

Abstract
While recognized as a therapeutic target, the spliceosome may offer a robust vector to improve established therapeutics against other protein targets. Here, we describe how modulating the spliceosome using small molecule splice modulators (SPLMs) can prime a cell for sensitivity to a target-specific drug. Using the cell cycle regulators aurora kinase and polo-like kinase as models, this study demonstrates how the combination of SPLM treatment in conjunction with kinase inhibition offers synergy for anti-tumor activity using reduced, sub-lethal levels of SPLM and kinase inhibitors. This concept of splice modulated drug attenuation suggests a possible approach to enhance therapeutic agents that have shown limited applicability due to high toxicity or low efficacy.
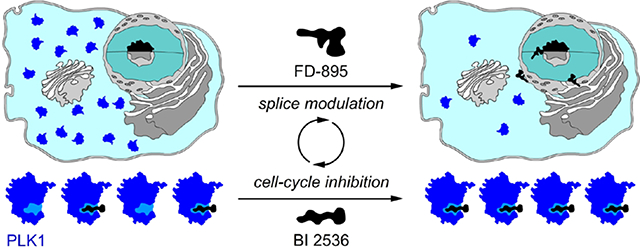
Graphical Abstract
Splicing is an essential eukaryotic biological process that is frequently mis-regulated in cancer.1 Since their discovery in the early 1990s, splice-modulating polyketide natural products FD-8952 (1a, Figure 1), pladienolide B3 (1b, Figure S1), or herboxidiene4 (1c, Figure S1), and FR9014645 (1d, Figure S1), have been proposed as new anticancer therapeutics and used to investigate the impact of spliceosome inhibition in healthy or tumor cells.6,7 These SPLMs share a common mode of action (MOA) by targeting the splicing factor 3b (SF3B) unit of the human spliceosome, leading to splicing inhibition and changes in alternative splicing patterns.8–10 Many tumors depend on aberrant use of splicing machinery for expansion and metastasis, but interruption of splicing by SPLMs limits the expression of genes necessary for tumor survival, ultimately resulting in apoptotic cell death.11 Because SPLMs regulate the splicing of several genes that are overexpressed in cancer, they have been identified for their potential in anti-cancer therapy.6,12–14 To date two SPLMs have entered the clinic, including the most recent entrance of H3B-880015 (1f, Figure S1) for acute myeloid leukemia (AML), and E-710716 (1e, Figure S1) against solid tumors. Recent efforts in medicinal chemistry, RNA biology, and structural biology have allowed for better understanding of SPLM activity and enhanced pharmaceutical access to SPLMs.17,18
Figure 1.

Concept of splice synergy of cell cycle inhibitors. Pre-treatment of tumor cells with a SPLM such FD-895 (1a) induces alternate splicing of a RNA associated with a cell cycle response. Rendered unproductive, the resulting mis-spliced RNA is no longer translated into protein leading to net loss in the levels of the given cell cycle regulatory protein, ultimately resulting in a synergistic enhancement of inhibitors of that cell cycle protein, as demonstrated by AURK inhibitors 2a-2b or a PLK-1 inhibitor 3a.
Despite recent progress towards the development of anticancer chemotherapeutics, desensitization, chemoresistance, and patient relapse remain challenges in the field. Advances in combination therapy indicate that splice modulators could serve as a potential way to overcome these issues. For instance, studies led by Mistelli suggest that splicing modulation can be used to combat vemurafenib-resistance.19 More recently, studies led by Yamano have shown that FR901464 (1d, Figure S1), the natural product precursor to spliceostatin A, synergistically improved efficacy of the PARP1 inhibitor olaparib.20 These studies suggest that splice modulatory combination therapy may offer a new approach to target many of the challenges associated protein-targeting chemotherapeutics.
To date, SPLMs have been found to interfere with the splicing of specific RNAs6 by targeting the branch point adenosine binding pocket defined by the PHF5A-SF3b complex,21 ultimately down-regulating expression of encoded proteins. The timing of this effect was demonstrated by examining the splice modulation of cell cycle regulatory proteins.22,23 We reasoned that that SPLMs could likewise sensitize tumor cells to cell cycle inhibitors, leading to synergistic anti-tumor effects (Figure 1). Here we describe these synergy studies to determine whether pre-treatment of cancer cells with SPLMs followed by administration of established kinase inhibitors could play a role in enhancing chemotherapeutics.
RESULTS AND DISCUSSION
Over the last decade, our laboratory has been exploring the multi-level effects of SPLMs upon the normal course of splicing.6 We have found that SPLMs regulate splicing at two levels: directly, through interactions with SF3b subunit within the spliceosome;17,18 and indirectly, by altering the expression of spliceosomal proteins, which in turn modifies the splicing of subsequent transcripts.22 As SPLMs such as FD-895 (1a, Figure 1) target the splicing process and result in aberrant splicing within tumor cells, we suspected that pre-treatment of cells with 1a could decrease the expression of cell cycle RNA, thereby decreasing cellular levels of a target protein for a specific inhibitor. Cell cycle RNA is only expressed at certain times in the cell cycle, such as the onset of mitosis, so FD-895 (1a) can be applied at this time for optimal regulation of cell cycle RNA. Therefore, tumor cells pre-treated with 1a would be more sensitive to inhibitors of a targeted protein, as its levels would be reduced by mis-splicing of its precursor RNA. In this way we hoped to achieve reduction of target protein levels, ultimately enhancing the efficacy of tumor cell death (Figure 1).
Using cell cycle regulation as a model, we sought to evaluate the effect of FD-895 (1a) treatment on the expression of critical cell cycle regulators (Figure 1). Using RNA-seq data previously collected on cancer cells treated with FD-895,6 we identified the oncogenic kinases aurora kinase A (AURKA), aurora kinase B (AURKB) and polo-like kinase 1 (PLK-1) as likely targets for SPLM modulation.24 These proteins are established chemotherapeutic targets. AURKA and AURKB regulate chromatin segregation during cell division. PLK-1 promotes centrosome development while activating the anaphase-promoting complex. A schematic representation of their role in the cell cycle has been provided in Supporting Figure S2.25,26 Despite much progress towards the development of cell cycle-inhibiting chemotherapeutics, the concentrations required for in vivo efficacy often lead to off-target activity.
We screened the cytotoxicity of FD-895 (1a) alone or in combination with the AURK inhibitors danusertib (2a)27 or PF-03814735 (2b).28 Consistent with the literature, the GI50 values were observed at 0.8±0.1 nM for 1a29 (Figure 2a,d), 4.3±0.4 μM for 2a27 (Figure 2a), and 2.1±0.2 μM for 2b28 (Figure 2d) in HCT-116 colorectal carcinoma cells. Comparable activities were observed in Caov3 ovarian adenocarcinoma or HeLa cervical adenocarcinoma cells with GI50 values at 2.0±0.1 nM for 1a (Caov3, Figure 2b), 3.7±0.4 nM for 1a (HeLa, Figure 2c), 5.5±0.5 μM for 2a (Caov3, Figure 2b), 10.1±1.0 μM for 2a (HeLa, Figure 2c), 6.0±0.6 μM for 2b (Caov3, Figure 2e) and 19.5±2.0 μM for 2b (HeLa, Figure 2f). Pre-treatment of either cell line with 1 nM 1a or 5 nM 1a was found to significantly decrease the associated GI50 values for each inhibitor (Figure 2a-f). Treatments with 1 nM 1a led to reductions up to 10-fold reduction in cell cycle inhibitor GI50 values, whereas 5 nM 1a led to reduction by up to 90,000-fold. At 1 nM 1a, the combination of 1a and the AURK inhibitors 2a and 2b was found to be antagonistic. Interestingly, in cells treated with 5 nM 1a, the combination was synergistic for both 2a and 2b. Similar trends were observed for combination treatment in Caov3 (Figure 2b,e) and HeLa (Figure 2c,f) cells, indicating that this effect was not cell line specific.
Figure 2.

Synergistic reduction in tumor cell viability. HCT-116, HeLa, or Caov3 cells were treated with FD-895 (1a) for 24 h, washed with PBS to remove 1a, and were treated with cell cycle inhibitors: a)-c) PF-03814735 (2a) or d)-f) danusertib (2b) or g)-i) BI 2536 (3a) for 72 h. Analysis of tumor cell viability showed pre-treatment with 1 nM 1a led to an antagonistic reduction in cell viability, whereas treatment with 5 nM 1a led to synergistic reductions in cell viability. Experiments were conducted in triplicate with GI50 values reported for each experiment. Statistical analyses and confidence limits are provided in Supporting Tables S2 and S3.
We then explored the effects of 1a in combination with the PLK-1 inhibitor BI 2536 (3a) (Figure 2g-i). In accordance with the literature, the GI50 values were 0.5 ± 0.1 nM for 1a28 and 160 ± 50 nM for 3a30 in HCT-116 cells. Comparable activities were observed in Coav3 or HeLa cells with GI50 values were observed at 2.0±0.2 nM for 1a (Caov3, Figure 2h), 4.0±0.4 nM for 1a (HeLa, Figure 2i), 25.9±3 nM for 3a (Caov3, Figure 2h), and 13.2±1 μM for 3a (HeLa, Figure 2i). Pre-treatment of either cell line with 1 nM 1a or 5 nM 1a was found to decrease the associated GI50 values for 3a (Figure 2g-i); 1 nM 1a led to reductions in cell cycle inhibitor GI50 values by a factor of up to 200-fold, whereas 5 nM 1a led to reduction by a factor of up to 60,000-fold. At 1 nM 1a, the combination of 1a and 3a was found to be antagonistic, but at the slightly higher concentration of 5 nM 1a, this combination was synergistic for 3a. Once again, similar trends were observed in HeLa and Caov3 cell lines.
To further investigate the synergistic relationship between 1a and the AURK inhibitors, we investigated the effects of FD-895 (1a) treatment on AURK gene expression at the RNA and AURK protein level. We treated HCT-116 cells with 1a and found that nanomolar concentrations could diminish the levels of AURKA (Figure 3a) and AURKB (Figure 3b). As depicted in Figure 3, SPLM 1a was found to decrease the expression of AURKA (Figure 3d) and AURKB (Figure 3e), inducing exon skipping in AURKA and AURKB (Figure S3), likely introducing a premature termination codon (PTC) and leading to nonsense mediated decay (NMD). These reductions in AURKA and AURKB also translated to decreases in AURKA and AURKB protein (Figure 3c,d).
Figure 3.

Demonstration of synergistic splice modulation of AURKA and AURKB. HCT-116 cells were treated with SPLM 1a for 24 h and then expression of a) AURKA RNA, b) AURKB RNA or c)-d) AURKA protein was analyzed. e)-g) For combination studies, synchronized HCT-116 cells were treated with 5 nM or 10 nM 1a for 6 h or 24 h, 200 nM 2a for 24 h, or a combination of 1a and 2a. Treatment efficacy was assessed by visualizing pAURKA and pAURKB levels by western blotting. pAURK values were expressed relative to the reference gene cofilin. The “+” sign above bars indicates synergy. Experiments were conducted in biological triplicate. Statistics were calculated using a standard one-way ANVOA; p-values were represented so that * signifies p<0.05, ** signifies p<0.01, *** signifies p<0.001, and **** signifies p<0.0001.
This observation was not limited to AURK alone. Similarly, SPLM 1a diminished PLK-1 RNA levels (Figure 4a). Previous studies have found that 1a induces utilization of an alternative 5’ splice site, introducing a PTC and leading to nonsense mediated decay (NMD) of PLK-1.22 Complete loss of PLK-1 was observed at treatments ≥ 20 nM 1a. As evident in Figure 4d, comparable results were also observed in phosphorylated PLK-1 where treatments ≥ 20 nM 1a demonstrated complete loss of PLK-1. Interestingly, treatment with ≤ 10 nM 1a resulted in an increase in PLK-1 protein (clearly evident at 2.5 nM in Figure 4b,c), presumably through feedback regulation. However, cells treated with ≥20 nM 1a underwent a loss in PLK-1 expression as expected from modulated splicing of its incipient PLK-1. We were able to confirm that this decreases in PLK-1 correlated with a reduction in PLK-1 protein (Figure 4e).
Figure 4.

Demonstration of synergistic splice modulation of PLK-1. HCT-116 cells were treated with SPLM 1a for 24 h and then expression of a) PLK-1 RNA or b)-c) PLK-1 protein was analyzed. d)-e) For combination studies, synchronized HCT-116 cells were treated with 5 nM or 10 nM 1a for 6 h or 24 h, 2 μM 3a for 24 h, or a combination of 1a and 3a. Treatment efficacy was assessed by visualizing PLK-1 levels via western blotting. The “+” sign above bars indicates synergy. Experiments were conducted in biological triplicate. Statistics were calculated using a standard one-way ANVOA; p-values were represented so that * signifies p<0.05, ** signifies p<0.01, *** signifies p<0.001, and **** signifies p<0.0001.
We also analyzed the effects of combination treatment on AURK protein expression. Because 2a inhibits AURK activation by blocking the kinase’s ATP binding site, AURK phosphorylation levels were used to assess the efficacy of this cell cycle inhibitor. Synchronized HCT-116 cells were treated with 1a, 2a, or a combination of 1a and 2a, and then phosphorylated AURKA (pAURKA) and phosphorylated AURKB (pAURKB) levels were examined by western blotting (Figure 3e-g). Although all treatments were found to reduce expression of pAURK to some extent, combination treatment most successfully decreased pAURK levels. In fact, in some treatment conditions, 1a and 2a were found to act synergistically to reduce pAURKA and pAURKB protein levels (Figure 3).
The effects of combination treatment were also evaluated for PLK-1 protein. Because 3a inhibits PLK-1 activation by targeting its ATP binding site, phosphorylated PLK-1 (pPLK-1) protein levels were used to measure efficacy of this inhibitor. Synchronized HCT-116 cells were treated with 1a, 3a, or a combination of 1a and 3a, followed by evaluating pPLK-1 levels by western blotting (Figure 3e). All treatments successfully reduced pPLK-1 expression, but combination treatment was found to be particularly effective in reducing pPLK-1. Synergy between 1a and 3a was observed for all conditions tested (Figure 4).
Interestingly, our studies suggest that SPLM response can be attenuated by exploring the structure activity relationships (SARs) within the SPLM. As shown in Figure S4, different SPLMs target AURK and PLK-1 to a different extent. These studies suggest the need to tailor the gene selectivity of splice modulation. While early evidence (Figure S4) suggest the potential to use medicinal chemical methods to encourage the mis-splicing of genes of therapeutic interest, the scope and potential of this interplay will require significant systems and gene-specific studies before the global regulatory network is revealed. While it is clear that SPLMs can play a role as tools to reduce the levels of a given target within a cell, and hence increase therapeutic potency, the development of methods that selectively engage splice modulation within a tumor cell would have profound implications, suggesting a means to specifically activate and target cells with a high-degree of cell specificity.
CONCLUSION
Nearly a decade ago, the concept of modulating the RNA levels of proteins associated with cell cycle regulatory proteins was suggested as a therapeutic option to treat diverse cancers.31 Studies such as that led by Ashihara32 demonstrate the potential for RNA interference of PLK-1 as a therapeutic approach for non-small cell lung cancers. Here, we show how the small molecule 1a can be used in a complementary approach to modulate the levels of properly spliced cell cycle regulators AURKA, AURKB and PLK-1 across a series of cell lines. This splice-induced loss resulted in a reduction in AURKA, AURKB and PLK-1 protein, attributing to a net improvement in efficacy of AURK inhibitors 2a-2b or PLK-1 inhibitor 3a, with marked enhancements up to 90000-fold (Figure 2).
Overall, this study suggests the potential to engage small molecule SPLM pre-treatment as a therapeutic tool to edit the levels of therapeutically targeted proteins by mis-splicing their RNAs. Here, one can envision the use of cell-specific SPLMs such as 1a for therapeutic intervention that begins with application of a SPLM to down-regulate the expression of a chemotherapeutic target (i.e., AURKA, AURKB or PLK-1) at the RNA level, resulting in a net loss of a targeted protein, followed by treatment with a target-selective inhibitor. Comparable to RNAi and RNAsi approaches, synergistic applications of SPLMs suggests an expanded potential for the use of splice modulation as a strategy for drug enhancement. SPLM combination therapy may be particularly useful for enhancing clinical agents that suffer from off-target effects or dose-limiting toxicity and could therefore allow for previously abandoned lead molecules (therapeutics) to re-enter the clinic. This suggestion was recently supported by studies in which FR901462, the natural product precursor to spliceostatin A (1d, Figure S1), synergistically-improved efficacy of the PARP1 inhibitor olaparib.20 Ongoing studies are now focused on exploring specificity of this SPLM combination therapy at a systems-wide level, with the overall goal of validating this strategy as mechanism-based approach to synergize chemotherapeutic treatment.
METHODS
Compounds.
FD-895 (1a) and 17S-FD-895 (1g) were prepared by total synthesis.28 PF-03814735 (2a), danusertib (2b) and BI 2536 (3a) were purchased from Millipore-Sigma, Adipogen Corporation, and Selleck Chemical, respectively. All oligonucleotides were purchased by custom synthesis (Integrated DNA Technologies). Unless stated otherwise, all reagents and media were purchased from VWR or Fisher Scientific.
Cell culture.
The HCT-116 cell line was cultured in McCoy’s 5a (Life Technologies) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, and 100 U mL−1 of penicillin and 100 μg mL−1 of streptomycin at 37 °C in an atmosphere of 5% CO2. Both the HeLa and Caov3 cell lines were maintained in DMEM (Life Technologies) supplemented with 10% FBS, 2 mM L-glutamine, and 100 U mL−1 of penicillin and 100 μg mL−1 of streptomycin at 37 °C in an atmosphere of 5% CO2.
Cellular drug treatments.
Compounds were dissolved in DMSO (MilliporeSigma). Cells were treated with 1a, 2a, 2b, or 3a in media with ≥0.5% DMSO for 24–72 h.
Cell viability assays.
HCT-116 cells were plated at 5 × 103 cells/well in McCoy’s 5a containing 10% FBS. Cell were cultured for 24 h and then pre-treated with 1a for 24 h, then washed twice with 100 μL PBS. Next, cells were treated with cell cycle inhibitors ranging from 0–10 μM of 2a, 2b, or 3a for 72 h. Then, the cells were washed twice with 100 μL PBS, and 100 μL of media was added to each well, followed by 20 μL of CellTiter Aqueous One Solution (Promega). After 2 h at 37 °C, absorbance readings were taken at 490 nm (test wavelength) and 690 nm (reference wavelength). GI50 values were calculated in Prism (GraphPad) using ≥ 3 biological replicates.
Analysis of drug effects.
CompuSyn (ComboSyn) was used to analyze cytotoxic effects of the combination of 1a with 2a, 2b, or 3a. The following equation was fitted to experimental data using nonlinear regression:
where Da is the concentration of drug A, Db in the concentration of drug B, GI50a is the median effective drug concentration, E is the fraction of cells surviving, and m is the slope parameter of the individual drug’s concentration-effect curve. When the combination index (CI) value > 1, antagonism is indicated, meaning that the observed efficacy is less than the expected additive effect. CI = 1 reflects additive effects, meaning the observed efficacy is within the range of expected additive effects. CI < 1 indicates synergy, meaning the observed efficacy is greater than the expected additive effects.33
Quantitative real time PCR (qPCR).
Cells were treated with 1a, 2a, 2b, or 3a in 0.5% DMSO for 24 h. Untreated cells were considered as a control. Total RNA was isolated using mirVana miRNA isolation kit (Life Technologies). A 1 μg sample of RNA was subjected to DNAseI from a TURBO DNA free kit (Life Technologies). The cDNA was prepared by using SuperScript III reverse transcriptase kit (Life Technologies). The amount of unspliced RNA for different genes was determined using Power SYBR Green PCR master mix (Applied Biosystems) by qPCR using specific primers for each gene (Supplementary Table 1). qPCR using 2.5 μM of each primer was performed on 5 ng of the obtained cDNA. qPCR conditions were as follows: 95 °C for 10 min for one cycle, then 95 °C for 30 s, 55 °C for 60 s, 72 °C for 60 s, for 40 cycles using the MXPro. Quantification cycle (Cq) values were identified for each sample, and then RNA levels were calculated using 2-ΔΔCT method.34 GAPDH was used as a control for normalization.21 At least three biological replicates were conducted. Statistics were calculated using a standard one-way ANVOA; p-values were represented so that * signifies p<0.05, ** signifies p<0.01, *** signifies p<0.001, and **** signifies p<0.0001.
Western blot analyses.
Cells were synchronized using a double thymidine block followed by treatment with 100 ng/mL nocodazole. Then cells were treated with 1a, 2a, 2b, or 3a in 0.5% DMSO for 6–24h. Untreated cells were considered as a control. Cells were washed twice with PBS, and lysed with modified RIPA buffer (Cell Signaling Technology) containing 1% of a human protease and phosphatase inhibitor cocktail (Cell Signaling Technology) at 4 °C. The protein content of the whole cell lysates was quantified using the Pierce BCA Assay (Thermo Fisher). Lysates in sample buffer comprised of 720 mM 2-mercaptoethanol, 0.001% bromophenol blue, 2% SDS, 10% glycerol, 80 mM Tris • HCl pH 6.8 were denatured at 95 °C for 5 min. Total cellular proteins (20–50 μg) were subjected to SDS PAGE using a 4–20% Criterion precast gel (Bio-Rad) followed by transfer to a 0.45 μm polyvinylidene difluoride (PVDF) membrane (Millipore). After blocking with 5% BSA for 1 h in 25 mL Tris-buffered saline with Tween 20 (TBST) comprised of 20 mM Tris • HCl, 137 mM NaCl, 0.1% Tween-20 pH 7.6, the membrane was incubated with a primary antibody overnight at 4 °C. The primary antibodies included a rabbit anti-PLK-1 (4513, Cell Signaling Technology), rabbit anti-phospho-PLK-1 (5472, Cell Signaling Technology), mouse anti-AURKA (610938, BD), rabbit anti-phospho-AURK (2914, Cell Signaling Technology), and mouse anti-cofilin (54532, Abcam). All primary antibodies were used at a 1:1000 dilution in TBST containing 5% BSA. After washing three times with 25 mL of TBST, the membranes were incubated with AP-labeled anti-rabbit (7054, Cell Signaling Technology) or AP-labeled anti-mouse (S372B, Promega) secondary antibodies with a dilution of 1:1000–7500 TBST containing 5% BSA for 60 min at rt. The membranes were washed three times with 25 mL TBST and protein-antibody complexes signals were detected using a BCIP/NBT (S3771, Promega) then stained blots were imaged on a conventional flatbed scanner (1260, Epson). Band signal was quantified using ImageStudio (LI-COR). At least three biological replicates were conducted. Statistics were calculated using a standard one-way ANVOA; p-values were represented so that * signifies p<0.05, ** signifies p<0.01, *** signifies p<0.001, and **** signifies p<0.0001.
Supplementary Material
ACKNOWLEDGEMENTS
K.A.T. was supported in part by a NIH Chemistry-Biology Interface Training Grant (T32 GM112584). J. J. L. and M. D. B. were supported in part by funding the CIRM Therapeutic Translational Research Project. We thank the laboratories of S. Panda (UC San Diego), N. Devaraj (UC San Diego), and J. O’Connor (UC San Diego) for providing starting cultures of HCT-116, HeLa, and Caov3 cells, respectively.
Footnotes
The authors declare no conflict of interest.
ASSOCIATED CONTENT
Supporting Information Available. Additional figures and tables are available free of charge via the Internet at http://pubs.asc.org.
References
- 1.Sveen A, Kilpinen S, Ruusulehto A, Lothe RA and Skotheim RI (2016) Aberrant RNA splicing in cancer expression changes and driver mutations of splicing factor genes. Oncogene 35, 2413–2427. [DOI] [PubMed] [Google Scholar]
- 2.Seki-Asano M, Okazaki T, Yamagishi M, Sakai N, Takayama Y, Hanada K, Morimoto S, Takatsuki A, and Mizoue K (1994) Isolation and characterization of a new 12-membered macrolide FD-895. J. Antibiot. (Tokyo) 47, 1395–1401. [DOI] [PubMed] [Google Scholar]
- 3.Sakai T, Sameshima T, Matsufuji M, Kawamura N, Dobashi K, and Mizui Y (2004) Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. I. Taxonomy, fermentation, isolation and screening. J. Antibiot. Tokyo 57, 173. [DOI] [PubMed] [Google Scholar]
- 4.Miller-Wideman M, Makkar N, Tran M, Isaac B, Biest N, and Stonard R (1992) Herboxidiene, a new herbicidal substance from Streptomyces Chromofuscus A7847. Taxonomy, fermentation, isolation, physico-chemical and biological properties. J. Antibiot. (Tokyo). 45, 914–921. [DOI] [PubMed] [Google Scholar]
- 5.Nakajima H, Hori Y, Terano H, Okuhara M, Manda T, Matsumoto S, and Shimomura K (1996) New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action. J. Antibiot. (Tokyo). 49, 1204–1211. [DOI] [PubMed] [Google Scholar]
- 6.León B, Kashyap MK, Chan WC, Krug KA, Castro JE, La Clair JJ, and Burkart MD (2017) A challenging pie to splice: Drugging the spliceosome. Angew. Chem. Int. Ed. 56, 12052–12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pham D, and Koide K (2016) Discoveries, target identifications, and biological applications of natural products that inhibit Splicing Factor 3B subunit 1. Nat. Prod. Rep. 33, 637–647. [DOI] [PubMed] [Google Scholar]
- 8.Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, Watanabe H, Kitahara T, Yoshida T, Nakajima H, Tani T, Horinouchi S, and Yoshida M (2007) Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-MRNA. Nat. Chem. Biol. 3, 576–583. [DOI] [PubMed] [Google Scholar]
- 9.Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, Ishihama Y, Iwata M, Mizui Y (2007) Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 3, 570–575. [DOI] [PubMed] [Google Scholar]
- 10.Hasegawa M, Miura T, Kuzuya K, Inoue A, Won Ki S, Horinouchi S, Yoshida T, Kunoh T, Koseki K, Mino K,. Sasaki R, Yoshida M, Mizukami T. (2011) Identification of SAP155 as the target of GEX1A (Herboxidiene), an antitumor natural product. ACS Chem. Biol. 6, 229–233. [DOI] [PubMed] [Google Scholar]
- 11.Martínez-Montiel N, Rosas-Murrieta NH, Martínez-Montiel M, Gaspariano-Cholula MP, Martínez-Contreras RD (2016) Microbial and natural metabolites that inhibit splicing: A powerful alternative for cancer treatment. Biomed Res. Int. 3681094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao Y, Vogt A, Forsyth CJ, Koide K (2013) Comparison of splicing factor 3b inhibitors in human cells. Chembiochem. 14, 49–52. [DOI] [PubMed] [Google Scholar]
- 13.Lee SC, Abdel-Wahab O (2016) Therapeutic targeting of splicing in cancer. Nat. Med, 22, 976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salton M, Misteli T (2016) Small molecule modulators of pre-mRNA splicing in cancer therapy. Trends Mol. Med. 22, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, Agrawal AA, Caleb B, Csibi A, Sean E, Fekkes P, Karr C, Klimek V, Lai G, Lee L, Kumar P, Lee SC, Liu X, Mackenzie C, Meeske C, Mizui Y, Padron E, Park E, Pazolli E, Peng S, Prajapati S, Taylor J, Teng T, Wang J, Warmuth M, Yao H, Yu L, Zhu P, Abdel-Wahab O, Smith PG, and Buonamici S (2018) H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 24, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong DS, Kurzrock R, Naing A, Wheler JJ, Falchook GS, Schiffman JS, Faulkner N, Pilat MJ, O’Brien J, and LoRusso P (2014) A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic Acid (pre-MRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest. New Drugs 32, 436–444. [DOI] [PubMed] [Google Scholar]
- 17.Cretu C, Agrawal AA, Cook A, Will CL, Fekkes P, Smith PG, Lührmann R, Larsen N, Buonamici S, Pena V (2018) Structural basis of splicing modulation by antitumor macrolide compounds. Mol. Cell 70, 265–273. [DOI] [PubMed] [Google Scholar]
- 18.Finci LI, Zhang X, Huang X, Zhou Q, Tsai J, Teng T, Agrawal A, Chan B, Irwin S, Karr C, Cook A, Zhu P, Reynolds D, Smith PG, Fekkes P, Buonamici S, Larsen NA (2018) The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action. Genes Dev. 2018, 32, 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salton M, Kasprzak WK, Voss T, Shapiro BA, Poulikakos PI, Misteli T (2015) Inhibition of vemurafenib-resistant melanoma by interference with pre-mRNA splicing. Nat. Commun. 6, 7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamano T, Kubo S, Yano A, Kominato T, Tanaka S, Ikeda M, Tomita N (2019) Splicing modulator FR901464 is a potential agent for colorectal cancer in combination therapy. Oncotarget. 10, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teng T, Tsai JH, Puyang X, Seiler M, Peng S, Prajapati S, Aird D, Buonamici S, Caleb B, Chan B, Corson L, Feala J, Fekkes P, Gerard B, Karr C, Korpal M, Liu X, Lowe T, J., Mizui Y, Palacino J, Park E, Smith PG, Subramanian V, Wu ZJ, Zou J, Yu L, Chicas A, Warmuth M, Larsen N, Zhu P. (2017) Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A-SF3b complex. Nat Commun. 8, 1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar D, Kashyap MK, La Clair JJ, Villa R, Spaanderman I, Chien S, Rassenti LZ, Kipps TJ, Burkart MD, and Castro JE (2016) Selectivity in small molecule splicing modulation. ACS Chem. Biol. 11, 2716–2723. [DOI] [PubMed] [Google Scholar]
- 23.Vanzyl EJ, Rick KRC, Blackmore AB, MacFarlane EM, and McKay BC (2018) Flow cytometric analysis identifies changes in S and M phases as novel cell cycle alterations induced by the splicing inhibitor isoginkgetin. PLoS One 13, e0191178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kashyap MK, Kumar D, Villa R, La Clair JJ, Benner C, Sasik R, Jones H, Ghia EM, Rassenti LZ, Kipps TJ, Burkart MD, and Castro JE (2015) Targeting the spliceosome in Chronic Lymphocytic Leukemia with the macrolides FD-895 and pladienolide-B. Haematologica 100, 945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salaun P, Rannou Y, and Prigent C (2008) Cdk1, Plks, Auroras, and Neks: The mitotic bodyguards. Adv. Exp. Med. Biol. 617, 41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carnero A (2002) Targeting the cell cycle for cancer therapy. Br. J. Cancer 87, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meulenbeld HJ, Mathijssen RH, Verweij J, de Wit R, and de Jonge MJ (2012) Danusertib, an aurora kinase inhibitor. Expert Opin. Investig. Drugs 21, 383–393. [DOI] [PubMed] [Google Scholar]
- 28.Schöffski P, Jones SF, Dumez H, Infante JR, Van Mieghem E, Fowst C, Gerletti P, Xu H, Jakubczak JL, English PA, Pierce KJ, Burris HA (2011) Phase I, open-label, multicentre, dose-escalation, pharmacokinetic and pharmacodynamic trial of the oral aurora kinase inhibitor PF-03814735 in advanced solid tumours. Eur. J. Cancer 47, 2256–2264. [DOI] [PubMed] [Google Scholar]
- 29.Villa R, Mandel AL, Jones BD, Clair J. J. La, and Burkart MD (2012) Structure of FD-895 revealed through total synthesis. Org. Lett. 14, 5396–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hofheinz R-D, Al-Batran S-E, Hochhaus A, Jäger E, Reichardt VL, Fritsch H, Trommeshauser D, and Munzert G (2010) An open-label, phase I study of the Polo-like Kinase-1 inhibitor, BI 2536, in patients with advanced solid tumors. Clin Cancer Res. 16, 4666–4674. [DOI] [PubMed] [Google Scholar]
- 31.Ashihara E, Kawata E, Maekawa T (2010) Future prospect of RNA interference for cancer therapies. Curr. Drug Targets. 11, 345–360. [DOI] [PubMed] [Google Scholar]
- 32.Kawata E, Ashihara E, Maekawa T (2011) RNA interference against polo-like kinase-1 in advanced non-small cell lung cancers. J. Clin. Bioinforma. 1, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foucquier J, and Guedj M (2015) Analysis of drug combinations: current methodological landscape. Pharmacol Res. Perspect. 3, e00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arocho A, Chen B, Ladanyi M, Pan Q (2006) Validation of the 2-DeltaDeltaCt calculation as an alternate method of data analysis for quantitative PCR of BCR-ABL P210 transcripts. Diagn. Mol. Pathol. 15, 56–61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.