

Abstract
Objectives
A better understanding of antitumor immunity will help predict the prognosis of gastric cancer patients and tailor the appropriate therapies in each patient. Therefore, we propose a novel immunological classification of gastric cancer.
Methods
We performed whole‐exome sequencing (WES), RNA‐Seq and flow cytometry in 29 gastric cancer patients who received surgery. The TCGA data set of 323 gastric cancer patients and RNA‐Seq data of 45 patients who received pembrolizumab (Kim et al. Nat Med 2018; 24: 1449–1458) were also analysed.
Results
Immunogram analysis of cancer–immunity interaction of gastric cancer revealed immune signatures of four main types, designated Hot1, Hot2, Intermediate and Cold. Immunologically hot tumors displayed a dysfunctional T‐cell signature, while cold tumors had an exclusion signature. Ex vivo tumor‐infiltrating lymphocyte analysis documented T‐cell dysfunction with the expression of checkpoint molecules and impaired cytokine production. The T‐cell function was more profoundly damaged in Hot1 than Hot2 tumors. Patients in Hot2 subtypes had better survival in our cohort and TCGA cohort. Although these immunological subtypes overlapped to some degree with the molecular subtypes in the TCGA, intratumoral immune responses cannot be predicted solely based on histological or molecular subtyping of gastric cancer. Molecular and immunological classifications complement each other to predict the responses to anti‐PD‐1 therapy and have the potential to be a biomarker for the treatment of gastric cancer.
Conclusion
The immunological classification of gastric cancer resulted in four subtypes. Hot tumors were further divided into two subtypes, between which the functional status of T cells was different.
Keywords: gastric cancer, immunogram, RNA‐Seq, T‐cell function, tumor immunity
In this study, an integrative immunogenomic analysis of gastric cancer was performed. The immunological classification of gastric cancer resulted in immune signatures of four main types, designated Hot1, Hot2, Intermediate and Cold, in which the functional status of T cells was different. Early administration of anti‐PD‐1 therapy might be recommended for patients with Hot1 tumors.

Introduction
Gastric cancer was the fifth most frequent cancer (over 1 000 000 new patients) and the third leading cause of cancer death (783 000 deaths) worldwide in 2018. 1 The introduction of checkpoint blockade was expected to change gastric cancer treatment regimens. 2 , 3 However, monotherapy response rates (RRs) are low, and a better understanding of antitumor immunity and immunosuppressive mechanisms active in the tumor microenvironment (TME) is required to facilitate the development of more effective immunotherapies.
Recently, it has been proposed that there are distinct molecular subtypes of gastric cancer. 4 , 5 , 6 , 7 , 8 Kim et al. reported that there was a dramatic overall RR to pembrolizumab in 86% of patients with microsatellite unstable tumors, and even 100% in Epstein–Barr virus‐(EBV) positive tumors. 9 However, predicting the clinical benefits of checkpoint inhibitors in the majority of gastric cancer patients remains an unmet need for treatment selection.
Antitumor immunity is a spatiotemporal process, where many cell types and molecules are positively and negatively regulated. 10 Besides, overcoming one obstacle in the cancer–immunity cycle with a currently available therapy will leave many other independent immunosuppressive regulatory systems operative in the TME, and adaptive or acquired resistance to therapy may be induced. This, together with considerable variation between patients in the induction of antitumor immunity, necessitates an assessment of each case for a better understanding of that individual's immune response to the tumor. Based on the concept of ‘immune contexture’ that consists of the combination of immune variables associating the nature, density, immune functional orientation and distribution of immune cell within the tumor, the first immune‐based, rather than a cancer‐based, classification was proposed as the immunoscore. 11 , 12 , 13 It is unlikely that a single biomarker will reflect the very complex TME and numerous host–tumor interactions. To this end, we generated an immunogram for the cancer–immunity cycle in which comprehensive profiling of the dynamic interactions between tumor and the immune system is depicted on a radar plot for each individual patient. 14 , 15 This contributes to understanding the immunobiology of each case and personalising the most suitable therapy for each patient.
In a previous immunogram study, we divided non‐small‐cell lung cancers into T‐cell‐rich and T‐cell‐poor phenotypes. 14 Parameters associated with T‐cell immunity and factors reflecting tumor biologies such as tumor antigens and antigen presentation machinery were separately clustered, suggesting that antitumor immunity is shaped by the interaction between tumor and the immune system. In the present study, we extended this type of analysis to 29 gastric cancer patients using a modified approach. Here, improved immunograms with nine axes consisting of innate immunity, priming and activation, T cells, IFN‐γ response, inhibitory molecules, regulatory T cells (Tregs), recognition of tumor cells, proliferation and glycolysis to reflect cancer–immunity interaction were employed to classify individual patients.
Here, we propose a novel immunological classification of gastric cancer using immunogram scores. We further applied it to 323 gastric cancer patients from the TCGA data set and 45 patients who received anti‐PD‐1 therapy reported by Kim et al. 9 A better understanding of antitumor immunity will help predict the prognosis of gastric cancer patients and tailor the appropriate therapies in each patient.
Results
Patient characteristics
Baseline characteristics of the 29 enrolled gastric cancer patients are listed in Table 1 and Supplementary table 1. All cases were of adenocarcinoma, two being oesophagogastric junction cancer and the other 27 gastric cancer. The median age was 71.0 years (range 57–88), and most patients were male (83%). Twenty‐five patients (Stages I–III) received radical surgery, and four (Stage IV) underwent palliative surgery. The median follow‐up after surgery was 14.8 months (range 1.6–51.2).
Table 1.
Characteristics of patients.
| Baseline characteristics | No. (%) |
|---|---|
| Sex | |
| Male | 24 (83) |
| Female | 5 (17) |
| Age | |
| Median (years) | 71 |
| Range (years) | 57–88 |
| pStage | |
| I | 4 (14) |
| II | 7 (24) |
| III | 14 (48) |
| IV | 4 (14) |
| pT | |
| 1b | 2 (7) |
| 2 | 2 (7) |
| 3 | 14 (48) |
| 4a | 8 (28) |
| 4b | 3 (10) |
| Histology | |
| Intestinal | 15 (52) |
| Diffuse | 11 (38) |
| Mixed | 3 (10) |
| Primary lesion location | |
| Oesophagogastric junction | 2 (7) |
| Upper | 9 (31) |
| Middle | 7 (24) |
| Lower | 11 (38) |
| Macroscopic type (Borrmann) | |
| 1 (Mass) | 2 (7) |
| 2 (Ulcerative) | 13 (41) |
| 3 (Infiltrative ulcerative) | 11 (38) |
| 4 (Diffuse infiltrative) | 2 (7) |
| 5 (Unclassifiable) | 1 (3) |
| HER2 status | |
| Positive | 6 (21) |
| Negative | 18 (62) |
| Unknown | 5 (17) |
| Helicobacter pylori infection | |
| Positive | 20 (69) |
| Negative | 9 (31) |
| Surgical treatment | |
| Total gastrectomy | 15 (52) |
| Distal gastrectomy | 13 (45) |
| Proximal gastrectomy | 1 (3) |
| Tumor infiltrative (INF) pattern | |
| INFa | 4 (3) |
| INFb | 18 (62) |
| INFc | 5 (17) |
| Unknown | 2 (7) |
| Lymphatic invasion (ly) | |
| ly0 | 10 (34) |
| ly1 | 5 (17) |
| ly2 | 8 (28) |
| ly3 | 5 (17) |
| Unknown | 1 (3) |
| Venous invasion (v) | |
| v0 | 9 (31) |
| v1 | 2 (7) |
| v2 | 5 (17) |
| v3 | 12 (41) |
| Unknown | 1 (3) |
Immunogram analysis
To evaluate the antitumor immune response in gastric cancer using RNA‐Seq data, we focused on the nine gene sets associated with antitumor immunity, proliferation and metabolism. A single‐sample gene set enrichment analysis (ssGSEA) of the tumors was performed, and the scores for these nine gene sets were converted into immunogram scores (IGSs) as described in the Methods (Supplementary table 2). As we reported elsewhere, 15 we selected an appropriate gene set for each axis of the immunogram by the Spearman correlation analysis. We modified our previous immunogram for the cancer–immunity cycle, which we had used for lung cancer patients. 14 Antigenicity was eliminated from the axes; glycolysis and proliferation were newly adopted in the current version in order to incorporate tumor cell factors into the evaluation of cancer–immune cell interactions. Nine IGSs for each patient were accordingly plotted onto a radar chart to generate individual immunograms (Figure 1), which differ substantially from patient to patient, suggesting that the immune responses in the tumor and TME are unique in each case.
Figure 1.
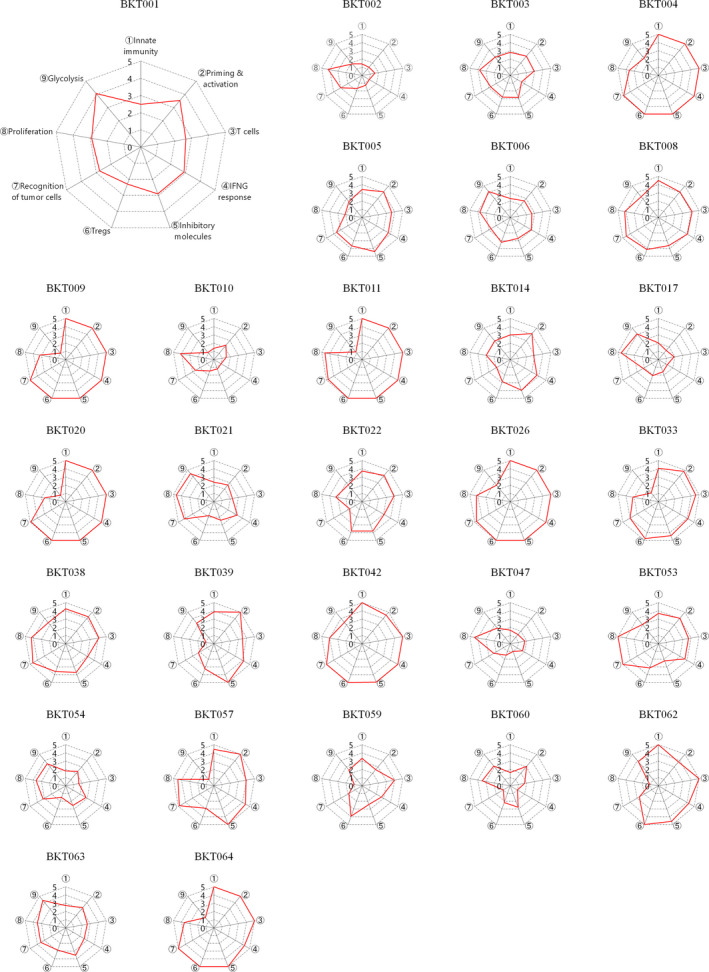
Immunograms for cancer–immunity interactions in 29 patients with gastric cancer. Immunograms were generated using RNA‐Seq data. We selected nine gene sets, including innate immunity (for immunogram score 1, IGS1), priming and activation (IGS2), T cells (IGS3), IFN‐γ response (IGS4), inhibitory molecules (IGS5), Tregs (IGS6), recognition of tumor cells (IGS7) proliferation (IGS8) and glycolysis (IGS9). We performed a single‐sample gene set enrichment analysis (ssGSEA). The ssGSEA scores for each IGS were assessed, normalised and scored onto these axes of the immunogram, which was generated for each patient by integration onto a radar chart.
Classification of the immune response in gastric cancer
Using the nine gene sets or the IGS, we performed a hierarchical cluster analysis and, as shown in the upper part of Figure 2, all 29 cases were initially classified into two clusters, one of which was characterised by high scores for innate immunity (IGS1), priming and activation (IGS2), T cells (IGS3), IFN‐γ response (IGS4), inhibitory molecules (IGS5) and Tregs (IGS6). This suggests that an active immune response is ongoing in the tumor. Therefore, we designated these tumors ‘Immune‐Hot’. In contrast, the IGS1–6 scores were low in the other cluster, designated ‘Immune‐Cold’. Immune‐Hot tumors were further subdivided into three clusters, ‘Hot1’, ‘Hot2’ and ‘Intermediate’. Low scores for recognition of tumor cells (IGS7) and proliferation (IGS8) characterised Intermediate tumors. Hot1 and Hot2 were distinguished from one another by glycolysis (IGS9), which was low in the latter. Overall, the immune responses in these 29 gastric cancers were classified into four signatures, namely Hot1, Hot2, Intermediate and Cold (Supplementary table 3).
Figure 2.
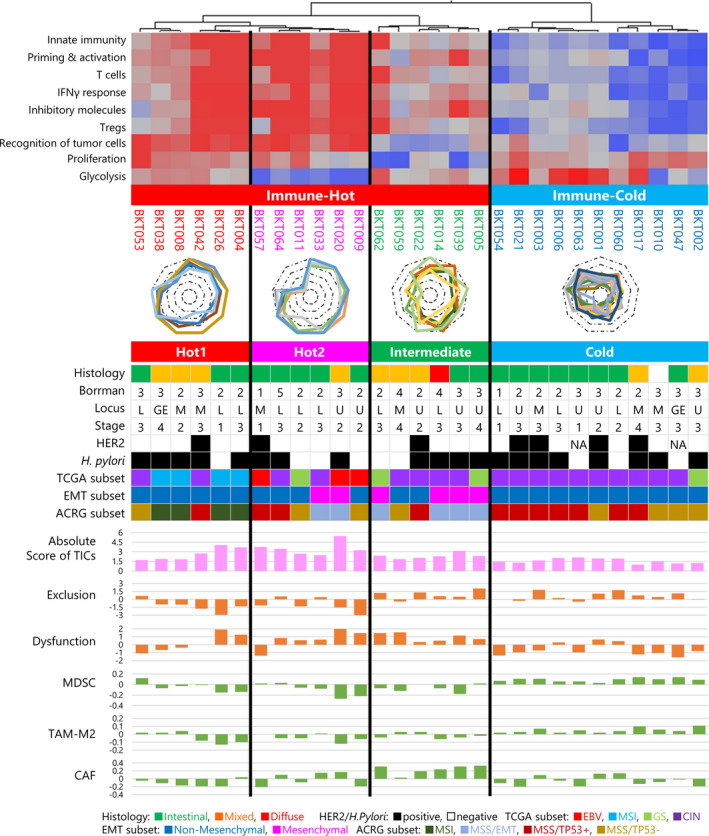
Immunological subtypes of gastric cancer. Immunogram analysis of 29 gastric cancer patients was performed. By hierarchical cluster analysis of the immunogram scores, gastric cancer cases were first divided into Immune‐Hot and Immune‐Cold subtypes. Then, Immune‐Hot subtype was further divided into Hot1, Hot2 and Intermediate subtypes. Clinical profiles with histology by the Lauren classification, macroscopic classification by the Borrmann classification, locus of the primary site, TNM clinical staging, overexpression of human epidermal growth factor receptor 2 (HER2) protein and the presence or absence of Helicobacter pylori infection are depicted. EBV, MSI, GS and CIN subtypes of molecular classification by TCGA 5 and MSI, MSS/EMT, MSS/TP53+ and MSS/TP53− subtypes of ACRG classification 6 are indicated by colour. Absolute scores of tumor‐infiltrating cells (TICs) were estimated by CIBERSORTx. 20 Scores for exclusion and dysfunction and signatures for MDSCs, TAM‐M2 and CAF were evaluated by TIDE. 21 The EMT subset was determined by a Mesenchymal or Non‐Mesenchymal signature. 19
Correlation between immune responses and clinical characteristics
The clinical characteristics of patients are depicted in the middle part of Figure 2 to visualise correlations between the immune response in the tumor and these variables. 16 Histology by the Lauren classification, 17 macroscopic classification according to Borrmann, 18 primary site location, TNM clinical stage, overexpression of human epidermal growth factor receptor 2 (HER2) protein and the presence or absence of Helicobacter pylori infection all failed to correlate with intratumoral immune responses. Recently, the TCGA project defined four molecular subtypes of gastric cancer, namely EBV, microsatellite instability (MSI), genomically stable (GS) and chromosomal instability (CIN). 5 As expected, EBV and MSI tumors were classified as Immune‐Hot with all four MSI tumors in Hot1, and all three EBV in Hot2 (Figure 2). Most tumors in the Cold and Intermediate groups were CIN in the TCGA classification, while the CIN subtype was present across all four immunological signatures. GS subtypes were present in the Hot2, Intermediate and Cold groups (Figure 2). Molecular classification according to Asian Cancer Research Group (ACRG) was also compared with these results. 6 According to the expression of 71 genes reported by the ACRG, gastric cancer is divisible into Mesenchymal or Non‐Mesenchymal subtypes (Supplementary figure 1). 19 Among our 29 gastric tumors, six (21%) were classified as Mesenchymal and the other 23 (79%) as Non‐Mesenchymal. The Hot2 group included two of the six Mesenchymal tumors, and the other four were Intermediate (Figure 2). MSI tumors were all in Hot1. MSS/TP53+ and MSS/TP53− subtypes were distributed over all four immunological categories. These results suggest that intratumoral immune responses cannot be predicted solely on the basis of histological or molecular subtyping of gastric cancer.
Tumor‐infiltrating cells
To evaluate immune responses in the tumor, the profiling of tumor‐infiltrating cells (TICs) was performed by CIBERSORTx. 20 Absolute TIC scores are incorporated into Figure 2, and subpopulations of TICs in individual patients are shown in Supplementary figure 2a. Absolute scores of TICs for Immune‐Hot tumors were higher than for Immune‐Cold tumors (P < 0.001, t‐test, Supplementary figure 2b). Absolute scores of TICs were highest in the Hot2 subtype, and lowest in the Cold subtype (P < 0.001, one‐way ANOVA test; Supplementary figure 2c). The absolute score of CD8+ T cells was high in Hot1 and Hot2 subtypes (P = 0.0011, one‐way ANOVA test; Supplementary figure 2d), whereas activated memory CD4+ T cells were high in Hot1 subtype (P < 0.001, one‐way ANOVA test; Supplementary figure 2e). There were no statistically significant differences between the four subtypes for any of the other cell populations (Supplementary figure 2f–k).
To investigate the immunosuppressive TME, expression signatures were evaluated by the TIDE web application (http://tide.dfci.harvard.edu/). 21 Consistent with data on TICs by CIBERSORTx, exclusion scores by TIDE were high in the Intermediate and Cold subtypes (P < 0.001, Figure 2 and Supplementary figure 3a). In Immune‐Hot tumors, dysfunction scores by TIDE were high (P = 0.0064, Figure 2 and Supplementary figure 3b). Both exclusion and dysfunction scores were high in the Intermediate group. MDSC and TAM‐M2 signatures were significantly higher in Cold tumors than in the others (P < 0.001, P < 0.001, one‐way ANOVA test, Supplementary figure 3c and d). The cancer‐associated fibroblast (CAF) signature was significantly higher in the Intermediate subtype (P < 0.001, one‐way ANOVA test, Supplementary figure 3e). These factors are thus considered to contribute to the immunosuppressive TME in the Intermediate and Cold subtypes.
Mutational analysis
Because high tumor mutational burden (TMB) could increase the chance to produce immunogenic neoantigens, 22 TMB was compared in the four immunological subtypes (Figure 3a). All MSI patients in the Hot1 subtype and one patient (BKT020) in the Hot2 subtype have high TMB. The BKT020 tumor had POLE exon19:c.A2151C:p.K717N mutation and POLD2 exon5:c.A614C:p.K205T and exon6:c.A509C:p.K170T mutations, which caused mismatch repair proficient, but hypermutated phenotype. 23 , 24
Figure 3.
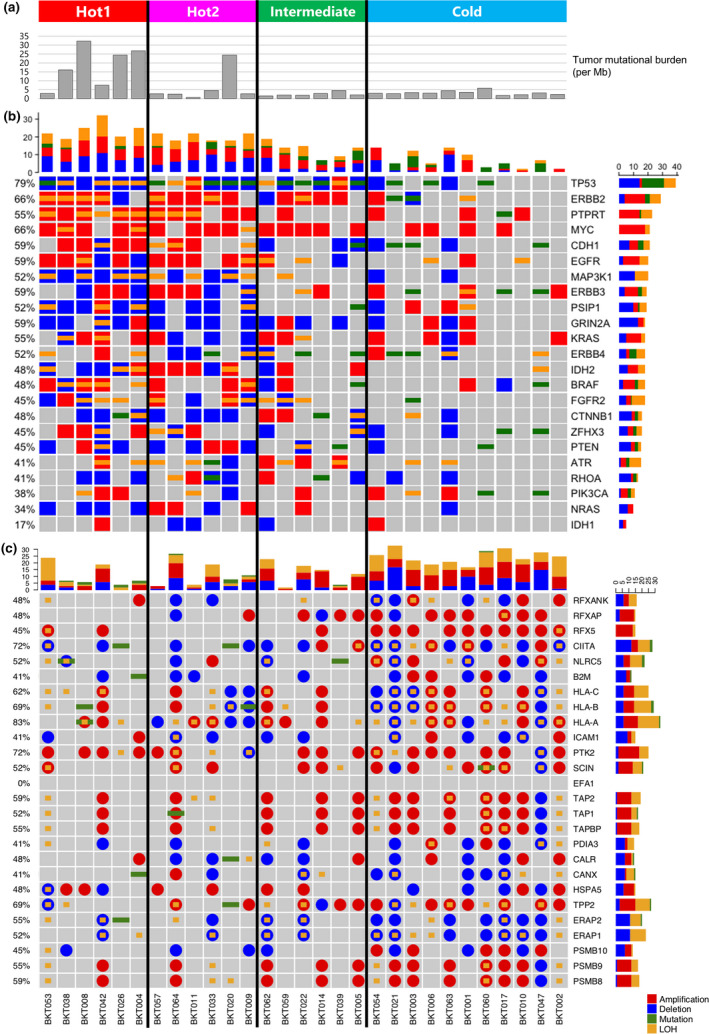
Mutational analysis. (a) Tumor mutational burden (TMB) was calculated as the total number of nonsynonymous mutations divided by the actual number of bases analysed (per Mb). (b) Heatmap representation of the distribution of gene alterations in known driver genes. (c) Nucleotide and copy‐number variants found in the antigen presentation pathway. Stacked bar plot summarising the total numbers of amplification, deletion, mutation and loss of heterozygosity (LOH) per patient (longitudinal) or per gene (horizontal). Different colours represent different types of nucleotide variants, red for amplification, blue for deletion, green for mutation and yellow for LOH.
There is increasing evidence that the activation of the different oncogenic pathways has a profound impact on the immune evasion. 25 Thus, driver gene alterations were investigated (Figure 3b). TP53 mutations were detected in 79% of the tumor. Amplification of Myc was observed in 66% of the tumor. Deletion and/or loss of heterozygosity (LOH) of PTEN were detected in seven of 29 patients. MAP3K1, PIK3CA, CTNNB1 and RHOA gene alterations were also detected. These driver gene alterations were detected more frequently in Hot tumors than Cold tumors.
Antigen presentation and sensitivity to IFN‐γ are major tumor cell‐intrinsic factors determining antitumor immunity. 26 , 27 , 28 Therefore, nucleotide and copy‐number variants in genes related to antigen presentation (Figure 3c) or the IFN‐γ pathway (Supplementary figure 4) were investigated. In contrast to driver gene alterations, more nucleotide and copy‐number variants in genes related to antigen presentation were detected in Cold tumors (Figure 3c and Supplementary figure 5). The same was true for the IFN‐γ pathway (Supplementary figures 4 and 5). A combination of LOH (yellow) and deletion (blue) or mutation (green) that resulted in losses of function (LOFs) was detected more often in Cold tumors (Supplementary figures 4 and 5). Because IFN‐γ is known to up‐regulate human leukocyte antigen (HLA) molecule expression, LOFs in the IFN‐γ pathway are also associated with reduced antigen presentation. These tumors might thus be invisible to the immune system, rendering them Cold. MSI tumors contained many mutations but few LOHs, and as a result, immune responses in these subtypes were maintained and tumors remain Hot.
Tumor antigens
To seek differences in tumor antigens in each subtype, we predicted neoantigens by estimating the MHC binding capacity of predicted mutated peptides using MHCflurry 1.4.0. 29 We considered mutated peptides as predicted neoantigens (pNeoAg) if their K D was < 500 nm. Because not all of them were expressed in the tumor, we considered them as expressed neoantigens (eNeoAg), only if their expression was confirmed by RNA‐Seq [transcripts per million (TPM) ≥ 1, variant allele frequency (VAF) ≥ 0.04]. As shown in Figure 4, MSI tumors contained many mutations, including missense single nucleotide variants (SNVs), insertions and deletions (indels) and fusion genes that resulted in many neoantigens. As a result, Hot1 tumors had more mutations, pNeoAg and eNeoAg than any others (P < 0.001, one‐way ANOVA, Figure 4, Supplementary figure 6b–d). In contrast, the number of CT antigens expressed in the tumor was low in Immune‐Hot and high in Immune‐Cold tumors (P = 0.030 using one‐way ANOVA or t‐test; Figure 4; Supplementary figure 6e). These results suggest that NeoAgs, rather than CT antigens, are the main targets of antitumor immunity in gastric cancer.
Figure 4.
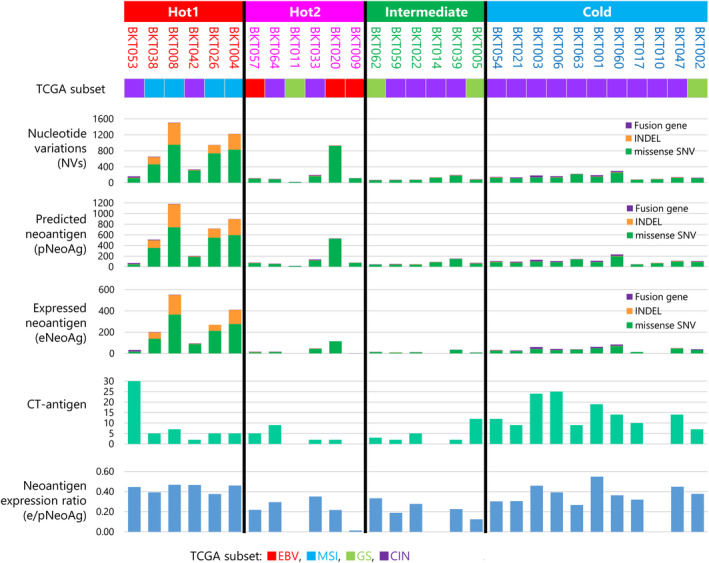
Tumor antigens. The number of single nucleotide variants (SNVs; green), indels (orange) and fusion genes (purple) is depicted in a stacked bar plot. The numbers of predicted neoantigens (pNeoAg), expressed neoantigens (eNeoAg), CT antigens (yellow) and neoantigen expression ratio (eNeoAg/pNeoAg) in each patient are depicted as bar graphs.
Because immune selection pressure affects the number and expression of neoantigens, we compared the ratio of eNeoAg to pNeoAg designated the ‘neoantigen expression ratio’ in the four immune‐related subtypes (Supplementary figure 6f). The neoantigen expression ratio was reduced in Hot2 and Intermediate tumors but not Hot1 (P = 0.0067), presumably because active immune selection pressures had eliminated tumor cells expressing those neoantigens. 30
Phenotypes and functions of TICs
Tumor‐infiltrating cells were isolated from the tumors and their phenotypes and functions analysed by flow cytometry (Figure 5; Supplementary figures 7, 8, 9, 10, 11, 12). More CD3+T cells were observed in Hot1 tumors than other subtypes (P = 0.046, Supplementary figure 7a). More CD4+ than CD8+ T cells were present in most of the tumors, but their percentages did not differ between the four subtypes (Figure 5a and Supplementary figure 7b and c). Infiltrations of Tregs in the tumors were similar among four subtypes (Supplementary figure 7d–f). CD8+ TICs exhibited exhausted phenotypes, with about 70% expressing PD‐1 in most patients. They expressed TIM3 and LAG3, though a lesser extent to PD‐1 (Figure 5a and Supplementary figure 8). The activation markers were also expressed on these cells, while there were no statistically significant differences in the levels of expression among subtypes.
Figure 5.
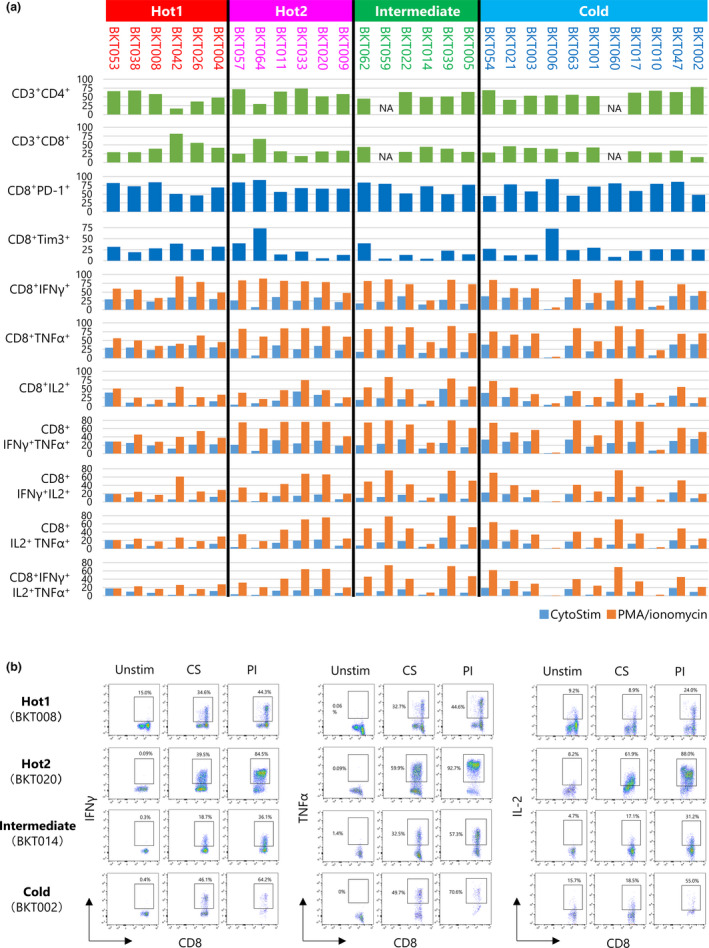
Summary of T‐cell phenotypes and functions. (a) Tumor‐infiltrating cells (TICs) were analysed by flow cytometry. Bar graphs show the percentage of CD3+CD4+ T cells, CD3+CD8+ T cells, CD8+PD‐1+ T cells, CD8+Tim‐3+ T cells, CD8+IFN‐γ+ T cells, CD8+IL‐2+ T cells, CD8+TNF‐α+ T cells, CD8+IFN‐γ+ T cells, CD8+IFN‐γ+IL‐2+ T cells, CD8+IFN‐γ+TNF‐α+ T cells, CD8+IL‐2+ TNF‐α+ T cells and CD8+ IFN‐γ+IL‐2+ TNF‐α+ T cells. TICs were stimulated with CytoStim (Blue) or PMA/ionomycin (orange); the percentages of cytokine‐producing cells were measured by the intracellular cytokine staining. (b) TICs were unstimulated (Unstim) or incubated with CytoStim (CS) or PMA/ionomycin (PI) for 4 h. Examples of staining patterns are shown. CD45+CD3+CD8+ T cells were gated. The percentage of IFN‐γ‐, TNF‐α‐ and IL‐2‐producing cells are shown.
To determine the capacity of these CD8+ T cells to produce cytokines, we applied two different stimulation procedures, namely CytoStim, which activates T cells via TCR signalling, and PMA together with ionomycin that bypasses TCR‐mediated signalling and directly activates several intracellular signalling pathways (Figure 5; Supplementary figures 9 and 10). The T‐cell stimulation by CytoStim was under the influence of inhibitory molecules, while forced cytokine production by PMA/ionomycin indicated their maximal capacity of cytokine production. As examples, FACS data of one patient from each subtype are shown in Figure 5b. More IFN‐γ, TNF‐α or IL‐2 was produced by PMA/ionomycin stimulation than CytoStim. Similarly, the production of TNF‐α and IL‐2 upon stimulation with CytoStim was lower than that of those after PMA/ionomycin stimulation (Figure 5b).
Although there were no statistical differences between the four subtypes regarding the presence of CD8+ T cells producing one of the cytokines IFN‐γ, TNF‐α or IL‐2, TNF‐α‐producing CD8+ T cells were lower in Hot1 than in Hot2 (Supplementary figure 9). IFN‐γ and TNF‐α double producers were also low in Hot1 tumors (Supplementary figure 10). These results indicate the severe CD8+ T‐cell dysfunction in Hot1. Cytokine production of CD4+ TICs was also compared; there were no differences between the four subtypes (Supplementary figures 11 and 12).
Immunological subtypes of gastric cancer and association with survival
We explored the prognostic implications of these immunological subtypes for survival after surgery. Although there were no statistically significant differences between the patients in these groups regarding OS (P = 0.18) and PFS (P = 0.13), both OS and PFS of Hot2 tended to be longer (Figure 6a and b). All patients with tumors of the Hot2 subtype survived over the follow‐up period.
Figure 6.
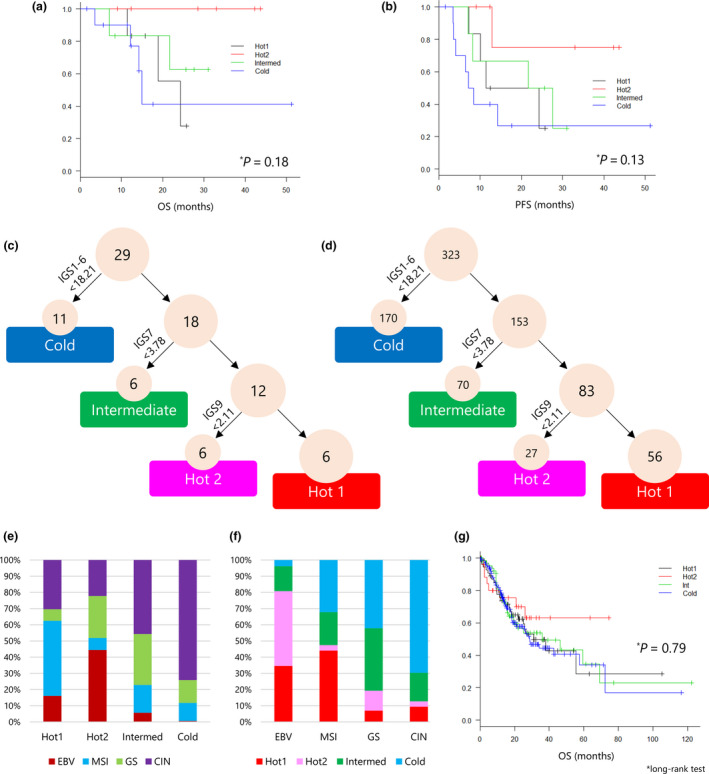
Immunological subtypes of gastric cancer and associations with survival. (a, b) The association between immunological subtypes and overall survival (OS) and progression‐free survival (PFS) was analysed by the Kaplan–Meier method, and the log‐rank test was used to determine the statistical significance of the differences. (c) A decision tree for the immunological subtypes of gastric cancer was applied to 29 gastric cancer patients. The sum of IGS1 to IGS6 of < 18.21 was taken as the cut‐off value to identify the Cold subtype. Similarly, IGS7 < 3.78 was determined as the cut‐off value for Intermediate and IGS9 < 2.11 was used to discriminate Hot1 from Hot2 tumors. (d) A decision tree for the immunological subtypes of gastric cancer was applied to 323 gastric cancer patients in the TCGA cohort. (e) The frequency of molecular subtypes of TCGA in each immunological subtype. (f) The frequency of immunological subtypes in each molecular subtype of TCGA. (g) Kaplan–Meier analysis and log‐rank test for OS of 311 gastric cancer patients for the four immunological subtypes.
To develop a simple approach for immunological subtyping of gastric cancer without cluster analysis, a decision tree was constructed (Figure 6c). Immune‐Hot and Immune‐Cold tumors were distinguished by the sum of immunogram scores of innate immunity (IGS1), priming and activation (IGS2), T cells (IGS3), IFN‐γ response (IGS4), inhibitory molecules (IGS5) and Tregs (IGS6) being < or > 18.21 (Supplementary figure 13a). Intermediate tumors were then defined by the recognition of tumor cells (IGS7) being < or > 3.78 (Supplementary figure 13b). Finally, Hot1 and Hot2 were determined by glycolysis (IGS9) being < or> 2.11 (Supplementary figure 13c). These cut‐off values and the decision tree classified 29 gastric cancers into six Hot1, six Hot2, six Intermediate and 11 Cold subtypes (Figure 6c).
We applied this decision tree to 323 gastric cancer patients from the TCGA data set (Figure 6d). They were stratified into 56 Hot1, 27 Hot2, 70 Intermediate and 170 Cold subtypes. Consistent with the results from our cohort, Hot1 contained MSI subtypes, and EBV subtypes were classified into Hot2. The Cold subtype included mainly CIN (Figure 6e and f). We analysed the OS of 311 patients whose survival data were available. Again, consistent with our cohort, OS of Hot1 and Hot2 was different, and as in our cohort, although not significant, the OS of patients with Hot2 tumors was the best (Figure 6g).
Immunological subtypes of gastric cancer and response to anti‐PD‐1 therapy
Kim et al. 9 reported the molecular features of 61 gastric cancer patients associated with responses to pembrolizumab. For the 45 patients for whom RNA‐Seq data were available, immunogram analysis was performed (Supplementary table 4) and the association of immunological subtypes with responses to anti‐PD‐1 therapy was examined. As shown in Figure 7a, 22, 5, 2 and 16 patients were classified into Hot1, Hot2, Intermediate and Cold subtype, respectively. MSI and EBV patients were determined as immunological Hot, though one MSI patient (EP‐43) was immunological Cold (Figure 7b and c). All three CR patients, two MSI and one CIN subtype, were classified in Hot1(Figure 7d and e). Nine PR patients were classified seven in Hot1, one in Hot2 and one in Cold, respectively. The RR of Hot1 subtypes was 45% and higher than other subtypes (P = 0.033, Fisher's exact test).
Figure 7.
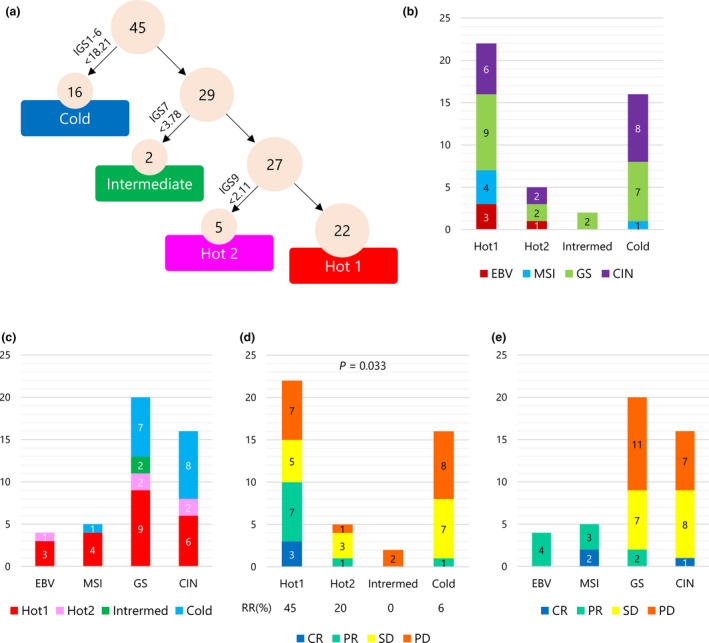
Immunological subtypes of gastric cancer and responses to anti‐PD‐1 therapy. (a) A decision tree for the immunological subtypes of gastric cancer was applied to 45 gastric cancer patients who received anti‐PD‐1 therapy from Kim et al. 9 (b) The number of patients by molecular subtypes of TCGA in each immunological subtype. (c) The number of patients by immunological subtypes in each molecular subtype of TCGA. (d) The number of patients by responses to anti‐PD‐1 therapy in each immunological subtype. (e) The number of patients by responses to anti‐PD‐1 therapy in each molecular subtype of TCGA.
Discussion
We generated an immunogram for each patient to visualise the state of cancer–immune system interactions and have investigated the tumor–immune microenvironment in gastric cancer. Our results demonstrate that the antitumor immune response in gastric cancer is heterogeneous (Figure 1). Based on IGSs, the hierarchical clustering of gastric cancers from 29 patients resulted in four novel immunological subtypes (Figure 2). We also examined the phenotypes and functions ex vivo of tumor‐infiltrating T cells by flow cytometry (Figure 5), integrated them together with next‐generation sequencing (NGS)‐based immunological subtyping and defined the intratumoral immunological status of gastric cancer.
The immunogram approach conceptually proposed by Blank et al., 31 which has been applied to lung cancer 14 and urothelial cancer, 32 integrates multiple parameters that influence cancer–immune system interactions and facilitates a comprehensive evaluation of patients' intratumoral immune status. It has the potential to overcome the limitations of a single biomarker approach. 33 Although not exactly matching each other, our immunological classification and the TCGA and ACRG molecular classifications do have something in common. Four of six Hot1 tumors were MSI in the TCGA subtype, while two were CIN. Three of six tumors in Hot2 were EBV in the TCGA subtype, while two CIN and one GS subtype were also included. The Intermediate subtype included four tumors in the Mesenchymal subtype according to the ACRG classification, and a further two ACRG Mesenchymal subtypes were classified as Hot2 (Figure 2). These results suggest that there is still missing information that determines the antitumor immune response in the tumor and further investigation is necessary to achieve a complete understanding of the immunological status of each patient.
Our immunological subtypes identified the different T‐cell dysfunctional status between Hot1 and Hot2. As shown in Figure 5, maximal cytokine‐producing capacity induced by PMA/ionomycin was lower in Hot1 than in Hot2 tumors despite many CD3+ T cells in the Hot1 tumors (Supplementary figure 7a). Consistently, the T‐cell dysfunctions in Hot1 tumors resulted in the low immune selection pressure and maintaining the neoantigen expression (Supplementary figure 6f). Rooney et al. 34 reported that high cytolytic activity was associated with a modest but significant pan‐cancer survival benefit. However, in our gastric cancer cohort, prognosis after surgery differed between Hot1 and Hot2 subtypes (Figure 6). Both OS and PFS of patients with Hot1 subtype tumors were shorter than those of patients with Hot2, albeit not significantly so. These results might be explained by the fact that the more profound dysfunction of CD8+ T cells was observed in Hot1 than in Hot2 (Figure 5), despite many CD8+ T cells infiltrating the former. There are two major differences distinguishing between Hot1 and Hot2 tumors, one being glycolysis (Figure 2) and the other tumor antigen expression (Figure 3). Considering that neoantigens and viral antigens are likely to drive cytolytic activity, 34 the difference in antigens is not likely to explain impaired T‐cell functions in Hot1 tumors. Recently, metabolic competition in the tumor has been acknowledged as one of the mechanisms for the immunosuppressive microenvironment, because cancer cells take up large amounts of glucose that are also required for T‐cell activity. 35 Ottensmeier et al. reported that tumor glucose metabolism was negatively correlated with tumor‐infiltrating T cells in squamous cell carcinoma. 36 Active glycolysis in Hot1 tumors might explain more severe T‐cell dysfunction in the Hot1 subtype.
To examine the association between immunological subtypes and the responses to anti‐PD‐1 therapy, we re‐analysed the Korean cohort reported by Kim et al. 9 Unlike our cohort and TCGA cohort in which patients undergoing surgery were analysed, patients in the Korean cohort had metastatic and/or recurrent gastric adenocarcinomas. They already received a couple of regimens prior to anti‐PD‐1 therapy. This might explain that more patients were classified into Hot1 or Cold subtype than Hot2 or Intermediate subtype (Figure 7a). Nevertheless, 45% of Hot1 patients responded to anti‐PD‐1 treatment. Therefore, it is recommended that gastric cancer patients with Hot1 subtype receive anti‐PD‐1 therapy as early as possible.
Intermediate subtype tumors are characterised by a moderate immune response in the tumor with positive signatures for both exclusion and dysfunction (Figure 2). Low IGSs for recognition of tumor cells impair the T‐cell response. Abundant CAF and the Mesenchymal phenotype create an immunosuppressive microenvironment. 6 , 19 The relatively slow growth of tumor cells with low IGSs in proliferation balances out the lower immune response in this subtype. As shown in Supplementary figure 6f, a low neoantigen expression ratio in this subtype suggests that immunoediting had operated in the tumors of Intermediate subtype. These results suggest pre‐existing antitumor immune responses in the Intermediate subtype. 30
Gastric cancer is a type of tumor with a high rate of HLA mutation. 34 Consistently, IGS for recognition of tumor cells was low in Intermediate and Cold subtypes; Cold tumors were characterised by an exclusion signature (Figure 2). Davoli et al. 37 reported that elevated expression of cell cycle and cell proliferation markers and reduced expression of immune signatures were associated with tumor aneuploidy. Furthermore, high somatic copy‐number alterations in tumors correlated with reduced response to immunotherapy. Consistently, Cold tumors in our cohort displayed high IGS for proliferation and copy‐number variations (CNVs) in genes associated with antigen presentation and IFN‐γ signalling pathways. These results suggest that the treatment of Cold gastric tumors by current checkpoint blockade immunotherapy alone might be difficult. Combination therapies with other modalities are warranted.
There were no significant differences in OS and PFS between immunological subtypes. This may be because of the small study cohort and different postoperative treatments, even though patients were treated according to the current guidelines. Additionally, our study was not designed to develop a biomarker for postoperative gastric cancer patients. However, it is encouraging that our classification is also supported by the analysis of the TCGA cohort.
In conclusion, antitumor immunity in gastric cancer was quite heterogeneous. We have developed a novel immunological classification of gastric cancer based on comprehensive molecular analysis. Incorporation of factors related to T‐cell immunity together with tumor cell factors, including antigen presentation, proliferation and glycolysis, identified four immunological subtypes of gastric cancer. Intratumoral immune responses cannot be predicted solely on the basis of histological or molecular subtyping of gastric cancer. Molecular and immunological classifications complement each other to predict the responses to anti‐PD‐1 therapy and have the potential to be a biomarker for the treatment of gastric cancer. Among the immunological hot tumors, some patients with Hot 1 subtype are recommended to receive anti‐PD‐1 therapy earlier than is currently approved.
Methods
Patients and data sets
In all, 29 patients histologically diagnosed with gastric cancer at Tokyo Metropolitan Bokutoh Hospital from June 2014 to October 2015 were enrolled. Tumors, adjacent tissues and peripheral blood from the same patients were collected at the time of surgery. After surgery, patients received standard therapy according to Japanese gastric cancer treatment guidelines. 38 The clinicopathological data for individual patients are summarised in Table 1. All procedures in this study were performed following the ethical standards of the institutions and in conformity with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Besides, the TCGA data set of 375 gastric cancer patients was downloaded from the TCGA data portal site (https://portal.gdc.cancer.gov/). Because patients in our cohort and TCGA cohort went through surgery and did not receive immunotherapy, RNA‐Seq data of PRJEB25780 study 9 with 45 metastatic or recurrent gastric cancer patients who received anti‐PD‐1 therapy were downloaded from the European Nucleotide Archive (https://www.ebi.ac.uk/ena/data/view/PRJEB25780) and analysed.
DNA and RNA preparation
Tumors and adjacent normal tissues were obtained immediately after surgical resection and stored in RNAlater RNA Stabilization Reagent (Qiagen, Hilden, Germany). Peripheral blood (30 mL) was also obtained, and peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Lymphoprep™ (Axis‐Shield Poc AS, Oslo, Norway), and cryopreserved in Bambanker™ freezing medium (NIPPON Genetics, Tokyo, Japan) until use. Genomic DNA and total RNA samples from fresh‐frozen tissues and matched PBMCs were extracted using either AllPrep DNA/RNA Mini Kit or AllPrep DNA/RNA/miRNA Universal Kits (Qiagen) according to the manufacturer's instructions. Quality of DNA was assured by Qubit Assay Kit® (Thermo Fisher Scientific K.K., Tokyo, Japan); samples with a DNA concentration ≥ 12.5 ng μL−1 and a total amount ≥ 2.0 μg were used for NGS. RNAs were accepted for NGS when the concentration was ≥ 20.0 ng μL−1, total amount ≥ 0.4 μg and RNA integrity number ≥ 7.0 using the Agilent 2200 TapeStation (Agilent Technologies, Santa Clara, CA, USA).
Whole‐exome sequencing
Sequencing libraries of genomic DNA from tumors and matched normal tissue were prepared using the SureSelect XT Human All Exon V6 Kit (Agilent Technologies) following the manufacturer's protocols. The enriched libraries were sequenced as 150‐bp paired‐end reads using the HiSeq X or NovaSeq (Illumina, San Diego, CA, USA) at CHEMICAL DOJIN (Kumamoto, Japan) or BGI Japan K.K (Kobe, Japan). The mean coverage of all protein‐coding sequences (CDS) by WES was 197.2×. Exome reads were independently mapped to the human genome (GRCh38/hg38) using Burrows–Wheeler Aligner (v0.7.15). 39 Picard (version 2.1.1; Broad Institute, Cambridge, MA, USA) was used to remove duplicate reads. The Genome Analysis Tool Kit (version 3.7) was used for the realignment of reads around indels. 40 The average of total mapped reads in WES was 89.4 m for the tumor samples and 86.7 m for normal samples. Putative somatic variants in tumor DNA were called against DNA taken from normal tissue using VarScan2 (version 2.4.2) and/or MuTect (version 1.1.7). 41 , 42 Default parameters were used for variant calling. Nonsynonymous mutations and splice site mutations were annotated by Annovar. 43 TMB was calculated as the total number of nonsynonymous mutations divided by the actual number of bases analysed.
Copy‐number variations
Alignments in the BAM format for tumor–normal pairs were read simultaneously to detect copy‐number alterations using GATK (v 4.0.1.2). 40 The circular binary segmentation algorithm was used, and LOH was determined by VarScan (v2.3). Bedtools (v2.27.1) was used for detecting copy‐number alterations of the specific gene. 44
RNA‐Seq
An RNA‐Seq library was prepared using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (Agilent Technologies) following the manufacturer's protocols. The enriched libraries were sequenced as 150‐bp paired‐end reads using NovaSeq (Illumina) at CHEMICAL DOJIN or GENEWIZ Japan (Saitama, Japan). An average of 35.1 million reads of 150 base length per sample was obtained on each sample and mapped to the reference genome (GRCh38/hg38) using STAR (v.2.5.2b). 45 Expression values were calculated as fragments per kilobase of exon per million fragments mapped (FPKM) using HTSeq (v.0.6.1) 46 and R (version 3.4.3; https://www.r‐project.org/).
VAF of each missense mutation in the RNA read was calculated as the proportion of variant read count per depth using bam‐readcount (https://github.com/genome/bam‐readcount).
HLA typing and prediction of neoantigens by estimating MHC class I peptide binding
HLA types of the 29 gastric cancer patients were assigned from WES data of normal tissues using HLA‐HD. 47 WES data were subjected to DRAGEN Bio‐IT Platform 3.5.7 (Illumina) to detect SNVs and insertions and deletions (indels). Phasing (or haplotyping) of any combination of SNVs and indels was performed with HapCUT2 1.2 48 before evaluating the binding of the mutated peptide to MHC class I. Variants were annotated by SnpEff 4.3T. 49 RNA‐Seq data were also analysed with DRAGEN Bio‐IT Platform 3.5.7 8 (Illumina) to detect fusion genes. BLASTn 2.6.0+ was used to eliminate false‐positive gene fusions. 50 The binding affinities of 8‐mer to 11‐mer mutated peptides for the specific HLA alleles of each patient were predicted using MHCflurry 1.4.0. 29 In the case of fusion genes, peptides with upstream and downstream 10 amino acids from the breakpoint were evaluated. Mutated peptides with predicted K D values ≤ 500 nmol L−1 were considered candidate neoepitopes. We defined a mutated gene capable of generating one or more HLA‐A‐, HLA‐B‐ or HLA‐C‐restricted neoepitopes as a pNeoAg. The expression of pNeoAgs was checked by RNA‐Seq data, and they were considered to be eNeoAg only when their TPM value and VAF were ≥ 1 and ≥ 0.04, respectively.
Immunograms
To depict the immunological status of the tumor in each patient, we constructed an immunogram based on RNA‐Seq data. 14 , 15 We focused on factors related to the cancer–immunity cycle and tumor cell proliferation and metabolism using nine selected gene sets. These were innate immunity, priming and activation, T cells, IFN‐γ response, inhibitory molecules, Tregs, recognition of tumor cells, proliferation and glycolysis. We then ran ssGSEA. 51 Incorporated gene sets are listed in Supplementary table 2. Similar ssGSEA was applied to the TCGA mRNA data of 375 gastric cancer patients. We obtained the mean (M) and standard deviation (SD) of the ssGSEA score of these 375 gastric cancer patients for each gene set. The score for each axis of the immunogram in each patient was calculated as the IGS = 3 + 1.5 × (ssGSEA score − M)/SD. This formula was applied for all axes of the immunogram of a patient.
Molecular classification of gastric cancer
Molecular classification of gastric cancer was performed as previously reported; 5 29 gastric cancers were classified as being EBV, having MSI, genomic stability (GS) or chromosomal instability (CIN). EBV tumors were identified by the BAM file format of RNA‐Seq using BioBloom software. 52 MSI testing was performed on all tumor DNA using the MSI Analysis System (Promega KK, Tokyo, Japan). The remaining tumors were further grouped by the number of somatic copy‐number alterations defining GS or CIN subtypes. According to the ACRG project, these 29 gastric cancers were grouped as Mesenchymal or Non‐Mesenchymal subtypes by their 71‐gene Mesenchymal signature (Supplementary table 5). 19 To quantify TICs, we analysed RNA‐Seq data with the CIBERSORTx algorithm. 20 Signatures for immune evasion by T‐cell dysfunction or exclusion were examined by the TIDE web application (http://tide.dfci.harvard.edu/). 21
Tumor‐infiltrating cell isolation and flow cytometry
Tumor‐infiltrating cells were prepared using a tumor dissociation kit (Miltenyi Biotec Inc., Auburn, CA, USA) according to the manufacturer's instructions. Briefly, surgically resected tumors were cut into pieces, transferred to gentleMACS C Tubes containing an enzyme mix (Miltenyi) and then passed through a 70‐μm cell strainer (Fisher Scientific, Hampton, NH, USA) to obtain TICs, which were cryopreserved in BambankerTM freezing medium (NIPPON Genetics, Tokyo, Japan). Cryopreserved TICs were thawed in RPMI supplemented with 50 IU mL−1 BenzonaseTM Nuclease (Sigma‐Aldrich, St. Louis, MO, USA) , and then stained using a Zombie Aqua™ Fixable Viability Kit (BioLegend, San Diego, CA, USA) with Alexa Fluor® 700‐labelled anti‐CD45 (BioLegend) and Brilliant Violet 605™‐labelled CD3 (BioLegend), CD4 (Thermo Fisher Scientific K.K.), CD8 (BioLegend), IFN‐γ (Beckman Coulter, Brea, CA, USA), TNF‐α (BioLegend), IL‐2 (BioLegend), PD‐1 (BioLegend) or Tim‐3 (R&D Systems, Minneapolis, MN, USA). For intracellular cytokine staining, cells were stimulated with 10 ng mL−1 phorbol 12‐myristate 13‐acetate (PMA; Sigma‐Aldrich) together with 1 μg mL−1 ionomycin (Sigma‐Aldrich) or CytoStim (Miltenyi Biotec) in the presence of 10 µg mL−1 brefeldin A (Sigma: I0634) at 37ºC for 4 h. Cytokine‐producing cells were then evaluated by intracellular cytokine staining carried out according to the manufacturer's instructions (using IntraPrep Permeabilization Reagent; Beckman Coulter). Stained cells were analysed on a Gallios flow cytometer (Beckman Coulter) and data processed using Kaluza software (Beckman Coulter) and FlowJo (version 7.6.5; TreeStar, Ashland, OR, USA).
Statistics
Ward's hierarchical clustering method was used for generating a hierarchical cluster of the 29 gastric cancer patients. The Kaplan–Meier method was used to generate survival curves for the subtypes in each data set, and the log‐rank test was used to determine the statistical significance of differences. For comparisons of two groups, statistical significance for normally distributed variables was estimated by unpaired Student's t‐tests. For comparisons of more than two groups, one‐way analysis of variance (ANOVA) was used. All statistical analyses were performed with JMP Pro 14 (SAS Institute Japan, Tokyo, Japan) or EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan), 53 which is a graphical user interface for R (https://www.r‐project.org/). A value of P < 0.05 was considered significant.
Data repository and accession numbers
Data are deposited on the Japanese Genotype–Phenotype Archive (Accession no. JGAS00000000210) and DDBJ Sequence Read Archive (Accession no. DRADRA009379). 54
Author Contributions
Yasuyoshi Sato: Conceptualization; Data curation; Formal analysis; Investigation; Visualization; Writing‐original draft. Ikuo Wada: Data curation; Formal analysis; Investigation. Kosuke Odaira: Data curation; Formal analysis; Investigation; Methodology; Validation. Akihiro Hosoi: Data curation; Formal analysis; Investigation; Methodology. Yukari Kobayashi: Data curation; Formal analysis; Investigation; Methodology. Koji Nagaoka: Data curation; Formal analysis; Investigation; Methodology. Takahiro Karasaki: Data curation; Formal analysis; Investigation. Hirokazu Matsushita: Conceptualization; Formal analysis; Supervision; Validation. Koichi Yagi: Data curation; Investigation. Hiroharu Yamashita: Formal analysis; Formal analysis; Investigation; Investigation. Masashi Fujita: Data curation; Formal analysis; Investigation; Methodology; Software. Shuichi Watanabe: Data curation; Formal analysis; Investigation; Methodology; Software; Visualization. Takashi Kamatani: Formal analysis; Investigation; Software. Fuyuki Miya: Data curation; Funding acquisition; Investigation; Methodology; Software; Validation. Junichi Mineno: Data curation; Formal analysis; Investigation; Methodology. Hidewaki Nakagawa: Formal analysis; Investigation; Supervision; Validation. Tatsuhiko Tsunoda: Formal analysis; Investigation; Methodology; Software; Supervision. Shunji Takahashi: Investigation; Supervision; Validation. Yasuyuki Seto: Formal analysis; Supervision; Validation. Kazuhiro Kakimi: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Validation; Visualization; Writing‐original draft; Writing‐review & editing.
Conflict of Interest
Dr Kakimi reports grants from TAKARA BIO Inc. and grants from MSD, outside the submitted work. In addition, Dr Kakimi has a patent Immunogram pending and the Department of Immunotherapeutics, The University of Tokyo Hospital, is an endowed department by TAKARA BIO Inc. Dr Sato reports personal fees from ONO Pharmaceutical Co., Ltd, Bristol‐Myers Squibb Company, MSD KK and TAIHO Pharmaceutical Co., Ltd, outside the submitted work. Dr Takahashi reports grants and personal fees from Bristol‐Myers Squibb KK, grants and personal fees from ONO Pharmaceutical Co., Ltd, grants and personal fees from MSD, grants and personal fees from AstraZeneca, grants and personal fees from Chugai, and grants and personal fees from BAYER, outside the submitted work. Drs Kakimi and Mineno have a patent Immunogram pending. The other authors have no competing interests to disclose.
Ethical approval
This study was approved by the Research Ethics Committees of the University of Tokyo (No. G3545) and Tokyo Metropolitan Bokutoh Hospital (No. 25‐38‐02). Informed written consent was obtained from all patients included in the study.
Supporting information
Acknowledgments
The authors thank Nao Fujieda for excellent technical assistance. This work was conducted by the institutional support of RIKEN. This study was also supported in part by the Japan Agency for Medical Research and Development (AMED) Project for Cancer Research and Therapeutic Evolution (P‐CREATE). The funder had no role in study design, data collection and analysis, interpretation, decision to publish or preparation of the manuscript, or any aspect of the study.
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018; 68: 394–424. [DOI] [PubMed] [Google Scholar]
- 2. Muro K, Chung HC, Shankaran V et al Pembrolizumab for patients with PD‐L1‐positive advanced gastric cancer (KEYNOTE‐012): a multicentre, open‐label, phase 1b trial. Lancet Oncol 2016; 17: 717–726. [DOI] [PubMed] [Google Scholar]
- 3. Kang YK, Boku N, Satoh T et al Nivolumab in patients with advanced gastric or gastro‐oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO‐4538‐12, ATTRACTION‐2): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet 2017; 390: 2461–2471. [DOI] [PubMed] [Google Scholar]
- 4. Wong SS, Kim KM, Ting JC et al Genomic landscape and genetic heterogeneity in gastric adenocarcinoma revealed by whole‐genome sequencing. Nat Commun 2014; 5: 5477. [DOI] [PubMed] [Google Scholar]
- 5. Cancer Genome Atlas Research Network . Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014; 513: 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cristescu R, Lee J, Nebozhyn M et al Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med 2015; 21: 449–456. [DOI] [PubMed] [Google Scholar]
- 7. Chen K, Yang D, Li X et al Mutational landscape of gastric adenocarcinoma in Chinese: implications for prognosis and therapy. Proc Natl Acad Sci USA 2015; 112: 1107–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kakiuchi M, Nishizawa T, Ueda H et al Recurrent gain‐of‐function mutations of RHOA in diffuse‐type gastric carcinoma. Nat Genet 2014; 46: 583–587. [DOI] [PubMed] [Google Scholar]
- 9. Kim ST, Cristescu R, Bass AJ et al Comprehensive molecular characterization of clinical responses to PD‐1 inhibition in metastatic gastric cancer. Nat Med 2018; 24: 1449–1458. [DOI] [PubMed] [Google Scholar]
- 10. Chen DS, Mellman I. Oncology meets immunology: the cancer‐immunity cycle. Immunity 2013; 39: 1–10. [DOI] [PubMed] [Google Scholar]
- 11. Galon J. Type, Density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313: 1960–1964. [DOI] [PubMed] [Google Scholar]
- 12. Galon J, Angell HK, Bedognetti D, Marincola FM. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity 2013; 39: 11–26. [DOI] [PubMed] [Google Scholar]
- 13. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov 2019; 18: 197–218. [DOI] [PubMed] [Google Scholar]
- 14. Karasaki T, Nagayama K, Kuwano H et al An immunogram for the cancer‐immunity cycle: towards personalized immunotherapy of lung cancer. Thorac Oncol 2017; 12: 791–803. [DOI] [PubMed] [Google Scholar]
- 15. Kobayashi Y, Kushihara Y, Saito N, Yamaguchi S, Kakimi K. A novel scoring method based on RNA‐Seq immunograms describing individual cancer‐immunity interactions. Cancer Sci 2020. e‐pub ahead of print. 10.1136/jitc-2019-000375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Japanese Gastric Cancer Association . Japanese classification of gastric carcinoma: 3rd English edition. Gastric Cancer 2011; 14: 101–112. [DOI] [PubMed] [Google Scholar]
- 17. Laurén P. The two histological main types of gastric carcinoma: diffuse and so‐called intestinal‐type carcinoma. Acta Pathol Microbiol Scand 1965; 64: 31–49. [DOI] [PubMed] [Google Scholar]
- 18. Borrmann R. Geschwülste des magens und duodenums In: Borchardt H, Borrmann R, Christeller E, Dietrich A, Fischer W, Von Gierke E. eds. Verdauungsschlauch: Rachen und Tonsillen Speiseröhre Magen und Darm Bauchfell. Springer, Berlin Heidelberg: Berlin, Heidelberg, 1926: 812–1054. [Google Scholar]
- 19. Lee J, Cristescu R, Kim KM et al Development of mesenchymal subtype gene signature for clinical application in gastric cancer. Oncotarget 2017; 8: 66305–66315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Newman AM, Steen CB, Liu CL et al Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 2019; 37: 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang P, Gu S, Pan D et al Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 2018; 24: 1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348: 69–74. [DOI] [PubMed] [Google Scholar]
- 23. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014; 513: 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer 2018; 18: 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sharma P, Hu‐Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017; 168: 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zaretsky JM, Garcia‐Diaz A, Shin DS et al Mutations associated with acquired resistance to PD‐1 blockade in melanoma. N Engl J Med 2016; 375: 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gao J, Shi LZ, Zhao H et al Loss of IFN‐gamma pathway genes in tumor cells as a mechanism of resistance to anti‐CTLA‐4 therapy. Cell 2016; 167: 397 – 404.e399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. O'Donnell TJ, Rubinsteyn A, Bonsack M, Riemer AB, Laserson U, Hammerbacher J. MHCflurry: open‐source class I MHC binding affinity prediction. Cell Systems 2018; 7: 129–132.e124. [DOI] [PubMed] [Google Scholar]
- 30. Nejo T, Matsushita H, Karasaki T, et al Reduced neoantigen expression revealed by longitudinal multiomics as a possible immune evasion mechanism in glioma. Cancer Immunol Res 2019; 7: 1148–1161. [DOI] [PubMed] [Google Scholar]
- 31. Blank CU, Haanen JB, Ribas A, Schumacher TN. The “cancer immunogram”. Science 2016; 352: 658–660. [DOI] [PubMed] [Google Scholar]
- 32. van Dijk N, Funt SA, Blank CU, Powles T, Rosenberg JE, van der Heijden MS. The cancer immunogram as a framework for personalized immunotherapy in urothelial cancer. Eur Urol 2019; 75: 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Botling J, Sandelin M. Immune biomarkers on the radar‐comprehensive "immunograms" for multimodal treatment prediction. J Thorac Oncol 2017; 12: 770–772. [DOI] [PubMed] [Google Scholar]
- 34. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015; 160: 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chang CH, Qiu J, O'Sullivan D et al Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015; 162: 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ottensmeier CH, Perry KL, Harden EL et al Upregulated glucose metabolism correlates inversely with CD8+ T‐cell infiltration and survival in squamous cell carcinoma. Cancer Res 2016; 76: 4136–4148. [DOI] [PubMed] [Google Scholar]
- 37. Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017; 355: eaaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Japanese Gastric Cancer Association . Japanese gastric cancer treatment guidelines 2010 (ver. 3). Gastric Cancer 2011; 14: 113–123. [DOI] [PubMed] [Google Scholar]
- 39. Li H, Durbin R. Fast and accurate long‐read alignment with Burrows‐Wheeler transform. Bioinformatics 2010; 26: 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McKenna A, Hanna M, Banks E et al The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 2010; 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koboldt DC, Zhang Q, Larson DE et al VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012; 22: 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cibulskis K, Lawrence MS, Carter SL et al Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013; 31: 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 2010; 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010; 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dobin A, Davis CA, Schlesinger F et al STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 2013; 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high‐throughput sequencing data. Bioinformatics 2015; 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kawaguchi S, Higasa K, Shimizu M, Yamada R, Matsuda F. HLA‐HD: an accurate HLA typing algorithm for next‐generation sequencing data. Hum Mutat 2017; 38: 788–797. [DOI] [PubMed] [Google Scholar]
- 48. Edge P, Bafna V, Bansal V. HapCUT2: robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res 2017; 27: 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cingolani P, Platts A, le Wang L et al A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly (Austin) 2012; 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 215: 403–410. [DOI] [PubMed] [Google Scholar]
- 51. Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA‐P: a desktop application for gene set enrichment analysis. Bioinformatics 2007; 23: 3251–3253. [DOI] [PubMed] [Google Scholar]
- 52. Chu J, Sadeghi S, Raymond A et al BioBloom tools: fast, accurate and memory‐efficient host species sequence screening using bloom filters. Bioinformatics 2014; 30: 3402–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kanda Y. Investigation of the freely available easy‐to‐use software 'EZR' for medical statistics. Bone Marrow Transplant 2013; 48: 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kodama Y, Mashima J, Kosuge T et al The DDBJ Japanese Genotype‐phenotype archive for genetic and phenotypic human data. Nucleic Acids Res 2015; 43: D18–D22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials