

Abstract
Sepsis remains a major global concern and is associated with high mortality and morbidity despite improvements in its management. Markers currently in use have shortcomings such as a lack of specificity and failures in the early detection of sepsis. In this study, we aimed to identify key genes involved in the molecular mechanisms of sepsis and search for potential new biomarkers and treatment targets for sepsis using bioinformatics analyses. Three datasets (GSE95233, GSE57065, and GSE28750) associated with sepsis were downloaded from the public functional genomics data repository Gene Expression Omnibus. Differentially expressed genes (DEGs) were identified using R packages (Affy and limma). Functional enrichment of the DEGs was analyzed with the DAVID database. Protein-protein interaction networks were derived using the STRING database and visualized using Cytoscape software. Potential biomarker genes were analyzed using receiver operating characteristic (ROC) curves in the R package (pROC). The three datasets included 156 whole blood RNA samples from 89 sepsis patients and 67 healthy controls. Between the two groups, 568 DEGs were identified, among which 315 were upregulated and 253 were downregulated in the septic group. These genes were enriched for pathways mainly involved in the innate immune response, T-cell biology, antigen presentation, and natural killer cell function. ROC analyses identified nine genes—LRG1, ELANE, TP53, LCK, TBX21, ZAP70, CD247, ITK, and FYN—as potential new biomarkers for sepsis. Real-time PCR confirmed that the expression of seven of these genes was in accordance with the microarray results. This study revealed imbalanced immune responses at the transcriptomic level during early sepsis and identified nine genes as potential biomarkers for sepsis.
1. Introduction
Sepsis is defined as a life-threatening organ dysfunction caused by a dysregulated host response to infection. Despite advances in critical care management over the past few years, sepsis is still associated with high mortality and morbidity worldwide [1]. It has been reported that sepsis causes 30 million episodes and 6 million deaths per year globally. However, according to the WHO, the data have missed incidences in the low- and middle-income countries, which means that the true burden arising from sepsis is far more serious. Therefore, the early diagnosis of sepsis is necessary to provide timely treatment. Markers currently in use, for example, CRP, PCT, and IL-6, have intrinsic shortcomings such as a lack of specificity and failures in the early detection of sepsis [2]. Many researchers are committed to exploring new biomarkers for sepsis. For example, studies have found that serum levels of presepsin, soluble urokinase plasminogen activator receptor, and soluble triggering receptor expressed on myeloid cell 1, as well as the expression of CD64, are upregulated among sepsis patients. Newly identified classes of biomarkers such as microRNAs, long noncoding RNAs, and the human microbiome are also arousing general interest [3]. Despite the increase in different potential biomarkers, such efforts have not yet yielded satisfactory results, which warrants further validation.
It is not surprising that a large proportion of sepsis biomarkers still focuses on the inflammatory part of this condition. Sepsis is characterized by disrupted inflammatory responses. It has been proposed that following a major inflammatory insult, there are simultaneous inflammatory and immunosuppressive responses [4]. Pattern recognition receptors such as TLR recognize and elicit inflammatory responses against pathogenic factors, for example, by triggering leukocyte and complement activation [5]. Concurrent immune cell function impairment (e.g., neutrophil defects) and T-cell apoptosis also occur, leading to immune suppression in patients with sepsis [6]. This could trigger secondary infections and undermine the immune system. However, the complex inflammatory responses during sepsis have not been fully elucidated.
Bioinformatics analysis offers an ideal way to screen large gene expression datasets to comprehensively understand the mechanisms underlying sepsis. In this study, we integrated three datasets and used a bioinformatics analysis approach to detect key genes and potential new biomarkers involved in sepsis. Molecular mechanisms underlying the inflammatory responses during sepsis were also explored to search for possible new treatment targets for sepsis.
2. Materials and Methods
2.1. Data Sources
The three gene expression datasets analyzed in this study were downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) and used to identify DEGs. GSE95233 [7], GSE57065 [8], and GSE28750 [9] were taken as representative datasets of patients with sepsis. For each dataset, data from day 1 of sample collection were used to analyze the gene expression based on the GPL570 platform (HG-U133_Plus_2). All data were freely available online, and this study did not involve any human or animal experiments.
2.2. Identification of DEGs
Background expression value correction and data normalization of the raw data were carried out using the Affy package in R (Affy, version 1.64.0). Subsequently, the Linear Models for Microarray Analysis R package (limma; version 3.42.2) was applied for differential expression analysis. Volcano plots were generated using Bioconductor (http://bioconductor.org/biocLite.R). DEGs were identified as those with a t-test value of P < 0.05 and a [logFC] > 1.5.
2.3. Functional and Pathway Enrichment Analyses
Gene Ontology (GO) analyses were used for the exploration of functional roles of gene sets, while KEGG analyses were used to classify the pathways in which such genes might function. For comprehensive functional annotation, GO and KEGG analyses of the identified DEGs were conducted using the DAVID tool (https://david.ncifcrf.gov/). A false discovery rate (FDR) < 0.05 in both GO and KEGG analyses was set as the threshold for significant enrichment.
2.4. Protein-Protein Interaction (PPI) Network Construction and Hub Gene Analysis
DEGs were uploaded to Search Tool for the Retrieval of Interacting Genes (STRING, https://string-db.org/) to analyze interactions among the proteins encoded by the identified DEGs. Results with a minimum interaction score of 0.4 were visualized using Cytoscape. The PPI network for hub genes was computed with the maximal clique centrality (MCC) method and CytoHubba. Based on the MCODE plugin, the PPI network was divided into two clusters (clusters A and B).
2.5. Receiver Operating Characteristic (ROC) Curve Analysis
ROC curve analyses to determine the specificity, sensitivity, likelihood ratios, positive predictive values, and negative predictive values for all possible thresholds of the ROC curve were performed using the R package (pROC, version 1.16.2). The diagnostic values of the genes were predicted based on the ROC curve analysis.
2.6. Patient Enrollment
Patients diagnosed with sepsis in the emergency department of Shanghai Ruijin Hospital from July 31, 2020, to August 21, 2020, were enrolled. This part of the study was approved by the Ethics Committee of Ruijin Hospital (No. 2017119). Enrollment criteria were as follows: (1) age: 18–90 years old, (2) patients met sepsis 3.0 sepsis diagnostic criteria, and (3) hospital stay > 24 h. Exclusion criteria were as follows: (1) discharged or died within 24 h after admission, (2) participated in other clinical research, (3) needed emergency surgery after admission, (4) had a malignant tumor, (5) pregnant or lactating, and (6) lacked necessary clinical data.
2.7. Quantitative Real-Time PCR
RNA was extracted from whole blood using TRIzol reagent (12183-555, Invitrogen) following the manufacturer's instructions. cDNA was synthesized with a SuperScript™ III First-Strand Synthesis SuperMix for qRT-PCR (11752-050, Invitrogen). Power SYBR® Green PCR Master Mix (4367659, Applied Biosystems) was used for qRT-PCR to analyze mRNA expression. GAPDH was used as an internal control, and the relative mRNA expression levels were calculated using the 2–ΔΔCT method. The primer pairs used in the experiments are listed in data S1.
3. Results
3.1. Identification of DEGs
Datasets GSE57065, GSE95233, and GSE28750 were downloaded from the GEO database and analyzed using R packages (Affy, limma, and ggplot2). Volcano plots were generated to visualize fold changes of the DEGs. Of the 568 DEGs evaluated, 315 were upregulated and 253 were downregulated in the sepsis group (Figure 1).
Figure 1.
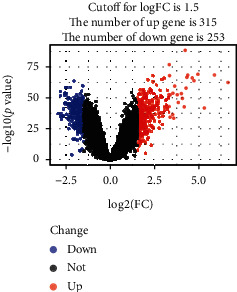
Identification of DEGs from three datasets using volcano plot analysis. Statistically significant DEGs were identified as those with a t-test P value < 0.05 and a [logFC] > 1.5. The blue dots represent downregulated DEGs, while the red dots represent upregulated DEGs.
3.2. Functional Enrichment of DEGs
Enrichment analysis techniques extract biological information from a set of genes or proteins. To identify key genes related to sepsis, gene functions were annotated using the DAVID online software database. GO annotation analysis showed enrichment of DEGs involved in inflammatory responses such as the innate response, T-cell receptor pathway, and antigen processing and presentation (Figure 2(a)). KEGG pathway analysis showed that the genes involved in sepsis were associated with different infections such as influenza, tuberculosis, and HTLV-1, further linking inflammation with sepsis (Figure 2(b)).
Figure 2.
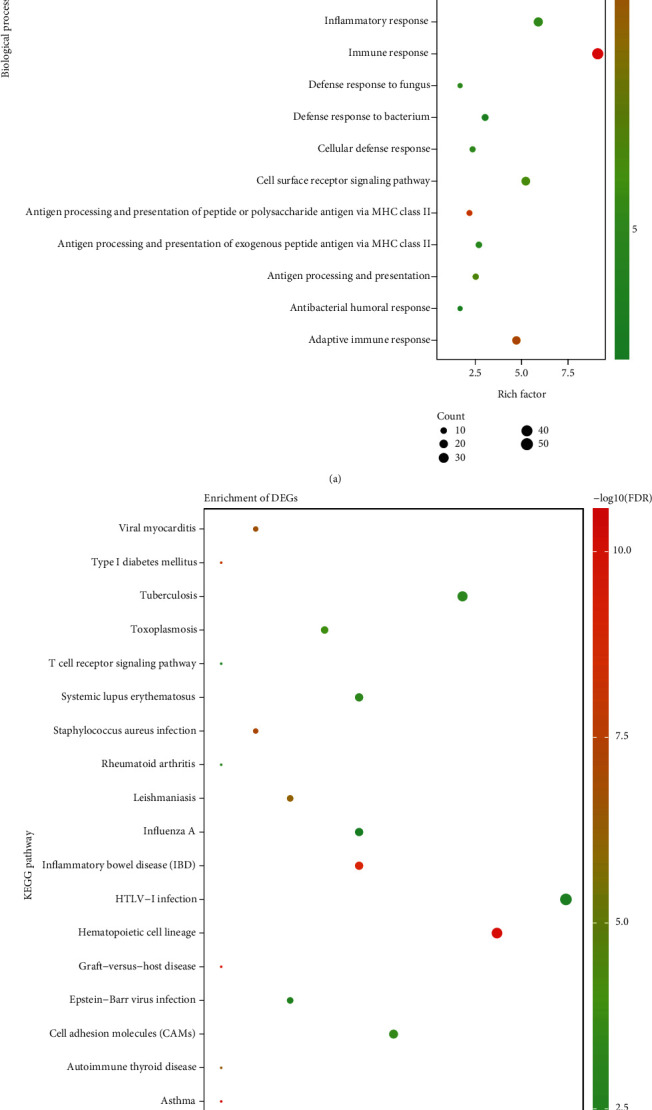
GO and KEGG enrichment analyses of DEGs. (a) Shows the results of biological process terms enriched by GO analysis. (b) Shows the enriched pathway by KEGG analysis. The colored dots represent the FDR for that term, with red representing greater significance. The size of the dots represents the number of involved genes. The rich factor represents the proportion of enriched genes for each term.
3.3. PPI Network and Hub Genes
To explore the key genes involved in sepsis, a PPI network with 443 nodes and 2470 edges was built for the 568 DEGs (Figure 3). Table 1(a) shows the top 20 genes with the highest degree rank, whereas the top 20 genes selected using the CytoHubba plugin according to the MCC method are sequentially ordered in Table 1(b). The PPI network was divided into two clusters (clusters A and B) using the MCODE plugin (Figures 4(a) and 4(b)). Interestingly, most of the top 40 genes identified using the different calculation methods, previously mentioned herein, were present in both clusters A and B, with the genes shown in Table 1(a) being in cluster A and genes in Table 1(b) in cluster B. These findings suggested that these genes play important roles in sepsis. Tables 2(a) and 2(b) show the functional annotation of the two clusters. Cluster A was enriched in genes involved in T-cell biology, antigen presentation, and natural killer (NK) cell function. Intriguingly, most genes in cluster A were downregulated, suggesting a possible immunosuppression process early in sepsis. In contrast, cluster B mainly contained genes related to innate immune responses such as neutrophil-mediated immunity and phagocytosis. These genes are closely related and cooperate with each other to respond to different types of insults. Overall, these 40 genes comprised the hub genes during sepsis. While some of these genes are well-characterized key elements in sepsis, others might represent new potential biomarkers for sepsis. Nine genes (LRG1, ELANE, TP53, LCK, TBX21, ZAP70, CD247, ITK, and FYN) were chosen for further investigation of their roles in and potential use as biomarkers for sepsis.
Figure 3.
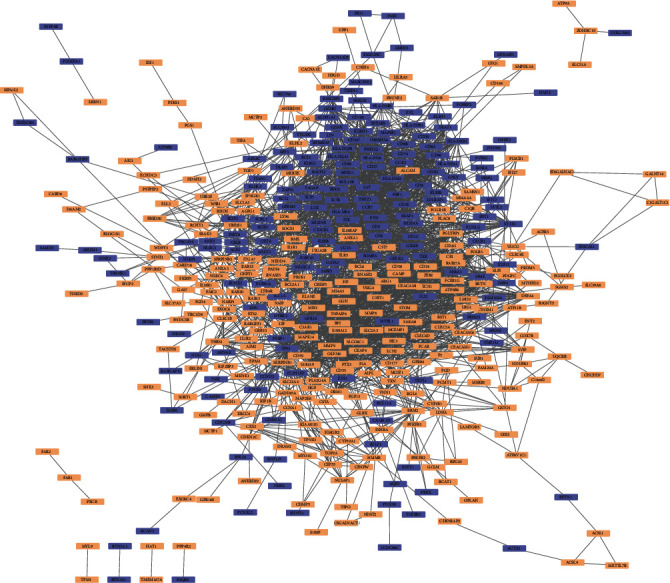
PPI network complex of DEGs. 568 DEGs with 443 nodes and 2470 edges were displayed by the PPI network, with upregulated genes shown in light orange and downregulated genes shown in bluish violet.
Figure 4.
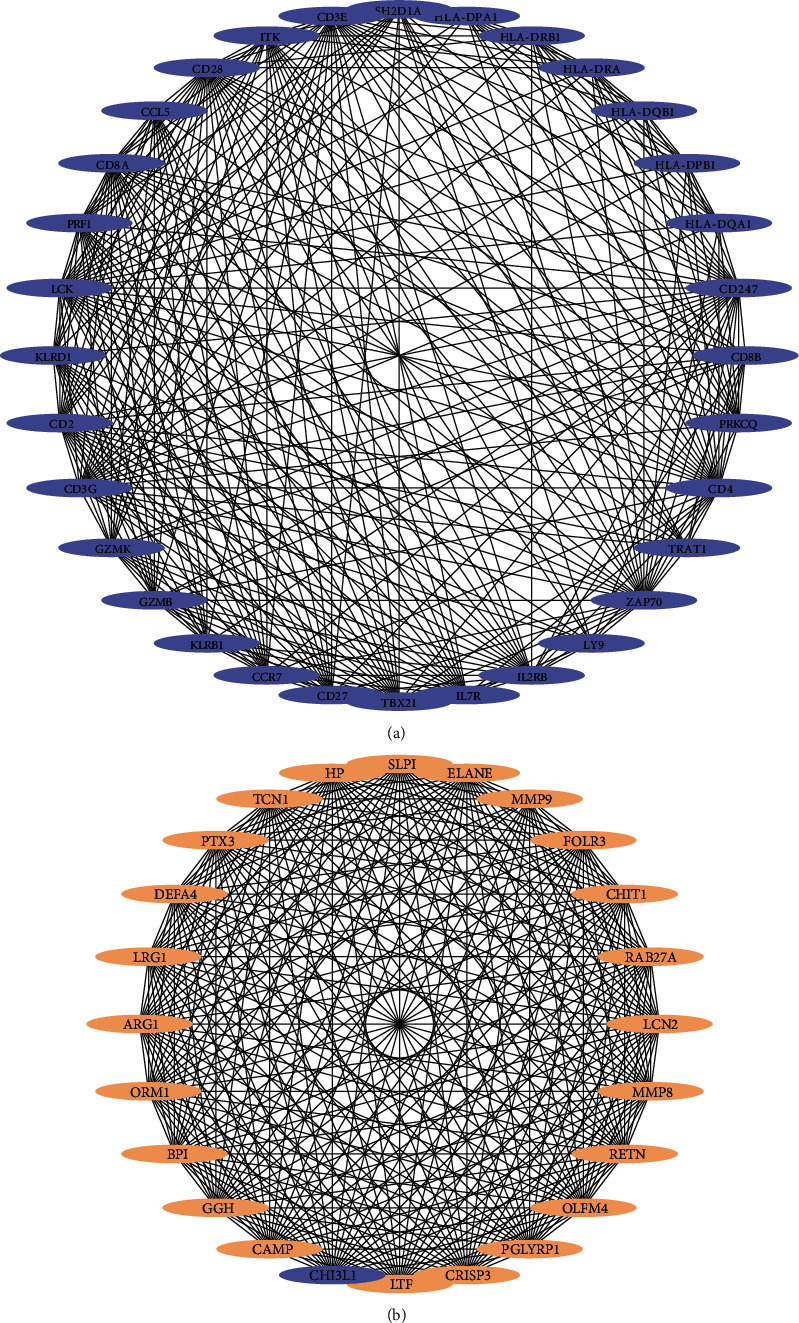
Clusters A and B identified by network analysis. The PPI network is divided into 2 clusters by the MCODE plugin. Light orange and bluish violet represent upregulated and downregulated genes separately.
3.4. ROC Curve
To identify new potential biomarkers for sepsis, ROC curves of data derived from healthy controls and patients with sepsis from datasets GSE57065, GSE95233, and GSE28750 were analyzed using the R package (Figure 5). ROC curves were generated, and the area under the curves was used to compare the different genes. This analysis demonstrated that the nine selected genes had a diagnostic role in sepsis. Thus, we chose these genes as candidates for further analysis and validation.
Figure 5.
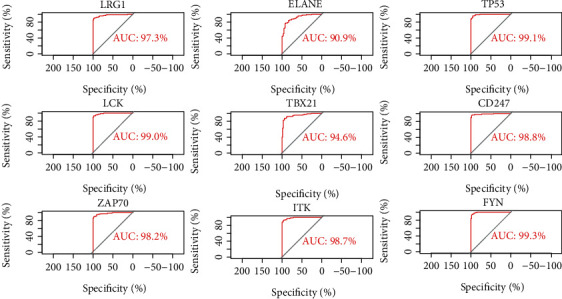
ROC curve analyses of 9 selected DEGs.
3.5. Validation of Selected Genes at the Transcriptional Level
The expression of nine key genes was compared between patients with sepsis (n = 5) and healthy controls (n = 5) using quantitative real-time PCR. The results showed that the expression of seven of these genes was consistent with the trend observed in the microarray analysis, whereas two genes, LRG1 and TP53, showed no significant difference in expression (Figure 6).
Figure 6.
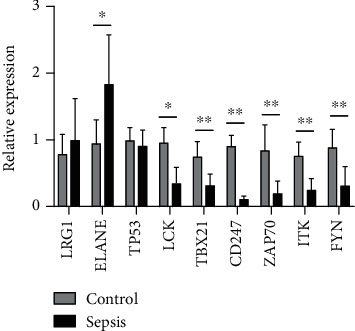
Expression of nine selected key genes was compared between healthy controls and sepsis patients by quantitative real-time PCR. Differences between two groups were analyzed using the t-test. ∗P < 0.05, ∗∗P < 0.01.
4. Discussion
A microarray study is an ideal way to comprehensively investigate sepsis. In this study, three gene datasets were integrated to search for potential biomarkers and explore molecular mechanisms of sepsis. Although sepsis is an inflammatory disease, it has recently been established that both proinflammatory and anti-inflammatory responses occur early during sepsis [10]. In our study, genes associated with both innate and adaptive immunity had altered expression patterns in patients with sepsis since the beginning of diagnosis. KEGG pathway enrichment analysis showed that different antigenic constituents from bacteria, viruses, and other insults can cause sepsis. Upon receptor contact with their cognate ligands, proinflammatory intermediates are recruited and intracellular signaling pathways such as NF-κB transduction are activated. The activation of NF-κB induces the expression of early activation genes such as IL1B, IFNG, and IL-6 to combat the insult. However, excessive cytokine release leads to an increase in the release of circulating immune cells [5]. We found that most genes in cluster B encoded proteins that participate in innate immune responses. All of these genes were upregulated, indicating their roles in innate immune responses. One of these genes is associated with OLFM4+ neutrophils, a subset of human neutrophils. Elevated levels of OLFM4+ neutrophils in patients with sepsis are associated with worse outcomes [11]. CHI3L1 is produced by several cells including macrophages and neutrophils. A recent study pointed out that the downregulation of CHI3L1 alleviates skeletal muscle stem cell injury, suggesting its therapeutic potential for sepsis [12]. In our study, we found that the genes in cluster B interact with one another to mediate inflammatory responses. Targeting these genes and corresponding pathways involved in innate immune responses might be one strategy to reduce inflammation and associated pathology during sepsis, although much work is still required, considering the previous unsatisfactory trials involving immune activation genes.
Previously, it was thought that the host immune response to sepsis is characterized by an initial hyperinflammatory response, followed by an immunosuppressive phase as the disease progresses. However, recent studies have shown that both proinflammatory and anti-inflammatory responses occur early and simultaneously in sepsis [13]. In our study, genes in cluster A were found to be mainly involved in T-cell biology, antigen processing and presentation, and NK cell function. Interestingly, all genes in cluster A were downregulated, suggesting immunosuppression during sepsis. In cluster A, genes encoding antigen presentation-related molecules, including HLA-DRA, HLA-DRB1, HLA-DPA1, HLA-DPB1, and CCR7, exhibited decreased expression. Studies have also shown that during sepsis, the number of dendritic cells (DCs), the major group associated with antigen presentation, is decreased in patients with sepsis. In addition, the surviving DCs also exhibit lower expression of HLA-DR. Moreover, endotoxin-tolerant macrophages express relatively low levels of HLA-DR on their surface, resulting in a lack of antigen presentation [14]. Similarly, NK cells are also underrepresented in patients with sepsis, and the remaining NK cells display defective cytotoxic functions [15, 16]. In our study, we found that genes involved in NK cell-mediated cytotoxicity (such as KLRB1, KLRD1, SH2D1A, and PRF1) showed decreased expression. In cluster A, LCK, ZAP70, CD2, CD247, CD27, CD28, CD3E, CD3G, CD4, CD8A, CD8B, ITK, LCK, TRAT1, TBX21, FYN, and IL-7R, which are related to T-cell biology, were represented, suggesting an important role for T-cell immunity during sepsis. Recently, the immunosuppressive phase has become a focus during sepsis treatment. It is inspiring that IL-7, a growth factor that stimulates the proliferation and maturation of many cell types, could continuously boost the absolute T lymphocyte counts of sepsis patients in a phase II clinical trial [17]. Our results also indicated that immunostimulatory therapy targeting immunosuppression-related genes and pathways could be a promising way to treat sepsis.
It should be noted that there were some DEGs that were not shared among all sepsis networks but have been proven to play indispensable roles in sepsis in recent years. For example, RETN encodes resistin, which is strikingly elevated in patients with sepsis and is associated with sepsis severity and outcomes [18]. Silswal et al. showed that resistin activated monocytes and macrophages as well as induced the release of proinflammatory cytokines [19]. Further, recent experimental data have highlighted resistin as a potential therapeutic target in sepsis [20]. These previous findings suggest a paradoxical role for resistin in sepsis. TCN1 encodes a member of the vitamin B12-binding protein family, transcobalamin I, which is elevated during infectious conditions. As a member of the cobalamin transport protein, transcobalamin elevation may contribute to the resolution of inflammation [21]. FOLR3 and GGH are associated with the folate pathway. These reports suggest that vitamin B12 and folate metabolism may also constitute an important part of sepsis. Nutritional therapy, including vitamin B12 and folate, may affect the pathogenesis of sepsis, which requires further research [22].
In this study, we also identified some genes for which the functions in sepsis have not been completely characterized, suggesting their potential as biomarkers for this disease. It should be noted that the expression levels of most of these genes demonstrated by real-time PCR corresponded to the patterns observed by microarray analyses, while two genes (LRG1, TP53) showed no significant difference. The inconsistency could be attributed to the different detection methods, sample size, patient heterogeneity, and course of the disease. Considering the small sample size of our study, it is not powerful enough to change the conclusions about the nine critical genes selected by bioinformatics analysis.
One of these genes, LRG1, encodes a highly conserved member of the leucine-rich repeat family of proteins, which has been reported to play a role in the inflammatory response. LRG1 is expressed by neutrophils and macrophages [23, 24]. Some studies found that circulating LRG1 mRNA and plasma LRG1 protein levels might together be helpful for diagnosing simple and complicated acute appendicitis in patients with acute abdominal pain [25]. However, the role of LRG1 in sepsis remains unclear to the best of our knowledge. Our microarray analyses showed that LRG1 mRNA levels are higher in patients with sepsis. Further study is needed to validate these results and investigate the roles of this marker in sepsis.
Another gene, ELANE, encodes neutrophil elastase (NE), a serine protease secreted by neutrophils into the extracellular milieu during the inflammatory response [26]. NE also participates in the formation of neutrophil extracellular traps [27]. A previous study found that NE is positively correlated with the severity of sepsis and organ dysfunction, suggesting its potential as a biomarker for sepsis [28]. This finding is consistent with our results.
LCK and FYN belong to the Src family of protein tyrosine kinases. LCK phosphorylates downstream signaling proteins, resulting in changes in the expression of genes that are essential for T-cell maturation and activation. FYN is involved in signal transduction pathways during the development and activation of T lymphocytes and NK cells under physiological conditions [29]. FYN and LCK are also essential for platelet production and activation [30]. However, the roles of LCK and FYN in sepsis have not been entirely elucidated. In our study, the expression of LCK and FYN was decreased in patients with sepsis, indicating possible suppression of T-cells and NK cells, as well as a role for these proteins in platelet functions during sepsis, which warrants further exploration. Our ROC curve analysis also showed that both genes have diagnostic value for sepsis.
ZAP70, a member of the Syk protein kinase family, is enriched in the TCR signaling pathway. This protein functions in the initial step of TCR-mediated signal transduction in combination with the Src family kinases LCK and FYN. Functional deletion of ZAP70 can lead to selective T-cell defects characterized by the selective absence of CD8-positive T-cells. Gomez-Rodriguez et al. [31] found that the downregulation of ZAP70 accelerates neonatal sepsis disease progression. The role of ZAP70 in adult sepsis warrants further investigation.
The gene product of CD247 (also known as CD3ζ) is T-cell receptor zeta, which is part of the T-cell receptor-CD3 complex. CD247 plays essential roles in coupling antigen recognition to several intracellular signal transduction pathways. CD3ζ chain expression is consistently reduced in T-cells from both the spleen and lymph nodes in sepsis [32]. Moreover, the downregulation of CD247 was reported to be accompanied by decreased expression of other T-cell-associated signal transduction molecules such as ZAP70, as well as T-cell apoptosis, in line with our data.
TP53 is a well-characterized gene for which the gene product induces cell cycle arrest, apoptosis, senescence, DNA repair, and metabolic changes [32]. TP53 may contribute to apoptosis in a tissue-dependent manner. A recent study revealed that p53 expression in T lymphocytes during sepsis could be responsible for enhancing both apoptosis and immune dysfunction in T-cells [33]. In our microarray analyses, TP53 expression was found to be downregulated in patients with sepsis, which is in accordance with the expression of genes involved in the T-cell signaling pathway, suggesting their possible interaction during sepsis.
TBX21, also known as T-bet, is the master regulator of effector T-cell activation. Many studies have shown that TBX21 controls the expression of IFNG, a hallmark Th1 cytokine, suggesting a role for this protein in initiating Th1 lineage development [34]. Recently, studies have also discovered T-bet expression in B-cells, CD8+ T-cells, and T-reg cells, suggesting variable functions under different circumstances [35–37]. The decrease in TBX21 expression might influence the expression of related immune cells, a notion that warrants further study.
Finally, ITK, which belongs to the Tec tyrosine kinase family, is involved in multiple aspects of T-cell development and functions such as T-cell activation and T-helper cell differentiation [31]. ITK is also known to be involved in the development of Th17 cells by regulating various transcription factors. For example, in ALI mice, ITK regulates the balance between inflammatory Th17 cells and anti-inflammatory T-reg cells [38]. However, the role of ITK in T-cell functions in patients with sepsis has not been fully evaluated. Our study showed that ITK was downregulated in patients, although further research is needed to explore the underlying mechanism.
5. Conclusions
Using a bioinformatics analysis of three gene datasets (GSE95233, GSE57065, and GSE28750), we identified the immune characteristics of sepsis. We found that DEGs in patients were enriched for pathways mainly involved in the innate immune response, T-cell biology, antigen presentation, and NK cell function. Focusing on the key genes and corresponding pathways involved in sepsis could provide new insights for sepsis treatment. Nine genes including LRG1, ELANE, TP53, LCK, TBX21, ZAP70, CD247, ITK, and FYN were also identified as potential new biomarkers for sepsis. The expression levels of most of these genes demonstrated by real-time PCR corresponded to the patterns observed by microarray analyses. Further investigations are needed to validate these preliminary findings.
Table 1.
(a) The top 20 genes in the network ranked by degrees
| Name | Degree | Change | Cluster |
|---|---|---|---|
| TP53 | 76 | Down | |
| LCK | 59 | Down | 1 |
| MMP9 | 56 | Up | 2 |
| CCL5 | 54 | Down | 1 |
| CD28 | 53 | Down | 1 |
| TBX21 | 50 | Down | 1 |
| CD2 | 48 | Down | 1 |
| ZAP70 | 48 | Down | 1 |
| CD3E | 46 | Down | 1 |
| CCR7 | 46 | Down | 1 |
| CD4 | 44 | Down | 1 |
| MPO | 43 | Up | |
| IL2RB | 43 | Down | 1 |
| CD247 | 42 | Down | 1 |
| ELANE | 42 | Up | 1; 2 |
| ITK | 42 | Down | 1 |
| GATA3 | 41 | Down | 1 |
| C3AR1 | 41 | Up | 1 |
| PRF1 | 40 | Down | 1 |
| FYN | 40 | Down | 1 |
(b) The top 20 genes in the network ranked by maximal clique centrality (MCC)
| Name | MCC | Change | Cluster |
|---|---|---|---|
| ELANE | 9.22E + 13 | Up | 1; 2 |
| SLPI | 9.22E + 13 | Up | 2 |
| HP | 9.22E + 13 | Up | 2 |
| PTX3 | 9.22E + 13 | Up | 2 |
| DEFA4 | 9.22E + 13 | Up | 2 |
| BPI | 9.22E + 13 | Up | 2 |
| CAMP | 9.22E + 13 | Up | 2 |
| ORM1 | 9.22E + 13 | Up | 2 |
| LCN2 | 9.22E + 13 | Up | 2 |
| MMP9 | 9.22E + 13 | Up | 2 |
| PGLYRP1 | 9.22E + 13 | Up | 2 |
| RAB27A | 9.22E + 13 | Up | 2 |
| MMP8 | 9.22E + 13 | Up | 2 |
| LTF | 9.22E + 13 | Up | 2 |
| TCN1 | 9.22E + 13 | Up | 2 |
| LRG1 | 9.22E + 13 | Up | 2 |
| RETN | 9.22E + 13 | Up | 2 |
| ARG1 | 9.22E + 13 | Up | 2 |
| OLFM4 | 9.22E + 13 | Up | 2 |
| CHI3L1 | 9.22E + 13 | Down | 2 |
Table 2.
(a) Functions enriched for the genes involved in cluster A
| Term | Description | Count | P value |
|---|---|---|---|
| GO:0050852 | T-cell receptor signaling pathway | 16 | 1.92E − 23 |
| GO:0006955 | Immune response | 14 | 1.76E − 13 |
| GO:0031295 | T-cell costimulation | 12 | 8.13E − 19 |
| GO:0007166 | Cell surface receptor signaling pathway | 11 | 3.72E − 11 |
| GO:0007169 | Transmembrane receptor protein tyrosine kinase signaling pathway | 8 | 3.76E − 10 |
| GO:0050776 | Regulation of immune response | 8 | 2.83E − 08 |
| GO:0042110 | T-cell activation | 7 | 2.41E − 10 |
| GO:0019882 | Antigen processing and presentation | 7 | 6.44E − 10 |
| GO:0042102 | Positive regulation of T-cell proliferation | 7 | 1.11E − 09 |
| GO:0002504 | Antigen processing and presentation of peptide or polysaccharide antigen via MHC class II | 6 | 9.31E − 11 |
| GO:0060333 | Interferon-gamma-mediated signaling pathway | 6 | 1.83E − 07 |
| GO:0019886 | Antigen processing and presentation of exogenous peptide antigen via MHC class II | 6 | 6.72E − 07 |
| GO:0002250 | Adaptive immune response | 6 | 7.02E − 06 |
| GO:0045087 | Innate immune response | 6 | 0.00105484 |
| hsa04660 | T-cell receptor signaling pathway | 11 | 2.82E − 12 |
| hsa04514 | Cell adhesion molecules (CAMs) | 11 | 9.77E − 11 |
| hsa04612 | Antigen processing and presentation | 10 | 8.8E − 12 |
| hsa05166 | HTLV-1 infection | 10 | 4.13E − 07 |
| hsa05332 | Graft-versus-host disease | 9 | 3.27E − 13 |
| hsa05330 | Allograft rejection | 9 | 8.99E − 13 |
| hsa04940 | Type I diabetes mellitus | 9 | 2.71E − 12 |
| hsa05320 | Autoimmune thyroid disease | 9 | 1.68E − 11 |
| hsa04640 | Hematopoietic cell lineage | 9 | 1.19E − 09 |
| hsa05416 | Viral myocarditis | 8 | 1.9E − 09 |
| hsa05323 | Rheumatoid arthritis | 8 | 4.19E − 08 |
| hsa05340 | Primary immunodeficiency | 7 | 3.19E − 09 |
| hsa04672 | Intestinal immune network for IgA production | 7 | 2.46E − 08 |
| hsa05321 | Inflammatory bowel disease (IBD) | 7 | 1.64E − 07 |
| hsa04650 | Natural killer cell-mediated cytotoxicity | 7 | 0.00000753 |
| hsa05322 | Systemic lupus erythematosus | 7 | 0.0000129 |
(b) Functions enriched for the genes involved in cluster B
| Term | Description | Count | P value |
|---|---|---|---|
| GO:0045087 | Innate immune response | 7 | 1.90E − 05 |
| GO:0006508 | Proteolysis | 5 | 0.004385255 |
| GO:0019731 | Antibacterial humoral response | 4 | 2.87E − 05 |
| GO:0006955 | Immune response | 4 | 0.019097644 |
| GO:0001878 | Response to yeast | 3 | 1.39E − 04 |
| GO:0050829 | Defense response to Gram-negative bacterium | 3 | 0.002549924 |
| GO:0071347 | Cellular response to interleukin-1 | 3 | 0.004210679 |
| GO:0022617 | Extracellular matrix disassembly | 3 | 0.004809135 |
| GO:0050830 | Defense response to Gram-positive bacterium | 3 | 0.005979232 |
| GO:0071356 | Cellular response to tumor necrosis factor | 3 | 0.009834724 |
| GO:0071222 | Cellular response to lipopolysaccharide | 3 | 0.010355235 |
| GO:0045766 | Positive regulation of angiogenesis | 3 | 0.010708943 |
| GO:0042742 | Defense response to bacterium | 3 | 0.016637646 |
| GO:0006032 | Chitin catabolic process | 2 | 0.009550291 |
| GO:0009635 | Response to herbicide | 2 | 0.009550291 |
| GO:0019732 | Antifungal humoral response | 2 | 0.01361651 |
| GO:0044130 | Negative regulation of growth of symbiont in host | 2 | 0.021701096 |
| GO:0070207 | Protein homotrimerization | 2 | 0.029722278 |
| GO:0002227 | Innate immune response in mucosa | 2 | 0.033709241 |
| GO:0035987 | Endodermal cell differentiation | 2 | 0.036358505 |
| GO:0032715 | Negative regulation of interleukin-6 production | 2 | 0.03768053 |
| GO:0050766 | Positive regulation of phagocytosis | 2 | 0.03900082 |
| GO:0010043 | Response to zinc ion | 2 | 0.048194457 |
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant number 81772107), the Shanghai Shenkang Hospital Development Center of China (grant number SHDC12017116), and the Program for Outstanding Medical Academic, Shanghai Municipal Committee of Science and Technology (grant number 18411950900).
Contributor Information
Ying Chen, Email: bichatlion@163.com.
Er-Zhen Chen, Email: rjchenerzhen@163.com.
Data Availability
The data used to support the findings of this study are available from the corresponding authors upon request.
Conflicts of Interest
The authors declare that there is no conflict of interest.
Supplementary Materials
Supplementary data 1: primers of 10 genes.
References
- 1.Rhodes A., Evans L. E., Alhazzani W., et al. Surviving Sepsis Campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Medicine. 2017;43(3):304–377. doi: 10.1007/s00134-017-4683-6. [DOI] [PubMed] [Google Scholar]
- 2.Pettila V., Hynninen M., Takkunen O., Kuusela P., Valtonen M. Predictive value of procalcitonin and interleukin 6 in critically ill patients with suspected sepsis. Intensive Care Medicine. 2002;28(9):1220–1225. doi: 10.1007/s00134-002-1416-1. [DOI] [PubMed] [Google Scholar]
- 3.Kim M. H., Choi J. H. An update on sepsis biomarkers. Infection & chemotherapy. 2020;52(1):1–18. doi: 10.3947/ic.2020.52.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delano M. J., Ward P. A. The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunological Reviews. 2016;274(1):330–353. doi: 10.1111/imr.12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rittirsch D., Flierl M. A., Ward P. A. Harmful molecular mechanisms in sepsis. Nature Reviews Immunology. 2008;8(10):776–787. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bone R. C., Grodzin C. J., Balk R. A. Sepsis: a new hypothesis for pathogenesis of the disease process. Chest. 1997;112(1):235–243. doi: 10.1378/chest.112.1.235. [DOI] [PubMed] [Google Scholar]
- 7.Tabone O., Mommert M., Jourdan C., et al. Endogenous Retroviruses Transcriptional Modulation After Severe Infection, Trauma and Burn. Frontiers in immunology. 2018;9:p. 3091. doi: 10.3389/fimmu.2018.03091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cazalis M. A., Lepape A., Venet F., et al. Early and dynamic changes in gene expression in septic shock patients: a genome-wide approach. Intensive Care Medicine Experimental. 2014;2(1):p. 20. doi: 10.1186/s40635-014-0020-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sutherland A., Thomas M., Brandon R. A., et al. Development and validation of a novel molecular biomarker diagnostic test for the early detection of sepsis. Critical care. 2011;15(3):p. R149. doi: 10.1186/cc10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hotchkiss R. S., Monneret G., Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nature Reviews Immunology. 2013;13(12):862–874. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alder M. N., Opoka A. M., Lahni P., Hildeman D. A., Wong H. R. Olfactomedin-4 is a candidate marker for a pathogenic neutrophil subset in septic shock. Critical Care Medicine. 2017;45(4):e426–e432. doi: 10.1097/CCM.0000000000002102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park K. R., Yun H. M., Yoo K., Ham Y. W., Han S. B., Hong J. T. Chitinase 3 like 1 suppresses the stability and activity of p53 to promote lung tumorigenesis. Cell communication and signaling : CCS. 2020;18(1):p. 5. doi: 10.1186/s12964-019-0503-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nedeva C., Menassa J., Puthalakath H. Sepsis: inflammation is a necessary evil. Frontiers in cell and developmental biology. 2019;7:p. 108. doi: 10.3389/fcell.2019.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Docke W. D., Randow F., Syrbe U., et al. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nature Medicine. 1997;3(6):678–681. doi: 10.1038/nm0697-678. [DOI] [PubMed] [Google Scholar]
- 15.Chiche L., Forel J. M., Thomas G., et al. Interferon-γ production by natural killer cells and cytomegalovirus in critically ill patients. Critical Care Medicine. 2012;40(12):3162–3169. doi: 10.1097/CCM.0b013e318260c90e. [DOI] [PubMed] [Google Scholar]
- 16.Giamarellos-Bourboulis E. J. Natural killer cells in sepsis: detrimental role for final outcome. Critical Care Medicine. 2014;42(6):1579–1580. doi: 10.1097/CCM.0000000000000352. [DOI] [PubMed] [Google Scholar]
- 17.Francois B., Jeannet R., Daix T., et al. Interleukin-7 restores lymphocytes in septic shock: the IRIS-7 randomized clinical trial. JCI insight. 2018;3(5) doi: 10.1172/jci.insight.98960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macdonald S. P. J., Bosio E., Neil C., et al. Resistin and NGAL are associated with inflammatory response, endothelial activation and clinical outcomes in sepsis. Inflammation research. 2017;66(7):611–619. doi: 10.1007/s00011-017-1043-5. [DOI] [PubMed] [Google Scholar]
- 19.Silswal N., Singh A. K., Aruna B., Mukhopadhyay S., Ghosh S., Ehtesham N. Z. Human resistin stimulates the pro-inflammatory cytokines TNF-alpha and IL-12 in macrophages by NF-kappaB-dependent pathway. Biochemical and Biophysical Research Communications. 2005;334(4):1092–1101. doi: 10.1016/j.bbrc.2005.06.202. [DOI] [PubMed] [Google Scholar]
- 20.Jang J. C., Li J., Gambini L., et al. Human resistin protects against endotoxic shock by blocking LPS-TLR4 interaction. Proceedings of the National Academy of Sciences of the United States of America. 2017;114(48):E10399–e10408. doi: 10.1073/pnas.1716015114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wheatley C. A scarlet pimpernel for the resolution of inflammation? The role of supra-therapeutic doses of cobalamin, in the treatment of systemic inflammatory response syndrome (SIRS), sepsis, severe sepsis, and septic or traumatic shock. Medical Hypotheses. 2006;67(1):124–142. doi: 10.1016/j.mehy.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 22.Manzanares W., Hardy G. Vitamin B12: the forgotten micronutrient for critical care. Current Opinion in Clinical Nutrition and Metabolic Care. 2010;13(6):662–668. doi: 10.1097/MCO.0b013e32833dfaec. [DOI] [PubMed] [Google Scholar]
- 23.O'Donnell L. C., Druhan L. J., Avalos B. R. Molecular characterization and expression analysis of leucine-rich alpha2-glycoprotein, a novel marker of granulocytic differentiation. Journal of Leukocyte Biology. 2002;72(3):478–485. [PubMed] [Google Scholar]
- 24.Serada S., Fujimoto M., Ogata A., et al. iTRAQ-based proteomic identification of leucine-rich α-2 glycoprotein as a novel inflammatory biomarker in autoimmune diseases. Annals of the Rheumatic Diseases. 2010;69(4):770–774. doi: 10.1136/ard.2009.118919. [DOI] [PubMed] [Google Scholar]
- 25.Rainer T. H., Leung L. Y., Chan C., et al. Circulating human leucine-rich α-2-glycoprotein 1 mRNA and protein levels to detect acute appendicitis in patients with acute abdominal pain. Clinical Biochemistry. 2017;50(9):485–490. doi: 10.1016/j.clinbiochem.2017.02.010. [DOI] [PubMed] [Google Scholar]
- 26.Young R. E., Thompson R. D., Larbi K. Y., et al. Neutrophil elastase (NE)-deficient mice demonstrate a nonredundant role for NE in neutrophil migration, generation of proinflammatory mediators, and phagocytosis in response to zymosan particles in vivo. Journal of immunology. 2004;172(7):4493–4502. doi: 10.4049/jimmunol.172.7.4493. [DOI] [PubMed] [Google Scholar]
- 27.Papayannopoulos V., Metzler K. D., Hakkim A., Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. The Journal of Cell Biology. 2010;191(3):677–691. doi: 10.1083/jcb.201006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar S., Gupta E., Kaushik S., et al. Quantification of NETs formation in neutrophil and its correlation with the severity of sepsis and organ dysfunction. Clinica chimica acta; international journal of clinical chemistry. 2019;495:606–610. doi: 10.1016/j.cca.2019.06.008. [DOI] [PubMed] [Google Scholar]
- 29.Thomas S. M., Brugge J. S. Cellular functions regulated by Src family kinases. Annual Review of Cell and Developmental Biology. 1997;13(1):513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 30.Senis Y. A., Mazharian A., Mori J. Src family kinases: at the forefront of platelet activation. Blood. 2014;124(13):2013–2024. doi: 10.1182/blood-2014-01-453134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomez-Rodriguez J., Kraus Z. J., Schwartzberg P. L. Tec family kinases Itk and Rlk / Txk in T lymphocytes: cross-regulation of cytokine production and T-cell fates. The FEBS Journal. 2011;278(12):1980–1989. doi: 10.1111/j.1742-4658.2011.08072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scumpia P. O., Delano M. J., Kelly-Scumpia K. M., et al. Treatment with GITR agonistic antibody corrects adaptive immune dysfunction in sepsis. Blood. 2007;110(10):3673–3681. doi: 10.1182/blood-2007-04-087171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang H., Xu C. F., Ren C., Wu T. T., Dong N., Yao Y. M. Novel role of p53 in septic immunosuppression: involvement in loss and dysfunction of CD4+ T lymphocytes. Cellular Physiology and Biochemistry : International Journal of Experimental Cellular Physiology, Biochemistry, and Pharmacology. 2018;51(1):452–469. doi: 10.1159/000495241. [DOI] [PubMed] [Google Scholar]
- 34.Szabo S. J., Kim S. T., Costa G. L., Zhang X., Fathman C. G., Glimcher L. H. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100(6):655–669. doi: 10.1016/S0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 35.Knox J. J., Myles A., Cancro M. P. T-bet(+) memory B cells: generation, function, and fate. Immunological Reviews. 2019;288(1):149–160. doi: 10.1111/imr.12736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levine A. G., Mendoza A., Hemmers S., et al. Stability and function of regulatory T-cells expressing the transcription factor T-bet. Nature. 2017;546(7658):421–425. doi: 10.1038/nature22360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xin A., Masson F., Liao Y., et al. A molecular threshold for effector CD8(+) T-cell differentiation controlled by transcription factors Blimp-1 and T-bet. Nature Immunology. 2016;17(4):422–432. doi: 10.1038/ni.3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagy Z., Mori J., Ivanova V. S., Mazharian A., Senis Y. A. Interplay between the tyrosine kinases Chk and Csk and phosphatase PTPRJ is critical for regulating platelets in mice. Blood. 2020;135(18):1574–1587. doi: 10.1182/blood.2019002848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data 1: primers of 10 genes.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding authors upon request.