

Abstract
Mixed Lineage Kinase 3 (MLK3), a MAP3K member has been envisioned as a viable drug target in cancer, yet its detailed function and signaling is not fully elucidated. We identified that MLK3 tightly associates with an oncogene, PAK1. Mammalian PAK1 being a Ste20 (MAP4K) member, we tested whether it is an upstream regulator of MLK3. In contrast to our hypothesis, MLK3 activated PAK1 kinase activity directly, as well as in the cells. Although, MLK3 can phosphorylate PAK1 on Ser133 and Ser204 sites, PAK1S133A mutant is constitutively active, whereas, PAK1S204A is not activated by MLK3. Stable overexpression of PAK1S204A in breast cancer cells, impedes migration, invasion and NFĸB activity. In vivo breast cancer cell tumorigenesis is significantly reduced in tumors expressing PAK1S204A mutant. These results suggest that mammalian PAK1 does not act as a MAP4K and MLK3-induced direct activation of PAK1 plays a key role in breast cancer tumorigenesis.
Keywords: Signaling, Breast Cancer, MAPKs, Oncogene, Phosphorylation
INTRODUCTION
Mixed Lineage Kinase 3 (MLK3) also known as MAP3K11 belongs to a large family of MAP3Ks, called the Mixed Lineage Kinases (MLKs) because their catalytic domains contain signature sequences of both Ser/Thr and Tyr kinases [1, 2]. The biochemical analyses hitherto, have shown that MLKs are functional Ser/Thr kinases and activate downstream MAPK pathways, however their tyrosine kinase activities if any, are still not known [1, 3]. There are nine mammalian MLK members and based on the functional domains and sequence similarities, they are classified in three sub-groups: the Mixed Lineage Kinases (MLKs), Dual-Leucine Zipper Kinases (DLKs) and Zipper sterile-α-motif kinases (ZAKs) [1]. MLK3 belongs to the MLK sub-group and has been implicated in various cancers [4-6] and neurodegenerative diseases [7, 8]. The pan-MLK inhibitor went to clinical trials for Parkinson’s disease, however the trial was abruptly stopped due to unknown reasons [8]. The role of MLK3 in cancer is an emerging area and earlier we reported that MLK3 kinase activity and transcripts were down-regulated by estrogen in breast cancer, providing a survival advantage to ER+ breast cancer cells [4]. We also reported that the kinase activity of MLK3 was downregulated by HER2 amplification and MLK3 activity was essential to promote cell death in HER2+ breast cancer cells by anti-HER2 therapies [9]. However, in triple-negative breast cancer cells, MLK3 activation promotes migration and invasion, and MLKs inhibitor blocked cancer cell migration/invasion [10]. Conceivably, MLK3 and other MLK family members are important therapeutic targets in various diseases, including cancer, yet, their upstream regulators and downstream signaling pathways are not fully elucidated.
Ste20 is a putative mitogen-activated kinase kinase kinase kinase (MAP4K) located upstream of Ste11 (MAP3K) in yeast [11], and mammalian p21-activated kinases (PAKs) are classified as Ste20 homologs [12]. Based on sequence homology, structure and activation mechanisms, six mammalian PAKs are classified into two groups: group I (PAK1-3) and group II (PAK4-6) [13]. The PAKs are not frequently mutated, yet overexpression and /or gene amplification of PAKs are common [13]. PAK1 and PAK4 genes are located on the chromosomal regions that are often amplified in cancer [13]. PAK1 is reported to be amplified and plays a critical role in cell growth, migration, invasion and apoptosis and functions as a major oncogene in various cancers, including breast cancer [14]. Several attempts have been made to specifically target PAK1 in cancer, however, its catalytic pocket is large and highly flexible, in addition to the highly mobile N-terminal lobe that presented a challenge in developing specific PAK inhibitors. Interestingly, PAK1 and MLK3 both are located on chromosome 11q13 that undergoes frequent amplification [15, 16]. Moreover, MLK3 contains a SH3 domain that is expected to interact with proline rich regions, as present in N- and C-terminal regions of PAK1[13]. All these shared features between MLK3 and PAK1, encouraged us to examine the possibility of any functional interaction between these two proteins, with an anticipation that PAK1 being a Ste20 member might act as an upstream regulator of MLK3.
Our results showed that indeed MLK3 and PAK1 proteins associate endogenously, however our initial hypothesis that PAK1 might be upstream regulator of MLK3, turned out to be incorrect. Instead, MLK3 overexpression and its agonists activated PAK1 kinase activity and a pan-MLK inhibitor, CEP-11004 inhibited agonist-induced PAK1 activity. The activation of PAK1 was via direct phosphorylation of Ser204 (S204) and the phospho-deficient PAK1 mutant (PAK1S204A) attenuated NFĸB activation. The colony formation, migration and invasion by breast cancer cells stably overexpressing phospho-deficient, PAK1S204A were significantly diminished. Orthotropic xenografts with breast cancer cells overexpressing PAK1S204A also presented smaller tumors with fewer proliferating cells and lower NFĸB activity. These data suggest that phosphorylation, and thus activation of PAK1 by MLK3 provides an excellent opportunity to therapeutically target MLK3 rather than PAK1 in breast cancer, and warrants further investigation. Our data also demonstrate that even though PAK1 being Ste20 member, is not located upstream of MLK3, a MAP3K member.
RESULTS
MLK3 specifically interacts with PAKs.
The presence of proline-rich regions in PAK1 with consensus binding sequences to the SH3 domain of MLK3 (Fig 1a) prompted us to determine any possible functional interaction between these two proteins. PAK1 and MLK3 were co-expressed in Human Embryonic Kidney (HEK-293) cells, and either GST-tagged PAK1 (Fig. 1b) or M2-tagged MLK3 (Fig. 1c) were immunoprecipitated and blotted for associated MLK3 or PAK1 respectively. GSH purification of GST-PAK1 brought down MLK3 (Fig. 1b), while immunoprecipitation of M2-MLK3 brought down PAK1 (Fig. 1c). To test whether MLK3 specifically interacts with PAK1 and not with other PAK family member(s), we examined the association between PAK2 and MLK3 in a similar co-transfection experiment. MLK3 immunoprecipitation also brought down PAK2 (Fig. Supplementary 1a). Since PAK2 isoform also interacted with MLK3, we examined the ability of other mammalian Ste20 members, GCK and MST1 to interact with MLK3. Co-transfection and co-immunoprecipitation with GCK and MST1 showed that MLK3 is quite specific for PAK1 and PAK2 and it does not interact, either with GCK or MST1 (Fig. Supplementary 1b).
Fig. 1.

MLK3 and PAK1 association. a The structure of MLK3 and PAK1 domains. The five proline-rich regions within PAK1 is marked by blue lines. b Total cell lysates were prepared from HEK-293, expressing indicated plasmids and mammalian GST-PAK1 was pulled-down and blotted for M2-tagged MLK3. c M2-tagged MLK3 was immunoprecipitated from HEK-293 cells expressing indicated plasmids and blotted for associated GST-PAK1 by anti-GST antibody. d Endogenous PAK1 was immunoprecipitated from Jurkat cell lysates and blotted with anti-MLK3 antibody. Anti-IRS1 and normal IgG were used as controls. e The direct association between MLK3 & PAK1 was determined by incubating purified proteins in solution and MLK3 was pulled down with anti-MLK3 antibody and blotted with PAK1 antibody. Anti-GAPDH antibody pull down was used as control. f GST-tagged (mammalian) MLK3 deletion mutants were created by PCR cloning and their association with Myc-tagged PAK1 was determined by pulling down GST-MLK3 mutants. Interacting domains are represented schematically. g GST-tagged PAK1 (mammalian) deletion mutants were created by PCR cloning and co-transfected in HEK-293 cells along with M2-tagged MLK3. The association between mutants and MLK3 was determined by M2-MLK3 pull down and blotted for GST associated PAK1. Since the molecular size of GST-PAK1 (268-545 aa), lane1 (denoted*), was similar to the size of IgG heavy chain, the GST-PAK1 associated with Flag-MLK3 was eluted off the beads after immunoprecipitation using Flag (M2 peptide). Interacting domains are represented schematically.
To further confirm that the association between PAK1 and MLK3 is not an artifact of over expression, and also occurs at the endogenous level, endogenous PAK1 protein was immunoprecipitated and blotted to determine interaction between MLK3 and PAK1 proteins. Endogenous PAK1 and MLK3 did interact with each other in Jurkat cells (Fig. 1d). The co-immunoprecipitation experiments do not rule out an indirect interaction between MLK3 and PAK1 and therefore, we determined direct interaction between PAK1 and MLK3 by incubating these two purified proteins, expressed in Baculovirus. The results clearly showed that these two proteins do interact directly (Fig. 1e). To map the binding region(s) to which MLK3 and PAK1 bind to each other, several MLK3 (Fig. 1f) and PAK1 (Fig. 1g) deletion mutants were generated and co-expressed in HEK-293 cells. The pull-down assays showed that the N-terminus of MLK3 (1-385 aa) that contains SH3 domain can interact with PAK1 (Fig 1f). However, the SH3 domain itself failed to interact with PAK1 (Fig 1f, lane 2), suggesting that probably the SH3 domain itself does not assume the optimal conformation for interaction. Similar co-transfection experiments with PAK1 deletion mutants showed that PAK1 encompasses at least three binding regions that can interact with MLK3 (Fig. 1g). Interestingly, the PAK1 kinase domain (268-545 aa) that lacks proline-rich region also interacted with MLK3 (Fig. 1g), suggesting that MLK3 could also interact with PAK1, independent of proline-rich regions.
MLK3 is an activator of PAK1.
Our initial assumption was that PAK1 being a mammalian homolog of yeast Ste20 [13] might interact with and act as a regulator of MLK3. To examine whether PAK1 can activate MLK3 or vice versa, HEK-293 cells were co-transfected with either a fixed amount of PAK1 (1μg) expression plasmid, along with two different concentrations (100 ng and 500 ng) of MLK3 expression plasmid. The co-transfected PAK1 was immunoprecipitated and its kinase activity was measured using histone H4 as the substrate [17]. The kinase activity of PAK1 was increased with increasing expression of MLK3 (Fig. 2a). However, when MLK3 expression was kept constant (1μg) and PAK1 expression was varied (100 ng and 500 ng), the MLK3 kinase activity was not altered by PAK1 (Fig. 2b). The co-expression experiments do not rule out an indirect effect of MLK3 on PAK1 activation and likelihood of PAK1 activation through protein: protein interaction, independent of MLK3 kinase activity is also a possibility. To determine that MLK3 kinase activity is essential for PAK1 activation, we transfected HEK293 cells with PAK1 along with either MLK3-WT or MLK3-KA (kinase dead). The PAK1 kinase activity was measured using histone H4 as the substrate. Our results showed that MLK3 kinase activity is required for PAK1 activation (Fig. Supplementary 2a). Interestingly, the PAK1 kinase activity was even lower in presence of MLK3-KA, compared to PAK1 alone (Fig. Supplementary 2a). Also, to determine a direct activation of PAK1 by MLK3, an in vitro couple kinase assay was done to determine whether purified MLK3 could directly activate PAK1 kinase activity. Inactive PAK1 is reported to be a homodimeric protein and binding of Cdc42/Rac1 to Cdc42/Rac1 binding domain (CRIB) relieves the inhibition by disrupting the PAK1 homodimer [18]. Monomeric PAK1 subsequently undergoes autophosphorylation/phosphorylation for full PAK1 activation. Therefore to determine direct activation of PAK1 by MLK3, first, the bacterially expressed Cdc42 protein was charged, either with -GTPγS or -GDPβS (as a control) and PAK1 protein was pre-incubated with charged Cdc42 proteins to disrupt the auto-inhibitory conformation. Subsequently, PAK1proteins, pre-bound to either Cdc42 –GTPγS, or -GDPβS were incubated with purified MLK3 enzyme, and the kinase activity of PAK1 was determined using histone H4 as the substrate [19]. As expected, the PAK1 was activated upon binding with Cdc42-GTPγS and its activity was significantly increased (about 65folds) in the presence of MLK3, thus proving that MLK3 directly activates PAK1 (Fig. 2c). To further validate that MLK3 is an activator of PAK1, we used MLK3 agonists, TNFα [20] and CD3/CD28 (our unpublished observation) in Jurkat cells, where we have observed a robust endogenous expression of MLK3 and PAK1 proteins. Both agonists of MLK3 activated PAK1 and this activation was blocked when the cells were pre-treated with a pan-MLK inhibitor, CEP-11004 (Fig. 2d), confirming further that MLK3 is an activator of endogenous PAK1. It was reported that CEP-1347, an analog of CEP-11004 also inhibits PAK1 [21] and therefore, we did additional experiments where CEP-11004 was added in in vitro kinase assays, where either PAK1 or MLK3 were immunoprecipitated from MLK3-agonist treated cells. Our results clearly showed that CEP-11004 is an MLK3 inhibitor and it does not inhibit PAK1 kinase activity directly (Fig. Supplementary 2b and c).
Fig. 2.

MLK3 activates PAK1. a Total cell lysates were prepared from HEK-293 cells, expressing constant amount of Myc-tagged PAK1 (1μg) with two different concentrations of M2-tagged MLK3 DNA (100 ng and 500 ng). PAK1 was immunoprecipitated by anti-Myc-tag specific antibody and kinase assay was done using histone H4 as the substrate of PAK1. b Similarly, cell lysates from HEK-293 cells expressing constant amount of MLK3 (1μg) and two different concentration of PAK1 (100 ng & 500 ng) DNA were prepared and MLK3 was immunoprecipitated (by anti-M2) and kinase assay was done using bacterially expressed SEK1(K-R) as a substrate. c Myc-PAK1 was immunoprecipitated from HEK-293 cells and pre-incubated, either with bacterially expressed GTP-Cdc42 or GDP-Cdc42 loaded proteins. Purified MLK3 enzyme was added in the reaction and PAK1 activity was measured with histone H4 as the substrate. d Jurkat cells were pre-treated with MLK inhibitor, CEP-11004 for 12 hours and then treated with either TNFα or CD3/CD28. The PAK1 was immunoprecipitated using anti-PAK1 antibody and kinase assay was done. These kinase assay data represent one of three similar experiments.
MLK3 activates PAK1 via direct phosphorylation.
PAK1 exists in an inactive homodimeric state and Cdc42/Rac1 binding disrupts the dimer, leading to autophosphorylation/phosphorylation and full activation of PAK1 [18], and therefore neither full length dimeric PAK1 nor Cdc42/Rac1 bound proteins are appropriate to determine direct phosphorylation by MLK3. To overcome these difficulties, two different fragments of PAK1 protein were expressed in bacteria and a direct phosphorylation assay was done using purified MLK3 enzyme. The MLK3 enzyme was able to directly phosphorylate PAK1 1-166 aa and 1-288 aa fragments (Fig. 3a). The phosphorylation sites on PAK1 were determined by mass spectrometry to be Ser133 and Ser204 (Figs. 3b and Supplementary 3a & b). To confirm that these are direct phosphorylation sites, Ser133 and Ser204 were replaced with a phospho-deficient amino acid, Alanine and direct phosphorylation of wild type and mutant PAK1 proteins was determined. Although, MLK3 enzyme was able to phosphorylate wild type PAK1, phosphorylation of S133A and S204A mutant proteins were significantly diminished in in vitro assays (Fig. 3c), indicating that these sites are direct targets of MLK3. To test whether these phosphorylation sites are necessary for PAK1 activation or inhibition, we replaced these two Serine residues individually or together with Alanine in full length mammalian PAK1 expression plasmid and co-expressed with MLK3 in HEK-293 cells. PAK1 kinase assay clearly showed that Ser204 phosphorylation is critical for PAK1 activation by MLK3. Interestingly, the PAK1S133A mutant had higher activity than full length PAK1 (Fig. 3d). The Ser133 site is located within the PAK1 auto-inhibitory domain and perhaps any perturbation within this domain could lead to PAK1 activation [22]. To further confirm that the Ser204 site phosphorylation is critical for PAK1 activation, Ser204 was replaced with phospho-mimetic, Aspartic acid (i.e. S204D). The PAK1 S204D mutant protein was constitutively active and no further activation was observed with MLK3 (Fig. 3e), indicating that phosphorylation of Ser204 by MLK3 is important for PAK1 activation.
Fig. 3.

Direct phosphorylation of PAK1 by MLK3 promotes PAK1 activation. a Bacterially expressed PAK1 fragments 1-166 aa and 1-288 aa were phosphorylated by purified MLK3 enzyme in vitro and sent for P-site determination by Mass spectrometry. b Showing location of S133 and S204 phosphorylation sites in PAK1. c Ser133 and Ser204 either alone or together were mutated to Alanine and in vitro phosphorylated with purified MLK3 enzyme. d Ser133 and Ser204, either alone or together mutated in PAK1 mammalian expression plasmid and co-transfected with M2-tagged MLK3. The PAK1 proteins were immunoprecipitated and kinase activity was measured using histone H4 as the substrate. e The Ser204 in PAK1 was mutated to Asp (D) and assayed for kinase activity. f A computational model of PAK1, based on the x-ray structure 1F3M. g A computational model of PAK1S204A. h A computational model of PAK1S133A,S204A. i A computational model of PAK1S204D. In all models, the kinase domain (249-545 aa) is colored dark gray, the auto-regulatory fragment 1 (70-149 aa) is colored red, and the auto-regulatory fragment 2 (150-220 aa) is colored blue. Asp389 and Asp407 are colored orange, Ser133 and S204 are shown in yellow, and Lys 141 is colored by atom. These kinase assay data represent one of three similar experiments.
To determine the mechanistic basis of PAK1 activation via MLK3-induced phosphorylation, we performed computational modelling of pSer133 and pSer204. The crystal structure of full length PAK1 is not available, however, the x-ray structure of the auto-inhibited conformation of PAK1 (PDB ID: 1F3M) that includes the auto-inhibitory fragment (70-149 aa) and the kinase domain (249-545 aa) is reported [18]. In this model, the kinase inhibitory segment (137-149 aa) stabilizes the inactive conformation of the catalytic domain and forms a triad, consisting of Lys141 in the auto inhibitory fragment and aspartates 389 and 407 in the catalytic domain. Using protein-protein docking, we found an energetically favorable position for the fragment (150-220 aa) that contains the Ser204 site and was located proximal to the auto-inhibitory Lys141 and to Ser133 (Fig. 3f). This unique position can influence PAK1 activity by either pulling Lys141 away from the active site or by forming an additional triad with Ser133. To test this, we used protein refinement tools from GalaxyRefineWeb [23], to map the possible Lys141 rotamer orientations and conformations affected by mutations of Ser133 or/and Ser204. Substitution of Ser204 to aspartate resulted in movement of Lys141 away from Asp389 and Asp407, disrupting the auto-inhibitory triad (Fig. 3i). However, single (S204A) or double (S133A, S204A) mutations had minimal effect on the Lys141-Asp389-Asp407 triad (Fig. 3g and h). Overall, our in silico results suggest that phosphorylation of Ser204 might alter the orientation of Lys141, liberating this residue from its inhibitory interaction with Asp389 and Asp407 and, subsequently, activating PAK1 kinase activity.
The downstream functions of PAK1 are regulated via MLK3-mediated phosphorylation.
PAK1 is a major oncogene amplified in about 17 % of breast cancers [24]. It is reported that PAK1 can activate NFĸB via unknown mechanisms [25]. Given that phosphorylation of PAK1 on Ser204 increases its kinase activity, we tested whether phosphorylation of Ser204 can regulate NFkB activation. NFĸB activity indeed was 200 folds less in a breast cancer stable cell line, expressing PAK1S204A, compared to cells expressing phosphomimetic PAK1 mutant (PAK1S204D) (Fig. 4a). Moreover, the NFĸB activity in cells expressing wild type PAK1 activity was almost 5 folds higher compared to cells expressing PAK1S204A (Fig. 4a). To further confirm that activation of NF-kB is through PAK1 phosphorylation by MLK3, and MLK3 does not activate NF-ĸB directly, we stably co-expressed doxycycline-regulated MLK3 in MDA-MB-468 cells, either expressing PAK1 -WT or -S204A mutant. The NF-ĸB was activated by PAK1-WT when MLK3 was expressed (i.e. +Dox) and PAK1 S204A mutant failed to activate, even in presence of MLK3 (i.e. +Dox). (Fig. Supplementary 4a). PAK1 is also reported to activate JNK [26], ERKs [27] and AKT [28] pathways. The co-expression of MLK3 and PAK1 did not activate JNK (Supplementary Fig. 4b) nor there was any effect of PAK1 -WT or -S204A on ERK or AKT activations (Fig. Supplementary 4c). Interestingly, the expression of PAK1S204A was significantly lower than wild type and phosphomimetic PAK1 (Fig. 4a), suggesting that perhaps phosphorylation of PAK1 by MLK3 helps in PAK1 protein stability.
Fig. 4.

The downstream functions of PAK1 is regulated via MLK3 phosphorylation. a MDA-MB-468 breast cancer cells were made stable, overexpressing either PAK1 -WT or -S204A, or -S204D and blotted with p-NFĸB p65 (pS536), total NFĸB p65 and anti-Myc (for Myc-tagged PAK1) antibodies. b Colony formation assay was done with MDA-MB-468 cells overexpressing either PAK1WT or PAK1S204A. Representative pictures for colony growth are shown. Plot shown as mean ± SEM by one-tailed Student t-test (n=3, t=42.14, df=2, P<0.0001). c The migration of MDA-MB-468 overexpressing either WT- or S204A-PAK1 was done by scratch assay and d Relative distance of wounds were depicted graphically as mean ± SEM by two-way ANOVA (n=3, df=2, F=241.6, P<0.001). Statistical testing was done with 95% confidence interval by Bonferroni posttests. e Invasion assay was done using Boyden chamber with MDA-MB-468 stable cells overexpressing either PAK1 -WT or -S204A, or -S204D and f quantified. Data represented as mean ± SEM by one-way ANOVA (n=3, df=2, F=1682, P<0.0001). Statistical testing was done with 95% confidence interval by Bonferroni multiple comparison test. g MDA-MB-468 stable cells, overexpressing either PAK1 -WT or -S204A were made stable, co-expressing doxycycline-regulated MLK3 and their invasive capacities were determined similarly as described in Fig. e, either in presence (+DOX) or absence (-DOX) of MLK3 expression and h quantified. Data represented as mean ± SEM by two-way ANOVA (n=3, df=3, F=10309, P<0.0001). Statistical testing was done with 95% confidence interval by Bonferroni Posttests. i MDA-MB-468 cells overexpressing PAK1WT or PAK1S204A were stained with anti-Myc-tag antibody and imaged using confocal microscope. j The Myc-tagged PAK1 was immunoprecipitated from MDA-MB-468 cells overexpressing either WT- or S204A-PAK1 and blotted for endogenous Paxillin.
PAK1 activation is known to promote cell growth, migration and invasion of breast as well as other cancer cell types [13, 29]. We determined, whether phospho-deficient, PAK1S204A could block any of the downstream effects of wild type PAK1. In fact, PAK1S204A mutant was able to attenuate the numbers of breast cancer colonies (Fig. 4b) and prevent migration (Figs. 4c and d). The PAK1 protein is reported to play significant roles in cell invasion and therefore, we determined the invasive capacity of MDA-MB-468 cells, stably expressing either PAK1 -WT or -S204A, or -S204D. The invasion of cells expressing PAK1 -S204D was highest compared to -WT and it was least in -S204A (Fig. 4e and f). MLK3 has also been reported to promote cell migration [6] and invasion [30] and therefore to determine the effect of MLK3 in PAK1-mediated cell invasion, MDA-MB-468 cell lines, stably expressing either PAK1 -WT or -S204A were made stable to co-express doxycycline-regulated MLK3. The invasion capacity of cells expressing PAK1-WT and MLK3 (i.e. +Dox) was about 5 folds higher compared to only PAK1-WT expressing cells (i.e. -Dox). (Fig. 4g and h). Interestingly the invasion of cells, expressing PAK1S204A was significantly lower, irrespective of MLK3 -expression (i.e. +Dox) or -no expression (-Dox) (Fig. 4g and h). We also determined the localization of PAK1 -WT and -S204A mutant in breast cancer cell line. Interestingly, the PAK1S204A mutant appeared to be localized at the focal adhesions-like structure (Fig. 4i). It is reported that PAK1 can phosphorylate cytoskeletal protein, Paxillin at the focal adhesion junction to control cell migration [31]. Since, the migration and invasion of breast cancer cell line expressing PAK1S204A was significantly attenuated, we determined interaction between PAK1S204A and Paxillin. Interestingly, the mutant PAK1 was tightly associated with Paxillin as compared to wild type PAK1, even though the expression of PAK1S204A was significantly lower than wild type PAK1 (Fig. 4j). It is reported that kinase active PAK1 transiently associates with Paxillin, and subsequently dissociates upon phosphorylating Paxillin and this dissociation is necessary for cell migration/invasion [31]. Therefore, it appears that phosphorylation of Ser204 on PAK1 by MLK3 is critical for PAK1 function in promoting cell migration and invasion, most likely by regulating interaction between Paxillin and PAK1.
Phosphorylation of PAK1 by MLK3 is necessary for breast tumorigenesis.
Activation of PAK1 has been reported to promote breast tumorigenesis [32]. We examined whether phosphorylation of PAK1S204 site by MLK3 has any role in breast tumorigenesis. Stable MDA-MB-468 breast cancer cell line, expressing luciferase along with, either PAK1 -WT or -S204A were orthotopically implanted in mammary fat pads of SCID mice. The tumors were imaged using IVIS and measured bioluminescence emission up to 34 days to determine tumor growth/volumes (Fig. 5a and b). Our data clearly showed that tumor volume and weight were significantly lower in cells expressing PAK1-S204A, compared to -WT (Figs. 5c and d). Cell proliferation as measured by Ki67 staining (Figs. 5e and f) and NFĸB activity (Figs. 5g and h) were diminished in tumors expressing PAK1S204A mutant. These data suggest that phosphorylation of PAK1 by MLK3 promotes breast tumorigenesis.
Fig. 5.

Phosphorylation of PAK1 by MLK3 promotes breast tumorigenesis. MDA-MB-468 cells stably expressing luciferase, either with PAK1WT or PAK1S204A was transplanted in 4th mammary fat pads and a tumors were imaged using IVIS imaging system. b Tumor volume was quantified by measuring bioluminescence emission (photonic flux) of ROI using Living Image software. Data denoted mean ± SEM by two-way ANOVA (n=4, df=2, F=107.6, P<0.001). Statistical testing was done with 95% confidence interval by Bonferroni posttests. c Tumor volume was measured using calipers and representative isolated tumors are presented. Plot presented as mean ± SEM by one-tailed Student t-test (n=4, t=3.876, df=3, P<0.001). d Tumor weight was determine and plotted with mean ± SEM by one-tailed Student t-test (n=4, t=15.50, df=3, P<0.0001). e The cell proliferation in tumors were determined by Ki67 staining and f Quantitative plot describing the percentage of Ki67-positive cells was estimated under microscope and presented as mean ± SEM by one-tailed Student t-test (n=4, t=28.04, df=3, P<0.0001). g The pNFĸB p65 (S536) and total NFĸB p65 in tumor were detected by immunohistochemistry (IHC) and h estimated under microscope as the percentage NFĸB -positive cells and presented as mean ± SEM by one-tailed Student t-test (n=4, t=20.79, df=3, P<0.0001).
DISCUSSION
MLK3, also called MAP3K11 is a family member of MLK group of kinases and most of our understanding about MLK family is centered on MLK3 [1]. Recently MLK3 and other MLK members have emerged as a therapeutic target in neurodegenerative diseases [8] and cancer [1], yet its upstream regulator(s) and downstream function(s) are not fully understood. Here we demonstrate that MLK3 associates with a mammalian homolog of yeast Ste20, PAK1. PAK1 being a Ste20 member [13], we expected it to be an upstream regulator of MLK3, however our observed results were opposite to our initial anticipation. Contrary to our initial assumption, MLK3 was able to phosphorylate and activate PAK1 kinase activity, whereas PAK1 was unable to phosphorylate or activate MLK3. MLK3 was able to directly phosphorylate PAK1 on two Serine residues of which Ser204 is critical for MLK3-induced PAK1 activation and downstream functions. The phosphorylation of PAK1Ser204 is important for PAK1 functions in breast cancer and promotes NFĸB activation, cell migration, invasion and tumorigenesis.
Most of the signaling pathways in yeast and mammals are evolutionary conserved and in yeast Ste20 members are reported to function as MAP4K [33]. PAK1 being one of the mammalian homologs of yeast Ste20 member reported to play a significant role in cancer, we initially envisioned that PAK1 might be an upstream regulator of MLK3. Our data clearly showed that endogenous or overexpressed MLK3 and PAK1 do associate with each other (Fig. 1b-d), but PAK1 was unable to modulate MLK3 kinase activity (Fig. 2b), whereas MLK3 activated PAK1 when co-expressed in mammalian cells (Fig. 2a). Since overexpression of PAK1 as well as MLK3 is reported to constitutively activate their kinase activities, one possibility remains that overexpressed MLK3 is super-active and it cannot be further activated by PAK1. We reported earlier that endogenous MLK3 was not constitutively active and can be activated by its agonist TNFα and Ceramides [20], and recently we have observed that MLK3 can also be activated upon T-cell receptor (TCR) engagement (unpublished data). To rule out the possibility of transfected MLK3 being super-active, we tested whether agonists of MLK3 were able to activate endogenous PAK1 kinase activity in Jurkat T-cells, where both proteins are abundantly expressed. Our results indicated that both MLK3 agonists, TNFα and CD3/CD28 (T-cell receptor engagement) were able to activate endogenous PAK1 and pan-MLK inhibitor was able to block MLK3’s agonist-induced PAK1 activation (Fig 2d). The pan-MLK inhibitor, CEP-1347 is also reported to be a PAK1 inhibitor, and CEP-11004 is an analogue of CEP-1347 and acts similar to CEP-1347 [21]. To rule out the possibility that CEP-11004 was inhibiting PAK1 directly and independent of MLK3, we determined its effect on PAK1 kinase activity in vitro. Interestingly, CEP-11004 was able to inhibit agonist-induced MLK3 kinase activity in vitro, however it failed to inhibit PAK1 kinase activity in same experiment (Supplementary Figs. 2b and c), indicating that MLK3 indeed is an activator of PAK1. The fact that PAK1 is a direct substrate of MLK3 was also supported by the experiment where MLK3 was able to activate PAK1 directly (in vitro) (Figs. 2a and c).
The process of PAK1 activation entails two major steps: 1) conversion of native dimeric PAK1 to monomeric form to relieve its auto inhibitory state, and 2) activation of PAK1 activity via autophosphorylation and phosphorylation by other kinases. We attempted several times to activate PAK1 by MLK3 in vitro, however MLK3 enzyme was unable to activate PAK1 directly, suggesting that perhaps MLK3 alone is not able to disrupt the homodimeric PAK1 in vitro. It is reported that binding of Cdc42/Rac1 to PAK1 CRIB domain disrupts the homodimers to generate monomeric forms that undergo autophosphorylation and phosphorylation for full activation [18]. The incubation of GTP-Cdc42 in vitro was able to activate PAK1 and addition of MLK3 enzyme further activated (more than 65 folds) PAK1 activity (Fig. 2c), suggesting that monomeric PAK1 is a substrate of MLK3 and perhaps MLK3 directly phosphorylates PAK1. Since full length PAK1 tend to form homodimer and disruption of PAK1 homodimer by GTP-Cdc42 leads to PAK1 kinase activation via autophosphorylation, therefore to identify any phosphorylation site(s) on PAK1, we used smaller fragments of PAK1 protein for direct phosphorylation assays. The PAK1 1-166 aa and 1-288 aa fragments were robustly phosphorylated by MLK3 (Fig. 3a) and phosphorylation site analyses identified Ser133 and Ser204, two phosphorylation sites on PAK1 (Fig. Supplementary 3). It was our expectation that phospho-deficient mutants of either Ser133 or Ser204 should be catalytically inactive, provided these sites are important for MLK3-induced PAK1 activation. To our surprise, the PAK1S133A mutant was constitutively active (Fig. 3d). Interestingly, the Ser133 is located within the auto-inhibitory domain of PAK1 [22], therefore it was not surprising to observe that the substitution of Ser133 by Ala was able to constitutively activate PAK1 kinase activity. The PAK1 Ser133 site is a new phosphorylation site and has never been reported before, and the significance of Ser133 in PAK1 biology is still not known. Nevertheless, when we measured kinase activity of PAK1S204A, with or without substituting Ala at Ser133 site, the PAK1 was completely catalytically inactive (Fig. 3d). The Ser204 has been reported as an auto-phosphorylation site [34, 35], however, our data clearly showed that Ser204 is a direct MLK3 target and important for MLK3-induced activation, which was confirmed by substituting phospho-mimetic aspartate on Ser204 (Fig. 3e). The in silico modelling showed that Ser204 site mutagenesis to aspartate substitution can influence Lys141 conformation leading to PAK1 activation (Fig. 3i). This is one of the plausible structural effects directly leading to activation of PAK1. Other structural changes caused by phosphorylation of Ser204 might indirectly enhance the kinase activity by binding the signaling partners, or/and influencing substrate affinity and specificity.
The activation of PAK1 has been shown to promote tumorigenesis, including breast tumorigenesis, by regulating signaling pathways that control cell proliferation, survival, migration, invasion, epithelial-mesenchymal transition (EMT) and anchorage-independent growth processes that are conceptualized to constitute the hall marks of cancer [13]. Previously, it was reported that activation of PAK1 promotes NFĸB activation and cell survival via some unknown mechanisms [25]. Since NFĸB relates to survival and our data showed that PAK1 phospho-deficient, Ser204A was able to inhibit NFĸB activation (Figs. 4a and 5g), therefore one of the mechanism by which PAK1 can activate NFĸB could be via PAK1 phosphorylation/activation by MLK3. In agreement with PAK1 function in cancer, we observed that PAK1S204A mutant was able to antagonize cell migration (Figs. 4c and d), invasion (Figs. 4e-h), proliferation (Fig. 4b) and breast tumorigenesis in animals (Fig 5a). PAK1 is reported to be in the nucleus and nuclear PAK1 is reported to promote Tamoxifen resistance [36]. We observed that PAK1 WT was partly localized in the nucleus, however S204A mutant was primarily in the cytoplasm in xenografts (Fig. 5e). Therefore, one possibility is that PAK1 phosphorylation by MLK3 might promote Tamoxifen or other therapeutic resistance, which needs to be further investigated.
Based on our current data and published results, we propose a model for MLK3-induced PAK1 phosphorylation and its impact on breast tumorigenesis (Fig. 6). Upon activation of MLK3 by stimuli, such as TNFα [20], or in breast cancer cells, loss of estrogen receptor (i.e. ER-negative) [4] or HER2 (i.e. HER2-negative) [9], that is reported to activate MLK3, the activated MLK3 could phosphorylate PAK1 on Ser204 site and promote activation of NFĸB, leading to cell survival, and thus tumorigenesis. Previously, we have reported that TNFα can activate MLK3 and it is reported that TNFα can also activate PAK1 to promote invasion via MMPs [37]. Therefore it is possible that MLK3 might mediate TNFα effect on cell invasion/migration through PAK1 phosphorylation/activation. Besides activating downstream NFĸB, the activated PAK1 can also phosphorylate cytoskeletal protein Paxillin to promote cell migration and therefore the net result of PAK1 activation by MLK3 will be to promote pathological function of PAK1 in cancer.
Fig. 6.
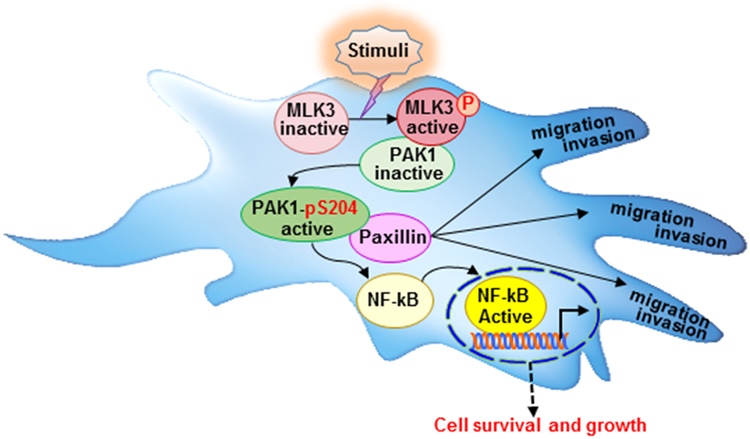
Model illustrating MLK3 can directly phosphorylate PAK1 on Ser204 site for its activation and promoting tumorigenesis, and migration/invasion.
In conclusion, our data provide a novel insight towards the role of MLK3 in PAK1 regulation via direct phosphorylation/activation that regulates the pathological functions of PAK1, including cell growth, migration, invasion and tumorigenesis. For more than two decades, efforts have been made to target PAK1 and other PAK members, but all attempts have failed due to the off target effects. Furthermore, caution approach needs to be considered because inhibiting all PAKs could prove detrimental. It is reported that knockdown of PAK1 or PAK4 inhibits proliferation of mutant KRas colon cancer cells independent of RAF/MEK/ERK and PI3K/AKT pathways [38], suggesting that alternative signaling pathways are involved. It is likely that MLK3-PAK1-NFĸB axis might be an alternative pathway, because ERK, JNK and AKT were not regulated by MLK3-PAK1 axis. Therefore, it is conceivable that MLK3 and perhaps other MLKs could serve as better upstream targets to prevent PAK1-induced tumorigenesis.
MATERIALS AND METODS
Cell lines and treatments.
The human breast cancer, MDA-MB-468, human embryonic kidney 293 (HEK-293) and Jurkat T-cell lines were purchased from American Type Cell Culture (ATCC, Manassas, VA), the HEK-293-FT cells were from Life Technologies, USA. The HEK-293, HEK-293-FT and MDA-MB-468 cell lines were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine, and 100 units/ml penicillin/streptomycin. Jurkat T-cells were maintained in RPMI medium containing 4.5 g/L glucose, 10 mM HEPES, 1.0 mM sodium pyruvate, 10% FBS and 50 μM 2-ME. The Jurkat T-cells were starved in RPMI medium containing 0.2% FBS for 12 hours, and then treated, either with 10 nM TNF-α (Roche, IN) or CD3/CD28 antibodies (BD Biosciences, CA). The CEP-11004 (KT 8138) was kindly provided by Cephalon, Inc. (USA), solubilized in DMSO at a concentration of 4 mM (stock solution) and stored at −20°C in small aliquots in amber glass vials. The CEP-11004 was freshly diluted in serum free DMEM containing 0.05% BSA, just before the treatments. The Cells were pretreated with 500 nM CEP-11004 for 12 hours and then treated with either TNF-α or CD3/CD28 antibodies as indicated. T-cell activation by CD3/CD28 antibodies was carried out as described [39]. Briefly, Jurkat T cells were treated on ice for 12 min with (5 mg/ml) of anti-CD3 (OKT3) and (2 mg/ml) anti-CD28 mAbs. The crosslinking of CD3/CD28 antibodies was done using 10 mg/ml goat anti-hamster IgG Ab and the cells were then immediately placed at 37 °C for 4 minutes.
Antibodies and reagents.
Antibodies against NFkB p65 (Cat#8242, dilution 1:2000), pAKTS473 (Cat#4060, dilution 1:1000), Akt (Cat#4691, dilution 1:2000), pERK (Cat#4370, dilution 1:1000), ERK (Cat#4695, dilution 1:2000), anti-Myc-Tag (Cat#2276, IHC 1:100) were purchased from Cell Signaling Technology, MA. Antibodies against pNFkB p65S536 (Cat#86988, dilution 1:1000) and Paxillin (Cat#32084, dilution 1:3000) were from Abcam. The anti-Myc Ab (Roche, Cat#10138400, dilution 1:5000), anti-M2 (Sigma, Cat#F1804, dilution 1:2000) and GAPDH (Proteintech, Cat#10494-1-AP, dilution 1:5000) antibodies were from Roche, Sigma-Aldrich, and Proteintech respectively. Protein A-Sepharose and GSH coated beads were purchased from GE Healthcare. Histone H4 was obtained from New England Biolab, MA; Guanosine 5′-diphosphate sodium salt (GDP) and GTP-γ-S tetralithium salt were from Sigma/Roche and all other chemicals or reagents including anti-c-Myc-magnetic beads were from Sigma-Aldrich, USA. The GST-MLK3 enzyme was obtained either from Invitrogen, USA or from SignalChem, Canada. The GST-PAK1 kinase was from Cell Signaling Technology, MA and Matrigel were from BD Biosciences USA. The D-luciferin was purchased from GoldBio, USA and Prolong-diamond antifade mount with DAPI was from Life Technologies, USA. Cytoselect 24-well wound healing assay kit was from Cell Biolab Inc., USA.
Expression vectors and mutagenesis.
The pRK5-Flag-MLK3 and kinase deficient pRK5-Flag-MLK3 (K-A) mammalian expression vectors are described earlier [3]. The GST versions of MLK3 deletion mutants were generated by PCR using pRK5-Flag-MLK3 wild type or kinase deficient as template and sub-cloned into mammalian expression vector, pEBG. Myc tagged PAK1 (WT), and pGEX-Cdc42 were used as described earlier[40].The GST versions of PAK1 were generated by PCR using pCMV5-Myc-PAK1 as a template and sub-cloned into pEBG vector. The pRK5-Myc-PAK2 was provided by Garry Bokoch (Scrips Research Institute), pEBG-GCK was provided by J. Kyriakis (Massachusetts general Hospital) and pEBG-MST1 by L. Zon (Children’s hospital, Boston). The mammalian and bacterial expression vectors of mutant PAK1 were generated using the QuikChange Lightning Site-Directed mutagenesis kit (Agilent Technologies, USA) and sequenced using Sanger’s method at UIC-RRC lab. For lentiviral vector construction, the human full-length Myc-PAK1 WT, Myc-PAK1 -S204A and -S204D coding sequences (CDS) were cloned into the pDONOR221 vector using BP Clonase (Life Technologies, USA), attB1 and attB2 primers. The LR recombination reaction was performed with the entry vector using LR Clonase (Life Technologies, USA) to generate lentiviral vector in the pLENTI 6.3-DEST backbone. The FuhLUCW plasmids that express luciferase was provided by Jithesh Pratap (RUSH University, Chicago, USA).
Virus production and cell transduction.
Lentiviruses were produced by co-transfecting the lentiviral vectors for Myc-PAK1 -WT, -S204A and -S204D, pFuHLUCW with psPAX2 and pMD2G packaging plasmids into HEK 293FT (Life Technologies, USA) cells using Lipofectamine 2000 (Life Technologies, USA). To achieve an MOI of 0.3–0.5, titrations were performed and the infection efficacy of was determined using western blotting. The MDA-MB-468 cells were infected with an optimal volume of virus in medium containing 5 μg/ml polybrene (Sigma-Aldrich, USA). Stable cell lines were selected using 2 μg/ml Balsticidin (GoldBio, USA) for 10 days.
Recombinant protein expression, GST pull-down, immunoprecipitation, immunoblotting kinase assay and phosphorylation site determination.
For in vitro assays, GST-SEK1 (K-R)/MKK4 (K-R), GST-Cdc42 and GST-PAK1 (1-166), GST-PAK1 (1-288) WT or mutants (either S133A, S204A or both) were expressed in bacteria (strain BL21; Pharmacia, NJ) and purified over GSH beads. Proteins were eluted with reduced glutathione, dialyzed and stored at −20°C in 50% glycerol. HEK-293 cells were lysed in NP-40 lysis buffer and cell lysates were prepared and Immunoblotting was performed using a standard method as described previously[3]. The recombinant MLK3 and PAK1 from HEK-293 cell extracts were immunoprecipitated using protein A-Sepharose pre-absorbed with either anti-Flag (M2) or anti-Myc (9E10) antibody or anti-c-Myc-magnetic beads. Kinase assay was performed as described earlier[3] in presence of either GST-SEK1 (K-R)/MKK4 (K-R) or histone H4 as the substrates. For in vitro kinase assays involving GST-Cdc42, bacterially expressed Cdc42 protein was charged either with GTPγS or -GDPβS and incubated with PAK1, prior to the assay. The proteins were separated on denaturing SDS/PAGE and transferred on to PVDF membrane and blotted with antibodies as indicated. The phosphorylation sites of PAK1 was determined upon incubating bacterially expressed PAK1 fragments, 1-166 and 1-288 aa with MLK3 enzyme. The phosphorylated PAK1 fragments were excised and sent out (UAMS Proteomics Core Lab, Little Rock, AR) to determine the p-sites by Mass Spectrometry.
Anchorage-independent growth in soft agar.
A volume of 1.5 ml of 0.6% agar (electrograde ultra-pure) supplemented with DMEM, 10% FBS, was plated in six-well culture dishes as bottom agar. Three thousand MDA-MB-468 cells stably expressing Myc-PAK1 WT or S204A were mixed with 1.5 ml of 0.3% agar supplemented with DMEM, 10% FBS, and plated on top of the bottom agar. A volume of 1ml of media was added on top of solidified agar layers and colonies were allowed to grow in incubator at 37ᴼC for 21 days. Colonies were stained with crystal violet and counted under bright-field microscope and imaged using Image Lab software in Bio-Rad imager system.
Immunohistochemistry, immunostaining and microscopy.
Formalin-fixed and paraffin-embedded MDA-MB-468 mouse xenograft breast cancer tumors were processed and prepared for tissue sectioning, using standard protocol at the UIC Histology Core laboratory. Paraffin-embedded tissue sections were deparaffinized in xylene, rehydrated through 100% ethanol to TBST wash solution. The antigenic epitope was retrieved using Sodium citrate buffer (pH 6.0) in decloaking chamber at 950C for 20 mins. Peroxidase quenching was performed using BLOXALL (Vector Lab, USA) and blocked in 5% goat serum for 30 min. Slides were then incubated with primary antibodies (anti-Myc-Tag Ab (CST, 1:200), anti-Ki67 (CST, 1:400), pNFĸB p65S536 (Abcam, 1:100), NFĸB p65 (CST, 1:100)) in 1X antibody diluent (DAKO, USA) for overnight at 4ᴼC in humidified chamber and washed three times with TBST. Slides were incubated with secondary-HRP Abs (Vector Lab, USA) and developed using DAB (Vector Lab, USA). The nuclei were counterstained with Hematoxylin (RICCA, USA) for 5 min, rinsed in deionized water and mounted with Krystalon mounting medium (Millipore Sigma, USA). Bright field microscopy was done using NIS-Elements imaging software in Nikon Eclipse Ti Microscope. For immunofluorescence (IF) analysis, stably myc-PAK1 WT / S204A overexpressing MDA-MB-468 cells were grown on the cover glass, after 48 hrs. cells were fixed with 4% paraformaldehyde and proceeded for the staining as described previously [5]. The Myc-tagged PAK1 and Paxillin were stained overnight at 4ᴼC with anti-Myc-Tag (CST, 1:200) and Paxillin (Abcam, 1:200) antibodies respectively. After extensive wash with TBST, slides were incubated with Alexa549-labeled anti-rabbit IgG (Abcam 1:1000) for Paxillin and Alexa488-labeled anti-mouse IgG (Abcam, 1:1000) for Myc-PAK1 for 45 min at room temperature. After washing with TBST, the nuclei were stained with DAPI using ‘prolong-diamond antifade mount with DAPI’ (Life technologies, USA) and pictures taken using confocal microscope (Zeiss LSM microscope 710 , Carl Zeiss, Inc.) equipped with Zeiss confocal scanning optics.
Cell migration and invasion assays.
The MDA-MB-468 cells, stably expressing either Myc-PAK1 WT or S204A were plated in Cytoselect 24-well wound healing assay plate and Cell migration was determined following manufacturer’s protocol (Cell Biolab Inc., USA). The images of specific wound areas were captured by phase contrast microscopy (Nikon Eclipse Ti inverted microscope) at 0, 48 and 72 hrs. The width of the wounds were measured using the NIS-Element software associated with the microscope. The cell invasion was determined using the 24-well Boyden chamber plate with 8-mm pore polycarbonate membranes pre-coated with Matrigel Basement Membrane Matrix (BD Biosciences). The chambers were rehydrated in serum-free medium and complete medium with 10% FBS was used as chemoattractant in the bottom chamber. The MDA-MB-468 (1×104) cells, stably expressing, either Myc-PAK1 -WT, -S204A or -S204D were incubated in the upper chamber for 36 hrs. in tissue culture incubator. In another experiment, MDA-MB-468 cells stably expressing either Myc-Pak1 -WT or -S204A were transduced with doxycycline-inducible MLK3-M2 WT and made stable for its expression. These stable cell lines were grown overnight either in presence or absence of doxycycline to regulate MLK3 expression. Cells were then trypsinized and seeded (1×104) in the upper Boyden chamber for 36 hrs. The non-invading cells from upper chamber and matrigel were wiped using a cotton swab and the membrane was stained with 0.05% crystal violet solution and imaged in a bright-field microscope.
In-silico modeling.
PAK1 Homology model:
The program Iterative Threading ASSEmbly Refinement (I-TASSER) [41-43] and I-TASSER server [44, 45] were used to build a homology model of the PAK1 region (150-220 aa). Five homology models were predicted using the settings and parameters recommended by the developers [41-45].
Docking:
ZDOCK server [46] was used for docking the generated models of PAK1 region (150-220 aa) to the x-ray structure of PAK1 (PDB ID: 1F3M) containing the kinase domain (249-545 aa) and the auto inhibitory fragment (70-149 aa). Each docking run generated 10 models. The model that showed the placement of PAK1 region in proximity of the auto inhibitory fragment was used for protein minimization/refinement calculations.
Protein minimization:
Based on the site mutagenesis studies and the biochemical functional assays, mutations were introduced to PAK1 region (150-220 aa) or/and the auto inhibitory fragment (70-149 aa) using Pymol 1.3 mutagenesis wizard (The PyMOL Molecular Graphics System, Version 1.3 Schrödinger, LLC). The resulting models were refined using GalaxyRefineComplex server [23]. For each mutation, ten models were generated. The resulting models were inspected for mutations effects on Lysine 141 rotamers orientation or/and conformations using Pymol 1.3.
Visualization:
The program Pymol 1.3 was used for visualization and production of PAK1 mutants’ models.
Mouse xenografts tumor model.
The female SCID mice (NOD.CB17-Prkdcscid/NCrCrl, strain code: 394) were purchased from the Charles River Laboratories. All SCID mice were housed in the specific pathogen free animal facility of the Biologic Resources Laboratory, University of Illinois at Chicago and experiments were conducted as per the approved protocol by IACUC. Randomly selected 4-5 weeks SCID mice (n=4) were anesthetized and MDA-MB-468 (1×106) cells, stably expressing luciferase along with either Myc-PAK1 -WT or -S204A were mixed with 100 μl matrigel and orthotopically inoculated into the mammary fat pad for tumor induction. The tumor growth was monitored upon intraperitoneal injection (150 mg/Kg. b.w.) of D-luciferin (GOLD BIO, USA). The mice were anesthetized by 2% isoflurane, 10 min after administrating luciferin, and imaged using the Xenogen IVIS Lumina system (Caliper Life Sciences, USA). Starting from the 3rd day of cells inoculation, the growth of xenografts were examined every week and euthanized on the 34th day. After scarifying these tumor-bearing mice, tumors were isolated, imaged and measured the isolated tumor volume & tumor weight, and processed for immunohistochemistry study.
Statistical analysis.
Unless otherwise indicated, all the experiments presented in the manuscript were repeated at least three times with minimum three replicates. A paired one-tail student t-test, one-way ANOVA followed by Bonferroni multiple comparison test, and two-way ANOVA followed by Bonferroni posttests was done using the GraphPad Prism (Version 5.03) software. The differences were considered significant when p values were ≤0.05.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge funding support from National cancer Institute to A.R. (CA 176846 and CA 216410), B.R. (CA 178063) and V.G. (CA 188427). Additional support came from Veteran Administration Merit Awards to A.R. (BX002703) and B.R. (BX003296). We also acknowledge the NIH shared instrument grant # S100D018445, awarded to U.A.M.S. Proteomics Core Lab, Little Rock, Arkansas, USA.
Competing Interests: The authors attest that we do not have any competing interests to declare. The work is supported through grants from National Cancer Institute to A.R. (CA 176846 and CA 216410), B.R. (CA 178063) and V.G. (CA 188427). Additional support came from Veteran Administration Merit Awards to A.R. (BX002703) and B.R. (BX003296).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
REFRENCES
- 1.Rana A, Rana B, Mishra R, Sondarva G, Rangasamy V, Das S et al. Mixed Lineage Kinase-c-Jun N-Terminal Kinase Axis: A Potential Therapeutic Target in Cancer. Genes & cancer 2013; 4: 334–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gallo KA, Johnson GL. Signalling: Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol 2002; 3: 663–72. [DOI] [PubMed] [Google Scholar]
- 3.Rana A, Gallo K, Godowski P, Hirai S, Ohno S, Zon L et al. The mixed lineage kinase SPRK phosphorylates and activates the stress-activated protein kinase activator, SEK-1. J Biol Chem 1996; 271: 19025–8. [DOI] [PubMed] [Google Scholar]
- 4.Rangasamy V, Mishra R, Mehrotra S, Sondarva G, Ray RS, Rao A et al. Estrogen suppresses MLK3-mediated apoptosis sensitivity in ER+ breast cancer cells. Cancer Res 2010; 70: 1731–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rangasamy V, Mishra R, Sondarva G, Das S, Lee TH, Bakowska JC et al. Mixed-lineage kinase 3 phosphorylates prolyl-isomerase Pin1 to regulate its nuclear translocation and cellular function. Proc Natl Acad Sci U S A 2012; 109: 8149–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mishra P, Senthivinayagam S, Rangasamy V, Sondarva G, Rana B. Mixed lineage kinase-3/JNK1 axis promotes migration of human gastric cancer cells following gastrin stimulation. Mol Endocrinol 2010; 24: 598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bodner A, Maroney AC, Finn JP, Ghadge G, Roos R, Miller RJ. Mixed lineage kinase 3 mediates gp120IIIB-induced neurotoxicity. J Neurochem 2002; 82: 1424–34. [DOI] [PubMed] [Google Scholar]
- 8.Parkinson Study Group PI. Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology 2007; 69: 1480–90. [DOI] [PubMed] [Google Scholar]
- 9.Das S, Sondarva G, Viswakarma N, Nair RS, Osipo C, Tzivion G et al. Human Epidermal Growth Factor Receptor 2 (HER2) Impedes MLK3 Kinase Activity to Support Breast Cancer Cell Survival. J Biol Chem 2015; 290: 21705–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rattanasinchai C, Llewellyn BJ, Conrad SE, Gallo KA. MLK3 regulates FRA-1 and MMPs to drive invasion and transendothelial migration in triple-negative breast cancer cells. Oncogenesis 2017; 6: e345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee BN, Elion EA. The MAPKKK Ste11 regulates vegetative growth through a kinase cascade of shared signaling components. Proc Natl Acad Sci U S A 1999; 96: 12679–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knaus UG, Bokoch GM. The p21Rac/Cdc42-activated kinases (PAKs). Int J Biochem Cell Biol 1998; 30: 857–62. [DOI] [PubMed] [Google Scholar]
- 13.Radu M, Semenova G, Kosoff R, Chernoff J. PAK signalling during the development and progression of cancer. Nat Rev Cancer 2014; 14: 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Letessier A, Sircoulomb F, Ginestier C, Cervera N, Monville F, Gelsi-Boyer V et al. Frequency, prognostic impact, and subtype association of 8p12, 8q24, 11q13, 12p13, 17q12, and 20q13 amplifications in breast cancers. BMC cancer 2006; 6: 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.French JD, Ghoussaini M, Edwards SL, Meyer KB, Michailidou K, Ahmed S et al. Functional variants at the 11q13 risk locus for breast cancer regulate cyclin D1 expression through long-range enhancers. Am J Hum Genet 2013; 92: 489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith CM, Ma NS, Nowak NJ, Shows TB, Gerhard DS. A 3-Mb contig from D11S987 to MLK3, a gene-rich region in 11q13. Genome Res 1997; 7: 835–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rider L, Oladimeji P, Diakonova M. PAK1 regulates breast cancer cell invasion through secretion of matrix metalloproteinases in response to prolactin and three-dimensional collagen IV. Mol Endocrinol 2013; 27: 1048–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parrini MC, Lei M, Harrison SC, Mayer BJ. Pak1 kinase homodimers are autoinhibited in trans and dissociated upon activation by Cdc42 and Rac1. Mol Cell 2002; 9: 73–83. [DOI] [PubMed] [Google Scholar]
- 19.Lu W, Mayer BJ. Mechanism of activation of Pak1 kinase by membrane localization. Oncogene 1999; 18: 797–806. [DOI] [PubMed] [Google Scholar]
- 20.Sathyanarayana P, Barthwal MK, Kundu CN, Lane ME, Bergmann A, Tzivion G et al. Activation of the Drosophila MLK by Ceramide Reveals TNF-alpha and Ceramide as Agonists of Mammalian MLK3. Mol Cell 2002; 10: 1527–33. [DOI] [PubMed] [Google Scholar]
- 21.Nheu TV, He H, Hirokawa Y, Tamaki K, Florin L, Schmitz ML et al. The K252a derivatives, inhibitors for the PAK/MLK kinase family selectively block the growth of RAS transformants. Cancer J 2002; 8: 328–36. [DOI] [PubMed] [Google Scholar]
- 22.Jha RK, Wu YI, Zawistowski JS, MacNevin C, Hahn KM, Kuhlman B. Redesign of the PAK1 autoinhibitory domain for enhanced stability and affinity in biosensor applications. J Mol Biol 2011; 413: 513–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heo L, Lee H, Seok C. GalaxyRefineComplex: Refinement of protein-protein complex model structures driven by interface repacking. Sci Rep 2016; 6: 32153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ong CC, Jubb AM, Haverty PM, Zhou W, Tran V, Truong T et al. Targeting p21-activated kinase 1 (PAK1) to induce apoptosis of tumor cells. Proc Natl Acad Sci U S A 2011; 108: 7177–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frost JA, Swantek JL, Stippec S, Yin MJ, Gaynor R, Cobb MH. Stimulation of NFkappa B activity by multiple signaling pathways requires PAK1. J Biol Chem 2000; 275: 19693–9. [DOI] [PubMed] [Google Scholar]
- 26.Qing H, Gong W, Che Y, Wang X, Peng L, Liang Y et al. PAK1-dependent MAPK pathway activation is required for colorectal cancer cell proliferation. Tumour Biol 2012; 33: 985–94. [DOI] [PubMed] [Google Scholar]
- 27.Wang Z, Fu M, Wang L, Liu J, Li Y, Brakebusch C et al. p21-activated kinase 1 (PAK1) can promote ERK activation in a kinase-independent manner. J Biol Chem 2013; 288: 20093–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mao K, Kobayashi S, Jaffer ZM, Huang Y, Volden P, Chernoff J et al. Regulation of Akt/PKB activity by P21-activated kinase in cardiomyocytes. J Mol Cell Cardiol 2008; 44: 429–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar R, Vadlamudi RK. Emerging functions of p21-activated kinases in human cancer cells. J Cell Physiol 2002; 193: 133–44. [DOI] [PubMed] [Google Scholar]
- 30.Misek SA, Chen J, Schroeder L, Rattanasinchai C, Sample A, Sarkaria JN et al. EGFR Signals through a DOCK180-MLK3 Axis to Drive Glioblastoma Cell Invasion. Mol Cancer Res 2017; 15: 1085–95. [DOI] [PubMed] [Google Scholar]
- 31.Nayal A, Webb DJ, Brown CM, Schaefer EM, Vicente-Manzanares M, Horwitz AR. Paxillin phosphorylation at Ser273 localizes a GIT1-PIX-PAK complex and regulates adhesion and protrusion dynamics. J Cell Biol 2006; 173: 587–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang RA, Mazumdar A, Vadlamudi RK, Kumar R. P21-activated kinase-1 phosphorylates and transactivates estrogen receptor-alpha and promotes hyperplasia in mammary epithelium. Embo J 2002; 21: 5437–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen RE, Thorner J. Function and regulation in MAPK signaling pathways: lessons learned from the yeast Saccharomyces cerevisiae. Biochim Biophys Acta 2007; 1773: 1311–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chong C, Tan L, Lim L, Manser E. The mechanism of PAK activation. Autophosphorylation events in both regulatory and kinase domains control activity. J Biol Chem 2001; 276: 17347–53. [DOI] [PubMed] [Google Scholar]
- 35.Mayhew MW, Jeffery ED, Sherman NE, Nelson K, Polefrone JM, Pratt SJ et al. Identification of phosphorylation sites in betaPIX and PAK1. J Cell Sci 2007; 120: 3911–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holm C, Rayala S, Jirstrom K, Stal O, Kumar R, Landberg G. Association between Pak1 expression and subcellular localization and tamoxifen resistance in breast cancer patients. J Natl Cancer Inst 2006; 98: 671–80. [DOI] [PubMed] [Google Scholar]
- 37.Zhou L, Yan C, Gieling RG, Kida Y, Garner W, Li W et al. Tumor necrosis factor-alpha induced expression of matrix metalloproteinase-9 through p21-activated kinase-1. BMC Immunol 2009; 10: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tabusa H, Brooks T, Massey AJ. Knockdown of PAK4 or PAK1 inhibits the proliferation of mutant KRAS colon cancer cells independently of RAF/MEK/ERK and PI3K/AKT signaling. Mol Cancer Res 2013; 11: 109–21. [DOI] [PubMed] [Google Scholar]
- 39.Sancho D, Montoya MC, Monjas A, Gordon-Alonso M, Katagiri T, Gil D et al. TCR engagement induces proline-rich tyrosine kinase-2 (Pyk2) translocation to the T cell-APC interface independently of Pyk2 activity and in an immunoreceptor tyrosine-based activation motif-mediated fashion. J Immunol 2002; 169: 292–300. [DOI] [PubMed] [Google Scholar]
- 40.Lu W, Katz S, Gupta R, Mayer BJ. Activation of Pak by membrane localization mediated by an SH3 domain from the adaptor protein Nck. Curr Biol 1997; 7: 85–94. [DOI] [PubMed] [Google Scholar]
- 41.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 2010; 5: 725–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods 2015; 12: 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang C, Mortuza SM, He B, Wang Y, Zhang Y. Template-based and free modeling of I-TASSER and QUARK pipelines using predicted contact maps in CASP12. Proteins 2018; 86 Suppl 1: 136–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 2008; 9: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang J, Zhang Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res 2015; 43: W174–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014; 30: 1771–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.