

Abstract
Vasculitis is an inflammatory process involving blood vessels of various sizes, including the small vessels in the kidneys to the large vessels, such as the aorta. This inflammatory condition is usually autoimmune in nature and is associated with involvement of many locations, such as the sinuses, lungs, kidneys, and even the heart. Specifically, eosinophilic granulomatosis with polyangiitis (EGPA) is a systemic vasculitis that may initially hide as asthma, allergic rhinitis, and/or sinusitis. However, it is known to become a lethal disease once progressed to include cardiovascular manifestations. It is important to remember EGPA as a differential for any patient with a history of asthma, allergic rhinitis, and/or sinusitis who also presents with cardiovascular complaints and eosinophilia. Treatment recommendations focus on immunosuppression in such cases. In this article, we discuss the case of a 62-year-old male, with a known history of asthma, who presented to the emergency department with concern for his chest pain and right-sided weakness. He was later diagnosed with EGPA with eosinophilic myocarditis. Diagnosis and treatment are described.
Keywords: EGPA, Churg-Strauss, eosinophilic granulomatosis with polyangiitis, EGPA myocarditis, eosinophilic myocarditis, vasculitis
Case Report
A 62-year-old Caucasian male with a history of well-controlled asthma on fluticasone-salmeterol presented to the emergency department with a chief complaint of chest pain and right-sided weakness.
Initial laboratory studies were notable for troponin-I of 0.044 ng/mL and elevated pro-brain natriuretic peptide of 5555 pg/mL. Initial chest X-ray demonstrated congestion at the lung bases and concern for congestive heart failure. Initial echocardiogram showed a 2-cm thrombus in the left atrial appendage along with an ejection fraction (EF) of 58%. Electrocardiogram exhibited pathological Q-waves, ST-segment depression, and inverted T-waves. Anterior myocardial infarction was suspected (Figures 1-3).
Figure 1.

Initial electrocardiogram on admission demonstrated pathological Q-waves, ST-segment depression, and inverted T-waves. Anterior myocardial infarction was suggested.
Figure 2.
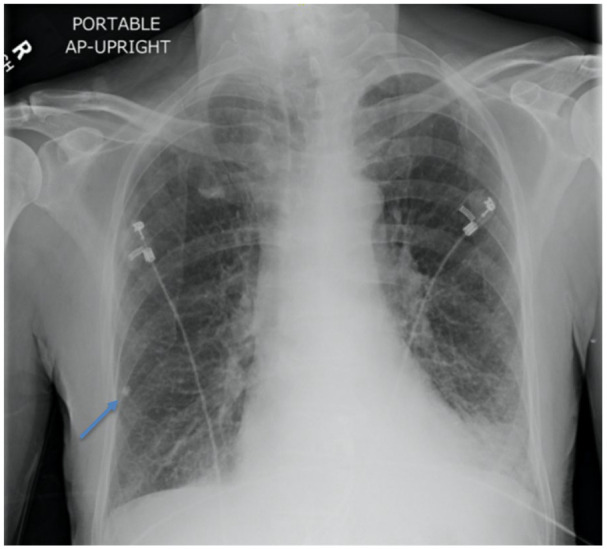
Initial chest X-ray in emergency department consistent with bilateral hyperinflation, congestion at lung bases, right lower lobe granuloma (blue arrow), and congestive heart failure.
Figure 3.
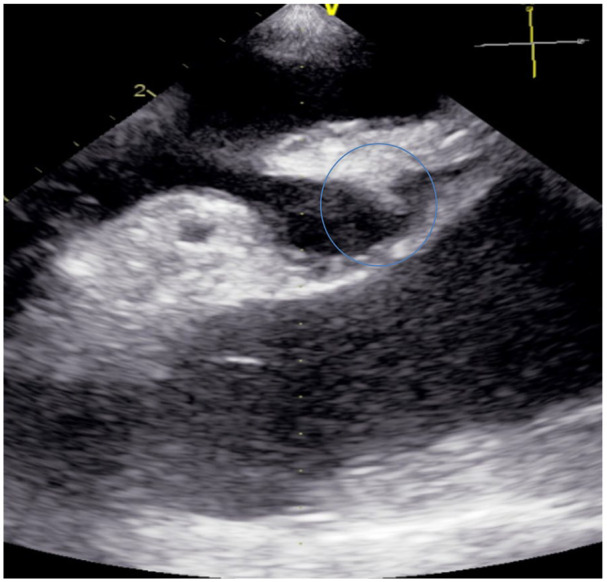
Initial echocardiogram in emergency department with evidence of 2-cm bilobed thrombus located at the left atrial appendage (blue circle).
Stroke workup continued with computed tomography (CT) scan of the head, which did not reveal hemorrhagic stroke or any ischemic changes. However, magnetic resonance imaging (MRI) demonstrated small, acute, non-hemorrhagic infarcts of the posterior left parietal lobe (Figure 4).
Figure 4.
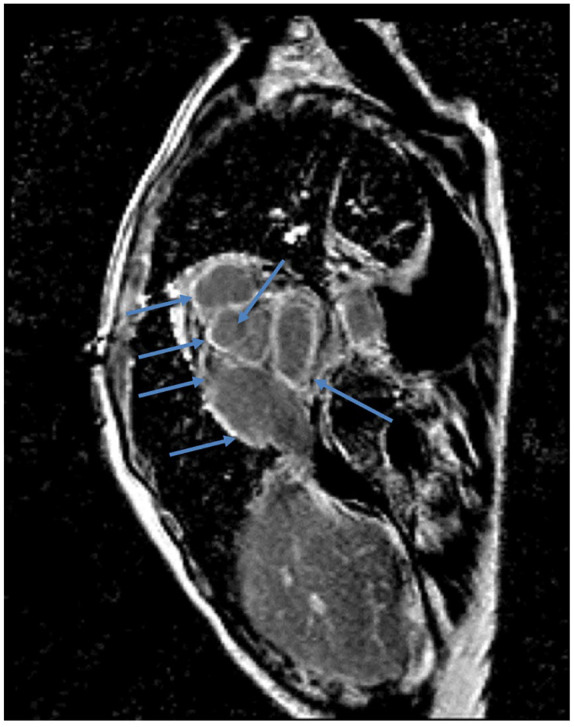
Cardiac magnetic resonance imaging showing functional sequence, T1-weighted, postcontrast, with diffuse gadolinium enhancement of myocardium (blue arrows).
Unfractionated heparin (4000 units intravenous [IV] bolus and 1000 units IV infusion) was given for concern of embolic stroke. Thrombolytics were not given, as the patient was outside of the therapeutic window. Aspirin (325 mg) was given in chewable form. The patient was admitted to the telemetry unit for acute ischemic stroke of the left middle cerebral artery and NSTEMI (non-ST-elevation myocardial infarction).
The patient continued to complain of persistent chest pain despite treatment. He was given IV morphine every 2 hours, nitroglycerin spray 400 µg (1 spray) every 5 minutes, and IV nitroprusside drip 40 µg/min, with holding parameters based on the mean arterial pressure.
On day 6 of his hospitalization course, the patient was noted to have a new onset of left-sided weakness and decreased alertness. Neurologically, his examination showed bilateral weakness, new onset of aphasia, and a decreased level of alertness. Physical examination revealed that other systems had also became involved, as noted by new onset of skin petechiae of his lower extremities and diffuse abdominal pain. Repeat chest imaging showed new concerns for pleural effusions and pericardial effusions. Repeat cardiac echocardiogram showed an EF of 18%. Further complications were identified that explained the onset of his diffuse, severe abdominal pain. A gastric perforation was identified on CT scan of the abdomen and he required emergent surgery with blood transfusions. In the intensive care unit, hemodialysis and a dopamine drip were also required. Neurological decline in the setting of the patient’s unstable condition required transfer to a tertiary center at this time.
On laboratory workup at the tertiary institution, hypereosinophilia was noted as prominent (Table 1). His absolute eosinophil count was 13 046 cells/mL. Inflammatory markers were also elevated (erythrocyte sedimentation rate 23 mm/h, C-reactive protein 16 mg/L). Chart history revealed that the patient was seen by his primary care physician 5 months earlier and was incidentally found to have an absolute eosinophil count of 700 cells/µL with an eosinophil percentage of 11% in the preceding 8 months before his initial emergency department visit (Table 2).
Table 1.
Tertiary Center Workup.
| All were negative except for rheumatoid factor (RF) | ||
|---|---|---|
| Rheumatologic | Clonal disorders/malignancy | Infectious |
| Lupus | Bone marrow cancer | Strongyloidiasis |
| EGPA | Lymphoid/myeloid abnormalities | Coccidioidomycosis |
| HES | Chronic eosinophilic leukemia | Histoplasmosis |
| Idiopathic hypereosinophilia | Myeloproliferative disorders | Tuberculosis |
| pANCA | Bone marrow biopsy | Cryptococcus |
| cANCA | IgG4 disease | Tuberculosis QuantiFERON gold |
| C3; C4 | Hematologic malignancy | Immunodiffusion coccidioidomycosis |
| ANA | Jak2, BCR/ABL, PDGFRα, FGFR 1, which includes chronic eosinophilic leukemia with F1P1L1/PDGFRα | (CM) IgG and IgM |
| RF: elevated | Complement fixation for CM | |
| MPO, PR3 | Histoplasmosis antigen | |
| Hepatitis B, C | ||
| Blood cultures and urine cultures | ||
| C-difficile toxin B | ||
| HIV antibody/antigen | ||
Abbreviations: EGPA, eosinophilic granulomatosis with polyangiitis; HES, hypereosinophilic syndrome; Ig, immunoglobulin; CM, coccidioidal meningitis.
Table 2.
Trended Laboratory Values of Entire Course From January to August.
| January to August | ||||||
|---|---|---|---|---|---|---|
| January at PCP | June ED hospital admission | June hospital day 6 | July tertiary center admission | August remission | Reference range | |
| WBC | 6.73 | 19.9 | 26.4 | 28.36 | 8.52 | 3.67-9.84 × 103/µL |
| Eosinophil % | 11% | 48.9% | 41.2% | 46% | 7.9% | 0% to 4.4% |
| Absolute eosinophil | 740 | 9408 | 108 777 | 13 046 | 673 | <500/µL |
| Bands | — | — | — | 2 | <11% | |
| Hemoglobin | 15.2 | 13.8 | 13.9 | 12.5 | 9.6 | 12.1-17.2 g/dL |
| Platelet(s) | 252 | 368 | 312 | 346 | 386 | 163-337 × 103/µL |
| Creatinine | 0.09 | 0.85 | 6.0 | 8.2 | 0.78 | 0.6-1.3 mg/dL |
| Troponin | — | 1.876 | 2.52 | — | — | <0.04 ng/mL |
| Pro-BNP | — | 5555 | 6998 | 418 | — | 0-125 pg/mL |
Abbreviations: PCP, primary care physician; ED, emergency department; WBC, white blood cell.
For his continued chest pain, a cardiac MRI was completed. The MRI was suggestive of an infiltrative disease of unknown etiology, indicating a need for an endomyocardial biopsy (EMB). EMB showed a right ventricle with evidence of eosinophilic myocarditis with myofiber necrosis, epicardial lympho-plasma cells, and eosinophil-rich inflammation with mesothelial hyperplasia. EMB was helpful in providing histological evidence of eosinophilic infiltration. It identified a reversible disease stage and helped exclude several rare differential diagnoses. IgG4 disease was specifically disproven with EMB. Collectively, malignancy and infectious etiologies were excluded.
Hypereosinophilic syndrome (HES) was diagnosed at the tertiary facility. His constellation of involved organ systems, plus the timeline of symptom manifestations, EMB findings and HES satisfied 6 out of the 6 established eosinophilic granulomatosis with polyangiitis (EGPA) criteria.
Treatment for EGPA was initiated. High-dose, 1-g IV methylprednisolone was administered. The patient’s condition remained refractory to high-dose IV steroids. The addition of cyclophosphamide to methylprednisolone normalized the patient’s EF within 1 week. His EF of 18% to 20% was subsequently restored to above 55%. A stable patient was discharged home after 1 week of cyclophosphamide and steroid therapy. His eosinophilia resolved within 3 months. The patient’s baseline of good health was nearly recovered, except for minor residual right-sided weakness. He returned to work and remained asymptomatic for the 12 months that he was followed. He was managed on dual therapy with cyclophosphamide and 5 mg of oral prednisone over the course of the 12 months. Cyclophosphamide was discontinued in the 12th month of therapy. Prednisone was continued as therapeutic maintenance.
Discussion
Eosinophilic myocarditis with thrombotic phenomena due to EGPA is a very rare condition.1 It presents as an embolic multisystemic phenomenon that mimics other diseases. It is difficult to diagnose for this reason.2,3 EGPA pathogenically lies at the crossroads between small vessel inflammation, eosinophil-driven tissue injury, and asthma. The incidence of EGPA is about 1 to 3 cases per 100 000 adults per year. The heart is a known preferential target for eosinophilic attack in up to one third of patients diagnosed with EGPA.2 In patients with no prior diagnosis of EGPA, histologically proven eosinophilic myocarditis presents with broad differentials. Idiopathic and hypersensitivity-associated eosinophilic myocarditis comprise the majority of reported cases (69%). Exclusion of known provocateurs such as amoxicillin, adalimumab, carbamazepine, clozapine, spironolactone, or tetracycline is important. Thirteen percent of histologically proven eosinophilic cases are due to EGPA.1
EGPA typically develops into 3 sequential phases. First there is the allergic phase, characterized by asthma, allergic rhinitis, and sinusitis. Second, there is the eosinophilic phase, in which the main pathological finding is the eosinophilic organ infiltrations (eg, lungs, heart, and gastrointestinal system). Third there is the vasculitic phase, portrayed by purpura, peripheral neuropathy, and constitutional symptoms.4
Hypereosinophilia, which is seen during the second phase is the hallmark of EGPA pathogenesis in the peripheral blood and/or tissues. This is the phase in which most diagnoses of EGPA are made. Absolute eosinophil counts >350 cells/µL are abnormal and is a marker for pathological interleukin (IL)-5 elevation. In cardiac manifestations due to EGPA from eosinophilic infiltration, a poor prognosis has been associated. Specifically, there is an increased risk for acute coronary syndrome, myocardial infarction, and acute decompensated heart failure.
According to the American College of Rheumatology (ACR), fulfillment of 4 or more criteria indicates a sensitivity of 85% and a specificity 99.7% for diagnosis of EGPA: (1) asthma; (2) eosinophilia, absolute eosinophils >10% on the differential white blood cell count; (3) mono- or polyneuropathy associated with systemic vasculitis; (4) migratory or transient pulmonary infiltrates; (5) paranasal sinus abnormalities; and (6) extravascular eosinophils on biopsy. Our case demonstrated all 6 criteria. See the Chapel Hill Conference Consensus 2012 Definition of EGPA for expanded diagnostic criteria.2
Cardiac imaging is performed in patients with myocarditis of unknown etiology and persistent symptoms. Cardiac transthoracic echocardiography with demonstration of the EF is initially recommended. CT and MRI with contrast are used but their role for diagnosis and response to treatment remains poorly defined.2,3,5
Endomyocardial biopsy is indicated when the inflammatory etiology remains unidentified and initial treatments have failed. EMB-confirmed eosinophilic myocardial infiltration identifies concern for drug-induced eosinophilic myocarditis (also known as hypersensitivity myocarditis), HES, EGPA, reactive eosinophilia, clonal disorders, and idiopathic HES.6,7
Eosinophilic myocarditis has 3 phases that deserve specific recognition: a necrotic phase, a thrombotic phase, and a fibrotic phase. These are distinguishable histopathologically by EMB, and diagnostically (ie, transient pericardial effusions) and clinically (ie, multisystem involvement and decompensation). A chronological course is typical for the necrotic, thrombotic, and fibrotic presentations.2,3,5 Histological findings on an EMB of necrosis indicates a reversible, early disease stage. The necrosis phase begins in a clinically silent patient with an average onset and duration of 5 weeks. A thrombotic phase occurs next. It develops within 10 months (on average) and clinical symptoms vary depending on thromboembolic location. Echocardiogram may be normal, as initially seen with our case. Intracardiac thrombi form and can embolize to induce strokes or ischemia in various parts of the body.8 The left atrial appendage is the most common site for thrombotic formation. The left ventricle is the second most common site.1 Histologically, necrosis, and rarely thrombosis is demonstrated.2,3,5 The fibrotic phase progresses after 2 years on average. There will be histologic evidence of fibrosis. Diagnostic imaging will demonstrate abnormalities in function and pathologic anatomy. Clinically, the patient will be highly symptomatic. This disease stage is irreversible. Valve replacement and/or cardiac transplant is indicated.2,5
Recommended treatment for EGPA are systemic glucocorticoids (prednisone) at a dose of 0.5 to 1 mg/kg per day and cyclophosphamide. For patients with severe vasculitis (impending respiratory failure, cardiac involvement, glomerulonephritis, and neuropathy) and acute multi-organ disease, IV glucocorticoids (methylprednisolone) at a dose of 1 g daily for 3 days is given followed by oral glucocorticoid therapy.9-11 If the treatment of glucocorticoids is stopped prematurely, a more severe disease exacerbation may be provoked. Pulsatile cycles of steroids and cyclophosphamide with intermittent drug holidays are administered based on symptomatic exacerbations, absolute eosinophil count, erythrocyte sedimentation rate, and C-reactive protein.5 See reference guidelines by the ACR.12
Mepolizumab, an IL-5 blocker, is a newly Food and Drug Administration–approved agent for acute and chronic management of EGPA dosed at 300 mg subcutaneously every 4 weeks. It is an alternative to cyclophosphamide, recommended as dual therapy with prednisone, and has demonstrated increased time/rates of remission and increased ability to taper steroids doses. At this time, mepolizumab for HES is available as part of a multicenter phase 3 placebo-controlled study in patients with severe HES (NCT02836496) but unfortunately for HES/overlap syndromes, it is not available outside of clinical trials or in the compassionate use program.13
Other EGPA treatments that are currently being reviewed are benralizumab (NCT03010436) or reslizumab (NCT02947945). These directly target eosinophils.
Conclusion
EGPA-induced myocarditis is challenging to diagnose but remission is achievable with prednisone and cyclophosphamide or prednisone and mepolizumab. Improved diagnostic markers and eosinophil-targeted therapies are needed for more direct pathways to diagnosis and overall better treatment options.2,3
Footnotes
Authors’ Note: This case was presented at the American Federation of Medical Research (AFMR) Western Conference (January 24, 2020), Carmel, CA, in the “Cardiovascular III” session. It was also presented at the AFMR South Regional Medical Conference (February 13, 2020), New Orleans, LA, in the “Case Reports in Cardiovascular Medicine” session and was merited the Cardiovascular Case Report Award.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval: The institution from which this case was derived does not require ethical approval for reporting individual cases or case series.
Informed Consent: Informed consent for patient information to be published in this article was not obtained because he was lost to follow-up after 1 year.
ORCID iDs: Valerie F. Civelli  https://orcid.org/0000-0002-1052-0144
https://orcid.org/0000-0002-1052-0144
Leila Moosavi
https://orcid.org/0000-0001-8790-910X
References
- 1. Brambatti M, Matassini MV, Adler ED, Klingel K, Camici PG, Ammirati E. Eosinophilic myocarditis. J Am Coll Cardiol. 2017;70:2363-2375. [DOI] [PubMed] [Google Scholar]
- 2. Ramirez GA, Yacoub MR, Ripa M, et al. Eosinophils from physiology to disease: a comprehensive review. Biomed Res Int. 2018;2018:1-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rothenberg ME. Eosinophilia. N Engl J Med. 1998;338:1592-1600. [DOI] [PubMed] [Google Scholar]
- 4. Gioffredi A, Maritati F, Oliva E, Buzio C. Eosinophilic granulomatosis with polyangiitis: an overview. Front Immunol. 2014;5:549. doi: 10.3389/fimmu.2014.00549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Butt NM, Lambert J, Ali S, et al. Guideline for the investigation and management of eosinophilia. Br J Haematol. 2017;176:553-572. [DOI] [PubMed] [Google Scholar]
- 6. Ullah MW, Berti A, Kane G, et al. Cardiac and cerebrovascular manifestations of eosinophilic granulomatosis with polyangiitis. J Am Coll Cardiol. 2019;73:2084. [Google Scholar]
- 7. Chauffaille MDLLF. Reactive eosinophilia, chronic eosinophilic leukemia and idiopathic hypereosinophilic syndrome. Rev Bras Hematol Hemoter. 2010;32:395-401. [Google Scholar]
- 8. Plastiras SC, Economopoulous N, Kelekis NL, Tzelepis GE. Magnetic resonance imaging of the heart in a patient with hypereosinophilic syndrome. Am J Med. 2006;119:130-132. [DOI] [PubMed] [Google Scholar]
- 9. Groh M, Pagnoux C, Baldini C, et al. Eosinophilic Granulomatosis with Polyangiitis (Churg–Strauss) (EGPA) Consensus Task Force Recommendations for evaluation and management. Eur J Intern Med. 2015;26:545-553. doi: 10.1016/j.ejim.2015.04.022 [DOI] [PubMed] [Google Scholar]
- 10. Sinico R, Paolo B. Churg–Strauss angiitis. Best Pract Res Clin Rheumatol. 2009;23:355-366. doi: 10.1016/j.berh.2009.02.004 [DOI] [PubMed] [Google Scholar]
- 11. Pagnoux C, Guilpain P, Guillevin L. Churg–Strauss syndrome. Curr Opin Rheumatol. 2007;19:25-32. doi: 10.1097/bor.0b013e3280119854 [DOI] [PubMed] [Google Scholar]
- 12. Chung S, Hoffman G, Langford C, et al. American College of Rheumatology (ACR) vasculitis guideline—ANCA-associated vasculitis. Accessed October 1, 2020 https://www.rheumatology.org/Portals/0/Files/Vasculitis-Guideline-Project-Plan.pdf
- 13. Wechsler ME, Akuthota P, Jayne D, et al. Mepolizumab or placebo for eosinophilic granulomatosis with polyangiitis. N Engl J Med. 2017;376:1921-1932. [DOI] [PMC free article] [PubMed] [Google Scholar]