

Abstract
Glioblastoma stem cells (GSCs) reprogram glucose metabolism by hijacking high-affinity glucose uptake to survive in a nutritionally dynamic microenvironment. Here, we trace metabolic aberrations in GSCs to link core genetic mutations in glioblastoma to dependency on de novo pyrimidine synthesis. Targeting the pyrimidine synthetic rate-limiting step enzyme carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, dihydroorotase (CAD) or the critical downstream enzyme dihydroorotate dehydrogenase (DHODH) inhibited GSC survival, self-renewal, and in vivo tumor initiation through the depletion of the pyrimidine nucleotide supply in rodent models. Mutations in EGFR or PTEN generated distinct CAD phosphorylation patterns to activate carbon influx through pyrimidine synthesis. Simultaneous abrogation of tumor-specific driver mutations and DHODH activity with clinically approved inhibitors demonstrated sustained inhibition of metabolic activity of pyrimidine synthesis and GSC tumorigenic capacity in vitro. Higher expression of pyrimidine synthesis genes portends poor prognosis of patients with glioblastoma. Collectively, our results demonstrate a therapeutic approach of precision medicine through targeting the nexus between driver mutations and metabolic reprogramming in cancer stem cells.
INTRODUCTION
Glioblastoma (World Health Organization grade IV glioma) is the most prevalent and lethal primary intrinsic tumor in the central nervous system (1). Current standard-of-care with maximal surgical resection followed by concurrent chemoradiation and adjuvant chemotherapy offers only palliation (2). Driven by genetic and epigenetic aberrations, intratumoral heterogeneity is an intrinsic and characteristic feature of glioblastoma (3). Glioblastoma stem cells (GSCs) enforce a pyramidal hierarchy of diverse cell populations through self-renewal and differentiation. The clinical significance of GSCs is supported by their relative resistance to conventional chemotherapy and radiation compared to differentiated glioblastoma cells (DGCs) (4, 5). GSCs have been implicated in tumor angiogenesis, invasion, and immune suppression, supporting the role of GSCs in tumor initiation and progression (6, 7). Understanding the regulation of GSCs may inform therapeutic approaches to improved clinical outcome for patients with glioblastoma.
Metabolic reprogramming, a hallmark of cancer, promotes tumor cell proliferation and survival (8). Genetic mutations and metabolic alterations interact bidirectionally (9, 10). Oncogenic signaling pathways, including phosphoinositide 3-kinases (PI3Ks) and epidermal growth factor receptor (EGFR), up-regulate anaerobic glycolysis, known as the Warburg effect, channeling carbon intermediates into anabolic synthesis to enable rapid cancer cell proliferation (10, 11). Metabolic reprogramming modulates the epigenome and global gene expression; for example, isocitrate dehydrogenase (IDH) mutations in gliomas generate the oncometabolite, 2-hydroxyglutarate, causing global histone and DNA hypermethylation to promote oncogenesis (12–14). Therefore, links between cancer genetics and metabolism offer potential synergistic targeting strategies for enhanced anticancer therapeutic efficacy (9, 15).
We previously showed that metabolic stress, such as hypoxia or low glucose, maintains the GSC self-renewal and proliferation (16, 17). GSCs up-regulate the high-affinity glucose transporter GLUT3 to compete for glucose and adapt to nutritional fluctuation in the tumor microenvironment (16, 18). Downstream of GLUT3, the influx of carbon is channeled into de novo purine synthesis to promote GSC self-renewal and tumorigenesis (19). The delineation of metabolic reprogramming uncovers potential GSC dependency as a promising therapeutic target.
Large-scale genomic studies of glioblastoma, such as The Cancer Genome Atlas (TCGA), demonstrate high variation of the genetic mutation spectrum across different patients (20, 21). In the era of precision medicine, the choice of appropriate targeted therapy based on the patient’s mutation profile has emerged as an attractive therapeutic approach (22). Personalized treatments may promote high value care by increasing therapeutic efficacy, avoiding unnecessary side effects, and lowering health care costs (23, 24). Here, we used in vitro analysis in GSCs and in vivo testing in rodent models to define the additional link between metabolic reprogramming and common genetic mutations in glioblastoma to discover therapeutic combinations to inform precision care.
RESULTS
GSCs up-regulate the de novo pyrimidine synthesis pathway
To discover metabolic pathways associated with increased malignancy in gliomas, we interrogated an in silico database with a comparative metabolomic analysis between bulk tumor specimens from grade II gliomas and glioblastoma (25), revealing enrichment of pyrimidine metabolites in glioblastoma (Fig. 1, A and B). Building on the connection between pyrimidine metabolism and increased tumor malignancy, we sought metabolic pathways specifically up-regulated in GSCs in an unbiased manner. Therefore, we profiled the genome-wide pattern of active promoters and enhancers using chromatin immunoprecipitation of histone 3 lysine 27 acetylation followed by deep sequencing (H3K27ac ChIP-seq) on two matched pairs (GSC23 and T3094) of patient-derived GSCs and DGCs. The differential quantities of H3K27ac at specific enhancer regions were compared between GSCs and DGCs (fig. S1). Unbiased enrichment analysis among all metabolic pathways identified specific up-regulation of pyrimidine synthesis pathway genes in both GSC models (Fig. 1C). We then performed focused analysis on the promoter regions of two key metabolic pathways: nucleotide and pyrimidine pathways. Active chromatin at the promoter regions of pyrimidine pathway genes, defined by higher amounts of H3K27ac, was increased in GSCs compared to matched DGCs (Fig. 1D). Collectively, these results demonstrate up-regulation of de novo pyrimidine synthesis in GSCs.
Fig. 1. Profiling reveals specific up-regulation of de novo pyrimidine synthesis pathway in GSCs.
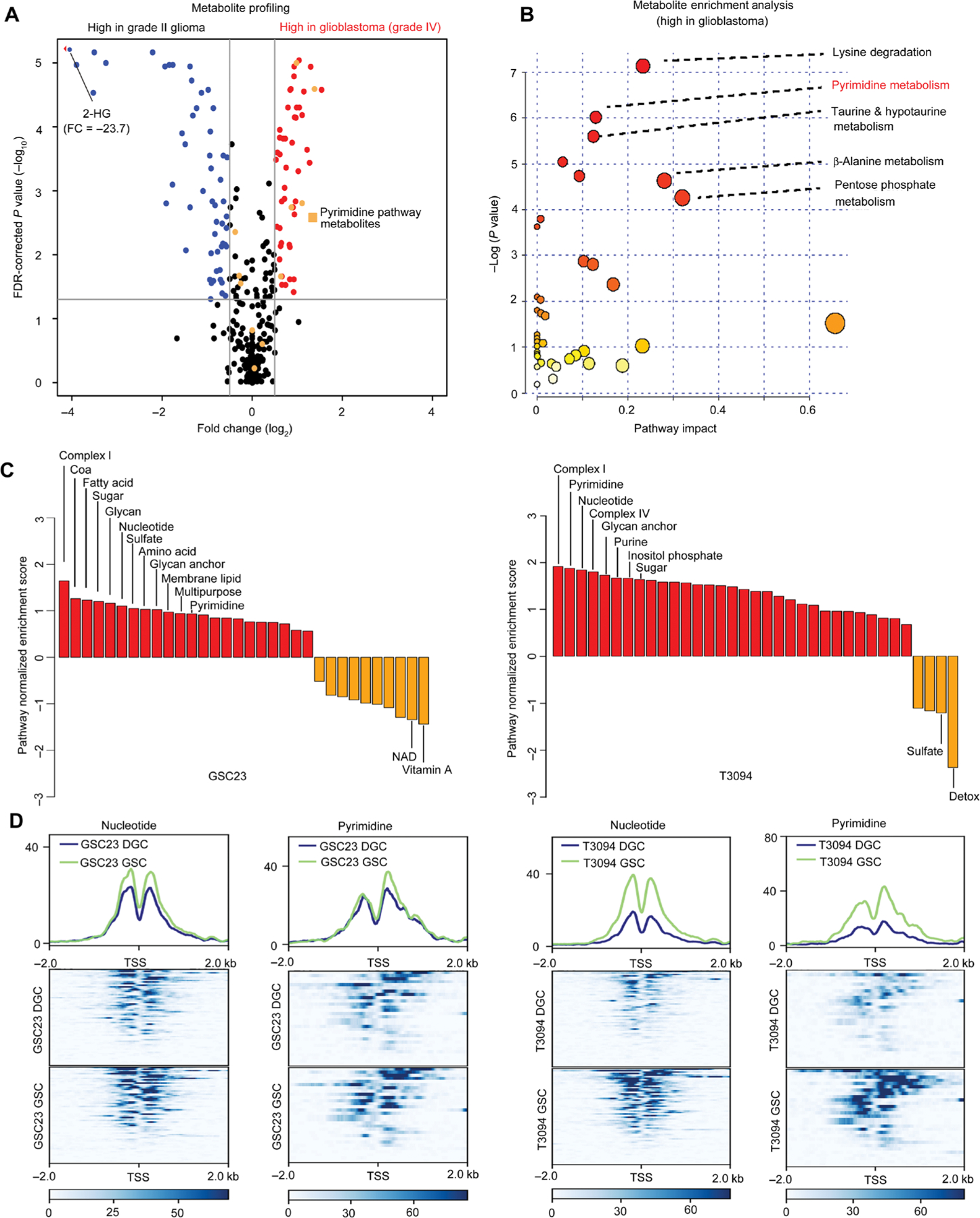
(A) Comparative metabolomic analysis of grade II glioma (n = 18) versus glioblastoma (n = 36). Volcano plot showing differential metabolite abundances from primary tumor samples between grade II glioma and glioblastoma (25). Red dots indicate metabolites that were increased [false discovery rate (FDR) P < 0.05] in glioblastoma compared to grade II glioma, whereas blue dots indicate those that were decreased (FDR P < 0.05). Orange dots indicate metabolites in the pyrimidine synthesis pathway. 2-HG, 2-hydroxyglutarate. (B) Metabolite pathway enrichment analysis of metabolites increased [FDR P < 0.05; fold change (FC) > 1] in glioblastoma compared to grade II glioma (25). Pathway impact refers to the importance of altered metabolites in the respective metabolic pathway, as calculated by Metabo-Analyst. (C) Enrichment analysis of all metabolic pathways up-regulated in glioblastoma stem cells (GSCs; red) versus differentiated glioblastoma cells (DGCs; orange) derived from differential H3K27ac in the GSC23 and T3094 glioblastoma models. NAD, nicotinamide adenine dinucleotide. (D) Patient-derived GSCs (GSC23 and T3094) were cultured under serum-free conditions to maintain their GSC state or induced into DGCs and then subjected to histone 3 lysine 27 acetyl chromatin immunoprecipitation followed by deep sequencing (H3K27ac ChIP-seq). Comparative coverage plots between matched GSCs and DGCs illustrate the specific promoters of matched GSC23 and T3094 GSCs and DGCs for focused metabolic pathways. Heat maps are shown to depict H3K27ac signal, normalized to read depth, for ±5 kb surrounding enhancer peaks. Color scale indicates reads per kilobase per million mapped reads (RPKM). The y axis is also normalized H3K27ac read depth (RPKM). Transcriptional start sites (TSS) for selected metabolic genes were mapped for nucleotide and pyrimidine metabolism. Pathway enrichment was assessed using single-sample gene set enrichment analysis (ssGSEA) comparing pathway enrichment scores between GSCs and DGCs [GSC23: P < 0.0001 (nucleotide) and P = 0.0178 (pyrimidine); T3094: P < 0.0001 (nucleotide) and P < 0.0001 (pyrimidine); sign test was used for statistical analysis].
Rate-limiting pyrimidine synthetic enzymes are essential for GSC maintenance
To determine the functional importance of de novo pyrimidine synthesis in GSCs, we interrogated the role of two rate-limiting steps in pyrimidine synthesis, catalyzed by carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, dihydroorotase (CAD) and dihydroorotate dehydrogenase (DHODH). The expressions of CAD and DHODH were up-regulated in GSCs compared to matched DGCs with statistical significance (Figs. 2, A and B, and 3, A and B). We inhibited the pathway using a lentivirus-based vector to express either a control short hairpin RNA (shRNA) sequence that does not target any known sequence in the mammalian genome (shCONT) or independent, nonoverlapping shRNAs targeting CAD (shCAD-1 and shCAD-2) or DHODH (shDHODH-1 and shDHODH-2). The efficacy of shRNAs on mRNA expression was confirmed using quantitative reverse transcription polymerase chain reaction (RT-PCR) (Figs. 2C and 3C). Knocking down either CAD or DHODH inhibited GSC growth and cell proliferation in two different GSC models (Figs. 2, D and E, and 3, D and E). Conversely, overexpression of wild-type CAD or DHODH enhanced GSC proliferation (figs. S2, A to C, and S3, A to D). Targeting CAD or DHODH also impaired GSC self-renewal, as measured by neurosphere formation using an in vitro limiting dilution assay (Figs. 2, F to H, and 3, F to H). The functional significance of CAD and DHODH was further confirmed in GSCs from two primary glioblastoma tumors (figs. S4 to S6).
Fig. 2. CAD regulates GSC growth and self-renewal.
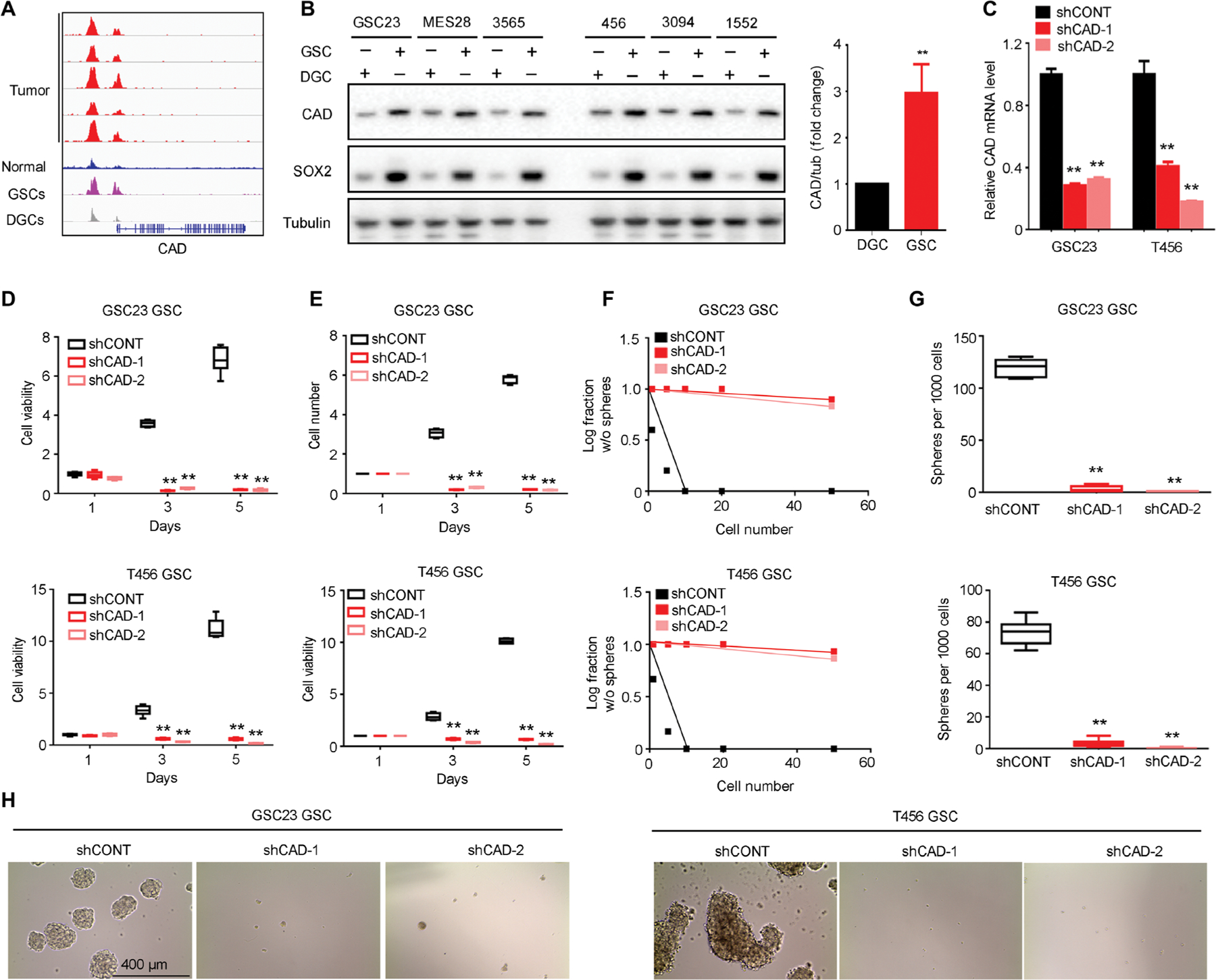
(A) H3K27ac ChIP-seq enrichment plot centered at the gene locus for CAD. Active chromatin was profiled by H3K27ac ChIP-seq for five primary glioblastoma tumors, five normal brain tissues, and three matched pairs of GSCs and DGCs from patient-derived glioblastoma specimens. Raw data from enhancer profiling of primary glioma tissues were downloaded from GSE101148. Matched pairs of GSCs and DGCs and normal tissues H3K27ac ChIP-seq data were downloaded from NCBI Gene Expression Omnibus GSE54047 and GSE17312. (B) Protein concentrations of CAD with normalized quantifications in matched pairs of GSCs and DGCs across human glioblastoma specimens GSC23, MES28, T3565, T456, T3094, and T1552 (n = 6 biological replicates; **P < 0.01, one-way ANOVA). (C) Quantitative RT-PCR assessment of CAD mRNA in GSC23 and T456 GSCs expressing a nontargeting control shRNA (shCONT), shCAD-1, or shCAD-2 (n = 6 independent experiments per group; **P < 0.01, one-way ANOVA). (D) Cell growth of GSC23 and T456 GSCs expressing shCONT, shCAD-1, or shCAD-2 was measured by CellTiter-Glo assay (n = 5 independent experiments per group; **P < 0.01, one-way ANOVA). (E) Growth of GSC23 and T456 GSCs expressing shCONT, shCAD-1, or shCAD-2 was measured by direct cell number count (n = 5 independent experiments per group; **P < 0.01, one-way ANOVA). (F) Sphere formation using an extreme limiting dilution assay (ELDA) was performed with GSC23 and T456 GSCs expressing shCONT, shCAD-1, or shCAD-2 (GSC23, P < 0.01; T456, P < 0.01, ELDA analysis). (G) The number of spheres formed using GSC23 and T456 GSCs expressing shCONT, shCAD-1, or shCAD-2 was determined with ELDA per 1000 cells seeded (n = 6 independent experiments per group; **P < 0.01, one-way ANOVA). (H) Representative images of neurospheres derived from GSC23 and T456 GSCs expressing shCONT, shCAD-1, or shCAD-2. Scale bar, 400 μm. Each image is representative of at least five similar experiments.
Fig. 3. DHODH promotes GSC growth and self-renewal.
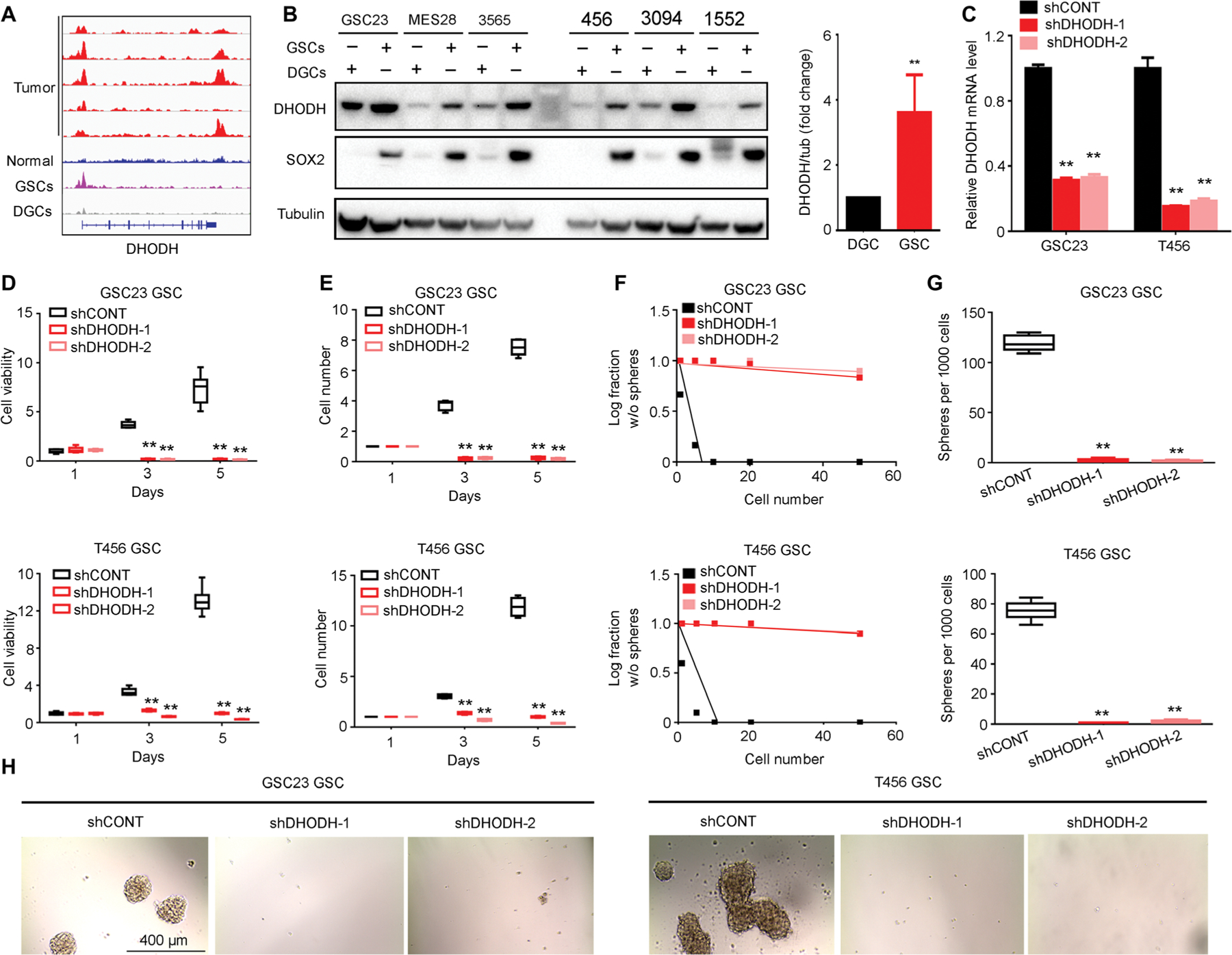
(A) H3K27ac ChIP-seq enrichment plot centered at the gene locus for DHODH. Active chromatin was profiled by H3K27ac ChIP-seq for five primary glioblastoma tumors, five normal brain tissue, and three matched pairs of GSCs and DGCs from patient-derived glioblastoma specimens. Raw data for enhancer profiling of primary glioma tissues were downloaded from GSE101148. Matched pairs of GSCss and DGCs and normal tissues H3K27ac ChIP-seq data were downloaded from NCBI Gene Expression Omnibus GSE54047 and GSE17312. (B) Protein concentrations of DHODH with normalized quantifications in matched pairs of GSCs and DGCs across human glioblastoma specimens GSC23, MES28, T3565, T456, T3094, and T1552 (n = 6 biological replicates; **P < 0.01, one-way ANOVA). (C) Quantitative RT-PCR assessment of DHODH mRNA in GSC23 and T456 GSCs expressing shCONT, shDHODH-1, or shDHODH-2 (n = 3 independent experiments per group; **P < 0.01, one-way ANOVA). (D) Cell growth of GSC23 and T456 GSCs expressing shCONT, shDHODH-1, or shDHODH-2 was measured by CellTiter-Glo assay (n = 6 independent experiments per group; **P < 0.01, one-way ANOVA). (E) Growth of GSC23 and T456 GSCs expressing shCONT, shDHODH-1, or shDHODH-2 was measured by direct cell number count (n = 5 independent experiments per group; **P < 0.01, one-way ANOVA). (F) Sphere formation using an ELDA was performed with GSC23 and T456 GSCs expressing shCONT, shDHODH-1, or shDHODH-2 (GSC23, P < 0.01; T456, P < 0.01, ELDA analysis). (G) The number of spheres formed using GSC23 and T456 GSCs expressing shCONT, shDHODH-1, or shDHODH-2 was determined with ELDA per 1000 cells seeded (n = 6 independent experiments per group; **P < 0.01, one-way ANOVA). (H) Representative images of neurospheres derived from GSC23 and T456 GSCs expressing shCONT, shDHODH-1, or shDHODH-2. Scale bar, 400 μm. Each image is representative of at least five similar experiments.
To interrogate the in vivo functions of CAD and DHODH in tumor propagation of GSCs, we transplanted two patient-derived GSC models transduced with specific shRNAs into the brains of immunocompromised mice. Mice bearing GSCs transduced with shCAD or shDHODH demonstrated increased survival compared to mice with respective GSCs transduced with shCONT [Fig. 4, A to D (for CAD) and E to H (for DHODH)]. The in vivo functional significance of CAD and DHODH was further confirmed in GSCs from two primary glioblastoma tumors (fig. S7). These results support the essential dependency of GSC on enzymes in the de novo pyrimidine synthesis pathway, specifically CAD and DHODH, which are required for GSC proliferation, self-renewal, and tumorigenesis.
Fig. 4. CAD and DHODH are essential for GSC maintenance with glioblastoma driver mutations differentially regulating pyrimidine synthesis.
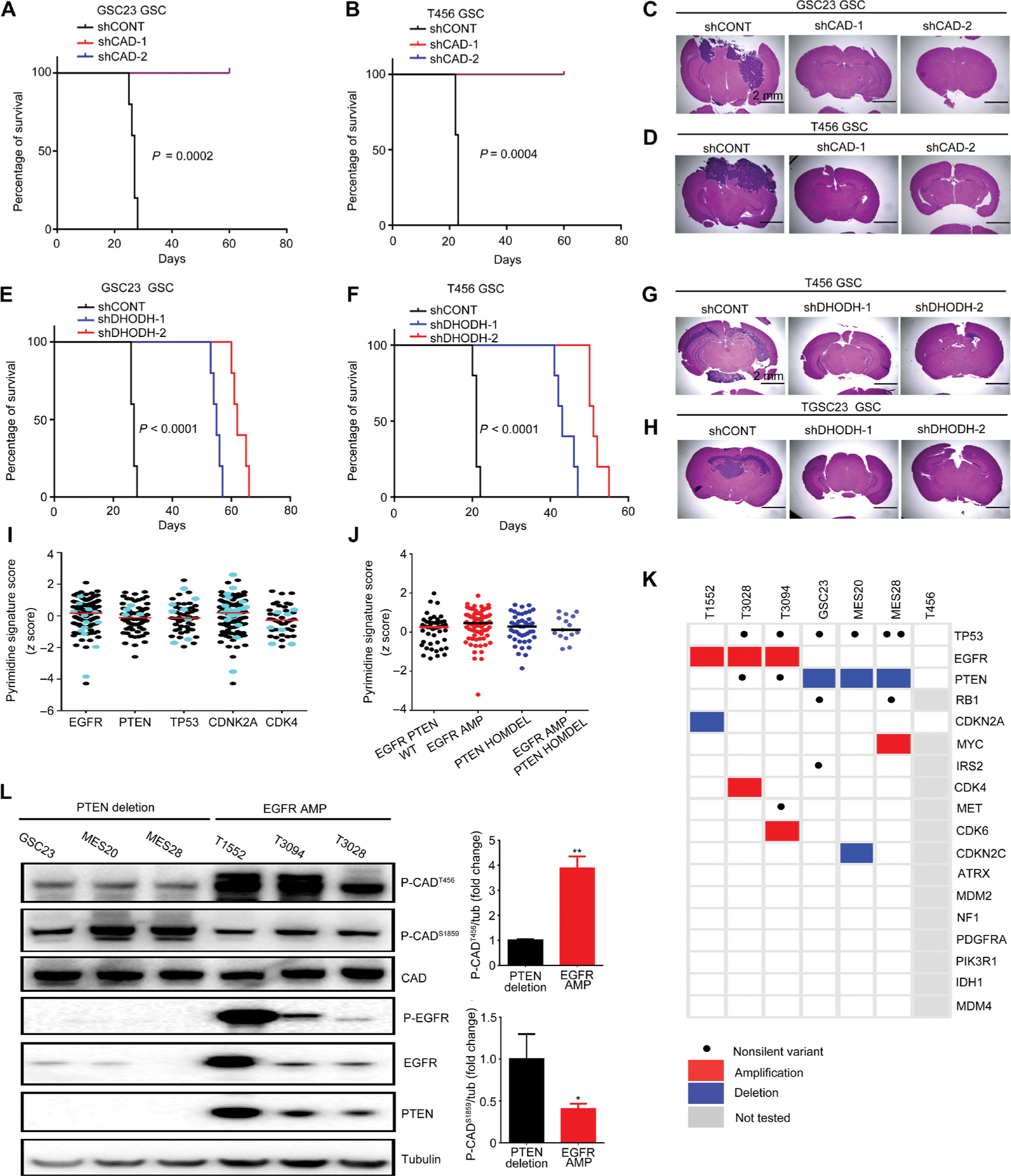
(A and B) Kaplan- Meier survival curves of immunocompromised mice bearing intracranial GSC23 (A) or T456 (B) GSCs transduced with either a control shRNA (shCONT) or one of two shRNAs targeting CAD (shCAD-1 or shCAD-2) [n = 5 for each group; P = 0.0002 (GSC23) and P = 0.0004 (T456) using log-rank test]. (C and D) Representative images of hematoxylin and eosin-stained sections of mouse brains collected on day 20 after transplantation of GSC23 (C) or T456 (D) GSCs expressing shCONT, shCAD-1, or shCAD-2. Scale bar, 2 mm. (E and F) Kaplan-Meier survival curves of immunocompromised mice bearing intracranial GSC23 (E) or T456 (F) GSCs expressing shCONT or one of two shRNAs targeting DHODH (shDHODH-1 or shDHODH-2) (n = 5 for each group; P < 0.0001 for GSC23 and T456, log-rank test). (G and H) Representative images of hematoxylin and eosin-stained sections of mouse brains collected on day 20 after transplantation of GSC23 (G) or T3094 (H) GSCs expressing shCONT, shDHODH-1, or shDHODH-2. Scale bar, 2 mm. (I and J) Overall expressions of pyrimidine metabolism genes across the TCGA glioblastoma dataset with different gene mutations/alterations, including EGFR, PTEN, TP53, CDKN2A, or CDK4. Black: Glioblastoma with mutation in the designated gene (may also have other gene mutations). Cyan: Glioblastoma with mutation only in the designated gene. WT, wild type; AMP, amplification; HOMDEL, homozygous deletion. (K) Genetic alterations in PTEN and EGFR were analyzed using exome sequencing for each GSC model used in this study. Black dot represented nonsilent variants including nonsynonymous mutations and/or splice site variants predicted to have moderate to high impact on protein structure. Red and blue represented focal amplifications (log2 copy number ratio > 2 or > 1, if focal) and deletions (log2 copy number ratio < −2 or < −1, if focal), respectively. (L) Immunoblot assessment and normalized quantifications of phosphorylation at CADT456 and CADS1859 across patient-derived GSC23, MES20, MES28, T1552, T3028, and T3094 GSCs. Tubulin was used as a loading control (n = 3 biological replicates; *P < 0.05 and **P < 0.01, one-way ANOVA).
Driver mutations differentially regulate pyrimidine synthetic enzyme function
Befitting its rate-limiting role in catalyzing the first three steps of de novo pyrimidine synthesis, CAD function is regulated at multiple layers, including allosteric regulation (both negative and positive), covalent regulation by phosphorylation, and metabolite channeling (26). Phosphorylation of CAD on the Ser1859 (CADS1859) site is regulated by mammalian target of rapamycin (mTOR)-S6K and promotes the oligomerization of CAD, leading to enhanced pyrimidine synthesis (27). In contrast, phosphorylation of on Thr456 (CADT456) is regulated by EGFR (28). As alterations in the PI3K-phosphatase and tensin homolog (PTEN)-mTOR and EGFR pathways are extremely prevalent in glioblastoma, we interrogated the relative activation of the pyrimidine metabolic pathway through a gene expression signature in the glioblastoma TCGA dataset. Pyrimidine metabolism, as determined by the pathway signature score, remained unchanged across glioblastoma specimens with different gene mutations/alterations, including EGFR, PTEN, TP53, CDKN2A, or CDK4 (Fig. 4I). Given the roles of EGFR and PI3K in CAD regulation, we parsed the association between pyrimidine metabolism and alterations of EGFR and PTEN. Pyrimidine metabolism varied little among glioblastomas with both wild-type EGFR and PTEN, EGFR mutation/amplification alone, PTEN deletion alone, or combined EGFR mutation/amplification and PTEN deletion (Fig. 4J). Furthermore, the expressions of both CAD and DHODH were largely invariant across tumors, regardless of genetic aberrations (fig. S8).
Given the unchanged expressions of pyrimidine metabolism genes across different genetic backgrounds, we speculated that CAD may be differentially regulated through control of protein phosphorylation by upstream kinases. We leveraged our panel of patient-derived GSC models, which we characterized by exome sequencing (Fig. 4K). To distinguish the roles of EGFR and PI3K-PTEN in pyrimidine synthesis, we selected GSC models with dichotomized genetic backgrounds: PTEN deletion with wild-type EGFR (GSC23, MES20, and MES28) and EGFR amplification with wild-type PTEN (T1552, T3094, and T3028). CAD phosphorylation measured by immunoblot segregated with GSC genetics, with elevated CADS1859 phosphorylation in PTEN-deleted GSCs and elevated CADT456 phosphorylation in GSCs with EGFR amplification (Fig. 4L). Functional significance of these two CAD phosphorylation sites was supported by the abrogated growth of PTEN-deleted GSCs overexpressing CADS1859A mutant and EGFR-amplified GSCs overexpressing CADT456A mutant (fig. S3). Thus, regulatory mechanisms of pyrimidine synthesis reflect tumor genetics.
EGFR regulates CADT456 phosphorylation through the MAPK-ERK pathway
To directly link EGFR activity to CADT456 phosphorylation in GSCs, we treated two EGFR-amplified GSCs (T3094 and T3028) with increasing concentrations of two EGFR inhibitors, lapatinib and gefitinib. Clinically achievable concentrations of lapatinib and gefitinib reduced phosphorylated CADT456, while phosphorylated CADS1859 remained unchanged (Fig. 5A and fig. S9A). Treatment with two specific inhibitors for downstream mediators of EGFR pathway, U0126 and GSK11202 for MAPK kinase (MEK), caused inhibition of CADT456 phosphorylation (fig. S9, B and C). Reciprocal gain-of-function studies were performed with the addition of epidermal growth factor (EGF) ligand, revealing elevated phosphorylated CADT456 (fig. S9D). Together, these results demonstrated EGFR as an upstream regulator of CADT456 phosphorylation.
Fig. 5. EGFR and PI3K/PTEN differentially activate CAD phosphorylation sites.
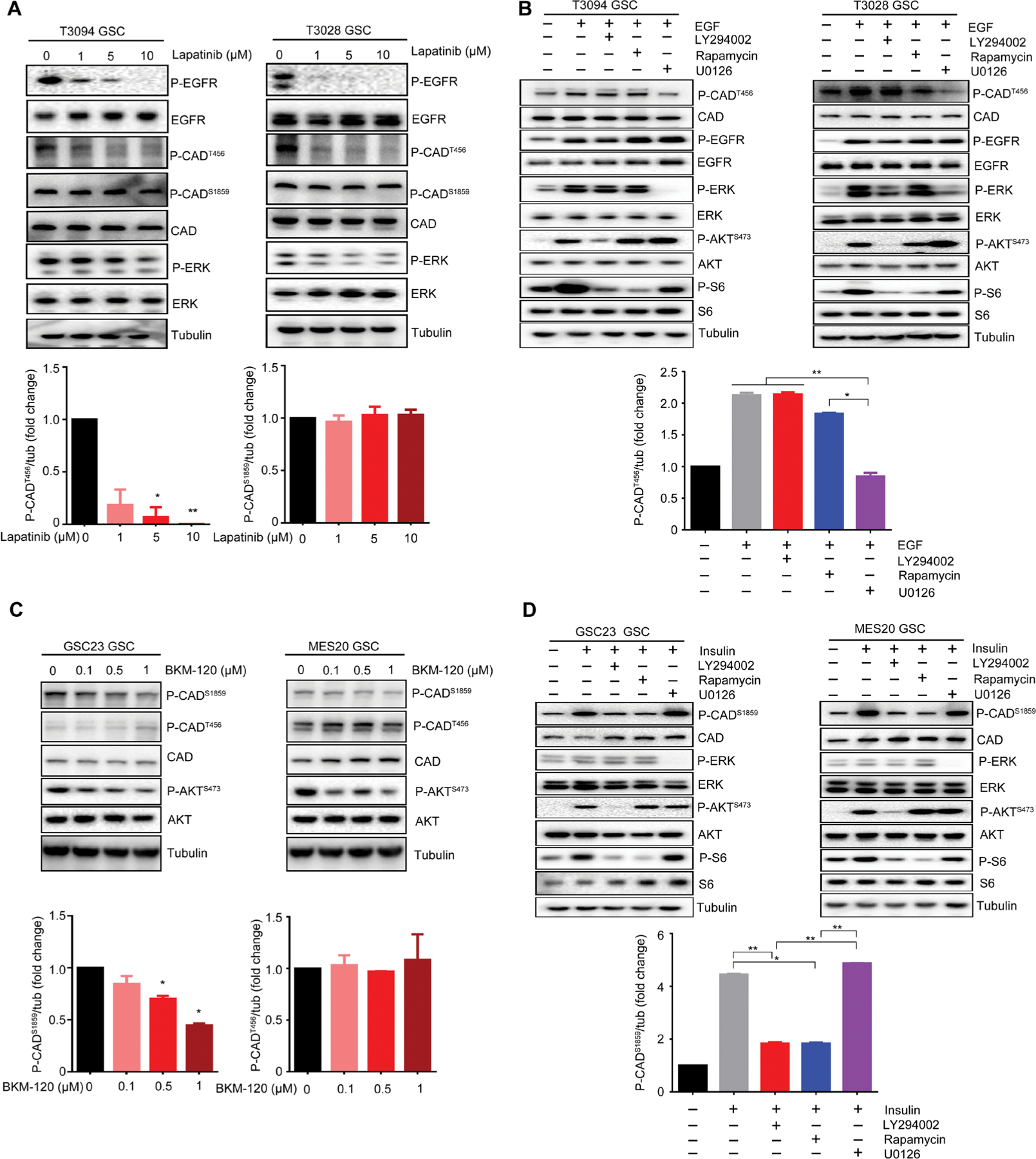
(A) Immunoblot assessment of phosphorylated EGFR, phosphorylated ERK, phosphorylated CADS1859, phosphorylated CADT456, and total CAD protein after treatment with the EGFR inhibitor lapatinib (1, 5, and 10 μM) for 48 hours in T3094 and T3028 GSCs. Normalized quantifications with tubulin as control were performed for phosphorylated CADS1859 and phosphorylated CADT456 (n = 2 biological replicates; *P < 0.05 and **P < 0.01, one-way ANOVA). (B) GSCs (T3094 and T3028) were treated with EGF (100 ng/ml) over a 30-min time course, together with inhibitors of the PI3K (LY294002), mTOR (rapamycin), or MEK (U0126) pathways. Amounts of phosphorylated CADT456, total CAD, phosphorylated EGFR, total EGFR, phosphorylated ERK, total ERK, phosphorylated AKTS473, total AKT, phosphorylated S6, and total S6 were assessed by immunoblot. Normalized quantifications with tubulin as control were performed for phosphorylated CADT456 (n = 2 biological replicates; *P < 0.05 and **P < 0.01, one-way ANOVA). (C) Immunoblot assessment of phosphorylated CADS1859, phosphorylated CADT456, total CAD, phosphorylated AKTS473, and total AKT after treatment with the PI3K inhibitor BKM120 (0.1, 0.5, and 1 μM) for 48 hours in GSC23 and MES20 GSCs. Normalized quantifications with tubulin as control were performed for phosphorylated CADS1859 and phosphorylated CADT456 (n = 2 biological replicates; *P < 0.05, one-way ANOVA). (D) GSCs (GSC23 and MES20) were treated with insulin (1 μM) over a 30-minute time course, together with inhibitors of the PI3K (LY294002), mTOR (rapamycin), or MEK (U0126) pathways. Amounts of phosphorylated CADS1859, total CAD, phosphorylated ERK, total ERK, phosphorylated AKTS473, total AKT, phosphorylated S6, and total S6 were assessed by immunoblot. Normalized quantifications with tubulin as control were performed for phosphorylated CADS1859 (n = 2 biological repli cates; *P < 60.05 and **P < 0.01, one-way ANOVA).
Downstream intracellular mediators of EGFR signaling include the mitogen-activated protein kinase (MAPK)-extracellular signal-regulated kinase (ERK) and AKT pathways, which were both activated immediately upon EGF treatment in our two GSC models (fig. S9D). To determine the relative contribution of the downstream pathways to CADT456 phosphorylation, GSCs were treated with EGF and inhibitors of the PI3K (LY294002), mTOR (rapamycin), and MEK (U0126) pathways. U0126 treatment inhibited the phosphorylation of MAPK-ERK and CADT456 after EGF treatment. In contrast, PI3K and mTOR inhibitors minimally altered CADT456 phosphorylation (Fig. 5B). Collectively, our data support EGFR as an upstream activator of CADT456 phosphorylation through the MAPK-ERK pathway, with minimal effects on CADS1859 phosphorylation.
PTEN deletion promotes CADS1859 phosphorylation through the PI3K-AKT pathway
Given the lack of regulation of CADS1859 by EGFR signaling, we interrogated patient-derived glioblastoma models with PTEN deletion without EGFR amplification, revealing elevated amounts of CADS1859 phosphorylation (Fig. 4L). Prior studies demonstrated that S6K1 directly phosphorylates CADS1859, which may promote the oligomerization and activation of the multienzyme CAD complex (27, 29). We, therefore, interrogated the upstream regulation of CADS1859 in two GSCs with PTEN deletions, GSC23 and MES20. Treatment with the PI3K inhibitor, BKM120 (buparlisib), decreased CADS1859 phosphorylation, without change in CADT456 phosphorylation in both GSC models (Fig. 5C). Another PI3K inhibitor, LY294002, demonstrated the same effect on the two phosphorylation sites on CAD (fig. S10A). Treatment with two specific inhibitors for downstream mediators of PI3K pathway, rapamycin for mTOR and PF-4708671 for S6K1, inhibited CADS1859 phosphorylation (fig. S10, B and C). Furthermore, insulin treatment of two GSC models harboring PTEN deletion without EGFR amplification induced phosphorylation of CADS1859, associated with activation of both AKT and MAPK-ERK (fig. S10D). Targeting PI3K (with LY294002) or mTOR (with rapamycin) reversed insulin-induced CADS1859 phosphorylation, in contrast to the MEK inhibitor, U0126 (Fig. 5D). Torin1, the active site inhibitor of mTOR (both mTORC1 and mTORC2), inhibited CADS1859 phosphorylation, although knocking down RICTOR, a key component of mTORC2, showed no effect (fig. S11, A to C). This is consistent with previous report that mTORC1 coordinates nucleotide synthesis with nucleotide demand in cancer cells (30). Collectively, these results suggest that PTEN deletion stimulates pyrimidine synthesis by activating CAD through the phosphorylation of CADS1859, mediated by the PI3K-AKT-mTOR-S6K pathway.
DHODH inhibitor teriflunomide combines with kinase inhibitors to block de novo pyrimidine synthesis in GSCs in a genetic context-dependent manner
To directly interrogate pyrimidine synthesis, we determined the amounts of metabolites in the de novo pyrimidine synthesis pathway in matched GSCs and DGCs harboring either PTEN mutations (MES20 and GSC23) or EGFR amplification (T3094). Consistently, concentrations of cytidine triphosphate (CTP), uridine monophosphate (UMP), and dihydroorotate were elevated in GSCs compared to DGCs (fig. S12A). Treatment of EGFR-amplified GSCs with the EGFR inhibitor, lapatinib, resulted in only partial reduction of the products of pyrimidine synthesis (CTP and UMP) and the immediate product of CAD (dihydroorotate) (fig. S12, B and D). Similarly, treatment of PTEN-deleted GSCs with the PI3K inhibitor, BKM120, caused partial inhibition of pyrimidine synthesis (fig. S12, C and E). Direct inhibition of pyrimidine synthesis has also been used for the treatment of multiple sclerosis, with the DHODH inhibitor, teriflunomide (31). Teriflunomide is a particularly attractive choice of pyrimidine targeting strategy as it is orally available, well tolerated, and used to treat a central nervous system disorder (multiple sclerosis), supporting its delivery into the brain. Teriflunomide treatment of GSCs harboring either EGFR amplification or PTEN deletion partially inhibited pyrimidine intermediates (UMP and CTP), as expected, and elevation in the concentrations of dihydroorotate, which is the immediate substrate for DHODH (fig. S12, D and E). On the basis of the hypothesis that targeting the intrinsic pyrimidine synthetic enzymatic activity could cooperate with extrinsic enzymatic control, we interrogated the efficacy of combining a DHODH inhibitor with targeting therapies against extrinsic control nodes for pyrimidine synthesis (EGFR or PI3K). Supporting this hypothesis, teriflunomide displayed combinatorial benefit with either lapatinib or BKM120 in reduction of pyrimidine synthesis, as demonstrated by reduction of UMP and CTP (fig. S12, D and E). Together, our data demonstrated that upstream EGRF or PI3K inhibition only partially inhibits pyrimidine synthesis, even in the correct genetic context, but combined targeting of intrinsic synthetic enzymatic activity (DHODH) demonstrated combinatorial effects in pyrimidine synthesis.
Combined targeting of pyrimidine synthesis inhibits GSC tumor growth
Given the combined effect of teriflunomide and selective upstream signaling inhibitor (lapatinib or BKM120) in the inhibition of pyrimidine synthesis within the corresponding genetic context, we investigated the selective therapeutic efficacy of these agents. As anticipated, EGFR inhibitor therapy was more effective against EGFR-amplified GSCs, whereas PI3K inhibition was more effective against PTEN-mutated GSCs, with relatively similar efficacy of the DHODH inhibitor (fig. S13, A to C). When mixed populations of EGFR-amplified GSCs and PTEN-mutated GSCs were used, the therapeutic efficacy of EGFR inhibitor or PI3K inhibitor was partially negated (fig. S13, D and E). In addition, triple targeting of CAD, DHODH, and EGFR demonstrated combinatorial benefit for GSC inhibition (fig. S13F).
To determine the combinatorial benefit in vivo, we transplanted GSCs with PTEN deletion (GSC23) transduced with luciferase into the brains of immunocompromised mice to interrogate the effect of targeting DHODH and PI3K in tumor initiation in vivo. Mice bearing GSC23 GSCs were treated with one of the four arms: vehicle control [dimethyl sulfoxide (DMSO)], teriflunomide monotherapy (50 mg/kg every 2 days), BKM120 monotherapy (50 mg/kg every 2 days), or the combination of teriflunomide (50 mg/kg every 2 days) and BKM120 (50 mg/kg every 2 days). The in vivo efficacy of teriflunomide and BKM120 on the respective targets was confirmed with immunoblot (fig. S14A). As shown in Fig. 6A and fig. S14B, compared to DMSO control, tumor volume at 30 days as assessed by luciferase assay was minimally decreased in the teriflunomide group, moderately decreased in the BKM120 group, and markedly decreased in the group receiving the combination of teriflunomide and BKM120, associated with an extension in overall survival (P < 0.0001, log-rank test; Fig. 6, B and C). The improved overall survival was also found in the group receiving the combination of teriflunomide and BKM120 using GSCs from primary glioblastoma tumors (fig. S15A).
Fig. 6. Combined targeting of pyrimidine synthesis inhibits GSC tumorigenesis in vivo.
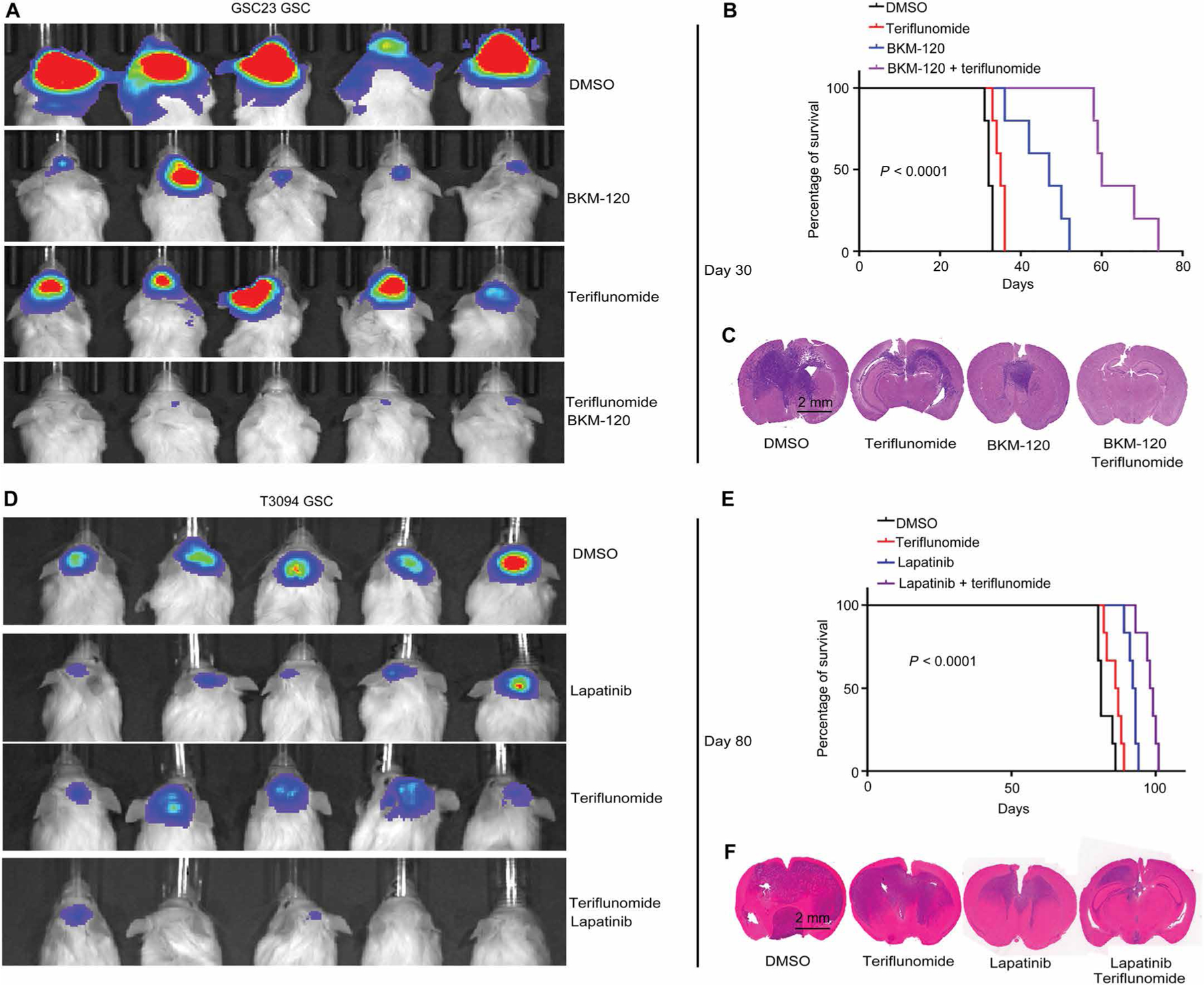
(A) GSCs (GSC23) were transduced with firefly luciferase before implantation into NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) immunocompromised mice. In vivo bioluminescence imaging was performed on NSG mice bearing intracranial xenografts derived from PTEN-deleted GSC23 treated with vehicle control (DMSO), teriflunomide monotherapy (50 mg/kg per day), BKM120 monotherapy (50 mg/kg per day), or the combination of teriflunomide (50 mg/kg per day) and BKM120 (50 mg/kg per day) (n = 5 independent experiments per group). (B) Kaplan-Meier survival curves of immunocompromised mice bearing intracranial tumors derived from PTEN-deleted GSC23 from the four treatment groups in (A) (n = 5 for each group; P < 0.0001, log-rank test). (C) Representative images of hematoxylin and eosin-stained sections of mouse brains from six independent experiments per group collected on day 35 after transplantation of PTEN-deleted GSC23 from the four treatment groups in (A). Scale bar, 2 mm. (D) In vivo bioluminescence imaging was performed on NSG mice bearing intracranial xenografts derived from EGFR-amplified T3094 GSCs treated with vehicle (DMSO), teriflunomide monotherapy (50 mg/kg per day), lapatinib monotherapy (50 mg/kg per day), or the combination of teriflunomide (50 mg/kg per day) and lapatinib (50 mg/kg per day) (n = 6 independent experiments per group). (E) Kaplan-Meier survival curves of immunocompromised mice bearing intracranial tumors derived from EGFR-amplified T3094 from the four treatment groups in (D) (n = 6 for each group; P < 0.0001, log-rank test). (F) Representative images of hematoxylin and eosin-stained sections of mouse brains from six independent experiments per group collected on day 35 after transplantation of EGFR-amplified T3094 GSCs from the four treatment groups in (D). Scale bar, 2 mm.
We next performed parallel studies with EGFR targeting in combination with DHODH inhibition. EGFR-amplified, PTEN wild-type T3094 GSCs were transduced with luciferase and implanted into the brains of immunocompromised mice, which were then treated with vehicle control (DMSO), teriflunomide monotherapy (50 mg/kg every 2 days), lapatinib monotherapy (50 mg/kg every 2 days), or the combination. The in vivo efficacy of teriflunomide and lapatinib on the respective targets was confirmed with immunoblot (fig. S14C). Treatment with teriflunomide or lapatinib each reduced tumor volume and extended survival, with greater effects from lapatinib than teriflunomide (Fig. 6D and fig. S14D). The combination of lapatinib and teriflunomide yielded the greatest efficacy in tumor control and overall survival (P < 0.0001, log-rank test; Fig. 6, E and F). The improved overall survival was also found in the group receiving the combination of teriflunomide and lapatinib using GSCs from primary glioblastoma tumors (fig. S15B). Collectively, our data demonstrated that combined targeting of pyrimidine synthesis with DHODH inhibitor and an upstream kinase inhibitor pertinent to the genetic context inhibited in vivo GSC tumor growth.
Pyrimidine metabolism informs poor outcome in glioblastoma
To determine the clinical significance of pyrimidine synthesis in patients with glioblastoma, we used a Kyoto Encyclopedia of Genes and Genomes (KEGG) pyrimidine metabolism signature to quantify the overall expression of genes involved in pyrimidine synthesis. Within this signature, we performed a pairwise correlation and determined concordantly regulated biological processes in the TCGA glioblastoma dataset, revealing positive correlation of the pyrimidine metabolic signature with regulation of innate immune, apoptotic regulation and metabolism and negative correlation with neuronal development, supporting an association with a stem-like state (fig. S16). The single-sample gene set enrichment analysis (ssGSEA) score of pyrimidine metabolism signature was elevated in all TCGA glioblastoma specimens compared to nontumor brains in the same dataset (Fig. 7A). We then profiled the ssGSEA score of the pyrimidine metabolism signature across different pathological glioma grades in a combined low-grade glioma (LGG) and glioblastoma TCGA dataset, revealing a strong correlation between tumor grade and pyrimidine metabolism (Fig. 7B). Molecular classification of gliomas has identified strongly divergent outcomes for patients with IDH1 or IDH2 mutations co-occurring with either codeletion of chromosomes 1p and 19q (classified as oligodendrogliomas) or ATRX mutations (classified as diffuse astrocytomas) (32, 33). We interrogated the TCGA glioma database, mapping these and other mutations against expressions of pyrimidine synthetic enzymes and the KEGG pyrimidine metabolism signature. Concordant with the differential expression amounts in lower tumor grades, pyrimidine synthetic pathway genes were elevated in IDH wild-type gliomas (Fig. 7C). To determine a role in patient outcome, we found higher scores of KEGG pyrimidine metabolism associated with poorer overall survival of patients with glioma across six different datasets: TCGA glioblastoma (P = 0.0401; Fig. 7D) (34), TCGA glioblastoma-LGG RNA sequencing (RNA-seq) (P < 0.0001; Fig. 7E) (35), REMBRANDT (P < 0.0001; Fig. 7F) (36), Gravendeel (P < 0.0001; Fig. 7G) (37), Phillips (P = 0.0064; Fig. 7H) (38), Freije (P = 0.1191; Fig. 7I) (39). Collectively, these results strongly link pyrimidine metabolism to poor survival for patients with glioma.
Fig. 7. Pyrimidine metabolism informs poor clinical outcome in glioblastoma.
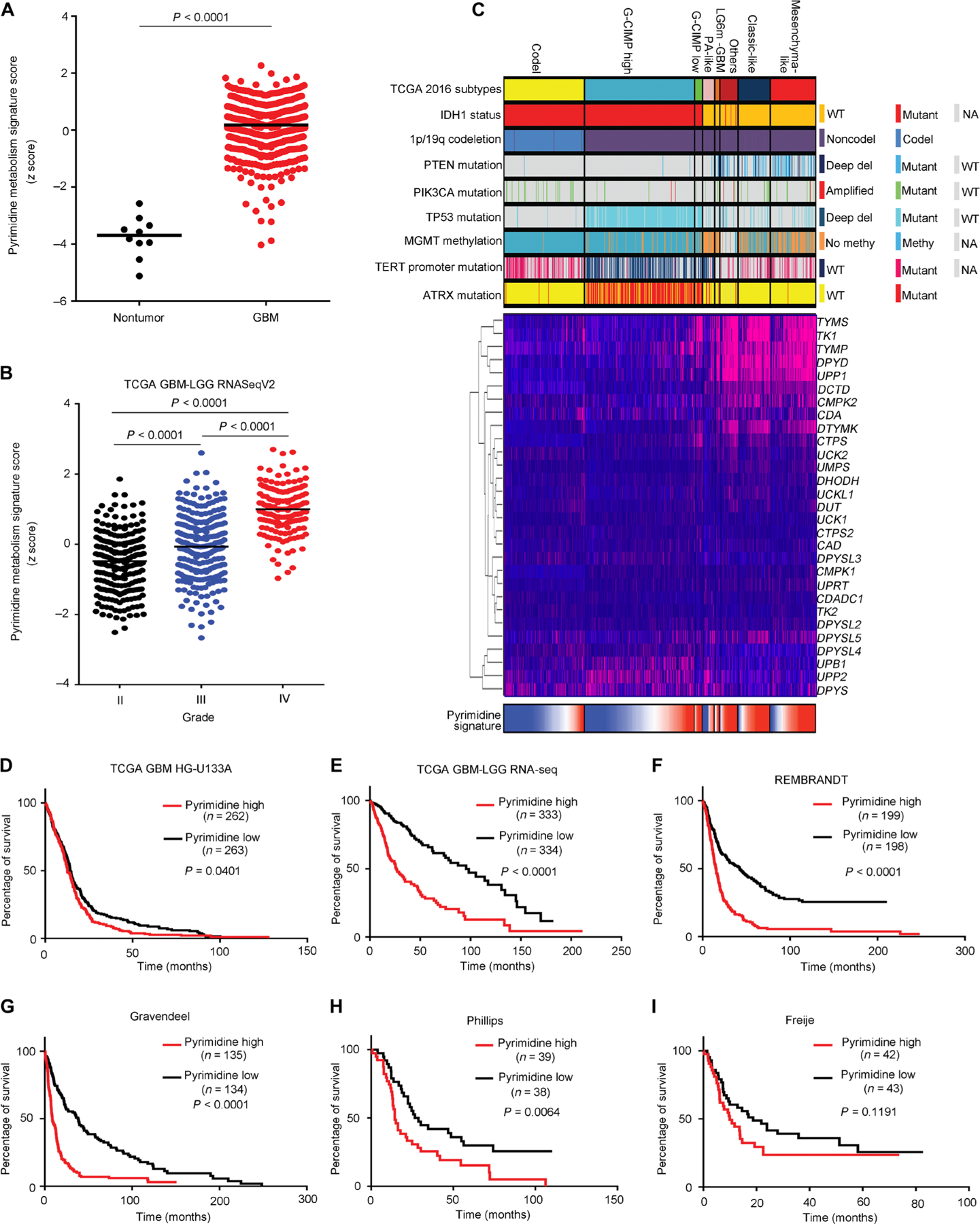
(A and B) KEGG pyrimidine metabolism signature ssGSEA score distribution between nontumor and tumor specimens in TCGA glioblastoma (GBM) HG-U133A (A) and among different grades in TCGA glioblastoma-LGG RNA-seq V2 datasets (B). (C) mRNA expression pattern of genes comprising the KEGG pyrimidine metabolism signature and corresponding ssGSEA score distribution in TCGA glioblastoma-LGG cohort (n = 667) stratified by TCGA DNA methylation cluster groups and associated molecular markers. G-CIMP, glioma CpG island methylator phenotype. (D to I) Kaplan-Meier survival analysis based on KEGG pyrimidine metabolism signature ssGSEA scores stratified by the median of six different glioma datasets: (D) TCGA glioblastoma (34), (E) TCGA glioblastoma-LGG RNA-seq (35), (F) REMBRANDT (36), (G) Gravendeel (37), (H) Phillips (38), and (I) Freije (39).
DISCUSSION
Metabolic reprogramming has long been recognized in cancer, but more recently, the altered molecular drivers of cancer metabolism have been elucidated and the discovery of cross-talk between metabolism and cancer epigenetics broadens the contributions of metabolic derangements to tumor initiation and maintenance. We and others have found that cancer stem cells, an epigenetically defined tumor cell population, display striking differences in their metabolism relative to the tumor bulk, suggesting points of fragility in this tumor cell population (18, 19, 40–45). As GSCs display preferential resistance to conventional therapies, understanding of the regulation between signaling pathways and metabolic reprogramming may inform the design of precision treatment for patients with glioblastoma and improve outcome. Given the failed efficacy of targeting single genetic alterations in glioblastoma, the approach of targeted combination therapy based on patient-specific molecular profile has gained more attention in the era of precision medicine (46). Using unbiased genomic profiling, we found that GSCs up-regulate de novo pyrimidine synthesis to maintain self-renewal, proliferation, and tumorigenesis. Disrupting key pyrimidine synthetic enzymes, either CAD or DHODH, attenuated GSC maintenance. As the expression of pyrimidine synthesis genes remains grossly unchanged across glioblastomas with different mutation profiles, post-transcriptional regulation, including the phosphorylation pattern and activity of CAD, reflected differential regulation from oncogenic drivers, including EGFR and PTEN, which revealed genotype-specific molecular regulation amenable for precision-based therapy design. Simultaneous targeting of an altered upstream signaling pathway (such as lapatinib for EGFR amplification or BKM120 for PTEN deletion) and downstream pyrimidine synthesis resulted in combinatorial inhibition of GSC tumorigenesis in vivo (modeled in fig. S17).
The multifunctional enzyme CAD is a single polypeptide with three distinct enzymatic domains for the first three steps of pyrimidine synthesis: CPSase (carbamoyl-phosphate synthetase) domain, ATCase (aspartate transcarbamylase) domain, and DHOase (dihydroorotase) domain (47, 48). The first step, catalyzed by CPSase domain of CAD, is the rate-limiting step of de novo pyrimidine synthesis (49). In the resting state, the enzymatic activity of CPSase is subject to end product inhibition by uridine 5′-triphosphate (UTP) and allosteric activation by phosphoribosyl pyrophosphate (PRPP). Through the MAPK kinase pathway, EGFR phosphorylates CPSaseT456, abolishing UTP inhibition and enhancing PRPP activation on CAD (50), with the net effect as EGFR-regulated influx of metabolites through pyrimidine synthesis (49). CAD assembles into hexamer or higher oligomers to facilitate the concerted action of its three enzymatic domains (26, 51). The phosphorylation of Ser1859, mediated by PI3K-PTEN-mTOR-S6K pathway, has been shown to promote the oligomerization and activation of CAD (27, 29). Consistent with the distinct regulatory mechanism of EGFR and PI3K on the activation of CAD, we showed that neither treatment with an EGFR inhibitor nor PI3K inhibitor alone is able to completely inhibit the metabolic activity of pyrimidine synthesis, partially explaining the limited clinical efficacy of single targeted therapy in the treatment of glioblastoma (52–54).
Immediately downstream of CAD, dihydroorotate is oxidized to orotate by DHODH (48, 49). Unlike other enzymes in de novo pyrimidine synthesis, DHODH is located on the outside surface of the inner mitochondrial membrane, linking nucleotide synthesis to mitochondrial electron transport chain (55). The inhibitors of DHODH, including leflunomide and its active metabolite, teriflunomide, are widely used as immunomodulatory medications in the management of autoimmune diseases, including rheumatoid/psoriatic arthritis and multiple sclerosis, respectively (31, 56). In acute myeloid leukemia, inhibition of DHODH induces myeloid differentiation, establishing the role of DHODH in maintaining self-renewing status of myeloblasts (57). Similar effects of DHODH on blocking differentiation were also seen in multipotent neural crest cells, an important precursor involved in melanoma pathogenesis (58). In GSCs, we showed that DHODH is essential for the maintenance of stem-like phenotype including self-renewal, differentiation, and in vivo tumorigenesis. Targeting DHODH using teriflunomide caused reduction in GSC biosynthesis of pyrimidine. Several recent studies have demonstrated the unique benefit of targeting pyrimidine synthesis in tumors with reprogrammed nucleotide metabolism, including melanomas carrying BRAF(V600E) mutation treated with BRAF(V600E) inhibitor (58), triple-negative breast cancer treated with doxorubicin (59), and breast cancer with PTEN deletion (60). In glioblastoma, we observed combinatorial inhibition of pyrimidine synthesis using teriflunomide combined with an upstream kinase inhibitor in a context-dependent manner, such as lapatinib for GSCs with EGFR amplification or BKM120 for GSCs with PTEN deletion. This would reflect basal activity in pyrimidine synthesis independent of upstream EGFR mutation or PTEN deletion that is targeted by DHODH inhibitor teriflunomide. Such metabolic combination was further supported by the superior overall survival of combined treatment compared to single treatment using in vivo tumorigenesis assay.
Although our study offers a promising translational direction in treatment of a lethal cancer, there are potential limitations to combined targeting strategies. To date, glioblastoma xenografts have been relatively modest in predicting patient responses to therapy, but effective inhibition of primary glioblastoma cells was achieved with combined inhibitors at concentrations achievable in patients, suggesting that these paradigms can be translated into clinical trial. In the clinical management of patients with glioblastoma, alternate concentrations of inhibitors may be required to achieve therapeutic doses in the brain. Furthermore, in vivo tumor metabolism and epigenetic regulation are distinct from findings in culture. Last, increasing evidence supports the critical role of the immune system in mediating responses to therapy, including metabolic dependencies. Given the need for an immunosuppressive growth environment, patient-derived xenografts lack interactions with a fully functional immune system in the mouse model, which may not recapitulate the immune surveillance during therapeutic treatment.
In summary, we propose a novel therapeutic approach to delivering precision treatment to patients with glioblastoma. GSCs reprogram their pyrimidine metabolism to maintain the stem-like status to promote self-renewal and tumorigenesis. The exact mechanism used to up-regulate pyrimidine synthesis in GSCs depends on genetic context. Targeted approaches against both the upstream genetic mutation and metabolic enzyme critical in pyrimidine synthesis demonstrated sustained effect of tumor inhibition. Given the relative resistance of GSCs to chemotherapy, radiation, and immunotherapy, our genetics-based therapeutic approach may yield improved clinical outcome.
MATERIALS AND METHODS
Study design
The overall objectives were to define the link between metabolic reprogramming and common genetic mutations in glioblastoma and to develop optimal combinatorial therapies to inform precision care. We aimed to identify metabolic pathway with preferential up-regulation in GSCs using genomic and metabolomic approaches. We found that GSCs up-regulate pyrimidine synthesis and evaluated the mechanism of genotype-specific regulation by upstream genetic mutations. We interrogated the efficacy of combined inhibition of GSCs through simultaneous targeting of upstream signaling pathway and downstream pyrimidine synthesis.
Patient-derived xenografts were generated and maintained as a recurrent source of tumor cells in our study to prevent culture-induced cell population drift, as previously described (19, 45). Briefly, tumor dissociation was performed using a papain dissociation system (Worthington Biochemical) immediately after xenograft removal from mice. Neurobasal medium (Life Technologies) supplemented with B27, L-glutamine, sodium pyruvate, basic fibroblast growth factor (10 ng/ml), and EGF (10 ng/ml; R&D Systems) was used to culture dissociated tumor cells for more than 6 hours to recover surface antigen expression. A combination of functional criteria to validate GSCs was used in our study because no marker is uniformly informative for GSCs. Both GSCs and DGCs were derived using prospective sorting followed by confirmatory assays for expression of stem cell marker, sphere formation, and secondary tumor initiation. Prospective sorting was performed using magnetic beads conjugated with CD133/1 antibody (Miltenyi Biotec). Validation of GSC phenotype was performed using expression of stem cell markers including OLIG2 and SOX2, self-renewal capacity with serial neurosphere passage, and tumor initiation through in vivo limiting dilution.
Statistical analysis
The choice of sample sizes is similar to those reported in previous studies (4, 16, 19). All grouped data were presented as box plot in figures. Two-sided Student’s t test was used to assess differences between two groups. One-way analysis of variance (ANOVA) with post hoc analysis was used to compare differences among more than two groups. Kaplan-Meier survival curves were generated using Prism software, and log-rank test was performed to assess statistical significance between groups. Correlation between gene expression and patient survival was performed through analysis of TCGA, Freije, Phillips, and REMBRANDT brain tumor datasets, downloaded from TCGA data portal or National Center for Biotechnology Information Gene Expression Omnibus (GEO) database.
Supplementary Material
Fig. S1. Identification of differential enhancer activation between GSCs and DGCs.
Fig. S2. DHODH promotes GSC growth.
Fig. S3. CADS1859 and CADT456 are important for GSC proliferation.
Fig. S4. Validation of knockdown efficacy of shRNAs directed against CAD.
Fig. S5. CAD regulates primary GSC growth and self-renewal.
Fig. S6. DHODH regulates primary GSC growth and self-renewal.
Fig. S7. CAD and DHODH are essential for primary GSC maintenance.
Fig. S7. CAD and DHODH are essential for primary GSC maintenance.
Fig. S9. EGFR regulates CADT456 phosphorylation through the MAPK-ERK pathway.
Fig. S10. PTEN deletion promotes CADS1859 phosphorylation through the PI3K-AKT pathway.
Fig. S11. PTEN deletion promotes CADS1859 phosphorylation through the PI3K-AKT-TORC1 but not the TORC2 pathway.
Fig. S12. Combinatorial blockade of de novo pyrimidine synthesis in GSCs by the DHODH inhibitor teriflunomide and EGFR or PI3K inhibition.
Fig. S13. Sensitivity of GSCs to inhibitor treatments.
Fig. S14. Impact of targeted therapies on signal transduction pathways in vivo.
Fig. S15. Combined targeting of pyrimidine synthesis inhibits GSC tumorigenesis in vivo.
Fig. S16. Pairwise correlation analysis of pyrimidine pathway genes and gene enrichment analysis.
Fig. S17. Proposed model of combined targeting of pyrimidine synthesis in GSCs.
Data file S1. Individual subject-level data (Excel file).
Acknowledgments:
We appreciate the assistance from R. Zhang for mass spectrometry analysis. We thank T. Roberts, P. Wen, J. Suh, and members of the Rich laboratory for insightful discussions.
Funding: This work was supported by the National Institutes of Health grants CA197718, CA154130, CA169117, CA171652, NS087913, and NS089272 to J.N.R.; CA184090, NS091080, and NS099175 to S. Bao; and NS073831 to P.S.M.
Footnotes
SUPPLEMENTARY MATERIALS
stm.sciencemag.org/cgi/content/full/11/504/eaau4972/DC1
Materials and Methods
Competing interests: P.S.M. is a cofounder of Pretzel Therapeutics Inc., for which he owns equity and serves as a consultant. All other authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or in the Supplementary Materials. Raw sequencing data files are deposited in the GEO database (ChIP-seq, GSE129438; exome-seq, PRJNA531753).
REFERENCES AND NOTES
- 1.Ostrom QT, Gittleman H, de Blank PM, Finlay JL, Gurney JG, McKean-Cowdin R, Stearns DS, Wolff JE, Liu M, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS, American Brain Tumor Association adolescent and young adult primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neuro Oncol. 18 (suppl. 1), i1–i50 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO; European Organisation for Research and Treatment of Cancer Brain, Tumuour amd Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials, Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suva ML, Regev A, Bernstein BE, Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN, Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS, Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 5, 67 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, Shi Q, McLendon RE, Bigner DD, Rich JN, Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 66, 7843–7848 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, Fang X, Sloan AE, Mao Y, Lathia JD, Min W, McLendon RE, Rich JN, Bao S, Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 153, 139–152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ward PS, Thompson CB, Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 21, 297–308 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeBerardinis RJ, Chandel NS, Fundamentals of cancer metabolism. Sci. Adv 2, e1600200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Venneti S, Thompson CB, Metabolic reprogramming in brain tumors. Annu. Rev. Pathol 12, 515–545 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Lunt SY, Vander Heiden MG, Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol 27, 441–464 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O’Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB, IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, Thompson CB, Kaufman A, Guryanova O, Levine R, Heguy A, Viale A, Morris LG, Huse JT, Mellinghoff IK, Chan TA, IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luengo A, Gui DY, Vander Heiden MG, Targeting metabolism for cancer therapy. Cell Chem. Biol 24, 1161–1180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bobrovnikova-Marjon E, Hurov JB, Targeting metabolic changes in cancer: Novel therapeutic approaches. Annu. Rev. Med 65, 157–170 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, Weil RJ, Nakano I, Sarkaria JN, Stringer BW, Day BW, Li M, Lathia JD, Rich JN, Hjelmeland AB, Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci 16, 1373–1382 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, Hjelmeland AB, Rich JN, Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15, 501–513 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cosset E, Ilmjarv S, Dutoit V, Elliott K, von Schalscha T, Camargo MF, Reiss A, Moroishi T, Seguin L, Gomez G, Moo JS, Preynat-Seauve O, Krause KH, Chneiweiss H, Sarkaria JN, Guan KL, Dietrich PY, Weis SM, Mischel PS, Cheresh DA, Glut3 addiction is a druggable vulnerability for a molecularly defined subpopulation of glioblastoma. Cancer Cell 32, 856–868.e855 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Yang K, Xie Q, Wu Q, Mack SC, Shi Y, Kim LJY, Prager BC, Flavahan WA, Liu X, Singer M, Hubert CG, Miller TE, Zhou W, Huang Z, Fang X, Regev A, Suva ML, Hwang TH, Locasale JW, Bao S, Rich JN, Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat. Neurosci 20, 661–673 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O’Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L; TCGA Reserach Network, The somatic genomic landscape of glioblastoma. Cell 155, 462–477 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cancer Genome Atlas Research Network, Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Byron SA, Tran NL, Halperin RF, Phillips JJ, Kuhn JG, de Groot JF, Colman H, Ligon KL, Wen PY, Cloughesy TF, Mellinghoff IK, Butowski NA, Taylor JW, Clarke JL, Chang SM, Berger MS, Molinaro AM, Maggiora GM, Peng S, Nasser S, Liang WS, Trent JM, Berens ME, Carpten JD, Craig DW, Prados MD, Prospective feasibility trial for genomics-informed treatment in recurrent and progressive glioblastoma. Clin. Cancer Res 24, 295–305 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garraway LA, Genomics-driven oncology: Framework for an emerging paradigm. J. Clin. Oncol 31, 1806–1814 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Stern AD, Alexander BM, Chandra A, How economics can shape precision medicines. Science 355, 1131–1133 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Reznik E, Luna A, Aksoy BA, Liu EM, La K, Ostrovnaya I, Creighton CJ, Hakimi AA, Sander C, A landscape of metabolic variation across tumor types. Cell Syst. 6, 301–313. e303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moreno-Morcillo M, Grande-GarcÍa A, Ruiz-Ramos A, Del Caño-Ochoa F, Boskovic J, Ramón-Maiques S, Structural insight into the core of CAD, the multifunctional protein leading de novo pyrimidine biosynthesis. Structure 25, 912–923.e915 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Moes S, Prescianotto-Baschong C, Sauer U, Jenoe P, Hall MN, Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 339, 1320–1323 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Makinoshima H, Takita M, Matsumoto S, Yagishita A, Owada S, Esumi H, Tsuchihara K, Epidermal growth factor receptor (EGFR) signaling regulates global metabolic pathways in EGFR-mutated lung adenocarcinoma. J. Biol. Chem 289, 20813–20823 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ben-Sahra I, Howell JJ, Asara JM, Manning BD, Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339, 1323–1328 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valvezan AJ, Turner M, Belaid A, Lam HC, Miller SK, McNamara MC, Baglini C, Housden BE, Perrimon N, Kwiatkowski DJ, Asara JM, Henske EP, Manning BD, mTORC1 couples nucleotide synthesis to nucleotide demand resulting in a targetable metabolic vulnerability. Cancer Cell 32, 624–638.e625 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Connor P, Wolinsky JS, Confavreux C, Comi G, Kappos L, Olsson TP, Benzerdjeb H, Truffinet P, Wang L, Miller A, Freedman MS; Group TT, Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N. Engl. J. Med 365, 1293–1303 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, Pekmezci M, Rice T, Kosel ML, Smirnov IV, Sarkar G, Caron AA, Kollmeyer TM, Praska CE, Chada AR, Halder C, Hansen HM, McCoy LS, Bracci PM, Marshall R, Zheng S, Reis GF, Pico AR, O’Neill BP, Buckner JC, Giannini C, Huse JT, Perry A, Tihan T, Berger MS, Chang SM, Prados MD, Wiemels J, Wiencke JK, Wrensch MR, Jenkins RB, Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N. Engl. J. Med 372, 2499–2508 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, Rodriguez FJ, Rosemberg S, Oba-Shinjo SM, Nagahashi Marie SK, Bettegowda C, Agrawal N, Lipp E, Pirozzi C, Lopez G, He Y, Friedman H, Friedman AH, Riggins GJ, Holdhoff M, Burger P, McLendon R, Bigner DD, Vogelstein B, Meeker AK, Kinzler KW, Papadopoulos N, Diaz LA, Yan H, Frequent ATRX CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 3, 709–722 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN; Cancer Genome Atlas Research Network, Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, Anjum S, Wang J, Manyam G, Zoppoli P, Ling S, Rao AA, Grifford M, Cherniack AD, Zhang H, Poisson L, Carlotti CG Jr., Tirapelli DP, Rao A, Mikkelsen T, Lau CC, Yung WK, Rabadan R, Huse J, Brat DJ, Lehman NL, Barnholtz-Sloan JS, Zheng S, Hess K, Rao G, Meyerson M, Beroukhim R, Cooper L, Akbani R, Wrensch M, Haussler D, Aldape KD, Laird PW, Gutmann DH; TCGA Research Network, Noushmehr H, Iavarone A, Verhaak RG, Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164, 550–563 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Madhavan S, Zenklusen JC, Kotliarov Y, Sahni H, Fine HA, Buetow K, Rembrandt: Helping personalized medicine become a reality through integrative translational research. Mol. Cancer Res 7, 157–167 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gravendeel LA, Kouwenhoven MC, Gevaert O, de Rooi JJ, Stubbs AP, Duijm JE, Daemen A, Bleeker FE, Bralten LB, Kloosterhof NK, De Moor B, Eilers PH, van der Spek PJ, Kros JM, Sillevis Smitt PA, van den Bent MJ, French PJ, Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 69, 9065–9072 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG, Aldape K, Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9, 157–173 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Freije WA, Castro-Vargas FE, Fang Z, Horvath S, Cloughesy T, Liau LM, Mischel PS, Nelson SF, Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 64, 6503–6510 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, Popescu R, Della Donna L, Evers P, Dekmezian C, Reue K, Christofk H, Mischel PS, Pajonk F, Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. U.S.A 108, 16062–16067 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mao P, Joshi K, Li J, Kim SH, Li P, Santana-Santos L, Luthra S, Chandran UR, Benos PV, Smith L, Wang M, Hu B, Cheng SY, Sobol RW, Nakano I, Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. U.S.A 110, 8644–8649 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC, Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med 2, 31ra34 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Kathagen A, Schulte A, Balcke G, Phillips HS, Martens T, Matschke J, Gunther HS, Soriano R, Modrusan Z, Sandmann T, Kuhl C, Tissier A, Holz M, Krawinkel LA, Glatzel M, Westphal M, Lamszus K, Hypoxia and oxygenation induce a metabolic switch between pentose phosphate pathway and glycolysis in glioma stem-like cells. Acta Neuropathol. 126, 763–780 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Jung J, Kim LJY, Wang X, Wu Q, Sanvoranart T, Hubert CG, Prager BC, Wallace LC, Jin X, Mack SC, Rich JN, Nicotinamide metabolism regulates glioblastoma stem cell maintenance. JCI Insight 2, 90019 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X, Huang Z, Wu Q, Prager BC, Mack SC, Yang K, Kim LJY, Gimple RC, Shi Y, Lai S, Xie Q, Miller TE, Hubert CG, Song A, Dong Z, Zhou W, Fang X, Zhu Z, Mahadev V, Bao S, Rich JN, MYC-regulated mevalonate metabolism maintains brain tumor-initiating cells. Cancer Res. 77, 4947–4960 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prados MD, Byron SA, Tran NL, Phillips JJ, Molinaro AM, Ligon KL, Wen PY, Kuhn JG, Mellinghoff IK, de Groot JF, Colman H, Cloughesy TF, Chang SM, Ryken TC, Tembe WD, Kiefer JA, Berens ME, Craig DW, Carpten JD, Trent JM, Toward precision medicine in glioblastoma: The promise and the challenges. Neuro Oncol. 17, 1051–1063 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coleman PF, Suttle DP, Stark GR, Purification from hamster cells of the multifunctional protein that initiates de novo synthesis of pyrimidine nucleotides. J. Biol. Chem 252, 6379–6385 (1977). [PubMed] [Google Scholar]
- 48.Lane AN, Fan TW, Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 43, 2466–2485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Evans DR, Guy HI, Mammalian pyrimidine biosynthesis: Fresh insights into an ancient pathway. J. Biol. Chem 279, 33035–33038 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Graves LM, Guy HI, Kozlowski P, Huang M, Lazarowski E, Pope RM, Collins MA, Dahlstrand EN, Earp III HS, Evans DR, Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature 403, 328–332 (2000). [DOI] [PubMed] [Google Scholar]
- 51.Lee L, Kelly RE, Pastra-Landis SC, Evans DR, Oligomeric structure of the multifunctional protein CAD that initiates pyrimidine biosynthesis in mammalian cells. Proc. Natl. Acad. Sci. U.S.A 82, 6802–6806 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wen PY, Lee EQ, Reardon DA, Ligon KL, Alfred Yung WK, Current clinical development of PI3K pathway inhibitors in glioblastoma. Neuro Oncol. 14, 819–829 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reardon DA, Groves MD, Wen PY, Nabors L, Mikkelsen T, Rosenfeld S, Raizer J, Barriuso J, McLendon RE, Suttle AB, Ma B, Curtis CM, Dar MM, de Bono J, A phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma. Clin. Cancer Res 19, 900–908 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Uhm JH, Ballman KV, Wu W, Giannini C, Krauss JC, Buckner JC, James CD, Scheithauer BW, Behrens RJ, Flynn PJ, Schaefer PL, Dakhill SR, Jaeckle KA, Phase II evaluation of gefitinib in patients with newly diagnosed grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int. J. Radiat. Oncol. Biol. Phys 80, 347–353 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones ME, Pyrimidine nucleotide biosynthesis in animals: Genes, enzymes, and regulation of UMP biosynthesis. Annu. Rev. Biochem 49, 253–279 (1980). [DOI] [PubMed] [Google Scholar]
- 56.Strand V, Cohen S, Schiff M, Weaver A, Fleischmann R, Cannon G, Fox R, Moreland L, Olsen N, Furst D, Caldwell J, Kaine J, Sharp J, Hurley F, Loew-Friedrich I; Leflunomide Rheumatoid Arthritis Investigators Group, Treatment of active rheumatoid arthritis with leflunomide compared with placebo and methotrexate. Arch. Intern. Med 159, 2542–2550 (1999). [DOI] [PubMed] [Google Scholar]
- 57.Sykes DB, Kfoury YS, Mercier FE, Wawer MJ, Law JM, Haynes MK, Lewis TA, Schajnovitz A, Jain E, Lee D, Meyer H, Pierce KA, Tolliday NJ, Waller A, Ferrara SJ, Eheim AL, Stoeckigt D, Maxcy KL, Cobert JM, Bachand J, Szekely BA, Mukherjee S, Sklar LA, Kotz JD, Clish CB, Sadreyev RI, Clemons PA, Janzer A, Schreiber SL, Scadden DT, Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell 167, 171–186.e115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.White RM, Cech J, Ratanasirintrawoot S, Lin CY, Rahl PB, Burke CJ, Langdon E, Tomlinson ML, Mosher J, Kaufman C, Chen F, Long HK, Kramer M, Datta S, Neuberg D, Granter S, Young RA, Morrison S, Wheeler GN, Zon LI, DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature 471, 518–522 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brown KK, Spinelli JB, Asara JM, Toker A, Adaptive reprogramming of de novo pyrimidine synthesis is a metabolic vulnerability in triple-negative breast cancer. Cancer Discov. 7, 391–399 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mathur D, Stratikopoulos E, Ozturk S, Steinbach N, Pegno S, Schoenfeld S, Yong R, Murty VV, Asara JM, Cantley LC, Parsons R, PTEN regulates glutamine flux to pyrimidine synthesis and sensitivity to dihydroorotate dehydrogenase inhibition. Cancer Discov. 7, 380–390 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG; Exome Aggregation Consortium, Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR, A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kuilman T, Velds A, Kemper K, Ranzani M, Bombardelli L, Hoogstraat M, Nevedomskaya E, Xu G, de Ruiter J, Lolkema MP, Ylstra B, Jonkers J, Rottenberg S, Wessels LF, Adams DJ, Peeper DS, Krijgsman O, CopywriteR: DNA copy number detection from off-target sequence data. Genome Biol. 16, 49 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM, Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem 273, 18623–18632 (1998). [DOI] [PubMed] [Google Scholar]
- 65.Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, Rominger CM, Erskine S, Fisher KE, Yang J, Zappacosta F, Annan R, Sutton D, Laquerre SG, GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin. Cancer Res 17, 989–1000 (2011). [DOI] [PubMed] [Google Scholar]
- 66.Burger MT, Pecchi S, Wagman A, Ni ZJ, Knapp M, Hendrickson T, Atallah G, Pfister K, Zhang Y, Bartulis S, Frazier K, Ng S, Smith A, Verhagen J, Haznedar J, Huh K, Iwanowicz E, Xin X, Menezes D, Merritt H, Lee I, Wiesmann M, Kaufman S, Crawford K, Chin M, Bussiere D, Shoemaker K, Zaror I, Maira SM, Voliva CF, Identification of NVP-BKM120 as a potent, selective, orally bioavailable class I PI3 kinase inhibitor for treating cancer. ACS Med. Chem. Lett 2, 774–779 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vlahos CJ, Matter WF, Hui KY, Brown RF, A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem 269, 5241–5248 (1994). [PubMed] [Google Scholar]
- 68.Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL, A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369, 756–758 (1994). [DOI] [PubMed] [Google Scholar]
- 69.Rusnak DW, Lackey K, Affleck K, Wood ER, Alligood KJ, Rhodes N, Keith BR, Murray DM, Knight WB, Mullin RJ, Gilmer TM, The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol. Cancer Ther 1, 85–94 (2001). [PubMed] [Google Scholar]
- 70.Ciardiello F, Caputo R, Bianco R, Damiano V, Pomatico G, De Placido S, Bianco AR, Tortora G, Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin. Cancer Res 6, 2053–2063 (2000). [PubMed] [Google Scholar]
- 71.Oh J, O’Connor PW, An update of teriflunomide for treatment of multiple sclerosis. Ther. Clin. Risk Manag 9, 177–190 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schneider CA, Rasband WS, Eliceiri KW, NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Identification of differential enhancer activation between GSCs and DGCs.
Fig. S2. DHODH promotes GSC growth.
Fig. S3. CADS1859 and CADT456 are important for GSC proliferation.
Fig. S4. Validation of knockdown efficacy of shRNAs directed against CAD.
Fig. S5. CAD regulates primary GSC growth and self-renewal.
Fig. S6. DHODH regulates primary GSC growth and self-renewal.
Fig. S7. CAD and DHODH are essential for primary GSC maintenance.
Fig. S7. CAD and DHODH are essential for primary GSC maintenance.
Fig. S9. EGFR regulates CADT456 phosphorylation through the MAPK-ERK pathway.
Fig. S10. PTEN deletion promotes CADS1859 phosphorylation through the PI3K-AKT pathway.
Fig. S11. PTEN deletion promotes CADS1859 phosphorylation through the PI3K-AKT-TORC1 but not the TORC2 pathway.
Fig. S12. Combinatorial blockade of de novo pyrimidine synthesis in GSCs by the DHODH inhibitor teriflunomide and EGFR or PI3K inhibition.
Fig. S13. Sensitivity of GSCs to inhibitor treatments.
Fig. S14. Impact of targeted therapies on signal transduction pathways in vivo.
Fig. S15. Combined targeting of pyrimidine synthesis inhibits GSC tumorigenesis in vivo.
Fig. S16. Pairwise correlation analysis of pyrimidine pathway genes and gene enrichment analysis.
Fig. S17. Proposed model of combined targeting of pyrimidine synthesis in GSCs.
Data file S1. Individual subject-level data (Excel file).