

Abstract
Background
Breast cancer subtypes are statistically associated with prognosis. The search for markers of breast tumor heterogeneity and the development of precision medicine for patients are the current focuses of the field.
Methods
We used a bioinformatic approach to identify key disease-causing genes unique to the luminal A and basal-like subtypes of breast cancer. First, we retrieved gene expression data for luminal A breast cancer, basal-like breast cancer, and normal breast tissue samples from The Cancer Genome Atlas database. The differentially expressed genes unique to the 2 breast cancer subtypes were identified and subjected to Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses. We constructed protein–protein interaction networks of the differentially expressed genes. Finally, we analyzed the key modules of the networks, which we combined with survival data to identify the unique cancer genes associated with each breast cancer subtype.
Results
We identified 1114 differentially expressed genes in luminal A breast cancer and 1042 differentially expressed genes in basal-like breast cancer, of which the subtypes shared 500. We observed 614 and 542 differentially expressed genes unique to luminal A and basal-like breast cancer, respectively. Through enrichment analyses, protein–protein interaction network analysis, and module mining, we identified 8 key differentially expressed genes unique to each subtype. Analysis of the gene expression data in the context of the survival data revealed that high expression of NMUR1 and NCAM1 in luminal A breast cancer statistically correlated with poor prognosis, whereas the low expression levels of CDC7, KIF18A, STIL, and CKS2 in basal-like breast cancer statistically correlated with poor prognosis.
Conclusions
NMUR1 and NCAM1 are novel key disease-causing genes for luminal A breast cancer, and STIL is a novel key disease-causing gene for basal-like breast cancer. These genes are potential targets for clinical treatment.
Keywords: Luminal a breast cancer, Basal-like breast cancer, Neoplasm genes, Bioinformatics
Background
Breast cancer comprises malignant tumors originating from mammary epithelial tissue. It is one of the most common cancers in women and the leading cause of cancer-related deaths in women globally [1]. In 2000, Perou et al. proposed the molecular classification of breast cancer to facilitate accurate diagnosis and treatment [2]. For the first time, breast cancer was classified into the following types: luminal-like, Her-2-positive, basal-like, and normal breast-like. In 2001, Sørlie et al. further classified luminal-like breast cancer into luminal A and luminal B subtypes and demonstrated that different subtypes of breast cancer were statistically associated with prognosis [3]: the luminal A subtype was associated with the best prognosis, followed by the luminal B subtype, whereas the Her-2-positive and basal-like subtypes were associated with the worst prognosis. Therefore, identifying the regulatory mechanisms of the breast cancer subtypes to develop targeted therapies is essential to achieve optimal results for individual patients.
Luminal A is the most common molecular subtype of breast cancer with a relatively good prognosis. Endocrine therapy is the preferred treatment option for luminal A breast cancer since the tumor is hormone receptor-positive [4]. However, several genetic factors determine the efficacy of endocrine therapy for luminal A breast cancer. FOXA1 expression is associated with estrogen receptor (ER) positivity in luminal A breast cancer [5, 6]. Prognostic testing by Thangavelu et al. revealed that CENPI overexpression is a strong independent marker for ER-positive breast cancer that can be used to predict patient prognosis and survival [7]. They further demonstrated that CENPI is an E2F target gene. Karn and Emerson suggested that mutations in GATA3 resulted in differential gene expression in ER-positive breast tumors, which affected prognosis [8]. In addition, Alfarsi et al. found that high KIF18A expression exhibited prognostic significance and could predict the adverse effects of endocrine therapy in patients with ER-positive breast cancer [9]. Thus, KIF18A testing of patients with ER-positive breast cancer prior to treatment could guide clinicians’ decision-making on whether the patients would benefit from endocrine therapy.
Basal-like breast cancer is associated with a poor prognosis. Due to the lack of effective therapeutic targets, the primary clinical treatment modality for basal-like breast cancer remains chemotherapy [10, 11]. Several groups have investigated the genetic profile of basal-like breast cancer to identify novel, specific targets to improve patient outcomes. Komatsu et al. identified cell cycle regulators, ASPM, and CENPK as potential disease-causing genes for basal-like breast cancer and utilized them as therapeutic targets in vitro [12]. Rodriguez-Acebes et al. demonstrated that the cell cycle gene CDC7 may represent an effective, highly specific anticancer target in triple-negative breast cancer (TNBC) overexpressing Her-2 [13]. Song et al. used miRNA microarray to analyze 2 BRCA1-mutated TNBC cell lines [14]. They found that the addition of a PARP inhibitor to the carboplatin plus gemcitabine therapy regimen led to increased expression of miR-664b-5p and that CCNE2 is a novel functional target of miR-664b-5p. Ye et al. showed that CDCA7 upregulated EZH2 transcripts and played a key role in TNBC progression, making CDCA7 a potential prognostic factor and therapeutic target for TNBC [15].
Most of the aforementioned studies analyzed general breast cancer samples or a single subtype of breast cancer. Few comparative analyses of breast cancer subtypes have been reported. In the present study, we aimed to identify novel, more accurate targets for clinical treatment based on the comparative analysis of the genetic profiles and prognoses of patients with luminal A and basal-like breast cancer. We used bioinformatics to comprehensively analyze the gene expression data for each subtype and determine uniquely differentially expressed genes. Then, we utilized protein–protein interaction (PPI) network analysis to identify the key genes for each subtype. The key genes were used as specific markers for luminal A breast cancer and basal-like breast cancer for receiver operating characteristic (ROC) curve and survival analyses to identify prognosis-associated candidate genes for targeted breast cancer treatment.
Methods
Data
Gene expression data from 439 samples—comprising 255 luminal A breast cancer samples, 87 basal-like breast cancer samples, and 97 normal breast tissue samples—were downloaded from The Cancer Genome Atlas (TCGA) database [16].
Differentially expressed gene analysis
We used R package limma [17] to compare the gene expression data from the luminal A breast cancer samples and normal breast tissue samples and between the basal-like breast cancer samples and normal breast tissue samples. The screening threshold was set to |logFC| > [mean(|logFC|) + 2sd(|logFC|)] with P value < 0.05, and the expression data were normalized based on Trimmed Mean of M value (TMM) in R package edgeR [18]. We used the R packages clusterProfiler [19] and org.H.eg.db to convert the IDs of the differentially expressed genes into gene names. Finally, the R package plot was used to create volcano plots of the differentially expressed genes.
To identify the common and unique differentially expressed genes in luminal A and basal-like breast cancer, we compared the differentially expressed genes identified in the 2 subtypes with R package dplyr [20]. We selected the common differentially expressed genes in the 2 subtypes with opposite modes of expression.
Enrichment analyses of differentially expressed genes
To analyze the unique biological processes in the pathogenesis of luminal A and basal-like breast cancer, we performed enrichment analyses with R package clusterprofiler. Gene ontology (GO) enrichment analysis categorized genes as related to biological processes, cellular components, or molecular functions, and Kyoto Encyclopedia of Genes and Genomes (KEGG) [21] analysis categorized genes based on pathway enrichment. We generated bubble charts of the functions and pathways of the differentially expressed genes to facilitate the interpretation of the biological significance of the genes.
Construction of PPI networks of differentially expressed genes
We used the online tool STRING [22] to obtain the PPI networks of the differentially expressed genes unique to luminal A and basal-like breast cancer. Isolated protein nodes with no interactions with other proteins were eliminated. We used Cytoscape software to visualize the PPI networks and the plugin MCODE to screen for the functional modules of the networks. The screening criterion was an MCODE score of ≥ 5. The cytoHubba plugin was used to identify key genes with the following settings: Hubba_nodes=8, Ranking Method=“DMNC.” We used the online tool DAVID [23] for KEGG pathway enrichment analysis of the key genes. A difference with a P value < 0.05 was considered significant.
Analysis of prognostic value
We used the key genes as specific markers for luminal A and basal-like breast cancer in ROC curve analyses. The key genes were further analyzed for associations with survival using the online tool PROGgeneV2 [24]. The settings were SURVIVAL MEASURE=“MEDIAN” and SURVIVAL MEASURE=“DEATH.” We obtained survival curves for patients with luminal A and basal-like breast cancer subtypes who had different expression levels of the unique key genes. A difference with a P value < 0.05 was considered significant.
Results
Identification of differentially expressed genes
We performed a differential analysis of 255 samples of luminal A breast cancer and 97 samples of normal breast tissue. Based on the criteria |logFC| > 2.199 and P value < 0.05, we identified 1114 differentially expressed genes, including 453 upregulated genes and 661 downregulated genes, from a total of 20,531 genes (Fig. 1). We also performed a differential analysis of 87 samples of basal-like breast cancer and 97 samples of normal breast tissue. Based on the criteria |logFC| > 2.799 and P value < 0.05, we selected 1042 differentially expressed genes, including 435 upregulated genes and 607 downregulated genes, from a total of 20,531 genes (Fig. 1).
Fig. 1.

Differentially expressed genes in luminal A and basal-like breast cancer versus normal breast tissue. a Volcano plot of the differentially expressed genes identified by comparison of 255 luminal A breast cancer samples and 97 normal breast tissue samples. The 453 upregulated genes are shown in red (Up), and the 661 downregulated genes are shown in green (Down). b Volcano plot of the differentially expressed genes identified by comparison of 87 basal-like breast cancer samples and 97 normal breast tissue samples. The 435 upregulated genes are shown in red (Up), and the 607 downregulated genes are shown in green (Down). Genes that were not differentially expressed are shown in black (Equal)
We compared the differentially expressed genes in the 2 breast cancer subtypes and found that 614 differentially expressed genes were unique to the luminal A breast cancer samples and 542 were unique to basal-like breast cancer samples (Fig. 2). The subtypes shared 500 differentially expressed genes. We identified 15 differentially expressed genes with opposite expression patterns in the luminal A and basal-like breast cancer samples. The relationships of the differentially expressed genes are shown in Fig. 2.
Fig. 2.

Expression patterns of the differentially expressed genes in luminal A and basal-like breast cancer. a Luminal A breast cancer had 614 unique differentially expressed genes. b Basal-like breast cancer had 542 unique differentially expressed genes. c The subtypes shared 15 common differentially expressed genes with opposite expression patterns (updownoppo)
Function and pathway enrichment analyses
GO enrichment analysis (Fig. 3) revealed that the 614 differentially expressed genes unique to luminal A breast cancer were mainly involved in biological processes, including the antimicrobial humoral response, epidermis development, glial cell differentiation, and hormone metabolic process. The differentially expressed genes with a relationship to cellular components were significantly associated with multiple components, such as the sarcolemma, apical plasma membrane, ion channel complex, transmembrane transporter complex, and neuronal cell body. The significantly enriched molecular functions of the differentially expressed genes included cation channel activity, substrate-specific channel activity, metal ion transmembrane transporter activity, and passive transmembrane transporter activity. In addition, the significantly enriched KEGG pathways comprised the oxytocin signaling pathway, neuroactive ligand–receptor interaction, ovarian steroidogenesis, vascular smooth muscle contraction, dopaminergic synapses, Staphylococcus aureus infection, and estrogen signaling pathway (Fig. 3).
Fig. 3.

Enrichment analyses of the differentially expressed genes in luminal A breast cancer. a GO enrichment analysis of biological processes. b GO enrichment analysis of cellular components. c GO enrichment analysis of molecular functions. d KEGG pathway analysis. P.adjust is the adjusted value of P value
GO enrichment analysis (Fig. 4) revealed that the 542 differentially expressed genes unique to basal-like breast cancer were mainly involved in biological processes, including organelle fission, nuclear division, nuclear chromosome segregation, sister chromatid segregation, mitotic nuclear division, chromosomal segregation, and DNA-dependent DNA replication. The differentially expressed genes were significantly associated with multiple cell components, such as collagen-containing extracellular matrix, postsynaptic membrane, collagen trimer, and chromosomal and centromeric regions. The significantly enriched molecular functions of the differentially expressed genes included aromatase activity, RNA polymerase II-specific DNA-binding transcription activation activity, oxidoreductase activity, and G protein-coupled peptide receptor activity. In addition, the significantly enriched KEGG pathways were cell cycle, neuroactive ligand–receptor interaction, oocyte meiosis, melanoma, and Cushing syndrome. The detailed results of the analyses are shown in Fig. 4.
Fig. 4.

Enrichment analyses of the differentially expressed genes in basal-like breast cancer. a GO enrichment analysis of biological processes. b GO enrichment analysis of cellular components. c GO enrichment analysis of molecular functions. d KEGG pathway analysis. P.adjust is the adjusted value of P value
PPI network construction
Next, we sought to further understand the functional modules in the PPI networks of the differentially expressed genes unique to luminal A and basal-like breast cancer to identify the key genes for each disease. The MCODE Cytoscape plugin was used to construct the functional modules in the PPI network of the differentially expressed genes unique to luminal A breast cancer. Functional modules with scores > 5 were selected. The module in Fig. 5 has a score of 6.182 and contains 12 nodes and 24 edges.
Fig. 5.
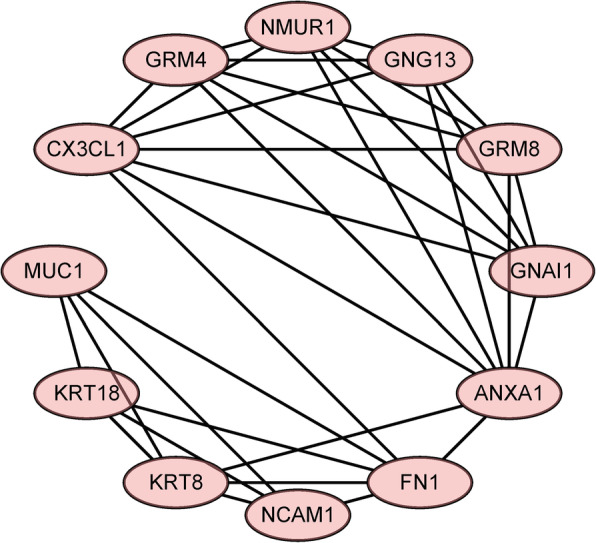
Luminal A breast cancer module
We similarly constructed functional modules in the PPI network of the differentially expressed genes unique to basal-like breast cancer. Module 1 has a score of 25.812 and contains 33 nodes and 413 edges; module 2 has a score of 5.818 and contains 12 nodes and 32 edges (Fig. 6).
Fig. 6.

Modules in the PPI network of differentially expressed genes in the basal-like breast cancer subtype. a PPI network module 1. b PPI network module 2
We further analyzed the above modules to find the hub genes. We used the cytoHubba Cytoscape plugin (settings: Hubba_nodes=8, Ranking Method=“DMNC”) to screen for 8 key genes respectively among the luminal A breast cancer module and the basal-like breast cancer module 1 (Fig. 7). The key genes identified for luminal A breast cancer were GRM4, GRM8, KRT18, NMUR1, MUC1, CX3CL1, GATA3, and NCAM1. The neuroactive ligand–receptor interaction pathway was enriched for GRM4, NMUR1, and GRM8 (P < 0.05). The metabotropic glutamate receptors were a family of G protein-coupled receptors that had been divided into 3 groups on the basis of sequence homology, putative signal transduction mechanisms, and pharmacologic properties. GRM8 and GRM4 belong to group III that receptors were linked to the inhibition of the cyclic AMP cascade but differed in their agonist selectivities [25]. KRT18 encoded the type I intermediate filament chain keratin 18. Keratin 18, together with its filament partner keratin 8, were perhaps the most commonly found members of the intermediate filament gene family. They were expressed in single layer epithelial tissues of the body [26]. Neuromedin U Receptor 1 (NMUR1) was a protein coding gene. Among its related pathways were RET signaling and signaling by GPCR. Gene Ontology (GO) annotations related to this gene included G protein-coupled receptor activity and neuromedin U receptor activity. An important paralog of this gene was NMUR2 [27]. The key genes identified for basal-like breast cancer were CENPI, CENPK, CDC7, CCNE2, KIF18A, STIL, CDCA7, and CKS2. The small cell lung cancer pathway was enriched for CCNE2 and CKS2, and the cell cycle pathway was enriched for CDC7 and CCNE2 (P < 0.05). CENPI encoded a centromere protein that was a component of the CENPA-NAC (nucleosome-associated) complex. This protein regulated the recruitment of kinetochore-associated proteins that were required to generate the spindle checkpoint signal. The product of this gene was involved in the response of gonadal tissues to follicle-stimulating hormone [28]. CENPK was a subunit of a CENPH-CENPI-associated centromeric complex that targeted CENPA to centromeres and was required for proper kinetochore function and mitotic progression [29]. The protein encoded by CCNE2 belongs to the highly conserved cyclin family, whose members were characterized by a dramatic periodicity in protein abundance through the cell cycle. Cyclins function as regulators of CDK kinases [30]. CDC7 encoded a cell division cycle protein with kinase activity that was critical for the G1/S transition. Overexpression of this gene product might be associated with neoplastic transformation for some tumors [31].
Fig. 7.

Key subtype-specific genes. a Key genes for luminal A breast cancer. b Key genes for basal-like breast cancer
Analysis of prognostic value
We created ROC curves for the 2 sets of key genes. ROC curve analysis showed that these genes exhibited good prognostic value for their associated cancer subtypes. The areas under the ROC curves were greater than 90% for all genes, as shown in Fig. 8.
Fig. 8.

ROC curves of the key genes. a ROC curves of the key genes, including GRM4, GRM8, KRT18, NMUR1, MUC1, CX3CL1, GATA3, and NCAM1, for luminal A breast cancer. b ROC curves of the key genes, including CENPI, CENPK, CDC7, CCNE2, KIF18A, STIL, CDCA7, and CKS2, for basal-like breast cancer
The prognostic values of the selected key genes unique to luminal A breast cancer were analyzed using the PROGgeneV2 online tool. We retrieved the survival curves of the patients from the TCGA database with the corresponding breast cancer subtype and analyzed survival by the expression levels of the key genes (Fig. 9). Of the key genes unique to the luminal A breast cancer subtype, the expression levels of only NMUR1 and NCAM1 were associated with patient survival time (P < 0.05). Survival analysis showed that higher expression levels of the prognosis-related key genes were associated with shorter survival time of luminal A breast cancer patients.
Fig. 9.

Survival of patients with luminal A breast cancer by expression of key genes. a NMUR1. b NCAM1. Survival and gene expression data were retrieved from TCGA. The cohort was divided at the median gene expression (P < 0.05)
Next, we used the same methodology to analyze the prognostic values of the key genes unique to basal-like breast cancer (Fig. 10). Of the key genes unique to basal-like breast cancer, the expression levels of only CDC7, KIF18A, STIL, and CKS2 were associated with patient survival time (P < 0.05). Lower than median expression levels of the prognosis-related key genes were associated with better prognosis .
Fig. 10.

Survival of patients with basal-like breast cancer by expression of key genes. a CDC7. b KIF18A. c STIL. d CKS2. Survival and gene expression data were retrieved from TCGA. The cohort was divided at the median gene expression (P < 0.05)
Discussion
Breast cancer is the most common malignancy, which with high heterogeneity in terms of the underlying molecular alterations. Different subtypes exhibit distinct biological behavior, prognosis and treatment. Although the treatment of breast cancer has improved, the prognosis of patients is still poor. Herein, it is urgently needed to identify precise therapeutic targets for different subtypes of breast cancer [3].
In our studies, we identify new molecular targets of luminal A breast cancer and basal-like breast cancer using bioinformatic analysis. We identified NMUR1 and NCAM1 as novel key genes associated with the development, progression, and prognosis of luminal A breast cancer, and STIL as a novel key gene associated with the prognosis of basal-like breast cancer.
NMUR1, which is associated with the poor prognosis of luminal A breast cancer patients, encodes the neuromedin U receptor 1. NMUR1 is broadly expressed in human tissues, with the highest expression in the adipose tissue, intestine, spleen, and lymphocytes. It likely possesses physiological effects that remain to be elucidated [32]. NCAM1 encodes neural cell adhesion molecule 1. It is broadly used as a marker of minimal residual disease and is expressed in most acute myeloid leukemia molecular subgroups with high levels of heterogeneity. Sasca et al. used complementary genetic strategies to demonstrate the important role of NCAM1 in the regulation of cell survival and stress resistance [33]. The roles of NMUR1 and NCAM1 in breast cancer have not been reported. We found that luminal A breast cancer patients with high expression of NMUR1 and NCAM1 had statistically worse overall survival than patients with below-median levels of expression. Thus, we predict that inhibition of NMUR1 and NCAM1 could represent a novel strategy to improve the treatment of luminal A breast cancer. STIL, also known as STIL centriolar assembly protein, encodes a cytoplasmic protein involved in the regulation of mitotic spindle checkpoints [34]. Ouyang et al. found that STIL expression was upregulated in various human tumor tissues and that higher expression of STIL was associated with shorter survival [35]. Our survival analysis showed that low expression of STIL statistically affects the overall survival of patients with basal-like breast cancer. Thus, we predict that promoting STIL expression could represent a novel modality to improve the treatment of basal-like breast cancer.
There are also several limitations in our study. We preliminarily explored the molecular mechanisms of these genes. The accuracy of new molecular targets needed for further experiments confirmed. In the future, we will investigate the expression of these oppositely regulated genes with the aim of developing specific therapies for the breast cancer subtypes.
Conclusions
In summary, we identified NMUR1 and NCAM1 as novel key genes associated with the development, progression, and prognosis of luminal A breast cancer, and STIL as a new target with the prognosis of basal-like breast cancer.
Acknowledgements
We would like to thank Editage (www.editage.cn) for the English language editing.
Abbreviations
- ER
Estrogen receptor
- TNBC
Triple-negative breast cancer
- TMM
Trimmed Mean of M value
- PPI
Protein–protein interaction
- ROC
Receiver operating characteristic
- TCGA
The Cancer Genome Atlas
- GO
Gene Ontology
- KEGG
Kyoto Encyclopedia of Genes and Genomes
Authors’ contributions
PN conceived and designed the study. RJ and ZL performed the statistical analyses. WL, YJ, YW, and YL contributed in collecting and interpreting data as well as in drafting and critically revising the manuscript. All authors approved the final version of the manuscript and are accountable for the accuracy and integrity in all aspects of the study.
Authors’ information
Rong Jia and Zhongxian Li contributed equally to this work.
Funding
None
Availability of data and materials
The data analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rong Jia and Zhongxian Li contributed equally to this work.
References
- 1.Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144:1941–1953. doi: 10.1002/ijc.31937. [DOI] [PubMed] [Google Scholar]
- 2.Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 3.Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shemin KMZ, Smitha NV, Jojo A, Vijaykumar DK. Molecular classification and prognostication of 300 node-negative breast cancer cases: a tertiary care experience. South Asian J Cancer. 2015;4:160–162. doi: 10.4103/2278-330X.175952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mehta RJ, Jain RK, Leung S, Choo J, Nielsen T, Huntsman D, et al. FOXA1 is an independent prognostic marker for ER-positive breast cancer. Breast Cancer Res Treat. 2012;131:881–890. doi: 10.1007/s10549-011-1482-6. [DOI] [PubMed] [Google Scholar]
- 6.Badve S, Turbin D, Thorat MA, Morimiya A, Nielsen TO, Perou CM, et al. FOXA1 expression in breast cancer--correlation with luminal subtype a and survival. Clin Cancer Res. 2007;13:4415–4421. doi: 10.1158/1078-0432.CCR-07-0122. [DOI] [PubMed] [Google Scholar]
- 7.Thangavelu PU, Lin CY, Vaidyanathan S, Nguyen THM, Dray E, Duijf PHG. Overexpression of the E2F target gene CENPI promotes chromosome instability and predicts poor prognosis in estrogen receptor-positive breast cancer. Oncotarget. 2017;8:62167–62182. doi: 10.18632/oncotarget.19131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karn R, Emerson IA. Breast cancer mutation in GATA3 zinc finger 1 induces conformational changes leading to the closer binding of ZnFn2 with a wrapping architecture. J Biomol Struct Dyn. 2020;38:1810–1821. doi: 10.1080/07391102.2019.1620635. [DOI] [PubMed] [Google Scholar]
- 9.Alfarsi LH, Elansari R, Toss MS, Diez-Rodriguez M, Nolan CC, Ellis IO, et al. Kinesin family member-18A (KIF18A) is a predictive biomarker of poor benefit from endocrine therapy in early ER+ breast cancer. Breast Cancer Res Treatment. 2019;173:93–102. doi: 10.1007/s10549-018-4978-5. [DOI] [PubMed] [Google Scholar]
- 10.Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer registry. Cancer. 2007;109:1721–1728. doi: 10.1002/cncr.22618. [DOI] [PubMed] [Google Scholar]
- 11.Weigelt B, Horlings HM, Krieke B, Hayes MM, Hauptmann M, Wessels LF, et al. Refinement of breast cancer classification by molecular characterization of histological special types. J Pathol. 2008;216:141–150. doi: 10.1002/path.2407. [DOI] [PubMed] [Google Scholar]
- 12.Komatsu M, Yoshimaru T, Matsuo T, Kiyotani K, Miyoshi Y, Tanahashi T, et al. Molecular features of triple negative breast cancer cells by genome-wide gene expression profiling analysis. Int J Oncol. 2013;42:478–506. doi: 10.3892/ijo.2012.1744. [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez-Acebes S, Proctor I, Loddo M, Wollenschlaeger A, Rashid M, Falzon M, et al. Targeting DNA replication before it starts: Cdc7 as a therapeutic target in p53-mutant breast cancers. Am J Pathol. 2010;177:2034–2045. doi: 10.2353/ajpath.2010.100421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song W, Tang L, Xu Y, Xu J, Zhang W, Xie H, et al. PARP inhibitor increases chemosensitivity by upregulating miR-664b-5p in BRCA1-mutated triple-negative breast cancer. Sci Rep. 2017;7:42319. doi: 10.1038/srep42319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye L, Li F, Song Y, Yu D, Xiong Z, Li Y, et al. Overexpression of CDCA7 predicts poor prognosis and induces EZH2-mediated progression of triple-negative breast cancer. Int J Cancer. 2018;143:2602–2613. doi: 10.1002/ijc.31766. [DOI] [PubMed] [Google Scholar]
- 16.Tomczak K, Czerwińska P, Wiznerowicz M. The cancer genome atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn) 2015;19(1A):A68–A77. doi: 10.5114/wo.2014.47136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smyth GK. Limma: linear models for microarray data. Bioinformatics and Computational Biology Solution Using R and Bioconductor. 2005;23:397–420.
- 18.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu G, Wang LG, Han Y. QY he. clusterProfiler: an R package for comparing biological themes among gene clusters. Journal of. Integr Biol. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riemondy KA, Sheridan RM, Gillen A, Yu Y, Bennett CG, Hesselberth JR. Valr: reproducible genome interval analysis in R. F1000Res. 2017;6:1025. doi: 10.12688/f1000research.11997.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44(D1):D457–D462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–D368. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang B, Dai JX, Pan YB, Ma YB, Chu SH. Identification of biomarkers and construction of a microRNA-mRNA regulatory network for ependymoma using integrated bioinformatics analysis. Oncol Lett. 2019;18(6):6079–6089. doi: 10.3892/ol.2019.10941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goswami CP, Nakshatri H. PROGgeneV2: enhancements on the existing database. BMC Cancer. 2014;14:970. doi: 10.1186/1471-2407-14-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z, Lu B, Sun L, Yan X, Xu J. Identification of candidate genes or microRNAs associated with the lymph node metastasis of SCLC. Cancer Cell Int. 2018;18:161. doi: 10.1186/s12935-018-0653-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korbut E, Janmaat VT, Wierdak M, Hankus J, Wójcik D, Surmiak M, et al. Molecular profile of Barrett’s esophagus and gastroesophageal reflux disease in the development of translational physiological and pharmacological studies. Int J Mol Sci. 2020;3:21(17). doi: 10.3390/ijms21176436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Filosi M, Kam-Thong T, Essioux L, Muglia P, Trabetti E, Spooren W, et al. Transcriptome signatures from discordant sibling pairs reveal changes in peripheral blood immune cell composition in autism spectrum disorder. Transl Psychiatry. 2020;10(1):106. doi: 10.1038/s41398-020-0778-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Basit S, Al-Edressi HM, Sairafi MH, Hashmi JA, Alharby E, Safar R, et al. Centromere protein I (CENPI) is a candidate gene for X-linked steroid sensitive nephrotic syndrome. J Nephrol. 2020;33(4):763–769. doi: 10.1007/s40620-019-00692-1. [DOI] [PubMed] [Google Scholar]
- 29.Wang J, Li H, Xia C, Yang X, Dai B, Tao K, et al. Down regulation of CENPK suppresses hepatocellular carcinoma malignant progression through regulating YAP1. Onco Targets Ther. 2019;12:869–882. doi: 10.2147/OTT.S190061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee C, Fernandez KJ, Alexandrou S, Sergio CM, Deng N, Rogers S, et al. Cyclin E2 promotes whole genome doubling in breast cancer. Cancers (Basel) 2020;12(8):2268. doi: 10.3390/cancers12082268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rainey MD, Bennett D, O'Dea R, Zanchetta ME, Voisin M, Seoighe C, et al. ATR restrains DNA synthesis and mitotic catastrophe in response to CDC7 inhibition. Cell Rep. 2020;32(9):108096. doi: 10.1016/j.celrep.2020.108096. [DOI] [PubMed] [Google Scholar]
- 32.Kojima M, Haruno R, Nakazato M, Date Y, Murakami N, Hanada R, et al. Purification and identification of neuromedin U as an endogenous ligand for an orphan receptor GPR66 (FM3). Biochem Biophys Res Commun. 2000;276:435–8. [DOI] [PubMed]
- 33.Sasca D, Szybinski J, Schüler A, Shah V, Heidelberger J, Haehnel PS, et al. NCAM1 (CD56) promotes leukemogenesis and confers drug resistance in AML. Blood. 2019;133:2305–19. [DOI] [PubMed]
- 34.Rasool S, Baig JM, Moawia A, Ahmad I, Iqbal M, Waseem SS, et al. An update of pathogenic variants in ASPM, WDR62, CDK5RAP2, STIL, CENPJ, and CEP135 underlying autosomal recessive primary microcephaly in 32 consanguineous families from Pakistan. Mol Genet Genomic Med. 2020;8(9):e1408. doi: 10.1002/mgg3.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ouyang Y, Jin YB, Chen XP, Zhang GY, Mao SL, Ling F, et al. STIL is upregulated in nasopharyngeal carcinoma tissues and promotes nasopharyngeal carcinoma proliferation, migration, and invasion. Neoplasma. 2020;67:37–45. doi: 10.4149/neo_2019_190306N192. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data analyzed during the current study are available from the corresponding author on reasonable request.