding site. Consistent with this hypothesis, mutation of Leu75 of SARS-CoV-1 PLpro to serine resulted in a fivefold reduction in binding of K48 diUb with no apparent effect on monoUb (13). In combination with other changes in SARS-CoV-2 PLpro such as E179D, there appears to be a cumulative effect of several relatively minor changes between SARS-CoV-1 and 2 PLpro at the S2 binding site that together have a substantial effect on their ability to process K48 polyUb. Whether these changes also account for the apparent preference of SARS-CoV-2 PLpro for ISG15 over Ub and whether these intriguing differences in the function of SARS-CoV-1 and 2 PLpro have any effect on the biology of the viruses remain to be seen. Notably, our conclusion regarding the deISGylating activity and diminished processing of K48-Ub linkages by CoV-2 PLpro relative to CoV-1 PLpro has been independently corroborated by two preprints (27, 28) and one recent manuscript (29).
DISCUSSION AND CONCLUSIONS
The outbreak of the current coronavirus pandemic leading to COVID-19 disease has markedly accelerated research into effective drugs and a vaccine to treat this disease. SARS-CoV-2 PLpro is an excellent candidate for antiviral drug development, as they are not only blocking virus replication but also inhibiting the dysregulation of signaling cascades in infected cells (9). A detailed molecular understanding of CoV-2 PLpro substrate specificity, structure, and mechanism would greatly facilitate development of effective PLpro inhibitors by enabling rational design and research on drug retargeting, and this was the major focus of our study. In this study, we examined SARS-CoV-2 PLpro substrate preferences at positions P4-P2 and compare them directly with the well-known SARS virus 2002/03 protein, SARS-CoV PLpro. For this purpose, we used positional scanning technology using natural and nonproteinogenic amino acids (HyCoSuL) (24). Library screening revealed that both enzymes recognize only Gly in P2 and have broad in P3 and rather narrow substrate specificity at the P4 position. Moreover, direct analysis of the preferences of both enzymes demonstrates that the architecture of S4-S2 pockets is almost identical, because they recognize natural and nonproteinogenic amino acids practically in a very similar way. The differences in activity for a given amino acid between the two enzymes observed in some positions are very small, and there are no amino acids that are recognized by one enzyme only. This is also confirmed by the analysis of amino acids building S4-S2 pockets in both enzymes, which is identical (Fig. 1B and fig. S1). This is critically important information in the aspect of using information from research on inhibitors or retargeting of drugs conducted in the past for SARS-CoV PLpro for immediate application to SARS-CoV-2 PLpro. Analysis of kinetic parameters for tetrapeptide substrates for both enzymes shows a high degree of similarity in terms of kcat/Michaelis constant (KM) values, proving that the catalytic yields of both enzymes are also similar. The sequences containing nonproteinogenic amino acids at P4-P3 positions were recognized only by both SARS-PLpro, not MERS-PLpro and the human DUB UCH-L3. This open the doors to the potential application of specific SARS-PLpro substrates developed in our work for use in cell culture studies such as localization of the targeted proteases and virus.
We next leveraged the information we gained regarding the molecular rules governing substrate selectivity by SARS-CoV-2 PLpro to develop covalent inhibitors VIR250 and VIR251. These inhibitors proved to be active and selectively inhibited the SARS-CoV-1 and -2 PLpro, but exhibited much weaker activity toward MERS-PLpro and practically no activity toward human DUB UCH-L3. This is valuable information in terms of conducting research toward the search for peptide antiviral compounds targeted to this enzyme. Our crystal structures of VIR250 and VIR251 in complex with SARS-CoV-2 PLpro reveal their inhibitory mechanisms and provide a structural basis for the observed substrate specificity profiles. Furthermore, the unexpected findings that the P4 amino acids of VIR250 and VIR251 occupy opposite sides of the broad S4 pocket of SARS-CoV-2 PLpro and that there are additional regions of this pocket that are unengaged by either inhibitor raise the possibility that our structures will inform future drug discovery efforts aimed at generating more potent inhibitors. Comparative analysis of the substrate specificity of SARS-CoV-2-Mpro and SARS-CoV-2 PLpro indicates that they have markedly different substrate specificity (30). This indicates that for peptidic inhibitors, it will be impossible to design an inhibitor that will act on both enzymes simultaneously. However, if peptidic inhibitors were found for both proteases separately, then it would probably be possible to use them as a cocktail. Another possible approach is searching for a small-molecule inhibitor that would promiscuously inhibit both Mpro and PLpro. Such an inhibitor would certainly be very beneficial in the therapeutic treatment of COVID-19, but it should be remembered that it could cross-react with human cysteine proteases, which could lead to undesirable side effects (31–33).
Furthermore, our substrate specificity studies conducted for SARS-CoV-2-Mpro (30) and here for SARS-CoV-2 PLpro indicate that both enzymes have virtually identical substrate specificity as their homologs from the previous SARS. Thus, the shapes of the binding pockets are virtually unchanged. This is valuable information from the standpoint of designing inhibitors as drugs for these enzymes. For the next SARS-type coronavirus that emerges in the future, there will definitely be a need to create a new vaccine, which is a time-consuming process. On the other hand, antiviral drugs developed on the basis of knowledge obtained from studies on SARS-1 and SARS-2 proteases will have a chance for immediate use in treatment through drug repurposing. This further indicates the high potential of both proteases as medical targets. Another possible application of our inhibitors is their use as selective ABPs to visualize SARS-CoV-2 PLpro activity in cells or even in COVID-19 diagnostics. Similar studies have already been conducted toward the use of ABPs for smallpox K7L protease, ZIKA, WNW, or dengue viruses proteases (34–36).
Last, we examined processing of Ub and Ubl variants by SARS-CoV-1 and -2 PLpro and found that SARS-CoV-2 PLpro harbors deISGylating activities similar to SARSCoV-1 PLpro but its ability to hydrolyze K48-linked Ub chains is substantially diminished. This was an unexpected result considering the very high sequence identity between SARS-CoV-1 and -2 PLpro; however, our structure analysis revealed subtle structural and sequence variations in the S2 Ub binding site of SARS-CoV-2 PLpro that we posit collectively diminish the ability of the S2 Ub of K48 polyUb to bind and subsequently be processed. Furthermore, analysis of the enzyme kinetics of the Ub-ACC substrate indicates that it is efficiently processed by the enzyme, but the difference between the tetrapeptide substrate and Ub is only about 10 times, when in the case of SARS-CoV-1 PLpro, this difference is around 60 times (Fig. 4C). This indicates some differences between both enzymes in the aspect of interaction in the exosite binding region related to amino acids identity and similarity. Given the role of Ub and ISG15 conjugation in evasion of the host innate immune responses, whether these intriguing differences in the function of SARS-CoV-1 and -2 PLpro have any effect on the biology of the viruses remains to be seen and will be the topic of future studies. Notably, two preprints (27, 28) and one recent manuscript (29) have all independently come the same conclusion as us regarding the deISGylating activity and diminished processing of K48-Ub linkages of CoV-2 PLpro relative to CoV-1 PLpro and have validated CoV-2 PLpro as a viable target for antiviral development.
Collectively, our work has revealed the molecular rules governing PLpro substrate specificity and reveals a very high level of sequence and structural similarity between SARS-CoV-1 and -2 PLpro in the substrate binding pocket. These findings signal that previously discovered information on SARS-CoV-1 PLpro can immediately be applied to the search for effective antiviral molecules and retargeting of known drugs for the inhibition of SARS-CoV-2 PLpro. Furthermore, structures of the novel inhibitors VIR250 and VIR251 in complex with SARS-CoV-2 PLpro provides a framework for rational development of inhibitors with improved potency and ABPs. It is worth noting that a flurry of preprint publications have conducted SARS-CoV-1 PLpro drug-repurposing studies against SARS-CoV-2 PLpro, showing that existing compounds can inhibit it (27–29, 37). We believe our profiling and crystallographic studies open up additional avenues in developing inhibitors with improved properties. Together, our data also provide hope for design of a drug that can act as a pan-selective inhibitor against both SARS-CoV-1 PLpro and SARS-CoV-2 PLpro and may have some universal value against emerging coronaviruses in the near future.
MATERIALS AND METHODS
Plasmids
For biochemical assays, the cDNA for PLpro corresponding to amino acids 745 to 1061 of SARS-CoV-2 NSP3 was codon-optimized for Escherichia coli expression, synthesized, and cloned into pGEX6P-1 (GE Healthcare, UK) using the Bam HI and Not I sites by Gene Universal (USA) for expression as a PreScission protease cleavable N-terminally glutathione S-transferase (GST)–tagged protein (table S3). For crystallization studies, the codon optimized SARS-CoV-2 cDNA was cloned into the Nde I and Xho I sites for expression as a C-terminally uncleavable 6× His tag (table S3). The plasmids were transformed into BL21 (DE3) codon and E. coli strain for protein expression.
Protein expression and purification
SARS-CoV PLpro, UCH-L3, and MERS-PLpro were obtained as described earlier (14, 25). SARS-CoV-2 PLpro transformed cells were grown in LB broth at 37°C with shaking until the optical density at 600 nm reached 1.5. Isopropyl-β-d-thiogalactopyranoside (0.1 mM) and ZnSO4 (0.1 mM) were added to induce protein expression overnight at 18°C. Cell pellet was resuspended in lysis buffer [20 mM tris-Cl (pH 8.0), 350 mM NaCl, and 2 mM β-mercaptoethanol) and lysed using sonication. The lysate was cleared by centrifugation at 35,000g for 30 min at 4°C. The lysate was passed onto Glutathione Sepharose 4B (GE) followed by washing with lysis buffer. The GST-tagged PLpro was eluted in lysis buffer supplemented with 20 mM reduced glutathione (pH 8.0). The fusion protein was cleaved using GST-PreScission protease at 4°C overnight followed with desalting and passing through fresh glutathione beads to remove cleaved GST and PreScission protease. The sample was further purified using Superdex 200-pg size-exclusion columns (GE) equilibrated with 20 mM tris-Cl (pH 8.0), 40 mM NaCl, and 2 mM dithiothreitol (DTT). The purified protein was then concentrated to ~10 mg/ml and snap-frozen in liquid nitrogen for later use.
Reagents
The reagents used for the solid-phase peptide synthesis (SPPS) were as follows: Rink amide (RA) resin (particle size 100 to 200 mesh; loading 0.74 mmol/g), 2-chlorotrityl chloride resin (particle size 100 to 200 mesh, loading 0.97 mmol/g), all 9-fluorenyl methoxycarbonyl–amino acids, O-benzotriazole-N,N,N`,N`-tetramethyl-uronium-hexafluoro-phosphate, 2-(1-H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluranium hexafluorophosphate (HATU), piperidine, diisopropylcarbodiimide, and trifluoroacetic acid (TFA), purchased from Iris Biotech GmbH (Marktredwitz, Germany); anhydrous N-hydroxybenzotriazole (HOBt) from Creosauls Louisville, KY, USA; 2,4,6-collidine (2,4,6-trimethylpyridine), high-performance liquid chromatography–grade acetonitrile, triisopropylsilane (TIPS), tBu-N-allyl carbamate, toluene, methyl acrylate, dichlorophenylborane, and second generation Grubbs catalyst from Sigma-Aldrich (Poznan, Poland); and N,N-diisopropylethylamie from VWR International (Gdansk, Poland). N,N-dimethylformamide (DMF), dichloromethane (DCM), methanol (MeOH), diethyl ether (Et2O), acetic acid (AcOH), and phosphorus pentoxide (P2O5) were obtained from Avantor (Gliwice, Poland). Individual substrates, Ub-ACC and B-Ub-VME, were purified by HPLC on a Waters M600 solvent delivery module with a Waters M2489 detector system using a semipreparative Wide Pore C8 Discovery column and Jupiter 10-μm C4 300-Å column (250 mm × 10 mm). The solvent composition was as follows: phase A (water/0.1% TFA) and phase B (acetonitrile/0.1% TFA). The purity of each compound was confirmed with an analytical HPLC system using a Jupiter 10-μm C4 300 Å column (250 × 4.6 mm). The solvent composition was as follows: phase A (water/0.1% TFA) and phase B (acetonitrile/0.1% TFA); gradient, from 5 to 95% B over a period of 15 or 20 min. The molecular weight of each substrate and B-Ub-VME was confirmed by high-resolution mass spectrometry (HRMS) on a High-Resolution Mass Spectrometer Waters LCT premier XE with electrospray ionization and a time-of-flight module.
Combinatorial and defined substrate library synthesis.
Detailed protocol of combinatorial and defined tetrapeptide fluorogenic substrate library synthesis was described elsewhere (26).
Determination of SARS-CoV and SARS-CoV-2 PLpro substrate specificity
Library screening was performed using a spectrofluorometer (Molecular Devices SpectraMax Gemini XPS) in 96-well plates containing substrates and enzymes. Assay conditions were 1 μl of substrate in dimethyl sulfoxide (DMSO) and 99 μl of enzyme, which had been incubated for 15 min at 37°C in assay buffer [150 mM NaCl, 20 mM tris, and 5 mM DTT (pH 8.0) for SARS-CoV PLpro; 5 mM NaCl, 20 mM tris, and 5 mM DTT (pH 8.0) for SARS-CoV-2 PLpro]. The final substrate concentration in each well was 200 μM combinatorial library and 100 μM defined P2 library. The final enzyme concentration was 1 μM SARS-CoV PLpro and 0.5 μM SARS-CoV-2 PLpro for P3 and P4 sublibraries and 0.1 μM SARS-CoV PLpro and 75 nM SARS-CoV-2 PLpro for Ac-Leu-Arg-P2-Gly-ACC. The release of ACC was measured continuously for 45 min (λex = 355 nm, λem = 460 nm). SARS-CoV and SARS-CoV-2 PLpro substrate specificity profiles were established by setting the highest relative fluorescence unit per second for the best substrate as to 100% and adjusting other results accordingly.
Synthesis of tetrapeptide fluorogenic substrates and Ub-ACC
Individual fluorogenic substrates were synthesized on a solid support using the SPPS method as previously described (24, 38). Each substrate was purified by HPLC and analyzed using analytical HPLC and HRMS. The purity of each compound was ≥95%. The individual substrates were dissolved at 20 mM in DMSO and stored at −80°C until use.
Kinetic studies of individual tetrapeptide substrates and Ub-ACC
Individual substrate hydrolysis was measured in the same assay conditions as for library screening. The final substrate concentration was 10 μM, SARS-CoV and SARS-CoV-2 PLpro concentration was 0.1 μM, MERS-CoV PLpro was 2.5 μM, and UCH-L3 was 8.8 μM. MERS-CoV PLpro and UCH-L3 were incubated for 30 min at 37°C in assay buffer [MERS-CoV PLpro: 150 mM NaCl, 20 mM tris, 5 mM DTT (pH 8.0); UCH-L3: 50 mM Hepes, 0.5 mM EDTA, 5 mM DTT (pH 7.5)] before add into the wells on plate. The measurements were repeated at least three times, and the results were presented as mean values with SDs. Kinetic parameters were determined for selected tetrapeptide substrates and Ub-ACC toward SARS-CoV and SARS-CoV-2 PLpro. Wells contained 20 μl of substrate in assay buffer at eight different concentrations (0.88 to 20 μM) and 80 μl of enzyme (0.5 μM SARS-CoV and SARS-CoV-2 PLpro for tetrapeptide substrates and 80 nM SARS-CoV-2 PLpro and 10 nM SARS-CoV PLpro for Ub-ACC). Substrate hydrolysis was measured for 30 min at the appropriate wavelength (λex = 355 nm, λem = 460 nm). Each experiment was carried out at least three times and the results reported as averages with SD. Due to the precipitation of tetrapeptide substrates at high concentrations, only the specificity constant (kcat/KM) was determined. When [S0] < < KM, the plot of vi (the initial velocities) versus [S0] yields a straight line with slope representing Vmax/KM, kcat/KM = slope/E (E, total enzyme concentration). Kinetic parameter for ISG15-AMC toward SARS-CoV-2 PLpro was determined in the same manner as described above. The final enzyme concentration was 1 nM, and the final substrate concentration was ranging from 0.3 to 5 μM.
SARS-CoV and SARS-CoV-2 PLpro inhibitor and B-Ub-VME synthesis
Inhibitor synthesis was performed in the three sequential stages. In the first step, vinyl methyl ester as a reactive group was synthesized according to published protocol (39). tBu-N-allyl carbamate (500 mg, 3.2 μmol) was dissolved in 10 ml of anhydrous toluene. Methyl acrylate (580 μl, 6.4 μmol), dichlorophenylborane (42 μl, 0.32 μmol), and second generation Grubbs catalyst (50 mg) were added. The reaction was carried out under reflux at 40°C with stirring overnight. After 12 hours, the solvent was evaporated under reduced pressure, and the mixture was purified by column chromatography on silica gel (Hex/EtOAc, 5:1). The crude product was obtained as a yellowish oil. tBu group deprotection was performed by adding TFA/DCM/TIPS [4.2 ml, 3/1/0.2 (v/v/v)] cleavage mixture for 45 min with stirring. TFA*H2N-Gly-VME was then crystallized in cold Et2O and stored at −20°C. In the second step, Ac-P4-P3-Gly-OH fragments were synthesized using 2-chlorotrityl chloride resin as previously described (40). In the last step, Ac-P4-P3-Gly-OH fragment (1.2 equiv.) was coupled to the reactive group (1 equiv.) using HATU (1.2 equiv.) and 2,4,6-collidine (3 equiv.) as a coupling reagents in DMF. The reaction was carried out at room temperature (RT) with stirring for 2 hours. The reaction mixture was diluted in ethyl acetate; washed once with 5% citric acid, once with 5% NaHCO3, and once brine; dried over MgSO4; and concentrated under reduce pressure. To remove side-chain amino acid protecting groups, Ac-P4-P3-Gly-Gly-VME was added to a mixture of TFA/DCM/TIPS [% (v/v/v), 70:27:3]. After 30 min, solvents were removed and inhibitor was purified on HPLC. B-Ub-VME was synthesized according to synthetic protocol described elsewhere (41, 42).
Determination of DUB inhibition
To assess activity and selectivity of designed SARS-CoV and SARS-CoV-2 PLpro inhibitors DUBs were incubated with inhibitors at eight different concentrations (2.3 to 300 μM) for 30 min at 37°C in assay buffers. DUB residual activity was estimated using Ac-Leu-Arg-Gly-Gly-ACC (50 μM). Assay conditions were 20 μl of inhibitor, 60 μl of DUB (0.3 μM SARS-CoV PLpro, 0.1 μM SARS-CoV-2 PLpro, 2.5 μM MERS-CoV PLpro, and 8 μM UCH-L3), and 20 μl of substrate (50 μM). Inhibition assays were measured for 40 min and repeated at least three times. The results were established as mean values with SDs.
Crystallization
SARS-CoV-2 PLpro (3 μM) was reacted with 30 μM peptide inhibitor in 5 mM NaCl, 20 mM tris-HCl (pH 8.0) at 37°C for 20 min. Protein was concentrated using a 30-kD cutoff Amicon Ultra Filter and desalted into 5 mM NaCl, 20 mM tris-HCl (pH 8.0), and 10 mM DTT. Final protein concentration was 5 to 10 mg/ml. VIR250 complex crystals were grown by mixing 0.4-μl protein and 0.4-μl well solution containing 0.2 M lithium citrate tribasic, and 20% polyethylene glycol, molecular weight 3350 on a 96-well sitting plate at 18°C. Crystals were cryo-protected by well solution plus 25% (v/v) ethylene glycol and snap-frozen in liquid nitrogen. VIR251 complex crystals were grown by mixing 0.2-μl protein sample with 0.2-μl well solution containing 0.8 M potassium sodium tartrate tetrahydrate, 0.1 M tris-HCl (pH 8.5), and 0.5% w/v polyethylene glycol monomethyl ether 5000 on a 96-well sitting plate at 18 degrees. Crystals were cryo-protected by 25% (v/v) ethylene glycol and flash-frozen with liquid nitrogen.
Structure determination and refinement
A complete data set was collected from the SARS-CoV-2 PLpro/VIR250 crystals to 2.79 Å resolution at the Advanced Photon Source, NE-CAT beamline 24-IDC at a wavelength of 0.979 Å. Dataset was indexed, integrated, and scaled using HKL2000. Crystal belongs to space group P21 with unit cell dimensions a = 58.4, b = 189.7, c = 63.1, and β = 98.7°. There are four SARS-CoV-2 PLpro/VIR250 complexes per asymmetric unit. The structure was solved by molecular replacement using the program PHASER. The search model was apo SARS-CoV-2 PLpro structure (PDB: 6W9C). Apparent ligand density for both Fo-Fc and 2Fo-Fc maps was observed projecting off Cys111 after first round of refinement. Model and restraints for VIR250 was prepared using Phenix.Elbow. Model of SARS-CoV-2 PLpro/VIR250 was subjected to iterative rounds of refinement and rebuilding using PHENIX (43) and COOT (44).
For SARS-CoV-2 PLpro/VIR251 crystals, data were collected and processed as described above for VIR250 to a resolution of 1.65 Å. The crystal belongs to space group I222 with unit cell dimensions a = 44.9, b = 113.5, and c = 151.1. There is one SARS-CoV-2 PLpro/VIR251 complex per asymmetric unit. The structure was determined by molecular replacement with Phaser and the search model was SARS-CoV-2 PLpro/VIR250 structure above (PDB: 6WUU). Structure with ligand was refined as described above for the VIR250 structure.
The final two models for PLpro-VIR250 and PLpro-VIR251 complexes have R/Rfree values of 0.195/0.230 and 0.170/0.196, respectively. The two structures also have excellent geometry as assessed using Molprobity: favored (95.3%), allowed (4.6%), and outliers (0.1%) for the PLpro/VIR250 structure and favored (97.0%), allowed (3.0%), and outliers (0%) for the PLpro/VIR251 structure.
PLpro-Ub/Ubl ABP panel assay
The probes used in this experiment (fig. S3, 1 to 4) were generous gifts of UbiQ. Development of the probes have been previously described: Probe 1 (45, 46), Probe 2 (46), Probe 3 (47), Probe 4 (48, 49). Plpro (3 μM) was incubated with 30 μM inhibitor or DMSO at 37 for 20 min and put on ice. Reaction buffer contains 5 mM NaCl, 20 mM tris-HCl (pH 8.0). Then, the indicated Ub/Ubl ABPs were mixed with PLpro at 4.5 and 2.7 μM, respectively, at RT for 2 min. Reactions were terminated by adding SDS sample buffer, subjected to SDS-PAGE sypro staining.
SARS-CoV and SARS-CoV-2 PLpro labeling by B-Ub-VME
Enzymes (200 nM) were incubated with different B-Ub-VME concentrations (100, 200, 400, 800, and 1000 nM) in assay buffer [150 mM NaCl, 20 mM tris, and 5 mM DTT (pH 8.0) for SARS-CoV PLpro; 5 mM NaCl, 20 mM tris, and 5 mM DTT (pH 8.0) for SARS-CoV-2 PLpro] for 45 min at 37°C. Then, 3× SDS/DTT was added, and the samples were boiled for 5 min at 95°C and resolved on 4 to 12% bis-tris Plus 12-well gels. Electrophoresis was performed at 200 V for 29 min. Next, the proteins were transferred to a nitrocellulose membrane (0.2 μm, Bio-Rad) for 60 min at 10 V. The membrane was blocked with 2% bovine serum albumin (BSA) in tris-buffered saline with 0.1% (v/v) Tween 20 (TBS-T) for 60 min at RT. B-Ub-VME was detected with a fluorescent streptavidin Alexa Fluor 647 conjugate (1:10,000) in TBS-T with 1% BSA using an Azure Biosystems Sapphire Biomolecular Imager and Azure Spot Analysis Software.
Gel-based Ub chain cleavage assays and Ub-VS labeling
Tetra-Ub chains (K48- and K63-linked; Boston Biochem) were cleaved in a reaction volume of 10 μl [in 20 mM tris (pH 7.5), 150 mM NaCl, and 5 mM DTT] with 25 to 500 nM PLpro or USP2 catalytic domain (Boston Biochem), as indicated in figures. Ub-Vinyl Sulfone-labeling was performed in 10 μl [in 20 mM tris (pH 7.5), 150 mM NaCl, and 5 mM DTT] with 1.5 μM Ub-VS (Boston Biochem) and 0.25 μM PLpro or USP2 catalytic domain. Reactions were incubated at 37°C for 30 min, terminated with sample loading buffer (4X LDS, Invitrogen), and analyzed by SDS-PAGE (4 to 12% bis-tris, NuPAGE) and SYPRO Ruby staining. Gels were imaged using an Azure Biosystems c500 imager.
HeLa lysate assay
HeLa cells were cultured in DMEM supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and antibiotics (100 U/mL penicillin, 100 µg/mL streptomycin) in a humidified 5% CO2 atmosphere at 37°C. Approximately 1 200 000 cells were harvested and washed three times with PBS. The cell pellet was lysed in buffer containing 20 mM Tris, 150 mM NaCl, and 5 mM DTT, pH 8.0, using a sonicator. The cell lysate was centrifuged for 10 min, and the supernatant was collected. Cell lysates were incubated with or without of inhibitors (Ac-Abu(Bth)-Dap-Gly-Gly-VME and Ac-hTyr-Dap-Gly-Gly-VME) at four different concentrations (25, 50, 100, and 200 µM) for 30 min at 37°C. Next 300 nM of B-Ub-VME was added and the samples were incubated for 30 min at 37°C. Then the samples were combined with 3xSDS/DTT, boiled, and run on a gel. Electrophoresis, protein transfer to a nitrocellulose membrane, and probe visualization were conducted in the same manner as described above.
Supplementary Material
Acknowledgments
We thank S. Toelle for administrative support. Funding: This project was supported by the National Science Center grant 2015/17/N/ST5/03072 (Preludium 9) in Poland (W.R.) and the “TEAM/2017-4/32” project, which is carried out within the TEAM program of the Foundation for Polish Science, cofinanced by the European Union under the European Regional Development Fund (M.D.). W.R. is a beneficiary of a START scholarship from the Foundation for Polish Science. This work is based on research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the NIH (P30 GM124165). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. The x-ray crystallography facility used for this work is supported by the Office of the Vice President for Research at the Medical University of South Carolina. The liquid handling robot used was purchased via an NIH Shared Instrumentation Award (S10 RR027139-01). Research reported in this publication was supported by CPRIT RR200030 and NIH R01 GM115568 (S.K.O.), ES025166 (T.T.H.), and GM099040 (S.J.S.). Z.L. is a Hollings Cancer Center Postdoctoral Fellow, and S.P. is an American Cancer Society Postdoctoral Fellow (PF-18-235-01-RMC). The content of this study is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Author contributions: W.R., M.B., S.K.O., and M.D. conceived the project; M.D., W.R., T.T.H., S.K.O., and M.Z. designed the research; W.R., M.Z., Z.L., S.P., and D.N. performed the research and collected data; D.N., Z.L., S.J.S., M.B., and T.T.H. contributed enzymes; F.E.O. contributed activity-based probes; M.D., W.R., T.T.H., M.B., S.K.O., and M.Z. analyzed and interpreted the data; Z.L. and S.K.O. performed structural experiments including crystallization, x-ray data collection/processing, model building/refinement, and structural analyses; W.R., S.K.O., and M.D. wrote the manuscript and all authors critically revised the manuscript. Competing interests: F.E.O. declares competing financial interests as cofounder and shareholder of UbiQ Bio BV. Although M.B. is an employee and shareholder of Arvinas Inc., Arvinas Inc. was not involved in this study. The remaining authors declare no competing interests. Data and materials availability: Atomic coordinates and structure factors are deposited in the RCSB with accession codes 6WUU (SARS-CoV-2 PLpro/VIR250) and 6WX4 (SARS-CoV-2 PLpro/VIR251). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/42/eabd4596/DC1
REFERENCES AND NOTES
- 1.Zhong N. S., Zheng B. J., Li Y. M., Poon, Xie Z. H., Chan K. H., Li P. H., Tan S. Y., Chang Q., Xie J. P., Liu X. Q., Xu J., Li D. X., Yuen K. Y., Peiris, Guan Y., Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People's Republic of China, in February, 2003. Lancet 362, 1353–1358 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Wit E., van Doremalen N., Falzarano D., Munster V. J., SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 14, 523–534 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zaki A. M., van Boheemen S., Bestebroer T. M., Osterhaus A. D. M. E., Fouchier R. A. M., Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367, 1814–1820 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Wang C., Horby P. W., Hayden F. G., Gao G. F., A novel coronavirus outbreak of global health concern. Lancet 395, 470–473 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B., Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Báez-Santos Y. M., St John S. E., Mesecar A. D., The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antiviral Res. 115, 21–38 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han Y.-S., Chang G.-G., Juo C.-G., Lee H.-J., Yeh S.-H., Hsu J. T.-A., Chen X., Papain-like protease 2 (PLP2) from severe acute respiratory syndrome coronavirus (SARS-CoV): Expression, purification, characterization, and inhibition. Biochemistry 44, 10349–10359 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Harcourt B. H., Jukneliene D., Kanjanahaluethai A., Bechill J., Severson K. M., Smith C. M., Rota P. A., Baker S. C., Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol. 78, 13600–13612 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barretto N., Jukneliene D., Ratia K., Chen Z., Mesecar A. D., Baker S. C., The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 79, 15189–15198 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barretto N., Jukneliene D., Ratia K., Chen Z., Mesecar A. D., Baker S. C., Deubiquitinating activity of the SARS-CoV papain-like protease. Adv. Exp. Med. Biol. 581, 37–41 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindner H. A., Lytvyn V., Qi H., Lachance P., Ziomek E., Ménard R., Selectivity in ISG15 and ubiquitin recognition by the SARS coronavirus papain-like protease. Arch. Biochem. Biophys. 466, 8–14 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindner H. A., Fotouhi-Ardakani N., Lytvyn V., Lachance P., Sulea T., Ménard R., The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 79, 15199–15208 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ratia K., Kilianski A., Baez-Santos Y. M., Baker S. C., Mesecar A., Structural basis for the ubiquitin-linkage specificity and deISGylating activity of SARS-CoV papain-like protease. PLOS Pathog. 10, e1004113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Békés M., Rut W., Kasperkiewicz P., Mulder M. P. C., Ovaa H., Drag M., Lima C. D., Huang T. T., SARS hCoV papain-like protease is a unique Lys(48) linkage-specific di-distributive deubiquitinating enzyme. Biochem. J. 468, 215–226 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Békés M., van der Heden van Noort G. J., Ekkebus R., Ovaa H., Huang T. T., Lima C. D., Recognition of Lys48-linked di-ubiquitin and deubiquitinating activities of the SARS coronavirus papain-like protease. Mol. Cell 62, 572–585 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calistri A., Munegato D., Carli I., Parolin C., Palù G., The ubiquitin-conjugating system: Multiple roles in viral replication and infection. Cells 3, 386–417 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mielech A. M., Kilianski A., Baez-Santos Y. M., Mesecar A. D., Baker S. C., MERS-CoV papain-like protease has deISGylating and deubiquitinating activities. Virology 450, 64–70 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Devaraj S. G., Wang N., Chen Z., Chen Z., Tseng M., Barretto N., Lin R., Peters C. J., Tseng C.-T. K., Baker S. C., Li K., Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 282, 32208–32221 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clementz M. A., Chen Z., Banach B. S., Wang Y., Sun L., Ratia K., Baez-Santos Y. M., Wang J., Takayama J., Ghosh A. K., Li K., Mesecar A. D., Baker S. C., Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 84, 4619–4629 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frieman M., Ratia K., Johnston R. E., Mesecar A. D., Baric R. S., Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-κB Signaling. J. Virol. 83, 6689–6705 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S., Chaudhuri R., Fu W., Prabhakar B. S., Johnson M. E., Baker S. C., Ghosh A. K., Mesecar A. D., A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. U.S.A. 105, 16119–16124 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hilgenfeld R., From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 281, 4085–4096 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin M.-H., Moses D. C., Hsieh C.-H., Cheng S.-C., Chen Y.-H., Sun C.-Y., Chou C.-Y., Disulfiram can inhibit MERS and SARS coronavirus papain-like proteases via different modes. Antiviral Res. 150, 155–163 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poreba M., Salvesen G. S., Drag M., Synthesis of a HyCoSuL peptide substrate library to dissect protease substrate specificity. Nat. Protoc. 12, 2189–2214 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Drag M., Mikolajczyk J., Bekes M., Reyes-Turcu F. E., Ellman J. A., Wilkinson K. D., Salvesen G. S., Positional-scanning fluorigenic substrate libraries reveal unexpected specificity determinants of DUBs (deubiquitinating enzymes). Biochem. J. 415, 367–375 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rut W., Zmudzinski M., Snipas S. J., Bekes M., Huang T. T., Drag M., Engineered unnatural ubiquitin for optimal detection of deubiquitinating enzymes. Chem. Sci. 11, 5058–6069 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klemm T., Ebert G., Calleja D. J., Allison C. C., Richardson L. W., Bernardini J. P., Lu B. G. C., Kuchel N. W., Grohmann C., Shibata Y., Gan Z. Y., Cooney J. P., Doerflinger M., Au A. E., Blackmore T. R., Geurink P. P., Ovaa H., Newman J., Riboldi-Tunnicliffe A., Czabotar P. E., Mitchell J. P., Feltham R., Lechtenberg B. C., Lowes K. N., Dewson G., Pellegrini M., Lessene G., Komander D., Mechanism and inhibition of SARS-CoV-2 PLpro. bioRxiv 2020.06.18.160614 , (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D. Shin, R. Mukherjee, D. Grewe, D. Bojkova, Inhibition of papain-like protease PLpro blocks SARS-CoV-2 spread and promotes anti-viral immunity (2020); 10.21203/rs.3.rs-27134/v1. [DOI]
- 29.Freitas B. T., Durie I. A., Murray J., Longo J. E., Miller H. C., Crich D., Hogan R. J., Tripp R. A., Pegan S. D., Characterization and Noncovalent Inhibition of the deubiquitinase and deISGylase activity of SARS-CoV-2 papain-like protease. ACS Infect. Dis. 6, 2099–2109 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Rut W., Groborz K., Zhang L., Sun X., Zmudzinski M., Pawlik B., Młynarski W., Hilgenfeld R., Drag M., Substrate specificity profiling of SARS-CoV-2 main protease enables design of activity-based probes for patient-sample imaging. bioRxiv 2020.03.07.981928 , (2020). [Google Scholar]
- 31.Drag M., Salvesen G. S., Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 9, 690–701 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kasperkiewicz P., Poreba M., Groborz K., Drag M., Emerging challenges in the design of selective substrates, inhibitors and activity-based probes for indistinguishable proteases. FEBS J. 284, 1518–1539 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turk B., Targeting proteases: Successes, failures and future prospects. Nat. Rev. Drug Discov. 5, 785–799 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Aleshin A. E., Drag M., Gombosuren N., Wei G., Mikolajczyk J., Satterthwait A. C., Strongin A. Y., Liddington R. C., Salvesen G. S., Activity, specificity, and probe design for the smallpox virus protease K7L. J. Biol. Chem. 287, 39470–39479 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rut W., Zhang L., Kasperkiewicz P., Poreba M., Hilgenfeld R., Drąg M., Extended substrate specificity and first potent irreversible inhibitor/activity-based probe design for Zika virus NS2B-NS3 protease. Antiviral Res. 139, 88–94 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Rut W., Groborz K., Zhang L., Modrzycka S., Poreba M., Hilgenfeld R., Drag M., Profiling of flaviviral NS2B-NS3 protease specificity provides a structural basis for the development of selective chemical tools that differentiate Dengue from Zika and West Nile viruses. Antiviral Res. 175, 104731 (2020). [DOI] [PubMed] [Google Scholar]
- 37.B. J. Anson, M. E. Chapman, E. K. Lendy, S. Pshenychnyi, R. T. D’Aquila, K. J. F. Satchell, A. D. Mesecar, Broad-spectrum inhibition of coronavirus main and papain-like proteases by HCV drugs. 10.21203/rs.3.rs-26344/v1 (2020).
- 38.Maly D. J., Leonetti F., Backes B. J., Dauber D. S., Harris J. L., Craik C. S., Ellman J. A., Expedient solid-phase synthesis of fluorogenic protease substrates using the 7-amino-4-carbamoylmethylcoumarin (ACC) fluorophore. J. Org. Chem. 67, 910–915 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Vedrenne E., Dupont H., Oualef S., Elkaim L., Grimaud L., Dramatic effect of boron-based Lewis acids in cross-metathesis reactions. Synlett, 670–672 (2005). [Google Scholar]
- 40.Rut W., Poręba M., Kasperkiewicz P., Snipas S. J., Drąg M., Selective substrates and activity-based probes for imaging of the human constitutive 20S proteasome in cells and blood samples. J. Med. Chem. 61, 5222–5234 (2018). [DOI] [PubMed] [Google Scholar]
- 41.El Oualid F., Merkx R., Ekkebus R., Hameed D. S., Smit J. J., de Jong A., Hilkmann H., Sixma T. K., Ovaa H., Chemical synthesis of ubiquitin, ubiquitin-based probes, and diubiquitin. Angew. Chem. Int. Ed. Engl. 49, 10149–10153 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Jong A., Merkx R., Berlin I., Rodenko B., Wijdeven R. H. M., Atmioui D. E., Yalçin Z., Robson C. N., Neefjes J. J., Ovaa H., Ubiquitin-based probes prepared by total synthesis to profile the activity of deubiquitinating enzymes. Chembiochem 13, 2251–2258 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Mulder M. P., El Oualid F., ter Beek J., Ovaa H., A native chemical ligation handle that enables the synthesis of advanced activity-based probes: Diubiquitin as a case study. Chembiochem 15, 946–949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flierman D., van der Heden van Noort G. J., Ekkebus R., Geurink P. P., Mevissen T. E. T., Hospenthal M. K., Komander D., Ovaa H., Non-hydrolyzable diubiquitin probes reveal linkage-specific reactivity of deubiquitylating enzymes mediated by S2 pockets. Cell Chem. Biol. 23, 472–482 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ekkebus R., van Kasteren S. I., Kulathu Y., Scholten A., Berlin I., Geurink P. P., de Jong A., Goerdayal S., Neefjes J., Heck A. J. R., Komander D., Ovaa H., On terminal alkynes that can react with active-site cysteine nucleophiles in proteases. J. Am. Chem. Soc. 135, 2867–2870 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Basters A., Geurink P. P., Röcker A., Witting K. F., Tadayon R., Hess S., Semrau M. S., Storici P., Ovaa H., Knobeloch K.-P., Fritz G., Structural basis of the specificity of USP18 toward ISG15. Nat. Struct. Mol. Biol. 24, 270–278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hewings D. S., Heideker J., Ma T. P., Ah Young A. P., Oualid F. E., Amore A., Costakes G. T., Kirchhofer D., Brasher B., Pillow T., Popovych N., Maurer T., Schwerdtfeger C., Forrest W. F., Yu K., Flygare J., Bogyo M., Wertz I. E., Reactive-site-centric chemoproteomics identifies a distinct class of deubiquitinase enzymes. Nat. Commun. 9, 1162 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/42/eabd4596/DC1