

Abstract
Coronaviruses have been reported previously due to their association with the severe acute respiratory syndrome (SARS). After SARS, these viruses were known to be causing Middle East respiratory syndrome (MERS) and caused 35% evanescence amid victims pursuing remedial care. Nowadays, beta coronaviruses, members of Coronaviridae, family order Nidovirales, have become subjects of great importance due to their latest pandemic originating from Wuhan, China. The virus named as human-SARS-like coronavirus-2 contains four structural as well as sixteen nonstructural proteins encoded by single-stranded ribonucleic acid of positive polarity. As there is no vaccine available to treat the infection caused by these viruses, there is a dire need for taking necessary steps against this virus. Herein, we have targeted two nonstructural proteins of SARS-CoV-2, namely, methyltransferase (nsp16) and helicase (nsp13), respectively, due to their substantial activity in viral pathogenesis. A total of 2035 compounds were analyzed for their pharmacokinetics and pharmacological properties. The screened 108 compounds were docked against both targeted proteins and were compared with previously reported known compounds. Compounds with high binding affinity were analyzed for their reactivity through DFT analysis, and binding was analyzed using molecular dynamics simulations. Through the analyses performed in this study, it is concluded that EryvarinM, Silydianin, Osajin, and Raddeanine can be considered potential inhibitors for MTase, while TomentodiplaconeB, Osajin, Sesquiterpene Glycoside, Rhamnetin, and Silydianin for helicase after these compounds are validated thoroughly using in vitro and in vivo protocols.
1. Introduction
Coronaviruses have become a source of multiple systematic infections in various animals [1]. These viruses have previously caused respiratory tract infections in humans, including severe acute respiratory syndrome and the Middle East respiratory syndrome [2]. As there are no specific therapeutic agents for SARS-CoV, so the use of face mask, hand washing, and careful disposal of medical equipment and other materials infected with nasal secretions are preliminary preventive measures which must be followed to avoid the spread of the pandemic virus [3]. The initial clinical manifestations in the SARS-CoV infected patients observed were fever, cough, headache, myalgia, diarrhoea, dyspnea, leucopenia, lymphopenia, thrombocytopenia, hypoxaemia, pulmonary infiltration, and disturbed hepatic and renal function leading to death in some individuals [4].
The genome of SARS-CoV is 30 kb encoding 27 proteins and has 14 ORFs. These proteins are further classified as structural proteins and nonstructural proteins. The structural proteins like nucleocapsid protein (N), pike surface glycoprotein (S), a small envelope protein (E), and matrix protein (M) are known to play a role in binding to host receptors. The genome of the virus encodes 16 nonstructural proteins like Helicase (nsp13), RNA-dependent RNA polymerase (nsp12), Papain-like protease (nsp3), main protease (nsp5) also known as 3C-like protease (3CLpro), and 2′O methyltransferase (nsp16). The nonstructural proteins are likely to be involved in transcription, replication, and pathogenesis and play a vital role in the life cycle of the pathogen [5–9]. Owing to the critical role of nonstructural proteins of the virus in virulence, these proteins are potential targets for antiviral drugs [9].
Helicase of coronavirus is a member of superfamily-I helicase comprising of seven conserved regions. This protein winds down partially duplex ribonucleic acid in a 5′ to 3′ way to make it open [10]. The helicase (nps13) of SARS-CoV belongs to superfamily 1 (SF1) of the six helicase superfamilies; the division is based upon various specified regions. This enzyme can open up both ribo- and deoxyribonucleic acid duplexes in 5′ to 3′ direction [11]. The associated NTPase action can target all-natural nucleotides and deoxynucleotides as substrates [12]. It is also reported that SARS-CoV-nsp12 can improve the helicase action of SARS-nsp13 by cumulative action on nucleic acid (dsRNA or dsDNA) unwinding by 2-folds [13].
There are almost a dozen conserved motifs in SF1 helicases, those involved in direct binding with nucleic acid triphosphates and nucleic acids. Two motifs known as Walker A and B are common in all of members of this super family. By investigating the structure of the enzyme, it is revealed that the two RecA-like domains (1A and 2A) constitute the catalytic site of SF1 helicases. This enzyme is known to be engaged in synthesis of viral RNA and also manipulates the DNA of host [13–15]. 2′-O-MTase (2'O methyltransferase) of coronaviruses is highly conserved and is known to play an indispensable role in viral replication and evasion from innate immunity [16]. It is also reported that the methyltransferases are vital for viral replication in cell cultures. Another protein nsp10 is also crucial for the proper functioning of MTase activity in the host cells for proliferation [17, 18]. Due to the conserved nature of this enzyme and its role in virulence, this enzyme is an ideal target for potential antiviral agents and vaccines against SARS-CoV-2, SARS-CoV, MERS-CoV, or other RNA and DNA viruses [19].
Phytochemicals have been known from ancient times for their immense potential and beneficial properties against several infectious diseases and health-related complications [20]. The phytochemicals constitute magnificent potentials, specifically antiviral properties, and can be used in treating viral infections. With time, in silico approaches are gaining much attention around the world for their advanced strategies and effective techniques related to the field of medical sciences. They provide an operative platform, where scientists can analyze a wide range of biological phenomena, different pathways, and molecular interactions. These methods are primarily cost-effective and consist of authentic methods which predict the results with the highest accuracy [21, 22].
The present study aims at the in silico analysis of two nonstructural proteins of novel coronavirus (SARS-CoV-2), i.e., helicase (nsp13) and methyltransferase (nsp16). The study is performed opting standard protocols of computer-aided drug discovery, as previously mentioned in a series of publication [23–35]. The analyses performed included ADMET analysis of phytochemicals, molecular docking of phytochemicals with targeted proteins of a novel coronavirus, binding analysis through molecular dynamics (MD) simulations, and reactivity analysis through density functional theory (DFT) calculations.
2. Materials and Methods
Herein, the MTase and helicase of the novel coronavirus were targeted for the identification of candidate inhibitors; thus, a series of analyses were performed, and the flowchart is shown in Figure 1.
Figure 1.

Flowchart of methodology.
2.1. Obtaining the Phytochemicals
The datasets comprising of a wide range of phytochemicals (2035) were collected by using two different databases [36, 37] and also have been reported previously in [35] (Table S1). The set comprised of a variety of phytochemical groups, i.e., 322 alkaloids, 113 terpenoids, 105 aurones, 101 chalcones, 378 flavonoids, 211 lignans, 255 carboxylic acids, 301 polyphenols, and 249 quinones [35]. The 3D structures for the phytochemicals were obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov/). The pharmacological and pharmacokinetic properties based on parameters of ADMET (Absorption, Distribution, Metabolism, Excretion and Toxicity) were evaluated with the help of the SwissADME web server [38] and PreADMET server [39], as reported in [23, 26, 28, 29, 31]. SwissADME server was utilized for the determination of ADME properties of the phytochemicals, while PreADMET was utilized to assess the druglikeness features and toxicity level of the drug. The structural file (.SDF) of the 2035 phytochemicals was utilized for the prediction purposes.
2.2. Homology Modelling
The crystal structure of MTase was available at RCSB with PDB ID: 6W61; however, helicase structure was unavailable. For protein structure prediction and homology modelling, the polypeptide sequence of SARS-CoV-2 (GenBank ID: QHD43415.1) was used, comprising 7096 residues. For mapping helicase sequence, multiple sequence alignment was performed between targeted sequence and SARS-CoV sequence (UniProt Acc ID: P0C6X7). Later on, homology modelling was performed. PSI-BLAST method was performed for template structure which was homologous to the helicase. Thus, it was observed that the sequence of helicase showed 96.08% similarity with 6YJT. The Modeller 9.18 was used to perform the homology modelling with help of 6YJT structure [40]. Opting homology modelling, 100 models were predicted and evaluated based on Discrete Optimized Protein Energy (DOPE) score. The structure quality assessment was performed by the Ramachandran Plot using RAMPAGE tool [41]. For identifying pocket, POVME 2.0 was used [42].
2.3. Molecular Docking and Binding Analysis
The possible inhibitory effects of these naturally occurring compounds were evaluated by the docking method against the targeted both proteins. AutoDock Tools and AutoDock Vina were used for molecular docking of proteins and ligands [43–45]. Binding energies were estimated, and interactions were analyzed. Using binding affinities, Ki values were also calculated by equation (1)
| (1) |
where ΔG represents binding energy, T depicts the temperature which is 298.15 k, and R depicts gas constant with value 1.9872036 kcal/mol. To find out the binding affinity of inhibitors with targeted proteins, docking was performed. A grid box was generated with the help of AutoDock Tools, and sizes of x, y, and z were determined. Grid box dimensions are provided in Table 1. Binding affinity values of these ligands were determined to evaluate how well they interact with the protein of interest. The docking was performed with 6 different exhaustiveness heuristics which were E = 4, E = 8, E = 16, E = 32, E = 64, and E = 128. However, no deviation was observed in the values of binding affinity after E = 8. The output files obtained from docking were used for structural analysis in the Discovery Studio 2.5 [46]; thus, 2D and 3D structural images for binding were generated.
Table 1.
Grid box dimensions for receptors (Å3).
| Receptor | Grid box dimensions |
|---|---|
| MTase | 26 × 20 × 18 |
| Helicase | 20 × 18 × 18 |
Constant temperature MD simulations were performed to study the stability in the binding of phytochemicals with MTase and helicase using Groningen Machine for Chemical Simulations (GROMACS) v 5.0 [47]. Only those complexes were analyzed, where phytochemicals showed high binding affinity. For all those protein-ligand complexes, the optimized potential for liquid simulation (OPLS-AA) was applied, and the system was solvated with spc216 water molecules. This solvated system was neutralized by adding counter ions of Na+ and Cl−. At the next step, this system was subjected to energy minimization with the steepest descent method, keeping step limit as 50000. Later on, constant Number Volume and Temperature (NVT) and constant Number Pressure and Temperature (NPT) equilibrations were performed with 1 atm pressure and at 300 K. Explicit water molecules were also added in the phosphoserine sites, and for all simulations, standard pH of 7.0 was considered. This set of constraints was selected due to keeping the simulations similar to the human biological system. The duration for both equilibrations was 1 ns whereas the force field used in both equilibrations was Particle Mesh Ewald (PME) with a cubic interpolation implementation [48]. The hydrogen bonds were readjusted with the help of Linear Constraint Solver (LINCS) technique while performing equilibrations [49]. The final production MD simulation was performed for 50 ns, keeping the method same as equilibrations [50].
2.4. Reactivity Studies through DFT
Density functional theory (DFT) analysis was performed to study the reactivity of ligands with MTase and helicase. For analysis, HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) energies were computed. The ΔE (band energy gap) calculation was performed using the expression ELUMO − EHOMO. These descriptors are based on quantum mechanics and its computations and were performed using the program named ORCA [51]. B3LYP exchange-correlation functional was employed for calculations, which is a hybrid exchange-correlation functional. Generally, the hybrid exchange-correlation is a combination of Hartree-Fock exact exchange functional and any other density functional. However, the targeted correlation, i.e., B3LYP, is defined as:
| (2) |
where a0 = 0.20, ax = 0.72, and ac = 0.81. ExGGA is the generalized gradient approximation for the Becke 88 functional while the EcGGA reflects the correlation functional of Lee-Yang-Parr. With this hybrid functional, local density approximation is added in the form of EcLDA [52].
3. Results and Discussion
Human CoVs are zoonotic pathogens, originating from animals, infecting humans leading to respiratory complications. All HCoVs provide substantial evidence of its origin from bats [53]. Phylogenetically related CoVs in bats and humans suggest host switches. SARS-CoV switched hosts from Rhinolophid bats to palm civets and human [54]. The evolutionary studies also showed that bats also host MERS-like virus suggesting bats as MERS-CoV source; infecting humans had evidence that camels might also be its likely source [55]. These viruses can transmit to humans, directly or indirectly [56].
Currently, there is no suitable vaccine available which can be used to cure infections caused by the novel coronavirus currently named as COVID-19. It is highly needed to design a drug or a vaccine to treat this virus as well as to reduce the viral deportment so that its spread could be limited. Repurposing of drugs is a remarkable approach to opt antiviral compounds as COVID-19 drug candidates [57]. Recently, a study has been reported targeting the main protease of SARS-CoV-2, using a set of phytochemicals [30]. However, herein, another set of phytochemicals has been used to target MTase and helicase.
The criteria for evaluating the compounds based on their ADMET profiles was “Violations from Lipinski's rule = Zero; Soluble = High/Very High; Absorption in Gastrointestinal (GI) tract = Efficient or Moderate; Blood-brain barrier (BBB) permeability = No; and Toxicity/carcinogenicity =0” [58]. Through the screening criteria for their ADMET properties, 108 phytochemicals out of 2035 qualified for performing the further analysis (Table S2). These selected phytochemicals were prepared for molecular docking with our targeted proteins.
3.1. Structure Evaluation for Helicase
The structure of helicase was modelled using homology modelling due to unavailability of X-ray crystallographic structure. As the predicted model with the lowest DOPE score was used further, it was observed to be -24913.17, and the predicted model is shown in Figure 2.
Figure 2.

Predicted 3D model for helicase. Red depicts α-helices; β-purple depicts the strands while green depicts random coil.
For quality assessment of the structure, the Ramachandran plot was generated for the predicted model using RAMPAGE tool [41]. As per the analysis of possible conformations of φ and ψ angles for individual amino acid residues in helicase model, it was observed that 98.0% residues were in the favoured region, and 1.9% residues were in the allowed region while only 0.1% residues were in outlier region (Figure 3).
Figure 3.

Ramachandran plot for predicted helicase model.
3.2. Molecular Docking and Binding Stability
Initially, Remdesivir, Prulifloxacin, and Nelfinavir were docked with MTase and helicase, and the binding affinities of these compounds were used as a threshold for screening phytochemicals (Tables 2 and 3). Based on the results, it was observed that threshold for screening phytochemicals for MTase was -8.2 kcal/mol, and for helicase, it was -8.1 kcal/mol. Using these thresholds, phytochemicals were screened and further analyzed for stability and reactivity.
Table 2.
Results of molecular docking of Remdesivir, Prulifloxacin, and Nelfinavir with MTase of SARS-CoV-2.
| Sr. no | Compound name | Binding affinity (kcal/mol) | K i value (μM) | Interactions with MTase of SARS-CoV-2 |
|---|---|---|---|---|
| 1 | Nelfinavir | -8.2 | 1.138 |

|
| 2 | Prulifloxacin | -7.6 | 2.648 |

|
| 3 | Remdesivir | -7.0 | 7.299 |

|
Table 3.
Results of molecular docking of Remdesivir, Prulifloxacin, and Nelfinavir with helicase of SARS-CoV-2.
| Sr. no | Compound name | Binding affinity (kcal/mol) | K i value (μM) | Interactions with helicase of SARS-CoV-2 |
|---|---|---|---|---|
| 1 | Prulifloxacin | -8.1 | 1.138 |

|
| 2 | Remdesivir | -6.8 | 10.234 |

|
| 3 | Nelfinavir | -6.2 | 28.205 |

|
Herein, we selected Nelfinavir, Prulifloxacin, and Remdesivir from three different drug repurposing studies and docked them as controls in the present study [59, 60]. A recent paper reported an inhibitor effect of Remdesivir (a new antiviral drug) on the growth of SARS-CoV-2 in vitro, and an early clinical trial conducted in SARS-CoV-2 Chinese patients [61]. When Remdesivir interacted with MTase and helicase, it showed -7.0 kcal/mol and -6.8 kcal/mol binding affinity, respectively. Recent studies proposed a few drugs that target COVID-19 and suggested these compounds could be used to treat COVID-19 [62]. Prulifloxacin gave -7.6 kcal/mol with MTase and -8.1 kcal/mol with helicase. Nelfinavir, a repurposed drug, is previously known for its selectively inhibitory properties against HIV protease. This enzyme performs the posttranslational treatment of HIV propeptides. Undeveloped, noninfectious viral particles form in the cells containing this drug [63]. When Nelfinavir was docked against MTase, it showed binding affinity of -8.2 kcal/mol, while for helicase, it gave binding affinity value of -6.2 kcal/mol.
3.2.1. Results of Molecular Docking of MTase of SARS-CoV-2
All 108 compounds, initially screened through ADMET, were docked with MTase of SARS-CoV-2, and the threshold used for screening phytochemicals was -8.2 kcal/mol. Thus, by applying the threshold, it was observed that only four compounds passed the threshold which were EryvarinM, Silydianin, Osajin, and Raddeanine and showed similar or better results than those of Remdesivir, Prulifloxacin, and Nelfinavir (Table 4).
Table 4.
Results of molecular docking of MTase of SARS-CoV-2.
| Sr. no | Compound name | Binding affinity (kcal/mol) | K i value (μM) | Interactions with MTase of SARS-CoV-2 |
|---|---|---|---|---|
| 1 | EryvarinM | -8.6 | 0.489 |

|
| 2 | Silydianin | -8.5 | 0.579 |

|
| 3 | Osajin | -8.2 | 0.961 |

|
| 4 | Raddeanine | -8.2 | 0.961 |

|
EryvarinM docked with MTase with a binding affinity of -8.6 kcal/mol with a Ki value of 0.489 μM while forming a conventional hydrogen bond with ASN6841, ASP6873, ASN6899, CYS6913, and LYS6968. Formation of pi-cation and pi-anion was observed with LYS6844 and ASP6897. Furthermore, along with these interactions, pi-alkyl interactions with MET6929 and LEU6898 were observed (Figure 4).
Figure 4.

3D interaction model for EryvarinM docked with MTase.
Silydianin docked with binding affinity -8.5 kcal/mol and Ki value of 0.579 μM while interacting with GLY6911, ASP6928, CYS6913, and ASN6899, by forming conventional hydrogen bonds. Furthermore, it formed a carbon-hydrogen bond and pi-acceptor-acceptor interactions with MET6929 and ASP6897, while pi-alkyl and alkyl interactions with LEU6898, PHE6947, and LYS6944 were observed.
Osajin and Raddeanine docked with binding affinity -8.2 kcal/mol and Ki value of 0.961 μM. Osajin formed a conventional hydrogen bond with ASP6928; while Raddeanine formed conventional hydrogen bonds with ASP6912, CYS6913, LEU6898, and ASN6841, however various other interactions were observed as shown in Table 4.
3.2.2. Results of Molecular Docking of Helicase of SARS-CoV-2
After analyzing the ligand interactions with the binding pocket of MTase, selected phytochemicals were auto docked with helicase of SARS-CoV-2. All 108 compounds, initially screened through ADMET, were docked with helicase of SARS-CoV-2, and the threshold used for screening phytochemicals was -8.1 kcal/mol. Thus, by applying the threshold, it was observed that only five compounds passed the threshold which was TomentodiplaconeB, Osajin, Sesquiterpene Glycoside, Rhamnetin, and Silydianin and showed similar or better results than those of Remdesivir, Prulifloxacin, and Nelfinavir (Table 5).
Table 5.
Results of molecular docking of helicase of SARS-CoV-2.
| Sr. no | Compound name | Binding affinity (kcal/mol) | K i value (μM) | Interactions with helicase of SARS-CoV-2 |
|---|---|---|---|---|
| 1 | TomentodiplaconeB | -8.4 | 0.685 |

|
| 2 | Osajin | -8.2 | 0.961 |

|
| 3 | Sesquiterpene Glycoside | -8.2 | 0.961 |
|
| 4 | Rhamnetin | -8.1 | 1.138 |

|
| 5 | Silydianin | -8.1 | 1.138 |

|
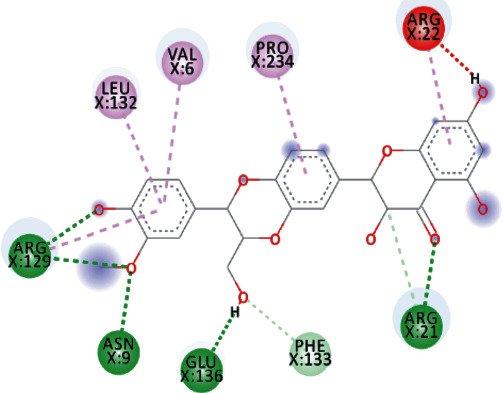
TomentodiplaconeB formed carbon-hydrogen bond with ARG129, amide pi-stacked with GLU128, and alkyl and pi-alkyl interactions with PRO23, 234, and 238, LEU132, VAL6, PHE24, and ARG1. As in result, the binding affinity was -8.4 kcal/mol while Ki was 0.685 μM (Figure 5).
Figure 5.
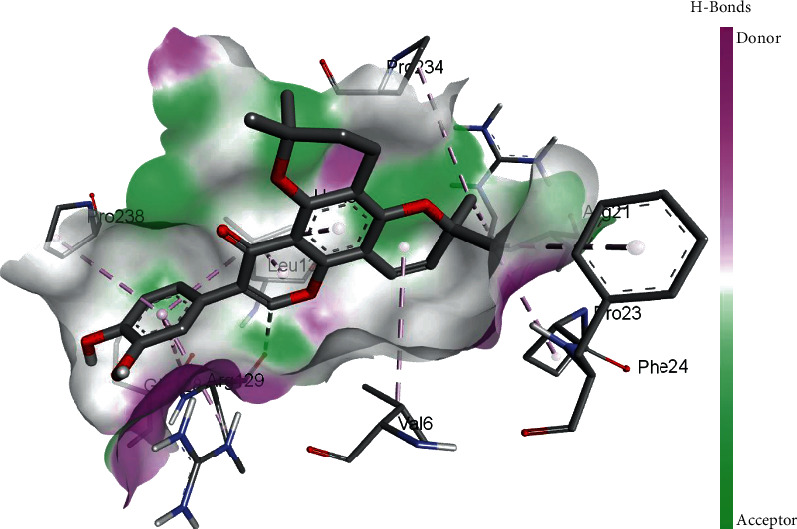
3D interaction model for TomentodiplaconeB docked with helicase.
Both, Sesquiterpene Glycoside and Osajin docked with a binding affinity of -8.2 kcal/mol and Ki of 0.961 μM. Sesquiterpene Glycoside formed conventional hydrogen bonds with ARG21 and ARG129, ASN9, and GLU136; donor interactions with ARG22; carbon-hydrogen bond with PHE133; and pi-alkyl interactions with LEU132, VAL6, and PRO234. Osajin formed conventional hydrogen bonds with ARG21 and ARG129 and alkyl and pi-alkyl interactions with LEU132 and VAL6.
Both, Rhamnetin and Silydianin docked with a binding affinity of -8.1 kcal/mol and Ki of 1.138 μM. Rhamnetin formed conventional hydrogen bonds with ASP179, ARG178, and SER310; pi-cation and anion with ASP534 and ARG560; carbon-hydrogen bond and pi-sigma interactions with PRO408; and amide pi-stacked with ALA407 as well as pi-alkyl with PRO406 and ARG409. Silydianin formed conventional hydrogen bonds with ARG129 and ASN9. Furthermore, it formed pi-alkyl and alkyl interactions with PHE133, LEU132, VAL6, and PHE24.
3.2.3. Stability in Complexes Analyzed through MD Simulations
After analyzing the interactions of ligands with targeted receptors, the MD simulations were performed to analyze the stability in complexes and binding of ligands with the proteins. These MD simulations helped in estimating the stability of binding of screened ligands with SARS-CoV-2 proteins. The radius of gyrations (Rg) was plotted to analyze the stability in complexes, while the root means square deviation (RMSD) values were also observed for whole simulations. Average RMSD values are reported in Table 6, while graphs of the radius of gyration are shown in Figure 3.
Table 6.
Average RMSD values for all complexes.
| Complex | Average RMSD (Å) |
|---|---|
| MTase-EryvarinM | 1.95 |
| MTase-Silydianin | 2.05 |
| MTase-Osajin | 2.99 |
| MTase-Raddeanine | 3.12 |
| Helicase-TomentodiplaconeB | 2.11 |
| Helicase-Osajin | 2.32 |
| Helicase-Sesquiterpene Glycoside | 2.46 |
| Helicase-Rhamnetin | 2.76 |
| Helicase-Silydianin | 3.31 |
According to the results shown in Table 6, the RMSD values were observed to have mere changes, while looking in complexes of same receptors; however, these values were very low, i.e., less than 3.50 Å. Furthermore, the graphs shown in Figure 6 depicted fewer fluctuations in the radius of gyration. These values depict high stability, compactness, and stable folding of protein tertiary structure, as well as stability in protein-ligand complexes. The changes and fluctuations were observed in all complexes and not in any specific complex. The complexes with less binding affinity showed a decrease in stability and compactness, as these compounds were not bound strongly. These trends and results are also in accordance with various previously reported results [33, 60].
Figure 6.

MD simulations based Rg graphs of complexes for strongly binding phytochemicals: (a) complexes of helicase; (b) complexes of MTase.
3.3. Reactivity Studies for Phytochemicals and Targeted Proteins' Complexes
Reactivity of bound phytochemicals with helicase and MTase was analyzed through density functional theory- (DFT-) based computations, which works on the principles of quantum mechanics and its descriptors. The result showed that the phytochemicals with the broad range of chemical diversity exhibited good binding interactions with both the proteins. EryvarinM, Silydianin, Osajin, and Raddeanine, which mainly exhibited best docking results for MTase, were further selected for reactivity analysis, while for helicase, TomentodiplaconeB, Osajin, Sesquiterpene Glycoside, Rhamnetin, and Silydianin were selected. For studying reactivity, band energy gap was computed using molecular orbital energy descriptors, and the results are shown in Table 7.
Table 7.
Reactivity of phytochemicals with helicase and MTase depicted by band energy gaps.
| Complexes | E LUMO (kcal/mol) | E HOMO (kcal/mol) | Band energy gap (ΔE) (kcal/mol) |
|---|---|---|---|
| MTase-EryvarinM | -0.274 | -0.391 | 0.112 |
| MTase-Silydianin | -0.236 | -0.351 | 0.115 |
| MTase-Osajin | -0.271 | -0.400 | 0.129 |
| MTase-Raddeanine | -0.242 | -0.375 | 0.133 |
| Helicase-TomentodiplaconeB | -0.280 | -0.395 | 0.116 |
| Helicase-Osajin | -0.218 | -0.335 | 0.117 |
| Helicase-Sesquiterpene Glycoside | -0.198 | -0.321 | 0.123 |
| Helicase-Rhamnetin | -0.303 | -0.428 | 0.125 |
| Helicase-Silydianin | -0.118 | -0.246 | 0.128 |
Lower band energy gaps show high reactivity; thus, these results exhibited in Table 7 exhibited high reactivity of phytochemicals with targeted receptors. The band energy gap values for these phytochemicals ranged from 0.112 kcal/mol to 0.133 kcal/mol and 0.116 kcal/mol to 0.128 kcal/mol, for MTase and helicase, respectively, showing narrow energy gaps and proving their high reactivity properties. B3LYP function from DFT was applied to analyze the molecular orbital energies, and it is well established in the literature that the lower band energy gap depicts higher reactivity of compounds. The reason is that band energy gaps are computed through molecular descriptors, and these descriptors are responsible for the charges transferred in a chemical reaction [64]. These energies can characterize the electrophilic or nucleophilic nature of a compound. Therefore, the screened phytochemicals illustrated the higher reactivity of these phytochemicals, as reported in various previous studies [23–25, 27, 30–33, 35, 52, 65].
4. Conclusion
The novel coronavirus is causing the COVID-19 worldwide, a disease which has high morbidity and a significant mortality rate. The virus, sweeping across the whole world, is pandemic as declared by the World Health Organization, and the disease was declared as a Public Health Emergency of International Concern on 30 January 2020. However, still, there exists no remedy, drug, or vaccine for the treatment of COVID-19. This study provides insights into the mechanism of selective phytochemicals, when docked against two main targets of a novel coronavirus, MTase and helicase, by showing their pharmacological properties, binding and its stability, and the reactivity. Through analysis, it is concluded that phytochemicals such as EryvarinM, Raddeanine, TomentodiplaconeB, Osajin, Sesquiterpene Glycoside, Rhamnetin, and Silydianin can be considered as candidate inhibitors for targeted proteins and as drugs, after their in vitro and in vivo examinations.
Data Availability
The data used to support the findings of this study are included within the supplementary information files.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Supplementary Materials
Table S1: list of 2035 phytochemicals and their plants. Table S2: ADMET of Screened 108 Phytochemicals.
References
- 1.Weiss S. R., Navas-Martin S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiology and Molecular Biology Reviews. 2005;69(4):635–664. doi: 10.1128/mmbr.69.4.635-664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chu C., Cheng V., Hung I., et al. Role of lopinavir/ritonavir in the treatment of SARS: initial virological and clinical findings. Thorax. 2004;59(3):252–256. doi: 10.1136/thorax.2003.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Song Z., Xu Y., Bao L., et al. From SARS to MERS, thrusting coronaviruses into the spotlight. Viruses. 2019;11(1):p. 59. doi: 10.3390/v11010059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sizun J., Arbour N., Talbot P. J. Comparison of immunofluorescence with monoclonal antibodies and RT-PCR for the detection of human coronaviruses 229E and OC43 in cell culture. Journal of Virological Methods. 1998;72(2):145–152. doi: 10.1016/S0166-0934(98)00013-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan J. F.-W., Kok K.-H., Zhu Z., et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerging Microbes & Infections. 2020;9(1):221–236. doi: 10.1080/22221751.2020.1719902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Y., Hou Y., Shen J., Huang Y., Martin W., Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discovery. 2020;6(1):1–18. doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gordon D. E., Jang G. M., Bouhaddou M., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:1–13. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.C S G of the International. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nature Microbiology. 2020;5(4):p. 536. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cascella M., Rajnik M., Cuomo A., Dulebohn S. C., Di Napoli R. Statpearls. StatPearls Publishing; 2020. Features, evaluation and treatment coronavirus (COVID-19) [PubMed] [Google Scholar]
- 10.Adedeji A. O., Lazarus H. Biochemical characterization of Middle East respiratory syndrome coronavirus helicase. mSphere. 2016;1(5, article e00235) doi: 10.1128/mSphere.00235-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fairman-Williams M. E., Guenther U.-P., Jankowsky E. SF1 and SF2 helicases: family matters. Current Opinion in Structural Biology. 2010;20(3):313–324. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanner J. A., Watt R. M., Chai Y.-B., et al. The severe acute respiratory syndrome (SARS) coronavirus NTPase/helicase belongs to a distinct class of 5′ to 3′ viral helicases. Journal of Biological Chemistry. 2003;278(41):39578–39582. doi: 10.1074/jbc.C300328200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adedeji A. O., Marchand B., te Velthuis A. J., et al. Mechanism of nucleic acid unwinding by SARS-CoV helicase. PLoS One. 2012;7(5) doi: 10.1371/journal.pone.0036521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seybert A., Hegyi A., Siddell S. G., Ziebuhr J. The human coronavirus 229E superfamily 1 helicase has RNA and DNA duplex-unwinding activities with 5′-to-3′ polarity. RNA. 2000;6(7):1056–1068. doi: 10.1017/S1355838200000728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seybert A., Van Dinten L. C., Snijder E. J., Ziebuhr J. Biochemical characterization of the equine arteritis virus helicase suggests a close functional relationship between arterivirus and coronavirus helicases. Journal of Virology. 2000;74(20):9586–9593. doi: 10.1128/JVI.74.20.9586-9593.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y., Sun Y., Wu A., et al. Coronavirus nsp10/nsp16 methyltransferase can be targeted by nsp10-derived peptide in vitro and in vivo to reduce replication and pathogenesis. Journal of Virology. 2015;89(16):8416–8427. doi: 10.1128/JVI.00948-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gorbalenya A., Baker S., Baric R., et al. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nature Microbiology. 2020;2020:03–04. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu A., Peng Y., Huang B., et al. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host & Microbe. 2020;27(3):325–328. doi: 10.1016/j.chom.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krafcikova P., Silhan J., Nencka R., Boura E. Structural analysis of the SARS-CoV-2 methyltransferase complex involved in RNA cap creation bound to sinefungin. Nature Communications. 2020;11(1):1–7. doi: 10.1038/s41467-020-17495-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dillard C. J., German J. B. Phytochemicals: nutraceuticals and human health. Journal of the Science of Food and Agriculture. 2000;80(12):1744–1756. doi: 10.1002/1097-0010(20000915)80:12<1744::AID-JSFA725>3.0.CO;2-W. [DOI] [Google Scholar]
- 21.Ma X. H., Shi Z., Tan C., et al. In-silico approaches to multi-target drug discovery. Pharmaceutical Research. 2010;27(5):739–749. doi: 10.1007/s11095-010-0065-2. [DOI] [PubMed] [Google Scholar]
- 22.Mukhtar M., Arshad M., Ahmad M., Pomerantz R. J., Wigdahl B., Parveen Z. Antiviral potentials of medicinal plants. Virus Research. 2008;131(2):111–120. doi: 10.1016/j.virusres.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akhtar A., Amir A., Hussain W., Ghaffar A., Rasool N. In silico computations of selective phytochemicals as potential inhibitors against major biological targets of diabetes mellitus. Current Computer-Aided Drug Design. 2019;15(5):401–408. doi: 10.2174/1573409915666190130164923. [DOI] [PubMed] [Google Scholar]
- 24.Akhtar A., Hussain W., Rasool N. Probing the pharmacological binding properties, and reactivity of selective phytochemicals as potential HIV-1 protease inhibitors. Universitas Scientiarum. 2019;24(3):441–464. doi: 10.11144/Javeriana.SC24-3.artf. [DOI] [Google Scholar]
- 25.Arif N., Subhani A., Hussain W., Rasool N. In silico inhibition of BACE-1 by selective phytochemicals as novel potential inhibitors: molecular docking and DFT studies. Current Drug Discovery Technologies. 2019;17 doi: 10.2174/1570163816666190214161825. [DOI] [PubMed] [Google Scholar]
- 26.Hussain W., Ali M., Sohail Afzal M., Rasool N. Penta-1,4-diene-3-one oxime derivatives strongly inhibit the replicase domain of tobacco mosaic virus: elucidation through molecular docking and density functional theory mechanistic computations. Journal of Antivirals & Antiretrovirals. 2018;10(3) [Google Scholar]
- 27.Hussain W., Amir A., Rasool N. Computer-aided study of selective flavonoids against chikungunya virus replication using molecular docking and DFT-based approach. Structural Chemistry. 2020;1:1–12. [Google Scholar]
- 28.Hussain W., Qaddir I., Mahmood S., Rasool N. In silico targeting of non-structural 4B protein from dengue virus 4 with spiropyrazolopyridone: study of molecular dynamics simulation, ADMET and virtual screening. Virus Disease. 2018;29:1–10. doi: 10.1007/s13337-018-0446-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qaddir I., Rasool N., Hussain W., Mahmood S. Computer-aided analysis of phytochemicals as potential dengue virus inhibitors based on molecular docking, ADMET and DFT studies. Journal of Vector Borne Diseases. 2017;54(3):p. 255. doi: 10.4103/0972-9062.217617. [DOI] [PubMed] [Google Scholar]
- 30.Rasool N., Akhtar A., Hussain W. Insights into the inhibitory potential of selective phytochemicals against Mpro of 2019-nCoV: a computer-aided study. Structural Chemistry. 2020;31(5):1777–1783. doi: 10.1007/s11224-020-01536-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rasool N., Ashraf A., Waseem M., Hussain W., Mahmood S. Computational exploration of antiviral activity of phytochemicals against NS2B/NS3 proteases from dengue virus. Turkish Journal of Biochemistry. 2019;44(3):261–277. doi: 10.1515/tjb-2018-0002. [DOI] [Google Scholar]
- 32.Rasool N., Bakht A., Hussain W. Analysis of inhibitor binding combined with reactivity studies to discover the potentially inhibiting phytochemicals targeting Chikungunya viral replication. Current Drug Discovery Technologies. 2020;17 doi: 10.2174/1570163817666200312102659. [DOI] [PubMed] [Google Scholar]
- 33.Rasool N., Hussain W. Three major phosphoacceptor sites in HIV-1 capsid protein enhances its structural stability and resistance against inhibitor: explication through molecular dynamics simulation, molecular docking and DFT analysis. Combinatorial Chemistry & High Throughput Screening. 2019;23 doi: 10.2174/1386207323666191213142223. [DOI] [PubMed] [Google Scholar]
- 34.Rasool N., Hussain W., Mahmood S. Prediction of protein solubility using primary structure compositional features: a machine learning perspective. Journal of Proteomics & Bioinformatics. 2017;10(12):324–328. [Google Scholar]
- 35.Rasool N., Jalal A., Amjad A., Hussain W. Probing the pharmacological parameters, molecular docking and quantum computations of plant derived compounds exhibiting strong inhibitory potential against NS5 from Zika virus. Brazilian Archives of Biology and Technology. 2018;61 [Google Scholar]
- 36.Ashfaq U. A., Mumtaz A., ul Qamar T., Fatima T. J. B. MAPS Database: medicinal plant activities, phytochemical and structural database. Bioinformation. 2013;9(19):p. 993. doi: 10.6026/97320630009993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mumtaz A., Ashfaq U. A., ul Qamar M. T., et al. MPD3: a useful medicinal plants database for drug designing. 2017;31(11):1228–1236. doi: 10.1080/14786419.2016.1233409. [DOI] [PubMed] [Google Scholar]
- 38.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports. 2017;7(1, article 42717) doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cruz J. V., Serafim R. B., da Silva G. M., et al. Computational design of new protein kinase 2 inhibitors for the treatment of inflammatory diseases using QSAR, pharmacophore-structure-based virtual screening, and molecular dynamics. Journal of Molecular Modeling. 2018;24(9):p. 225. doi: 10.1007/s00894-018-3756-y. [DOI] [PubMed] [Google Scholar]
- 40.Webb B., Sali A. Protein Structure Prediction. Springer; 2014. Protein structure modeling with MODELLER; pp. 1–15. [Google Scholar]
- 41.Prisant M., Richardson J., Richardson D. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50, article 437450 doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 42.Durrant J. D., Votapka L., Sørensen J., Amaro R. E. POVME 2.0: an enhanced tool for determining pocket shape and volume characteristics. Journal of Chemical Theory and Computation. 2014;10(11):5047–5056. doi: 10.1021/ct500381c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koebel M. R., Schmadeke G., Posner R. G., Sirimulla S. AutoDock VinaXB: implementation of XBSF, new empirical halogen bond scoring function, into AutoDock Vina. Journal of Cheminformatics. 2016;8(1):p. 27. doi: 10.1186/s13321-016-0139-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morris G. M., Huey R., Lindstrom W., et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. Journal of Computational Chemistry. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trott O., Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Studio D. Version 2.5. San Diego, CA, USA: Accelrys Inc.; 2009. [Google Scholar]
- 47.Abraham M. J., Murtola T., Schulz R., et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. [Google Scholar]
- 48.Cheatham T. I., Miller J., Fox T., Darden T., Kollman P. Molecular dynamics simulations on solvated biomolecular systems: the particle mesh Ewald method leads to stable trajectories of DNA, RNA, and proteins. Journal of the American Chemical Society. 1995;117(14):4193–4194. doi: 10.1021/ja00119a045. [DOI] [Google Scholar]
- 49.Hess B., Bekker H., Berendsen H. J. C., Fraaije J. G. E. M. LINCS: a linear constraint solver for molecular simulations. Journal of Computational Chemistry. 1997;18(12):1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- 50.Turner P. XMGRACE, version 5.1. 19. Beaverton, OR: Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology; 2005. [Google Scholar]
- 51.Neese F. The ORCA program system. Wiley Interdisciplinary Reviews: Computational Molecular Science. 2012;2(1):73–78. [Google Scholar]
- 52.Rasool N., Iftikhar S., Amir A., Hussain W. Structural and quantum mechanical computations to elucidate the altered binding mechanism of metal and drug with pyrazinamidase from Mycobacterium tuberculosis due to mutagenicity. Journal of Molecular Graphics & Modelling. 2017;80:126–131. doi: 10.1016/j.jmgm.2017.12.011. [DOI] [PubMed] [Google Scholar]
- 53.Drexler J. F., Corman V. M., Drosten C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Research. 2014;101:45–56. doi: 10.1016/j.antiviral.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Balboni A., Palladini A., Bogliani G., Battilani M. Detection of a virus related to betacoronaviruses in Italian greater horseshoe bats. Epidemiology and Infection. 2011;139(2):216–219. doi: 10.1017/S0950268810001147. [DOI] [PubMed] [Google Scholar]
- 55.Reusken C. B., Haagmans B. L., Müller M. A., et al. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. The Lancet Infectious Diseases. 2013;13(10):859–866. doi: 10.1016/S1473-3099(13)70164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Snijder E. J., Decroly E., Ziebuhr J. The nonstructural proteins directing coronavirus RNA synthesis and processing. Advances in Virus Research. 2016;96:59–126. doi: 10.1016/bs.aivir.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Colson P., Rolain J.-M., Raoult D. Chloroquine for the 2019 novel coronavirus SARS-CoV-2. International Journal of Antimicrobial Agents. 2020;55(3):p. 105923. doi: 10.1016/j.ijantimicag.2020.105923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Singh A. N., Baruah M. M., Sharma N. Structure based docking studies towards exploring potential anti-androgen activity of selected phytochemicals against prostate cancer. Scientific Reports. 2017;7(1, article 1955) doi: 10.1038/s41598-017-02023-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li Y., Zhang J., Wang N., et al. Therapeutic drugs targeting 2019-nCoV main protease by high-throughput screening. bioRxiv. 2020 [Google Scholar]
- 60.ul Qamar M. T., Alqahtani S. M., Alamri M. A., Chen L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. Journal of Pharmaceutical Analysis. 2020;10(4):313–319. doi: 10.1016/j.jpha.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang M., Cao R., Zhang L., et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Research. 2020;30(3):269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu Z., Peng C., Shi Y., et al. Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, Mmolecular docking and binding free energy alculation. bioRxiv. 2020 [Google Scholar]
- 63.Jarvis B., Faulds D. Nelfinavir. Drugs. 1998;56(1):147–167. doi: 10.2165/00003495-199856010-00013. [DOI] [PubMed] [Google Scholar]
- 64.Eroglu E., Türkmen H. A DFT-based quantum theoretic QSAR study of aromatic and heterocyclic sulfonamides as carbonic anhydrase inhibitors against isozyme, CA-II. Journal of Molecular Graphics and Modelling. 2007;26(4):701–708. doi: 10.1016/j.jmgm.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 65.Rasool N., Husssain W., Khan Y. D. Revelation of enzyme activity of mutant pyrazinamidases from Mycobacterium tuberculosis upon binding with various metals using quantum mechanical approach. Computational Biology and Chemistry. 2019;83, article 107108 doi: 10.1016/j.compbiolchem.2019.107108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: list of 2035 phytochemicals and their plants. Table S2: ADMET of Screened 108 Phytochemicals.
Data Availability Statement
The data used to support the findings of this study are included within the supplementary information files.