

Abstract
Type I protein arginine methyltransferases (PRMTs) catalyze asymmetric dimethylation of various proteins, and their dysregulations often correlate with tumorigenesis or developmental deficiency. Recent studies have focused on the in vivo substrate identification and the enzyme mechanism with peptide substrates. However, how PRMTs recognize substrates at the protein level remains unknown. PRMT8 is one of the least characterized type I PRMTs, and its crystal structure has not been reported. Here, we report the crystal structure of the PRMT8:SAH complex, identify a new non-histone protein substrate NIFK, and uncover a previously unknown regulatory region specifically required for recognizing NIFK. Instead of the canonical dimeric structure for other type I PRMTs, PRMT8 exists as a tetramer in solution. Using X-ray crystallography in combination with small-angle X-ray scattering experiments, the dimer of dimers architecture in which two PRMT8 dimers are held together mainly by β strand interactions was proposed. Mutation of PRMT8-β15 impedes the methylation of NIFK but still allows methylation of the histone H2A/H2B dimer or a peptide substrate, suggesting a possible structural basis for recognition of protein substrates. Lastly, we observed two PRMT8 dimer orientations resulting in open (without SAH) and closed (with SAH bound) conformations. The comparison between open and closed conformations may provide useful information for PRMT1/8 inhibitor design.
Graphical Abstract
Arginine methylation is a widespread protein posttranslational modification in eukaryotic organisms. Approximately 2% of the arginine residues in rat liver proteins are methylated.1 Methylation of arginine residues is mediated by a family of enzymes known as protein arginine methyltransferase (PRMTs).2 PRMTs catalyze the transfer of the methyl group from S-adenosyl-l-methionine (SAM) to the side-chain nitrogen atoms of arginine residue to form the methylated arginine and the coproduct, S-adenosyl-l-homocysteine (SAH).2 To date, nine PRMTs have been found in humans, all of which consist of at least one conserved catalytic core for the methyltransferase activity flanked by distinguishable N- and C-terminal sequences or domains.3–5 The members of the PRMTs can be classified into three types on the basis of the pattern of modification of the arginine residues. Type I PRMTs (including PRMT1, −3, −4, −6, and −8) generate asymmetric dimethylarginine (ADMA) as the final product.
Most PRMTs prefer highly positively charged glycine-arginine rich motifs as substrates.6 Structural studies of the catalytic core in complex with peptides reveal how the PRMT active site recognizes the arginine residue and the flanking residues.7–10 In addition, it was proposed that the acidic grooves on the surface surrounding the active site facilitate the binding to the positively charged peptide substrate.7,8 However, no direct evidence of how the PRMT catalytic core recognizes substrates at the protein level has been published. PRMT catalytic cores are flanked by various sequences, which often possess the architecture of a protein-protein interaction module, such as the SH3 domain in PRMT2, the zinc finger domain in PRMT3, the pleckstrin homology domain in PRMT4, and the TIM barrel domain in PRMT5.9,11–14 One study has shown that deletion of the zinc finger of PRMT3 reduces methyltransferase activity toward hnRNPA1 up to 4-fold relative to that of full length PRMT3.15 Hence, it is generally agreed that these flanking regions have an important role in protein substrate recognition.16 However, little evidence has shown that protein substrate specificity can be altered through the PRMT catalytic core.15,17
All type I PRMT catalytic cores adopt a characteristic head-to-tail homodimeric architecture that is essential for PRMT activity, and the deletion of the dimerization arm produces an inactive PRMT monomer.7,15,17,18 Crystallographic studies show that all type I PRMTs reveal a homodimer with the exception of yeast PRMT1 (Hmt1).7,15,18–20 Recent studies suggest that the substrate binding and methylation events occur complementarily and intermolecularly within the dimer (the cross-dimer mechanism), confirming the importance of head-to-tail dimer formation.17,21 However, larger oligomers of type I PRMTs are often observed by size exclusion chromatography, suggesting PRMTs can exist in assemblies larger than dimers.9,12,22–26 Because of the lack of structural information, this higher-order oligomeric state of type I PRMTs has been rarely discussed. However, the higher-order oligomeric state may be relevant to the heterointeractions among type I PRMT members. For example, the heteromeric interaction of PRMT1 and PRMT2 increases PRMT1 activity, and PRMT8 can recruit PRMT1 to the plasma membrane.27,28 A heterotetrameric model with intermember interactions between two type I PRMT dimers was suggested on the basis of this study.
Herein, we have determined the crystal structure of the human PRMT8 catalytic core in complex with SAH at 3.5 Å resolution and have identified a new protein substrate, an RNA binding protein called the nucleolar protein interacting with the forkhead-associated domain of Ki-67 (NIFK). We further demonstrated that strand β15 of the catalytic core is specifically required for NIFK methylation. In combination with small-angle X-ray scattering (SAXS) experiments, we propose a homotetrameric architecture of PRMT8, which may apply to other type I PRMT members and suggest a heterotetramer model for intermember interactions. Lastly, two dimer conformations upon SAH binding were observed. The dimer without SAH (open state) has a hinge angle of the dimerization arm wider than that of the SAH bound dimer (closed state). In the recent developments, PRMT3-specific inhibitors bind allosterically at the hinge region, resulting in a slightly wider hinge angle.29,30 Hence, our open conformation of PRMT8 may represent the allosterically inactive state. Compounds that occupy the hinge region may lock PRMT8 in the open conformation and function as allosteric inhibitors.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification.
The full length human PRMT8 (PRMT8) containing an N-terminal His6 tag was expressed in baculovirus using multibac vectors.31 Cells were pelleted and suspended in cold lysis buffer [50 mM HEPES (pH 7.4) and 100 mM sodium chloride] with 20 mM imidazole and lysed by sonication followed by centrifugation. The supernatant was loaded onto a Ni-NTA resin (GE Healthcare) and washed with the lysis buffer with 50–75 mM imidazole. The PRMT8 protein was eluted with the lysis buffer with 250 mM imidazole and immediately dialyzed to the desired buffer, purified by size exclusion chromatography, and concentrated to the required concentration.
The N-terminally truncated human PRMT8, tPRMT8 (residues 61–394), was subcloned into a pET-21 plasmid encoding an N-terminal His6 tag. Escherichia coli BL21-(DE3)-RIPL cells were transformed with the resulting tPRMT8 plasmid and grown at 37 °C in Terrific Broth. The culture was induced by 0.5 mM IPTG at an OD600 of 0.6 and incubated overnight at 18 °C. The purification procedure was similar to that for PRMT8.
On the basis of the sequence alignment (Figure S1), we engineered a chimera tPRMT8C by replacing β15 of tPRMT8 (GQLCETSVSNDYKMR) with Hmt1 (IDGNSRSRKNEGSY-LMH). Sequential polymerase chain reaction was performed to replace the sequence, and the final construct was confirmed by DNA sequencing. tPRMT8C was expressed and purified under the same condition as tPRMT8. The full length human NIFK and histone dimer H2A/H2B were obtained as previously described.32,33
Crystallization, Data Collection, and Structural Determination.
The tPRMT8 was concentrated to 7 mg/mL with 0.5 mM SAH under 50 mM ADA (pH 6.5), 100 mM sodium chloride, 20 mM arginine, 10 mM β-ME, and 10% glycerol. tPRMT8 was crystallized under 150 mM d-glucose, 100 mM HEPES/MOPS (pH 7.5), and 40% glycerol/PEG 4000 using microseeding techniques and the hanging-drop vapor diffusion method at 20 °C. The crystal was cryocooled in a stream of nitrogen at 100 K. X-ray diffraction data were collected at beamline 15A1 of the National Synchrotron Radiation Research Center (NSRRC) on an MX300HE detector at 100 K and processed using the HKL2000 program suite.34 The structure was determined using the molecular replacement method to calculate phases using the structure of mPRMT1 [Protein Data Bank (PDB) entry 1ORI] as the search model with the program Molrep in the CCP4 suite.35 The structure was iteratively rebuilt in COOT and refined in Refmac5.36,37 Data processing and refinement statistics are summarized in Table S1. The electron density map of the Arg229–Trp238 region (part of the dimerization arm) of the SAH-bound form is poor, and that region was modeled mostly on the basis of geometric restraints.
Characterizing PRMT8 Oligomers by Size Exclusion Chromatography.
The tPRMT8 was concentrated to ~1 mg/mL and loaded on a Superdex-200 column (GE Healthcare) with the buffer pH ranging from 6.5 to 8.5 (ADA at pH 6.5, HEPES at pH 7.5, and Tris at pH 8.5). In addition, the sample was also separately eluted with a buffer comprising 50 mM HEPES (pH 7.5) but with 100–1000 mM sodium chloride.
Characterizing PRMT8 Oligomers by Analytical Ultracentrifugation.
After tPRMT8 and tPRMT8C had been subjected to size exclusion chromatography, the samples were concentrated to the desired concentration using Centricon centrifugal filter devices. Sedimentation velocity ultracentrifugation was performed using an XL-A analytical ultracentrifuge (Beckman Coulter, Fullerton, CA) with absorption optics and an AnTi60 rotor with cells containing quartz windows. Radial absorbance data at 280 nm were acquired at 1 min intervals with a rotor speed of 60000 rpm at 20 °C. The buffer density and viscosity were calculated with SEDNTERP,38 and the data were analyzed with SEDFIT.39
Small-Angle X-ray Scattering.
The sample of tPRMT8 was purified as described above and concentrated to 5 mg/mL in the same buffer condition that was used for crystallization. The SEC-SAXS experiments were performed on Bio-SAXS beamline 4–2 at Stanford Synchrotron Radiation Lightsource (SSRL).40 All data were collected on a Rayonix (Evanston, IL) MX225-HE CCD detector with a 2.5 m sample-to-detector distance, and a beam energy of 11 keV (wavelength λ = 1.127 Å) was used. The momentum transfer (scattering vector) q was defined as q = 4π sin(θ)/λ, where 2θ is the scattering angle. The q scale was calibrated by silver behenate powder diffraction,41 and all data were collected up to a maximum q of 0.46 Å−1. The details of the SEC-SAXS experiment at BL4–2 were described previously.42–44 For the SEC step, a 100 μL sample of 5.0 mg/mL tPRMT8 was applied to a Superdex 200PC3.2/30 instrument (GE Healthcare). The data acquisition program Blu-ICE45,46 was employed for data collection, and the data processing program SasTool (http://ssrl.slac.stanford.edu/~saxs/analysis/sastool.htm) was used for scaling, azimuthal integration, and averaging of individual scattering images after inspection for any variations potentially caused by radiation damage. The early parts of the void volume were averaged and used as a buffer scattering profile for background subtraction. The scattering profiles at the last half of an elution peak were averaged, and pairwise distribution functions P(r) were calculated up to q = 0.18 using the program GNOM in the ATSAS package.47 The program DAMMIF was employed for ab initio modeling.48 The 20 independent DAMMIF calculations were performed with P2 symmetry, and the results were averaged by the program DAMAVER.49 The results were reproducible with an average normalized spatial discrepancy (NSD) of 0.849. Curve fitting with the tPRMT8 tetramer model was performed using FOXS.50,51 Data collected and parameters are summarized in Table S2.
Electrophoresis of EGFP-Tagged PRMT8 Proteins.
The 293T cell line was maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Life Technologies). The 293T cells were transfected with the pECFP-C1 vector encoding GFP-tPRMT8. The cleared lysates were subjected to immunoprecipitation procedures with an anti-GFP antibody (Roche) that was covalently cross-linked on Dynabeads (Life Technologies). The beads were then washed with RIPA lysis buffer, and the immunoprecipitated proteins were eluted with 1 M NaCl/PBS followed by electrophoresis and immunoblotting. The Native-Mark unstained protein standard (Thermo Fisher Scientific) was used for the molecular weight estimation in the native-PAGE.
MS Analysis for Methylation.
The methods for in vivo NIFK methylation detection are similar to those described previously.32 The RGG peptide (based on the nucleolin sequence) was incubated with PRMT8 with SAM (Sigma-Aldrich) for MADL-MS analysis. MADL-MS analyses were conducted with an Autoflex III MALDI-TOF/TOF mass spectrometer equipped with a 200 Hz SmartBeam Laser (Bruker Daltonik, Bremen, Germany) in the positive ionization and linear mode in the m/z range of 4000–20000. A protein mixture of insulin, ubiquitin, cytochrome c, and myoglobin (Protein calibration Mix I, Bruker Daltonik) was used for external calibration of the mass spectra. Data were acquired manually with FlexControl 3.4, and data processing was performed with Flex-Analysis 3.4 (both from Bruker Daltonik).
In Vitro Methylation Activity Assay.
The recombinant H2A/His-tagged H2B dimer and NIFK are produced on the basis of previously reported protocols.32,33 After incubation of the NIFK and histone H2A/H2B with PRMTs in the presence of [3H]AdoMet in 50 mM Tris (pH 8) and 2.5 mM DTT at 37 °C, the samples were separated by electrophoresis. The methylation is detected by fluorogram using EN3HANCE (PerkinElmer).
RESULTS
Overall Structure of PRMT8 and SAM Binding Site.
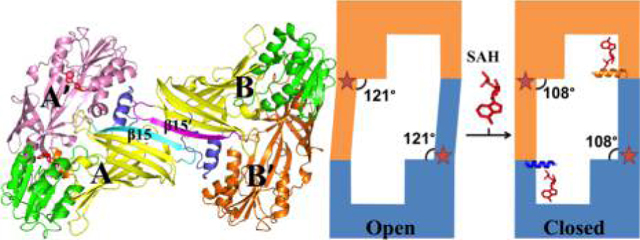
For this study, two constructs were generated: full length PRMT8 (PRMT8) and a version with the first 60 amino acids truncated, PRMT861–394 (tPRMT8, PRMT8 catalytic core). tPRMT8 was pursued because the N-terminal sequence was predicted to be flexible and unfavorable to protein crystallization. The crystal structure of tPRMT8 was determined at 3.5 Å resolution (PDB entry 4X41). The structure revealed that the PRMT8 catalytic core adopted a conserved N-terminal Rossmann fold domain and C-terminal β barrel domain where the dimerization arm is located (Figure 1A). The PRMT8 structure is highly similar to the well-studied PRMT1 structure, so the same nomenclature is used for the secondary structure elements, except that strand β12 that breaks into two short strands (β12 and β12′) and helices αE and αF are missing.7 Two monomers are present in the crystallographic asymmetric unit and share a similar conformation, but with the exception that molecule A is in complex with SAH while molecule B is in the apo form (Figure 1B). The application of crystallographic symmetry can generate two characteristic PRMT head-to-tail dimers (1, A/A′; 2, B/B′) with an average dimer interface area of 1163 Å2 [calculated by the PISA server52 (Figure 1B,C)]. In our crystal structure analysis, the two dimer subunits can be packed to form a tetramer through strand-strand interaction by β15 with a buried surface area of 936 Å2 (Figure 1B,C). The proposed tetramer structure was further validated by SAXS experiments (discussed below). This is the first reported structural observation of type I PRMTs in a tetrameric state.
Figure 1.
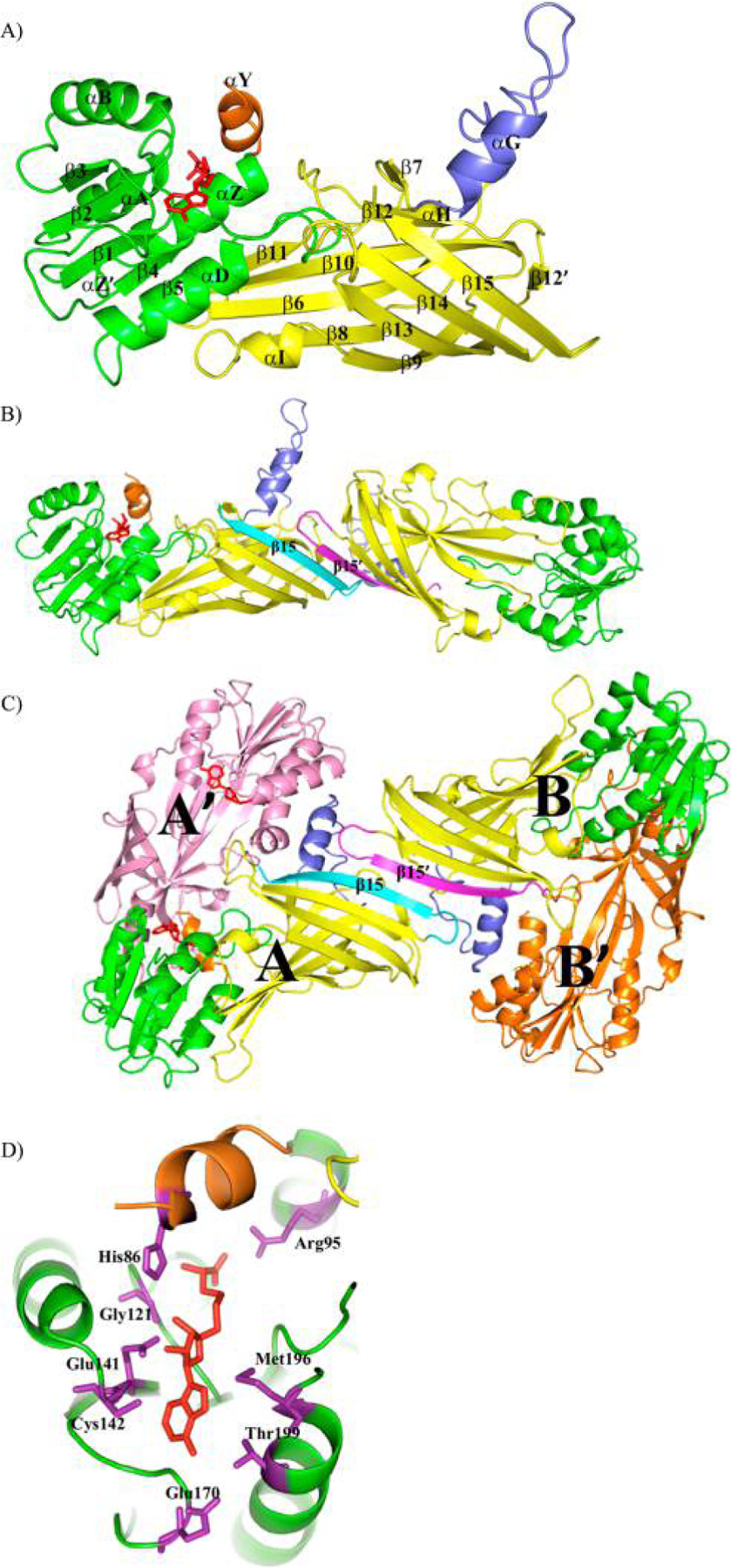
Structure of tPRMT8. (A) Overall structure of the tPRMT8 monomer. Each α helix and each β strand are labeled accordingly. The SAH is shown as red sticks to show the active site region. The Rossmann fold and the β barrel domain are colored green and yellow, respectively. The dimerization arm is colored blue, and the N-terminal α helix is colored brown. (B) Asymmetric unit of tPRMT8 containing two monomers. Each monomer is colored the same as in panel A and connected by β15, which is colored cyan or magenta. The SAH (in red stick) is present in monomer A but absent in monomer B. (C) Proposed tetrameric architecture of tPRMT8. Molecules A and B are colored the same as in panel B. Molecules A and A′ (pink) form a dimer with SAH (red) in the active site, whereas molecules B and B′ (orange) are free of SAH. The two β15 strands that connect two dimers are labeled. Molecules A′ and B′ are generated by crystallographic symmetries. (D) Focused view of the SAH binding sites. The color scheme is the same as in panel A, but the key residues that are in contact with SAH are labeled and shown as purple sticks.
Our structure also delineates how the conserved residues are coordinated with SAM in PRMT8. Helix αY is observed only in the presence of SAH (Figure 1A). The corresponding region (residues 84–91) of the apo form is not visible in the electron density map. Helix αY functions coherently with other key residues for methylation activities as demonstrated in other PRMTs.7,18,19,53 Like His45 in PRMT1, residue His86 on helix αY together with other residues presents a conserved SAM binding site (Figure 1D). These residues are highly conserved and can be grouped on the basis of the previous studies of PRMT1:7 (1) His86 (helix αY), Arg95 (helix αZ), and Gly121 (strand β1) contact the L-homocysteine of SAH; (2) Glu141 (strand β2) is in close contact with the ribose hydroxyl groups; and (3) Cys142 (strand β2), Glu170 (after β3), Met196 (helix αD), and Thr199 (helix αD) are in the proximity of the adenine.
Conformational Change upon SAH Binding.
Our crystal structure displays two different forms of PRMT8. One is in complex with SAH, and the other is free of SAH. Two significant differences between these two PRMT8 forms are observed. The first difference is the formation of helix αY upon SAH binding. N-Terminal helix formation upon SAH binding was also observed previously in structural studies of PRMT4.12 Helix αY is required to create a solvent-excluded active site pocket for catalysis, and mutations of helix αY can abolish PRMT activity.54 It was proposed that SAH-induced rearrangement for catalysis might be a common feature among type I PRMTs.19 Our observation is consistent with the previous studies that SAM (or SAH) binding induces a conformational change in this N-terminal region, a step toward active state formation to a catalytically competent conformation.19,54
The other significant difference is that the dimerization arm shifts by 13° resulting in two different dimer orientations upon SAH binding (Figure 2A,B). A similar phenomenon of a 15° shift in the dimerization arm upon SAH binding was observed with PRMT4.12 The dimer interface consists of an outer hydrophobic surface near helix αY and two dimerization arm residues (Tyr243 and Phe245) from the adjacent monomer (Figure 2C). In the absence of helix αY (no SAH binding), Tyr243 and Phe245 from the adjacent molecule are in close contact with Thr96, Ile124, Met127, and Phe128 (residues involved in the outer hydrophobic surface). Upon SAH binding, the formation of helix αY (residues 84–91) allows the backbone of Leu90 to hydrogen bond with Tyr243 (Figure 2C). Although Thr96, Ile124, Met127, and Phe128 are still in close contact with Tyr243 and Phe245, the dimerization arm is reoriented and the PRMT8 dimer conformation is spatially rearranged (Figure 2C). From our structural observation, the “open” and “closed” forms (based on the hinge angle of the dimerization arm) of PRMT8 upon SAM binding are proposed (Figure 2D). We may rule out the possibility that the conformational rearrangement is caused by the crystal packing because a similar type of conformational rearrangement is also observed in PRMT4.
Figure 2.
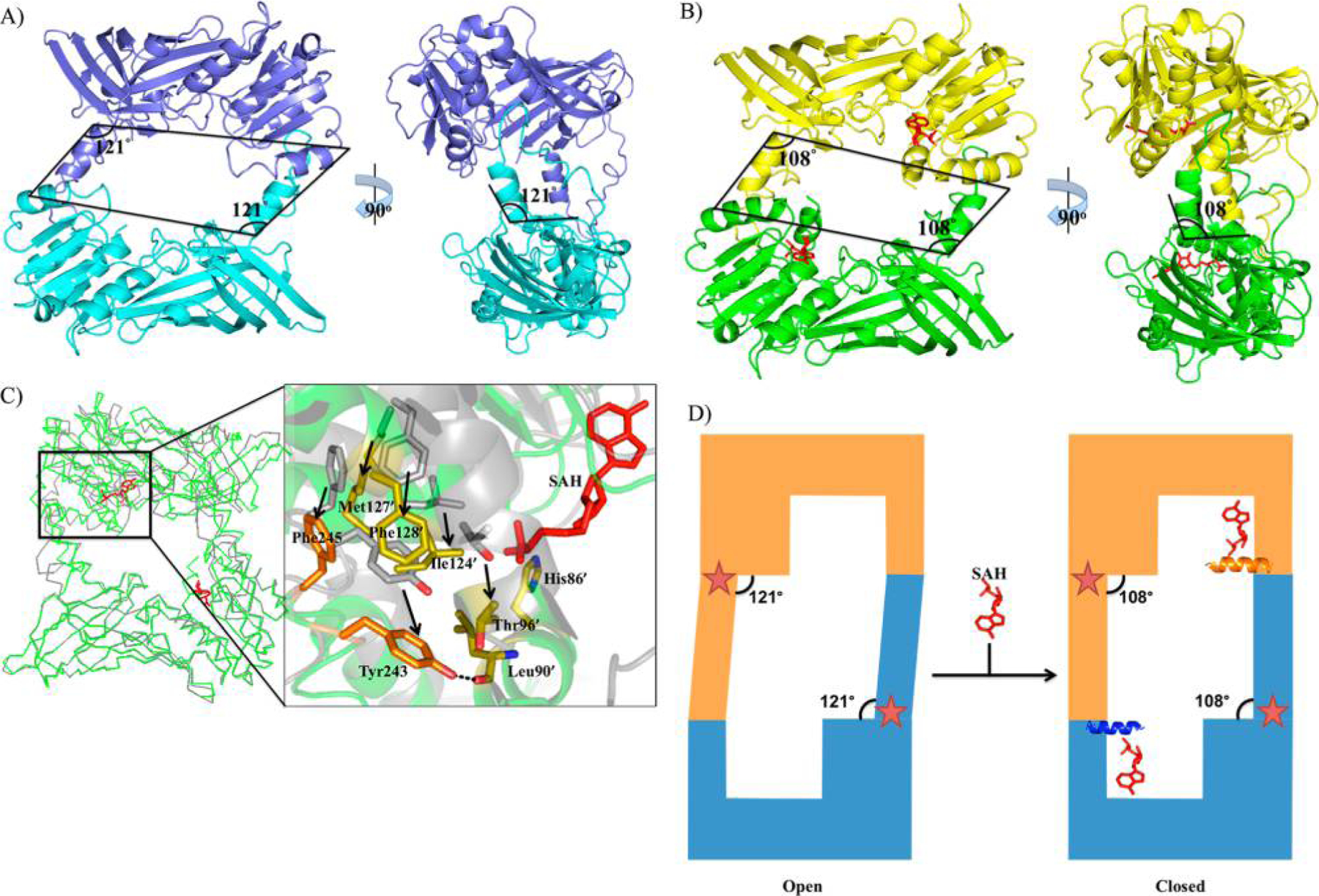
Conformational change upon SAH binding. (A) Open conformation of the tPRMT8 dimer (apo). The crystallographic symmetry was applied to generate the tPRMT8 dimer, and each molecule is colored cyan and blue. The angle of the dimerization arm in both views is 121°. (B) Closed conformation of the tPRMT8 dimer (SAH-bound). The dimer of tPRMT8 is shown as a green and yellow cartoon with SAH shown as red sticks. The angle of the dimerization arm in both views is 108°. The SAH molecules are shown as red sticks. (C) Reorientation of the dimerization interface upon SAH binding. The open and closed conformations are shown as gray and green ribbons, respectively. The SAH molecules are shown as red sticks. The key residues in the dimer interface of the closed conformation are labeled and shown as orange sticks. The key residues from the adjacent PRMT8 monomer in the dimer interface of the closed conformation are labeled with an apostrophe and shown as yellow sticks. The hydrogen bond between Tyr243 and the backbone of Leu90 is shown as a dotted line. The corresponding residues in the open conformation are labeled in gray and shown as gray sticks. Arrows indicate the movements of the residues. (D) Schematic of the dimerization arm movement. In the presence of SAH, the conformation of the dimer changes from open to closed where an additional α helix is observed upon SAH binding and provides additional contacts to the dimerization arm as the result of a bending of the dimerization arm. The proposed pocket (the hinge region) for allosteric inhibitors is represented by the stars.
tPRMT8 Homotetramerization.
The characteristic PRMT head-to-tail dimer is essential for enzymatic activity and is observed in our crystal structure.7,15,18 However, on several occasions, higher-order oligomerization states of PRMTs were also observed in solution.9,12,22–26 Our PRMT1 catalytic core construct behaves as a tetramer during size exclusion chromatography (data not shown). Previously, the only structural evidence of higher-order oligomerization in type I PRMTs is in the yeast PRMT1 (Hmt1) that reveals a concentration-dependent hexamer (a trimer of dimers), but the function of hexamer formation remains unclear.18 In the case of the tPRMT8 studied here, a single peak was observed via size exclusion chromatography (SEC) and the major species of tetrameric tPRMT8 was confirmed by analytical ultracentrifugation (AUC) (Figure S2 and Figure 3A). Unlike the hexameric Hmt1, which can be disrupted by a high salt concentration, our tPRMT8 tetramer is observed in up to 1 M sodium chloride and at various pH values [pH 6.5–8.5 (Figure S2)].
Figure 3.
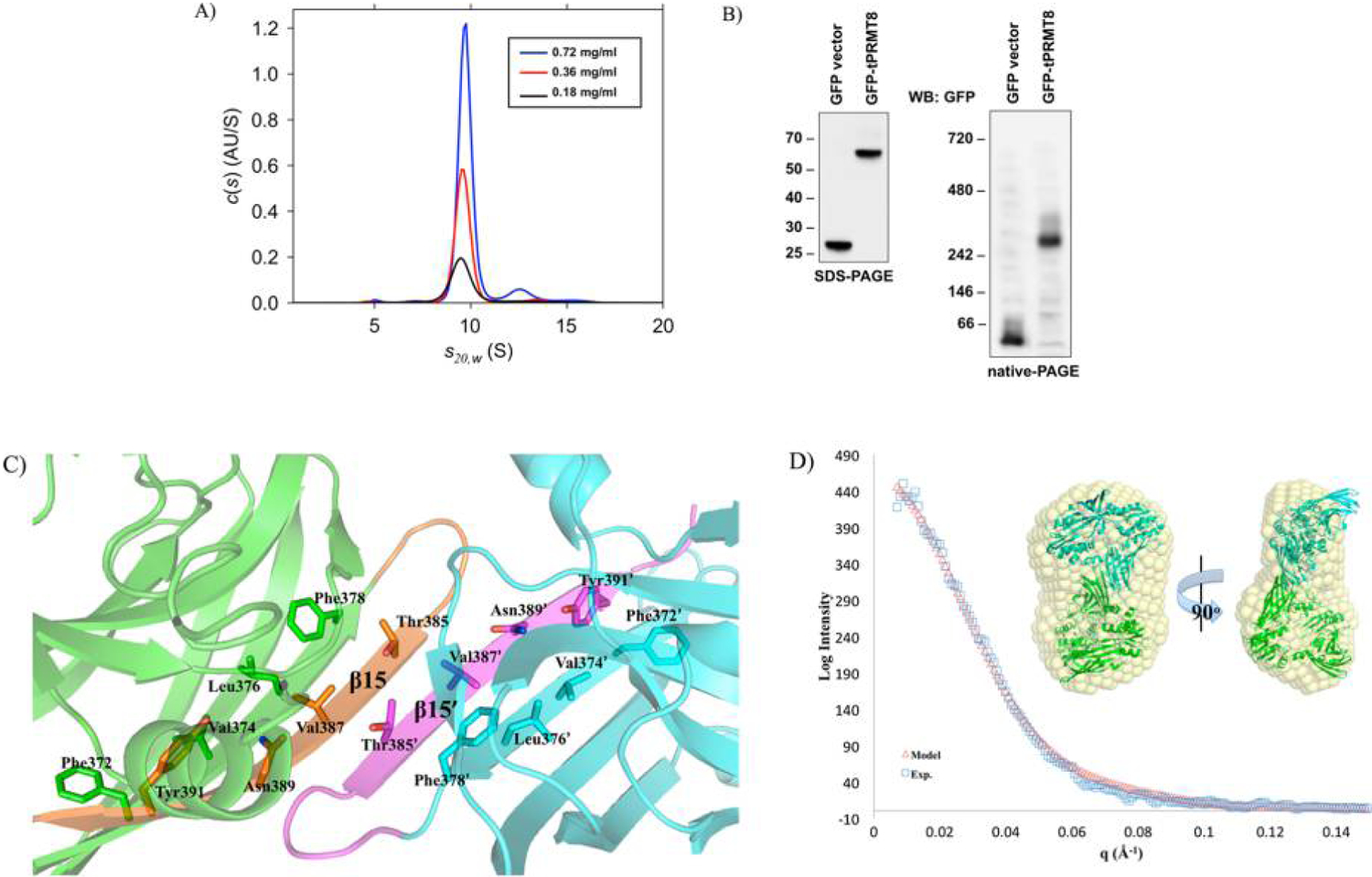
Homotetramerization of tPRMT8. (A) Formation of the tPRMT8 tetramer. The AUC data indicate the presence of tetrameric tPRMT8 in the concentration range of 0.18–0.72 mg/mL. (B) tPRMT8 tetramerization in vivo. The purified EGFP and EGFP-tPRMT8 proteins from 293T cells were separated by (left) SDS-PAGE and (right) native-PAGE and detected by Western blotting with an anti-GFP antibody. EGFP-tPRMT8 shows one major oligomerization state (tetramer). (C) The β15–β15′ interactions connect hydrophobic cores of two β barrels. Two PRMT monomers are shown as blue and green cartoons. The corresponding β15 strands are colored magenta and orange. The residues of β14 and β15 that are oriented into the interior of the β barrel are shown as sticks and labeled. (D) Homotetramer model of tPRMT8 and the SAXS profile. The theoretical SAXS curve of the tPRMT8 tetramer from crystallography is calculated and matches the experimental SAXS curve. The homotetramer model is built with each tPRMT8 dimer colored cyan and green and docked with the most populated volume in solution generated by DAMMIF and DAMAVER (yellow sphere).
To investigate the tetramerization of tPRMT8 in vivo, we used electrophoresis to evaluate the size of tPRMT8 with an EGFP tag that is expressed in 293T cells. The result shows that GFP-tPRMT8 is slightly less than 70 kDa as determined by SDS-PAGE and migrates to a slightly above 242 kDa under native-PAGE conditions, consistent with the expected size of a tPRMT8 tetramer (Figure 3B).
Tetrameric Architecture of tPRMT8.
In our crystal structure, two tPRMT8 dimer subunits can be packed together to form a homotetramer via β15–β15′ interactions (Figure 1C). The β15 strand is the last β strand of the conserved β barrel where hydrophobic residues are oriented into the interior to form a hydrophobic core. The β15–β15′ interactions connect two hydrophobic cores (Figure 3C). In addition, we identify 13 hydrogen bonds within the tetrameric interface. Most of the residues contributing to those hydrogen bonds are located on the β15 strand, but a few hydrogen bonds are formed by residues located on strand β10, strand β13, and the dimerization arm. To show that the proposed tetramer is not an artifact of the crystal packing, we performed small-angle X-ray scattering (SAXS) analysis. The theoretical Bio-SAXS curve matches with the experimental data with a χ score of 1 (Figure 3D). An ab initio model calculated with DAMMIF also agrees with the tetramer model, indicating that our tPRMT8 tetramer model is a genuine representative of the solution state of the protein.
Identification of a New PRMT1/8 Substrate.
RNA binding proteins are frequently methylated by PRMTs; in particular, Ewing sarcoma protein (EWS) was previously reported as a protein substrate of PRMT8.55–58 The “nucleolar protein interacting with the forkhead-associated (FHA) domain of Ki-67” (NIFK) is a RNA binding protein, which is required for large subunit ribosomal RNA maturation.32,59,60 Sequence analysis shows several arginine rich regions in NIFK, which indicate a potential PRMT substrate. Our in vitro methylation studies demonstrated that NIFK is a substrate of PRMT8, tPRMT8, and PRMT1 (Figure 4A,B). We postulate that full length PRMT8 has methyltransferase activities lower than that of tPRMT8 because the N-terminus inhibits activity.53 Mass spectrometry analysis (64% sequence coverage) showed that arginine residues Arg114, Arg244/Arg245, and Arg284 in NIFK are methylated by tPRMT8 (Figure S3). We further confirmed that ectopic NIFK R244 is methylated in HEK293 cells (Figure 4C). Thr234 and Thr238 phosphorylation in NIFK is critical for interaction of FHA with Ki-67, and Arg244 is in the proximity of Thr238.61 Although the correlation of methylation and phosphorylation of NIFK has yet to be explored, a study has suggested a significant influence between these two posttranslational modifications.62
Figure 4.
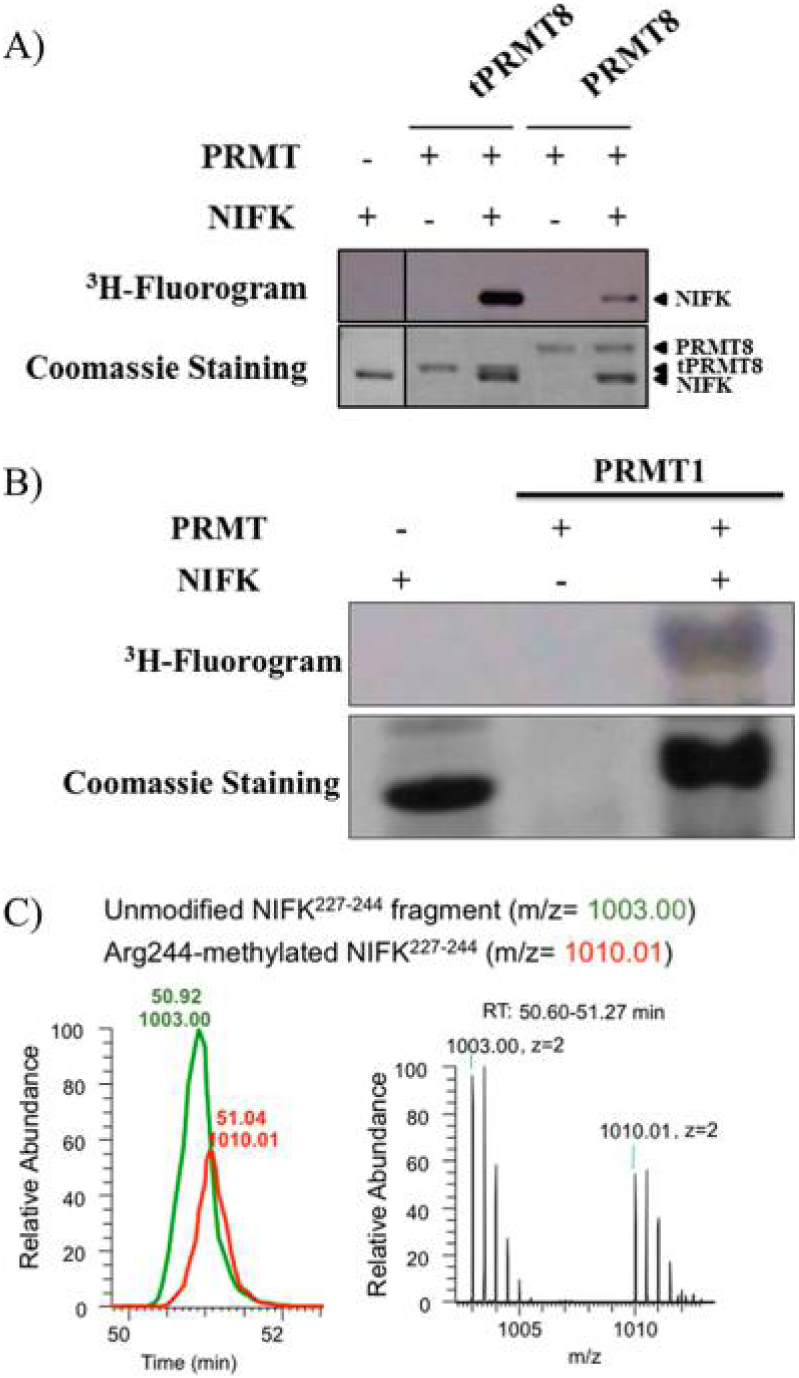
NIFK as the protein substrate of PRMT1/8. (A) In vitro NIFK methylation by PRMT8 and tPRMT8. 3H fluorography indicates that NIFK is a methylation target of recombinant PRMT8 and tPRMT8. (B) In vitro NIFK methylation by PRMT1. 3H fluorography indicates that NIFK is a methylation target of the recombinant PRMT1. (C) LC-MS trace of tryptic NIFK that is ectopically expressed in HEK293. Monoisotopic peaks extracted from ion chromatograms (left, with a 15 ppm tolerance) reveal the relative level of the unmodified tryptic NIFK227–244 (TVDSQGPTPVCTPTFLER, m/z 1003.0) fragment and the Arg244 methylated fragment (m/z 1010.01). The corresponding precursor mass spectral scans (right) from 50.60 to 51.27 min of retention time (RT) are illustrated at the right.
A Novel Regulatory Site for PRMT8 Substrate Selectivity.
The catalytic core is highly conserved within type I PRMTs and the yeast homologue Hmt1. Notably, the last β strand (β15) in PRMT1, PRMT8, and Hmt1 displays a rather diverse sequence (Figure S1). β15 is located away from the PRMT active site (Figure 1A). It was reported that the deletion of the last β strand of Hmt1 reduces the level of methylation of Npl3.17,18 The last β strand is part of the conserved β barrel of PRMTs, so the deletion may cause global structural rearrangement or protein instability that impedes PRMT activity. To test the role of the β15 strand, we engineered the chimera tPRMT8C by replacing β15 of PRMT8 with that of Hmt1 to minimize the structural rearrangement. The major species of tPRMT8C remains a tetramer, and the AUC spectrum overlaid well with that of the wild type (Figure S4A). Circular dichroism confirmed similar secondary structure content between tPRMT8C and tPRMT8 but with lower temperature stability for tPRMT8C (Figure S4B). In addition, we observed that the replacement of β15 still allowed the methylation of the RGG peptide and histone H2A/H2B dimer, suggesting the active site of tPRMT8C remains intact (Figure 5A,B). Surprisingly, we did not observe NIFK methylation by tPRMT8C (Figure 5C).
Figure 5.
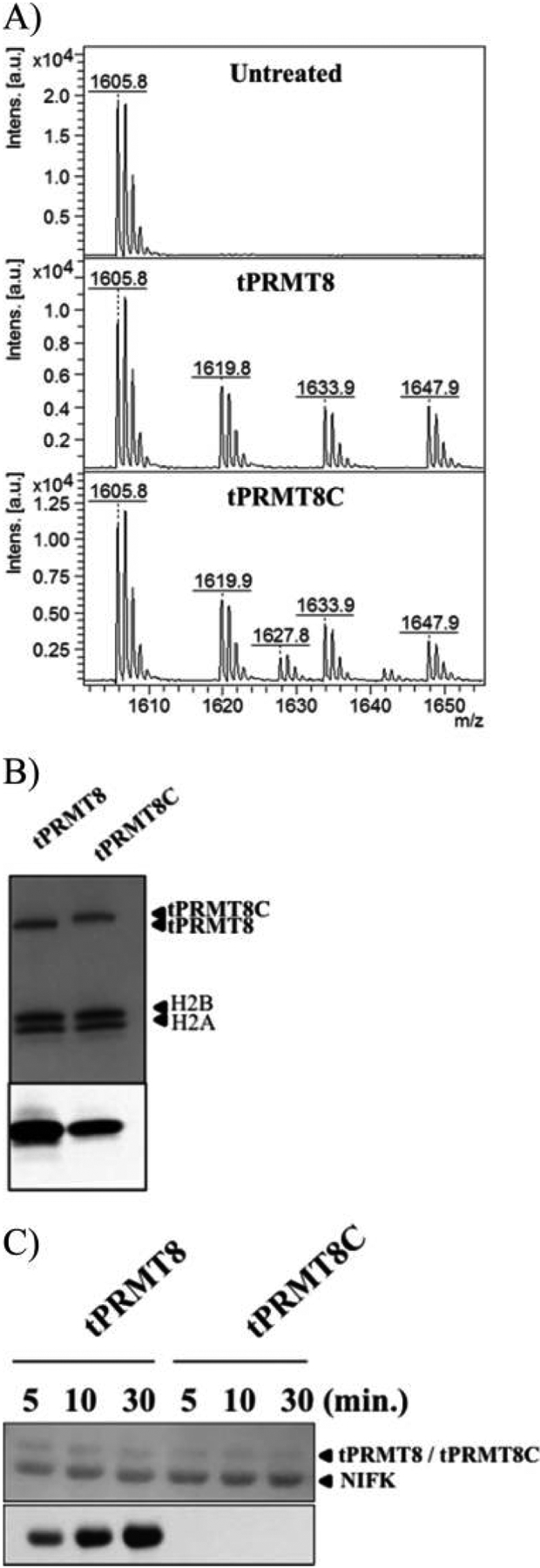
Substrate specificity of PRMT8 methylation activity. (A) RGG peptide methylation of tPRMT8 and tPRMT8C. The RGG peptide (GRGGFGGRGGFRGGRGG) has three arginine residues. The MALDI-MS data indicate the shift of the methylated peptide is in multiples of 14, which is the molecular weight of a methyl group. (B) Methylation of the H2A/H2B dimer by tPRMT8 and tPRMT8C. The SDS-PAGE Coomassie staining (top) and 3H fluorogram (bottom) demonstrate that PRMT8 and tPRMT8 methylate H2A/H2B in a similar fashion. (C) Methylation assay of tPRMT8 and tPRMT8C against NIFK. The SDS-PAGE Coomassie staining and 3H fluorogram demonstrate that tPRMT8 can methylate NIFK and increases with time but tPRMT8C failed to methylate NIFK in vitro.
DISCUSSION
In this study, we show that tPRMT8 exists as a homotetramer in solution and provide a dimer-of-dimers model. Although type I PRMTs are widely considered to be dimers, larger type I PRMT oligomers in solution have been reported previously.9,12,22–26 The PRMT8 catalytic core constructed and presented herein behaved as a tetramer during size exclusion chromatography and AUC. In our proposed dimer-of-dimers model, β15 plays a vital role in the assembly of the tetramer. Because β15 is structurally conserved among type I PRMTs, this PRMT8 tetramer model may apply to other type I PRMTs.7,12,15,63 In addition, it has been reported that PRMT1 can interact with other isoforms. For example, the recruitment of PRMT2 can increase PRMT1 activity and PRMT8 can bring PRMT1 to the plasma membrane.27,28,64 Our tetramer architecture may provide a novel perspective of the PRMT heteromolecular interactions. In our proposed model, two different type I PRMT dimers form a heterotetramer via the β15–β15′ interactions.
Previously, the deletion of β15 of Hmt1 weakens its methylation activities.18 To minimize the structural alternation caused by the β15 deletion, we replaced tPMRT8 β15 with yeast PRMT1 (Hmt1) β15. Size exclusion chromatography, analytic ultracentrifugation, and circular dichroism suggest the chimeric PRMT8 (tPRMT8C) is structurally similar to wild-type tPRMT8. In addition, tPRMT8C was still able to methylate the RGG peptide and histone H2A/H2B dimer, suggesting the strand swap does not cause major changes in PRMT8 catalytic activity. Surprisingly, the mutation causes a dramatic reduction in the level of methylation of the protein substrate, NIFK, suggesting that β15 is important in NIFK recognition. PRMTs are known to methylate various protein substrates in vivo despite structural differences. The mechanism of how PRMTs interact with protein substrates and how this relates to target selectivity remains an enigma. In the case of PRMT5 (a type II PRMT), several protein cofactors are involved in protein substrate recognition and/or selectivity. However, no protein cofactors have yet been identified for type I PRMTs. This exposes a question of how type I PRMTs interact with target proteins, and more importantly how the protein substrate specificity is regulated. It is generally hypothesized that the unique flanking region of type I PRMTs that usually contains a protein interaction module is involved in protein recognition.16 Our findings lend credence to our hypothesis that β15 of the PRMT catalytic core is a critical motif involved in substrate recognition at the protein level.
With the promise of PRMTs being cancer targets, a plethora of inhibitors have been developed. Most PRMT inhibitors target the structurally conserved SAM binding site, often resulting in nonspecific inhibition. Targeting alternative sites may be a better approach. The PRMT3 allosteric inhibitors that target the hinge of the dimerization arm, a recent development, are isoform-specific because of the diversity of sequence and structure at the hinge region.29,30 PRMT3 reveals a relatively open conformation upon allosteric inhibitor binding similar to the conformer observed by us for tPRMT8.29,30 Hence, the open conformation of tPRMT8 observed in the study herein may represent the allosteric state of the catalytically incompetent PRMT8. PRMT3 allosteric inhibitors cannot inhibit PRMT8.30 The superimposition of the two isoforms shows steric clash between the PRMT3 inhibitor and the side chain of Tyr305 in PRMT8 (Figure 6). A structure-based inhibitor design approach is being conducted currently to discover PRMT8-specific inhibitors that can occupy the hinge region. Given the high level of similarity between PRMT1 and PRMT8 and the fact that the residues located at the hinge region are conserved, the PRMT8 inhibitors developed by structural approaches may also inhibit PRMT1, one of the most important targets for anticancer therapeutics.
Figure 6.
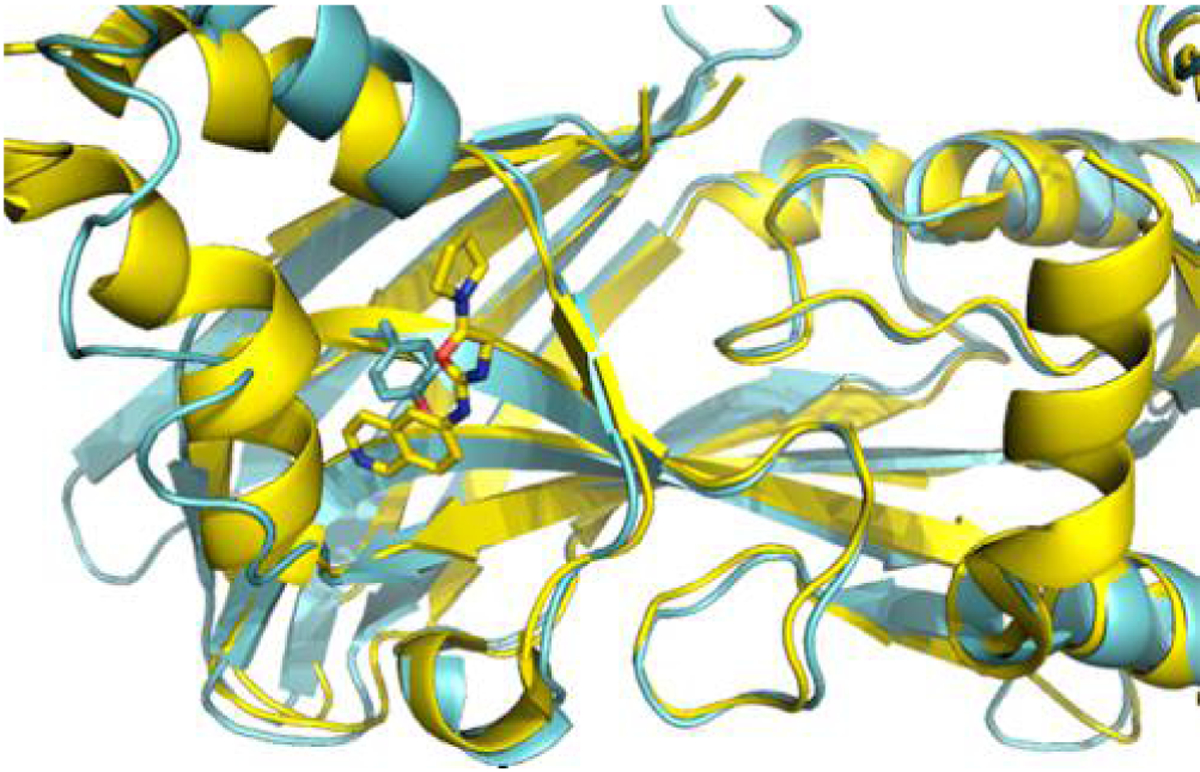
Superimposition of PRMT3 and PRMT8. The tPRMT8 monomer is superimposed with PRMT3 (PDB entry 4RYL in the presence of the allosteric inhibitor, 3ZG). tPRMT8 and PRMT3 are shown as blue and yellow cartoons, respectively. The side-chain Tyr305 and the PRMT3 allosteric inhibitor, 3ZG, are shown as sticks. There is a steric clash between PRMT8 Tyr305 and the PRMT3 allosteric inhibitor.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Academia Sinica Common Mass Spectrometry Facilities, the IMB Mass spectrometer facility, and NSRRC Beamline 23A for their help with LC-ESI-MS, MALDI-MS, and SAXS experiments, respectively. We thank the Biophysical Instrumentation Laboratory at the Institute of Biological Chemistry, Academia Sinica, Taiwan, for their assistance with AUC service. Portions of this research were conducted at the National Synchrotron Radiation Research Center, a national user facility supported by the National Science Council of Taiwan, ROC. The Synchrotron Radiation Protein Crystallography Facility is supported by the National Core Facility Program for Biotechnology. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including Grant P41GM103393).
Funding
This work was supported by the Ministry of Science and Technology, ROC (Grants 102-2628-B-001-008-MY3 and 103-2811-B-001-095) and partly by Academia Sinica (Thematic Project AS-101-TP-B02 to M.-D.T.). Y.G.Z. acknowledges financial support by National Institutes of Health Grant R01GM086717.
ABBREVIATIONS
- PRMT
protein arginine methyltransferase
- SAH
S-adenosyl-l-homocysteine
- SAM
S-adenosyl-l-methionine
- ADMA
asymmetric dimethylarginine
- NIFK
nucleolar protein interacting with the forkhead-associated domain of Ki-67
- SAXS
small-angle X-ray scattering
- SEC
size exclusion chromatography
- tPRMT8
N-terminally truncated human PRMT8
- MS
mass spectrometry
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- PBS
phosphate-buffered saline
- RIPA buffer
radioimmunoprecipitation assay buffer
- AUC
analytical ultracentrifugation
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-chem.5b00995.
Tables and additional figures (PDF)
Additional materials and methods (PDF)
REFERENCES
- (1).Boffa LC, Karn J, Vidali G, and Allfrey VG (1977) Distribution of NG, NG,-dimethylarginine in nuclear protein fractions. Biochem. Biophys. Res. Commun 74, 969–976. [DOI] [PubMed] [Google Scholar]
- (2).Wolf SS (2009) The protein arginine methyltransferase family: an update about function, new perspectives and the physiological role in humans. Cell. Mol. Life Sci 66, 2109–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bedford MT, and Clarke SG (2009) Protein arginine methylation in mammals: who, what, and why. Mol. Cell 33, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Yang Y, and Bedford MT (2012) Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 13, 37–50. [DOI] [PubMed] [Google Scholar]
- (5).Osborne TC, Obianyo O, Zhang X, Cheng X, and Thompson PR (2007) Protein arginine methyltransferase 1: positively charged residues in substrate peptides distal to the site of methylation are important for substrate binding and catalysis. Biochemistry 46, 13370–13381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bedford MT (2007) Arginine methylation at a glance. J. Cell Sci 120, 4243–4246. [DOI] [PubMed] [Google Scholar]
- (7).Zhang X, and Cheng X (2003) Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure 11, 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hasegawa M, Toma-Fukai S, Kim JD, Fukamizu A, and Shimizu T (2014) Protein arginine methyltransferase 7 has a novel homodimer-like structure formed by tandem repeats. FEBS Lett. 588, 1942–1948. [DOI] [PubMed] [Google Scholar]
- (9).Antonysamy S, Bonday Z, Campbell RM, Doyle B, Druzina Z, Gheyi T, Han B, Jungheim LN, Qian Y, Rauch C, Russell M, Sauder JM, Wasserman SR, Weichert K, Willard FS, Zhang A, and Emtage S (2012) Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. U. S. A 109, 17960–17965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kolbel K, Ihling C, Bellmann-Sickert K, Neundorf I, Beck-Sickinger AG, Sinz A, Kuhn U, and Wahle E (2009) Type I Arginine Methyltransferases PRMT1 and PRMT-3 Act Distributively. J. Biol. Chem 284, 8274–8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Frankel A, and Clarke S (2000) PRMT3 is a distinct member of the protein arginine N-methyltransferase family. Conferral of substrate specificity by a zinc-finger domain. J. Biol. Chem 275, 32974–32982. [DOI] [PubMed] [Google Scholar]
- (12).Troffer-Charlier N, Cura V, Hassenboehler P, Moras D, and Cavarelli J (2007) Functional insights from structures of coactivator-associated arginine methyltransferase 1 domains. EMBO J. 26, 4391–4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Meyer R, Wolf SS, and Obendorf M (2007) PRMT2, a member of the protein arginine methyltransferase family, is a coactivator of the androgen receptor. J. Steroid Biochem. Mol. Biol 107, 1–14. [DOI] [PubMed] [Google Scholar]
- (14).Krause CD, Yang ZH, Kim YS, Lee JH, Cook JR, and Pestka S (2007) Protein arginine methyltransferases: evolution and assessment of their pharmacological and therapeutic potential. Pharmacol. Ther 113, 50–87. [DOI] [PubMed] [Google Scholar]
- (15).Zhang X, Zhou L, and Cheng X (2000) Crystal structure of the conserved core of protein arginine methyltransferase PRMT3. EMBO J. 19, 3509–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Schapira M, and Ferreira de Freitas R (2014) Structural biology and chemistry of protein arginine methyltransferases. MedChemComm 5, 1779–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Li HT, Gong T, Zhou Z, Liu YT, Cao X, He Y, Chen CD, and Zhou JQ (2015) Yeast Hmt1 catalyzes asymmetric dimethylation of histone H3 arginine 2 in vitro. Biochem. J 467, 507–515. [DOI] [PubMed] [Google Scholar]
- (18).Weiss VH, McBride AE, Soriano MA, Filman DJ, Silver PA, and Hogle JM (2000) The structure and oligomerization of the yeast arginine methyltransferase, Hmt1. Nat. Struct. Biol 7, 1165–1171. [DOI] [PubMed] [Google Scholar]
- (19).Wang C, Zhu Y, Chen J, Li X, Peng J, Chen J, Zou Y, Zhang Z, Jin H, Yang P, Wu J, Niu L, Gong Q, Teng M, and Shi Y (2014) Crystal structure of arginine methyltransferase 6 from Trypanosoma brucei. PLoS One 9, e87267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Milliman EJ, Hu Z, and Yu MC (2012) Genomic insights of protein arginine methyltransferase Hmt1 binding reveals novel regulatory functions. BMC Genomics 13, 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ho MC, Wilczek C, Bonanno JB, Xing L, Seznec J, Matsui T, Carter LG, Onikubo T, Kumar PR, Chan MK, Brenowitz M, Cheng RH, Reimer U, Almo SC, and Shechter D (2013) Structure of the arginine methyltransferase PRMT5-MEP50 reveals a mechanism for substrate specificity. PLoS One 8, e57008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lin WJ, Gary JD, Yang MC, Clarke S, and Herschman HR (1996) The mammalian immediate-early TIS21 protein and the leukemia-associated BTG1 protein interact with a protein-arginine N-methyltransferase. J. Biol. Chem 271, 15034–15044. [DOI] [PubMed] [Google Scholar]
- (23).Tang J, Gary JD, Clarke S, and Herschman HR (1998) PRMT 3, a type I protein arginine N-methyltransferase that differs from PRMT1 in its oligomerization, subcellular localization, substrate specificity, and regulation. J. Biol. Chem 273, 16935–16945. [DOI] [PubMed] [Google Scholar]
- (24).Rho J, Choi S, Seong YR, Cho WK, Kim SH, and Im DS (2001) Prmt5, which forms distinct homo-oligomers, is a member of the protein-arginine methyltransferase family. J. Biol. Chem 276, 11393–11401. [DOI] [PubMed] [Google Scholar]
- (25).Lim Y, Kwon YH, Won NH, Min BH, Park IS, Paik WK, and Kim S (2005) Multimerization of expressed protein-arginine methyltransferases during the growth and differentiation of rat liver. Biochim. Biophys. Acta, Gen. Subj 1723, 240–247. [DOI] [PubMed] [Google Scholar]
- (26).Herrmann F, Pably P, Eckerich C, Bedford MT, and Fackelmayer FO (2009) Human protein arginine methyltransferases in vivo-distinct properties of eight canonical members of the PRMT family. J. Cell Sci 122, 667–677. [DOI] [PubMed] [Google Scholar]
- (27).Pak ML, Lakowski TM, Thomas D, Vhuiyan MI, Husecken K, and Frankel A (2011) A protein arginine N-methyltransferase 1 (PRMT1) and 2 heteromeric interaction increases PRMT1 enzymatic activity. Biochemistry 50, 8226–8240. [DOI] [PubMed] [Google Scholar]
- (28).Lee J, Sayegh J, Daniel J, Clarke S, and Bedford MT (2005) PRMT8, a new membrane-bound tissue-specific member of the protein arginine methyltransferase family. J. Biol. Chem 280, 32890–32896. [DOI] [PubMed] [Google Scholar]
- (29).Kaniskan HU, Szewczyk MM, Yu Z, Eram MS, Yang X, Schmidt K, Luo X, Dai M, He F, Zang I, Lin Y, Kennedy S, Li F, Dobrovetsky E, Dong A, Smil D, Min SJ, Landon M, Lin-Jones J, Huang XP, Roth BL, Schapira M, Atadja P, Barsyte-Lovejoy D, Arrowsmith CH, Brown PJ, Zhao K, Jin J, and Vedadi M (2015) A Potent, Selective and Cell-Active Allosteric Inhibitor of Protein Arginine Methyltransferase 3 (PRMT3). Angew. Chem., Int. Ed 54, 5166–5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Siarheyeva A, Senisterra G, Allali-Hassani A, Dong A, Dobrovetsky E, Wasney GA, Chau I, Marcellus R, Hajian T, Liu F, Korboukh I, Smil D, Bolshan Y, Min J, Wu H, Zeng H, Loppnau P, Poda G, Griffin C, Aman A, Brown PJ, Jin J, Al-Awar R, Arrowsmith CH, Schapira M, and Vedadi M (2012) An allosteric inhibitor of protein arginine methyltransferase 3. Structure 20, 1425–1435. [DOI] [PubMed] [Google Scholar]
- (31).Trowitzsch S, Bieniossek C, Nie Y, Garzoni F, and Berger I (2010) New baculovirus expression tools for recombinant protein complex production. J. Struct. Biol 172, 45–54. [DOI] [PubMed] [Google Scholar]
- (32).Pan WA, Tsai HY, Wang SC, Hsiao M, Wu PY, and Tsai MD (2015) The RNA recognition motif of NIFK is required for rRNA maturation during cell cycle progression. RNA Biol. 12, 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Luger K, Rechsteiner TJ, Flaus AJ, Waye MM, and Richmond TJ (1997) Characterization of nucleosome core particles containing histone proteins made in bacteria. J. Mol. Biol 272, 301–311. [DOI] [PubMed] [Google Scholar]
- (34).Otwinowski Z, and Minor W (1997) Processing of X-ray Diffraction Data Collected in Oscillation Mode Methods in Enzymology. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (35).Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, and Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Murshudov GN, Vagin AA, and Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr 53, 240–255. [DOI] [PubMed] [Google Scholar]
- (37).Emsley P, and Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (38).Hayes D, Laue T, and Philo J (1995) Program Sednterp: Sedimentation interpretation program, University of New Hampshire, Durham, NH. [Google Scholar]
- (39).Schuck P (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J 78, 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Smolsky IL, Liu P, Niebuhr M, Ito K, Weiss TM, and Tsuruta H (2007) Biological small-angle X-ray scattering facility at the Stanford Synchrotron Radiation Laboratory. J. Appl. Crystallogr 40, S453–S458. [Google Scholar]
- (41).Huang TC, Toraya H, Blanton TN, and Wu Y (1993) X-ray powder diffraction analysis of silver behenate, a possible low-angle diffraction standard. J. Appl. Crystallogr 26, 180–184. [Google Scholar]
- (42).West AL, Evans SE, Gonzalez JM, Carter LG, Tsuruta H, Pozharski E, and Michel SL (2012) Ni(II) coordination to mixed sites modulates DNA binding of HpNikR via a long-range effect. Proc. Natl. Acad. Sci. U. S. A 109, 5633–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Edwards AL, Matsui T, Weiss TM, and Khosla C (2014) Architectures of whole-module and bimodular proteins from the 6-deoxyerythronolide B synthase. J. Mol. Biol 426, 2229–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Matsui T, Gu S, Lam KH, Carter LG, Rummel A, Mathews II, and Jin R (2014) Structural basis of the pH-dependent assembly of a botulinum neurotoxin complex. J. Mol. Biol 426, 3773–3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, and Kuhn P (2002) Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat 9, 401–406. [DOI] [PubMed] [Google Scholar]
- (46).Martel A, Liu P, Weiss TM, Niebuhr M, and Tsuruta H (2012) An integrated high-throughput data acquisition system for biological solution X-ray scattering studies. J. Synchrotron Radiat 19, 431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HD, Konarev PV, and Svergun DI (2012) New developments in the program package for small-angle scattering data analysis. J. Appl. Crystallogr 45, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Franke D, and Svergun DI (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr 42, 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Volkov VV, and Svergun DI (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr 36, 860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Schneidman-Duhovny D, Hammel M, Tainer JA, and Sali A (2013) Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys. J 105, 962–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Schneidman-Duhovny D, Hammel M, and Sali A (2010) FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 38, W540–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Krissinel E, and Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- (53).Dillon MB, Rust HL, Thompson PR, and Mowen KA (2013) Automethylation of protein arginine methyltransferase 8 (PRMT8) regulates activity by impeding S-adenosylmethionine sensitivity. J. Biol. Chem 288, 27872–27880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Wang C, Zhu Y, Caceres TB, Liu L, Peng J, Wang J, Chen J, Chen X, Zhang Z, Zuo X, Gong Q, Teng M, Hevel JM, Wu J, and Shi Y (2014) Structural determinants for the strict mono-methylation activity by trypanosoma brucei protein arginine methyltransferase 7. Structure 22, 756–768. [DOI] [PubMed] [Google Scholar]
- (55).Kim JD, Kako K, Kakiuchi M, Park GG, and Fukamizu A (2008) EWS is a substrate of type I protein arginine methyltransferase, PRMT8. Int. J. Mol. Med 22, 309–315. [PubMed] [Google Scholar]
- (56).Burd CG, and Dreyfuss G (1994) Conserved structures and diversity of functions of RNA-binding proteins. Science 265, 615–621. [DOI] [PubMed] [Google Scholar]
- (57).Najbauer J, Johnson BA, Young AL, and Aswad DW (1993) Peptides with sequences similar to glycine, arginine-rich motifs in proteins interacting with RNA are efficiently recognized by methyltransferase(s) modifying arginine in numerous proteins. J. Biol. Chem 268, 10501–10509. [PubMed] [Google Scholar]
- (58).Gary JD, and Clarke S (1998) RNA and protein interactions modulated by protein arginine methylation. Prog. Nucleic Acid Res. Mol. Biol 61, 65–131. [DOI] [PubMed] [Google Scholar]
- (59).Takagi M, Sueishi M, Saiwaki T, Kametaka A, and Yoneda Y (2001) A novel nucleolar protein, NIFK, interacts with the forkhead associated domain of Ki-67 antigen in mitosis. J. Biol. Chem 276, 25386–25391. [DOI] [PubMed] [Google Scholar]
- (60).Maris C, Dominguez C, and Allain FH (2005) The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 272, 2118–2131. [DOI] [PubMed] [Google Scholar]
- (61).Byeon IJ, Li H, Song H, Gronenborn AM, and Tsai MD (2005) Sequential phosphorylation and multisite interactions characterize specific target recognition by the FHA domain of Ki67. Nat. Struct. Mol. Biol 12, 987–993. [DOI] [PubMed] [Google Scholar]
- (62).Sims RJ 3rd, Rojas LA, Beck D, Bonasio R, Schuller R, Drury WJ 3rd, Eick D, and Reinberg D (2011) The C-terminal domain of RNA polymerase II is modified by site-specific methylation. Science 332, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Bonnefond L, Stojko J, Mailliot J, Troffer-Charlier N, Cura V, Wurtz JM, Cianferani S, and Cavarelli J (2015) Functional insights from high resolution structures of mouse protein arginine methyltransferase 6. J. Struct. Biol 191, 175–183. [DOI] [PubMed] [Google Scholar]
- (64).Huang BW, Ray PD, Iwasaki K, and Tsuji Y (2013) Transcriptional regulation of the human ferritin gene by coordinated regulation of Nrf2 and protein arginine methyltransferases PRMT1 and PRMT4. FASEB J. 27, 3763–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.