

Abstract
With the global epidemic of the COVID-19 virus, extensive and rapid research on drug therapy is underway around the world. In this regard, one of the most widely studied drugs is Favipiravir. Our aim in this paper is to conduct comprehensive research based on the Density Functional Theory (DFT) on the potential of metallofullerenes as suitable drug carriers. The surface interaction of Favipiravir with organometallic compound resulted by doping of the five transition metals of the first row of the periodic table (Ti, Cr, Cr, Fe, Ni, and Zn) was examined in depth to select the most suitable metallofullerenes. First, the adsorption geometries of Favipiravir drug onto each metallofullerene were deeply investigated. It was found that Cr-, Fe-, and Ni-doped fullerenes provide the excellent adsorbent property with adsorption energies of −148.2, −149.6, and −146.6 kJ/mol, respectively. The Infrared spectroscopy (IR) study was conducted to survey the stretching vibration of bonds involving in the systems, specialty the C O in the drug, C—M in the metallofullerene, and M—O in the metallofullerene-drug complex. Finally, the UV–vis analysis showed that the absorption spectra for the studied systems may be attributed to the transition from π-π* and/or n-π*.
Keywords: COVID-19, Favipiravir, Metallofullerene, Adsorption, Water solvent, Density Functional Theory
Graphical abstract

Highlights
-
•
The surface interaction of Favipiravir with different metallofullerenes was deeply investigated.
-
•
The formation of transition metal‑oxygen bonding vibrations was found.
-
•
The transfer of charge from Favipiravir molecule to the metallofullerenes refers to the p-type semiconductivity of metallofullerenes.
-
•
UV–vis analysis showed that the absorption spectra may attribute to the transition from π-π* and/or n-π*.
-
•
The doping of fullerene with Cr, Fe, and Ni results oustanding adsorbents for Favipiravir, as a potential drug COVID-19 treatment.
1. Introduction
The widespread coronavirus infection 2019 (COVID-19) produced by SARS-CoV-2 that begun in Dec. 2019, is one of those worldwide challenges that transcends territorial, political, ideological, religious, cultural, and definitely academic borders [1]. No established medical effectiveness of antiviral substrate for COVID-19 has been discovered by this time, whereas specific drugs, including Favipiravir, Remdesivir, Chloroquine, and Arbidol are presently under intensive investigation for the treatment of COVID-19 [2,3]. Favipiravir is an approved antiviral medicine in Japan for influenza [4]. Comparing the effectiveness of Favipiravir with Arbidol [2] suggest that Favipiravir may be a prospective nominee to cure COVID-19. According to the results of Chen et al. [2], it can be theorized that Favipiravir could be a suitable medicine for the treatment of COVID-19 based on improving clinical recovery rate on Day 7 and lessening pyrexia, cough, and ARDS. Very recently, it has been found that Favipiravir as a prodrug, excellently prevents the COVID-19 infection in Vero E6 cells (ATCC-1586) [5,6]. Additionally, other investigations display that Favipiravir is an effective drug in keeping mice safe against the Ebola virus trial [7]. So, clinical researches are immediately required to assess the usefulness and security of this antiviral nucleoside against the COVID-19 cure.
Various nanostructures were established extensive applications in drug delivery systems owing to high surface/volume ratio that is noticeably superior compared to that of the conventional microstructures [[8], [9], [10], [11], [12], [13]]. Among all nanostructures, carbon nanotubes, graphene and fullerenes are prevalent because of their straightforward method of functionalizing and surface improvement, so that they have been considered extensively as drug carriers [11,[13], [14], [15], [16], [17], [18], [19], [20]].
Fullerene and its derivatives have been used as potential carriers towards anticancer medicines owing to have outstanding features such as high loading capability [21] and protecting impact on the heart and liver versus chronic poisonousness resulted from chemotherapeutics [22]. In addition, it was found that fullerene can pass the cell membrane to arrive at the tumor cells, concentrating in the nucleus, lysosomes, and cytoplasm [23,24]. Among fullerenes, the C20 is the tiniest structure that has a dodecahedral cage structure. The C20 was synthesized for the first time by Prinzbach et al. [25] via the gas-phase production method. Further, this kind of fullerene was synthesized through ion-beam irradiation [26] and laser-ablation [27] methods.
The main limiting factor of fullerene family for biological application is their intrinsic hydrophobicity. Towards overcoming this problem, different investigations have been carried out, aiming to find suitable approaches for the production of water-soluble fullerene. These techniques comprise, construction of host-guest complexes that are water-soluble [28], addition of surfactant [29], alcoholization [30], and chemical modification [[31], [32], [33]]. Among these procedures, chemical modification by inserting impure atoms to fullerene showed promising results [[34], [35], [36]].
Doping through an exterior atom is one of the current approaches for changing the electronic properties of a nano-system. Exo-hedral, endo-hedral and substitutional doping are three different methods by which an exterior atom is hosted. The substitution doping that encompasses substitution of an atom of fullerene through a dopant (transition metal in this study) is comparatively less investigated than exo-hedral and endo-hedral doping. It has been concluded that substituting a carbon atom of fullerene by a metal atom (resulting in a metallofullerene molecule) is a suitable tactic to advance the drug delivery property by increasing its adsorption potential [37]. For instance, Rad et al. [34,35] investigated the Cr- and Ni-doped fullerenes and found significant adsorption of adenine, thymine, and uracil nucleotides on the surface of metal-doped fullerene. In another work conducted by the same group, they found that the potential of cytosine and guanine nucleobases adsorption onto fullerene substantially increases by metal doping [36].
The ideal carrier provides a moderate and controllable release of Favipiravir drug in order to prolong the spread time of the drug and shielding it from abolition by phagocytic cells or early destruction. The idea of the present research of using metallofullerene carrier for Favipiravir is raised from the successful applications of metallofullerene as a drug carrier [[34], [35], [36], [37]]. In the following research, we aimed to investigate if certain kinds of metallofullerenes are potentially appropriate for the adsorption and keeping of the Favipiravir drug. The experimental investigation on new materials as a drug carrier is essential; however, they are costly, time-consuming and also they cannot provide enough fundamental information in the atom-scale environment. Therefore, computational approaches have been progressively employed to help to understand the mechanism and the nature of the molecule interactions [38].
In the following research, for the first time we investigated the adsorption property of Favipiravir drug onto different metallofullerenes resulted by substitution doping of fullerene C20 by one of the five selected elements in the first row of the transition metals in the periodic table; titanium (Ti), chromium (Cr), iron (Fe), nickel (Ni), and zinc (Zn). The reason for choosing the stated transition metals for doping on C20 is based on the intrinsic tendency of these atoms to form complexes with organic compounds [39]. It has been revealed that density functional theory (DFT) provides great information on the molecular structures and their interactions [40]. So, it was employed in this study to survey the potential of the selected metallofullerenes for carrying Favipiravir drug by focusing on the molecular interactions. Different geometries were investigated and the most energetically favorable ones were selected for further studies. The FMO analysis was conducted to see any possible hybridizing of the drug molecule and each metallofullerene. The Natural bond orbital (NBO) analysis was carried out to see any changes in the electronic structure of metallofullerenes upon drug adsorption. The vibrational spectra corresponding to the adsorption of Favipiravir molecule on metallofullerene is computed to study the change in the IR intensity and vibrational frequencies. Time-dependent DFT (TD-DFT) method was used for the study of change in the UV–vis spectra and properties of excited states of each system upon adsorption of Favipiravir molecule.
2. Simulation method
The initial geometry optimization of all structures in the ground state was carried out in the water solvent by B3LYP/6-31G level of theory, but to find more accuracy geometry, the output structures were used for next optimization at ωb97xd/6-31 + G(d,p) level of theory. Both levels of theory were implemented in the Gaussian 09 suite of the program [41]. The DFT technique was used owing to the accuracy associated. Vibration frequency calculation was performed for each resulted structure to find out thermodynamical parameters, including enthalpy change (ΔH) and Gibbs free energy change (ΔG) of Favipiravir molecule adsorption process. The frequency calculation revealed that there was no imaginary value in the spectrum, implying the structures were really minima.
The ωb97xd/6-31 + G(d,p) has been found as one of the most favorable levels of theory for responsible and precise calculation at minimum cost for fullerene system [42]. The electronic properties, including the density of states (DOSs), FMO, and NBO charge analysis were calculated at the same level of theory. Following the geometry optimization, the TD-DFT formalism from the ground state at the TD-ωb97xd/6-31++G(d,p) level of theory was used to calculate the excited states to achieve theoretical UV–vis spectra.
To investigate the stability of each metallofullerene (C19M, M = Ti, Cr, Fe, Ni, and Zn), the cohesive energies (Ec) was calculated through the following equation [43]:
| (1) |
where EC and EM, and EC19M are the energies of a single C atom, a single M atom, and metallofullerene molecule, correspondingly. N is the total numbers of atoms involved in system (here, N = 20).
The adsorption energy (Ead) of Favipiravir molecule onto each metallofullerene (C19M, M = Ti, Cr, Fe, Ni, and Zn) was calculated by Eq. (2):
| (2) |
where EC19M/F is the total energy of C19M-Favipiravir complex, EC19M is the total energy of metallofullerene, EF is the total energy of the Favipiravir, and EBSSE is the basis set superposition error (BSSE) corrected for all interactions. The enthalpy change (ΔH) and Gibbs free energy change (ΔG) were calculated at T = 298 K and P = 1 atm by the Eqs. (3), (4), respectively [44].
| (3) |
| (4) |
where H(G)C19M/F, H(G)C19M, and H(G)F are the sum of electronic and thermal enthalpies (the sum of electronic and thermal free energies) of C19M-Favipiravir complex, C19M metallofullerene, and Favipiravir molecule, respectively.
The energy gap between HOMO and LUMO (Eg) was well-defined as
| (5) |
whereas ELUMO and EHOMO are energy of HOMO and LUMO.
The energy of Fermi level (EFL) that lies in the middle of the HOMO and LUMO was calculated through the Eq. (6) [45]:
| (6) |
3. Results and discussion
3.1. Geometry properties of pure metallofullerenes
It was known that C20 fullerene has a symmetric structure. It can be anticipated that substitution of a carbon atom of C20 with a transition metal changes symmetric geometry owing to the lengthening of the M—C bond (M = Ti, Cr, Fe, Ni, and Zn). The optimized geometries of metallofullerenes revealed that the stated metal elements protrude out of the fullerene skeleton. The length of the M—C bond (d) strongly depends on the kind of transition metal. Furthermore, the M—C bond lengths in a metallofullerene are not the same. According to Fig. 1 and Table 1 , the average M—C bond length for each system was calculated as 2.05, 1.93, 1.89, 1.88, and 2.05 Å using B3LYP and 2.03, 1.95, 1.89, 1.86, and 2.04 Å using ωB97XD functional for Ti—C, Cr—C, Fe—C, Ni—C, and Zn—C, respectively. Despite the fact that the calculated values by two functionals were slightly different but both calculations showed the same decrease in the M—C bond length from Ti to Ni (lessening while moving from left to right in the same row) but then an increase in Zn. Such a trend in the M—C bond length attributes to the electron configuration and the effect of d-orbital splitting on the radii of the studied elements. The obtained trend is in agreement with the experimental results reported by Jin et al. on the complex formation of the first-row transition metals [46].
Fig. 1.

The overall geometry of metallofullerene (C19X, X = Ti, Cr, Fe, Ni, and Zn).
Table 1.
The Cohesive Energy (Ec, eV) of all metallofullerenes, and the M—C bond distance (d, Å), and the dimension (D, Å) of metallofullerenes before and after adsorption of Favipiravir drug.
| System |
C19Ti |
C19Cr |
C19Fe |
C19Ni |
C19Zn |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Method | B3lyp | ωb97xd | B3lyp | ωb97xd | B3lyp | ωb97xd | B3lyp | ωb97xd | B3lyp | ωb97xd |
| Ec | – | −7.96 | – | −7.95 | – | −7.96 | – | −7.95 | – | −7.74 |
| d1 | 2.05 | 2.03 | 1.93 | 1.95 | 1.89 | 1.89 | 1.88 | 1.86 | 2.05 | 2.04 |
| d1 after adsorption | – | 2.03 | – | 1.99 | – | 1.91 | – | 1.90 | – | 2.05 |
| D | 5.04 | 4.99 | 4.94 | 4.98 | 5.01 | 5.05 | 4.91 | 4.89 | 5.13 | 5.13 |
| D after adsorption | – | 4.95 | – | 5.15 | – | 5.08 | – | 4.97 | – | 5.16 |
An average of three X-C bond lengths has been calculated.
As depicted in Table 1, the dimension (D) of the metallofullerene also did not follow any regular pattern. The b3lyp (ωB97XD) calculated diameter of each metallofullerene were 5.04 (4.99), 4.94 (4.98), 5.01 (5.05), 4.91 (4.89), and 5.13(5.13) Å for C19 —Ti, C19 —Cr, C19 —Fe, C19 —Ni, and C19 —Zn, respectively.
The cohesive energies of all metallofullerenes were calculated using the ωB97XD method to compare the stability of all the studied metallofullerenes. As is presented in Table 1, the Ec values show no regular pattern, but they revealed that all the metallofullerenes have stable structures owing to the negative value of cohesive energies. However, they all were relatively less stable than the pure C20 fullerene (−8.01 eV) as investigated by our group [47]. One can conclude that the decreasing or increasing trend of Ec depends on the electrostatic interaction of the transition metal and carbon atoms of the metallofullerene, as the evidence from the charge analysis (vide infra).
3.2. The structure of Favipiravir molecule
It was found that three possible isomers for a Favipiravir molecule can be expected. Fig. S1 depicted all these possible isomers. The computational approach was used to study at the above-mentioned level of theory for all these isomers (named I, II, and III) and to evaluate the more energetically favorable isomer. The calculation showed that the isomer (II) was the most stable isomer among all three possible isomers and therefore it was further used for adsorption study on the surface of metallofullerenes.
Fig. 2 depicts the relaxed structure of the Favipiravir molecule along with the active positions to interact with the electro-active part of metallofullerenes. The metal part (Ti, Cr, Fe, Ni, and Zn) of each metallofullerene was placed near different possible parts of the Favipiravir molecule to find the most electroactive part of the molecule. The results showed that for each transition metal, there are two active positions on the Favipiravir molecule. The star-shown places in Fig. 2 are the most active areas of the molecule that were considered for geometry optimization of each metallofullerene-Favipiravir complex (vide infra).
Fig. 2.

The relaxed structure of Favipiravir molecule (left) and the NBO charge distribution (right) achieved by 6-31 + G(d,p)/ωb97xd method in the water solvent. The most active interaction areas of the molecule are shown as stars 1 and 2.
3.3. Adsorption geometry of Favipiravir molecule onto metallofullerenes
Given that the metal part of metallofullerene is the most electroactive place of adsorbent towards interaction with molecules [[34], [35], [36]], for each metallofullerene, the Favipiravir molecule was places near to the metal part through the two star-shown positions in Fig. 2. After making the initial geometries of Favipiravir-metallofullerene complex, they were allowed to be fully optimized first through 6-31G/B3LYP and then via 6-31 + G(d,p)/ωb97xd methods, both in the water as the solvent. For each Favipiravir-metallofullerene complex, two relaxed structures were achieved (P1 = position 1, and P2 = position 2) depending on their initial geometries. The results revealed that for Favipiravir adsorption on C19M (M = Ti, Cr, Fe, and Ni), position 1 (P1) was more energetically favorable geometry than P2. The reverse result was achieved for C19Zn in which the P2 was more energetically favorable than P1. The superior geometry of each complex was given in Fig. 3 that were selected for further investigation, while the inferior energetically favorable geometry of each complex were shown in Fig. S2.
Fig. 3.

Tube (right) and ball & bond (left) types presentation of superior optimized geometries of adsorbed Favipiravir molecule on different metallofullerenes (C19X, X = Ti, Cr, Fe, Ni, and Zn) using 6-31 + G(d,p)/ωb97xd in the water solvent.
As shown in Fig. 3, for all C19M systems except C19Zn, the transition metal atom made two new bonds with Favipiravir molecule while the complex formation of C19Zn was a result of one bond formation. The calculated bond lengths were 2.05 and 2.22 Å for C19Ti, 2.00 and 2.10 Å for C19Cr, 2.00 and 2.07 Å for C19Fe, 1.96 and 2.04 Å for C19Ni, and 1.97 Å for C19Zn, respectively. One reason for the superior geometry of P1 (for all metallofullerenes complex except C19Zn) than P2 attributes to the possibility of two bonds formation at P1 while there is only the possibility of one bond formation at P2 (See Fig. 3 and Fig. S2). For the C19Zn complex, both geometries of P1 and P2 provided only one bond formation, and the superior geometry of P2 than P1 may attribute to the stronger orbital hybridizing of P2.
The values of calculated bonds length suggest that the connection of Favipiravir molecule with all the stated metallofullerenes is considerable. It also can be found that for each metallofullerene, the Favipiravir molecule had the unique orientation and configuration that was a result of the different orbital hybridizing between them (vide infra).
The Favipiravir adsorption energy values calculated from different geometries are shown in Fig. 4 to illustrate the differences between each case.
Fig. 4.

The values of adsorption energy calculated for P1 and P2 geometries of all metallofullerene-Favipiravir (C19M-F) complexes through 6-31G/B3LYP and 6-31 + G(d,p)/ωb97xd levels of theory in water as the solvent.
As is shown in Fig. 4, the calculated adsorption energy values of Favipiravir at P1 (P2) were −153.9 (−119.1), −157.7 (−115.9), −144.9 (−87.7), −138.5 (−115.0), and −88.6 (−91.3) kJ/mol for Ti-, Cr-, Fe-, Ni-, and Zn-metallofullerene using 6-31G/B3LYP method. On the other hand, these values were calculated as −134.2 (−116.1), −148.2 (−112.3), −149.6 (−92.8), −146.6 (−122.2), and −94.6 (−97.6) kJ/mol in the same order through 6-31 + G(d,p)/ωb97xd method.
It is not surprising if there was a slight difference between the values calculated through two methods, as the 6-31G/B3LYP was not the prominent method for calculation and it has been used solely to obtain the initial estimation of geometry. As mentioned before, all the structures obtained from 6-31G/B3LYP were optimized again through the more precise method of 6-31G + (d,p)/ωb97xd. However, similar trends were noticed in the calculated values of the two methods. For instance, both methods agreed that P1 for C19M (M = Ti, Cr, Fe, and Ni) and P2 for C19Zn were superior positions owing to the higher values of adsorption energies. Also, both methods confirmed that the adsorption energy value of C19Zn was much lower than the rest of the metallofullerenes.
By considering only the superior positions P1 for C19M (M = Ti, Cr, Fe, and Ni), and P2 for (C19Zn), the results of 6-31 + G(d,p)/ωb97xd revealed that moving around from left to right in the first row of transition metals in the periodic table, the value of adsorption energy raises from Ti to Fe and then gradually decreases from Fe to Zn. Therefore, based on the calculation the C19Fe was the best metallofullerene to use for adsorption of Favipiravir. But it should be noted that the difference of energy values between the three metals situated at the middle part of first-row of the periodic table (i.e., Cr, Fe, and Ni) are insignificant (ΔE ≈ 1.4–3 kJ/mol), which means that depending on which metallofullerene synthesis is more economical, it can be considered for Favipiravir adsorption without substantial reductions in its adsorption energy. One can find that the Favipiravir adsorption on each of the stated metallofullerenes is neither too weak nor too strong. So, it can be expected that the rate of drug release in our system is a time-dependent process and it can be expected that longer times will lead to more drug release in the target site. Table 1 shows that the adsorption of Favipiravir made considerable changes not only on M—C bond length but also on the dimension of metallofullerene. The M—C bond length remained constant upon Favipiravir adsorption on 2.03 Å for C—Ti (2.03 to 2.03 (0% change)), but changed from 1.95 to 1.99 Å (2.05% change) for C—Cr, 1.89 to 1.91 Å (1.06% change) for C—Fe, 1.86 to 1.90 Å (2.15% change) for C—Ni, and 2.04 to 2.05 Å (0.49% change) for C—Zn. In addition, by adsorption of Favipiravir on each metallofullerene, the dimension of each C19M changed from 4.99 to 4.95 Å (−0.80%), 4.98 to 5.15 Å (3.41%), 5.05 to 5.08 Å (0.59%),4.89 to 4.97 Å (1.63%) and 5.13 to 5.16 Å (0.58%) for Ti-, Cr-, Fe-, Ni-, and Zn—C19 metallofullerenes, respectively. So it can be expected that the stretching vibration of each bond in the metallofullerene was subjected to change upon Favipiravir adsorption. These results prompted the need of check IR spectra of each metallofullerene before and after drug uptake (vide infra).
3.4. Infrared vibrational study
Frequency calculations for the pure metallofullerenes and their complexes with Favipiravir drug at the most stable geometries were carried out at the same basis set/functional to investigate the dynamical stability and IR spectra. The obtained IR spectra of the pure C19M and C19M-Favipiravir complexes were depicted in Fig. 5, Fig. 6 , respectively. Dynamical stability of the studied systems showed no imaginary value in the spectra, which confirms all structures were real minima. It should be mentioned that the main goal of this part of the study is to evaluate the changes in the bonds stretching vibration of each metallofullerene relative to the other ones and detailed examination of the vibrational frequency of all the bonds present in each metallofullerene was not targeted. Previous studies have shown that for Fullerenes, including C20, a wide range of C—C vibrational frequencies appeared, which is due to the different kinds of C—C bonds [48]. These vibrations were reported in the range of 500 to 1500 cm−1 [[48], [49], [50]]. Therefore, the vibration peaks in this range (Fig. 5) were all related to different C—C vibrational frequencies. It should be noted that the C20 fullerene has a symmetrical structure and if one of its atoms is replaced by a metal, the symmetry structure will be disturbed. Therefore, it can be expected that the number of vibration peaks for metallofullerene is much higher than those for pure fullerene [50]. As expected, the location of each vibration for a metallofullerene differs compared to the other, because of the presence of different metal atoms (Ti—Zn) in the structure of the C19M had different effects on the C—C vibrational frequencies.
Fig. 5.
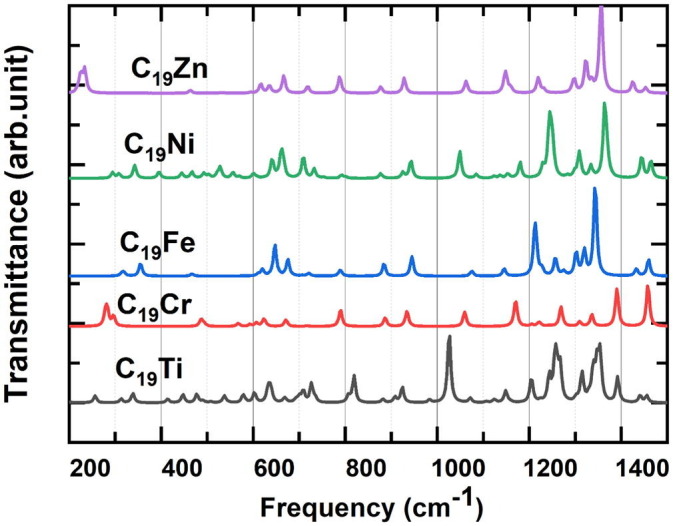
The IR spectra of different metallofullerenes.
Fig. 6.
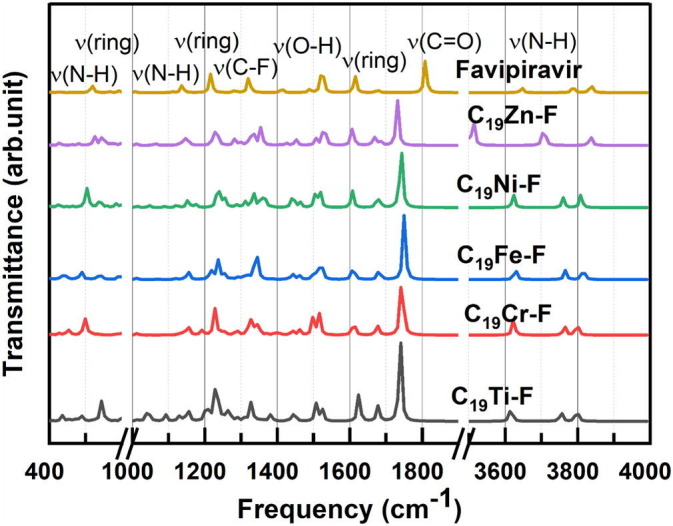
IR spectra of Favipiravir molecule and its complexes with different metallofullerenes.
Literature review revealed that for Ti—C, Cr—C, Fe—C, Ni—C, and Zn—C bonds the vibration frequency are experimentally found around 580 cm−1 [51], 484 cm−1 [52], 496 cm−1 [53], 400 ± 5 cm−1 [54], and 470 cm−1 [55], respectively. According to Fig. 5, the vibration peaks in the above defined range can be related to the metal‑carbon bonding vibrations. It can be concluded that there is a great agreement between the calculation results and the experimental reports. Of course, the difference between the exact location of the peaks obtained by the calculation method and what was experimentally reported can be attributed to the operational conditions and the effect of the type of adjacent atoms and solvent types.
Fig. 6 shows the IR spectra of a Favipiravir as a free molecule a complex form with the stated C19M. Since the vibrational frequencies associated with each of the metallofullerenes were already discussed, then only vibrational frequencies related to the Favipiravir drug molecule upon adsorption was elaborated in more details (Fig. 6).
The first examination of the structure of the Favipiravir molecule indicated that among all possible vibrations, the following vibration frequencies were strongly expected: ν(C O) and ν(N—H) bonds of amid functional group and ν(O—H) bond of the phenolic group. The most important associated vibrations were completely in accordance with what was reported for the Favipiravir molecule in the literature [56]. The computations specified that all the raw values for stretching vibrations were within the estimated range. When Favipiravir get adsorbed on the surface of metallofullerene, the value of IR intensity, as well as vibrational frequencies, were also altered. Fig. 6 shows that not only the new vibrations bond appeared upon adsorption of this molecule on C19M, but also some peaks were shifted. For instance, the ν(C O) of the drug shifted to the low values depending on the kind of metallofullerene. Based on the prior claim on the geometry of the adsorbed drug on any of the metallofullerenes (Fig. 3 ) in which the drug molecule bonds from the oxygen side with the transition metal, the stretching vibration of the metal‑oxygen bond must be observed within the IR spectrum. It is noteworthy to know that a low-intensity vibrational frequency associated with the formation of a new metal‑oxygen bond was observed for each spectrum of C19M-Favipiravir complex. Literature reviews revealed that the stretching vibration of Ti—O, Cr—O, Fe—O, Ni—O, and Zn—O is about 450 cm−1 [57], 500–563 cm−1 [58], 568 cm−1 [59], 422 cm−1 [60], and 433 cm− [61], respectively that are quite in agreement with the calculated values. Comparing the IR spectrums of pure C19M (M = Ti, Cr, Fe, Ni, and Zn) with those of C19M-Favipiravir concludes that the M—O bond was formed, and therefore, the calculated geometries were correct.
3.5. Thermodynamic of drug adsorption
Calculation of vibrational frequency determines if the adsorption of Favipiravir molecule on the stated metallofullerenes is thermodynamically favorable at the ambient temperature and pressure (T = 298.14 K, and P = 1 atm). The change in the enthalpy (ΔH) and the Gibbs free energy (ΔG) for the superior positions were calculated using Eqs. (3), (4) and the results are depicted in Fig. 7 . In addition to the thermodynamic parameters, the corresponding values of adsorption energy are also shown to demonstrate the changes in ΔH and ΔG and their relationship to the absorption value (Fig. 7).
Fig. 7.
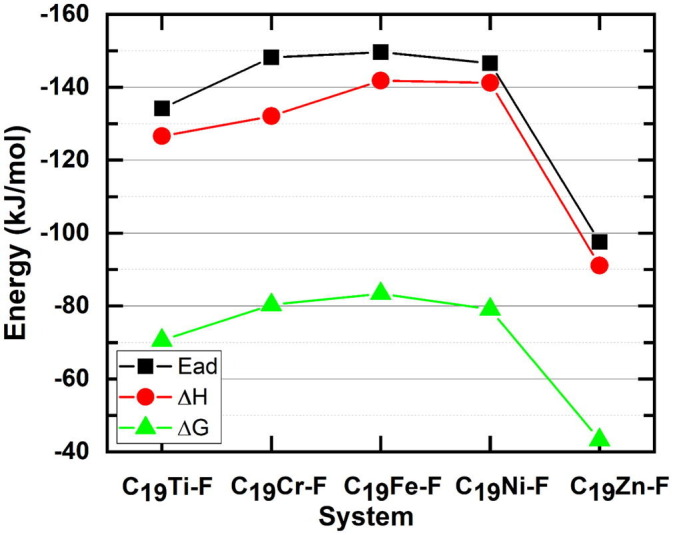
The calculated adsorption parameters for all C19M-F complexes (F represents the Favipiravir molecule).
By moving from left to right of the first-row of the periodic table, the calculated values of ΔH and ΔG for the adsorption of drug molecule on C19M were −126.6 and −70.6 (M = Ti), −132.1 and −80.3 (M = Cr), −141.8 and −83.4 (M = Fe), −141.2 and −79.1 (M = Ni), and −91.1 and −43.3 (M = Zn) kJ/mol, respectively. It should be noted that the negative value of ΔH indicates that the adsorption process is exothermic, and therefore the adsorption of the drug on all the suggested metallofullerenes is associated with the release of heat. On the other hand, the negativity of ΔG specifies that the adsorption process is spontaneous. Therefore, based on the calculated values, it was confirmed that the studied metallofullerenes were completely suitable for Favipiravir drug absorption, but the degree to which the conditions are favorable for different metallofullerenes is different. Fig. 7 clearly shows that there is a perfect agreement between the thermodynamic parameters and the absorption values. Considering the transition metals of metallofullerenes, moving from the left to the right of the periodic table, ΔH and ΔG first become more negative (more desirable) and after passing the maximum value, shifted to less negative values (less desirable). Similar to the results of adsorption energy, the three middle elements of the table, namely chromium, iron, and nickel, provided the most suitable thermodynamic conditions compared to the other elements.
3.6. Frontier molecular orbital analysis
Given that the orbital hybridizing corresponds to the bond formation of metallofullerene and Favipiravir molecule, the frontier molecular orbitals (FMO) were analyzed for pure metallofullerenes and their equivalent complexes at the superior position (vide supra). Fig. 8 represents the HOMO and LUMO of the stated metallofullerenes before and after complexation with the drug molecule.
Fig. 8.
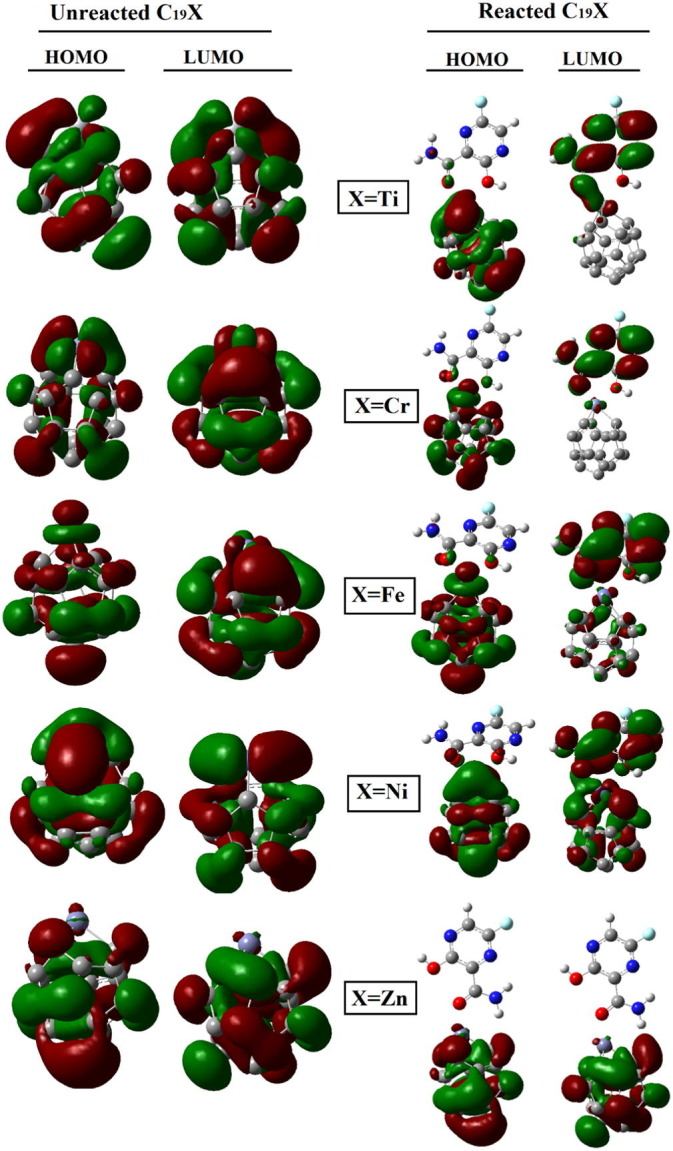
The HOMO and LUMO distributions of each pure (unreacted) metallofullerene along with it reacted form with Favipiravir molecule.
Fig. 8 shows that the orbital distribution of HOMO and LUMO in each metallofullerene was not uniform, which is due to the effect of doped transition metal on its electronic distribution. Because these effects vary from metal to metal, the orbital distribution of each metallofullerene was different. For instance, the density of HOMO orbitals on the metal part of C19M was more pronounced for the middle transition metals of the periodic table, namely chromium, iron, and nickel, but in the case of zinc metal, it was the lowest. Regarding the distribution of LUMO orbitals, it appeared to be less affected by the doped metal atom, but it was quite clear that the LUMO density on the zinc atom was as low as the HOMO density on it, which may explain why C19Zn was less reactive than other metallofullerenes.
The results of the changes in the distribution of HOMO and LOMO on each of the metallofullerenes after the absorption of the Favipiravir drug on them are informative. As can be seen from Fig. 8, after drug adsorption, both HOMO and LUMO distributions on metallofullerenes were affected, but it is obvious that the effect of drug absorption on the LUMO distribution was far greater than the effect on HOMO. For example, in the case of C19Ti and C19Cr, the LUMO orbitals were almost completely transferred to the drug. On the other hand, for C19Fe and C19Ni, the displacement of the LUMO orbitals and the shifting towards the drug molecule was also noticeable, although the two metallofullerenes still retained a small amount of LUMO distribution after drug adsorption. The HOMO or LUMO orbitals of C19Zn, did not show significant changes, which again showed that the low reactivity of the later metallofullerene compared to others may be attributed to the lower orbital hybridization compared to the other studied systems. Given the shift in the distribution of LUMO orbitals to the drug molecule, it can be hypothesized that the transfer of electrical charge from the drug to the metallofullerenes may have occurred during the drug adsorption process. To investigate this hypothesis, the electrical charge was analyzed and the results were disused in section NBO charge analysis (vide infra). The graphical presentation of the calculated energy values of HOMO and LUMO along with the energy of Fermi level (EFL) and the H-L gap of energy (Eg) was provided in Fig. 9 and the numerical values were collected in Table S1.
Fig. 9.

The graphical presentation of calculated values of HOMO, LUMO, Fermi level (Efl), and HOMO-LUMO energy gap (Eg) of all pure metallofullerenes and theirs corresponding complex with Favipiravir drug.
The calculated energy values of HOMO and LUMO, were −6.18 and −0.75 eV for C19Ti, and −6.12 and −1.18 eV for C19Ti-F complex, −6.69 and −0.94 eV for C19Cr, and −6.19 and −0.98 eV for C19Cr-F complex, and −6.74 and −0.86 eV for C19Fe, and −6.55 and −0.87 eV for C19Fe-F complex, and −6.71 and −1.49 eV for C19Ni, and −6.39 and −1.02 eV for C19Ni-F, and finally −7.65 and −1.02 eV for C19Zn and −7.61 and −1.01 for C19Zn-F complex. It can be concluded from Fig. 9 and Table S1 that due to the Favipiravir drug adsorption, the change in the electronic structure of each metallofullerene was different, and as a result, the difference in energy levels of HOMO and LUMO were different for one system than the other. Looking at the energy changes of HOMO and LUMO, it can be seen that in all cases, by Favipiravir drug adsorption, the HOMO level shifted to less negative (or more positive) values, but the LUMO level in some systems such as C19Ti, C19Cr, and C19Fe shifted to the more negative values but for some systems, such as C19Ni and C19Zn, shifted to less negative values. These differences in HOMO and LUMO energy levels caused different changes in Eg and EFL as presented in Fig. 9. Moreover, the density of states (DOS) of each system before and after drug adsorption can be found in Figs. S2, S3, S4, S5, S6, and S7 respectively for metallofullerene achieved by doping of Ti, Cr, Fe, Ni, and Zn. The displacement of the HOMO and LUMO peaks in the DOS pictogram and the change in their height, as well as the changes in the Eg value of each system due to drug adsorption, were practically displayed in each of the mentioned systems. The examination of the extent of the changes in the DOS curves, highlight the sensitivity of each system that can be useful to make the electrochemical sensor for Favipiravir drug [62].
3.7. NBO charge analysis
Natural bond orbital (NBO) analysis which is one of the various existing choices for converting computational solutions of Schrödinger's wave equation into the conversant language of chemical bonding theories [63] was performed to track changes in the electronic structure of each system due to Favipiravir adsorption. It is expected that the electronic structure of both the drug molecule and metallofullerene change upon adsorption. Fig. 2 shows the electronic charge distribution of the unreacted Favipiravir molecule. The electronic charge distribution on each metallofullerene before and after the complex formation with the stated drug is shown in Fig. 10 . By comparing the charge distribution of atoms on the unreacted molecule with those of the adsorbed molecule, the significant changes in the electron charge allocation of each atom were noticeable. These changes are more pronounced in areas of the molecule that were directly involved in adsorption. For instance, the charge on the oxygen of the carbonyl group (C O) and the hydroxyl group (O—H) was −0.662 and −0.684 e, respectively. Due to the adsorption of the drug molecule, these charge allocations change to −0.674 and −0.700 e for C19Ti, −0.652 and −0.691 e for C19Cr, −0.649 and −0.692 e for C19Fe, −0.646 and −0.685 e for C19Ni, and −0.687 and −0.687 e for C19Zn, respectively.
Fig. 10.


The NBO charge distribution of each metallofullerene before and after Favipiravir drug adsorption: a, b, c, d, e, f, g, h, i, and j represents C19Ti, C19Ti-F, C19Cr, C19Cr-F, C19Fe, C19Fe-F, C19Ni, C19Ni-F, C19Zn, and C19Zn-F.
The algebraic sum of the electric charges of the atoms for an unreacted drug molecule is zero (Fig. 2). However, the algebraic sum of the electric charge of the atoms of the absorbed drug molecule (Fig. 10), was no longer zero, which indicates the movement of the electric charge between adsorbent and drug molecule. The algebraic sum of the electric charge of the atoms of the adsorbed drug molecule for the studied systems was +0.359, +0.310, +0.266, and +0.329 e, upon adsorption on C19Ti, C19Cr, C19Fe, C19Ni, and C19Zn, respectively (Table 2 ). The positive values indicated that for all the studied systems, the transfer of charge (Qt) was from the drug molecule towards metallofullerene. It means that the drug molecule lost some charges and the metallofullerene molecule gained some charges. This result illustrates the p-type semiconductor property of metallofullerenes. This confirms that one of the reasons for the binding of the Favipiravir drug with each metallofullerene is the electrostatic attraction between them (connection of positive-negative moieties). The results also fully justify the FMO results as to why the LUMO distribution (which actually depicts areas with electron deficiencies) of a system was transferred to the drug molecule after drug molecule adsorption. Similarly, one can track changes in the electron charge of metallofullerene molecule atoms upon drug molecule adsorption. As shown in Fig. 10, the specific electric charge of all atoms of each metallofullerene changed due to adsorption. The major change attributes to the transition metals. Fig. 10 and Table 2 show that the electric charge of transition metal changes from 1.855 to 1.373 e for Ti, 1.325 to 0.993 e for Cr, 1.170 to 0.858 e for Fe, 1.091 to 0.777 e for Ni, and 1.385 to 1.103 e for Zn upon drug molecule adsorption. For all the metal atoms, a decrease in the positive charge upon drug molecule adsorption was observed. This result confirms the accuracy of the transfer of charge from the drug molecule to the metallofullerene molecule. In fact, as a result of this transfer, the metal atom compensates part of its electron deficiency by taking some of this donated electron. The dipole moment (DM) results of the studied systems are shown in Fig. S8. Due to the specific geometry of the metallofullerene molecule, in which the metal atom has a positive charge, the DM direction for each metallofullerene was from the center of the cluster to the metal atom. Table 2 shows the calculated values of DM for the pure C19M are 15.31, 9.61, 10.09, 7.56, and 7.62 Debye, in which M = Ti, Cr, Fe, Ni, and Zn, respectively. The magnitude of each DM is directly related to the positive charge of the metal atom of metallofullerene, so that the more positive the charge of the metal atom, the larger the DM. Fig. S8 and Table 2, show that as the drug molecule was adsorbed on the metallofullerene, the DM values were significantly increased as expected. Part of the reason for this increase is due to the positive charge of the drug molecule (due to the displacement of the electric charge) and the other part was due to the change in the center of gravity of the complex due to the specific geometry of adsorption. The later reason corresponds to a slight shift in the direction of DM from what was before the drug adsorption.
Table 2.
The calculated values of dipole moment (Debye), transition metal charge (Qm, e), and charge transfer (Qt, e) for all the studied systems.
| System | DM | QM | Qt |
|---|---|---|---|
| C19Ti | 15.31 | +1.855 | – |
| C19Ti-F | 24.78 | +1.373 | 0.359 |
| C19Cr | 9.61 | +1.325 | – |
| C19Cr-F | 21.37 | +0.933 | 0.310 |
| C19Fe | 10.09 | +1.170 | – |
| C19Fe-F | 16.70 | +0.858 | 0.266 |
| C19Ni | 7.56 | +1.091 | – |
| C19Ni-F | 16.58 | +0.777 | 0.329 |
| C19Zn | 7.62 | +1.385 | – |
| C19-Zn-F | 13.57 | +1.103 | 0.167 |
3.8. UV–vis study
The time-dependent functional theory (TD-DFT) [64] was used to calculate the UV–vis spectra and features such as the electronic transitions, the excitation energy, the absorbances, and the oscillator strength of water solvent optimized structures through TD-ωb97xd/6-31++G(d,p) method. The absorption spectra for the studied metallofullerenes were observed due to transitions from in the range of 400–600 nm (see Fig. 11 ) in which this area of wavelengths may be attributed to the transition from π-π* and/or n-π* [65]. Table 3 lists the main molecular characteristics of UV–vis parameters including maximum wavelength (λmax), excitation energy (Ee), oscillator strength (f), and major contributions of all studied systems. According to Fig. 11 and Table 3, the calculated λmax is about 542, 456, 530, 481, and 459 nm for Ti-, Cr-, Fe-, Ni-, and Zn-doped fullerenes, respectively. After the adsorption of Favipiravir molecule on a metallofullerene, the λmax shifted to about 448 (Δ = −94 nm), 522 (Δ = +66 nm), 537 (Δ = +7 nm), 501(Δ = +20 nm), 458 (Δ = −1 nm) nm, for the above-mentioned order of metallofullerenes. Therefore, the blueshift for Ti- and Zn-doped fullerenes but redshift for Cr-, Fe-, and Ni-doped fullerenes was noticed. By knowing this fact that the UV extends from 100 to 400 nm and the visible spectrum from 400 to 700 nm [66], it can be concluded that all absorption values were in the area of visible. This behavior proved that the suggested systems could be used for nonlinear optical (NLO) applications [67]. Table 3 also lists the major contribution for each λmax that may be useful to deep study of the spectroscopic properties of the studied systems. For instance, one can find that the minimum effect of Favipiravir adsorption on the UV–vis spectra of metallofullerene was for Zn-doped fullerene in which resulted in lest shift in its λmax (Δ = −1 nm) with those same major contributions of H-1 → L + 1 and H-2 → L. This result confirmed again that Zn is not a suitable choice as a drug carrier given that Zn electronic structure was less affected by drug adsorption.
Fig. 11.

Computed UV–visible spectra of each metallofullerene and its complex with Favipiravir molecule through TD-ωb97xd/6-31++G(d,p) in water solvent.
Table 3.
The calculated UV–vis parameters: maximum wavelength (λmax), excitation energy (Ee), oscillator strength (f), and major contributions of all studied systems.
| System | λmax (nm) | Ee (eV) | f | Major contributions |
|---|---|---|---|---|
| C19Ti | 542.10 | 2.287 | 0.0139 | H-1 → L (88.2%), H-3 → L + 2 (2.6%), H → L + 5 (2.1%) |
| C19Ti-F | 447.93 | 2.768 | 0.0419 | H → L (64.3%), H → L + 7 (14.6%), H → L + 4 (7.0%) |
| C19Cr | 456.30 | 2.717 | 0.0195 | H-3 → L(36.8%), H-2 → L + 1 (13.5%), H-1 → L (13.3%) |
| C19Cr-F | 522.13 | 2.375 | 0.0142 | H-1 → L + 8(12.8%), H-1 → L + 5(11.3%), H-1 → L + 2(10.3%) |
| C19Fe | 530.30 | 2.338 | 0.0021 | H → L + 7(28.6%), H → L (18.4%), H-5 → L (9.7%) |
| C19Fe-F | 537.93 | 2.305 | 0.0113 | H-2 → L + 2(16.4%), H-2 → L (7.0%), H-2 → L + 1 (6.3%) |
| C19Ni1 | 805.15 | 1.540 | 0.0084 | H → L + 1 (82.1%), H-2 → L (7.2%), H-4 → L + 1 (2.2%) |
| C19Ni2 | 480.72 | 2.579 | 0.0051 | H-1 → L + 3 (31.9%), H-1 → L + 2 (20.0%), H-8 → L (11.9%) |
| C19Ni-F1 | 848.59 | 1.461 | 0.0197 | H → L + 2 (66.8%), H → L + 1 (20.7%), H → L (2.4%) |
| C19Ni-F2 | 500.82 | 2.476 | 0.0113 | H → L (37.6%), H → L + 5 (12.5%), H → L + 1 (12.0%) |
| C19Zn | 458.76 | 2.703 | 0.0305 | H-1 → L + 1 (40.0%), H-2 → L (40.0%), H-6 → L (5.3%) |
| C19Zn-F | 458.10 | 2.706 | 0.0428 | H-1 → L + 1 (32.0%), H-2 → L (29.3%), H-1 → L (5.4%) |
Peak no. 1.
Peak no. 2.
4. Conclusion
The superior geometry of different (C19M (M = Ti, Cr, Fe, and Ni) metallofullerene-Favipiravir complex was investigated. Based on the reported results, there are two active positions on the metal part of metallofullerenes for Favipiravir adsorption. For all C19M systems except C19Zn, the transition metal atom made two new bonds with Favipiravir molecule while the complex formation of C19Zn with Favipiravir molecule was a result of one bond formation. The metallofullerene achieved by the middle part of the first row of the periodic table (i.e. Cr, Fe, and Ni) were best candidates for drug carriers based on the higher adsorption energies. Results of thermodynamic parameters confirmed that the three stated metals provide the most suitable thermodynamic conditions for metallofullerene molecules for Favipiravir adsorption. Infrared vibrational calculations were carried to investigate the dynamical stability and IR spectra of the systems. The calculated data were verified by the experimentally reported data in the literature. The comparison of the calculated vibration peaks with those experimentally reported in the literature showed the formation of transition metal‑oxygen bonding vibrations which confirmed the accuracy of calculated adsorption geometries. Results of FMO and NBO confirmed the transfer of charge from Favipiravir molecule to the metallofullerenes that refers to the p-type semiconductivity of metallofullerenes. The results of the UV–vis study showed that the absorption spectra for the studied metallofullerenes were due to transitions from in the range of 400–600 nm that may be attributed to the transition from π-π* and/or n-π*. The results confirmed that metallofullerenes showed promising properties to be used as a carrier of Favipiravir drug. But the type of metal element is the critical point. The doping of fullerene with the middle elements of the first row of the periodic table, namely chromium, iron, and nickel, would be the best structure to adsorb Favipiravir, as a potential drug COVID-19 treatment.
Associated content
The Supporting Information is available free of charge via the Internet at http://pubs.acs.org.
CRediT authorship contribution statement
Ali Shokuhi Rad: Conceptualization, Methodology, Data curation. Mehdi Ardjmand: Investigation, Data curation. Milad Rabbani Esfahani: Writing – original draft, Writing – review & editing. Bahareh Khodashenas: Validation.
Declaration of competing interest
The authors of this manuscript certify that they have NO affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript. The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
The authors gratefully acknowledge use of the resources of the Alabama Water Institute and the Department of Chemical and Biological Engineering at The University of Alabama.
Biographies
Ali Shokuhi Rad is an associate professor of IAU, Qaemshahr branch. His research activities include nanoscience, drug delivery, surface chemistry, Quantum-mechanic calculations, and adsorption.
Mehdi Ardjmand is the associate professor of IAU, South Tehran Branch, where he was the head of the chemical engineering department for more than two decades. His works focus on the bioprocess engineering and controlled release of medicines and the modeling of drug delivery systems.
Milad Rabbani Esfahani is an assistant professor at the University of Alabama, Alabama, USA. His research area is in the field of nanotechnology, polymer science and adsorption.
Bahareh Khodashenas just received a Ph.D. in chemical engineering from IAU, South Tehran Branch. Her works focus on the application of nanomaterials for drug delivery.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.saa.2020.119082.
Appendix A. Supplementary data
Supplementary material
References
- 1.Huang H., Fan C., Li M., Nie H.L., Wang F.B., Wang H., Wang R., Xia J., Zheng X., Zuo X., Huang J. COVID-19: a call for physical scientists and engineers. ACS Nano. 2020;14(4):3747–3754. doi: 10.1021/acsnano.0c02618. [DOI] [PubMed] [Google Scholar]
- 2.Chen C., Huang J., Cheng Z., Wu J., Chen S., Zhang Y., Chen B., Lu M., Luo Y., Zhang J., Yin P. Favipiravir versus arbidol for COVID-19: a randomized clinical trial. MedRxiv. 2020 doi: 10.1101/2020.03.17.20037432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dong, L.; Hu, S.; Gao, J.; Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discov Ther. 2020, 14, 58–60. [DOI] [PubMed]
- 4.Goldhill D.H., Te Velthuis A.-J., Fletcher R.A., Langat P., Zambon M., Lackenby A., Barclay W.-S. The mechanism of resistance to favipiravir in influenza. Proc. Natl. Acad. Sci. U. S. A. 2018;115:11613–11618. doi: 10.1073/pnas.1811345115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai Q., Yang M., Liu D., Chen J., Shu D., Xia J., Liao X., Gu Y., Cai Q., Yang Y., Shen C. Experimental treatment with favipiravir for COVID-19: an open-label control study. Engineering. 2020 doi: 10.1016/j.eng.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30(3):269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oestereich L., Lüdtke A., Wurr S., Rieger T., Muñoz-Fontela C., Günther S. Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model. Antivir. Res. 2014;105:17–21. doi: 10.1016/j.antiviral.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 8.Yadava S.K., Basu S.M., Chauhan M., Sharma K., Pradhan A., Remya V., Giri J. Low temperature, easy scaling upmethod for development of smart nanostructure hybrid lipid capsulesfor drug delivery application. Colloids Surf. B Biointerfaces. 2020;110927 doi: 10.1016/j.colsurfb.2020.110927. [DOI] [PubMed] [Google Scholar]
- 9.Hill R. Enhancing the development of therapeutics against SARS-CoV-2 by exploring the properties of therapeutic nano-structures. Precis. Nanomed. 2020;3(2):525–532. [Google Scholar]
- 10.Khodashenas B., Ardjmand M., Baei M.S., Rad A.S., Akbarzadeh A. Conjugation of pectin biopolymer with Au-nanoparticles as a drug delivery system: experimental and DFT studies. Appl. Organomet. Chem. 2020;34(6) [Google Scholar]
- 11.Jafari Z., Baharfar R., Rad A.S., Asghari S. Potential of graphene oxide as a drug delivery system for Sumatriptan: a detailed density functional theory study. J. Biomol. Struct. Dynam. 2020:1–10. doi: 10.1080/07391102.2020.1736161. [DOI] [PubMed] [Google Scholar]
- 12.Khodashenas B., Ardjmand M., Sharifzadeh Baei M., Shokuhi Rad A., Akbarzadeh Khiyavi A. Bovine serum albumin/gold nanoparticles as a drug delivery system for Curcumin: experimental and computational studies. J. Biomol. Struct. Dyn. 2019:1–11. doi: 10.1080/07391102.2019.1683073. [DOI] [PubMed] [Google Scholar]
- 13.Mirmotahari M., Sani E., Shokuhi Rad A., Khalilzadeh M.-A. Calcium-doped single-wall nanotubes (Ca/SWCNTs) as a superior carrier for atropine drug delivery: a quantum-chemical study in gas and solvent phases. J. Biomol. Struct. Dyn. 2019;37(16):4267–4273. doi: 10.1080/07391102.2018.1546233. [DOI] [PubMed] [Google Scholar]
- 14.Alipour E., Alimohammady F., Yumashev A., Maseleno A. Fullerene C60 containing porphyrin-like metal center as drug delivery system for ibuprofen drug. J. Mol. Model. 2020;26(1):7. doi: 10.1007/s00894-019-4267-1. [DOI] [PubMed] [Google Scholar]
- 15.Zaibaq N.G., Pollard A.C., Collins M.J., Pisaneschi F., Pagel M.D., Wilson L.J. Evaluation of the biodistribution of serinolamide-derivatized C60 fullerene. Nanomaterials. 2020;10(1):143. doi: 10.3390/nano10010143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nalepa P., Gawecki R., Szewczyk G., Balin K., Dulski M., Sajewicz M., Mrozek-Wilczkiewicz A., Musioł R., Polanski J., Serda M. A [60] fullerene nanoconjugate with gemcitabine: synthesis, biophysical properties and biological evaluation for treating pancreatic cancer. Cancer Nanotechnol. 2020;11(1):1–21. [Google Scholar]
- 17.Daniyal M., Liu B., Wang W. Comprehensive review on graphene oxide for use in drug delivery system. Curr. Med. Chem. Number 22, 2020;27:3665–3685. doi: 10.2174/13816128256661902011296290. [DOI] [PubMed] [Google Scholar]
- 18.Moradi M., Nouraliei M., Moradi R. Theoretical study on the phenylpropanolamine drug interaction with the pristine, Si and Al doped [60] fullerenes. Phys. E Low-dimen. Syst. Nanostruct. 2017;87:186–191. [Google Scholar]
- 19.Baei M.-T., Peyghan A.-A., Moghimi M. Theoretical study of cyano radical adsorption on (6, 0) zigzag single-walled carbon nanotube. Monat. Chem.-Chem. Monthly. 2012;143(11):1463–1470. [Google Scholar]
- 20.Mauro N., Scialabba C., Agnello S., Cavallaro G., Giammona G. Folic acid-functionalized graphene oxide nanosheets via plasma etching as a platform to combine NIR anticancer phototherapy and targeted drug delivery. Mater. Sci. Eng. C. 2020;107:110201. doi: 10.1016/j.msec.2019.110201. [DOI] [PubMed] [Google Scholar]
- 21.Chaudhuri P., Paraskar A., Soni S., Mashelkar R.-A., Sengupta S. Fullerenol–cytotoxic conjugates for cancer chemotherapy. ACS Nano. 2009;3(9):2505–2514. doi: 10.1021/nn900318y. [DOI] [PubMed] [Google Scholar]
- 22.Injac R., Perse M., Cerne M., Potocnik N., Radic N., Govedarica B., Djordjevic A., Cerar A., Strukelj B. Protective effects of fullerenol C60 (OH) 24 against doxorubicin-induced cardiotoxicity and hepatotoxicity in rats with colorectal cancer. Biomaterials. 2009;30(6):1184–1196. doi: 10.1016/j.biomaterials.2008.10.060. [DOI] [PubMed] [Google Scholar]
- 23.Raoof M., Mackeyev Y., Cheney M.-A., Wilson L.-J., Curley S.-A. Internalization of C60 fullerenes into cancer cells with accumulation in the nucleus via the nuclear pore complex. Biomaterials. 2012;33(10):2952–2960. doi: 10.1016/j.biomaterials.2011.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tong J., Zimmerman M.-C., Li S., Yi X., Luxenhofer R., Jordan R., Kabanov A.V. Neuronal uptake and intracellular superoxide scavenging of a fullerene (C60)-poly (2-oxazoline) s nanoformulation. Biomaterials. 2011;32(14):3654–3665. doi: 10.1016/j.biomaterials.2011.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prinzbach H., Weiler A., Landenberger P., Wahl F., Wörth J., Scott L.-T., Gelmont M., Olevano D., Issendorff B.V. Gas-phase production and photoelectron spectroscopy of the smallest fullerene, C20. Nature. 2000;407(6800):60–63. doi: 10.1038/35024037. [DOI] [PubMed] [Google Scholar]
- 26.Wang Z., Ke X., Zhu Z., Zhu F., Ruan M., Chen H., Huang R., Zheng L. A new carbon solid made of the world’s smallest caged fullerene C20. Phys. Lett. A. 2001;280(5–6):351–356. [Google Scholar]
- 27.Iqbal Z., Zhang Y., Grebel H., Vijayalakshmi S., Lahamer A., Benedek G., Bernasconi M., Cariboni J., Spagnolatti I., Sharma R., Owens F.J. Evidence for a solid phase of dodecahedral C20. Eur. Phys. J. B-Condens. Matter Complex Syst. 2003;31(4):509–515. [Google Scholar]
- 28.Priyadarsini K.-I., Mohan H., Tyagi A.-K., Mittal J.-P. Inclusion complex of gamma-cyclodextrin-C60: formation, characterization, and photophysical properties in aqueous solutions. J. Phys. Chem. 1994;98(17):4756–4759. [Google Scholar]
- 29.Ungurenasu C., Airinei A. Highly stable C60/poly (vinylpyrrolidone) charge-transfer complexes afford new predictions for biological applications of underivatized fullerenes. J. Med. Chem. 2000;43(16):3186–3188. doi: 10.1021/jm991175q. [DOI] [PubMed] [Google Scholar]
- 30.Dugan L.-L., Gabrielsen J.-K., Shan P.-Y., Lin T.-S., Choi D.-W. Buckminsterfullerenol free radical scavengers reduce excitotoxic and apoptotic death of cultured cortical neurons. Neurobiol. Dis. 1996;3(2):129–135. doi: 10.1006/nbdi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 31.Nierengarten J.-F. Chemical modification of C60 for materials science applications. New J. Chem. 2004;28(10):1177–1191. [Google Scholar]
- 32.Hasegawa S., Clever G.-H. Metallo-supramolecular shell enables regioselective multi-functionalization of fullerenes. Chem. 2020;6(1):5–7. [Google Scholar]
- 33.Liu Y., Pu Y., Sun L., Yao H., Zhao B., Zhang R., Zhang Y. Folic acid functionalized γ-cyclodextrin C60, a novel vehicle for tumor-targeted drug delivery. J. Biomed. Nanotechnol. 2016;12(7):1393–1403. doi: 10.1166/jbn.2016.2275. [DOI] [PubMed] [Google Scholar]
- 34.Rad A.-S., Aghaei S.-M., Aali E., Peyravi M. Study on the electronic structure of Cr-and Ni-doped fullerenes upon adsorption of adenine: a comprehensive DFT calculation. Diam. Relat. Mater. 2017;77:116–121. [Google Scholar]
- 35.Rad A.-S., Aghaei S.-M., Aali E., Peyravi M., Jahanshahi M. Application of chromium-doped fullerene as a carrier for thymine and uracil nucleotides: comprehensive density functional theory calculations. Appl. Organometall. Chem. 2018;32(2):e4070. [Google Scholar]
- 36.Rad A.-S., Aghaei S.-M. Potential of metal–fullerene hybrids as strong nanocarriers for cytosine and guanine nucleobases: a detailed DFT study. Curr. Appl. Phys. 2018;18(2):133–140. [Google Scholar]
- 37.Hazrati M.-K., Hadipour N.-L. Adsorption behavior of 5-fluorouracil on pristine, B-, Si-, and Al-doped C60 fullerenes: a first-principles study. Phys. Lett. A. 2016;380(7–8):937–941. [Google Scholar]
- 38.Gallo M., Favila A., Glossman-Mitnik D. DFT studies of functionalized carbon nanotubes and fullerenes as nanovectors for drug delivery of antitubercular compounds. Chem. Phys. Lett. 2007;447(1–3):105–109. [Google Scholar]
- 39.Zoberbier T., Chamberlain T.-W., Biskupek J., Kuganathan N., Eyhusen S., Bichoutskaia E., Kaiser U., Khlobystov A.-N. Interactions and reactions of transition metal clusters with the interior of single-walled carbon nanotubes imaged at the atomic scale. J. Am. Chem. Soc. 2012;134(6):3073–3079. doi: 10.1021/ja208746z. [DOI] [PubMed] [Google Scholar]
- 40.Khalilzadeh M.-A., Hosseini S., Rad A.-S., Venditti R.-A. Synthesis of grafted nano-fibrillated cellulose-based hydrogel and study of its thermodynamic, kinetic, and electronic properties. J. Agric. Food Chem. 2020 doi: 10.1021/acs.jafc.0c03500. [DOI] [PubMed] [Google Scholar]
- 41.Frisch M.-J., Clemente F.-R. In: Gaussian 09, Revision A. 01. Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., Nakatsuji H., Caricato M., Li X., Hratchian H.P., Izmaylov A.F., Bloino J., Zhe G., editors. 2009. [Google Scholar]
- 42.Pal A., Wen L.-K., Jun C.-Y., Jeon I., Matsuo Y., Manzhos S. Comparative density functional theory–density functional tight binding study of fullerene derivatives: effects due to fullerene size, addends, and crystallinity on band structure, charge transport and optical properties. Phys. Chem. Chem. Phys. 2017;19(41):28330–28343. doi: 10.1039/c7cp05290a. [DOI] [PubMed] [Google Scholar]
- 43.Rad A.-S., Ayub K. Substitutional doping of zirconium-, molybdenum-, ruthenium-, and palladium: an effective method to improve nonlinear optical and electronic property of C20 fullerene. Comput. Theor. Chem. 2017;1121:68–75. [Google Scholar]
- 44.Roy G., Chattopadhyay A.-P. Dissociation of methane on Ni4 cluster—a DFT study. Comput. Theor. Chem. 2017;1106:7–14. [Google Scholar]
- 45.Lee L.H., editor. Fundamentals of Adhesion. Springer Science & Business Media; 2013. p. 354. [Google Scholar]
- 46.Jin R.-F., Yang S.-Y., Li H.-M., Long L.-S., Huang R.-B., Zheng L.-S. Effect of ionic radius on the assemblies of first row transition metal–5-tert-butylisophthalates–(2, 2′-bipyridine or phenanthroline) coordination compounds. CrystEngComm. 2012;14(4):1301–1316. [Google Scholar]
- 47.Rad A.-S., Ayub K. Change in the electronic and nonlinear optical properties of fullerene through its incorporation with Sc-, Fe-, Cu-, and Zn transition metals. Appl. Phys. A. 2019;125(6):430. [Google Scholar]
- 48.Małolepsza E., Witek H.-A., Irle S. Comparison of geometric, electronic, and vibrational properties for isomers of small fullerenes C20−C36. J. Phys. Chem. A. 2007;111(29):6649–6657. doi: 10.1021/jp068529r. [DOI] [PubMed] [Google Scholar]
- 49.Zamani M., Motahari A., Dabbagh H.-A., Farrokhpour H. IR and UV spectroscopic analysis of C20 carbon nanostructures. J. Nano. Anal. 2014;1(1):31–40. [Google Scholar]
- 50.Guha S., Nakamoto K. Electronic structures and spectral properties of endohedral fullerenes. Coord. Chem. Rev. 2005;249(9–10):1111–1132. [Google Scholar]
- 51.Chen X., Sun X., Xu W., Pan G., Zhou D., Zhu J., Wang H., Bai X., Dong B., Song H. Ratiometric photoluminescence sensing based on Ti3C2MXene quantum dots as an intracellular pH sensor. Nanoscale. 2018;10(3):1111–1118. doi: 10.1039/c7nr06958h. [DOI] [PubMed] [Google Scholar]
- 52.Adams D.-M., Christopher R.-E., Stevens D.-C. Vibrational analysis of benzenetricarbonylchromium and its deuterated analog. Inorg. Chem. 1975;14(7):1562–1566. [Google Scholar]
- 53.Carson S.-D., Constantinidis I., Satterlee J.-D., Ondrias M.-R. A resonance Raman study of ligand binding geometry in Glycera dibranchiata carbonmonoxyhemoglobin. J. Biol. Chem. 1985;260(15):8741–8745. [PubMed] [Google Scholar]
- 54.Hansma P.-K., Kaska W.-C., Laine R.-M. Inelastic electron tunneling spectroscopy of carbon monoxide chemisorbed on alumina-supported transition metals. J. Am. Chem. Soc. 1976;98(19):6064–6065. [Google Scholar]
- 55.Chang S.-C., Hauge R.-H., Kafafi Z.-H., Margrave J.-L., Billups W.-E. Isolation and characterization of ZnCH2 by Fourier transform IR matrix isolation spectroscopy and its photolytic rearrangement to HZnCH. J. Chem. Soc. Chem. Commun. 1987;22:1682–1684. [Google Scholar]
- 56.Rhyman L., Tursun M., Abdallah H.-H., Choong Y.-S., Parlak C., Kharkar P., Ramasami P. Theoretical investigation of the derivatives of favipiravir (T-705) as potential drugs for Ebola virus. Phys. Sci. Rev. 2018;3(9) [Google Scholar]
- 57.Peng G.-W., Chen S.-K., Liu H.S. Infrared absorption spectra and their correlation with the Ti-O bond length variations for TiO2 (rutile), Na-titanates, and Na-titanosilicate (natisite, Na2TiOSiO4) Appl. Spectrosc. 1995;49(11):1646–1651. [Google Scholar]
- 58.Barbu M., Stoia M., Stefanescu O., Stefanescu M. Thermal and FT-IR studies on the interaction between Cr(NO3)3·9H2O and some diols. Chem. Bull. 2010;55(69):180–185. [Google Scholar]
- 59.Nakamoto K., Paeng I.-R., Kuroi T., Isobe T., Oshio H. Metal ion effect on vibrational frequencies of bound dioxygen in model compounds of heme proteins: Fe (II) versus Co (II) J. Mol. Struct. 1988;189(3–4):293–300. [Google Scholar]
- 60.Ramos J.-M., Versiane O., Felcman J., Soto C.-A.-T. FT-IR vibrational spectrum and DFT: B3LYP/6-31G structure and vibrational analysis of guanidinoaceticserinenickel (II) complex: [Ni (GAA)(Ser)] Spectrochim. Acta A Mol. Biomol. Spectrosc. 2007;67(3–4):1037–1045. doi: 10.1016/j.saa.2006.07.058. [DOI] [PubMed] [Google Scholar]
- 61.Uysal I., Severcan F., Evis Z. Characterization by Fourier transform infrared spectroscopy of hydroxyapatite co-doped with zinc and fluoride. Ceram. Int. 2013;39(7):7727–7733. [Google Scholar]
- 62.Parlak C., Alver Ö., Şenyel M. Computational study on favipiravir adsorption onto undoped-and silicon-decorated C60 fullerenes. J. Theor. Comput. Chem. 2017;16(02):1750011. [Google Scholar]
- 63.Weinhold F., Landis C.R., Glendening E.D. What is NBO analysis and how is it useful? Int. Rev. Phys. Chem. 2016;35(3):399–440. [Google Scholar]
- 64.Runge E., Gross E.K.U. Density-functional theory for time-dependent systems. Phys. Rev. Lett. 1984;52:997–1000. [Google Scholar]
- 65.Omar S., Shkir M., Khan M.A., Ahmad Z., AlFaify S. A comprehensive study on molecular geometry, optical, HOMO-LUMO, and nonlinear properties of 1,3-diphenyl-2-propen-1-ones chalcone and its derivatives for optoelectronic applications: a computational approach. Optik. 2020;204:164172. [Google Scholar]
- 66.Penner M.H. Food Analysis. Springer; Boston, MA: 2010. Ultraviolet, visible, and fluorescence spectroscopy; pp. 387–405. [Google Scholar]
- 67.Felscia U.R., Rajkumar B.J., Sankar P., Philip R., Mary M.B. Theoretical and experimental investigations of nitropyrene on silver for nonlinear optical and metal ion sensing applications. Mater. Chem. Phys. 2020;243:122466. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material