

Abstract
Phellinus noxius is a pathogenic fungus that causes brown root rot disease, resulting in a widespread tree and crop mortality in the tropics and subtropics. Early stages of this disease are largely asymptomatic, hindering early diagnosis and effective treatment. We hypothesized that P. noxius infection would alter the rhizosphere microbiome of infected trees, based on which diagnostic biomarkers could be developed. Here, we examined for the first time the bacterial, archaeal, and fungal rhizosphere microbiome in four species of healthy and P. noxius‐infected trees (Ficus microcarpa, Celtis sinensis, Mallotus paniculatus, and Cinnamomum camphora) using high‐throughput amplicon sequencing. Results revealed the dominance of Proteobacteria and Actinobacteria in bacteria, Crenarchaeota and Euryarchaeota in archaea, and Ascomycota and Basidiomycota in fungi. Phellinus noxius infection did not affect the alpha diversity of the bacterial rhizosphere microbiome in all four tree species but affected that of archaea and fungi in a tree species‐dependent manner. Infection with P. noxius only affected the bacterial rhizosphere composition in M. paniculatus but not the other three tree species. By contrast, P. noxius infection affected the composition of the archaeal and fungal rhizosphere microbiome in all four tree species. Collectively, these results suggest that potential diagnostic biomarkers for brown root rot disease are tree species‐specific and should be developed based on different taxonomic groups. Our study has provided insights into the rhizosphere microbiome in healthy and P. noxius‐infected trees and laid a solid foundation for future comprehensive studies.
Phellinus noxius is a pathogenic fungus that causes the devastating brown root rot disease. We examined the bacterial, archaeal, and fungal rhizosphere microbiome in healthy and P. noxius‐infected trees of four species commonly found in Hong Kong using high‐throughput amplicon sequencing. Our results revealed distinct effects of P. noxius infection on the diversity and composition of the rhizosphere microbiome across host tree species, suggesting that potential diagnostic biomarkers for brown root rot disease are tree species‐specific.

1. INTRODUCTION
Brown root rot is a devastating disease prevalent in many tropical and subtropical countries and is caused by the pathogenic white‐rot basidiomycete fungus Phellinus noxius. This fungus has a wide host range of over 260 species of trees (Ann, Chang, & Ko, 2002) and crops (Ann, Lee, & Huang, 1999; Ann, Lee, & Tsai, 1999; Farid, Lee, Maziah, & Patahayah, 2009). It causes irrevocable and fatal damage to the plant hosts by targeting their water‐transport system, culminating in root mortality and compromising stability (Hodges & Tenorio, 1984). The life cycle of P. noxius is similar to other root‐rotting basidiomycetes—a new infection starts from previously infected plants or colonized wood debris, from which the mycelium of P. noxius grows to infect the lateral and taproots of the host tree (Ann et al., 2002).
Phellinus noxius is difficult to eradicate due to its ability to survive on decayed root tissue in the soil for over 10 years (Chang, 1996). Early stages of brown root rot disease are largely asymptomatic, hindering early diagnosis and effective treatment and resulting in a high mortality rate of infected plants. Visible symptoms such as chlorosis and crown dieback can only be seen at the late stages of infections, in which the majority of roots have already been destroyed (Ann et al., 2002; Sahashi, Akisa, Ishihara, Abe, & Morita, 2007). To date, there is no standard curative measure for this disease and most research has been focused on its management, such as the use of biocontrol agents or fumigants (Chang & Chang, 1999; Gohet, Van Canh, Louanchi, & Despreaux, 1991; Prasad & Naik, 2002; Schwarze, Jauss, Spencer, Hallam, & Schubert, 2012). New methods for early diagnosis of the disease are needed.
The rhizosphere microbiome is important to plant health. Microorganisms in the rhizosphere form a symbiotic relationship with the plant hosts, aiding phosphorus and nitrogen uptake, and weathering of minerals, among others (Berendsen, Pieterse, & Bakker, 2012; Trivedi, Van Nostrand, Albrigo, Zhou, & Wang, 2012). Plant exudates can alter the rhizosphere microbiome composition by recruiting specific microorganisms for defense against invasive pathogens (Gu et al., 2016; Pascale, Proietti, Pantelides, & Stringlis, 2020; Wei et al., 2018; Weston et al., 2012; Zhang et al., 2011). These beneficial microbes recruited, for example, Pseudomonas, Bacillus, and Trichoderma, can produce different elicitors and trigger induced systemic resistance (ISR) of the plant hosts via a complex network of defense‐related hormone signaling pathways and thereby making them resistant against pathogenic threats (Pascale et al., 2020). To date, the majority of rhizosphere microbiome studies have focused on healthy plants and mostly on the bacterial communities (e.g. Chaparro, Badri, & Vivanco, 2013; Chapelle, Mendes, Bakker, & Raaijmakers, 2016). Rhizosphere microbiome studies on archaea and fungi are limited. These three kingdoms interact with each other and play important roles in nutrient cycling and soil upkeep, and are therefore important to be studied together (Kirk et al., 2004). Until now, there were no studies of the rhizosphere microbiome concerning P. noxius infection.
In this study, we examined the bacterial, archaeal, and fungal rhizosphere microbiome in healthy and P. noxius‐infected trees of four species commonly found in Hong Kong. Our aims were (a) to characterize and compare the diversity and composition of the rhizosphere microbiome in healthy and P. noxius‐infected trees and (b) to examine whether the changes in the rhizosphere microbiome due to P. noxius infections are consistent across host tree species. We hypothesized that there would be clear differences in the rhizosphere microbiome between trees with different health status.
2. MATERIALS AND METHODS
2.1. Soil sampling
Rhizosphere soil samples of P. noxius‐infected trees belonging to Ficus microcarpa (n = 3), Celtis sinensis (n = 1), Mallotus paniculatus (n = 1), and Cinnamomum camphora (n = 1) were collected around Hong Kong (22°18′N, 114°12′E) (Table 1). These tree species, especially F. microcarpa, are commonly found in Hong Kong and are vulnerable to P. noxius infection. Phellinus noxius‐infected trees were initially identified by visual symptoms of chlorosis, crown dieback, the presence of basidiocarps, and the characteristic brown webbing throughout the roots after they were cut open. Rhizosphere samples were collected in triplicate around each tree, 5 cm below the soil surface to avoid surface contamination. Large roots were exposed carefully and soil attached to the roots was sampled with a small shovel. For each sample, a small portion of roots was also taken back to the laboratory for confirmatory tests of P. noxius infection. Rhizosphere samples were also collected from one healthy tree for each of the four tree species from proximal areas for comparison purposes. In total, 30 rhizosphere soil samples were examined in this study. In the laboratory, ~2 g of soil subsamples from each sample were oven‐dried at 105 °C overnight and then used for total carbon and total nitrogen content analysis in triplicate on the vario MICRO cube elemental analyzer (Elementar, Langenselbold, Hesse, Germany).
TABLE 1.
Collection locations and physicochemical parameters of rhizosphere samples of healthy and Phellinus noxius‐infected trees
| Tree species | Health status | Collection location in Hong Kong | Total C (%) | Total N (%) |
|---|---|---|---|---|
| Mallotus paniculatus | Diseased | Sau Nga Road Playground (22°19′19″N, 114°13′37″E) | 1.00 ± 0.08 | 0.07 ± 0.02 |
| Mallotus paniculatus | Healthy | Sau Nga Road Playground (22°19′19″N, 114°13′37″E) | 1.59 ± 0.54 | 0.12 ± 0.06 |
| Celtis sinensis | Diseased | King's Park (22°18′45″N, 114°10′27″E) | 2.59 ± 1.75 | 0.23 ± 0.16 |
| Celtis sinensis | Healthy | King's Park (22°18′45″N, 114°10′27″E) | 1.17 ± 0.48 | 0.07 ± 0.03 |
| Cinnamomum camphora | Diseased | The Chinese University of Hong Kong (22°25′10″N, 114°12′24″E) | 1.21 ± 0.02 | 0.09 ± 0.02 |
| Cinnamomum camphora | Healthy | The Chinese University of Hong Kong (22°25′10″N, 114°12′24″E) | 1.58 ± 0.43 | 0.11 ± 0.04 |
| Ficus microcarpa | Diseased | Kowloon Park (22°17′58″N, 114°10′14″E) | 1.54 ± 0.61 | 0.12 ± 0.05 |
| Ficus microcarpa | Diseased | Kowloon Park (22°17′58″N, 114°10′14″E) | 1.62 ± 0.52 | 0.12 ± 0.04 |
| Ficus microcarpa | Diseased | Kowloon Park (22°17′58″N, 114°10′14″E) | 1.31 ± 0.20 | 0.08 ± 0.01 |
| Ficus microcarpa | Healthy | Kowloon Park (22°17′58″N, 114°10′14″E) | 2.51 ± 1.27 | 0.15 ± 0.07 |
The values of total carbon and total nitrogen contents are in mean ± standard deviation.
2.2. Phellinus noxius infection confirmation
Portions of tree roots were washed in distilled water, placed on 2% malt extract agar amended with gallic acid, streptomycin, benomyl, and dichloran (Chang, 1995), and incubated in the dark at 28°C. Pure cultures were then obtained and DNA extracted using DNeasy Plant Mini Kit (Qiagen, Germantown, MD, USA) following the manufacturer's instructions. PCR amplifications were performed using two P. noxius‐specific primer sets: G1F (5′‐GCC CTT TCC TCC GCT TAT TG‐3′) and G1R (5′‐ CTT GAT GCT GGT GGG TCT CT‐3′) (Wu et al., 2009), and PN‐1F (5′‐AGT TTG CGC TCA TCC ATC TC‐3′) and PN‐2R (5′‐AGCCGACTTACGCCAGCAG‐3′) (Tsai, Hsieh, Ann, & Yang, 2007). Each of the 25 μl PCR mixture consisted of 1× of Green GoTaq Reaction Buffer, 0.8 mM of dNTP, 0.5 μM of primers, 1.25 U of GoTaq DNA Polymerase, and ~0.1 μg of DNA. The PCR regime contained an initial denaturation at 95°C for 2 min, followed by 30 cycles of denaturation at 95°C for 1 min, annealing at 55°C for 30 s and extension at 72°C for 30 s, and a final extension step at 72°C for 5 min. PCR products were then visualized with electrophoresis on 1.5% agarose gels.
2.3. DNA extraction, PCR, and amplicon sequencing
Total DNA was extracted from the rhizosphere samples using MoBio PowerSoil DNA Extraction Kit (MO BIO Laboratories, Carlsbad, CA, USA) according to the manufacturer's instructions. The procedure was slightly modified with the additional use of TissueLyser (Qiagen, Germantown, MD, USA) at 30 Hz for 1 min per side to improve cell lysis (Cheung, Wong, Chu, & Kwan, 2018). The V1‐V2 hypervariable region of the bacterial 16S rRNA gene was amplified using primers 28F (5′‐GAG TTT GAT CNT GGC TCA G‐3′) (Handl, Dowd, Garcia‐Mazcorro, Steiner, & Suchodolski, 2011) and 338R (5′‐GCT GCC TCC CGT AGG AGT‐3′) (Amann, Ludwig, & Schleifer, 1995). For archaea, the V1‐V2 region of the 16S rRNA gene was amplified using primers 21F (5′‐TTC CGG TTG ATC CYG CCG GA‐3′) (DeLong, 1992) and Pro341R (5′‐CTG STG CVN CCC GTA GG‐3′) (Takahashi et al., 2014). For fungi, the internal transcribed spacer 1 (ITS1) region was amplified using primers ITS1F (5′‐CTT GGT CAT TTA GAG GAA GTA A‐3′) (Gardes & Bruns, 1993) and ITS2R (5′‐ GCT GCG TTC TTC ATC GAT GC‐3′) (White, Bruns, Lee, & Taylor, 1990). Each PCRmixture consisted of 1× Phusion HF Buffer, 0.5 μM of primers, 200 μM of dNTPs, 1 U of Phusion High‐Fidelity DNA polymerase (New England Biolabs, Ipswich, MA, USA), and 50 ng of DNA. Bacterial 16S rRNA amplifications were performed with an initial denaturation of 98°C for 3 min, followed by 33 cycles of denaturation at 98°C for 10 s, annealing at 61°C for 30 s and extension at 72°C for 30 s, and a final extension step at 72°C for 10 min. For archaea, an annealing temperature of 67°C was used, and for fungi, an annealing temperature of 55°C with 35 cycles was used. PCR products were quality‐checked on 1.5% agarose gels, purified with QIAquick Gel Extraction Kit (Qiagen, Germantown, MD, USA) and sequenced from the forward primer end on an Ion Torrent PGM (318 chip v2) at the Core Facilities of The Chinese University of Hong Kong.
2.4. Sequence analysis
Raw sequence reads were demultiplexed, filtered for quality, and analyzed using QIIME 1.9.1 (Caporaso et al., 2010) as previously described (Cheung et al., 2015). Chimeric sequences were identified and removed using USEARCH 6.1 (Edgar, 2010) against the “Gold” reference dataset for bacteria and archaea, and against the UNITE dynamic ITS1 reference dataset (2016–01–01 release) (Kõljalg et al., 2013) for fungi. Sequence reads from the same kingdom were clustered into operational taxonomic units (OTUs) at 97% similarity using uclust with the open‐reference OTU picking method. Representative OTUs were aligned to the Greengenes reference dataset (13_8 release) (DeSantis et al., 2006) for bacteria and archaea, and the UNITE dynamic reference dataset (2016–11–20 release) for fungi, and taxonomically assigned using the RDP naïve Bayesian Classifier (Wang, Garrity, Tiedje, & Cole, 2007). Sequence reads of plant origin were removed from further analysis.
2.5. Statistical analysis
Before diversity analyses, the bacterial, archaeal, and fungal sequence datasets were rarefied to 13,055, 13,821, and 16,386 reads, respectively. Alpha diversity was estimated with the Shannon index and the number of observed OTUs. Beta diversity was estimated using principal coordinate analysis (PCoA) with the unweighted UniFrac distance for bacteria and archaea and the Bray–Curtis dissimilarity for fungi. All the above statistical tests were conducted using scripts in QIIME. Linear regression analysis was performed using GraphPad Prism 8 (GraphPad Software Inc., San Diego, CA). Differences were considered to be statistically significant when p < 0.05.
3. RESULTS
3.1. Physicochemical parameters
All rhizosphere samples shared a similar total carbon and total nitrogen contents among tree species and between healthy and diseased samples of the same species (Table 1).
3.2. Alpha diversity
After quality filtering, a total of 710,349 bacterial 16S rRNA reads (13,055–32,347 per sample), 1,327,491 archaeal 16S rRNA reads (13,821–109,396 per sample), and 1,343,825 fungal ITS reads (16,386–101,826 per sample) were obtained for the 30 rhizosphere soil samples. The Shannon diversity and the number of observed OTUs calculated ranged, respectively, from 11.28–11.85 and 5858–6959 for bacteria, 4.72–6.16, and 384–608 for archaea, and 3.49–5.34 and 550–844 for fungi (Table 2). There was no obvious difference in the Shannon diversity of bacteria between healthy and P. noxius‐infected trees for all tree species. The archaeal Shannon diversity was also similar between healthy and diseased samples of C. sinensis and F. microcarpa but was higher in diseased samples of M. paniculatus and C. camphora. The fungal Shannon diversity was lower in diseased samples of all tree species apart from F. microcarpa.
TABLE 2.
Alpha diversity of the bacterial, archaeal, and fungal rhizosphere microbiome in healthy and Phellinus noxius‐infected trees
| Tree species | Health status | Bacteria | Archaea | Fungi | |||
|---|---|---|---|---|---|---|---|
| Shannon | Observed OTUs | Shannon | Observed OTUs | Shannon | Observed OTUs | ||
| Mallotus paniculatus | Healthy | 11.28 ± 0.23 | 5857.56 ± 49.65 | 5.44 ± 0.48 | 517.63 ± 44.81 | 5.14 ± 0.91 | 646.40 ± 91.20 |
| Mallotus paniculatus | Diseased | 11.74 ± 0.12 | 6367.70 ± 309.14 | 6.16 ± 0.51 | 440.67 ± 71.72 | 3.83 ± 0.84 | 616.23 ± 77.70 |
| Celtis sinensis | Healthy | 11.85 ± 0.03 | 6864.50 ± 104.34 | 5.23 ± 0.59 | 607.97 ± 182.90 | 5.34 ± 0.23 | 756.47 ± 98.02 |
| Celtis sinensis | Diseased | 11.60 ± 0.02 | 6439.00 ± 54.64 | 4.86 ± 0.15 | 442.67 ± 73.38 | 3.79 ± 0.59 | 695.27 ± 52.40 |
| Ficus microcarpa | Healthy | 11.83 ± 0.06 | 6862.00 ± 112.48 | 4.99 ± 0.89 | 383.50 ± 29.50 | 3.55 ± 0.63 | 621.46 ± 92.84 |
| Ficus microcarpa | Diseased | 11.77 ± 0.06 | 6857.81 ± 304.80 | 4.72 ± 0.10 | 445.33 ± 164.08 | 3.49 ± 0.58 | 549.50 ± 108.00 |
| Cinnamomum camphora | Healthy | 11.83 ± 0.05 | 6958.77 ± 99.03 | 4.82 ± 0.14 | 446.33 ± 5.25 | 5.24 ± 0.17 | 844.30 ± 1.10 |
| Cinnamomum camphora | Diseased | 11.70 ± 0.05 | 6651.20 ± 69.00 | 6.16 ± 0.35 | 573.63 ± 165.30 | 4.79 ± 0.13 | 738.30 ± 32.72 |
Values are in mean ± standard deviation.
3.3. Beta diversity
PCoA revealed that the rhizosphere microbiome structure of samples collected around the same tree were in general similar to each other (Figure 1). For bacteria, samples from the same tree species clustered together, except for M. paniculatus (Figure 1a). Samples from healthy and P. noxius‐infected trees also formed distinct clusters for all tree species apart from F. microcarpa. For archaea, PCoA revealed distinct clusters for M. paniculatus and C. camphora, and between healthy and diseased samples of M. paniculatus, C. sinensis, and C. camphora (Figure 1b). For fungi, no clear clusters were formed according to tree species, and healthy and diseased samples were only separated in C. sinensis (Figure 1c).
FIGURE 1.

Principal coordinate analysis plots of the bacterial (a), archaeal (b), and fungal (c) rhizosphere microbiome
3.4. Taxonomic composition in healthy trees
For bacteria, the eight major (>1% average relative abundance) phyla recovered were Proteobacteria (36%), Actinobacteria (22%), Acidobacteria (12%), Planctomycetes (6%), Chloroflexi (6%), Bacteroidetes (3%), Gemmatimonadetes (3%), and Nitrospirae (2%), which together accounted for ~90% of all bacterial 16S rRNA reads (Figure 2a). All four tree species were dominated by Proteobacteria, Acidobacteria, and Actinobacteria. At the order level, all four tree species were dominated by Rhizobiales, Rhodospirillales, Actinomycetales, and Gaiellales (Figure 2b). Besides, there was an abundant amount of Acidobacteriales and Ellin6513 in M. paniculatus; iii1‐15 in C. sinensis; and iii1‐15 and Solirubrobacterales in C. camphora and F. microcarpa. At the family level, all four tree species were dominated by Hyphomicrobiaceae, Gaiellaceae, and Rhodospirillaceae (Figure 2c). Besides, there was an abundant amount of Koribacteraceae in M. paniculatus.
FIGURE 2.
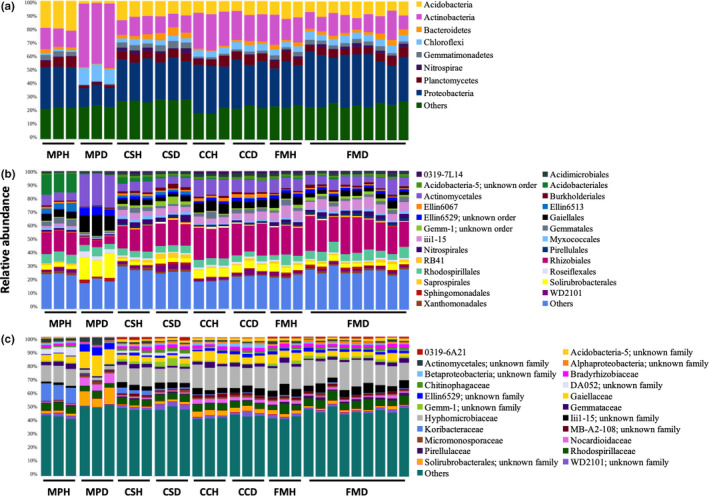
The relative abundance of dominant (>1%) bacterial phyla (a), orders (b), and families (c) in the rhizosphere microbiome of four species of trees. MP: Mallotus paniculatus, CS: Celtis sinensis, CC: Cinnamomum camphora, FM: Ficus microcarpa; H: healthy, D: diseased
For archaea, all four tree species were dominated by Crenarchaeota and Euryarchaeota at the phylum level (Figure 3a). At the order level, M. paniculatus was dominated by NRP‐J, whereas C. sinensis, C. camphora, and F. microcarpa were dominated by Nitrososphaerales (Figure 3b). Besides, Cenarchaeales was dominant in M. paniculatus and C. sinensis. At the family level, all tree species apart from M. paniculatus were dominated by Nitrososphaeraceae (Figure 3c). Besides, SAGMA‐X was dominant in M. paniculatus and C. sinensis.
FIGURE 3.

The relative abundance of dominant (>1%) archaeal phyla (a), orders (b), and families (c) in the rhizosphere microbiome of four species of trees. MP: Mallotus paniculatus, CS: Celtis sinensis, CC: Cinnamomum camphora, FM: Ficus microcarpa; H: healthy, D: diseased
Over 65% of the fungal ITS reads could not be identified to a known fungal phylum. Ascomycota and Basidiomycota were the most abundant fungal phyla for all four tree species (Figure 4a). At the class level, M. paniculatus was dominated by Sordariomycetes, whereas C. camphora and F. microcarpa were dominated by Dothideomycetes (Figure 4b). Besides, C. sinensis and F. microcarpa contained an abundant amount of Agaricomycetes. At the genus level, M. paniculatus was dominated by Fusarium and Pochonia, whereas C. sinensis, C. camphora, and F. microcarpa were dominated by Candida, Archaeorhizomyces, and Phoma, respectively (Figure 4c).
FIGURE 4.
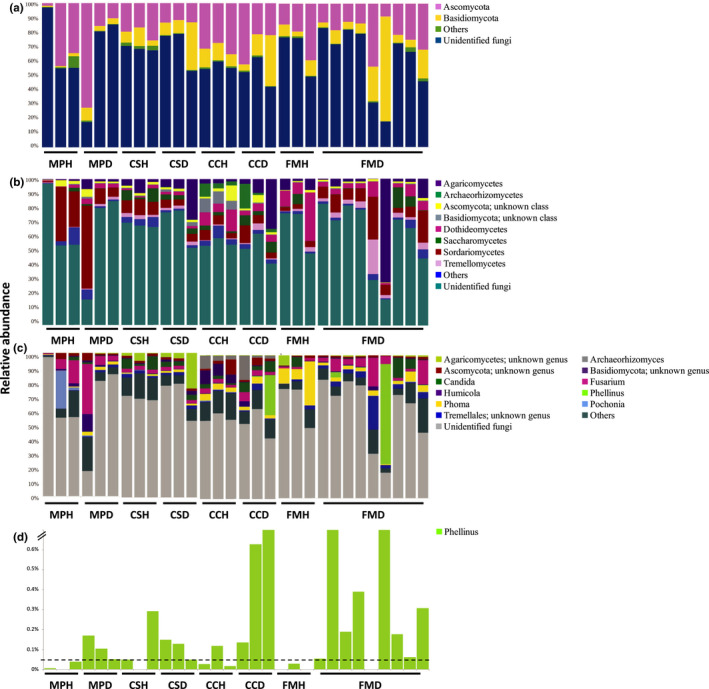
The relative abundance of dominant (>1%) fungal phyla (a), classes (b), and genera (c) as well as the Phellinus genus (d) in the rhizosphere microbiome of four species of trees. MP: Mallotus paniculatus, CS: Celtis sinensis, CC: Cinnamomum camphora, FM: Ficus microcarpa; H: healthy, D: diseased. The horizontal dotted line in panel (d) denotes the relative abundance at 0.05%
3.5. Taxonomic composition in Phellinus noxius‐infected trees
There was no obvious difference in the taxonomic composition of major bacterial phyla, orders, or families between healthy and P. noxius‐infected trees of C. sinensis, C. camphora, and F. microcarpa (Figure 2). By contrast, in P. noxius‐infected trees of M. paniculatus, there was more Actinobacteria and Chloroflexi and fewer Acidobacteria, Proteobacteria, and Planctomycetes at the phylum level (Figure 2a); more Actinomycetales, Gaiellales, and Solirubrobacterales and fewer Acidobacteriales, Ellin6513, Gemmatales, and Rhizobiales at the order level (Figure 2b); and more Gaiellaceae and Nocardioidaceae and fewer Gemmataceae and Hyphomicrobiaceae at the family level (Figure 2c).
For archaea, there was no obvious difference in the taxonomic composition of major phyla between healthy and P. noxius‐infected trees of C. sinensis and F. microcarpa (Figure 3a). By contrast, there was more Euryarchaeota and fewer Crenarchaeota in P. noxius‐infected trees of M. paniculatus and C. camphora. At the order level, there was more E2, B10, and pGrfC26, and fewer Cenarchaeales, NPR‐J in diseased trees of M. paniculatus; fewer Cenarchaeales and NRP‐J in diseased trees of C. sinensis; more pGrfC26 and fewer Nitrososphaerales in diseased trees of C. camphora; and more pGrfC26 in diseased trees of F. microcarpa (Figure 3b). At the family level, there was more DHVEG‐1 and fewer SAGMA‐X in diseased trees of M. paniculatus; fewer SAGMA‐X in diseased trees of C. sinensis; and more DHVEG‐1 and fewer Nitrososphaeraceae in diseased trees of C. camphora (Figure 3c).
For fungi, there was more Basidiomycota in diseased trees of M. paniculatus and fewer Ascomycota in diseased trees of C. sinensis (Figure 4a). At the class level, there was more Agaricomycetes and Dothideomycetes in diseased trees of M. paniculatus; more Agaricomycetes and fewer Sordariomycetes in diseased trees of C. sinensis; more Agaricomycetes and Saccharomycetes and fewer Dothideomycetes in diseased trees of C. camphora; and more Sordariomycetes and Tremellomycetes and fewer Dothideomycetes in diseased trees of F. microcarpa (Figure 4b). At the genus level, there was more Phoma and fewer Pochonia in diseased trees of M. paniculatus, and more Fusarium and fewer Phoma in diseased trees of F. microcarpa (Figure 4c). Moreover, sequences from the Phellinus genus were detected in all except one P. noxius‐infected rhizosphere samples, with a relative abundance ranging from 0.05% to 70.1% (Figure 4c,d). By contrast, most healthy samples contained <0.05% of Phellinus reads. Nonetheless, it should be reminded that all trees in this study were selected according to visible symptoms, with the health status of diseased trees further confirmed with P. noxius‐specific PCRs (Figure A1).
Linear regression analysis revealed no significant correlation between the relative abundance of the Phellinus genus and the dominant bacterial families Hyphomicrobiaceae, Gaiellaceae, and Rhodospirillaceae, the dominant archaeal families Nitrososphaeraceae, DHVEG‐1, and SAGMA‐X, or the dominant fungal genera Fusarium, Phoma, and Candida (p > 0.05, F test) (Figure A2).
4. DISCUSSION
In this study, we have examined for the first time the effects of P. noxius on the structure and diversity of the bacterial, archaeal, and fungal rhizosphere microbiome in trees. Until now, only a few rhizosphere microbiome studies have simultaneously investigated these three taxonomic groups, and the majority of them have studied only the healthy rhizosphere of trees (Uroz et al., 2016; Veach et al., 2019). By contrast, studies involving diseased samples usually focus on agricultural crop plants (Filion, Hamelin, Bernier, & St‐Arnaud, 2004; Han et al., 2017; Li, Ren, Jia, & Dong, 2014; Wei et al., 2018; Zhang et al., 2011), but none of them has examined the archaeal communities. Information on the rhizosphere microbiome across multiple kingdoms is essential due to their integral roles in nutrient cycling and the ability to maintain symbiotic and antagonistic relationships with the plant hosts. Our results have demonstrated that P. noxius can alter the rhizosphere microbiome of healthy trees but the effects depend on the species of trees. However, a major limitation of this study is the lack of true biological replicates for each tree species, which has hindered statistical testing on the differences observed.
All rhizosphere samples examined in this study were abundant in Proteobacteria. This is not unexpected as members of this bacterial phylum are fast‐growing (Fierer, Bradford, & Jackson, 2007). Similar results have also been reported in other soil microbiome studies of trees (Uroz et al., 2016) and crops (Fu et al., 2017; Li et al., 2014). Comparing healthy and diseased samples in all four tree species together, no clear clustering between samples of healthy and P. noxius‐infected trees was observed. This suggests that the effect of host tree species on the rhizosphere composition is larger than that resulting from P. noxius infection. Comparing healthy and diseased samples in each tree species separately, only M. paniculatus showed compositional changes in the bacterial microbiome; M. paniculatus and C. camphora showed changes in the archaeal microbiome, and C. camphora and F. microcarpa showed changes in the fungal microbiome. The profiles of all three kingdoms were not different between healthy and diseased samples of C. sinensis. This suggests that the effects of P. noxius infection on the rhizosphere microbiome vary across kingdoms and depend on the host tree species. Notably, members from the phylum Chloroflexi were more abundant in diseased samples of M. paniculatus (Figure 2a). This could represent a direct response to the invasion as Chloroflexi members are often found in disease suppressive soils and have been suggested to be part of the host plant defense system due to their ability to prevent iron uptake and root colonization by fungal plant pathogens (Lemanceau & Alabouvette, 1993; Liu et al., 2016; Rodriguez & Fraga, 1999). However, it is also possible that some Chloroflexi members are opportunistic pathogens that were enriched in the diseased samples. The archaeal order E2 was enriched in the diseased samples of M. paniculatus (Figure 3b). E2 comprises uncultured members of potential methanogens and its enrichment in the diseased samples might be related to an increase of detritus and carbon sources resulting from root decay (Gannes, Eudoxie, Bekele, & Hickey, 2015; Iino et al., 2013). For fungi, the genus Fusarium was enriched in the diseased samples of F. microcarpa (Figure 4c). Fusarium comprises plant growth‐promoting members that can trigger ISR for enhancing the defense of the plant host upon pathogen attack (Pascale et al., 2020).
Infection with P. noxius did not affect the bacterial diversity in the rhizosphere of all four tree species examined here. This differs from other studies on diseased microbiomes in crops (Han et al., 2017; Li et al., 2014; Shang et al., 2016; Wei et al., 2018), tree seedlings (Filion et al., 2004), and shrubs (Zhang et al., 2011). For example, wilted Lanzhou Lily has a higher rhizosphere bacterial diversity (Shang et al., 2016), whereas diseased black spruce (Picea mariana) seedlings (Filion et al., 2004) and cotton plants (Zhang et al., 2011) have a lower rhizosphere bacterial diversity. This suggests that, in contrast to the general belief that microbial diversity can act as a biomarker for plant health (Berg et al., 2017), for P. noxius infection, the rhizosphere bacterial diversity is not a reliable indicator of plant health. Moreover, the fact that the effects of P. noxius infection on the archaeal and fungal diversity are different across host tree species suggests that the diversity of different microbial taxonomic groups should be examined for different host tree species to indicate the status of P. noxius infection.
Differences in the microbiome composition between healthy and diseased rhizospheres have been reported in some previous studies of crop plants (Li et al., 2014; Shang et al., 2016; Wei et al., 2018). However, no obvious differences are observed for most tree species in this study. The differences in responses could be explained by species‐dependent microbial recruitment (Turner et al., 2013) or disparities in the stages of infection (Zhang et al., 2011). A previous study on apple replant disease also shows no changes in the bacterial and fungal rhizosphere microbiome in diseased apple trees (Jiang et al., 2017). A possible reason for the lack of changes in some of our samples is the physical difference between trees examined in this study and plants in previous ones. Compared to smaller crop plants that have a simple root system, all trees examined here are over seven meters tall with an extensive root system, comprising both fresh and decaying roots even when the infection is serious. Among the four tree species examined here, M. paniculatus showed the most significant changes in its bacterial and archaeal rhizosphere communities. The trunk diameter, height, and root radius of M. paniculatus were the smallest among all four tree species examined in this study. The smallest and simplest root system of M. paniculatus might explain why P. noxius infection affected its rhizosphere microbiome more than the other three tree species. In particular, all F. microcarpa trees examined in this study are with a diameter at breast height (DBH) of >1000 mm (Leisure & Cultural Services Department, 2016). Ficus microcarpa is known for its large root network and abundance of aerial roots, which constantly produce fresh roots (Zimmerman, Wardrop, & Tomlinson, 1968). Therefore, P. noxius infection may exert a smaller effect on its rhizosphere microbiome as roots unaffected by the infection may produce exudates that diffuse to the nearby rhizosphere of the affected roots. Nonetheless, further studies are needed to understand more clearly how the tree rhizosphere microbiome changes when a pathogen invades.
In conclusion, we have examined here the effects of P. noxius on the diversity and composition of the bacterial, archaeal, and fungal rhizosphere microbiomes in four tree species. We showed that P. noxius infection did not affect the alpha diversity of the bacterial rhizosphere microbiome in all four tree species but affected that of archaea and fungi in a tree species‐dependent manner. The effects of P. noxius infection on the composition of the three kingdoms also varied with host tree species. Our study has provided insights into the rhizosphere microbiome in healthy and P. noxius‐infected trees and showed that potential diagnostic biomarkers for brown root rot disease are likely tree species‐specific. Future studies employing more host tree species and a larger sample size per species should revisit these preliminary findings with sophisticated statistical tests. With this, we anticipate the development of rhizosphere microbiome‐based diagnostic biomarkers to facilitate early detection of the devastating brown root rot disease in the near future.
CONFLICT OF INTERESTS
None declared.
AUTHOR CONTRIBUTIONS
Karen Sze Wing Tsang: Formal analysis (lead); funding acquisition (lead); investigation (lead); methodology (lead); visualization (equal); writing – original draft (equal). Man Kit Cheung: Funding acquisition (supporting); visualization (equal); writing – original draft (equal); writing – review & editing (equal). Regent Yau Ching Lam: Investigation (supporting); resources (supporting). Hoi Shan Kwan: Conceptualization (lead); funding acquisition (supporting); methodology (supporting); resources (lead); supervision (lead); writing – review & editing (equal).
ETHICS STATEMENT
None required.
ACKNOWLEDGMENTS
We would like to thank Mr. Tsz Kai Li and Miss. Lei Xing for their help with Ion Torrent sequencing and Mr. Mike Wing Keung Leung for providing advice on the tree locations. This work was supported by a research grant from the Environment and Conservation Fund (Project No: 27/2013) of the Hong Kong SAR Government.
Appendix A.
FIGURE A1.

Gel photographs of PCR products amplified using Phellinus noxius‐specific primer sets G1F/G1R (a) and PN‐1F/PN‐2R (b) on pure cultures obtained from the roots of Phellinus noxius‐infected trees. L: ladder, 1 to 3: Ficus microcarpa, 4: Mallotus paniculatus, 5: Cinnamomum camphora, and 6: Celtis sinensis. The expected amplicon size is indicated with arrows
FIGURE A2.

Correlation between the relative abundance of the Phellinus genus and the bacterial families Hyphomicrobiaceae (a), Gaiellaceae (b), and Rhodospirillaceae (c), the archaeal families Nitrososphaeraceae (d), DHVEG‐1 (e), and SAGMA‐X (f), and the fungal genera Fusarium (g), Phoma (h), and Candida (i). Samples with a relative abundance of Phellinus >3% were regarded as outliers and excluded from the analysis
Tsang KS, Cheung MK, Lam RY, Kwan HS. A preliminary examination of the bacterial, archaeal, and fungal rhizosphere microbiome in healthy and Phellinus noxius‐infected trees. MicrobiologyOpen. 2020;9:e1115 10.1002/mbo3.1115
Karen Sze Wing Tsang and Man Kit Cheung contributed equally to this work.
DATA AVAILABILITY STATEMENT
Raw sequence reads generated in this study are available in the NCBI Sequence Read Archive (SRA) under the accession PRJNA433610. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA433610.
REFERENCES
- Amann, R. I. , Ludwig, W. , & Schleifer, K. H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiological Reviews, 59(1), 143–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ann, P. J. , Chang, T. T. , & Ko, W. H. (2002). Phellinus noxius brown root rot of fruit and ornamental trees in Taiwan. Plant Disease, 86(8), 820–826. [DOI] [PubMed] [Google Scholar]
- Ann, P. J. , Lee, H. L. , & Huang, T. C. (1999). Brown root rot of 10 species of fruit trees caused by Phellinus noxius in Taiwan. Plant Disease, 83(8), 746–750. [DOI] [PubMed] [Google Scholar]
- Ann, P. J. , Lee, H. L. , & Tsai, J. N. (1999). Survey of brown root disease of fruit and ornamental trees caused by Phellinus noxius in Taiwan. Plant Pathology Bulletin, 8(2), 51–60. [Google Scholar]
- Berendsen, R. , Pieterse, C. , & Bakker, P. (2012). The rhizosphere microbiome and plant health. Trends in Plant Science, 17(8), 478–486. [DOI] [PubMed] [Google Scholar]
- Berg, G. , Köberl, M. , Rybakova, D. , Müller, H. , Grosch, R. , & Smalla, K. (2017). Plant microbial diversity is suggested as the key to future biocontrol and health trends. FEMS Microbiology Ecology, 93(5), fix050 10.1093/femsec/fix050 [DOI] [PubMed] [Google Scholar]
- Caporaso, J. G. , Kuczynski, J. , Stombaugh, J. , Bittinger, K. , Bushman, F. D. , Costello, E. K. , … Knight, R. (2010). QIIME allows analysis of high-throughput community sequencing data. Nature Methods, 7(5), 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, T. T. (1995). A selective medium for Phellinus noxius . European Journal of Forest Pathology, 25(4), 185–190. [Google Scholar]
- Chang, T. T. (1996). Survival of Phellinus noxius in soil and in the roots of dead host plants. Phytopathology, 86(3), 272–276. [Google Scholar]
- Chang, T. T. , & Chang, R. J. (1999). Generation of volatile ammonia from urea fungicidal to Phellinus noxius in infest wood in soil under controlled conditions. Plant Pathology, 48(3), 337–344. [Google Scholar]
- Chaparro, J. M. , Badri, D. V. , & Vivanco, J. M. (2013). Rhizosphere microbiome assemblage is affected by plant development. ISME Journal, 8(4), 790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapelle, E. , Mendes, R. , Bakker, P. A. H. M. , & Raaijmakers, J. M. (2016). Fungal invasion of the rhizosphere microbiome. ISME Journal, 10(1), 265–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung, M. K. , Wong, C. K. , Chu, K. H. , & Kwan, H. S. (2018). Community structure, dynamics and interactions of bacteria, archaea and fungi in subtropical coastal wetland sediments. Scientific Reports, 8(1), 14397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung, M. K. , Yip, H. Y. , Nong, W. , Law, P. T. , Chu, K. H. , Kwan, H. S. , & Hui, J. H. (2015). Rapid change of microbiota diversity in the gut but not the hepatopancreas during gonadal development of the new shrimp model Neocaridina denticulata . Marine Biotechnology (NY), 17(6), 811–819. [DOI] [PubMed] [Google Scholar]
- de Gannes, V. , Eudoxie, G. , Bekele, I. , & Hickey, W. (2015). Relations of microbiome characteristics to edaphic properties of tropical soils from Trinidad. Frontiers in Microbiology, 6, 1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong, E. F. (1992). Archaea in coastal marine environments. Proceedings of the National Academy of Sciences of the United States of America, 89(12), 5685–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis, T. Z. , Hugenholtz, P. , Larsen, N. , Rojas, M. , Brodie, E. L. , Keller, K. , … Andersen, G. L. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and Environmental Microbiology, 72(7), 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26(19), 2460–2461. [DOI] [PubMed] [Google Scholar]
- Farid, A. M. , Lee, S. S. , Maziah, Z. , & Patahayah, M. (2009). Pathogenicity of Rigidoporus microporus, and Phellinus noxius against four major plantation tree species in peninsular Malaysia. Journal of Tropical Forest Science, 21(4), 289–298. [Google Scholar]
- Fierer, N. , Bradford, M. A. , & Jackson, R. B. (2007). Toward an ecological classification of soil bacteria. Ecology, 88(6), 1354–1364. [DOI] [PubMed] [Google Scholar]
- Filion, M. , Hamelin, R. C. , Bernier, L. , & St-Arnaud, M. (2004). Molecular profiling of rhizosphere microbial communities associated with healthy and diseased black spruce (Picea mariana) seedlings grown in a nursery. Applied and Environment Microbiology, 70(6), 3541–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, L. , Benton, C. R. , Ruan, Y. , Shen, Z. , Xue, C. , & Rong, L. (2017). Inducing the rhizosphere microbiome by biofertilizer application to suppress banana Fusarium wilt disease. Soil Biology & Biochemistry, 104, 39–48. [Google Scholar]
- Gardes, M. , & Bruns, T. D. (1993). ITS primers with enhanced specificity for basidiomycetes –Application to the identification of mycorrhizae and rusts. Molecular Ecology, 2(2), 113–118. [DOI] [PubMed] [Google Scholar]
- Gohet, E. , Van Canh, T. , Louanchi, M. , & Despreaux, D. (1991). New developments in chemical control of white root disease of Hevea brasiliensis in Africa. Crop Protection, 10(3), 234–238. [Google Scholar]
- Gu, Y. , Wei, Z. , Wang, X. , Friman, V. , Huang, J. , Wang, X. , … Wei, Z. (2016). Pathogen invasion indirectly changes the composition of soil microbiome via shifts in root exudation profile. Biology and Fertility of Soils, 52(7), 997–1005. [Google Scholar]
- Han, Y. , Xu, L. , Liu, L. , Yi, M. , Guo, E. , Zhang, A. , & Yi, H. (2017). Illumina sequencing reveals a rhizosphere bacterial community associated with foxtail millet smut disease suppression. Plant and Soil, 410, 411–421. [Google Scholar]
- Handl, S. , Dowd, S. E. , Garcia-Mazcorro, J. F. , Steiner, J. M. , & Suchodolski, J. S. (2011). Massive parallel 16S rRNA gene pyrosequencing reveals highly diverse fecal bacterial and fungal communities in healthy dogs and cats. FEMS Microbiology Ecology, 76(2), 301–310. [DOI] [PubMed] [Google Scholar]
- Hodges, C. S. , & Tenorio, J. A. (1984). Root disease of Delonix regia and associated tree species in the Marina Islands caused by Phellinus noxius . Plant Disease, 68(4), 334–336. [Google Scholar]
- Iino, T. , Tamaki, H. , Tamazawa, S. , Ueno, Y. , Ohkuma, M. , Suzuki, K. , … Haruta, S. (2013). Candidatus Methanogranum caenicola: A novel methanogen from the anaerobic digested sludge, and proposal of Methanomassiliicoccaceae fam Nov and Methanomassiliicoccales ord Nov, for a methanogenic lineage of the class thermoplasmata. Microbes and Environments, 28(2), 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, J. , Song, Z. , Yang, X. , Mao, Z. , Nie, X. , Guo, H. , & Peng, X. (2017). Microbial community analysis of apple rhizosphere around Bohai Gulf. Scientific Reports, 7, 8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk, J. L. , Beaudette, L. A. , Hart, M. , Moutoglis, P. , Klironomos, J. N. , Lee, H. , & Trevors, J. T. (2004). Methods of studying soil microbial diversity. Journal of Microbiological Methods, 58(2), 169–188. [DOI] [PubMed] [Google Scholar]
- Kõljalg, U. , Nilsson, R. H. , Abarenkov, K. , Tedersoo, L. , Taylor, A. F. , Bahram, M. , … Larsson, K. H. (2013). Towards a unified paradigm for sequence-based identification of fungi. Molecular Ecology, 22(21), 5271–5277. [DOI] [PubMed] [Google Scholar]
- Leisure and Cultural Services Department (2016). Register of old and valuable trees. [Google Scholar]
- Lemanceau, P. , & Alabouvette, C. (1993). Suppression of Fusarium wilts by fluorescent pseudomonads: Mechanisms and applications. Biocontrol Science and Technology, 3(3), 219–234. [Google Scholar]
- Li, J. G. , Ren, G. D. , Jia, Z. J. , & Dong, Y. H. (2014). Composition and activity of rhizosphere microbial communities associated with healthy and diseased greenhouse tomatoes. Plant and Soil, 380, 337–347. [Google Scholar]
- Liu, X. , Zhang, S. , Jiang, Q. , Bai, Y. , Shen, G. , Li, S. , & Ding, W. (2016). Using community analysis to explore bacterial indicators for disease suppression of tobacco bacterial wilt. Scientific Reports, 6, 36773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascale, A. , Proietti, S. , Pantelides, I. S. , & Stringlis, I. A. (2020). Modulation of the root microbiome by plant molecules: The basis for targeted disease suppression and plant growth promotion. Frontiers in Plant Science, 10, 1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad, M. , & Naik, S. T. (2002). Management of root rot and heart rot of Acacia mangium Willd. Karnataka Journal of Agricultural Sciences, 15(2), 321–326. [Google Scholar]
- Rodriguez, H. , & Fraga, R. (1999). Phosphate solubilizing bacteria and their role in plant growth promotion. Biotechnology Advances, 17(4–5), 319–339. [DOI] [PubMed] [Google Scholar]
- Sahashi, N. , Akisa, M. , Ishihara, M. , Abe, Y. , & Morita, S. (2007). First report of the brown root rot disease caused by Phellinus noxius, its distribution and newly recorded host plants in the Amami Islands, southern Japan. Forest Pathology, 37(3), 167–173. [Google Scholar]
- Schwarze, F. W. M. R. , Jauss, F. , Spencer, C. , Hallam, C. , & Schubert, M. (2012). Evaluation of an antagonistic Trichoderma strain for reducing the rate of wood decomposition by the white rot fungus Phellinus noxius . Biological Control, 61(2), 160–168. [Google Scholar]
- Shang, Q. , Yang, G. , Wang, Y. , Wu, X. , Zhao, X. , Hao, H. , … Wang, R. (2016). Illumina-based analysis of the rhizosphere microbial communities associated with healthy and wilted Lanzhou lily (Lilium davidii var unicolor) plants grown in the field. World Journal of Microbiology & Biotechnology, 32(6), 95. [DOI] [PubMed] [Google Scholar]
- Takahashi, T. , Sutherland, S. C. , Chipman, D. W. , Goddard, J. G. , Ho, C. , Newberger, N. , … Munro, D. R. (2014). Climatological distributions of pH, pCO2, total CO2, alkalinity, and CaCO3 saturation in the global surface ocean, and temporal changes at selected locations. Marine Chemistry, 164, 95–125. [Google Scholar]
- Trivedi, P. , Van Nostrand, J. D. , Albrigo, G. , Zhou, J. , & Wang, N. (2012). Huanglongbing alters the structure and functional diversity of microbial communities associated with citrus rhizosphere. ISME Journal, 6(2), 363–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, J. N. , Hsieh, W. H. , Ann, P. J. , & Yang, C. M. (2007). Development of specific primers for Phellinus noxius . Plant Pathology Bulletin, 16(4), 193–202. [Google Scholar]
- Turner, T. R. , Ramakrishnan, K. , Walshaw, J. , Heavens, D. , Alston, M. , Swarbreck, D. , … Poole, P. S. (2013). Comparative metatranscriptomics reveals kingdom level changes in the rhizosphere microbiome of plants. ISME Journal, 7(12), 2248–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uroz, S. , Oger, P. , Tisserand, E. , Cebron, A. , Turpault, M. P. , Buee, M. , … Frey-Klett, P. (2016). Specific impacts of beech and Norway spruce on the structure and diversity of the rhizosphere and soil microbial communities. Scientific Reports, 6, 27756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veach, A. M. , Morris, R. , Yip, D. Z. , Yang, Z. K. , Engle, N. L. , Cregger, M. A. , … Schadt, C. W. (2019). Rhizosphere microbiomes diverge among Populus trichocarpa plant-host genotypes and chemotypes, but it depends on soil origin. Microbiome, 7(1), 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Garrity, G. M. , Tiedje, J. M. , & Cole, J. R. (2007). Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology, 73(16), 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, Z. , Hu, J. , Gu, Y. , Ying, S. , Xu, Y. , Jousset, A. , … Friman, V.-P. (2018). Ralstonia solanacearum pathogen disrupts bacterial rhizosphere microbiome during an invasion. Soil Biology & Biochemistry, 118, 8–17. [Google Scholar]
- Weston, D. J. , Pelletier, D. A. , Morrell-Falvey, J. L. , Tschaplinski, T. J. , Jawdy, S. S. , Lu, T. Y. , … Tuskan, G. A. (2012). Pseudomonas fluorescens induces strain-dependent and strain-independent host plant responses in defense networks, primary metabolism, photosynthesis and fitness. Molecular Plant-Microbe Interactions, 25(6), 765–778. [DOI] [PubMed] [Google Scholar]
- White, T. J. , Bruns, T. D. , Lee, S. B. , & Taylor, J. W. (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics In Innis M. A., Gelfand D. H., Sninsky J. J., & White T. J. (Eds.), PCR protocols: A guide to methods and applications (pp. 315–322). Cambridge, MA: Academic Press. [Google Scholar]
- Wu, M. L. , Chang, T. T. , Jaung, L. M. , Hung, T. H. , Chen, C. H. , & Lin, L. D. (2009). Establishment of PCR rapid detection technique for tree brown root rot disease. Quarterly Journal of Chinese Forestry., 42(2), 239–247. [Google Scholar]
- Zhang, Y. , Du, B. , Jin, Z. , Li, Z. , Song, H. , & Ding, Y. (2011). Analysis of bacterial communities in rhizosphere soil of healthy and diseased cotton (Gossypium sp.) at different plant growth stages. Plant and Soil, 339, 447–455. [Google Scholar]
- Zimmerman, M. H. , Wardrop, A. B. , & Tomlinson, P. B. (1968). Tension wood in aerial roots of Ficus benjamina L. Wood Science and Technology, 2, 95–104. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw sequence reads generated in this study are available in the NCBI Sequence Read Archive (SRA) under the accession PRJNA433610. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA433610.