

Abstract
In this study, we investigated the role of the transcription factor Six2 in palate development. Six2 was selected using the SysFACE tool to predict genes from the 2p21 locus, a region associated with clefting in humans by GWAS, that are likely to be involved in palatogenesis. We functionally validated the predicted role of Six2 in palatogenesis by showing that 22% of Six2 null embryos develop cleft palate. Six2 contributes to palatogenesis by promoting mesenchymal cell proliferation and regulating bone formation. The clefting phenotype in Six2−/− embryos is similar to Pax9 null embryos, so we examined the functional relationship of these two genes. Mechanistically, SIX2 binds to a PAX9 5’ upstream regulatory element and activates PAX9 expression. In addition, we identified a human SIX2 coding variant (p.Gly264Glu) in a proband with cleft palate. We show this missense mutation affects the stability of the SIX2 protein and leads to decreased PAX9 expression. The low penetrance of clefting in the Six2 null mouse combined with the mutation in one patient with cleft palate underscores the potential combinatorial interactions of other genes in clefting. Our study demonstrates that Six2 interacts with the developmental gene regulatory network in the developing palate.
INTRODUCTION
Cleft lip with or without cleft palate (CL/P) is among the most common human birth defects, affecting between 1 in 500 and 1 in 2,500 newborns each year, depending on the population examined (Conway et al., 2015; Dixon et al., 2011). Isolated, non-syndromic CLP (NSCL/P), a subset of CL/P, is classified in patients who exhibit clefting of the lip with or without palatal clefting that occurs without any other obvious developmental defects (Stuppia et al., 2011). NSCL/P makes up about 70% of clefting cases (Dixon et al., 2011). The complex etiology of the condition includes both environmental and genetic predisposing factors (Leslie et al., 2017). Developmental studies into the genetic underpinnings of clefting have revealed a number of genes that are required for palatogenesis. Many of these genes are transcription factors that are expressed in the neural crest derived mesenchymal tissue in the developing craniofacial region, for example Pax9, which is essential for palatogensesis as well as tooth morphogenesis (Peters et al., 1998; Zhou et al., 2013). More work is required to comprehensively identify all the genes which participate in the development of the palate, and to understand how they interact with each other to guide this complex process.
GWAS studies have been essential for the identification of genes that contribute to the risk of NSCL/P, and over 15 different genomic loci have been implicated to date (Birnbaum et al., 2009; Butali et al., 2018; Cox et al., 2018; Grant et al., 2009; Leslie et al., 2016; Ludwig et al., 2012; Mangold et al., 2010; Sun et al., 2015; Wolf et al., 2015; Yu et al., 2017). After genomic loci are identified, studies must be employed to identify functional variants that are causative for clefting. Several of these studies have been successfully performed to date (Leslie et al., 2015; Liu et al., 2017; Rahimov et al., 2008; Zhang et al., 2015). However, more work is required to streamline the identification and functional validation of variants found within genomic loci implicated by GWAS studies. In this work, we use a new approach, the web-based tool SysFACE (Systems tool for craniofacial expression-based gene discovery), to identify high-priority candidate genes for palate development from regions identified by GWAS, and confirmed this prediction by functional analysis in vivo and in vitro (Liu et al., 2017).
Previous reports have indicated a role for Six2 in craniofacial development, but these efforts did not identify or examine the etiology of clefting in Six2−/−embryos and mice (He et al., 2010; Liu et al., 2019; Okello et al., 2017). In the present study, we observed that 22% of Six2−/− embryos examined had oral clefts. This re-examination of the Six2−/− mouse line was prompted by the SysFACE tool, which identified Six2 as a relevant transcription factor for craniofacial development because it is highly expressed and enriched in craniofacial tissues at different developmental stages when palatogenesis is occurring. Further, we find that a role for human SIX2 in clefting is also evident from the enrichment of a variant SIX2 (p.Gly264Glu) in a cleft palate patient, compared with control populations.
There are six members in the sine oculus (Six) family named Six1-Six6 (Nonomura et al., 2010). In humans, SIX1 and SIX2 regulate one another to control nephrogenesis (O’Brien et al., 2016). Each of the six members in the Six family has a DNA-binding homeodomain (HD) and a Six domain, which contributes to the DNA binding and protein-protein interaction function. There are three homologous C-termini shared among the Six family members, allowing the division of the proteins into groups (Six1/2, Six3/6, and Six4/5) (Brodbeck et al., 2004; Kawakami et al., 1996).
The deletion of SIX2 in humans causes frontonasal dysplasia syndrome, suggesting a role for SIX2 in craniofacial development (Hufnagel et al., 2016). Six2−/− mice die shortly after birth. Six2 also has been found to play an important role in kidney development. In undifferentiated nephron progenitors, a Six2/Lef/Tcf complex prevents the expression of targets to maintain progenitor self-renewal. In differentiating nephron progenitor cells, Six2 expression is diminished and ß-catenin expression is increased, initiating Wnt target gene transcription including Fgf8 and Wnt4 (Park et al., 2012). However, the function of Six2 in palatogenesis is not well studied.
It is known that Pax9 regulates palatogenesis through Bmp, Fgf and Shh signaling; knocking out Pax9 causes decreased cell proliferation in mouse palatal shelves (Kist et al., 2005; Peters et al., 1998; Zhou et al., 2013). Recently, Pax9 has also been reported to regulate Wnt signaling in palate development by directly regulating the transcription of Dkk1 and Dkk2 (Jia et al., 2017). Both Six2 and Pax9 are expressed on the palatal mesenchyme at E13.5 (Jia et al., 2017; Okello et al., 2017). However, little is known about the regulation between Six2 and Pax9 during palatal development. In our study, we found PAX9 is a target of SIX2. We further show that SIX2 directly binds to the PAX9 upstream promoter and activates PAX9 expression.
MATERIALS AND METHODS
Gene identification using SysFACE (Systems tool for craniofacial expression-based gene discovery
SysFACE was used to prioritize candidate genes in the list from the GWAS meta-analysis according to their expression in craniofacial (CF) tissues. This tool has been previously applied to prioritize several important cleft palate candidate genes (Butali et al., 2018; Cox et al., 2018; Liu et al., 2017). While the tool itself will be described in detail elsewhere, in summary, SysFACE is based on a unique processing protocol of microarray-based genome-level gene CF expression profiles from publicly available resources such as FaceBase. Specifically, expression datasets from the following mouse CF tissues (Mandible, Palate, Frontonasal, and Maxillary) obtained from FaceBase (FB00000467.01, FB00000468.01, FB00000474.01) and NCBI Gene Expression Omnibus (GEO) (GSE35091, GSE7759, GSE11400, GSE31004) were analyzed. In this analysis, we estimate the enriched expression of individual genes in specific CF-tissues based on their fold-change upregulation in these tissues compared to their expression in mouse whole-embryo body (WB) tissue at E10.5, E11.5, and E12.5 (GSE32334) in an approach termed “WB in silico subtraction” as previously described (Anand et al., 2018; 2018; Lachke et al., 2012). The obtained SysFACE-based CF-tissue enrichment as well CF-tissue expression scores are presented in a heatmap figure, created using in-house python script. From this analysis, we identified Six2 as being highly expressed and enriched in specific CF tissues.
Mouse Lines and embryonic staging
Mice were housed and cared for by the standards and suggestions of The Program of Embryo Resources at the University of Iowa. Six2+/− mice were obtained from Jackson Laboratories (stock number 009600). This mouse line was maintained on a C57BL/6 background. In order to time pregnancies, females from breeder cages were checked for the presence of a vaginal plug in the morning of the day after the male was introduced. Upon finding a plug, the breeding pair were separated. The day of plug observation is noted as E0.5.
Tissue fixation and slide preparation
Embryos were harvested from pregnant females on the designated day and dissected in cold 1X PBS. A non-essential tissue sample was taken for genotyping purposes. The tissues were subsequently fixed in 4% PFA (ChemCruz) for 0.5–4 hr and then dehydrated through an ethanol gradient (70%–100%, each step 4 hr to overnight). Tissues were precleared in xylene and embedded in paraffin. After embedding, 7 μm sections were cut using the microtome (Thermo, HM325), floated out and attached to slides (VWR VistaVision Microscope Slides). Slides used for immunofluorescence were baked no longer than 2 hr at 65 °C; slides for other applications were baked overnight to affix the tissue section to the slide.
Immunofluorescence staining and confocal imaging
Slides were re-hydrated and subjected to antigen retrieval by citrate boiling for 20 minutes. Slides were washed 3 times with 1X PBS and blocked with 20% donkey serum(sigma, D9663A). After blocking, the primary antibody for Six2 (Proteintech, 11562–1-AP), Osx (abcam, ab22552), BrdU (Invitrogen 00–0103) was applied and incubated overnight at 4°C. Slides were washed 3 times with 1X PBS and in a dark box, incubated with secondary antibody (Life Technologies) for 30 minutes at room temperature. The remainder of the procedure was carried out in the dark. Slides were washed 3 times in 1X PBS and incubated with DAPI solution (Thermo Scientific). Slides were washed 3 times in 1X PBS and a hydroseal buffer was used to mount a cover slip. Slides were dried for 1–2 hr at room temperature and subjected to confocal imaging. Images were captured using the ZEISS 700 confocal microscope and Zen imaging software. The different channels were combined using ImageJ software prior to publication.
BrdU assay
Pregnant females were injected 2 hr prior to sacrifice to label proliferative cells with BrdU (10ul/g body weight, Invitrogen 00–0103) and detected using a rat monoclonal anti-BrdU antibody (Abcam, ab6326, 1:250). Medial tip regions of the extending palatal shelves were analyzed by counting the number of BrdU+ cells in a given area, represented by a white dotted circle.
Hematoxylin and Eosin staining
Slides were de-parrafinized and rehydrated using a reverse ethanol gradient. After rehydration, slides were incubated in Hematoxylin for 4 minutes. Slides were de-stained using 9 dips in 95%EtOH, 1M HCl and bluing was carried out using 0.1% Sodium Bicarbonate for 30 seconds. Slides were washed in water and dehydrated in an ethanol gradient to 95% EtOH and stained in Eosin for 1 minute. Images were taken using an inverted Nikon microscope.
Whole skeletal staining
Skin and internal organs were removed from P0 mice. The mice were scalded in 65°C H2O to fully remove the skin. The carcass was then fixed overnight in 95% EtOH. Alcian Blue staining proceeded overnight (800 ml 95% EtOH, 200 ml acetic acid, Alcian Blue 8GX sigma A3175 150mg. Carcasses were washed in 95% EtOH for 5 hr and cleared using 2% KOH. A final dissection step was then performed to remove the remaining fat and skin. Alizarin red staining (1% KOH, 0.015% Alizarin red Sigma A3757) staining was performed overnight. Sections were cleared for an additional 3 days in 1%KOH, 20% glycerol for 3 days, and stored in 1:1 glycerol and 95% EtOH. Images were taken using a dissecting microscope.
Alizarin Red Staining of Tissue Sections
Slides were re-hydrated to water and stained with an alizarin red solution (Sigma, 2g, in 100ml H2O PH 4.2) for 5 minutes. Slides were dehydrated in acetone and then in acetone-xylene, and mounted in xylene.
RT-qPCR
Total RNA was extracted using the RNeasy Mini Kit from Qiagen. Total RNA was reverse transcribed into cDNA by iScript Select cDNA Synthesis kit (BioRad). cDNAs were used in qPCR assays and loading was adjusted by measuring the housekeeping β-actin gene. The Real-time PCR was performed with different probes (Table 1).
Table 1.
Primers used in the manuscript.
| ChIP Primers | ||||
| Negtive control | F | 5’ | CCTGGTTTGTTCCCACTATAGTG | 3’ |
| R | 5’ | GCGCGCTCTCAGAGCATC | 3’ | |
| SIX2 BS | F | 5’ | GCGACTCCGACGCTAACCC | 3’ |
| R | 5’ | AGCTGCGTTTCCAGCCTG | 3’ | |
| qRT-PCR Primers | ||||
| SIX2 | F | 5’ | AGGCCAAGGAAAGGGAGAAC | 3’ |
| R | 5’ | GAGCTGCCTAACACCGACTT | 3’ | |
| β-actin | F | 5’ | CTCTTCCAGCCTTCCTTC | 3’ |
| R | 5’ | ATCTCCTTCTGCATCCTGTC | 3’ | |
ChIP
The ChIP protocol was modified from our previous publication (Sun et al., 2016) to detect interactions of the Six2 protein with chromatin. The primers used for ChIP assay are listed in Table 1.
DNA Extraction and Primer Design
Saliva samples were collected using the Oragene DNA collection kits (http://www.dnagenotek.com) from participants in Ghana, Ethiopia and Nigeria after ethical approval at all the recruitment sites. We used the optimized Oragene saliva processing protocol available in the Butali laboratory to extract DNA from both saliva and cheek swab samples. Then, we determined the concentration of double stranded DNA in each DNA sample using Quibit Assay and Quibit 2.0 Fluorometer (http://www.invitrogen.com/site/us/en/home/brands/Product-Brand/Qubit.html). Quality control for all samples was also done by checking the gender of each sample by conducting XY-genotyping using Taqman Assay kits and real time PCR.
We designed two primer sets to cover the two coding exons and the untranslated (UTRs) 5’ and 3’ of SIX2 gene. Primers were designed with Primer 3 software (http://biotools.umassmed.edu/bioapps/primer3_www.cgi) and UCSC genome browser (https://genome.ucsc.edu/) to ascertain whether these primer sets annealed to the targeted genomic region. Primer sets were designed based on SIX2 Ref Seq number NM_016932 of genome assembly number GRCh37/hg19, 2009 (http://genome.ucsc.edu). Gradient PCR was performed to determine the annealing temperatures of each of the primer sets. Before DNA sequencing, we amplified the coding exon of SIX2 using initial PCR. A 9 μl of master mix (comprising 10X NH4 buffer, 5% DMSO, 200 μM DNTPs, 50 μM MgCl, water, 20 μM of forward and reverse primers as well as 5 U/μl Taq polymerase) was added to 1 μl of each DNA sample at a concentration of 4 ng/μl. In addition to the case samples on each 96-well PCR plate, 2 HAPMAP Yoruba and two water samples were added to each plate to serve as template and non-template controls respectively. A total of 270 samples were sequenced. The PCR conditions and primer sequences are available from the Butali laboratory upon request.
The success of the initial PCR was confirmed by running part of the PCR product on 2% agarose gel and size markers at 100A and 220V for 20 minutes. All gel electrophoretic products were viewed using ultraviolet light. Successful initial PCR products were shipped to Functional Biosciences, Madison, Wisconsin (http://order.functionalbio.com/seq/index) for sequencing using the ABI 3730XL DNA sequencer.
Bioinformatics analyses of DNA sequence results
Chromatograms generated from the DNA sequence data was transferred to a Unix workstation and the bases were called using PHRED (v. 0.961028) (www.phrap.org/phredphrapconsed.html). All sequences were assembled to form the contigs using PHRAP (v. 0.960731), scanned with POLYPHRED (v. 0.970312), and viewed with CONSED (v. 4). We aligned variants to human genome assembly number GRCh37/hg19, 2009 (http://genome.ucsc.edu) to ascertain the genomic location or co-ordinates of variants. Novel variants are those not reported in 1000 Genomes (http://www.1000genomes.org/), Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org/), gnomad and Exome Variant Server (EVS) (http://snp.gs.washington.edu/EVS/) databases as well as literature on orofacial clefts. We carried out segregation analyses by sequencing DNA from relatives of individuals with the novel or rare variants (MAF<1%) to ascertain whether these variants were limited to cases or if they were inherited from a parent. Functional effects of identified variants on SIX2 was predicted using in-silico bioinformatics tools such as polyphen 2 (http://genetics.bwh.harvard.edu/pph2/) (Adzhubei et al., 2010), SIFT (http://sift.jcvi.org/) (Kumar et al., 2009) and HOPE (http://www.cmbi.ru.nl/hope) (Venselaar et al., 2010).
Statistical Analysis for Experiments
For each condition, three experiments were performed and the results are presented as the mean ± SEM. The differences between two groups of conditions were analyzed using an independent, two-tailed t-test.
RESULTS
Six2 is identified as a high-priority candidate in craniofacial development by SysFACE
We mined the GWAS significant loci reported in the first meta-analysis for cleft lip with or without cleft palate (Ludwig et al., 2012). This meta-analysis data was from two previous GWAS (Beaty et al., 2010; Mangold et al., 2010). The bioinformatics resource Systems tool for craniofacial expression-based gene discovery (SysFACE) was used to prioritize potential cleft-linked candidate genes. We used SysFACE to analyze gene expression in craniofacial tissues- maxilla, mandible, frontonasal and palate (Fig. 1). This analysis showed that Six2 exhibits high expression in craniofacial tissues- maxilla, frontonasal and palate. Furthermore, Six2 showed enriched expression (fold change difference of individual craniofacial tissues compared to the whole embryo body reference dataset – please see methods for details) at various embryonic developmental stages, which suggests a potential function in craniofacial morphogenesis (Fig. 1).
Fig. 1. SysFACE identifies Six2 as a high-priority candidate in craniofacial development.

Mouse craniofacial expression data was analyzed by SysFACE (Systems tool for craniofacial expression-based gene discovery). SysFACE analysis reveals that Six2 has significant expression (absolute expression in fluorescent intensity units >100, p<0.05) and significant enrichment (craniofacial tissue compared to whole-embryo body tissue reference dataset >1.5-fold change, p<0.05) in several mouse craniofacial tissues at various embryonic stages. Other gene candidates are shown from the 2p21 analyses.
Six2 is highly expressed during embryonic stages of palate development and Six2 null mice have cleft palate
To test the SysFACE prediction, we next carefully examined Six2 expression during palatal development using immunofluorescence to detect Six2 expressing cells at E12.5, E13.5, E14.5, E15.5 and E16.5 embryonic stages. We found that Six2 was highly expressed in the developing palatal shelves at different developmental stages (Fig. 2A–E). Six2 was highly expressed at the E12.5 and E13.5 stages of palate shelf (PS) development, and expressed at lower levels in E16.5 palatal mesenchyme. We stained for Six2 expression in Six2−/− embryos (Fig. 2A’–E’) and did not detect Six2 expression in the Si2−/− embryos confirming successful knockout of Six2.
Fig. 2. Six2 is expressed in the palatal mesenchyme and undetectable in Six2−/− embryos.
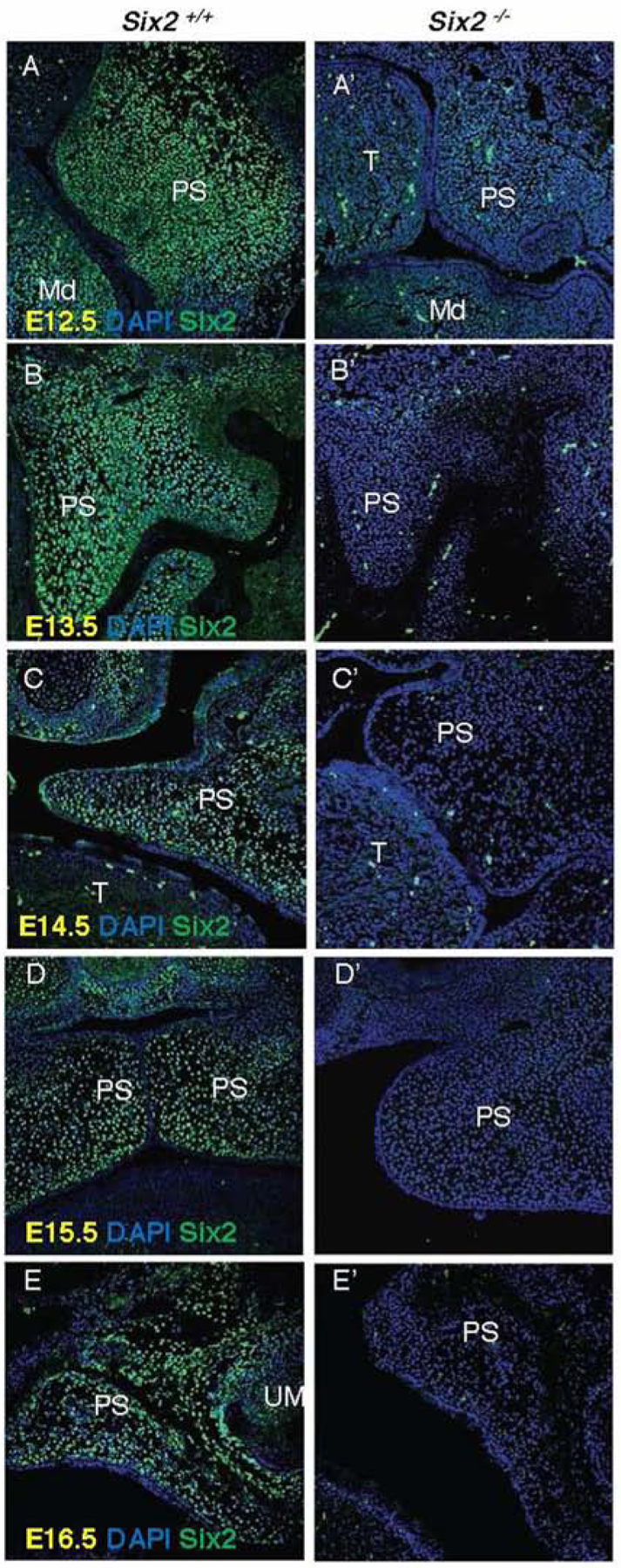
A-E. Six2 is expressed in the palatal mesenchyme from E12.5 to E16.5 developmental stages. A’-E’. Six2 is not detectable in Six2−/− embryos at any developmental stage. PS, palate shelf; Md, mandible; T, tongue; UM, upper molar.
Six2 is associated with nephrogenesis and craniofacial development and is expressed during palatogenesis, but Six2−/− mice had not been shown to develop oral clefts (He et al., 2010; Okello et al., 2017; Self et al., 2006). The Six2−/− mice die shortly after birth at P0. These mice exhibit several craniofacial defects, including a domed skull and a short snout (Fig. 3A, A’), consistent with previous reports (He et al., 2010). Gross examination of a large number of Six2−/− embryos was performed during palatal development (n=183 from E15.5-E18.5), and 22% of Six2−/− embryos were found to exhibit cleft palate (Fig. 3B).
Fig. 3. Six2−/− neonatal mice have craniofacial phenotypes.

A-A’. Six2+/+and Six2−/− P0 mice were collected after birth. Six2 −/− mice die and exhibit a domed skull and a short snout. B. Examining the palate of P0 mice revealed a cleft in the secondary palate of Six2−/− embryos.
Palate development starts from E11.5 to E17, and includes outgrowth, elevation, extension and fusion (Bush and Jiang, 2012). In order to determine which processes of palate development were defective in Six2−/− mice, we examined palate development at different embryonic stages (Fig. 4). Embryos were sectioned (coronal) and H&E staining used to examine palate formation. In wildtype (WT) embryos, the palate develops through a series of stages that begins with the outgrowth of the palatal shelves (E13.5), which occurred normally in WT and Six2−/− embryos. The clefting phenotype of the Six2−/− embryos becomes evident at E14.5 when palate shelf elevation is delayed (Fig 4D–F, D’–F’) compared with WT embryos. At E15.5, the palate shelves have extended in WT embryos and made contact with each other forming the mid-epithelial seam (Fig. 4G–I). In Six2−/− embryos at E15.5, the process of palate shelf elevation has not been completed (Fig. 4G’–I’). In WT embryos, palate shelves are fused with each other at E16.5 and the mid-epithelial seam is lost; the mesenchymal tissues of the left and right palate shelves are merged (Fig. 4J–O). In the Six2−/− embryos, the palate shelves fail to extend at E16.5, creating a cleft at this stage and later stages (Fig. 4J’–O’).
Fig. 4. Six2−/− embryos develop cleft palate.

A-C. WT embryo coronal sections at E13.5. A’-C’. Six2−/− coronal sections at E13.5. D-F. WT craniofacial morphology at E14.5. D’-F’. Six2−/− palate shelves fail to elevate at E14.5. G-I. WT craniofacial morphology at E15.5. G’-I’. Six2−/− palate shelves fail to extend to the midline. J-L. WT palate formation is complete at E16.5. J’-L’. Six2−/− embryos have cleft palate at E16.5. M-O. WT palates are well formed in P0 mice. M’-O’. Six2−/− mice have cleft palate at P0. Scale Bars, 100 μm. 1, mouse embryos’ anterior region; 2, mouse embryos’ medial region; 3, mouse embryos’ posterior region.
Six2 promotes mesenchymal cell proliferation in the early stages of palate development
We sought to determine the mechanism resulting in the failure of palate shelf extension. We reasoned that the shorter length of the palate shelves might be a direct result of a defect in cell proliferation. To test the hypothesis, BrdU was injected into pregnant mice 2 hours before sacrificing to harvest E14.5 embryos. Subsequently, BrdU immunofluorescence staining was performed (Fig. 5). Interestingly a decrease in BrdU positive cells in the medial tip portion of Six2−/− palate shelves (approximately 17% reduction, P<0.01, n=3), compared with WT, was identified through quantifying the proliferating BrdU positive cells (Fig. 5C). E14.5 Six2−/− embryos that did not exhibit cleft palate were also compared with WT using Ki67 staining to quantify cell proliferation, and no notable difference was found (Sup. Fig. 1A–B).
Fig. 5. Six2−/− embryos have decreased mesenchymal cell proliferation in palate shelves at E14.5.
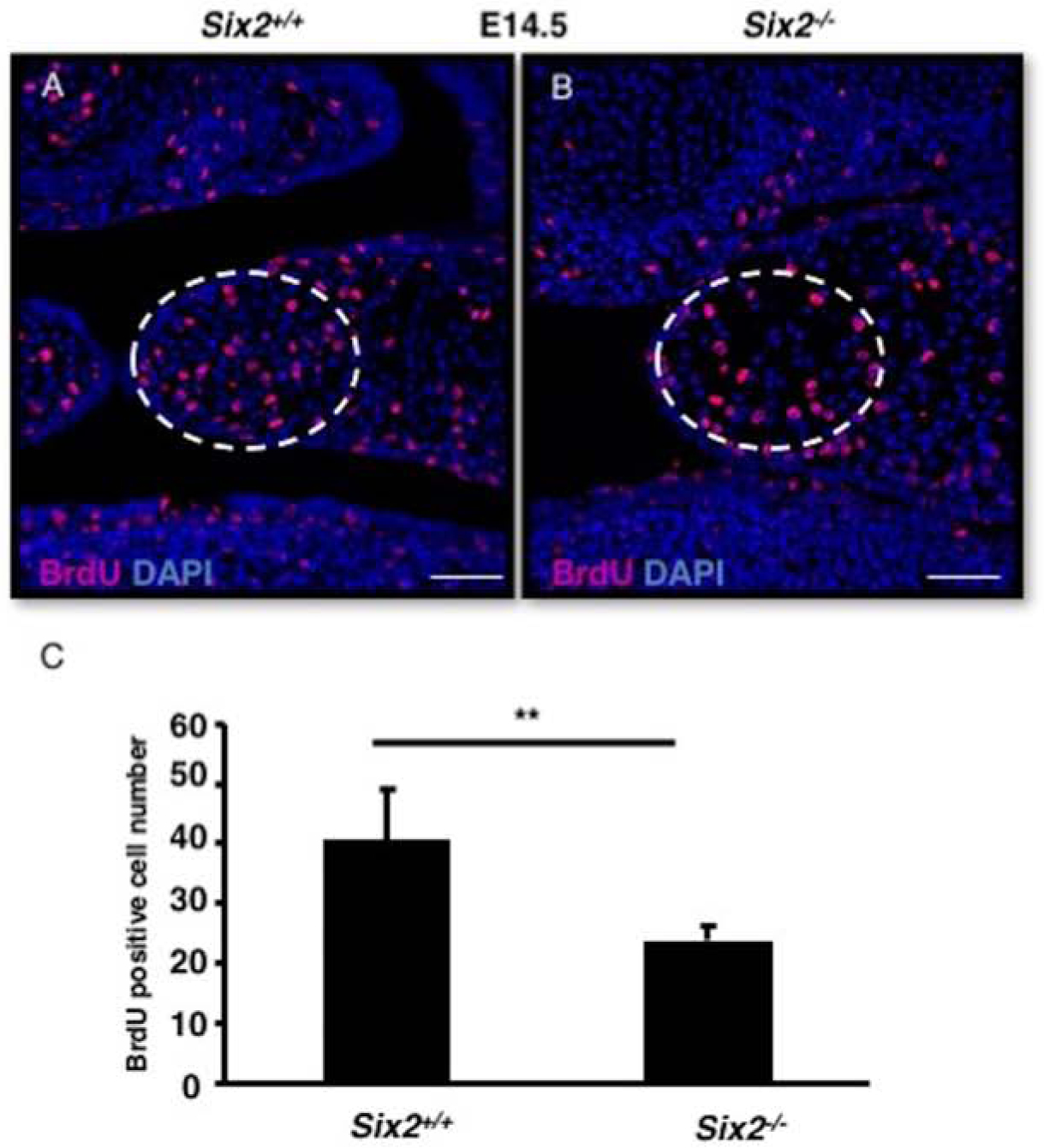
A. In order to label dividing cells, BrdU was injected into an E14.5 stage pregnant female and embryos were harvested 2 hours later. Immunostaining was used to label BrdU+ cells on the palate shelves of WT and Six2−/− embryos. C. The number of proliferative cells found in E14.5 WT and Six2−/− palate mesenchyme were counted, and a statistically significant decrease in proliferation was observed in the Six2−/−. Scale Bars, 50 μm. Error bars show SEM. **: p-values<0.01.
Six2−/−embryos and neonate mice have defects associated with palate bone development and Six2 is a negative regulator of ossification
The mouse secondary palate is composed of the palatine bone as well as the maxillary processes (Bush and Jiang, 2012), so we wanted to determine if the development of these ossified structures was defective in Six2−/− embryos. In order to better understand how bone development was affected in Six2−/− embryos, we used alizarin red staining to label mineralized tissue in coronal sections at E16.5 (Fig. 6). Interestingly, we saw a premature accumulation of mineralized tissue in the palate shelves of E16.5 Six2−/− embryos compared to WT (Fig. 6A–B, A’–B’). Further investigation revealed higher expression of the osteogenic differentiation markers Osx and Runx2 in palate shelves isolated from E15.5 Six2−/− embryos with CP compared to WT, (Sup. Fig. 2A–B) and there is an expansion of Osx protein on the palatal shelves in Six2−/− embryos (Sup. Fig. 2C–D).
Fig. 6. An increase in ossification accompanies defective palatine bone formation in Six2−/− embryos and mice.
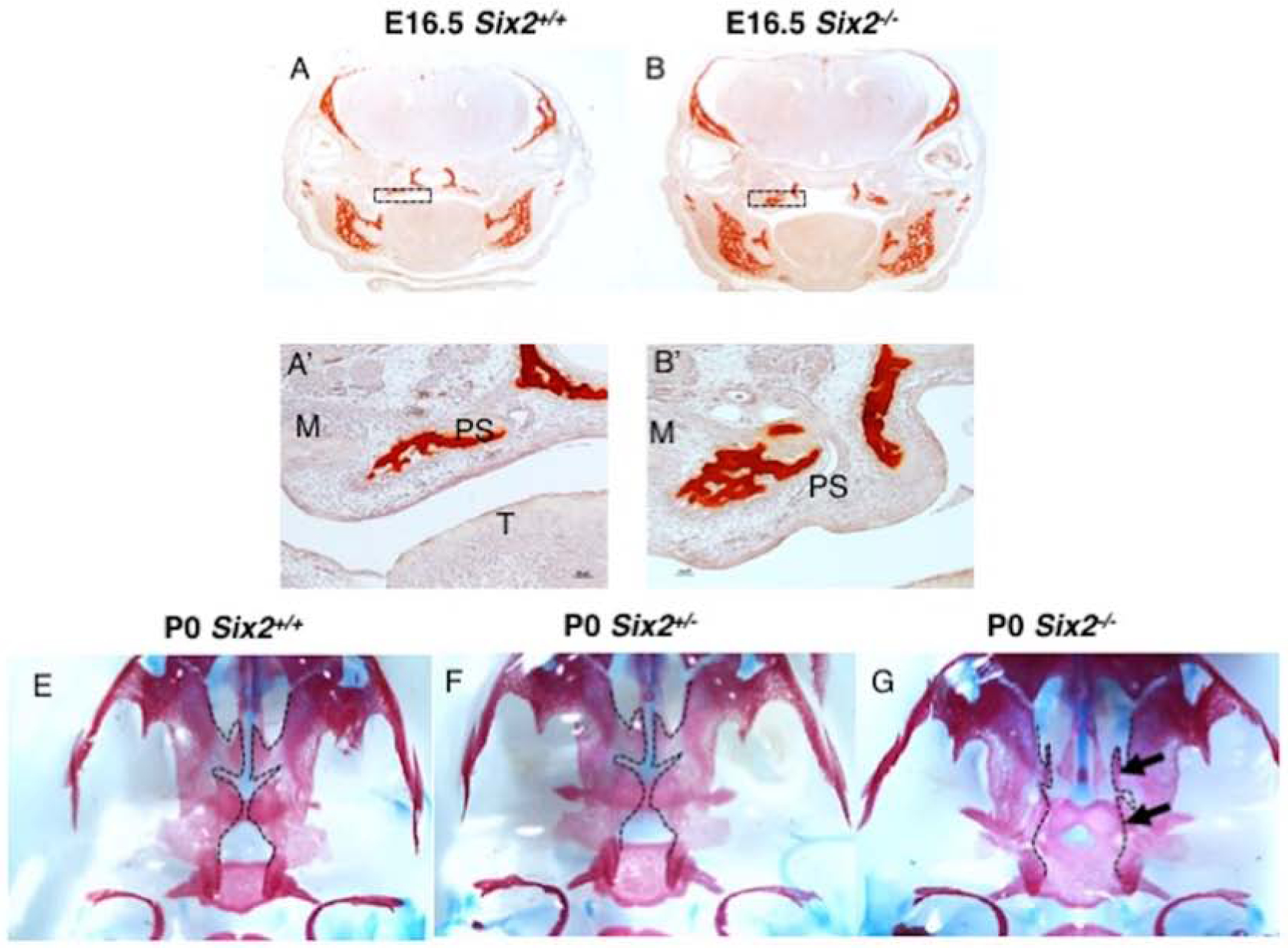
A-B. An increase in alizarin red staining demonstrates an increase in ossification in E16.5 Six2−/− embryos compared to the WT. A’-B’. A magnification of the boxed regions in A and B. E-G. The inferior view of the oral cavity. Arrows point to the malformation of the maxillary process and abnormal palatine bone in Six2−/− mice. PS, palate shelf; M, maxilla; T, tongue.
We next examined P0 skulls using alizarin red and alcian blue staining. We found that the palate formed normally in WT embryos. However, the Six2−/− palatine process of the maxilla and palatine bone failed to extend to the midline, forming a mineralized structure on each non-extended palate (Fig. 6E–G). Taken together, these results demonstrate Six2 is involved with regulating the ossification of the palatal mesenchyme and deleting Six2 results in premature ossification of the maxillary process and palatine bone.
Interestingly, the increase in ossification we observed in Six2−/− embryos was specific for those with cleft palate. We examined WT and Six2−/− E17.5 embryos and found no difference in the amount of ossification of the palate shelves (Sup. Fig. 3).
SIX2 binds the PAX9 promoter and directly regulates PAX9 expression
Pax9−/− mice have similar clefting phenotypes with Six2−/− mice, and these genes are co-expressed in the palatal mesenchyme at E13.5 (Zhou et al., 2013), suggesting these genes are involved in a common pathway. These genes are co-expressed in the developing palatal mesenchyme, and the ablation of Six2 results in decreased Pax9 expression (Sup. Fig. 4), suggesting that Six2 may directly regulate Pax9 expression. The SIX2 binding motif has been previously identified, so we examined the PAX9 promoter for the potential interaction with SIX2 (Park et al., 2012) and discovered a potential SIX2 binding site closely resembling the consensus binding sequence (Fig. 7A).
Fig. 7. SIX2 binds the PAX9 promoter to directly promote PAX9 expression.

A. A schematic of the PAX9 promoter highlighting a consensus SIX2 binding site and a negative control (NC) region lacking a consensus SIX2 binding site. B. To test the identified binding site, ChIP was performed using either an antibody specific for SIX2 or a non-specific IgG in HEK293t cells. Primers flanking the SIX2 binding site were used to successfully amplify a product from input chromatin used in ChIP reactions as well as from chromatin isolated from SIX2 ChIP, but failed to amplify a product from the IgG reaction. C. Primers flanking an NC region of the PAX9 promoter not containing a SIX2 binding site amplified products from input samples but not from ChIP reactions. D. SIX2 and PAX9 expression were assayed by Western blot in HEPM cells overexpressing empty vector, SIX2, or SIX2 (p.Gly264Glu).
In order to test the potential SIX2 binding site for functionality, a ChIP assay using IgG or a SIX2 antibody was performed in HEK293t cells, which endogenously express SIX2. Primers flanking the SIX2 binding site on the PAX9 promoter were used to amplify chromatin resulting from inputs and from each ChIP reaction. Another set of primers which do not flank a potential SIX2 binding site were designed as a negative control. PCR was performed using the two sets of primers to determine the enrichment of chromatin in the ChIP sample (Fig. 7B–C). The SIX2 binding site ChIP PCR product was amplified and indicated by an asterisk (Fig. 7B). The negative control primers did not amplify a detectable product (Fig. 7C). Therefore, SIX2 is enriched in the PAX9 promoter region containing the SIX2 binding site, but not in the region lacking the binding site.
To verify the regulation of PAX9 by SIX2, Western blot was used to examine the protein level of SIX2 and PAX9. Not surprisingly, when SIX2 was overexpressed, endogenous PAX9 expression was increased as well (Fig. 7D).
Identification of the p.Gly264Glu SIX2 variant in a family with isolated cleft palate
We sequenced SIX2 exons in 270 cases with non-syndromic CL/P from Africa (Ghana, Ethiopia and Nigeria). We identified a rare variant in patients that has a MAF of 0.00013 (16 / 121,358 alleles) in the ExAc database of more than 100,000 whole exomes (accessed on 10/30/2018) and gnomAD databases (37/281,676 i.e. MAF 0.00013) that was accessed on 01/14/2019. This variant was only found in African and Hispanic individuals in both databases. It is absent in 1000 genomes and EVS (Fig. 8A). The variant segregates in the family since the unaffected mother has the variant suggesting incomplete penetrance of the phenotype (Fig. 8B and 8C).
Fig. 8. Family pedigree and individual with a human SIX2 variant.

The figure shows the pedigree of the family and segregation of the p.Gly264Glu SIX2 variant inherited by the affected child from an unaffected mother. A. Shows the chromatograms with nucleotide changes in the affected child, as well as the mother and father. The mother and child both carry the risk allele. B-C. Shows the pedigree of the family and segregation of the p.Gly264Glu SIX2 variant inherited by the affected child from an unaffected mother. HGVS= Human genome variation society, g= genomic reference sequence, p= protein level, 1kg= 1000 genomes, EVS= Exome variant server, ExAC= Exome aggregate consortium, P= Polyphen (B-benign) and S= Sift (T-tolerated).
The SIX2 (p.Gly264Glu) variant protein is labile and unstable
The rare SIX2 variant was identified in CL/P patients and contains a glycine to glutamate substitution in the SIX2 C-terminal domain, which has been reported to be important for transcriptional regulation (Brodbeck et al., 2004). To examine if the SIX2 (p.Gly264Glu) variant affects the function of SIX2, we expressed both proteins to determine their stability in cells. The RT-qPCR results show that both SIX2 and the variant are overexpressed with similar transcript levels (Fig. 9A). We next checked the WT SIX2 and SIX2 variant protein levels by Western blotting. Both SIX2 and the SIX2 (p.Gly264Glu) are overexpressed, however the level of SIX2 (p.Gly264Glu) protein detected was much lower than the SIX2 WT protein level (Fig. 9B). Taken together, the results of these two assays suggests that the SIX2 (p.Gly264Glu) might affect protein stability (Fig. 9B). To confirm these findings, cells transfected with either the wildtype or variant SIX2 expression plasmids were treated with actinomycin D to inhibit transcription, and the levels of SIX2 transcripts were identified on the initial day and the following three days (Fig. 9C). Similar levels of the two transcripts were detected over the four different time points (D= days after actinomycin D treatment) and both transcript levels as well as total mRNA levels in the cells were decreased on D2 and D3 compared with D0 and D1 as expected. SIX2 protein levels were also measured at the four time points (Fig. 9D). After SIX2 transcription was inhibited, degradation of the wildtype and variant peptides was observed at D3, however much less SIX2 variant protein remained on D3, confirming the SIX2 (p.Gly264Glu) is indeed more labile (see asterisk Fig. 9D).
Fig. 9. The SIX2 (p.Gly264Glu) protein is unstable and degraded compared to WT.

A. Empty vector, SIX2 or SIX2 (p.Gly264Glu) expression plasmids were transfected into 293T cells and after 48 hr; cells were harvested for RT-qPCR. SIX2 expression (normalized to β-actin) is reported. B. A Western blot experiment to measure SIX2 protein levels from the same conditions in A. C. HEK293t cells were transfected with either SIX2 or SIX2(G264E) and treated with actinomycin D 24 hr later (D0). RT-PCR from RNA isolated on day 0 and the following days were performed to detect SIX2 and β-actin transcripts. D. Western blotting of SIX2 and the GAPDH proteins from the same lysates examined in C.
Furthermore, the transfection of the SIX2 (p.Gly264Glu) variant resulted in less PAX9 endogenous expression compared to WT SIX2 (Fig. 7D). Thus, our results show that SIX2 can directly regulate PAX9 expression, and this activation is compromised by the p.Gly264Glu variant.
DISCUSSION
NSCL/P is one of the most common birth defects. Patients who are afflicted by these severe craniofacial defects will require multiple surgeries during the course of their development. In addition to physical adjustments, NSCL/P patients also require speech therapy and potentially other types of treatment in order to deal with the psychosocial burdens of their condition. In all, the total cost of all types of care these patients require may total well above 200,000 dollars. There are both genetic and environmental underpinnings for NSCL/P, and the identification of these factors using different technologies including GWAS, SysFACE and mouse models, is currently a hot research topic. A small number of genes have been identified so far, suggesting there are many more genes waiting to be discovered.
Mouse models are the most commonly selected animal model used to study the genetic underpinnings of NSCL/P, due to the conservation of the biological processes underlying palatogenesis in mice and humans. Previous studies have identified different requirements for the outgrowth, elevation, extension and fusion of the palate shelves; the temporospatial regulation of each of these processes is required for complete palate development. The palatine process of the maxilla and palatine bone form the anterior and posterior secondary palate, respectively. Palate shelves meet at the midline and form the medial epithelium seam (MES) at E15.5. The anterior two-thirds of the palate, which forms the hard palate, undergoes an intramembranous ossification process after E15.5. Proper timing of this process is essential for palate development; both premature and delayed bone formation on palate shelves could cause cleft palate (Baek et al., 2011; Mori-Akiyama et al., 2003). For example, the expression of Runx2 and Osx transcription factors are increased in Sox9−/− embryos leading to premature ossification and clefting. Lack of osteogenesis also can cause cleft palate. In Bmpr1a mutant mice, there is decreased osteogenesis of the palatine process and a loss of mesenchymal condensation, causing cleft palate. Besides cell differentiation, correct cell proliferation is also important to maintain palatogenesis successfully. Sonic hedgehog (Shh) signaling is important for epithelial-mesenchymal interaction and controlling epithelial and mesenchymal cell proliferation during palatogenesis. Errors in the Shh pathway can also cause cleft palate.
A recent study showed how Six1 and Six2 regulate frontonasal development partly through the expression of Alx genes (Liu et al., 2019). Six1 and Six2 are expressed in the neural crest-derived frontonasal mesenchyme and a double knockout of Six1/Six2 resembles defects in Alx family knockout mice (Beverdam et al., 2001; Qu et al., 1999). Furthermore, it was reported that Six2 complements Six1 function in maxillary and mandibular development through regulating BMP signaling (Liu et al., 2019). It is clear from these studies that both Six1 and Six2 are required for frontonasal development and have some redundant functions during craniofacial development.
Six2 regulates cell proliferation and a lack of Six2 causes premature ossification of the palate shelves
Cell differentiation and cell proliferation are important to maintain palatogenesis. Cell proliferation of the palatal mesenchyme drives the outgrowth of the palatal shelves that will form the roof of the oral cavity. BrdU staining at E14.5 identified a decrease in cell proliferation of the palatal shelves in Six2−/− embryos. At E16.5, the non-extended palatal shelves of Six2−/− embryos were more ossified than their WT littermates. In Six2−/− embryos, we found delays in the elevation of the palatal shelves. However, Six2−/− palate shelves were able to elevate, but much later during embryonic development. Even after Six2−/− palatal shelf elevation had completed by E16.5, palate shelf extension failed to occur. Together, the decrease in the proliferation of the palatal mesenchyme leading to a lack of palate shelf extension, combined with premature ossification, were likely responsible for the clefting observed in Six2−/− embryos and mice.
A SIX2 human variant is associated with clefting
A human variant of SIX2 was identified and associated with one family whose proband presented clinically with cleft palate. While this variant was predicted by polyphen to be benign and tolerated by sorting intolerant from tolerant (SIFT) we show that this variant caused the mutant protein to be relatively unstable compared to the WT SIX2 protein. We also speculate that this variant may affect protein interactions which decrease the transcriptional activity of the SIX2 variant. Clearly protein interactions regulate transcriptional activation and palate development as we have shown for TBX1 and PITX2 (Gao et al., 2015). While there are several human syndromic gene variants associated with clefting, this newly identified non-syndromic SIX2 variant provides new mechanisms for the role of SIX2 and transcription factors in cleft palate morphogenesis. We note that the mother with the variant does not have overt clefts. This is not surprising, and it is very possible that the mother has a subclinical form of cleft only observable using advanced imaging techniques (Neiswanger et al., 2007). Another consideration is that the cleft phenotype observed in the child could be the outcome of genetic risk shared with the mother combined with environmental exposures during the periconceptional period (Little et al., 2004). Interestingly, the low penetrance of clefting observed in humans correlates with the low penetrance we observed in the Six2 null embryos. The low penetrance of the clefting phenotype in the Six2 null mice could also be due to the expression of Six1 as they have overlapping expression domains and function (Liu et al., 2019).
Six2 regulates Pax9 expression
Pax9 has been identified as an essential master regulator of palate development in humans, and also in mice (Ichikawa et al., 2006; Jia et al., 2017). When Pax9 is ablated in the developing mouse embryonic palate shelves, defects in palate shelf elevation and elongation are observed, resulting in the loss of palatal fusion (Zhou et al., 2013). Interestingly, these are the same defects we characterized in Six2−/− embryos. Pax9 is expressed in the developing palate epithelium and mesenchyme, and is therefore partially colocalized with Six2 (Zhou et al., 2013). Pax9 has been demonstrated to interact with many different mesenchymal transcription factors and signaling pathways required for palate development such as Wnt, β-catenin, Msx1, Osr2, Bmp4, Fgf10, and Fgf8 to regulate cell differentiation and cell proliferation during palatogenesis. (Hilliard et al., 2005; Ichikawa et al., 2006; Jia et al., 2017; Zhou et al., 2013). Our work contributes to this understanding by uncovering a Six2-Pax9 axis that regulates the proliferation and differentiation of the palatal mesenchyme during the process. Six2 directly regulates PAX9 expression. Disrupting this axis results in cleft palate due to the failure of palate shelf extension, an outcome resultant of the loss of cell proliferation and premature ossification.
Supplementary Material
Supplemental Fig. 1. Six2−/− embryos lacking cleft palate have normal levels of proliferating mesenchymal cells. A-A’. Coronal sectioning and Ki67 IF staining of WT E14.5 embryos. A’ is an enlargement of the boxed region of A. B-B’. Ki67 of null embryos that do not exhibit cleft reveal similar levels of mesenchymal cell proliferation.
Supplemental Fig. 2. The expression of Runx2 and Osx is increased in Six2−/− embryos. A-B. Runx2 and OSX mRNAs were quantified using RT-qPCR from RNA isolated from palate shelves in Six2+/+ and Six2−/− embryos at E15.5. The levels of these transcripts were increased in Six2−/− embryos compared with control littermates. C-C’. Staining for Osx in WT embryos at E16.5 in the palate shelves. C’ is an enlargement of the boxed region shown in C. D-D’. Osx staining in E16.5 Six2−/− embryos, where a greater area of cells are expressing the differentiation marker.
Supplemental Fig. 3. Six2−/− embryos without cleft palate have regular palatine bone formation. A-A’. Sectioning and alizarin red staining of E17.5 WT embryos labels the developing palatine bone. A’ is an enlargement of the boxed region in A. B-B’. Sectioning and alizarin red staining of E17.5 Six2−/− embryos that do not have a cleft palate revealed similar amounts of ossification of the palatine bone.
Supplemental Fig. 4. Six2 promotes Pax9 expression in the medial edge of the developing palate. A. Staining of Six2 and Pax9 in E14.5 embryos demonstrates that these proteins are co-localized at the tip region of the palate shelf. B. In Six2−/− embryos, Six2 protein is not detectable and Pax9 protein expression is strongly diminished. C. Using RT-qPCR to quantitate the relative amount of Pax9 mRNA demonstrates a decrease in null embryos compared with WT control littermates.
Highlights.
SysFACE (Systems tool for craniofacial expression-based gene discovery) was used to identify Six2 as a possible gene for clefting.
Six2 null mice have cleft palate caused by decreased cell proliferation and premature ossification of the palate shelves.
Mechanistically, SIX2 regulates PAX9 to control palate development.
Incomplete penetrance of clefting in the Six2 null mice and associated SIX2 mutation in a family with cleft palate demonstrate that SIX2 is part of a gene regulatory network controlling palatogenesis.
ACKNOWLEDGMENTS
We thank members of the Amendt laboratory for helpful discussions, Christine Blaumueller for editorial expertise and the Carver Trust for support of the microCT scanner. We thank Dr. Rulang Jiang for sharing unpublished data and critical comments. This research project was support by funds from the University of Iowa Carver College of Medicine and College of Dentistry and NIDCR grants R03DE024776 to S.L., I.S.; R01DE026172 to I.S.; DE026433 to B.A.A and DE023520 to B.A.A.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors have no conflicts of interest to report.
REFERENCES
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR, 2010. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand D, Kakrana A, Siddam AD, Huang H, Saadi I, Lachke SA, 2018. RNA sequencing-based transcriptomic profiles of embryonic lens development for cataract gene discovery. Human Genetics 137, 941–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek J-A, Lan Y, Liu H, Maltby KM, Mishina Y, Jiang R, 2011. Bmpr1a signaling plays critical roles in palatal shelf growth and palatal bone formation. Dev. Biol 350, 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaty TH, Murray JC, Marazita ML, Munger RG, Ruczinski I, Hetmanski JB, Liang KY, Wu T, Murray T, Fallin MD, Redett RA, Raymond G, Schwender H, Jin S-C, Cooper ME, Dunnwald M, Mansilla MA, Leslie E, Bullard S, Lidral AC, Moreno LM, Menezes R, Vieira AR, Petrin A, Wilcox AJ, 2010. A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat. Genet 42, 525–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beverdam A, Brouwer A, Reijnen M, Korving J, Meijlink F, 2001. Severe nasal clefting and abnormal embryonic apoptosis in Alx3/Alx4 double mutant mice. Development 128, 3975–3986. [DOI] [PubMed] [Google Scholar]
- Birnbaum S, Ludwig KU, Reutter H, Herms S, Steffens M, Rubini M, Baluardo C, Ferrian M, Almeida de Assis N, Alblas MA, Barth S, Freudenberg J, Lauster C, Schmidt G, Scheer M, Braumann B, Berge SJ, Reich RH, Schiefke F, Hemprich A, Potzsch S, Steegers-Theunissen RP, Potzsch B, Moebus S, Horsthemke B, Kramer FJ, Wienker TF, Mossey PA, Propping P, Cichon S, Hoffmann P, Knapp M, Nothen MM, Mangold E, 2009. Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nat. Genet 41, 473–477. [DOI] [PubMed] [Google Scholar]
- Brodbeck S, Besenbeck B, Englert C, 2004. The transcription factor Six2 activates expression of the Gdnf gene as well as its own promoter. Mech. Dev 121, 1211–1222. [DOI] [PubMed] [Google Scholar]
- Bush JO, Jiang R, 2012. Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development 139, 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butali A, Mossey PA, Adeyemo WL, Eshete MA, Gowans LJJ, Busch TD, Jain D, Yu W, Huan L, Laurie CA, Laurie CC, Nelson S, Li M, Sanchez-Lara PA, Magee WP 3rd, Magee KS, Auslander A, Brindopke F, Kay DM, Caggana M, Romitti PA, Mills JL, Audu R, Onwuamah C, Oseni GO, Owais A, James O, Olaitan PB, Aregbesola BS, Braimah RO, Oginni FO, Oladele AO, Bello SA, Rhodes J, Shiang R, Donkor P, Obiri-Yeboah S, Arthur FKN, Twumasi P, Agbenorku P, Plange-Rhule G, Oti AA, Ogunlewe OM, Oladega AA, Adekunle AA, Erinoso AO, Adamson OO, Elufowoju AA, Ayelomi OI, Hailu T, Hailu A, Demissie Y, Derebew M, Eliason S, Romero-Bustillous M, Lo C, Park J, Desai S, Mohammed M, Abate F, Abdur-Rahman LO, Anand D, Saadi I, Oladugba AV, Lachke SA, Amendt BA, Rotimi CN, Marazita ML, Cornell RA, Murray JC, Adeyemo AA, 2018. Genomic analyses in african populations identify novel risk loci for cleft palate. Hum. Mol. Genet [DOI] [PMC free article] [PubMed]
- Conway JC, Taub PJ, Kling R, Oberoi K, Doucette J, Jabs EW, 2015. Ten-year experience of more than 35,000 orofacial clefts in Africa. BMC Pediatrics 15, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox LL, Cox TC, Moreno Uribe LM, Zhu Y, Richter CT, Nidey N, Standley JM, Deng M, Blue E, Chong JX, Yang Y, Carstens RP, Anand D, Lachke SA, Smith JD, Dorschner MO, Bedell B, Kirk E, Hing AV, Venselaar H, Valencia-Ramirez LC, Bamshad MJ, Glass IA, Cooper JA, Haan E, Nickerson DA, van Bokhoven H, Zhou H, Krahn KN, Buckley MF, Murray JC, Lidral AC, Roscioli T, 2018. Mutations in the Epithelial Cadherin-p120-Catenin Complex Cause Mendelian Non-Syndromic Cleft Lip with or without Cleft Palate. Am, J. Hum. Genet 102, 1143–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon MJ, Marazita ML, Beaty TH, Murray JC, 2011. Cleft lip and palate: understanding genetic and environmental influences. Nat. Rev. Genet 12, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Moreno M, Eliason S, Cao H, Li X, Yu W, Bidlack FB, Margolis HC, Baldini A, Amendt BA, 2015. TBX1 Protein Interactions and microRNA-96–5p Regulation Controls Cell Proliferation During Craniofacial and Dental Development: Implications for 22q11.2 Deletion syndrome. Hum. Mol. Genet 24, 2330–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant SF, Wang K, Zhang H, Glaberson W, Annaiah K, Kim CE, Bradfield JP, Glessner JT, Thomas KA, Garris M, Frackelton EC, Otieno FG, Chiavacci RM, Nah HD, Kirschner RE, Hakonarson H, 2009. A genome-wide association study identifies a locus for nonsyndromic cleft lip with or without cleft palate on 8q24. J. Pediatr 155, 909–913. [DOI] [PubMed] [Google Scholar]
- He G, Tavella S, Hanley KP, Self M, Oliver G, Grifone R, Hanley N, Ward C, Bobola N, 2010. Inactivation of Six2 in mouse identifies a novel genetic mechanism controlling development and growth of the cranial base. Dev. Biol 344, 720–730. [DOI] [PubMed] [Google Scholar]
- Hilliard SA, Yu L, Gu S, Zhang Z, Chen YP, 2005. Regional regulation of palatal growth and patterning along the anterior-posterior axis in mice. J. Anat 207, 655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufnagel RB, Zimmerman SL, Krueger LA, Bender PL, Ahmed ZM, Saal HM, 2016. A new frontonasal dysplasia syndrome associated with deletion of the SIX2 gene. Am. J. Med. Genet. Part A 170a, 487–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa E, Watanabe A, Nakano Y, Akita S, Hirano A, Kinoshita A, Kondo S, Kishino T, Uchiyama T, Niikawa N, Yoshiura K, 2006. PAX9 and TGFB3 are linked to susceptibility to nonsyndromic cleft lip with or without cleft palate in the Japanese: population-based and family-based candidate gene analyses. J. Hum. Genet 51, 38–46. [DOI] [PubMed] [Google Scholar]
- Jia S, Zhou J, Fanelli C, Wee Y, Bonds J, Schneider P, Mues G, D’Souza RN, 2017. Small-molecule Wnt agonists correct cleft palates in Pax9 mutant mice in utero. Development 144, 3819–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakrana A, Yang A, Anand D, Djordjevic D, Ramachandruni D, Singh A, Huang H, Ho JWK, Lachke SA, 2018. iSyTE 2.0: a database for expression-based gene discovery in the eye. Nuc. Acids Res 46, D875–d885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K, Ohto H, Takizawa T, Saito T, 1996. Identification and expression of six family genes in mouse retina. FEBS Lett 393, 259–263. [DOI] [PubMed] [Google Scholar]
- Kist R, Watson M, Wang X, Cairns P, Miles C, Reid DJ, Peters H, 2005. Reduction of Pax9 gene dosage in an allelic series of mouse mutants causes hypodontia and oligodontia. Hum. Mol. Genet 14, 3605–3617. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC, 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc 4, 1073–1081. [DOI] [PubMed] [Google Scholar]
- Lachke SA, Ho JW, Kryukov GV, O’Connell DJ, Aboukhalil A, Bulyk ML, Park PJ, Maas RL, 2012. iSyTE: integrated Systems Tool for Eye gene discovery. Invest. Ophthal. & Vis. Sci 53, 1617–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie EJ, Carlson JC, Cooper ME, Christensen K, Weinberg SM, Marazita ML, 2017. Exploring Subclinical Phenotypic Features in Twin Pairs Discordant for Cleft Lip and Palate. The Cleft Palate-Craniofacial Journal 54, 90–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie EJ, Liu H, Carlson JC, Shaffer JR, Feingold E, Wehby G, Laurie CA, Jain D, Laurie CC, Doheny KF, McHenry T, Resick J, Sanchez C, Jacobs J, Emanuele B, Vieira AR, Neiswanger K, Standley J, Czeizel AE, Deleyiannis F, Christensen K, Munger RG, Lie RT, Wilcox A, Romitti PA, Field LL, Padilla CD, Cutiongco-de la Paz EMC, Lidral AC, Valencia-Ramirez LC, Lopez-Palacio AM, Valencia DR, Arcos-Burgos M, Castilla EE, Mereb JC, Poletta FA, Orioli IM, Carvalho FM, Hecht JT, Blanton SH, Buxó CJ, Butali A, Mossey PA, Adeyemo WL, James O, Braimah RO, Aregbesola BS, Eshete MA, Deribew M, Koruyucu M, Seymen F, Ma L, de Salamanca JE, Weinberg SM, Moreno L, Cornell RA, Murray JC, Marazita ML, 2016. A Genome-wide Association Study of Nonsyndromic Cleft Palate Identifies an Etiologic Missense Variant in GRHL3. Am. J. Hum. Genet 98, 744–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie EJ, Taub MA, Liu H, Steinberg KM, Koboldt DC, Zhang Q, Carlson JC, Hetmanski JB, Wang H, Larson DE, Fulton RS, Kousa YA, Fakhouri WD, Naji A, Ruczinski I, Begum F, Parker MM, Busch T, Standley J, Rigdon J, Hecht JT, Scott AF, Wehby GL, Christensen K, Czeizel AE, Deleyiannis FW, Schutte BC, Wilson RK, Cornell RA, Lidral AC, Weinstock GM, Beaty TH, Marazita ML, Murray JC, 2015. Identification of functional variants for cleft lip with or without cleft palate in or near PAX7, FGFR2, and NOG by targeted sequencing of GWAS loci. Am. J. Hum. Genet 96, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little J, Cardy A, Arslan MT, Gilmour M, Mossey PA, 2004. Smoking and orofacial clefts: a United Kingdom-based case-control study. The Cleft Palate-Craniofacial Journal 41, 381–386. [DOI] [PubMed] [Google Scholar]
- Liu H, Leslie EJ, Carlson JC, Beaty TH, Marazita ML, Lidral AC, Cornell RA, 2017. Identification of common non-coding variants at 1p22 that are functional for non-syndromic orofacial clefting. Nature Commun. 8, 14759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Li C, Xu J, Lan Y, Liu H, Li X, Maire P, Wang X, Jiang R, 2019. Crucial and Overlapping Roles of Six1 and Six2 in Craniofacial Development. J.Dent. Res 98, 572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig KU, Mangold E, Herms S, Nowak S, Reutter H, Paul A, Becker J, Herberz R, AlChawa T, Nasser E, Bohmer AC, Mattheisen M, Alblas MA, Barth S, Kluck N, Lauster C, Braumann B, Reich RH, Hemprich A, Potzsch S, Blaumeiser B, Daratsianos N, Kreusch T, Murray JC, Marazita ML, Ruczinski I, Scott AF, Beaty TH, Kramer F-J, ienker TF, Steegers-Theunissen RP, Rubini M, Mossey PA, Hoffmann P, Lange C, Cichon S, Propping P, Knapp M, Nothen MM, 2012. Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat. Genet 44, 968–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangold E, Ludwig KU, Birnbaum S, Baluardo C, Ferrian M, Herms S, Reutter H, de Assis NA, Chawa TA, Mattheisen M, Steffens M, Barth S, Kluck N, Paul A, Becker J, Lauster C, Schmidt G, Braumann B, Scheer M, Reich RH, Hemprich A, Potzsch S, Blaumeiser B, Moebus S, Krawczak M, Schreiber S, Meitinger T, Wichmann HE, Steegers-Theunissen RP, Kramer FJ, Cichon S, Propping P, Wienker TF, Knapp M, Rubini M, Mossey PA, Hoffmann P, Nothen MM, 2010. Genome-wide association study identifies two susceptibility loci for nonsyndromic cleft lip with or without cleft palate. Nat Genet 42, 24–26. [DOI] [PubMed] [Google Scholar]
- Mori-Akiyama Y, Akiyama H, Rowitch DH, d.C. B, 2003. Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc Natl Acad Sci U S A 100, 9360–9365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neiswanger K, Weinberg SM, Rogers CR, Brandon CA, Cooper ME, Bardi KM, Deleyiannis FW, Resick JM, Bowen A, Mooney MP, de Salamanca JE, Gonzalez B, Maher BS, Martin RA, Marazita ML, 2007. Orbicularis oris muscle defects as an expanded phenotypic feature in nonsyndromic cleft lip with or without cleft palate. Am. J. Med. Genet. Part A 143a, 1143–1149. [DOI] [PubMed] [Google Scholar]
- Nonomura K, Takahashi M, Wakamatsu Y, Takano-Yamamoto T, Osumi N, 2010. Dynamic expression of Six family genes in the dental mesenchyme and the epithelial ameloblast stem/progenitor cells during murine tooth development. J. Anatomy 216, 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien LL, Guo Q, Lee Y, Tran T, Benazet JD, Whitney PH, Valouev A, McMahon AP, 2016. Differential regulation of mouse and human nephron progenitors by the Six family of transcriptional regulators. Development 143, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okello DO, Iyyanar PPR, Kulyk WM, Smith TM, Lozanoff S, Ji S, Nazarali AJ, 2017. Six2 Plays an Intrinsic Role in Regulating Proliferation of Mesenchymal Cells in the Developing Palate. Frontiers Physiology 8, 955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Ma W, O’Brien LL, Chung E, Guo JJ, Cheng JG, Valerius MT, McMahon JA, Wong WH, McMahon AP, 2012. Six2 and Wnt regulate self-renewal and commitment of nephron progenitors through shared gene regulatory networks. Dev. Cell 23, 637–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters H, Neubuser A, Kratochwil K, Balling R, 1998. Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes & Dev 12, 2735–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu S, Tucker SC, Zhao Q, deCrombrugghe B, Wisdom R, 1999. Physical and genetic interactions between Alx4 and Cart1. Development 126, 359–369. [DOI] [PubMed] [Google Scholar]
- Rahimov F, Marazita ML, Visel A, Cooper ME, Hitchler MJ, Rubini M, Domann FE, Govil M, Christensen K, Bille C, Melbye M, Jugessur A, Lie RT, Wilcox AJ, Fitzpatrick DR, Green ED, Mossey PA, Little J, Steegers-Theunissen RP, Pennacchio LA, Schutte BC, Murray JC, 2008. Disruption of an AP-2alpha binding site in an IRF6 enhancer is associated with cleft lip. Nat. Genet 40, 1341–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self M, Lagutin OV, Bowling B, Hendrix J, Cai Y, Dressler GR, Oliver G, 2006. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. Embo J. 25, 5214–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuppia L, Capogreco M, Marzo G, La Rovere D, Antonucci I, Gatta V, Palka G, Mortellaro C, Tete S, 2011. Genetics of syndromic and nonsyndromic cleft lip and palate. J. Craniofacial Surg 22, 1722–1726. [DOI] [PubMed] [Google Scholar]
- Sun Y, Huang Y, Yin A, Pan Y, Wang Y, Wang C, Du Y, Wang M, Lan F, Hu Z, Wang G, Jiang M, Ma J, Zhang X, Ma H, Ma J, Zhang W, Huang Q, Zhou Z, Ma L, Li Y, Jiang H, Xie L, Jiang Y, Shi B, Cheng J, Shen H, Wang L, Yang Y, 2015. Genome-wide association study identifies a new susceptibility locus for cleft lip with or without a cleft palate. Nat. Commun 6, 6414. [DOI] [PubMed] [Google Scholar]
- Sun Z, Yu W, Navarro MS, Sweat M, Eliason S, Sharp T, Liu H, Seidel K, Zhang L, Moreno M, Lynch T, Holton NE, Rogers L, Neff T, Goodheart MJ, Michon F, Klein OD, Chai Y, Dupuy A, Engelhardt JF, Chen Z, Amendt BA, 2016. Sox2 and Lef-1 interact with Pitx2 to regulate incisor development and stem cell renewal. Development 143, 4115–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G, 2010. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics 11, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ZT, Brand HA, Shaffer JR, Leslie EJ, Arzi B, Willet CE, Cox TC, McHenry T, Narayan N, Feingold E, Wang X, Sliskovic S, Karmi N, Safra N, Sanchez C, Deleyiannis FWB, Murray JC, Wade CM, Marazita ML, Bannasch DL, 2015. Genome-Wide Association Studies in Dogs and Humans Identify ADAMTS20 as a Risk Variant for Cleft Lip and Palate. PLOS Genet. 11, e1005059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Zuo X, He M, Gao J, Fu Y, Qin C, Meng L, Wang W, Song Y, Cheng Y, Zhou F, Chen G, Zheng X, Wang X, Liang B, Zhu Z, Fu X, Sheng Y, Hao J, Liu Z, Yan H, Mangold E, Ruczinski I, Liu J, Marazita ML, Ludwig KU, Beaty TH, Zhang X, Sun L, Bian Z, 2017. Genome-wide analyses of non-syndromic cleft lip with palate identify 14 novel loci and genetic heterogeneity. Nat. Commun 8, 14364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang R, Liu Z, Hou C, Zong W, Zhang A, Sun X, Gao J, 2015. Loss of lysyl oxidase-like 3 causes cleft palate and spinal deformity in mice. Hum. Mol. Genet 24, 6174–6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Gao Y, Lan Y, Jia S, Jiang R, 2013. Pax9 regulates a molecular network involving Bmp4, Fgf10, Shh signaling and the Osr2 transcription factor to control palate morphogenesis. Development 140, 4709–4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1. Six2−/− embryos lacking cleft palate have normal levels of proliferating mesenchymal cells. A-A’. Coronal sectioning and Ki67 IF staining of WT E14.5 embryos. A’ is an enlargement of the boxed region of A. B-B’. Ki67 of null embryos that do not exhibit cleft reveal similar levels of mesenchymal cell proliferation.
Supplemental Fig. 2. The expression of Runx2 and Osx is increased in Six2−/− embryos. A-B. Runx2 and OSX mRNAs were quantified using RT-qPCR from RNA isolated from palate shelves in Six2+/+ and Six2−/− embryos at E15.5. The levels of these transcripts were increased in Six2−/− embryos compared with control littermates. C-C’. Staining for Osx in WT embryos at E16.5 in the palate shelves. C’ is an enlargement of the boxed region shown in C. D-D’. Osx staining in E16.5 Six2−/− embryos, where a greater area of cells are expressing the differentiation marker.
Supplemental Fig. 3. Six2−/− embryos without cleft palate have regular palatine bone formation. A-A’. Sectioning and alizarin red staining of E17.5 WT embryos labels the developing palatine bone. A’ is an enlargement of the boxed region in A. B-B’. Sectioning and alizarin red staining of E17.5 Six2−/− embryos that do not have a cleft palate revealed similar amounts of ossification of the palatine bone.
Supplemental Fig. 4. Six2 promotes Pax9 expression in the medial edge of the developing palate. A. Staining of Six2 and Pax9 in E14.5 embryos demonstrates that these proteins are co-localized at the tip region of the palate shelf. B. In Six2−/− embryos, Six2 protein is not detectable and Pax9 protein expression is strongly diminished. C. Using RT-qPCR to quantitate the relative amount of Pax9 mRNA demonstrates a decrease in null embryos compared with WT control littermates.