

Abstract
Plasmacytoid dendritic cells (pDCs) are professional type I IFN producers believed to promote lupus. However, questions exist about whether they function at the same level throughout the course of lupus disease. We analyzed high-purity pDCs sorted from lupus mice. Although pDCs produced a large amount of IFN-α during disease initiation, those sorted from late-stage lupus mice were found to be defective in producing IFN-α. These pDCs expressed an increased level of MHC, suggesting a functional drift to Ag presentation. We examined the potential mechanism behind the defect and identified a novel transcriptional factor, Foxj2, which repressed the expression of several genes in pDCs, but not IFN-α. Dysregulation in pDCs appears to be predisposed, because they exhibited an altered transcriptional profile before the onset of clinical signs. Our results suggest that pDCs do not function the same throughout the disease course and lose the ability to produce IFN-α in late-stage lupus mice.
It has been long recognized that type I IFNs, including IFN-α, facilitate the progression of systemic lupus erythematosus (SLE). Based on large amounts of supporting evidence obtained using human patient samples (1–6) and murine models (7–11), four Abs targeting IFN-α or type IIFNR have been tested in human clinical trials for SLE (12–16). A couple of them showed promising efficacy (13–15). A beneficial effect was observed in SLE patients with a low type I IFN signature (i.e., those with lower expression of IFN-responsive genes); IFN-α blockade partially inhibited such expression. This suggests that early intervention (when the IFN signature is still low) may be required for IFN-α–targeted therapies for SLE. Interestingly, IFN signatures returned to pretreatment levels in all patients by 6 mo after the last dose of anti–IFN-α Abs (17), suggesting that type I IFNs are constantly produced and pathogenic throughout disease progression. Because virtually all cells can produce IFN-α upon stimulation, identifying which types of cells play a predominant role in driving early- and late-stage SLE has become an interesting, but challenging, question.
Plasmacytoid dendritic cells (pDCs) were identified as professional IFN-producing cells (18, 19). They produce a large amount of IFN-α in response to TLR7/9 ligation (20). Studies of SLE patient samples revealed that immune complexes in the patient sera are also capable of inducing IFN-α in pDCs (21, 22), a process in which neutrophil extracellular traps are involved (23, 24). Recently, two groups of researchers independently showed that depletion of pDCs from mouse models of SLE ameliorates lupus-like disease (25, 26). Deletion of pDCs in predisease lupus-prone mice significantly reduced lymphadenopathy and improved kidney pathology later in life. Strikingly, the benefit was sustained even after the pDC population recovered (26). This suggests that pDCs contribute to disease early in SLE pathogenesis and that the recovered pDCs present in late-stage lupus mice might be less pathogenic.
In this study, we used a classical mouse model of SLE, MRL/Mp-Faslpr (lpr), as well as its parent strain, MRL, to explore pDC functions in lupus mice. Female lpr mice exhibit systemic autoimmunity and glomerulonephritis and die at an average age of 18 wk. Like SLE patients (27), diseased lpr mice show an elevated level of IFN-α in the circulation (10, 28), but the source of this cytokine remains unclear.
Materials and Methods
Mice and cell sorting
Mice were purchased from The Jackson Laboratory and bred in-house. All mice used were female. pDCs were sorted as CD11c+CD11b−PDCA1+B220+. Siglec-H inhibits IFN-α induction and was not used for sorting. Anti-mouse Abs were from eBioscience. RNA sequencing (RNA-seq) was performed, and the data can be found under BioProject accession PRJNA284002.
Cell culture and analyses
CpG (ODN1585, 5 μM) was used to stimulate sorted pDCs or total bone marrow cells. For gene knockdown, mouse Foxj2 small interfering RNA (siRNA) and negative control were purchased from QIAGEN. For quantitative PCR of cDNA, iTaq Universal SYBR Green Supermix and ABI 7500 Fast System were used, with ribosomal protein L32 as the housekeeping gene. For FACS, cells were blocked with Fc, stained, and analyzed with a BD FACSAria II. Mouse IFN-α levels were measured with a PBL ELISA kit.
Statistical analysis
Analysis of nonsequencing data was performed with the Student t test, one-way ANOVA, and the Tukey posttest. The results were considered statistically significant when p < 0.05. Error bars denote SEs.
Results and Discussion
IFN signatures in lupus pDCs
Early depletion of pDCs from lupus mice ameliorates disease (26). However, whether pDCs are the primary source of IFN-α for both early- and late-stage lupus remains unclear. We sorted pDCs (Fig. 1A) from the bone marrow of 6- and 16-wk-old female lpr mice, representing predisease and late-stage lupus disease, respectively, and analyzed the gene expression profile. As controls, pDCs were sorted from the bone marrow of age-matched female MRL mice. MRL mice carry wild-type Fas and display lupus much later than do lpr mice. They appear normal at 6 wk and are considered to be at predisease stage at 16 wk (29). Therefore, our mouse groups included 16-wk-old lpr (late-stage lupus), 6 wk-old lpr and 16-wk-old MRL (predisease), and 6-wk-old MRL mice (control). Although mouse pDCs are most enriched in the bone marrow compared with other organs, they are still a minor population in the bone marrow (29). Rigorous quality control measures were taken to ensure accurate processing of samples. Measurement of pDC-specific transcription factor E2-2 showed high purity of our sorted cells (Fig. 1B). Because B cell progenitors in the bone marrow share surface markers with pDCs, we measured the expression levels of B cell genes Cd20, Ebf1, Vpreb3, and Igha. Less than 5% of our sorted pDCs might have been contaminated with B cell progenitors (data not shown). With high-purity pDCs, we found that many IFN-responsive genes were upregulated when comparing 16 wk-old lpr mice with age-matched MRL mice (Fig. 1C, highlighted with yellow bars). This suggests that bone marrow pDCs in late-stage lpr mice exhibit both type I and II IFN signatures, indicating previous exposure to IFNs.
FIGURE 1.
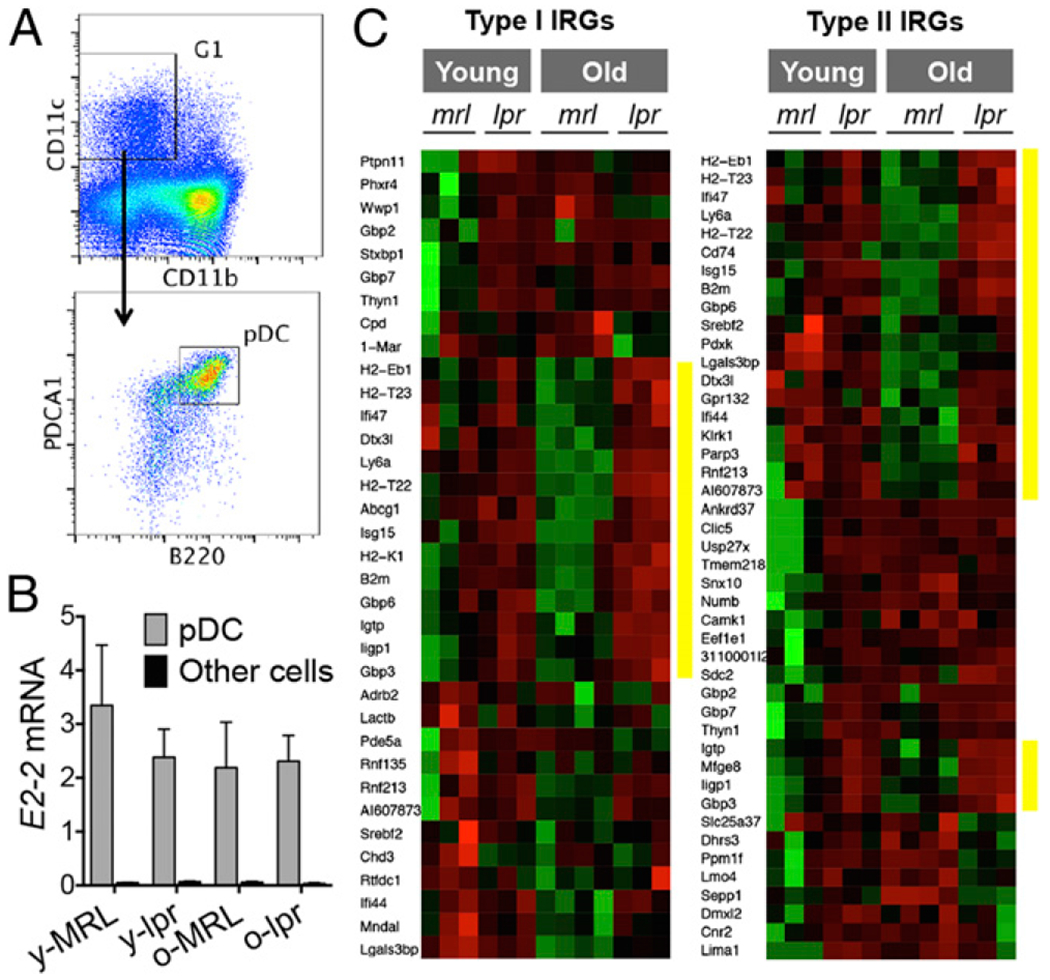
IFN signatures in pDCs sorted from lupus mice. (A) Sorting strategy. (B) Transcript level of pDC-specific gene E2-2 in sorted pDCs compared with pDC-depleted bone marrow cells (other cells). (C) Heat maps of types I and II IFN-responsive genes (IRGs). Red, upregulation. Green, downregulation. Yellow bars highlight the genes that were upregulated in older lpr mice versus older MRL mice.
pDCs from late-disease lupus mice are unable to produce IFN-α upon CpG stimulation
We next identified signaling pathways based on the downstream transcript signatures. Among five top canonical pathways identified, four were found in the comparison between 16-wk-old lpr mice and age-matched MRL mice (Fig. 2A). A common feature of the four pathways was upregulation of MHC molecules (data not shown). Detection of MHC class II (MHC-II) on the surface of pDCs confirmed its upregulation in lpr mice with late-stage disease (Fig. 2B). This suggests that pDCs from late-stage lupus mice might excel at Ag presentation, a phenomenon similar to human SLE in which patient pDCs were shown to be better APCs than normal pDCs (30).
FIGURE 2.
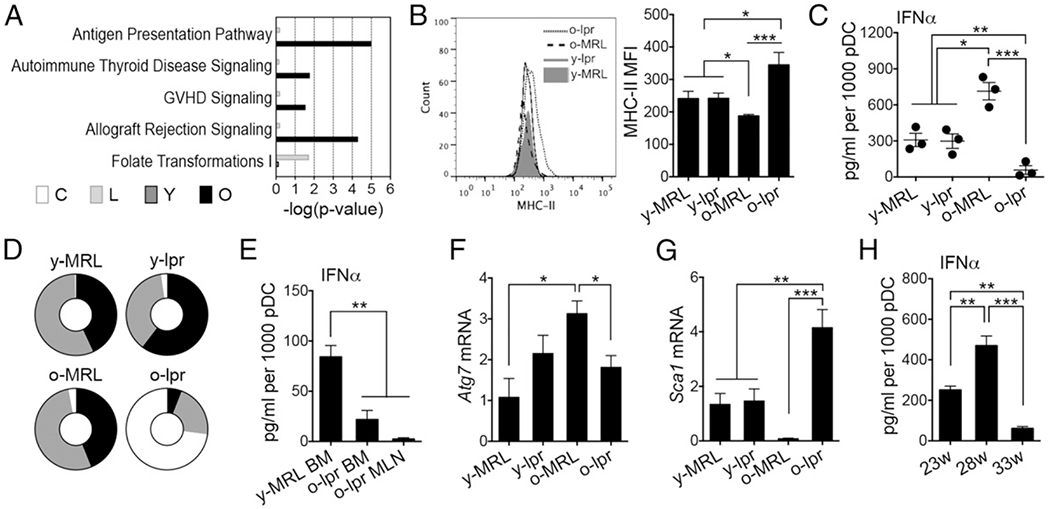
pDCs from late-stage lupus mice unable to produce IFN-α upon CpG stimulation. (A) Ingenuity pathway analysis showing the top canonical pathways. (B) MHC-II expression on bone marrow pDCs. (C) Production of IFN-α in sorted bone marrow pDCs upon CpG stimulation for 6 h. (D) Distribution of pDCs among bone marrow (black), spleen (gray), and mesenteric lymph node (white). (E) Production of IFN-α from sorted pDCs upon CpG stimulation for 6 h. (F) Transcript level of Atg7 in sorted pDCs. (G) Transcript level of Sca1 in sorted pDCs. (H) Production of IFN-α from bone marrow pDCs from 23-, 28-, and 33-wk-old NZB/W F1 female mice upon CpG stimulation for 6 h. *p < 0.05, **p < 0.01, ***p < 0.001, ANOVA, n ≥ 3 mice/group. BM, bone marrow; C, older versus younger MRL; L, older versus younger lpr; MFI, mean fluorescence intensity; MLN, mesenteric lymph node; o, older; O, older lpr versus older MRL; y, younger; Y, younger lpr versus younger MRL.
We next questioned whether pDCs isolated from older lpr mice, expressing a higher level of MHC-II, were still capable of producing IFN-α upon stimulation. A previous study showed some defect in IFN-α production from a mixed population of bone marrow dendritic cells from late-stage lupus mice (31). With sorted pDCs of high purity, we showed almost complete loss of CpG-mediated induction of IFN-α from late-stage lupus pDCs (Fig. 2C). Interestingly, pDCs from 16-wk-old MRL mice (predisease) showed enhanced IFN-α production, consistent with a significantly lower level of MHC-II on the surface of these cells (Fig. 2B). This suggests that pDCs produce a large amount of IFN-α to initiate the disease. Although the bone marrow has the highest percentage of pDCs (29), lpr mice with late-stage disease possess very large lymph nodes, increasing the absolute numbers of pDCs. Indeed, we found that pDCs were primarily found in lymph nodes in 16-wk-old lpr mice (Fig. 2D, Supplemental Fig. 1A). However, lymph node pDCs were also found to be defective in producing IFN-α (Fig. 2E). This suggests that the high level of IFN-α in late-stage lpr mice (10, 28) may not have come from pDCs. Additionally, we measured the transcript level of Atg7, a gene required for IFN-α induction by immune complexes (32) and found it be significantly lower in pDCs from 16-wk-old lpr mice compared with age-matched MRL mice (Fig. 2F). This suggests that immune complex–mediated IFN-α production from pDCs may be defective as well. Interestingly, we found a significantly higher level of Sca1 in late-stage lupus pDCs; in contrast, the expression of this gene was negligible in pDCs from 16-wk-old MRL mice (Fig. 2G). This observation is consistent with a previous report that Sca1 expression on pDCs correlated negatively with their ability to produce IFN-α (33). Lastly, we confirmed that the defective ability of pDCs to produce IFN-α in late-stage lupus mice was not restricted to the lpr model. In another classical SLE mouse model, NZB/W F1 pDCs exhibited a reduced ability to produce IFN-α during late-stage disease compared with early disease (Fig. 2H). These results suggest that pDCs from late-disease lupus mice lose their ability to produce IFN-α. Instead, they express more MHC molecules and may become better APCs.
Defeet in IFN-α production in pDCs is independent of the transcriptional repressor Foxj2
We next investigated the mechanism behind the functional loss of pDCs to produce IFN-α. Upstream receptors leading to IFN-α induction in pDCs, including TLR9 for DNA Ags and FcRs for immune complexes, were expressed in pDCs isolated from late-stage lupus mice (Fig. 3A, Supplemental Fig. 1B). We also measured the transcript level of another potential sensor, TLR7, and found it to be overexpressed in these pDCs (Supplemental Fig. 1C). This suggests that the defect in IFN-α production was not due to a lack of upstream receptors. However, the problem seemed to be related to transcription or RNA stability, because pDCs from late-stage lupus mice expressed little IFN-α mRNA upon CpG stimulation (Fig. 3B). Thus, we explored a novel strategy of using RNA-seq data to identify differentially expressed genes that followed the same or opposite pattern of CpG-induced IFN-α. We reasoned that common machineries might modulate IFN-α transcripts together with other gene transcripts. By studying regulatory elements of the other genes, we might be able to identify the mechanism behind the defect in IFN-α production. To identify genes with the same expression pattern as CpG-induced IFN-α (Fig. 2C), we used the following criteria: expression in pDCs of older MRL mice had to be ≥2-fold higher than that of younger MRL mice, and expression in pDCs of older lpr mice had to be ≥2-fold lower than that of older MRL mice. Similar criteria were applied to identify genes with the opposite pattern. Using this strategy, we identified seven genes (Fig. 3C). We searched for shared microRNAs using DIANA-microT but found no matches. However, promoter analysis revealed two shared cis-regulatory elements that were present in the promoter regions of all genes except one (Supplemental Fig. 1D). One of the two motifs, TGTTT, can be bound by three members of the Fox family of transcription factors (Supplemental Fig. 1E). Among them, only Foxj2 was expressed in pDCs. In lpr mice, Foxj2 was expressed significantly higher in pDCs than in other bone marrow cells (Fig. 3D). The other shared cis-regulatory element was an Rreb1 binding site, but Rreb1 was not preferentially expressed in pDCs (data not shown).
FIGURE 3.
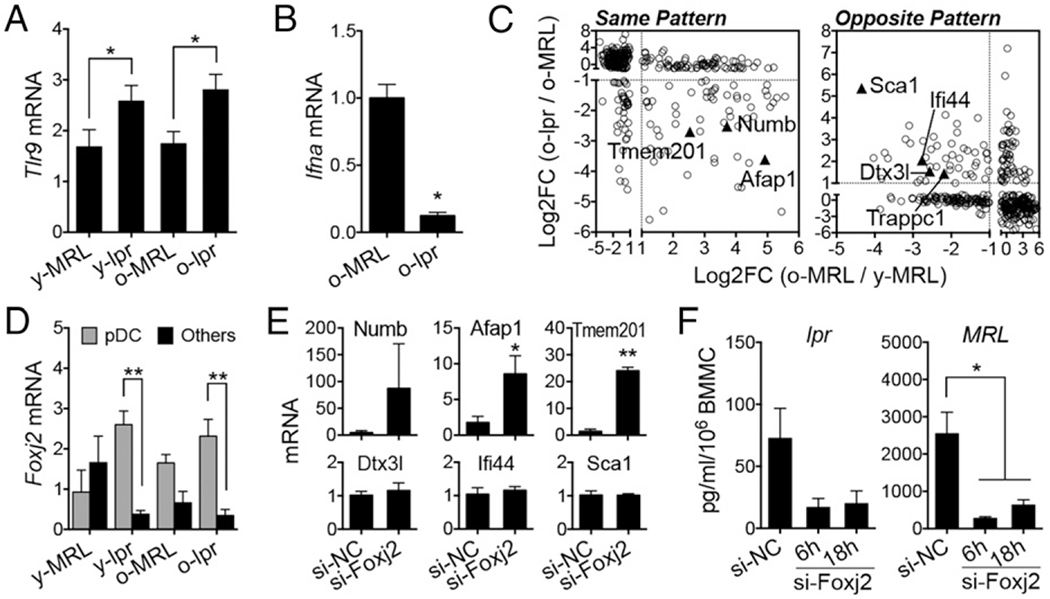
Defect in IFN-α production in pDCs independent of the transcriptional repressor Foxj2. (A) Transcript level of Tlr9 in sorted pDCs. (B) Transcript level of IFNα in pDCs from older mice stimulated with CpG for 6 h. (C) Differentially expressed genes following the same or opposite pattern of CpG-induced IFN-α. (D) Transcript level of Foxj2 in sorted pDCs and pDC-depleted bone marrow cells (Others). (E) Transcript levels of genes identified in (C) in bone marrow cells of 16-wk-old lpr mice after transfection with negative-control siRNA (si-NC) or Foxj2-specific siRNAs (si-Foxj2). Fold differences are shown. (F) Production of IFN-α in bone marrow cells (BMMC) of 4-mo-old lpr or MRL mice, transfected with siRNAs and stimulated with CpG for 6 or 18 h. *p < 0.05, **p < 0.01, Student t test (n ≥ 3 mice per group). Log2FC, log2-fold changes.
We hypothesized that Foxj2 overexpression in pDCs repressed the transcription of both IFN-α and genes with the same pattern (Afap1, Numb, Tmem201). To test this hypothesis, we used siRNA to knock down Foxj2 in pDCs from late-stage lupus mice (Supplemental Fig. 1F). Such knockdown increased the transcript levels of Afap1, Numb, and Tmem201 (Fig. 3E). Although little is known about the functions of these three genes in pDCs, our results suggest that Foxj2 is a functional transcriptional repressor involved in shaping the gene expression profile of pDCs in late-stage lupus mice. Unexpectedly, however, knocking down Foxj2 did not restore IFN-α production in pDCs from lpr mice (Fig. 3F). In fact, Foxj2 seems to be required for the production of IFN-α in pDCs, because Foxj2-specific siRNA decreased IFN-α levels for both lpr and MRL mice. In addition, siRNA of Foxj2 did not change the expression of Atg7 (Supplemental Fig. 1G), a gene required for IFN-α induction (32). These results suggest that the defect in IFN-α production in pDCs of late-stage lupus mice is independent of Foxj2. Other potential mechanisms that we will explore in the future to try to explain the defect include the involvement of lysosome-related organelle complexes, such as BLOC1 (Supplemental Fig. 1H), whose downregulation might hinder TLR7/9-induced IFN-α production in pDCs (34); a possible role for apoptosis, because pDCs from late-stage lupus mice appeared to express a decreased level of the antiapoptotic gene Bcl-xl; and upregulation in older lpr mice of Ptpn21 (also called PTPD1), a tyrosine phosphatase that interacts with Tec family kinases to increase the activation of Stat3 (35), and Stat3 can negatively regulate the type I IFN response (36).
Additional comparisons revealed that only 25 genes were differentially expressed when comparing pDCs of older versus younger lpr mice (Supplemental Fig. 2A, blue, false discovery rate < 0.1). Among these genes, only nine genes reached a p value of 0.05 (Supplemental Fig. 2B). Many of these are IFN-responsive genes, such as Ptpn11 and Pde5a, which were upregulated in pDCs of early diseased mice (Fig. 1C) but significantly downregulated in pDCs of older lpr mice. This is consistent with the observation that pDCs from late-stage lupus mice produced less IFN-α. In contrast to the identification of very few gene differences between pDCs of older versus younger lpr mice, 207 genes were differentially expressed when pDCs in older versus younger MRL mice were compared (Supplemental Fig. 2A, green). This suggests that there might be more changes in transcript abundance in pDCs prior to the onset of disease, whereas few genes change as lupus progresses from the predisease stage to the late stage. Thus, we hypothesized that an altered gene expression profile (i.e., a lupus-prone profile) might already be present in pDCs prior to disease onset. To identify lupus-prone genes independent of Faslpr mutation, because few SLE patients carry mutations of the Fas gene, we investigated whether pDCs from older MRL mice at the predisease stage of lupus shared differentially expressed genes with pDCs from younger lpr mice, which represents the predisease stage for the lpr mouse strain. We identified 54 such genes, including 44 protein-coding genes (Supplemental Fig. 2C, 2D) and 10 long unannotated transcripts (Supplemental Fig. 2E, 2F). The significance of the lupus-prone profile in pDCs remains to be explored. However, our findings raise an important question of whether pDCs in SLE patients or lupus mice are born with the predisposed, dysregulated gene expression profile. In future investigations, we will study pDCs from even younger mice to determine potential early defects, as well as whether triggers later in life might manifest the defects.
Supplementary Material
Acknowledgments
This work was supported by X.M.L.’s startup funds, as well as by the Virginia Bioinformatics Institute and Fralin Life Science Institute Small Grants Program (VBI/Fralin-GRL-01).
Abbreviations used in this article:
- MHC-II
MHC class II
- pDC
plasmacytoid dendritic cell
- RNA-seq
RNA sequencing
- siRNA
small interfering RNA
- SLE
systemic lupus erythematosus
Footnotes
The RNA sequence data presented in this article have been submitted to BioProject under accession number PRJNA284002.
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Rönnblom L, Eloranta ML, and Alm GV. 2006. The type I interferon system in systemic lupus erythematosus. Arthritis Rheum. 54: 408–420. [DOI] [PubMed] [Google Scholar]
- 2.Blanco P, Palucka AK, Gill M, Pascual V, and Banchereau J. 2001. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science 294: 1540–1543. [DOI] [PubMed] [Google Scholar]
- 3.Crow MK 2014. Advances in understanding the role of type I interferons in systemic lupus erythematosus. Curr. Opin. Rheumatol 26: 467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crow MK, Olferiev M, and Kirou KA. 2015. Targeting of type I interferon in systemic autoimmune diseases. Transl. Res 165: 296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elkon KB, and Stone VV. 2011. Type I interferon and systemic lupus erythematosus. J. Interferon Cytokine Res 31: 803–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elkon KB, and Wiedeman A. 2012. Type I IFN system in the development and manifestations of SLE. Curr. Opin. Rheumatol 24: 499–505. [DOI] [PubMed] [Google Scholar]
- 7.Nickerson KM, Cullen JL, Kashgarian M, and Shlomchik MJ. 2013. Exacerbated autoimmunity in the absence of TLR9 in MRL. Fas(lpr) mice depends on Ifnar1. J. Immunol 190: 3889–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agrawal H, Jacob N, Carreras E, Bajana S, Putterman C, Turner S, Neas B, Mathian A, Koss MN, Stohl W, et al. 2009. Deficiency of type I IFN receptor in lupus-prone New Zealand mixed 2328 mice decreases dendritic cell numbers and activation and protects from disease. J. Immunol 183: 6021–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braun D, Geraldes P, and Demengeot J. 2003. Type I Interferon controls the onset and severity of autoimmune manifestations in lpr mice. J. Autoimmun 20: 15–25. [DOI] [PubMed] [Google Scholar]
- 10.Hadj-Slimane R, Chelbi-Alix MK, Tovey MG, and Bobé P. 2004. An essential role for IFN-alpha in the overexpression of Fas ligand on MRL/lpr lymphocytes and on their spontaneous Fas-mediated cytotoxic potential. J. Interferon Cytokine Res 24: 717–728. [DOI] [PubMed] [Google Scholar]
- 11.Sriram U, Varghese L, Bennett HL, Jog NR, Shivers DK, Ning Y, Behrens EM, Caricchio R, and Gallucci S. 2012. Myeloid dendritic cells from B6.NZM Sle1/Sle2/Sle3 lupus-prone mice express an IFN signature that precedes disease onset. J. Immunol 189: 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petri M, Wallace DJ, Spindler A, Chindalore V, Kalunian K, Mysler E, Neuwelt CM, Robbie G, White WI, Higgs BW, et al. 2013. Sifalimumab, a human anti-interferon-α monoclonal antibody, in systemic lupus erythematosus: a phase I randomized, controlled, dose-escalation study. Arthritis Rheum. 65: 1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McBride JM, Jiang J, Abbas AR, Morimoto A, Li J, Maciuca R, Townsend M, Wallace DJ, Kennedy WP, and Drappa J. 2012. Safety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: results of a phase I, placebo-controlled, double-blind, dose-escalation study. Arthritis Rheum. 64: 3666–3676. [DOI] [PubMed] [Google Scholar]
- 14.Merrill JT, Wallace DJ, Petri M, Kirou KA, Yao Y, White WI, Robbie G, Levin R, Berney SM, Chindalore V, et al. ; Lupus Interferon Skin Activity (LISA) Study Investigators. 2011. Safety profile and clinical activity of sifalimumab, a fully human anti-interferon α monoclonal antibody, in systemic lupus erythematosus: a phase I, multicentre, double-blind randomised study. Ann. Rheum. Dis 70: 1905–1913. [DOI] [PubMed] [Google Scholar]
- 15.Yao Y, Richman L, Higgs BW, Morehouse CA, de los Reyes M, Brohawn P, Zhang J, White B, Coyle AJ, Kiener PA, and Jallal B. 2009. Neutralization of interferon-alpha/beta-inducible genes and downstream effect in a phase I trial of an anti-interferon-alpha monoclonal antibody in systemic lupus erythematosus. Arthritis Rheum. 60: 1785–1796. [DOI] [PubMed] [Google Scholar]
- 16.Kirou KA, and Gkrouzman E. 2013. Anti-interferon alpha treatment in SLE. Clin. Immunol 148: 303–312. [DOI] [PubMed] [Google Scholar]
- 17.Crow MK 2014. Type I interferon in the pathogenesis of lupus. J. Immunol 192: 5459–5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu YJ 2005. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol 23: 275–306. [DOI] [PubMed] [Google Scholar]
- 19.Colonna M, Trinchieri G, and Liu YJ. 2004. Plasmacytoid dendritic cells in immunity. Nat. Immunol 5: 1219–1226. [DOI] [PubMed] [Google Scholar]
- 20.Colonna M, Krug A, and Cella M. 2002. Interferon-producing cells: on the front line in immune responses against pathogens. Curr. Opin. Immunol 14: 373–379. [DOI] [PubMed] [Google Scholar]
- 21.Lövgren T, Eloranta ML, Kastner B, Wahren-Herlenius M, Alm GV, and Rönnblom L. 2006. Induction of interferon-alpha by immune complexes or liposomes containing systemic lupus erythematosus autoantigen- and Sjögren’s syndrome autoantigen-associated RNA. Arthritis Rheum. 54: 1917–1927. [DOI] [PubMed] [Google Scholar]
- 22.Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, and Luster AD. 2005. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J. Clin. Invest 115: 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, et al. 2011. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med 3: 73ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R, Riccieri V, et al. 2011. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med 3: 73ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sisirak V, Ganguly D, Lewis KL, Couillault C, Tanaka L, Bolland S, D’Agati V, Elkon KB, and Reizis B. 2014. Genetic evidence for the role of plasmacytoid dendritic cells in systemic lupus erythematosus. J. Exp. Med 211: 1969–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rowland SL, Riggs JM, Gilfillan S, Bugatti M, Vermi W, Kolbeck R, Unanue ER, Sanjuan MA, and Colonna M. 2014. Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J. Exp. Med 211: 1977–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL, and Notkins AL. 1979. Immune interferon in the circulation of patients with autoimmune disease. N. Engl. J. Med 301: 5–8. [DOI] [PubMed] [Google Scholar]
- 28.Sadanaga A, Nakashima H, Akahoshi M, Masutani K, Miyake K, Igawa T, Sugiyama N, Niiro H, and Harada M. 2007. Protection against autoimmune nephritis in MyD88-deficient MRL/lpr mice. Arthritis Rheum. 56: 1618–1628. [DOI] [PubMed] [Google Scholar]
- 29.Liao X, Ren J, Wei CH, Ross AC, Cecere TE, Jortner BS, Ahmed SA, and Luo XM. 2015. Paradoxical effects of all-trans-retinoic acid on lupus-like disease in the MRL/lpr mouse model. PLoS One 10: e0118176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jin O, Kavikondala S, Mok MY, Sun L, Gu J, Fu R, Chan A, Yeung J, Nie Y, and Lau CS. 2010. Abnormalities in circulating plasmacytoid dendritic cells in patients with systemic lupus erythematosus. Arthritis Res. Ther 12: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pau E, Cheung YH, Loh C, Lajoie G, and Wither JE. 2012. TLR tolerance reduces IFN-alpha production despite plasmacytoid dendritic cell expansion and anti-nuclear antibodies in NZB bicongenic mice. PLoS One 7: e36761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L, Sasai M, Latz E, Brinkmann MM, Iwasaki A, et al. 2012. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity 37: 986–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niederquell M, Kurig S, Fischer JA, Tomiuk S, Swiecki M, Colonna M, Johnston IC, and Dzionek A. 2013. Sca-1 expression defines developmental stages of mouse pDCs that show functional heterogeneity in the endosomal but not lysosomal TLR9 response. Eur. J. Immunol 43: 2993–3005. [DOI] [PubMed] [Google Scholar]
- 34.Blasius AL, Arnold CN, Georgel P, Rutschmann S, Xia Y, Lin P, Ross C, Li X, Smart NG, and Beutler B. 2010. Slc15a4, AP-3, and Hermansky-Pudlak syndrome proteins are required for Toll-like receptor signaling in plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. USA 107: 19973–19978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jui HY, Tseng RJ, Wen X, Fang HI, Huang LM, Chen KY, Kung HJ, Ann DK, and Shih HM. 2000. Protein-tyrosine phosphatase D1, a potential regulator and effector for Tec family kinases. J. Biol. Chem 275: 41124–41132. [DOI] [PubMed] [Google Scholar]
- 36.Ho HH, and Ivashkiv LB. 2006. Role of STAT3 in type I interferon responses. Negative regulation of STAT1-dependent inflammatory gene activation. J. Biol. Chem 281: 14111–14118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.