

Abstract
Marine mammals are an important part of ocean ecosystems, of which, whales play a vital role in the marine food chain. In this study, the mucosa and contents from different intestinal tract segments (ITSs) of a stranded dwarf minke whale (Balaenoptera acutorostrata) were analyzed. The gut microbiota were sequenced using high‐throughput sequencing technology, based on a 16S rRNA approach. The microbial composition of the intestinal mucosa and its contents were similar in every single ITS. Large intestine microbiota richness and diversity were significantly higher when compared to the duodenum and jejunum. The dominant bacteria in the gut were Firmicutes and Actinobacteria; the former was enriched in the large intestine, whereas the latter was more abundant in the duodenum and jejunum. Our findings provide novel insights for microbiota in B. acutorostrata.
Keywords: dwarf minke whale Balaenoptera acutorostrata, gut microbiota, high‐throughput sequencing, intestinal tract segment
This study has characterized the microbiota of five sampling sources (duodenum, jejunum, cecum, colon, and rectum) in a fresh stranded Balaenoptera acutorostrata carcass. Significant differences were observed in gut microbiota composition across different intestinal tract segments; the microbiomes of posterior compartments were more diverse than the foregut. The duodenum and jejunum were enriched for Erysipelotrichales, Selenomonadales, and Coriobacteriales, whereas Clostridiales were more abundant in the large intestine.

1. INTRODUCTION
Animal digestive tracts are complex microecosystems composed of host cells and intestinal microbiota (Backhed, 2005). Their symbiotic relationship not only influences host metabolism, energy production, growth, and immunity but also underlies several pathological conditions (Flint, Scott, Louis, & Duncan, 2012; Malmuthuge, Li, Goonewardene, Oba, & Guan, 2013). Marine mammals are important microbial reservoirs thanks to their constant body temperature and large sizes (Higgins, 2000), and in recent years, the commensal microbiota of various marine mammals have been elucidated (Chiarello, Villéger, Bouvier, Auguet, & Bouvier, 2017; Numberger, Herlemann, Jürgens, Dehnhardt, & Schulz‐Vogt, 2016). Monitoring the intestinal microbiota of marine mammals can help unravel adaptive mechanisms to different diets or habitats. For example, a metagenomic study reported high nutrient transport and cycling potential in the gut microbiota of the Australian sea lion, Neophoca cinerea (Lavery, Roudnew, Seymour, Mitchell, & Jeffries, 2012). Moreover, the gut microbiota of whales and terrestrial herbivores exhibit similar fermentative metabolism, while protein catabolic and amino acid synthesis pathways in the whale gut closely resemble terrestrial carnivores (Sanders et al., 2015).
The dwarf minke whale, Balaenoptera acutorostrata, is the most abundant species of baleen whale and is widely distributed in the North Atlantic and North Pacific regions (Walquist, Stormo, Jensen, Bjarne, & Eilertsen, 2017). It forages on fish such as capelin, herring, mackerel, and codfish, as well as crustaceans, such as krill (Jonsgård, 1982; Windsland, Lindstrom, Nilssen, & Haug, 2007), suggesting the whale's gut microbiota are adaptive to diverse food sources. Due to its size, B. acutorostrata has a long digestive tract, suggesting a varied intestinal microbiota composition from different intestinal tract segments (ITSs). However, these compositions are currently unknown. Therefore, intestinal microbiota comparisons between different ITSs of B. acutorostrate could provide novel insights into food digestion and metabolism.
A freshly dead B. acutorostrata was found on a beach in Dalian, China, and its gut mucosa and contents were subsequently obtained. We analyzed microbiota from different ITSs using high‐throughput sequencing technology based on 16S rRNA gene analysis. To the best of our knowledge, this is the first study to analyze microbiota of different ITSs from B. acutorostrata.
2. MATERIALS AND METHODS
2.1. Sample collection
The minke whale was found on a beach on ChangXing Island, Dalian, China (122°45′36.932″E, 39°1′24.872″N). As soon as local fishermen encountered the whale (which was still alive), they immediately contacted local authorities. By the time this personnel arrived, the whale had been dead for an hour. Thus, the stranded individual was considered a fresh stranding. Within two hours, it was transported to the laboratory (temperature = 10℃) in a sterilized plastic bag. The animal weighed 3350.0 kg and was dissected at the Anatomy Laboratory of Liaoning Ocean and Fisheries Research Institute. According to the clinical report of an attending veterinarian (Qualification Certificate No. C012015210021), there was no damage to the body surface and no pathological changes were observed, suggesting the animal was disease‐free. Species identification and dissection were approved by the Ministry of Agriculture and Rural Affairs of the People's Republic of China (No. 1376). The carcass was identified as a dwarf minke whale (B. acutorostrata). This identification was based on 256 pairs of yellow or milky white baleen in its mouth, a white transverse band on the lateral central part of the fin limb, and a distinct ridge on the back of the head. The body was dissected, and intestinal segments were harvested. Three samples of duodenum, jejunum, cecum, colon, and rectum contents and mucosa were collected and stored at −80°C (Figure 1).
Figure 1.

Different intestinal tract segments of Balaenoptera acutorostrata.
2.2. DNA extraction
Bacterial genomic DNA was extracted from 30 samples using the TIANamp Stool DNA Kit (Tiangen, China) according to the manufacturer's instructions. DNA was verified by electrophoresis on 1.5% agarose gels. DNA concentration and purity were measured on the Qubit™ 4 Fluorometer (Invitrogen, USA) and stored at −20°C until required.
2.3. 16S rRNA gene amplification and sequencing
V3–V4 regions of 16S rRNA were amplified using the 341F (5′‐CCTACGGRRBGCASCAGKVRVGAAT‐3′) and 806R (5'‐GGACTACNVGGGTWTCTAATCC‐3′) primers. All PCR reactions were performed in a 25 µl master mix, consisting of 2.5 µl TransStart Buffer, 2 µl dNTPs (2.5 mM), 0.5 µl TranstartTaq DNA polymerase (TransGene, China), 1 µl each of forward and reverse primers (100 µM), and 20 ng DNA template. Cycling parameters included an initial denaturation at 94°C for 3 min, followed by 24 denaturation cycles at 94°C for 5 s, annealing at 57°C for 90 s, and elongation at 72°C for 10 s. PCR products were purified and quantified, and amplicon libraries were generated. Libraries were sequenced on an Illumina Miseq platform using a 250 base pair (bp) paired‐end strategy.
2.4. Processing sequencing data
All read sequences were analyzed by Bcl2fastq v 2.17.1.14. Lower quality reads were removed, based on average Phred scores <20, ambiguous bases, homopolymer runs >8, primer mismatches, and sequence lengths <200 bp. Paired‐end reads with >20 bp overlaps, and without mismatches were assembled into tags using FLASH (Magoc & Salzberg, 2011). These tags were annotated to samples based on their unique barcodes, and then, the barcode and primer sequences were removed. Chimeric sequences were recognized and eliminated by the QIIME software package (Caporaso et al., 2010), and tags with ≥97% similarity were assigned to the same operational taxonomic unit (OTU) using VSEARCH v 1.9.6 (Edgar, 2010). Representative sequences of each OTU were chosen randomly and assigned taxonomy using the Ribosomal Database Project classifier (Wang, Garrity, Tiedje, & Cole, 2007) based on the SILVA database (Yilmaz et al., 2014). Finally, the relative abundances of different OTUs were calculated and normalized to the sample, with the least number of tags (Table S1).
2.5. Statistical analysis
Chao1 and Shannon indices representing alpha diversities were calculated, and rarefaction curves created using QIIME (Kemp & Aller, 2004). Indices from different ITSs were compared using Tukey's HSD tests. The unweighted pair‐group method, with arithmetic means (UPGMA) clustering, was used to analyze microbial composition similarities across the different ITSs. Species abundance at the phylum level was visualized using Circos software (Krzywinski et al., 2009).
3. RESULTS AND DISCUSSION
3.1. Gut microbiota diversity
We sequenced 30 samples from different ITSs of B. acutorostrata and generated an average of 70,288 tags per sample. Rarefaction curves for each sample were close to a straight line (Figure 2a), indicating that sequencing data represented the entire microbial community. These tags were clustered into 172 OTUs at the 97% similarity level, and each OTU was annotated from phylum to species level.
Figure 2.
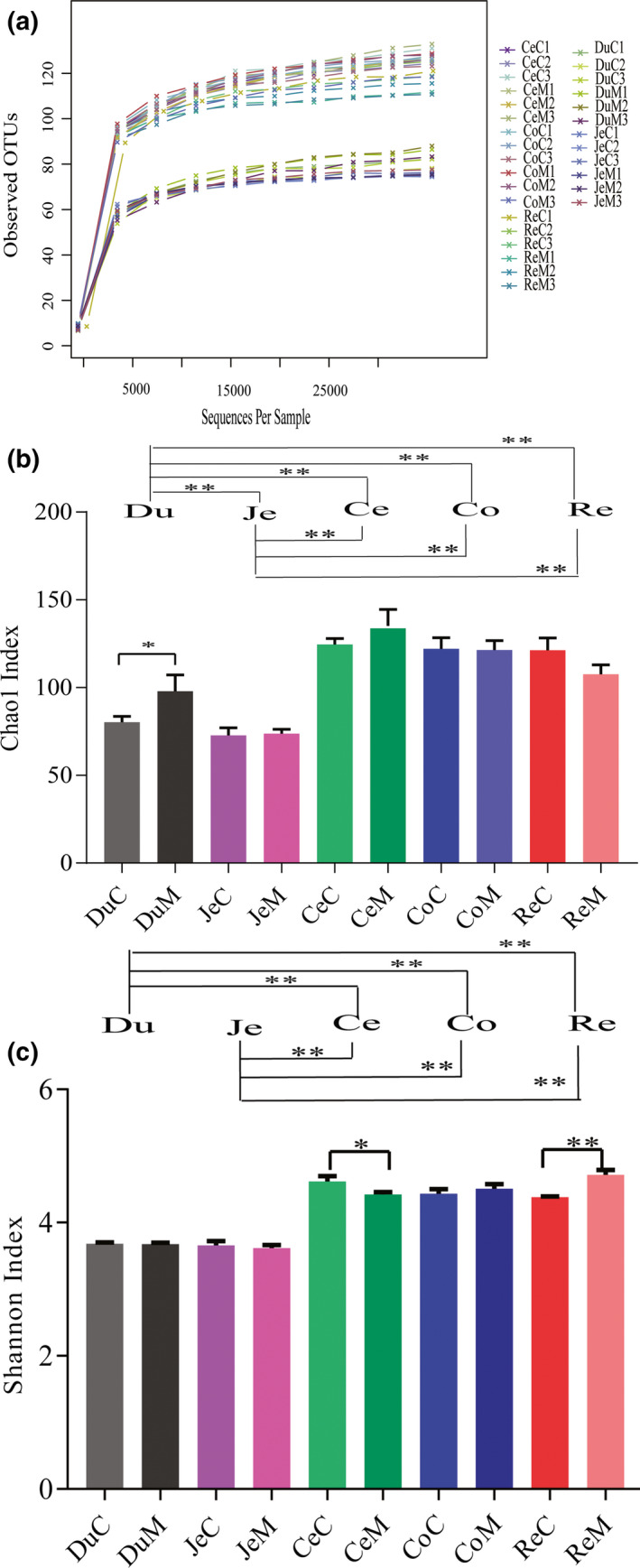
(a) Rarefaction curves for each sample from different ITSs of Balaenoptera acutorostrata. Comparison of Chao1 (b) and Shannon (c) indices of B. acutorostrata ITSs microbiota. Tukey's HSD test was used to determine significance among different ITSs samples. ** indicates a p‐value <0.05. The names of each sample are duodenum (Du), jejunum (Je), cecum (Ce), colon (Co), and rectum (Re) contents(C) or mucosa (M) with the serial number of 1 to 3.
We used alpha‐diversity indices to analyze the gut microbiota of different ITSs. The Chao1 index was used to evaluate species richness, that is, the number of species in microbial communities (Colwell & Levin, 2009). As shown (Figure 2b), the microbial richness of intestinal mucosa and contents form the same ITSs were similar, except the duodenum. Gut microbiota richness ranged from 70 to 130, which was similar (Chao1 = 84) to microbial communities from beluga whale feces (Sanders et al., 2015). In contrast, gut microbiota richness from smaller cetaceans, such as bottlenose dolphins (Chao1 = 41) and Yangtze finless porpoises (Chao1 = 30), were lower (McLaughlin, Chen, Zheng, Zhao, & Wang, 2012). Furthermore, >500 OTUs were detected in the gut microbiomes from humpback and right whales (Sanders et al., 2015), which are larger than B. acutorostrata. Therefore, cetacean gut microbiota richness may be proportional to body size, although more evidence is required to validate this observation.
The Shannon index estimates microbial community diversity in terms of species richness and evenness (Moreno & Rodríguez, 2010). Unlike the Chao1 index, the microbial diversity of cecal and rectal mucosa and contents, were significantly different in each segment (Figure 2c, p < 0.05). Also, microbial richness and diversity in the large intestine (cecum, colon, and rectum) were significantly higher than the duodenum and jejunum (Figure 2b and c, p < 0.05). Due to the comparative whale studies are not available, we used other animals as comparators. The result of this study was consistent with higher Shannon indices of the colon versus duodenum/jejunum microbiota reported in felines (Ritchie, Steiner, & Suchodolski, 2008). Furthermore, molecular fingerprinting technology revealed that bacterial diversity in the canine colon and rectum was significantly higher than the duodenum and jejunum (Suchodolski, Ruaux, Steiner, Fetz, & Williams, 2005). Moreover, bacterial communities in the midgut and hindgut of sea cucumbers had a significantly higher richness and diversity when compared with the foregut (Wang et al., 2018). Taken together, the posterior compartment of the intestinal tract could be more suitable for microbial colonization across diverse animal species.
3.2. Gut microbiota composition
Balaenoptera acutorostrata gut microbiota divergence was demonstrated by UPGMA clustering analysis, based on Bray–Curtis distances (Figure 3). The microbiota of mucosa and the contents of the same ITS were clustered together, indicating that attached and free bacteria across the B. acutorostrata gut were consistent. Furthermore, samples from the cecum, colon, and rectum formed a cluster, and the duodenum and jejunum microbiota were clustered, distinct to the large intestine. Thus, it appeared that gut microbiome composition in B. acutorostrata was significantly dependent on anatomical location.
Figure 3.
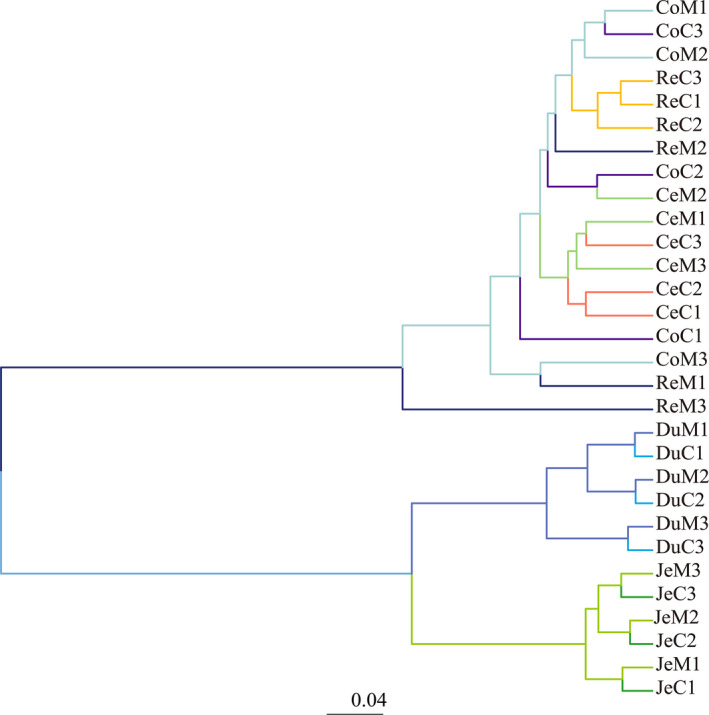
The unweighted pair‐group method with arithmetic means (UPGMA) clustering analysis of gut microbiota composition in different ITSs from Balaenoptera acutorostrata. The names of each sample are duodenum (Du), jejunum (Je), cecum (Ce), colon (Co), and rectum (Re) contents(C) or mucosa (M) with the serial number of 1 to 3.
The dominant phyla were Firmicutes and Actinobacteria, accounting for >90% of the total microbiome (Figure 4a). Firmicutes were the predominant group and comprised over 50% of the resident bacteria of the duodenum and jejunum and almost 90% of the large intestine microbiota. Also, Firmicutes primarily included the Clostridiales, Erysipelotrichales, and Selenomonadales orders (Figure 4b). The relative abundance of Clostridiales in cecum, colon, and rectum was significantly higher than the duodenum and jejunum. Clostridiales species ferment dietary fiber in the gut to generate short‐chain fatty acids (SCFAs) (Chinda et al., 2004). Erysipelotrichales was significantly enriched in the jejunum, when compared to other ITSs, and is abundant in canine fecal microbiota when fed carbohydrate‐rich diets (Hang et al., 2012). Erysipelotrichales is associated with chronic canine enteropathies due to its recognition by Toll‐like receptors (Allenspach et al., 2010). Selenomonadales was only detected in the duodenum and jejunum. Thai children on high‐fat diets exhibit a greater abundance of Selenomonadales in their gut when compared to children on largely vegetable‐based diets (Kisuse et al., 2018). The Actinobacteria phylum was more abundant in the duodenum (46.61%) and jejunum (30.86%), when compared to other ITSs (6.19% –12.49%), and consisted almost entirely of the Coriobacteriales order (Figure 4b). This latter order is saccharolytic and metabolizes a wide range of carbohydrates to lactate and other metabolites (Gupta, Anish, & Bijendra, 2017). Taken together, we observed distinct and varied bacterial compositions in different ITSs of B. acutorostrata, which could reflect different metabolic responses to different food types.
Figure 4.
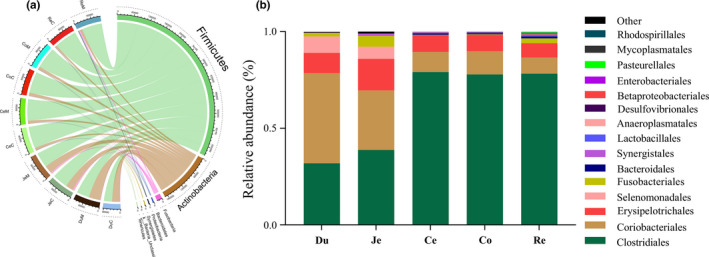
Bacterial distribution at phylum (a) and order (b) levels among different samples. The names of each sample are duodenum (Du), jejunum (Je), cecum (Ce), colon (Co), and rectum (Re) contents(C) or mucosa (M) with the serial number of 1 to 3.
4. CONCLUSIONS
In this study, the gut microbial composition of different ITSs from a minke whale was sequenced and compared. Significant differences were observed in gut microbiota composition across different ITSs; the microbiomes of posterior compartments were more diverse than the foregut. The duodenum and jejunum were enriched for Erysipelotrichales, Selenomonadales, and Coriobacteriales, whereas Clostridiales were more abundant in the large intestine.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTION
Jiashen Tian: Conceptualization (equal); Formal analysis (equal); Funding acquisition (equal); Methodology (equal); Writing‐original draft (equal). Jing Du: Conceptualization (equal); Data curation (equal); Funding acquisition (equal); Investigation (equal); Methodology (equal); Writing‐original draft (equal). Zhichuang Lu: Formal analysis (equal); Software (equal); Writing‐review & editing (equal). Jiabo Han: Conceptualization (equal); Resources (equal); Supervision (equal); Writing‐review & editing (equal). Zhen Wang: Investigation (equal); Methodology (equal); Writing‐original draft (equal). Duohui Li: Formal analysis (equal); Project administration (equal). Xiaoyan Guan: Formal analysis (equal); Project administration (equal). Zhaohui Wang: Investigation (equal).
ETHICS STATEMENT
Species identification and dissection were approved by the Ministry of Agriculture and Rural Affairs of the People's Republic of China (permit number: 1376). This study was conducted under a permit issued by the Liaoning Fisheries Administration Bureau, Liaoning Province, China (approval number: LSYXFZ20111105).
Supporting information
TableS1
ACKNOWLEDGEMENTS
This research was funded by the Foundation of Department of Ocean and Fisheries of Liaoning Province, China, grant number 201812 and 201822. The authors recognize the contribution of Mr. Zelong Zhao from the Dalian University of Technology for his technical assistance with gut microbiota function information of animals.
Tian J, Du J, Lu Z, et al. Distribution of microbiota across different intestinal tract segments of a stranded dwarf minke whale, Balaenoptera acutorostrata . MicrobiologyOpen. 2020;9:e1108 10.1002/mbo3.1108
DATA AVAILABILITY STATEMENT
Raw sequence reads were uploaded to the National Center for Biotechnology Information's Sequence Read Archive under the accession number PRJNA600130: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA600130
REFERENCES
- Allenspach, K. , House, A. , Smith, K. , Mcneill, F. M. , Hendricks, A. , Elson‐Riggins, J. , … Suchodolski, J. S. (2010). Evaluation of mucosal bacteria and histopathology, clinical disease activity and expression of toll‐like receptors in German shepherd dogs with chronic enteropathies. Veterinary Microbiology, 146, 326–335. 10.1016/j.vetmic.2010.05.025 [DOI] [PubMed] [Google Scholar]
- Backhed, F. (2005). Host‐bacterial mutualism in the human intestine. Science, 307, 1915–1920. [DOI] [PubMed] [Google Scholar]
- Caporaso, J. G. , Kuczynski, J. , Stombaugh, J. , Bittinger, K. , Bushman, F. D. , Costello, E. K. , … Knight, R. (2010). QIIME allows analysis of high‐throughput community sequencing data. Nature Methods, 7(5), 335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarello, M. , Villéger, S. , Bouvier, C. , Auguet, J. C. , & Bouvier, T. (2017). Captive bottlenose dolphins and killer whales harbor a species‐specific skin microbiota that varies among individuals. Scientific Reports, 7, 15269 10.1038/s41598-017-15220-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinda, D. , Nakaji, S. , Fukuda, S. , Sakamoto, J. , Shimoyama, T. , Nakamura, T. , … Sugawara, K. (2004). The fermentation of different dietary fibers is associated with fecal clostridia levels in men. Journal of Nutrition, 134, 1881–1886. 10.1093/jn/134.8.1881 [DOI] [PubMed] [Google Scholar]
- Colwell, R. K. (2009). Biodiversity: concepts, patterns and measurement In Levin S. A. (Ed.), The princeton guide to ecology, 2nd ed. (pp. 257–263). Princeton, NJ: Princeton University Press. [Google Scholar]
- Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26, 2460–2461. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- Flint, H. J. , Scott, K. P. , Louis, P. , & Duncan, S. H. (2012). The role of the gut microbiota in nutrition and health. Nature Review Gastroenterology & Hepatology, 9, 577–589. 10.1038/nrgastro.2012.156 [DOI] [PubMed] [Google Scholar]
- Gupta, R. S. , Anish, N. , & Bijendra, K. (2017). Novel molecular, structural and evolutionary characteristics of the phosphoketolases from bifidobacteria and coriobacteriales . PLoS One, 12, e0172176 10.1371/journal.pone.0172176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang, I. , Rinttila, T. , Zentek, J. , Kettunen, A. , Alaja, S. , Apajalahti, J. , … Sprillmann, T. (2012). Effect of high contents of dietary animal‐derived protein or carbohydrates on canine faecal microbiota. BMC Veterinary Research, 8, 90 10.1186/1746-6148-8-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins, R. (2000). Bacteria and fungi of marine mammals: A review. Canadian Veterinary Journal, 41, 105–116. 10.1080/03079450094216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsgård, A. (1982). The food of minke whales (Balaenoptera acutorostrata) in northern North Atlantic waters. Report of the International Whaling Commission, 32, 259–262. [Google Scholar]
- Kemp, P. F. , & Aller, J. Y. (2004). Bacterial diversity in aquatic and other environments: what 16S rDNA libraries can tell us. FEMS Microbiologyl Ecology, 47, 161–177. 10.1016/S0168-6496(03)00257-5 [DOI] [PubMed] [Google Scholar]
- Kisuse, J. , Laongkham, O. , Nakphaichit, M. , Therdtatha, P. , Momoda, R. , Tanaka, M. , … Nakayama, J. (2018). Urban diets linked to gut microbiome and metabolome alterations in children: a comparative cross‐sectional study in Thailand. Frontiers in Microbiology, 9, 1345 10.3389/fmicb.2018.01345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski, M. I. , Schein, J. E. , Birol, I. , Connors, J. , Gascoyne, R. , Horsman, D. , … Marra, M. A. (2009). Circos: An information aesthetic for comparative genomics. Genome Research, 19, 1639–1645. 10.1101/gr.092759.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavery, T. J. , Roudnew, B. , Seymour, J. , Mitchell, J. G. , & Jeffries, T. (2012). High nutrient transport and cycling potential revealed in the microbial metagenome of Australian sea lion (Neophoca cinerea) faeces. PLoS One, 7, e36478 10.1371/journal.pone.0036478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoc, T. , & Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics, 27, 2957–2963. 10.1093/bioinformatics/btr507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmuthuge, N. , Li, M. , Goonewardene, L. A. , Oba, M. , & Guan, L. L. (2013). Effect of calf starter feeding on gut microbial diversity and expression of genes involved in host immune responses and tight junctions in dairy calves during weaning transition. Journal of Dairy Science, 96, 3189–3200. 10.3168/jds.2012-6200 [DOI] [PubMed] [Google Scholar]
- McLaughlin, R. W. , Chen, M. , Zheng, J. , Zhao, Q. , & Wang, D. (2012). Analysis of the bacterial diversity in the fecal material of the endangered Yangtze finless porpoise, Neophocaena phocaenoides asiaeorientalis . Molecular Biology Reports, 39, 5669–5676. 10.1007/s11033-011-1375-0 [DOI] [PubMed] [Google Scholar]
- Moreno, C. E. , & Rodríguez, P. (2010). A consistent terminology for quantifying species diversity? Oecologia, 163, 279–282. 10.1007/s00442-010-1591-7 [DOI] [PubMed] [Google Scholar]
- Numberger, D. , Herlemann, D. P. R. , Jürgens, K. , Dehnhardt, G. , & Schulz‐Vogt, H. (2016). Comparative analysis of the fecal bacterial community of five harbor seals (Phoca vitulina). MicrobiologyOpen, 5, 782–792. 10.1002/mbo3.369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie, L. E. , Steiner, J. M. , & Suchodolski, J. S. (2008). Assessment of microbial diversity along the feline intestinal tract using 16s rRNA gene analysis. FEMS Microbiology Ecology, 66, 590–598. 10.1111/j.1574-6941.2008.00609.x [DOI] [PubMed] [Google Scholar]
- Sanders, J. G. , Beichman, A. C. , Roman, J. , Scott, J. J. , Emerson, D. , Mccarthy, J. J. , & Girguis, P. R. (2015). Baleen whales host a unique gut microbiome with similarities to both carnivores and herbivores. Nature Communications, 6, 8285 10.1038/ncomms9285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchodolski, J. S. , Ruaux, C. G. , Steiner, J. M. , Fetz, K. , & Williams, D. A. (2005). Assessment of the qualitative variation in bacterial microflora among compartments of the intestinal tract of dogs by use of a molecular fingerprinting technique. American Journal of Veterinary Research, 66, 1556–1562. 10.2460/ajvr.2005.66.1556 [DOI] [PubMed] [Google Scholar]
- Walquist, M. J. , Stormo, S. K. , Jensen, I. J. , Bjarne, O. , & Eilertsen, K. E. (2017). Antioxidant and anti‐inflammatory activities in extracts from minke whale (Balaenoptera acutorostrata) blubber. Mediators of Inflammation, 2017, 3835815–. 10.1155/2017/3835851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Zhao, X. W. , Xu, H. C. , Bao, X. Y. , Liu, X. J. , Chang, Y. Q. , & Ding, J. (2018). Characterization of the bacterial community in different parts of the gut of sea cucumber (Apostichopus japonicus) and its variation during gut regeneration. Aquaculture Research, 49, 1987–1996. 10.1111/are.13654 [DOI] [Google Scholar]
- Wang, Q. , Garrity, G. M. , Tiedje, J. M. , & Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology, 73, 5261–5267. 10.1128/aem.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windsland, K. , Lindstrom, U. , Nilssen, K. T. , & Haug, T. (2007). Relative abundance and size composition of prey in the common minke whale diet in selected areas of the northeastern Atlantic during 2000–04. Journal of Cetacean Research & Management, 9, 167–178. [Google Scholar]
- Yilmaz, P. , Parfrey, L. W. , Yarza, P. , Gerken, J. , Pruesse, E. , Quast, C. , … Glöckner, F. O. (2014). The SILVA and "All‐species Living Tree Project (LTP)" taxonomic frameworks. Nucleic Acids Reserach, 42, 643–648. 10.1093/nar/gkt1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TableS1
Data Availability Statement
Raw sequence reads were uploaded to the National Center for Biotechnology Information's Sequence Read Archive under the accession number PRJNA600130: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA600130