

Preproinsulin-specific CD8+ T cells residing in the exocrine pancreas become attracted to islets in type 1 diabetes.
Abstract
Preproinsulin (PPI) is presumably a crucial islet autoantigen found in patients with type 1 diabetes (T1D) but is also recognized by CD8+ T cells from healthy individuals. We quantified PPI-specific CD8+ T cells within different areas of the human pancreas from nondiabetic controls, autoantibody-positive donors, and donors with T1D to investigate their role in diabetes development. This spatial cellular quantitation revealed unusually high frequencies of autoreactive CD8+ T cells supporting the hypothesis that PPI is indeed a key autoantigen. To our surprise, PPI-specific CD8+ T cells were already abundantly present in the nondiabetic pancreas, thus questioning the dogma that T1D is caused by defective thymic deletion or systemic immune dysregulation. During T1D development, these cells accumulated in and around islets, indicating that an islet-specific trigger such as up-regulation of major histocompatibility complex class I might be essential to unmask beta cells to the immune system.
INTRODUCTION
Type 1 diabetes (T1D) is a chronic autoimmune disease. Before the clinical diagnosis of T1D, the presence of autoantibodies against beta cell autoantigens such as insulin can predict future disease development (1, 2). Autoreactive T cells recognizing islet autoantigens are then believed to infiltrate the pancreas and contribute to beta cell destruction (3).
During T1D development, CD8+ T cells are the principal T cell type infiltrating the pancreatic islets (4, 5). While some reports suggest that the numbers of islet-reactive CD8+ T cells in the blood are higher in patients with T1D than those without the disease (6, 7), more recent data question this observation (8–10). Moreover, preproinsulin (PPI) was recognized by peripheral blood CD8+ T cells (at low frequencies of 1:104 to 1:106) in healthy donors (11). It thus appears that the peripheral presence of PPI-reactive CD8+ T cells is the default state (i.e., not caused by a profound thymic defect) and that organ-specific rather than systemic tolerance mechanisms counterbalance this in nondiabetic individuals.
Because of limited access to human pancreas samples, autoreactive T cells have traditionally been studied in human blood and thus quantitative information on their frequency at the target organ remains unknown. Our group was the first to demonstrate the presence of antigen-specific CD8+ T cells recognizing autoantigens, including PPI, in the islets of donors with recent-onset and long-standing T1D (12). This particular epitope, shown to be naturally processed and presented by beta cells in vitro, facilitated the killing of beta cells by cytotoxic CD8+ T cells (13).
Little is known about the precise spatial distribution of autoreactive CD8+ T cells in the human pancreas. Therefore, we wanted to determine their exact location in pancreas tissue samples from donors diagnosed with T1D, autoantibody-positive (aab+; “at-risk”), and not-at-risk (aab−) healthy controls obtained through nPOD, the Network for Pancreatic Organ Donors with Diabetes (Table 1).
Table 1. General donor characteristics.
The donor demographic information including age, percentage of females and males, ethnicity, body mass index (BMI), disease duration, C-peptide levels, and time in the intensive care unit (ICU).
| No diabetes | aab+ |
Short-term TID, ≤2 years |
Long-term TID, >2–8 years |
Total | |
| Donors, n | 6 | 5 | 7 | 4 | 22 |
| Age (years) (mean ± SEM) | 19.83 (±5.87) | 25.52 (±7.17) | 17.08 (±3.24) | 21.15 (±4.00) | 20.44 (±2.47) |
| Female, n (%) | 2 (33.33) | 3 (60) | 5 (71.43) | 3 (75) | 13 (59.01) |
| Male, n (%) | 4 (66.67) | 2 (40) | 2 (28.57) | 1 (25) | 9 (40.1) |
| Ethnicity, n (%) | |||||
| African American | 2 | 1 | 2 | 0 | 5 (22.73) |
| Caucasian | 3 | 4 | 5 | 4 | 16 (72.73) |
| Multiracial | 1 | 0 | 0 | 0 | 1 (4.54) |
| BMI (mean ± SEM) | 23.28 (±1.85) | 20.14 (±1.88) | 23.41 (±1.68) | 25.1 (±1.23) | 22.94 (±0.90) |
| Disease duration (mean ± SEM) |
- | - | 1.19 (±0.30) | 5.75 (±1.11) | 2.85 (±0.80) |
| C-peptide (ng/ml) (mean ± SEM) |
10.57 (±2.35) | 3.75 (±1.57) | 1.62 (±1.45) | 0.05 (±0.00) | 4.26 (±1.20) |
| Time in ICU (days) (mean ± SEM) |
3.71 (±1.05) | 3.58 (±0.39) | 3.35 (±0.55) | 2.86 (±0.83) | 3.41 (±0.36) |
We show here that the relative frequencies of PPI-specific CD8+ T cells detected in the exocrine regions are similar irrespective of disease status. In contrast to previous observations documenting such cells within the peripheral blood, frequencies in the pancreas are much higher, indicating a high degree of selective accumulation within the target organ. Our study thus questions the prevailing dogma that T1D is fundamentally caused by defective thymic selection, as well as systemic defects in immune regulation. It rather identifies the loss of local, organ-specific control mechanisms, as the most probable key driver of T1D development. Last, we found most of the CD8+ T cells close to or infiltrating insulin-containing islets (ICIs) in donors with T1D to be specific for PPI. This enrichment of antigen-specific CD8+ T cells suggests that they become attracted to their key antigen in ICIs during disease development, possibly due to the previously documented up-regulation of major histocompatibility complex (MHC) class I (14) and accumulation of proinsulin (15), both hallmarks of T1D pathogenesis.
RESULTS
Many PPI15–24-specific CD8+ T cells reside in the human exocrine pancreas
On the basis of the evidence that CD8+ T cells are the predominant cell type in human T1D and can also be found in the exocrine pancreas of donors with T1D, we first analyzed a total of 261 exocrine regions from donor pancreas sections (Fig. 1, fig. S1, and table S1). Overall, CD8+ T cells were present in the exocrine pancreas in all donor groups. Among them, the density of CD8+ T cells was significantly higher in aab+ (P < 0.0001) and T1D (P = 0.0006) donors when compared with donors without diabetes (Fig. 1A). In addition, there was a strong trend in increased PPI-specific CD8+ T cells in exocrine regions comparing donors with T1D to healthy controls (P = 0.0564; Fig. 1B), and compared to previously reported frequencies (13) of such autoreactive CD8 cells in blood, this constituted a substantial enrichment. The mean ratios of PPI15–24-specific/total CD8+ T cells were higher than 40% in all donor groups (Fig. 1C). Many PPI15–24-specific CD8+ T cells are already present in the exocrine pancreas of nondiabetic, aab− donors (Fig. 1, D and E). We conclude that the abundant presence of PPI-reactive CD8+ T cells in the exocrine pancreas is considerably higher in frequency than previously documented within peripheral blood mononuclear cells (PBMCs) (7, 11). The presence of autoantigen-specific CD8+ T cells in the pancreas is, therefore, the default state in humans without T1D, and CD8+ T cell density increases with the advancement of T1D.
Fig. 1. Many PPI15–24-specific CD8+ T cells reside in the human exocrine pancreas.
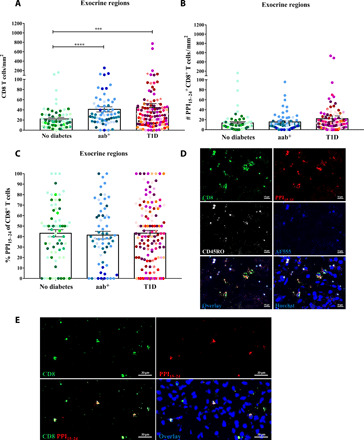
Exocrine regions (no diabetes, n = 69; aab+, n = 63; and T1D, n = 129) were randomly selected from pancreas tissue sections of all 22 donors. (A) A higher density of CD8+ T cells was found in donors with aab+ (P < 0.0001) and T1D (P = 0.0006) compared with donors without diabetes. (B) A strong tendency to higher numbers of PPI15–24+CD8+ T cells in donors with T1D compared to donors without diabetes was observed (P = 0.0564). (C) Frequencies of PPI15–24+CD8+ T cells among detected CD8 T cells are similar between all donor groups. Every dot represents an exocrine region. Bars represent the mean ± SEM values in different groups. Every color represents a donor. For statistical analysis, nonparametric Kruskal-Wallis test followed by Dunn multiple comparison test was used to determine significance: ***P < 0.001 and ****P < 0.0001. (D and E) PPI15–24-specific CD8+ T cells are present in the pancreas of healthy, aab− donors. Representative immunofluorescence images of exocrine regions from nondiabetic donors stained with PPI15-24−APC (allophycocyanin) tetramer, CD8-AF (Alexa Fluor) 488, and CD45RO-AF594 (pseudo-color white). PPI15–24-specific CD8+ T cells were counted manually, and the density was calculated per square millimeter. Images were taken with the AxioScan.Z1 slide scanner (Zeiss, ×20 magnification). (D) Donor #6271. To differentiate between staining and background signal or autofluorescence, an additional channel (AF555, pseudo-color blue) was added during image acquisition. (E) Donor #6232.
PPI15–24-specific CD8+ T cells are attracted to the proximity of and into the islets already in aab+ donors and later on in T1D
To study whether PPI15–24-specific CD8+ T cells might be preferentially infiltrating exocrine areas close to the islets, a total of 267 neighboring islet areas were analyzed (Fig. 2, fig. S1, and table S1). We detected a higher density of CD8+ T cells close to the islets in aab+ donors compared with donors without diabetes (P = 0.0004; Fig. 2A). The number of PPI15–24-specific CD8+ T cells close to the islets was significantly increased in aab+ donors as compared with those without diabetes (P = 0.089; Fig. 2, B and E). Moreover, in donors with T1D, the number of PPI15–24-specific CD8+ T cells was further enriched in neighboring islet areas compared with aab+ donors (P = 0.0123) and those without diabetes (P < 0.0001; Fig. 2B). Furthermore, the frequency of PPI15–24-specific CD8+ T cells was increased in neighboring islet areas from donors with T1D compared with nondiabetic donors (P = 0.0061; Fig. 2C). Collectively, these data suggest a progressive attraction of autoreactive T cells from the exocrine pancreas toward the islets during the development of T1D.
Fig. 2. PPI15–24-specific CD8+ T cells are attracted to the proximity of and into the islets.
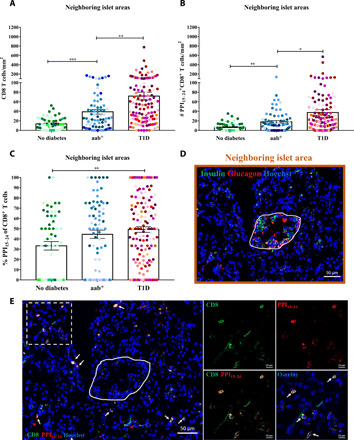
Neighboring islet areas (n = 267) were randomly selected from pancreas tissue sections of all 22 donors (no diabetes, n = 57; aab+, n = 73; and T1D, n = 137). (A) A higher density of CD8+ T cells close to the islets in aab donors compared with donors without diabetes (P = 0.0004). (B) The number of PPI15–24+CD8+ T cells is increased in donors with abb+ (P = 0.089) and T1D (P < 0.0001) compared with donors without diabetes. (C) Higher frequency of PPI15–24-specific CD8+ T cells in neighboring islet areas in donors with T1D compared with donors without diabetes (P = 0.0061). Every dot represents a neighboring islet area. Bars represent the mean ± SEM values in different groups. Every color represents a donor. PPI15–24-specific CD8+ T cells were counted manually, and the density was calculated per square millimeter. For statistical analysis, nonparametric Kruskal-Wallis test followed by Dunn multiple comparison test was used to determine significance: *P = 0.05, **P < 0.05, and ***P < 0.001. (D) Restaining of a pancreas tissue section from a donor with aab+ (#6154) for insulin and glucagon (see Supplementary Methods for more details). The image shows an insulin-containing islet. The neighboring islet area is demonstrated in brown. (E) PPI15–24-specific CD8+ T cells were found close to islets already in donors with aab+. White arrows indicate PPI15–24-specific CD8+ T cells.
PPI15–24-specific CD8+ T cells are found within the islets of donors with T1D
Next, a total of 302 islets were analyzed (Fig. 3). There were significantly more islet-infiltrating CD8+ T cells in donors with T1D (P < 0.0001) than in those without diabetes (Fig. 3A and table S2). Furthermore, the total numbers of islet-infiltrating CD8+ T cells were higher in donors with T1D compared with islets from aab+ donors, but this finding did not extend to a correlation with disease duration (fig. S3A). In addition, the percentage of infiltrated islets among all islets analyzed was lower in aab+ donors and donors without diabetes compared with those with T1D (P < 0.0001; fig. S3B). The number of PPI15–24-specific CD8+ T cells in the islets was significantly increased in donors with diabetes compared with those with aab+ (P < 0.0001) and without diabetes (P < 0.0001; Fig. 3B). Last, the mean frequency of CD8+ T cells recognizing PPI15–24 was higher in islets from donors with T1D compared with donors without T1D (P < 0.0001; Fig. 3C). Thus, PPI-specific CD8+ T cell infiltration in pancreatic islets is only occasionally found in aab+ donors and is most prevalent in individuals who have been diagnosed with T1D.
Fig. 3. PPI15–24-specific CD8+ T cells are found within the islets of donors with T1D.
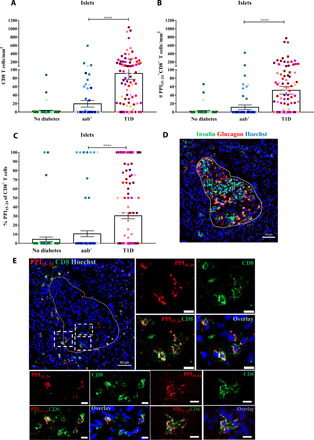
Islets were randomly selected from pancreas sections of donors without diabetes (n = 63), aab+ (n = 83), and T1D (n = 156). (A) A higher density of CD8+ T cells in donors with T1D (P < 0.0001) compared with donors with aab+. (B) High numbers of PPI15–24-specific CD8+ T cells in donors with T1D (P < 0.0001). (C) The percentage of PPI15–24-specific CD8+ T cells in the islets is higher in donors with T1D. Every dot represents an islet (n = 302). Bars represent the mean ± SEM values. For statistical analysis, nonparametric Kruskal-Wallis test followed by Dunn multiple comparison test was used to determine significance: ****P < 0.0001. (D and E) Representative immunofluorescence images of a pancreas section from a donor with T1D (#6052, 1 year of disease duration). (D) Restaining for insulin and glucagon (see Supplementary Methods for more details). The image shows an insulin-containing islet. (E) In situ PPI15–24 staining (red) combined with CD8 (green) and nuclear marker (blue, Hoechst). PPI15–24-specific CD8+ T cells are shown in yellow. Magnification ×20. Scale bars, 10 μm for cropped images.
PPI15–24-specific CD8+ T cell infiltration or retention in the islets depends on the presence of insulin
Given our results that CD8+ T cells infiltrate the islets of donors with T1D, we further studied the role of insulin in this process. We quantified the number of CD8+ T cells recognizing the PPI15–24 epitope within or close to ICIs. A total of 56 ICI neighboring islet areas and 81 insulin-deficient islet (IDI) neighboring areas from 11 donors with T1D were analyzed (Fig. 4). Among them, PPI15–24-specific CD8+ T cells were predominately found close to ICIs rather than IDIs (P < 0.0001; Fig. 4A), with a higher frequency of PPI15–24-specific CD8+ T cells in ICI neighboring areas compared to IDI neighboring areas (P = 0.0453; Fig. 4B). PPI15–24-specific CD8+ T cells infiltrated mostly islets that still contained insulin compared with islets without insulin (P = 0002; Fig. 4C). Furthermore, a significant difference was observed in the frequency of PPI15–24-specific CD8+ T cells in ICIs compared with IDIs (P = 0.0043; Fig. 4D). In addition, no significant differences in PPI15–24–CD8+ T cell density were found between donors with short and long disease duration (fig. S4). This marked decrease in PPI15–24-specific CD8+ T cell density in IDIs suggests that the presence of insulin is critical to attract or retain PPI15–24-specific CD8+ T cells to the islets.
Fig. 4. PPI15–24-specific CD8+ T cells infiltration or retention in the islets depends on the presence of insulin.
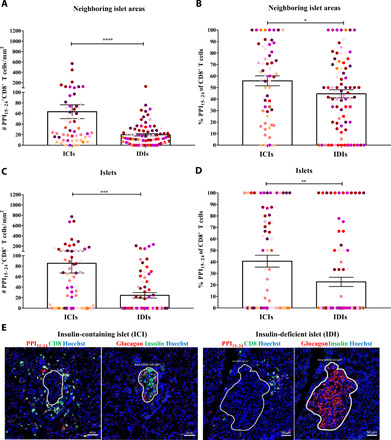
Insulin-containing islets (ICIs; n = 69) and insulin-deficient islets (IDIs; n = 87) from donors with T1D (n = 11) were analyzed for the presence of PPI15–24-specific CD8+ T cells. (A) A higher density (P < 0.0001) and (B) frequency (P = 0.045) of PPI15–24+CD8+ T cells was detected in close proximity to ICIs (n = 56) compared to IDIs (n = 81). The number (P = 0.0002) and percentage (P = 0.003) of PPI15–24+CD8+ T cells are increased in ICIs compared to IDIs. (A and B) Every dot represents a neighboring islet area (n = 267) or (C and D) an islet (n = 302). Bars represent the mean ± SEM numbers (A and C) and frequencies (B and D) of PPI15–24+CD8+ T cells per square millimeter. Every color represents a donor. For statistical analysis, the Mann-Whitney test was used to determine significance: *P = 0.05, **P < 0.05, ***P < 0.001, and ****P < 0.0001. (E) Representative immunofluorescence images of an ICI and IDI from a donor (#6052) with T1D (1 year of disease duration). PPI15–24-specific CD8+ T cells are shown in yellow.
Most PPI15–24-specific CD8+ T cells are memory cells
To identify the phenotype of PPI15–24-specific CD8+ T cells in the human pancreas, the expression of the memory marker CD45RO was analyzed (Fig. 5). In the exocrine regions of all donor groups, on average, 50 to 60% PPI15–24-specific CD8+ T cells were CD45RO+ (Fig. 5A). In contrast, the CD45RO frequency was lower in neighboring islet areas in donors without diabetes compared with donors with T1D (P = 0.02; Fig. 5B).
Fig. 5. Most PPI15–24-specific CD8+ T cells are memory cells.
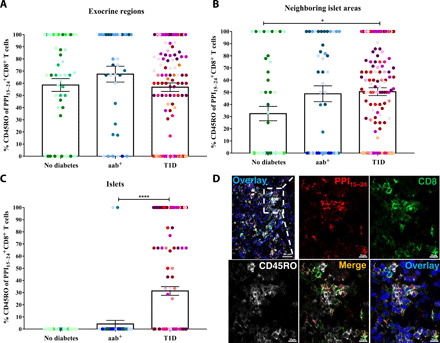
The percentages of CD45RO+ cells of PPI15–24-specific CD8+ T cells are shown for (A) exocrine regions (no diabetes, n = 59; aab+, n = 36; T1D, n = 123), (B) neighboring islet areas (no diabetes, n = 48; aab+, n = 44; T1D, n = 126), and (C) islets (no diabetes, n = 53; aab+, n = 48; T1D, n = 143). Bars represent the mean ± SEM percentages of PPI15–24-specific CD8+ CD45RO+ T cells per square millimeter for each group. Every dot represents a region or an islet. Every color represents a donor. For statistical analysis, nonparametric Kruskal-Wallis test followed by Dunn multiple comparison test was used to determine significance: *P = 0.05 and ****P < 0.0001. (D) Immunofluorescence images of PPI15–24-specific CD8+ T cells, including the CD45RO memory marker of a pancreas section from a donor with T1D (#6052, 1 year of disease duration). The image demonstrates an islet (complementary to Fig. 4E). Scale bar, 10 μm for images with higher magnification.
DISCUSSION
Some beta cell–specific T cells are detected in the blood of healthy individuals (6, 7). However, here, we provide the first evidence that PPI-specific CD8 T cells constitute an integral and large fraction of the pancreatic CD8 T cell population in nondiabetic individuals and can locally be detected in large numbers. This finding extends observations from peripheral blood, where autoreactive CD8+ T cells, particularly PPI-reactive CD8 T cells, had been identified (6, 11, 16), albeit at much lower relative frequencies compared to our current findings within the pancreas. This observation suggests that defective thymic selection may not constitute a primary cause for autoimmunity in T1D. Furthermore, it is unlikely that the failure of systemic peripheral tolerance mechanisms is a key pathogenic feature of T1D, because the differences between donors with and without T1D were mostly observed in the target organ (7). It is, therefore, likely that events leading to islet attraction of autoreactive CD8+ T cells already within the pancreas may be a crucial mechanism in T1D development.
T1D is considered an autoimmune disease that affects the endocrine pancreas. However, more studies have indicated a critical involvement of the exocrine pancreas in the pathogenesis of T1D (17–20). Consistent with previous studies (19), CD8+ T cells were detected in the exocrine pancreas of all donors. In addition, we found more CD8+ T cells in aab+ donors and donors with T1D than in donors without diabetes. This increase could be due to nonspecific inflammatory triggers related to viral infections (21) or, for example, a “leaky” gut (22), as both have a pathogenic role in T1D. The “pancreas-dwelling” CD8+ T cells have an antigen-experienced phenotype (23–25), and we detected the expression of the memory marker CD45RO on most PPI15–24-specific CD8+ T cells, representing the activation status of these cells (26–28). A substantial fraction of these cells were PPI (autoantigen) specific.
Our study shows that many of these cells are PPI reactive. While a CD8+ T cell–rich environment has been described in target organs in humans (29, 30), why are so many cells in the exocrine pancreas PPI specific? Is it due to an abundance of PPI antigen in the pancreas? Perhaps in healthy individuals, beta cells under physiological conditions are not visible, a status termed immunological “ignorance” (31). What then happens to these cells during disease development and during the aab+ state in the islet proinsulin area (15)? Single-cell resolution microscopy in the pancreas from diabetic mice revealed that islet-reactive T cells freely migrate through the exocrine tissue and do not arrest in the absence of their islet antigen (32). Foulis et al. (33) were the first to demonstrate that following a putative environmental trigger, islet cells from patients with T1D start hyperexpressing MHC class I and thus become visible targets to islet-reactive CD8+ T cells. This observation has since emerged as a histopathological hallmark of T1D (14). It might constitute a key reason for the PPI-specific T cells to spend more time during T1D pathogenesis at the site of antigenic presentation while progressively destroying beta cells.
We found significant enrichment of CD8+ T cells recognizing PPI15–24 in donors with T1D and in some aab+ donors compared with healthy donors. They predominantly infiltrated areas close to ICIs and islets that still contained beta cells, indicating that PPI is an important driver of autoreactivity. This finding supports the hypothesis that beta cells are, in part, responsible for their demise (34) by adopting a proinsulin-high, MHC class I–positive, “visible” phenotype that causes the exocrine T cells to arrest, form immune synapses, and execute killing. In the final phase of T1D immunopathology, islets lose insulin positivity and, in the absence of MHC I:antigen complexes, the T cells continue their random exocrine walk (32, 35). We show that PPI15–24-specific CD8+ T cell density is lower in IDIs versus ICIs irrespective of disease duration (up to 8 years), supporting the hypothesis that PPI15–24-specific CD8+ T cells leave the islets after beta cells are destroyed. These findings are in line with other studies of postmortem pancreas samples from recent-onset T1D that reveal that CD8+ T cells are primarily retained within the ICIs but are absent from IDIs (19, 36).
Another question is whether all subjects with T1D exhibit this marked increase of PPI-specific CD8+ T cells. We found that the numbers of PPI-specific CD8 T cells differ between donors and that some donors did not have this feature. Thus, it is possible that different “driver” autoantigens could exist for these individuals. However, based on our limited number of subjects, the proportion of patients harboring PPI reactivity could be at least 60 to 70%. In addition, while other islet-reactive specificities will probably contribute (12), a sizeable non–islet-specific “bystander” population may also be involved (37).
Collectively, our results document that PPI drives islet infiltration of CD8+ T cells during the development of human T1D. These antigen-specific CD8+ T cells are present in large numbers in the nondiabetic exocrine pancreas (with higher numbers in aab+ donors), arguing against a defective thymic selection of CD8 T cells and systemic regulatory mechanisms as fundamental disease drivers. Last, their increased accumulation in pancreatic islets after clinical diagnosis suggests mechanisms that selectively “unmask” beta cells to the immune system such as up-regulation of MHC class I (14) possibly mediated by a viral trigger (38) as well as accumulation of proinsulin, potentially caused by beta cell metabolic stress (15, 39, 40), resulting in their recognition, functional impairment, and ultimately demise (34). Our findings strongly argue the case for a more beta cell–centric rather than a systemic therapeutic approach to tackle the complicated problem of T1D (41).
MATERIALS AND METHODS
Study subjects
Human pancreas samples from 22 HLA-A*02:01-positive donors were obtained through nPOD (jdrfnpod.org). These were subdivided into six donors without diabetes, five aab+ donors, seven donors with short-term T1D (0 to 2 years), and four donors with long-standing T1D (>2 to 8 years). Each donor case was color-coded (table S2). Information on demographics and medical history were obtained from the nPOD DataShare (Table 1 and table S3). All experimental procedures were approved by the La Jolla Institute for Immunology Institutional Review Board, approved protocol number DI-054-0218.
In situ immunofluorescence staining
In situ immunofluorescence staining of antigen-specific CD8 T cells on frozen pancreatic tissue sections (6 μm) was performed using a PPI15–24 tetramer (ALWGPDPAAA) labeled with allophycocyanin (APC) from two different sources, the National Institutes of Health (NIH) Tetramer Core Facility, Atlanta, GA (1.4 mg/dl), and the Fred Hutchinson Cancer Research Center, Immune Monitoring Core, Seattle, WA (0.1 mg), to achieve a robust and reproducible staining approach. Unfixed, frozen tissue sections were first blocked with 10% goat serum for 30 min at 4°C and incubated with the PPI15–24 tetramer (1/1000 NIH and 1/500 Fred Hutchinson) diluted in phosphate-buffered saline containing 2% goat serum and a protease inhibitor cocktail tablet (Roche). After overnight incubation at 4°C and gentle washing, tissue sections were fixed with 1% paraformaldehyde for 20 min and incubated with mouse anti-APC antibody [1/1000; clone APC003, immunoglobulin G1 (IgG1), BioLegend] for 3 hours at 4°C. Afterward, the goat anti-mouse AF647 antibody (1/1000; Jackson ImmunoResearch Laboratories Inc.) was added for 1 hour at room temperature to detect the tetramer signal. After a second blocking step with 10% mouse serum for 20 min, mouse monoclonal anti-CD8 (1/300; clone RFT8, IgG1, SouthernBiotech) and mouse monoclonal anti-CD45RO (1/200; clone UCHL1, BioLegend) antibodies directly conjugated to Alexa Fluor 488 (AF488) and AF594, respectively, were used to detect T cells (overnight at 4°C) and to confirm the specificity of the tetramer staining (fig. S1). To confirm the reliability of the tetramer, we developed a second in situ staining strategy. Briefly, after the blocking step, the polyclonal rabbit anti-CD8 antibody (1/300; Abcam ab4055) was incubated with the tetramer overnight at 4°C. Following fixation and incubation with mouse anti-APC antibody as described above, goat anti-mouse IgG AF647 antibody (1/1000; Jackson ImmunoResearch Laboratories Inc.) and goat anti-rabbit IgG AF488 antibody (1/1000; Thermo Fisher Scientific, A11070) were added for 1 hour at room temperature. Sections were counterstained with Hoechst (1/5000; Life Technologies). Upon mounting with ProLong Gold antifade mounting medium (Invitrogen) and drying, sections were scanned (image acquisition) using a Zeiss AxioScan.Z1 slide scanner (×20 magnification). In addition, the PPI15–24 tetramer was tested and validated using PBMCs (fig. S2). Consecutive sections were included as a negative control and incubated without the tetramer (fig. S5).
Immunofluorescence restaining
The slide scanner allows for analysis of entire pancreatic sections and the use of only four different fluorophore combinations. After the acquisition of the tetramer, CD8, and CD45RO staining combination, tissue sections were incubated with 3% H2O2 for 1 hour at room temperature, to inactivate fluorophores chemically, as described previously (42). Subsequently, the same tissue section was restained using guinea pig anti-insulin (1/1000; Dako), rabbit antiglucagon (1/1000: clone EP3070, Abcam), and mouse anti–HLA-ABC (1/100; clone W6/32, IgG2a, Dako) antibodies. The secondary antibodies were incubated for 1 hour at room temperature and used as follows: anti-rabbit AF555 (1/1000; Invitrogen), anti–guinea pig AF488 (1/500, Jackson ImmunoResearch Laboratories Inc.), and anti-mouse AF647 (1/1000; Thermo Fisher Scientific). After washing and mounting, tissue sections were scanned using a Zeiss AxioScan.Z1 slide scanner (×20 magnification). This approach is essential for in situ studies to visualize the exact location of such autoreactive T cells in the pancreas relative to the islets (fig. S1).
Image analysis
Among all donors, a total of 302 islets, 267 neighboring islet areas, and 261 exocrine regions were analyzed (table S1). First, using the restained image (insulin, glucagon, and MHC class I), exocrine regions and islets were picked randomly across the tissue section based on the presence or absence of insulin (fig. S1). Islets with an area of ≥10,000 μm2 were chosen (fig. S6). The exocrine area surrounding the islets was defined as a neighboring islet area. The same area was picked and analyzed using the tetramer image (PPI15–24, CD8, and CD45RO) (fig. S1C). The number of PPI15–24-specific CD8+ T cells (cells/mm2) in the three defined pancreatic compartments (exocrine region/neighboring islet area/islet) was determined by manually counting the cells using the ZEN 2.5 lite software (Zeiss).
Statistical analysis
Islets, neighboring islet area, and exocrine regions were picked randomly and combined per group (Table 1 and table S1). In the nondiabetes group (n = 6), a total of 63 islets, 57 neighboring islet areas, and 69 exocrine regions were selected. Furthermore, in the aab+ group (n = 5), a total of 83 islets, 73 neighboring islet areas, and 63 exocrine regions were picked. Last, a total of 69 ICIs, 87 IDIs, 137 neighboring islet areas, and 129 exocrine regions were selected for the T1D group (n = 11). Group differences were analyzed using Kruskal-Wallis followed by a Dunn multiple comparison test. Differences between ICIs versus IDIs were analyzed with the Mann-Whitney test. The analysis was performed using GraphPad Prism version 7 (GraphPad Software, San Diego, CA). Data in graphs and tables are presented as mean ± SEM. Findings were assumed as statistically significant at P < 0.05.
Supplementary Material
Acknowledgments
We thank Z. Mikulski and S. McArdle of La Jolla Institute for Immunology for help and support with image acquisition. We acknowledge the NIH Tetramer Core Facility for the provision of human PPI15–24 tetramer. Suggestions given by Prof. Bjoern Peters of La Jolla Institute for Immunology have been a great help in data analysis and presentation. Funding: The study was performed with the support of nPOD, a collaborative type 1 diabetes research project sponsored by JDRF. We thank the donors and their families who have generously donated organs to support this research. In addition, we thank the Organ Procurement Organizations that partner with nPOD to recover organ donor tissues and the nPOD staff members for collecting data and providing pancreas tissue samples. Organ Procurement Organizations (OPO) partnering with nPOD to provide research resources are listed at http://www.jdrfnpod.org//for-partners/npod-partners/. The research was also supported by the National Institutes of Health (NIH), grant # R01 AI092453. C.B. received support from the German research funding organization (DFG). Author contributions: C.B. participated in the project design; performed experiments, data analysis, and interpretation; and wrote the manuscript. T.R.-C. was involved in project design and data interpretation and participated in writing the manuscript. N.A. performed experiments. K.T.C. participated in writing the paper. M.G.v.H. participated in study design, data interpretation, and writing the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/42/eabc5586/DC1
REFERENCES AND NOTES
- 1.Atkinson M. A., Eisenbarth G. S., Michels A. W., Type 1 diabetes. Lancet 383, 69–82 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Katsarou A., Gudbjörnsdottir S., Rawshani A., Dabelea D., Bonifacio E., Anderson B. J., Jacobsen L. M., Schatz D. A., Lernmark Å., Type 1 diabetes mellitus. Nat. Rev. Dis. Primers. 3, 17016 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Pugliese A., Autoreactive T cells in type 1 diabetes. J. Clin. Invest. 127, 2881–2891 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.In't Veld P., Insulitis in human type 1 diabetes: A comparison between patients and animal models. Semin. Immunopathol. 36, 569–579 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babon J. A. B., De Nicola M. E., Blodgett D. M., Crèvecoeur I., Buttrick T. S., Maehr R., Bottino R., Naji A., Kaddis J., Elyaman W., James E. A., Haliyur R., Brissova M., Overbergh L., Mathieu C., Delong T., Haskins K., Pugliese A., Campbell-Thompson M., Mathews C., Atkinson M. A., Powers A. C., Harlan D. M., Kent S. C., Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat. Med. 22, 1482–1487 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Velthuis J. H., Unger W. W., Abreu J. R. F., Duinkerken G., Franken K., Peakman M., Bakker A. H., Reker-Hadrup S., Keymeulen B., Drijfhout J. W., Schumacher T. N., Roep B. O., Simultaneous detection of circulating autoreactive CD8+ T-cells specific for different islet cell-associated epitopes using combinatorial MHC multimers. Diabetes 59, 1721–1730 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Culina S., Lalanne A. I., Afonso G., Cerosaletti K., Pinto S., Sebastiani G., Kuranda K., Nigi L., Eugster A., Østerbye T., Maugein A., Mc Laren J. E., Ladell K., Larger E., Beressi J.-P., Lissina A., Appay V., Davidson H. W., Buus S., Price D. A., Kuhn M., Bonifacio E., Battaglia M., Caillat-Zucman S., Dotta F., Scharfmann R., Kyewski B., Mallone R.; ImMaDiab Study Group , Islet-reactive CD8(+) T cell frequencies in the pancreas, but not in blood, distinguish type 1 diabetic patients from healthy donors. Sci. Immunol. 3, eaao4013 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skowera A., Ladell K., Mc Laren J. E., Dolton G., Matthews K. K., Gostick E., Kronenberg-Versteeg D., Eichmann M., Knight R. R., Heck S., Powrie J., Bingley P. J., Dayan C. M., Miles J. J., Sewell A. K., Price D. A., Peakman M., beta-cell-specific CD8 T cell phenotype in type 1 diabetes reflects chronic autoantigen exposure. Diabetes 64, 916–925 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiedeman A. E., Muir V. S., Rosasco M. G., De Berg H. A., Presnell S., Haas B., Dufort M. J., Speake C., Greenbaum C. J., Serti E., Nepom G. T., Blahnik G., Kus A. M., James E. A., Linsley P. S., Long S. A., Autoreactive CD8+ T cell exhaustion distinguishes subjects with slow type 1 diabetes progression. J. Clin. Invest. 130, 480–490 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Speake C., Bahnson H. T., Wesley J. D., Perdue N., Friedrich D., Pham M. N., Lanxon-Cookson E., Kwok W. W., Hansen B. S., von Herrath M., Greenbaum C. J., Systematic assessment of immune marker variation in type 1 diabetes: A prospective longitudinal study. Front. Immunol. 10, 2023 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu W., Jiang N., Ebert P. J. R., Kidd B. A., Müller S., Lund P. J., Juang J., Adachi K., Tse T., Birnbaum M. E., Newell E. W., Wilson D. M., Grotenbreg G. M., Valitutti S., Quake S. R., Davis M. M., Clonal deletion prunes but does not eliminate self-specific αβ CD8(+) T lymphocytes. Immunity 42, 929–941 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coppieters K. T., Dotta F., Amirian N., Campbell P. D., Kay T. W. H., Atkinson M. A., Roep B. O., von Herrath M. G., Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J. Exp. Med. 209, 51–60 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skowera A., Ellis R. J., Varela-Calviño R., Arif S., Huang G. C., Van-Krinks C., Zaremba A., Rackham C., Allen J. S., Tree T. I. M., Zhao M., Dayan C. M., Sewell A. K., Unger W. W., Drijfhout J. W., Ossendorp F., Roep B. O., Peakman M., CTLs are targeted to kill beta cells in patients with type 1 diabetes through recognition of a glucose-regulated preproinsulin epitope. J. Clin. Invest. 118, 3390–3402 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richardson S. J., Rodriguez-Calvo T., Gerling I. C., Mathews C. E., Kaddis J. S., Russell M. A., Zeissler M., Leete P., Krogvold L., Dahl-Jørgensen K., von Herrath M., Pugliese A., Atkinson M. A., Morgan N. G., Islet cell hyperexpression of HLA class I antigens: A defining feature in type 1 diabetes. Diabetologia 59, 2448–2458 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez-Calvo T., Zapardiel-Gonzalo J., Amirian N., Castillo E., Lajevardi Y., Krogvold L., Dahl-Jørgensen K., von Herrath M. G., Increase in pancreatic proinsulin and preservation of beta-cell mass in autoantibody-positive donors prior to type 1 diabetes Onset. Diabetes 66, 1334–1345 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Danke N. A., Koelle D. M., Yee C., Beheray S., Kwok W. W., Autoreactive T cells in healthy individuals. J. Immunol. 172, 5967–5972 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Summers K. L., Behme M. T., Mahon J. L., Singh B., Characterization of dendritic cells in humans with type 1 diabetes. Ann. N. Y. Acad. Sci. 1005, 226–229 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Meier J. J., Bhushan A., Butler A. E., Rizza R. A., Butler P. C., Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: Indirect evidence for islet regeneration? Diabetologia 48, 2221–2228 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez-Calvo T., Ekwall O., Amirian N., Zapardiel-Gonzalo J., von Herrath M. G., Increased immune cell infiltration of the exocrine pancreas: A possible contribution to the pathogenesis of type 1 diabetes. Diabetes 63, 3880–3890 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Damond N., Engler S., Zanotelli V. R. T., Schapiro D., Wasserfall C. H., Kusmartseva I., Nick H. S., Thorel F., Herrera P. L., Atkinson M. A., Bodenmiller B., A map of human type 1 diabetes progression by imaging mass cytometry. Cell Metab. 29, 755–768.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez-Calvo T., Sabouri S., Anquetil F., von Herrath M. G., The viral paradigm in type 1 diabetes: Who are the main suspects? Autoimmun. Rev. 15, 964–969 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Li X., Atkinson M. A., The role for gut permeability in the pathogenesis of type 1 diabetes—A solid or leaky concept? Pediatr. Diabetes 16, 485–492 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gonzalez-Duque S., Azoury M. E., Colli M. L., Afonso G., Turatsinze J.-V., Nigi L., Lalanne A. I., Sebastiani G., Carré A., Pinto S., Culina S., Corcos N., Bugliani M., Marchetti P., Armanet M., Diedisheim M., Kyewski B., Steinmetz L. M., Buus S., You S., Dubois-Laforgue D., Larger E., Beressi J.-P., Bruno G., Dotta F., Scharfmann R., Eizirik D. L., Verdier Y., Vinh J., Mallone R., Conventional and neo-antigenic peptides presented by beta cells are targeted by circulating naive CD8+ T cells in type 1 diabetic and healthy donors. Cell Metab. 28, 946–960.e6 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Kuric E., Seiron P., Krogvold L., Edwin B., Buanes T., Hanssen K. F., Skog O., Dahl-Jørgensen K., Korsgren O., Demonstration of tissue resident memory CD8 T cells in insulitic lesions in adult patients with recent-onset type 1 diabetes. Am. J. Pathol. 187, 581–588 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Chee J., Ko H.-J., Skowera A., Jhala G., Catterall T., Graham K. L., Sutherland R. M., Thomas H. E., Lew A. M., Peakman M., Kay T. W. H., Krishnamurthy B., Effector-memory T cells develop in islets and report islet pathology in type 1 diabetes. J. Immunol. 192, 572–580 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Gagnerault M. C., Luan J. J., Lotton C., Lepault F., Pancreatic lymph nodes are required for priming of beta cell reactive T cells in NOD mice. J. Exp. Med. 196, 369–377 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Graham K. L., Krishnamurthy B., Fynch S., Mollah Z. U., Slattery R., Santamaria P., Kay T. W., Thomas H. E., Autoreactive cytotoxic T lymphocytes acquire higher expression of cytotoxic effector markers in the islets of NOD mice after priming in pancreatic lymph nodes. Am. J. Pathol. 178, 2716–2725 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Morchoe C. C., Lymphatic system of the pancreas. Microsc. Res. Tech. 37, 456–477 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Weisberg S. P., Carpenter D. J., Chait M., Dogra P., Gartrell-Corrado R. D., Chen A. X., Campbell S., Liu W., Saraf P., Snyder M. E., Kubota M., Danzl N. M., Schrope B. A., Rabadan R., Saenger Y., Chen X., Farber D. L., Tissue-resident memory T cells mediate immune homeostasis in the human pancreas through the PD-1/PD-L1 pathway. Cell Rep. 29, 3916–3932.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thome J. J. C., Yudanin N., Ohmura Y., Kubota M., Grinshpun B., Sathaliyawala T., Kato T., Lerner H., Shen Y., Farber D. L., Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell 159, 814–828 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dissanayake D., Gronski M. A., Lin A., Elford A. R., Ohashi P. S., Immunological perspective of self versus tumor antigens: Insights from the RIP-gp model. Immunol. Rev. 241, 164–179 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Coppieters K., Amirian N., von Herrath M., Intravital imaging of CTLs killing islet cells in diabetic mice. J. Clin. Invest. 122, 119–131 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foulis A. K., Farquharson M. A., Meager A., Immunoreactive alpha-interferon in insulin-secreting beta cells in type 1 diabetes mellitus. Lancet 2, 1423–1427 (1987). [DOI] [PubMed] [Google Scholar]
- 34.Atkinson M. A., Bluestone J. A., Eisenbarth G. S., Hebrok M., Herold K. C., Accili D., Pietropaolo M., Arvan P. R., Herrath M. V., Markel D. S., Rhodes C. J., How does type 1 diabetes develop?: The notion of homicide or beta-cell suicide revisited. Diabetes 60, 1370–1379 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benechet A. P., Menon M., Khanna K. M., Visualizing T cell migration in situ. Front. Immunol. 5, 363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willcox A., Richardson S. J., Bone A. J., Foulis A. K., Morgan N. G., Analysis of islet inflammation in human type 1 diabetes. Clin. Exp. Immunol. 155, 173–181 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christoffersson G., Chodaczek G., Ratliff S. S., Coppieters K., von Herrath M. G., Suppression of diabetes by accumulation of non-islet-specific CD8(+) effector T cells in pancreatic islets. Sci. Immunol. 3, eaam6533 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Rodriguez-Calvo T., von Herrath M. G., Enterovirus infection and type 1 diabetes: Closing in on a link? Diabetes 64, 1503–1505 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Riahi Y., Israeli T., Cerasi E., Leibowitz G., Effects of proinsulin misfolding on β-cell dynamics, differentiation and function in diabetes. Diabetes Obes. Metab. 20 ( Suppl 2), 95–103 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Arunagiri A., Haataja L., Cunningham C. N., Shrestha N., Tsai B., Qi L., Liu M., Arvan P., Misfolded proinsulin in the endoplasmic reticulum during development of beta cell failure in diabetes. Ann. N. Y. Acad. Sci. 1418, 5–19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skyler J. S., Prevention and reversal of type 1 diabetes—Past challenges and future opportunities. Diabetes Care 38, 997–1007 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Lin J. R., Fallahi-Sichani M., Chen J. Y., Sorger P. K., Cyclic immunofluorescence (CycIF), a highly multiplexed method for single-cell imaging. Curr. Protoc. Chem. Biol. 8, 251–264 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/42/eabc5586/DC1