

Abstract
The quinoline ring system has long been known as a versatile nucleus in the design and synthesis of biologically active compounds. Currently, more than one hundred quinoline compounds have been approved in therapy as antimicrobial, local anaesthetic, antipsychotic, and anticancer drugs. In drug discovery, indeed, over the last few years, an increase in the publication of papers and patents about quinoline derivatives possessing antiproliferative properties has been observed. This trend can be justified by the versatility and accessibility of the quinoline scaffold, from which new derivatives can be easily designed and synthesized. Within the numerous quinoline small molecules developed as antiproliferative drugs, this review is focused on compounds effective on c-Met, VEGF (vascular endothelial growth factor), and EGF (epidermal growth factor) receptors, pivotal targets for the activation of important carcinogenic pathways (Ras/Raf/MEK and PI3K/AkT/mTOR). These signalling cascades are closely connected and regulate the survival processes in the cell, such as proliferation, apoptosis, differentiation, and angiogenesis. The antiproliferative biological data of remarkable quinoline compounds have been analysed, confirming the pivotal importance of this ring system in the efficacy of several approved drugs. Furthermore, in view of an SAR (structure-activity relationship) study, the most recurrent ligand–protein interactions of the reviewed molecules are summarized.
Keywords: quinoline, antiproliferative compounds, targeted therapy, SAR studies, carcinogenic pathways, kinases modulators, biological data
1. Introduction
Quinoline 1 is an important and versatile nucleus recurrent in several natural and pharmacologically active molecules. Due to the presence of nitrogen, which withdraws electrons by resonance, this fused aromatic scaffold is an electron-deficient ring system (Figure 1), displaying weak tertiary base properties. Quinoline reacts with similar behaviour of pyridine and allows both nucleophilic and electrophilic substitution reactions. From a biological point of view, the nontoxic effect to humans, via inhalation and oral absorption, makes the 1-aza-naphthalene an interesting scaffold to be studied for the development of more selective drugs [1].
Figure 1.

(a) Quinoline 1 or 1-aza-naphthalene and benzo[b]pyridine; (b) quinoline 3D structure; (c) quinoline electron density; (d) electron density encoded with the electrostatic potential.
Quinoline and its related derivatives exhibit a broad spectrum of pharmacological activities from antibacterial, antifungal, antimalarial, anthelmintic, local anaesthetic, antipsychotic, and anticancer [2]. Especially in the research of new anticancer agents, quinoline is one of the most important scaffolds in drug discovery. Indeed, over the last few years, an increase in the publication of papers about quinoline derivatives possessing antiproliferative properties has been observed. This trend can be justified by the “druggability”, versatility, and accessibility of the quinoline nucleus [3].
Furthermore, different quinoline small molecules, acting as protein kinases inhibitors, have been approved by the Food and Drug Administration (FDA) for clinical uses [4,5]. In this contest, bosutinib, a potent inhibitor of Abl, was approved in 2012 for the treatment of Philadelphia chromosome-positive chronic myelogenous leukaemia (CML) [6]; lenvatinib, an inhibitor of RET and VEGFR has been employed, since 2015, for the treatment of differentiated thyroid cancers [7]; and neratinib and cabozantinib were authorized for the clinical treatment respectively in 2017 and 2012 [8,9].
Due to the importance of quinoline nucleus in the medicinal chemistry field, the main purpose of this review is to provide an overview of the most relevant anticancer quinoline molecules described in the literature, in the last decade. In particular, the attention has been focused on quinoline drugs and experimental active derivatives, able to interfere with c-Met (mesenchymal-epithelial transition factor), VEGF, and EGF receptors and the correlated proteins of the intracellular signalling pathways. The interest on these targets rises from their crucial involvement in several carcinogenic degenerations. In fact, closely connected by each other, they regulate the survival mechanisms in the cell, such as proliferation, apoptosis, differentiation, and angiogenesis.
In this light, the evaluation of the antiproliferative data and the analysis of most frequent ligand–protein interactions of quinoline-based compounds could support, in a drug discovery process, the search for new and more efficacious multi-targets drugs.
EGFR, c-MET, and VEGFR Signal Pathways in Cancer Cell
Aberrant receptor tyrosine kinases (RTKs) are deeply involved in cancer progression, and for this reason represent the key oncology therapeutic targets. Among the most studied, in this review the attention has been focused on three crucial growth factor receptors: c-Met, EGFR, and VEGFR [10].
C-Met is a tyrosine kinase receptor belonging to the MET (MNNG HOS transforming gene) family, and is normally expressed on the surfaces of liver, pancreas, prostate, kidney, muscle, and bone marrow cells, where mediates tissue regeneration and wound healing [11,12]. The endogenous ligand of c-Met is HGF/SF (ligand hepatocyte growth factor/scatter factor), that induces signal transduction from the extracellular to the intracellular domain [13]. The kinase catalytic activity triggers the transphosphorylation of the tyrosines Tyr1234 and Tyr1235, turning on a whole signalling cascade of biological pathways correlated to the cellular growth and replication [14,15].
C-Met is strictly connected with the phosphoinositide 3-kinase (PI3K) a cytosolic family of kinases responsively to phosphorylate the 3’-hydroxyl group of phosphoinositides [16], (Figure 2). After activation by growth factors and/or cytokines, PI3K is able to turn on downstream proteins, such as AkT, a serine-threonine kinase, that consequently controls stimulation of mTOR complex 1 (mTORC1). AkT is responsible for the handling of the cell cycle, through genes transcription activation. Furthermore, another component of these pathways is mTOR complex 2 (mTORC2), that determinates activation of AkT by its phosphorylation. It has been studied that this signalling pathway is upregulated in many cancers and apparently drives the resistance to the typical cytotoxic anticancer drugs. For these reasons PI3K/AkT/mTOR appear attractive targets in cancer targeted therapy [17,18,19].
Figure 2.

Cross-talk between EGFR, VEGFR, and c-Met signalling pathways; in boxes are reported some quinoline based inhibitors developed up to date.
C-Met intracellular signalling cascade is also correlated to the carcinogenic pathways stimulated by EGFR and VEGFR (Figure 2). The equilibrium in the activation of these three growth factor receptors is at the basis of normal cell survival processes, such as proliferation, apoptosis, differentiation, and angiogenesis [11,20].
EGFR belong to a family of tyrosine kinases receptors and include four members: EGFR (ErbB1), ErbB2, ErbB3, and ErbB4. These receptors are widely distributed in mammalian epithelial cell membranes and normally modulate different processes such as cell proliferation, death, and differentiation. It has been detected that an anomalous over-expression of these proteins has an important role in the formation and development of many types of solid tumours, principally Non-Small Cell Lung (NSCLC), breast, colorectal, head, and neck cancers types [21,22,23].
VEGFR can be secreted by tumour cells and, through its interaction with one of the three tyrosine kinase receptors (VEGFR1-3), it is able to promotes vascular growth and permeability, with an antiapoptotic effect on newly formed cells [24,25].
EGFR and/or VEGFR recruits Rat sarcoma (RAS) protein family, with consequent activation of Raf/MEK/ERK and of PI3K/AkT/mTOR signalling cascade, directly activated also by c-Met [16,20].
With respect to the aforementioned intracellular effectors, Ras, a membrane-associated guanine nucleotide-binding protein, plays a central role in regulating the cytoplasmatic transduction cascades, especially those of Raf/MEK and PI3K, leading to cell growth (Figure 2) [26,27,28].
2. Quinolines as Inhibitors of Carcinogenic Pathways
2.1. Quinoline Based Inhibitors of c-Met Receptor
An alteration of the HGF/c-Met signalling has been detected in liver, pancreatic, breast, ovarian, gastric, and prostate cancers and it contributes to the development, progression, dissociation of cells from its primary site, and distant colonization. Therefore, c-Met is considered an attractive target for cancer therapy [11,12]. Currently, a lot of small molecules with a quinoline nucleus, classified as c-Met inhibitors, have been approved for therapeutic treatments. Among these, we could mention cabozantinib, a multikinase inhibitor administered in the treatment of advanced medullary thyroid cancers [8,29], and foretinib, a drug candidate under experimental evaluation (Figure 3) [3].
Figure 3.

C-Met quinoline inhibitors: cabozantinib and foretinib (in boxes structure analogies are highlighted).
From a molecular point of view, all the quinoline-based inhibitors of c-Met interact with the kinase domain of the receptor in the cytosolic side, involving amino acids residues from 1078 to 1345 [13,30]. As an example, in Figure 4a, the crystal structure of c-Met kinase domain in complex with foretinib (PDB id: 3LQ8) is reported: the ligand binds to c-Met in an extended conformation occupying the ATP-binding site and the adjacent deep hydrophobic pocket. The cyclopropane-1,1-dicarboxiamide framework interferes with the Phe1223 of the DFG motif, determining the disruption of the catalytic conformation (from DFG-in to DFG out) and the reorganization of the activation loop, which almost entirely envelops the ligand (Figure 4a). In the cavity the reoriented Phe1223 forms a π-π stacking with the central fluorophenyl ring, the quinoline nitrogen links with a hydrogen bond the backbone amide of Met1160 and also the dicarboxamide moiety forms additional hydrogen bonds with Lys1110 and Asp1222 (Figure 4b) [30,31,32].
Figure 4.

(a) crystal structure of kinase domain of c-Met in complex with foretinib (PDB ID: 3LQ8). In the box the binding cavity of the receptor occupied by the quinoline small molecule is illustrated (the activation loop is represented with transparent surface) [31]; (b) interactions of foretinib with amino acid residues of the c-Met active site [32].
Considering cabozantinib and foretinib as lead compounds, for the design of new quinoline derivatives selectively acting on c-Met, SAR studies highlight the 4-phenoxyquinoline and an aromatic moiety, connected each other by a linker, as the fundamental pharmacophoric portions in the modulation of c-Met tyrosine kinase activity (Figure 5). Generally, the linker should have two important features: (1) the distance of five atoms (the so-called “five atoms regulation”) between the 4-phenoxyquinoline moiety and the aromatic one; (2) the presence of donor and/or acceptor hydrogen-bond groups and at least one amide. These general features are also shown in the structure of cabozantinib and foretinib, in which there is a cyclopropane-1,1-dicarboxiamide framework as linker [33].
Figure 5.

General structure for quinoline c-Met inhibitors as analogues of cabozantinib and foretinib.
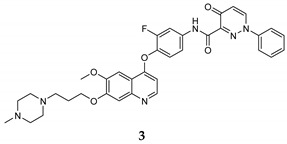
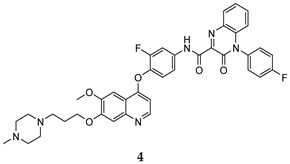
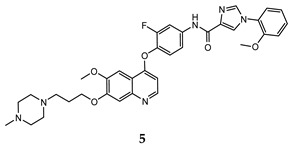
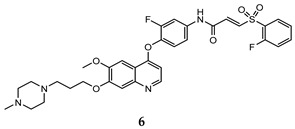
In the last few years, a lot of more active and selective quinoline derivatives have been synthesized and biologically evaluated by modifying lead compounds (Table 1). The most representative modifications are: (1) at position C-7 of quinoline ring system, generally with the introduction of appropriate substituents to improve solubility, (2) at the linker portion [34], that has been modified by different cyclic/acyclic 5-atoms-analogues with similar electronic features: pyridazinone-3-carboxyamide 3 [35], 3-oxo-3,4-dihydroquinoxaline 4 [36], 1H-imidazole-4-carboxamide or (E)-3-hydrosulfonylacrylamide 5,6 [37], 1,2,3-triazole-4-carboxamide 7,8 [38,39], 2-imidazolone-4-carboxamide 9 [40], acylhydrazone moiety 10 [41], pyridine/pyrimidine-2-carboxyamide 11 [42], 2-phenylquinoline-4-carboxamide 12 [33], 4-oxo-1,4-dihydrocinnoline-3-carboxamide moiety 13 [43], acyclic semicarbazones 14 [44,45], acylthiourea moiety [46], l,2,4-triazine-3,5-dione 15 [47], 1,8-naphthyridin-2-one 16 [48], 2-oxo-1,2-dihydropiridine-3-carboxamide 17 [49], 4-oxo-1,4-dihydroquinoline-3-carboxamide 18 [50], 5-(aminomethylene)pyrimidine-2,4,6-trione moiety 19 [51], 4-oxo-3,4-dihydrophthalazine-1-carboxamide 20 [52], pyrazolone 21 [53], and (thio)semicarbazone 22 [54].
Table 1.
Examples of some interesting quinoline based c-Met inhibitors and their corresponding biological activities.
| Quinoline Compound | Linker | IC50 (μM) Sensitive Cell Lines | IC50 on c-Met (nM) | Ref. |
|---|---|---|---|---|
|
pyridazinone-3- carboxamide | A549 (IC50 = 0.003
±
0.001) HepG2 (IC50 = 0.49 ± 0.003) MCF-7 (IC50 = 0.006 ± 0.001) |
0.6 | [35] |
|
3-oxo-3,4- dihydroquinoxaline |
HT-29 (IC50 = 0.06 ± 0.001) A549 (IC50 = 0.050 ± 0.006) H460 (IC50 = 0.18 ± 0.01) MKN-45 (IC50 = 0.023 ± 0.005) U87 MG (IC50 = 0.66 ± 0.09) |
0.9 | [36] |
|
1H-imidazole-4- carboxamide |
HT-29 (IC50 = 0.08 ± 0.02) MKN-45 (IC50 = 0.22 ± 0.03) A549 (IC50 = 0.07 ± 0.01) |
1.1 ± 0.21 | [37] |
|
(E)-3-hydrosulfonyl acrylamide |
HT-29 (IC50 = 0.15 ± 0.003) MKN-45 (IC50 = 0.28 ± 0.003) A549 (IC50 = 0.15 ± 0.05) |
17 ± 0.20 | [37] |
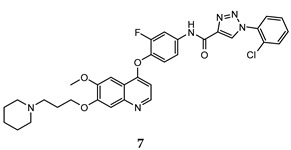
|
1,2,3-triazole-4- carboxamide |
HT-29 (IC50 = 0.10 ± 0.003) H460 (IC50 = 0.18 ± 0.03) A549 (IC50 = 0.07 ± 0.01) MKN-45 (IC50 = 0.03 ± 0.01) |
2.27 | [38] |
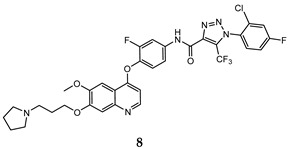
|
1,2,3-triazole-4- carboxamide |
A549 (IC50 = 0.08 ± 0.004) H460 (IC50 = 0.15 ± 0.03) HT-29 (IC50 = 0.12 ± 0.02) MKN-45 (IC50 = 0.021 ± 0.003) U87MG (IC50 = 0.85 ± 0.12) |
1.04 ± 0.09 | [39] |
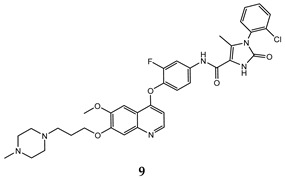
|
2-imidazolone-4- carboxamide |
A549 (IC50 = 0.25 ± 0.02) H460 (IC50 = 0.10 ± 0.008) HT-29 (IC50 = 0.086 ± 0.005) MKN-45 (IC50 = 0.014 ± 0.004) |
1.42 ± 0.14 | [40] |
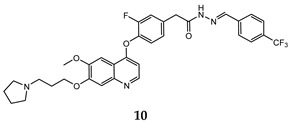
|
acylhydrazone | HT-29 (IC50 = 0.15 ± 0.03) H460 (IC50 = 0.031 ± 0.008) MKN-45 (IC50 = 0.37 ± 0.08) A549 (IC50 = 0.080 ± 0.01) U87MG (IC50 = 0.29 ± 0.08) |
1.86 | [41] |
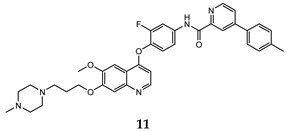
|
pyridine -2-carboxyamide | HT-29 (IC50 = 0.026 ± 0.003) H460 (IC50 = 0.037 ± 0.01) MKN-45 (IC50 = 0.073 ± 0.01) A549 (IC50 = 0.10 ± 0.02) U87MG (IC50 = 0.81 ± 0.19) |
1.39 | [42] |
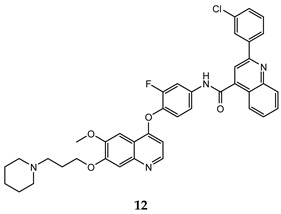
|
2-phenylquinoline-4- carboxamide |
H460 (IC50 = 0.011 ± 0.002) HT-29 (IC50 = 0.062 ± 0.01) MKN-45 (IC50 = 0.011 ± 0.003) U87MG (IC50 = 0.15 ± 0.03) SMMC-7721 (IC50 = 0.03 ± 0.004) |
1.32 | [33] |
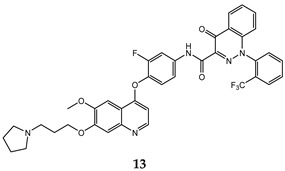
|
4-oxo-1,4-dihydrocinnoline-3-carboxamide | A549 (IC50 = 0.035 ± 0.0042) H460(IC50 = 0.055 ± 0.010) HT29 (IC50 = 0.11 ± 0.016) MKN-45 (IC50 = 0.022 ± 0.0035) U87MG (IC50 = 0.35 ± 0.060) SMMC-7721 (IC50 = 0.25 ± 0.072) |
0.59 ± 0.07 | [43] |
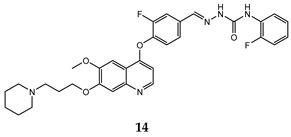
|
acyclic semicarbazone | HT-29 (IC50 = 0.059 ± 0.0039) MKN-45 (IC50 = 0.016 ± 0.0012) A549 (IC50 = 0.0090 ± 0.0012) MDA-MB-231 (IC50 = 0.77 ± 0.064) |
4.3 | [44] |
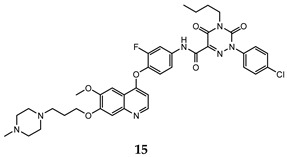
|
l,2,4-triazine-3,5-dione | H460 (IC50 = 0.14) HT-29 (IC50 = 0.21) MKN-45 (IC50 = 0.066) SMMC-7721 (IC50 = 0.73) |
6.3 | [47] |
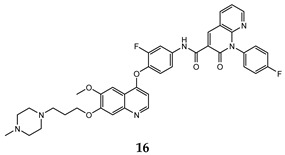
|
1,8-naphthyridin-2-one | HepG2 (IC50 = 0.23 ± 0.01) MCF-7 (IC50 = 0.42 ± 0.06) A549 (IC50 = 0.21 ± 0.02) |
2.36 | [48] |
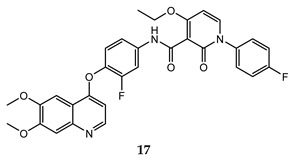
|
2-oxo-1,2-dihydropiridine-3-carboxamide | MKN45 (IC50 = 0.039 ± 0.009) BaF3/TRP-Met ((IC50 = 0.019 ± 0.0088) |
0.9 ± 0.1 | [49] |
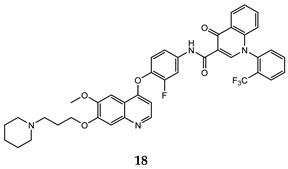
|
4-oxo-1,4-dihydroquinoline-3-carboxamide | H460 (IC50 = 0.075) HT-29 (IC50 = 0.051) MKN-45 (IC50 = 0.010) U87MG (IC50 = 0.53) SMMC-7721 (IC50 = 0.20) |
1.35 | [50] |
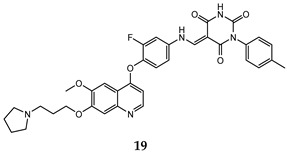
|
5-(aminomethylene) pyrimidine-2,4,6-trione moiety | HT-29 (IC50 = 0.13 ± 0.060) H460 (IC50 = 0.051 ± 0.0080) MKN-45 (IC50 = 0.057 ± 0.0040) A549 (IC50 = 0.072 ± 0.0060) U87MG (IC50 = 0.64 ± 0.045) |
1.15 | [51] |
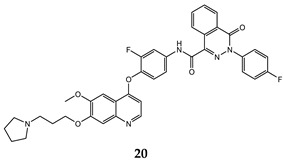
|
4-oxo-3,4-dihydrophthalazine-1-carboxamide | H460 (IC50 = 0.055 ± 0.010) MKN-45 (IC50 = 0.071 ± 0.011) HT-29 (IC50 = 0.13 ± 0.016) MDA-MB-231 (IC50 = 0.43 ± 0.050) |
1.63 | [52] |
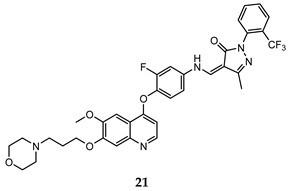
|
pyrazolone | HT-29 (IC50 = 0.14 ± 0.02) H460 (IC50 = 0.18 ± 0.03) A549 (IC50 = 0.09 ± 0.02) MKN-45 (IC50 = 0.03 ± 0.001) U87MG (IC50 = 1.06 ± 0.05) |
2.20 | [53] |
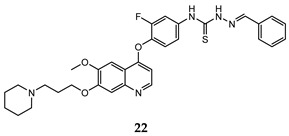
|
(thio)semicarbazones | A549 (IC50 = 0.33 ± 0.02) HT-29 (IC50 = 0.21 ± 0.02) MKN-45 (IC50 = 0.71 ± 0.08) MDA-MB-231 (IC50 = 1.2 ± 0.17) |
8.92 | [54] |
In this regard, after treatment with interesting quinoline compounds, in Table 1 are reported the IC50 values (concentration of the compound producing 50% of cell growth inhibition) evaluated against the more responsive human cancer cell lines and the IC50 values (concentration of the compound producing 50% of inhibition activity) against the c-Met receptor, (bold data show the values of target compounds lower than that of the positive control in the corresponding experiment).
In order to extrapolate important general information about the interactions between ligands and amino acid residues in the active site, the binding mode of the previous cabozantinib/foretinib-like derivatives has been explored through molecular docking simulations, (Figure 6). These studies pointed out π-π stacking interactions between the quinoline ring and Tyr1159 and the formation of a hydrogen bond between the lone pair of the quinoline nitrogen and Met1160, playing a pivotal role in the stabilization of the ligand-protein binding. Other important interactions occur through hydrogen bonds between the O and N atoms of the different linker regions and amino acids residues as Asp1222 and Lys1110 [35,36,38,40,41,48,51].
Figure 6.

Binding models with c-Met kinase domain of quinoline- based compounds (a) 3 [35]; (b) 4 [36]; (c) 7 [38]; (d) 9 [40]; (e) 10 [41]; (f) 16 [48]; (g) 19 [51].
In 2011, Wang and co-workers designed a relatively unexplored quinoline chemotype, identifying the lead compound 3,5,7-trisubstituted derivative (zgwatinib) and the quinoline 23 as novel anticancer agents with a potent and selective inhibition activity on c-Met (IC50 of 0.93 and 0.95 nM, respectively) (Figure 7a). In particular, compound 23 showed significant antiproliferative effects (IC50 in the range 1–5 μM) especially on cancer cell lines characterized by c-Met overexpression (ICMKN45, SNU-5 and H1993), exhibiting a promising PK profile and a remarkable in vivo tumour growth inhibition in human glioblastoma xenograft models [55]. The crystal structure of zgwatinib in complex with the kinase domain of c-Met was determined in a further study, to elucidate the binding mode of this class of compounds. In detail, quinoline nitrogen forms an H-bond with Met1160 in the hinge region of the kinase, the trifluoromethyl and the benzylamino groups occupy the hydrophobic pocket, and the nitro substituent forms H-bond with Asp1222 (Figure 7b) [56].
Figure 7.

(a) structures of a new cluster of 3,5,7-trisubstituted quinoline compounds with promising anticancer activity on c-Met [55,57]; (b) crystal structure of zgwatinib in complex with the kinase domain of c-Met (residues coloured in cyan form hydrophobic interactions with the ligand, whereas those coloured in yellow formed H-bonds, indicated with black dash lines; PDB id: 4GG5), [56].
Nevertheless, further toxicology studies of both derivatives demonstrated considerable cardiovascular safety risk in normal rats, arising from significant inhibition of hERG K+ channels (IC50 in the range 37–463 nM). For this reason, in the attempt to reduce the off-target effect, the same researcher group developed a new set of quinoline molecules, among which the compounds 24 and 25 were identified as the most active, (Figure 7a). The modification was directed towards the C-3 piperazinyl portion with the aim of improve the PK profile and reduce the hERG activity without influencing the key interactions. In detail, bearing a Michael acceptor moiety to the N-4 piperazinyl ring, these new quinoline compounds showed c-Met IC50 values comparable to the parent molecules (2.3 and 2.6 nM respectively for 24 and 25), and an appreciable lower activity against hERG [57].
Nishii and co-workers explored modifications at the C-3 and C-6 positions of quinoline scaffold. 3,6-disubstituted quinoline 26 (Figure 8), shows an inhibition effect selectivity against c-Met kinase among more than 20 kinases with IC50 = 9.3 nM, and a potent antiproliferative activity against MKN45 cancer cell line (IC50 = 0.093 μM), [58].
Figure 8.

4,6,7-substitued quinoline derivatives selectively active on c-Met [58,59].
In 2019, focusing the attention on C-6 substitution, Lien and co-workers designed and evaluated several 4,6,7-substituted quinolines, analogues of cabozantinib (Figure 8) [59]. In vitro antiproliferative assays highlight that derivatives 27 and 28 are more active than the parent compound on leukaemia, CNS, and breast cancer cell lines. The c-Met kinase inhibition assay denotes higher, or comparable activity than cabozantinib, with IC50 values of 19 and 64 nM respectively for 27 and 28 (IC50 cabozantinib = 40 nM). On this basis, from the point of view of structure–activity relationships, these derivatives demonstrated well fittings in the binding pocket of c-Met. Indeed, docking studies (Figure 9) demonstrated that, besides the interactions with crucial amino acids residues, above observed, these compounds could engage, through the p-aminophenyl moiety at C-6, additional hydrogen bonding with Ala1226 [59].
Figure 9.

Main interactions of compound 28 with the amino acid residues in c-Met active site [59].
Different studies on hybrid derivatives, with a quinoline scaffold linked at the C-6 position to another heterocyclic system, as triazolo-pyrazine or imidazo-pyrazine, demonstrate inhibition activity on c-Met and antiproliferative effects unless the 4-phenoxy substitution typical of the cabozantinib/foretinib-like compounds. In detail, Cui and co-workers described anticancer properties of 2-(4-(1-(quinolin-6-ylmethyl)-1H-[1,2,3]triazolo[4–b]pyrazin-6-yl)-1H-pyrazol-1-yl)-ethanol methanesulfonate (PF-04217903), (Figure 10), an extremely potent and selective c-Met inhibitor with good oral bioavailability and an acceptable safety profile in preclinical trials [60].
Figure 10.

Structures of some hybrid quinoline derivatives as c-Met inhibitors.
After SGX523 and JNJ-38877605 clinical failure [61,62,63], Zhang and co-authors synthetized 3-N-substituted quinoline triazolopyridine analogues to understand the electron-donating and steric hindrance effects on the metabolic activity of the aldehyde oxidase [64].
Furthermore, Zhao et al. have conducted some studies on both triazolo[4,5-b]pyrazine and imidazo[4,5-b]pyrazine quinoline derivatives 29, 30 (Figure 10), showing significant inhibitory effects on both enzymatic and cellular assay, as well as in vivo selectivity and metabolic stability [65,66].
2.2. Quinoline as Inhibitor of PI3k/AkT/mTOR Pathway
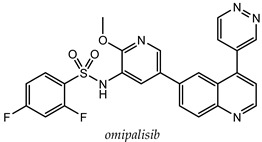
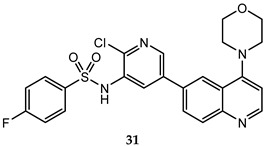
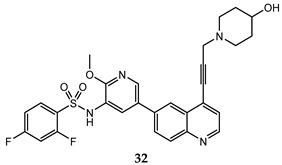
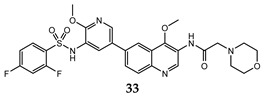
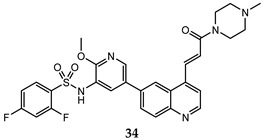
A lot of quinoline derivatives, basically substituted or condensed with other ring systems, exhibit inhibitory activity on PI3K/AkT/mTOR pathway and several molecules are currently in clinical trials. For example, omipalisib (GSK2126458) is a quinoline derivative, substituted at C-6 with a 2-(methoxy)-3-benzenesulfonamide pyridinyl moiety, at present under investigation in the first-in-human phase I study. This molecule shows the high inhibition effect of both PI3K and mTOR with an acceptable oral bioavailability [67,68]. Several omipalisib-like derivatives, characterized by a N-{5-[6-quinolinyl]-3-pyridinyl}benzenesulfonamide scaffold, have been synthesized and biologically evaluated for their PI3K/mTOR inhibition capability (biological data of omipalisib and compounds 31–34 are reported in Table 2) [69,70,71,72].
Table 2.
Omipalisib and analogues 31–34 selective active on PI3Kα and mTOR.
| Quinoline Compound | Inhibition Data (IC50 or GI50) In Antiproliferative Assays | Enzymatic Inhibition Data (IC50 or Ki) | Ref. |
|---|---|---|---|
|
T47D (IC50 = 3 nM) BT474 (IC50 = 2.4 nM) |
PI3Kα IC50 = 0.04 nM; mTORC1 Ki = 0.18 nM; mTORC2 Ki = 0.3 nM |
[67] |
|
U-87 MG (IC50 = 16 ± 5.8 nM) | PI3Kα (IC50 = 4.6 ± 3 nM; Ki = 0.6 ± 0.5 nM); mTOR IC50 = 3.9 ± 1 nM |
[69] |
|
PC-3 (IC50 = 0.37 μM); HCT-116 (IC50 = 2.47 μM) |
PI3Kα IC50 = 1.63 nM mTOR IC50 = 3.6 nM |
[70] |
|
PC-3 (IC50 = 0.08 μM)); HCT-116 (IC50 = 0.47 μM) |
PI3Kα IC50 = 1.7 nM mTOR IC50 = 10.0 nM |
[71] |
|
PC-3 (GI50 = 0.40 μM); HCT-116 (GI50 = 0.47 μM) |
PI3Kα IC50 = 0.50 nM mTOR IC50 = 1.3 nM |
[72] |
Crystallography and in silico studies highlight some recurrent and crucial interactions between these ligands and the ATP-binding site of PI3Kγ: the backbone NH of Val882 (Val851 in PI3Kα) forms a hydrogen bond with quinoline nitrogen; Lys883 interacts, through a charged bond, with the sulfonamide group; and the C-4 quinoline substituent frequently stabilizes the ligand-protein complex, accommodating itself in the ribose pocket physiologically occupied by ATP. In Figure 11 it is reported the co-crystal structure of omipalisib in complex with the catalytic subunit of PI3Kγ, [67,69,70,71,72].
Figure 11.

Crystal structure of omipalisib in complex with the catalytic subunit of PI3Kγ (PDB id: 3L08) and detailed interactions [67,73].
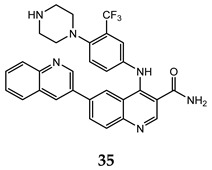
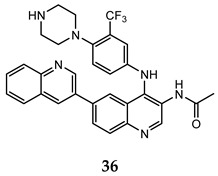
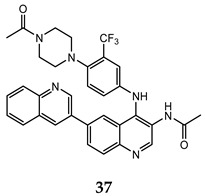
Ma and co-workers reported the synthesis and the biological activities of a series of quinoline derivatives with a substituted aniline at C-4 position, and linked to another quinoline moiety at the C-6 one (compound 35–37 in Table 3). These quinoline compounds showed interesting inhibitory activities on both mTORC1 and mTORC2, with an adequate stability in simulated gastric, intestinal fluids, and liver microsome. The docking studies for these derivatives into the mTOR catalytic cleft showed the importance of some recurrent interactions, that stabilize the complex: hydrogen bond between the nitrogen of central quinoline scaffold and the critical residue Val2240, π– π interaction between indole moiety of Trp2239 and quinoline scaffold, and hydrogen bond between quinoline substituent at C-6 and Tyr2225 [74,75].
Table 3.
Quinoline derivatives 35–37 with interesting activity on both mTORC1 and mTORC2.
| Compound | Antiproliferative Assays (IC50, μM) | mTOR Inhibition Data (IC50, nM) | Ref. |
|---|---|---|---|
|
HCT-116 (IC50 = 0.46 μM) PC-3 (IC50 = 0.61 μM) MCF-7 (IC50 = 0.24 μM) |
14 | [74] |
|
HCT-116 (IC50 = 0.36 μM) PC-3 (IC50 = 0.50 μM) MCF-7 (IC50 = 0.11 μM) |
22 | [75] |
|
HCT-116 (IC50 = 0.11 μM) PC-3 (IC50 = 0.17 μM) MCF-7 (IC50 = 0.05 μM) |
30 | [75] |
Zhao et al. published the synthesis and the biological activity of quinoline derivatives with dual PI3K/mTOR inhibitory effects. This new set of 4-aniline quinoline compounds containing phenylsulfonylurea are a combination of the benzensulfonamido and the anilino moieties, which are generally present in most of the quinoline active molecules reported in this review. Quinoline 38 (Figure 12) is the best inhibitor of the series, with IC50 values against PI3K and mTOR, respectively of 0.72 μM and 2.62 μM, and with an IC50 (half-maximal inhibitory concentration) against MCF-7 cell line comparable to the positive control [76].
Figure 12.

The most active derivative of 3-phenylsulfonylurea-4-aniline quinoline series [76].
The PI3K/mTOR inhibition activity is also conserved in numerous derivatives in which the quinoline scaffold is condensed with other heterocycles. Among these, imidazo[4,5-c]quinolines are the most important, such as investigational drugs like dactolisib (BEZ-235), panulisib (P-7170), LY-3023414, SHR8443, and BGT-226 (Figure 13) [3].
Figure 13.

Imidazo[4,5-c]quinoline derivatives as promising anticancer PI3K/mTOR inhibitors.
Dactolisib,2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1- -yl)-phenyl]-propionitrile, was patented by Novartis in 2006 [77]. It demonstrated high selectivity and potency in the nanomolar concentration range against PI3K/mTOR kinases, acceptable oral bioavailability, potent antiproliferative activity on acute lymphoblastic leukaemia T-cell and also in paclitaxel-resistant gastric cancer models. Moreover, it has been tested in clinical trials, alone or in combination with cytotoxic agents, in patients with advanced solid tumours, as for example nasopharyngeal carcinoma [78,79,80,81,82,83]. From a molecular point of view, in Figure it 14 is shown the binding mode of dactolisib in the catalytic site of PI3Kα as an example for all the imidazo[4,5-c]quinolines: the nitrogen of the quinoline core forms an hydrogen bond with the backbone of the conserved Val851, interaction observed also for the ATP; other two hydrogen bonds involved the -CN substituent and the nitrogen of the lateral quinoline scaffold respectively with the amino acids Ser774 and Asp933 (Figure 14) [78,84].
Figure 14.

Model for the interactions of dactolisib in the ATP-binding cleft of PI3Kα [78,84].
Panulisib (P-7170) was patented in 2012 [85]. This quinoline compounds demonstrated potent inhibition activity on mTORC1/mTORC2/ALK1, cell cycle arrest, induction of apoptosis, antiangiogenic activity. Moreover, it showed antitumor efficacy on both in vitro and in vivo studies against the erlotinib–sensitive and –insensitive models NSCLC and anti-estrogen models of ER+ breast cancer [86,87,88], reaching the clinical trials evaluation for patients with advanced refractory solid tumours (ClinicalTrials.gov number NCT01762410) [89].
LY-3023414, developed as a selective PI3K/mTOR dual inhibitor, was patented in 2012, with an absolute mTOR IC50 and PI3Kα IC50 values respectively of 0.165 μM and 0.00607 μM. In cancer cell human line panels, it showed broad antiproliferative activity [90,91]. In a first-in-human phase I study it demonstrated a tolerable safety profile and single-agent activity in patients with advanced tumours [92].
SHR8443 has been evaluated in vitro for the enzymatic inhibition assay of mTOR, showing IC50 value of 1 nM [93]. Preclinical studies evidenced pharmaceutical properties favourable for the clinical use in antitumor treatments [94].
BGT-226, 8-(6-Methoxy-pyridin-3-yl)-3-methyl-1-(4-piperazin-1-yl-3-trifluoromethyl-phenyl)- 1,3-dihydro-imidazo[4,5-c]quinolin-2-one, developed by Novartis [77], demonstrated effectiveness in reducing in vitro and in vivo the growth of several human cancer types: head and neck [95]; human pancreatic [96]; human acute leukaemia and myeloma cancer cell lines [97,98]; NSCLC cancer (in combination with gefitinib) [99]; normoxic and hypoxic hepatocarcinoma cells [100]. BGT-226 reached the phase I of clinical trials (ClinicalTrials.gov Identifier: NCT00742105) where it demonstrated good tolerability, but exhibited insufficient results for a clinically meaningful PI3K pathway inhibition [101].
Recently, new series of imidazo[4,5-c]quinoline derivatives have been reported in the literature as PI3K/mTOR inhibitors. Xiao et al. proved anticancer properties of a set of quinoline molecules, from which compound 39 (Figure 13) is the most active, with a mTOR IC50 value of 1.4 μM and PI3Kα IC50 of 0.9 μM [102].
Bahekar et al. synthesized quinoline 40, an orally available, selective, and effective inhibitor of PI3Kδ, with IC50 value of 1.9 nM [103].
Besides imidazo[4,5-c]quinolines, also some interesting thieno[3,2-c]quinoline derivatives show PI3K inhibitory activity; compound 41 (Figure 15), for example, exhibits a PI3K IC50 = 1 μM and IC50 values against K562 and DU145 cancer cell lines of 0.15 μM and 2.5 μM, respectively [104].
Figure 15.

Thieno[3,2-c]quinoline compound active as PI3K inhibitor [104].
Furthermore, recently, Guo et al. through high-throughput screening (HTS), selected a novel hit compound 42 (Figure 16a), containing pyrazino[2,3-c]quinolin-2(1H)-one scaffold, as mTOR inhibitor. This molecule demonstrated an mTOR IC50 = 31 nM, but weak antiproliferative activity in vitro. Further structural optimization led to compound 43 (Figure 16), that showed a better mTOR inhibitory activity than the parent compound (IC50 = 7 nM), good selectivity profile, potent antiproliferative activities against breast and cervical cancer lines, and significant tumor regression in the T-47D xenograft model after an oral once-daily dose [105].
Figure 16.

(a) mTOR inhibitors containing pyrazino[2,3-c]quinolin-2(1H)-one scaffold; (b) structures of torin 1 and torin 2, mTOR inhibitors containing benzo[h]-1,6-naphthyridin-2(1H)-one scaffold [105,106,107].
Remarkable interesting, are torin 1 and torin 2, two benzo[h][1,6]-naphthyridin-2(1H)-one compounds, containing a quinoline moiety and acting as selective mTOR inhibitors (Figure 16b). Developed by Liu and co-workers, these tricyclic pyridoquinolines derivatives are the result of two consecutive generations of quinoline-based compounds. They are active on the phosphorylation process of mTORC1/ mTORC2 substrates in the cell, exhibiting EC50 values in the range 0.25–7 nM. Furthermore, with respect to torin 1, second generation compound torin 2 demonstrates 800-fold selectivity over PI3K, improving the bioavailability (54%), metabolic stability, and plasma exposure [106,107].
2.3. Quinoline Derivatives as Inhibitors of Epidermal Growth Factor Receptors
Various quinoline derivatives, with EGFR inhibitory potential, have been studied and the 4-anilinoquinoline-3-carbonitrile class is one of the first group developed. These compounds have been designed by molecular modelling studies, considering the approved EGFR 4-anilinoquinazoline inhibitors (gefitinib, erlotinib, and afatinib) as lead compounds (Figure 17).
Figure 17.

4-anilinoquinoline-3-carbonitrile 44 and of the approved 4-anilinoquinazoline EGFR inhibitors gefitinib, erlotinib, afatinib.
Weissner et al. demonstrated that the N-3 of quinazoline ring system can be replaced by a C-X, where X is an electron withdrawing group (i.e., CN). It was found that 4-anilinoquinoline-3-carbonitriles are effective inhibitors of EGFR kinase with activity comparable to the 4-anilinoquinazoline-based inhibitors. Compound 44 (Figure 17) showed the interesting IC50 value of 7.5 nM [108].
The same authors developed a second generation of EGFR inhibitors, namely 4-anilinoquinoline-3-carbonitrile derivatives bearing Michael acceptor groups (such as 4-(dimethylamino)crotonamide) at the position C-6 (Figure 18).
Figure 18.

Structures of the irreversible EGFR inhibitors pelitinib (EKB-569), neratinib (HKI-272), and pyrotinib (SHR-1258).
These molecules act as EGFR irreversible inhibitors, forming a covalent bond with a conserved cysteine residue located in the ATP binding pocket of EGFR (Cys773 or Cys797) and HER-2 (Cys805) [109]. In particular, pelitinib (EKB-569; EGFR IC50 = 0.083 μM), currently under investigation in clinical trials, showed excellent oral in vivo activity [3,110].
Subsequently, Tsou et al., introducing a lipophilic substituent at the para position of the 4-arylamino ring, isolated the molecule HKI-272, more active than pelitinib, and approved in 2017 with the name neratinib (Figure 18) for the extended adjuvant therapy against early-stage HER2/ErbB2-amplified/overexpressed breast cancer [3,4,109,111,112]. Crystal structure of EGFR/T290M mutant kinase domain in complex with neratinib showed the main interactions recurrent in the binding pocket (Figure 19): non-covalent interactions (H-bond of quinoline nitrogen with residues of the hinge region; hydrophobic interactions between the 2-pyridinyl group and the Met766, Phe856 and Met790) and a covalent bond (Cys797 at the edge of the active site cleft attacked the crotonamide group of the ligand) [113].
Figure 19.

EGFR/T290M mutant kinase domain (PDB id: 2JIV) in complex with neratinib [113].
Starting from the lead compound neratinib, SHR1258 has been developed as irreversible EGFR/HER2 tyrosine kinase inhibitor, and afterwards named pyrotinib (Figure 18). Figure 20 shows a comparison between binding modes of neratinib and pyrotinib in the catalytic region of HER2 kinase. In this model the two molecules are completely superimposed (Figure 20b), except for the substituents on the Michael acceptor group. Both display the same stabilizing interactions: hydrogen bond between N-1 of the quinoline ring and the hinge region of Met801, a covalent bond between Cys805 and Michael acceptor group (double bond) [114].
Figure 20.

(a) binding mode of pyrotinib in the catalytic region of HER2 kinase; (b) overlap binding mode of neratinib (colored blue) and pyrotinib (colored yellow) [114].
In vitro preclinical studies show high potency of pyrotinib, comparable to neratinib, against HER2 dependent BT474 (breast cancer) and SK-OV-3 (ovarian cancer) cell lines and also in vivo efficacy in HER2-dependent mouse xenograft models. Moreover, pyrotinib displays high selectivity when tested against a panel of different kinases [114]. Clinical trials demonstrate a safety profiles, desirable pharmacokinetic properties, tolerability and promising antitumor activity in HER2-positive patients with metastatic breast cancer, especially in combination with capecitabine [115,116]. Currently, pyrotinib is also under trial evaluation for the use in HER2-positive gastric cancer and NSCLC [117].
Pannala et al. described the synthesis and the evaluation of EGFR inhibition of some 4-(2-aryl-cyclopropylamino)-quinoline-3-carbonitriles. These compounds possess an arylcyclopropylamino group at the C-4 position of the quinoline-3-carbonitrile core instead of 4-aniline one. Quinoline 45 (Figure 21) shows an interesting inhibition effect on EGFR with IC50 value of 5 nM [118].
Figure 21.

Structure of 4-(2-aryl-cyclopropylamino)-quinoline-3-carbonitrile as selective inhibitor of EGFR [118].
Aly et al. planned and synthesized a set of 4-anilino-3-carboxyamide derivatives. They based their studies on the high similarities between the 4-anilinoquinazoline, 4-anilinoquinoline-3-carbonitrile, and 4-anilinoquinoline-3-carboxamide nuclei. In a previous work, quinoline 46 (EGFR IC50 = 5.283 μM) was selected as a lead structure for the design of other more active derivatives [119]. In particular, compound 47 with a substituted-thiophene moiety at C-6 (Figure 22) exhibited selective activity on EGFR with IC50 value of 0.49 μM [120].
Figure 22.

4-anilino-3-carboxyamide derivatives as EGFR inhibitors [119,120].
In addition, 2-styrylquinolines 48 and 49, without a 4-anilino substitution (Figure 23), have been synthesized by El-Sayed et al., showing moderate inhibition effects towards EGFR with IC50 values of 2.23 and 1.11 μM respectively [121].
Figure 23.

2-styrylquinolines 48 and 49 as promising EGFR inhibitors [121].
In lasts years, several hybrids derivatives containing quinoline scaffold linked with other heterocyclic ring systems have been studied as EGFR inhibitors with interesting activity. Makawana et al. reported numerous Schiff’s base derivatives bearing nitroimidazole and quinoline nuclei as potential EGFR tyrosine kinase inhibitors. Compound 50 (Figure 24) showed an IC50 value on EGFR receptor of 0.12 ± 0.05 μM [122].
Figure 24.

Schiff’s base quinoline derivative as EGFR tyrosine kinase inhibitor [122].
George et al. described some new quinoline hybrid derivatives bearing either pyrazoline or pirazolinylthiazole heterocycles. Most of the tested compounds revealed promising anticancer activity especially against DLD1 cancer cell line (colorectal). Furthermore, compounds 51–53 (Figure 25) revealed inhibitory effect on EGFR at nanomolar concentration, with IC50 values of 31.80, 37.07, and 42.52 nM, respectively [123].
Figure 25.

Pyrazoline/pirazolinylthiazole quinoline hybrids [123].
The quinoline scaffold is frequently present in numerous polycondensed ring system compounds with proved selective inhibition activity on EGFR.
Abdelbaset et al., for example, designed several thieno[2,3-b]quinoline-2-carboxamide-chalcone derivatives, as molecule 54 (Figure 26a) that showed significant antiproliferative effects against tested cancer cell lines (IC50 values in the range 0.9–1.2 μM) and an EGFR IC50 = 0.5 μM. Molecular docking studies showed that the thienoquinoline moiety occupied the ATP-binding site of the receptor, whereas chalcone moiety was located in an allosteric pocket of the enzyme (Figure 26b) [124].
Figure 26.

(a) Thieno[2,3-b]quinoline-2-carboxamide-chalcone derivative 54; (b) orientation and interactions of 54 within the EGFR active site [124].
Afterwards, Abdelsalam and co-workers designed some benzo[h]quinoline derivatives, as molecule 55 (Figure 27), with good anticancer activity against MCF-7 cancer cell line (IC50 = 7.21 ± 0.43 μM) and also an EGFR IC50 = 0.14 μM [125].
Figure 27.
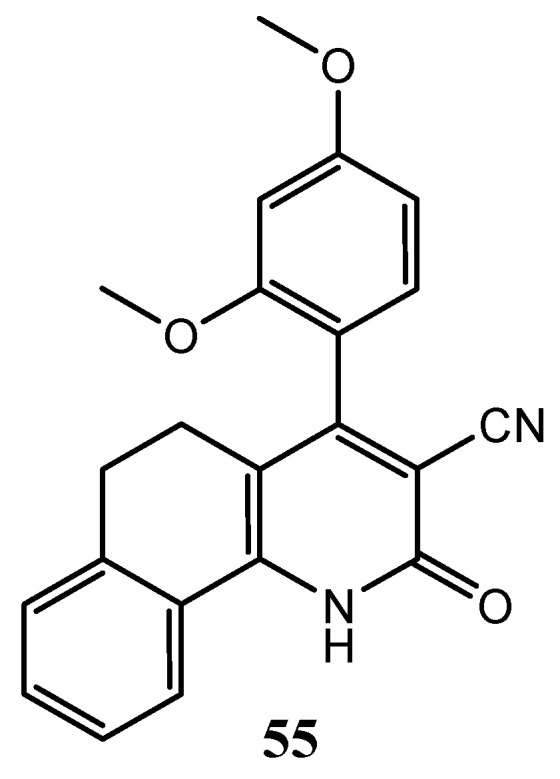
Structure of benzo[h]quinoline derivative [125].
2.4. Quinoline Derivatives as Inhibitors of Vascular Endothelial Growth Factor Receptor
VEGF-VEGFR abnormal signals play central roles in angiogenic processes in a variety of diseases, especially in cancer. Some studies demonstrated, that, although the VEGF binds more effectively the VEGFR-1, its major mitogenic and angiogenic effects seem to be mediated through the interaction with VEGFR of type 2. In recent years, a lot of selective angiogenesis and multi-receptor kinase inhibitors have been synthesized, and some of these have been approved for clinical use [24,25].
Lenvatinib, (Figure 28), approved in 2015 for the treatment of differentiated thyroid, hepatocellular cancers, and the second-line treatment of renal carcinoma, is a multikinase quinoline inhibitor with a selective effect against VEGFR [4,7].
Figure 28.

Structures of VEGFR inhibitors with quinoline-urea moiety lenvatinib, Ki8751, tivozanib.
Many other quinoline derivatives are reported to possess selective VEGFR inhibitor activity, especially against VEGFR-2. Kubo et al. reported the synthesis of some N-Phenyl-N’-{4-(4-quinolyloxy)phenyl}ureas, such as compound Ki8751 (Figure 28), which exhibited the best inhibition effect on VEGFR with IC50 value of 0.9 nM [126].
Nakamura and co-workers firstly reported the antitumor and anti-angiogenic activity of KEN951, after called tivozanib, (Figure 28). With a very similar structure to the previous studied compounds, this quinoline-urea inhibits VEGFR-1, VEGFR-2, and VEGFR-3 tyrosine kinases (IC50 = 30, 6.5, and 15 nM, respectively). Tivozanib potently inhibited VEGF-induced VEGFR-2 phosphorylation in endothelial cells at in vitro sub-nanomolar concentration (IC50 = 0.16 nM). In xenografts models, tivozanib demonstrated significant inhibition of tumor-induced angiogenesis and tumor vessel normalization and in phase I of clinical trial it exhibited safety and tolerability when orally administered [127,128,129].
Analysing the structural analogies between lenvatinib, Ki8751, and tivozanib, it is possible to underline the recurrent and important presence of the C-4 oxyphenyl urea substituted moieties and the C-6, C-7 substitutions on the quinoline scaffold. These structure features play a pivotal role for the stabilization of ligand-protein complex, as it is shown by the X-ray crystal structure of VEGFR-2 in complex with lenvatinib (Figure 29, PDB id: 3WZD): the ligand binds the receptor kinase domain in its DFG-in conformation, occupying the ATP-binding site through its quinoline moiety and the neighboring region via the cyclopropane ring. In particular, the key interactions are three H-bonds (quinoline nitrogen and Cys919; urea oxygen and Asp1046; urea nitrogen and Glu885), two hydrophilic interactions (the C-6 carboxamide with Asn923, bridged by water molecules) and several π-interactions (quinoline and C-4 phenoxy moiety with Leu840, Phe818 and Lys868) [130]. Quinoline core and urea moiety in tivozanib showed the same key interactions observed for the parent compound (PDB id: 4ASE) [131,132].
Figure 29.

(a) crystal structure of lenvatinib in complex with VEGFR-2 (PDB id: 3WZD); (b) binding pocket of VEGFR-2 occupied by lenvatinib; (c) scheme for interactions of lenvatinib in the VEGFR-2 binding pocket [130].
Yang et al. reported the synthesis of some quinoline amide derivatives with VEGFR-2 inhibition activity. Compound 56 (Figure 30) resulted to be the most active to inhibit VEGFR kinase (IC50 = 3.8 nM) and proliferation of HUEVEC cancer cells (IC50 = 5.5 nM); the docking analysis confirmed that this compound is suitable bonded to VEGFR-2 [133].
Figure 30.

Structure of the quinoline VEGFR-2 inhibitor 56 [133].
Through a combination of some structural elements present in molecules with antiproliferative activity, such as 4-piperazinoquinoline scaffold and aminoacyl chain, Aboul-Enein et al. planned and synthesized 7-Chloro-4-(piperazin-1-yl)quinoline derivatives as VEGFR-2 inhibitors. The most promising compound 57 (Figure 31), exhibited cytotoxicity higher than that of reference drug (doxorubicin) against MCF-7 cell line (6.502 μM vs. 6.774 μM) and a VEGFR-2 IC50 = 1.38 μM, but less potent than the reference drug sorafenib (IC50 of 0.33 μM). Docking analysis in the ATP-binding site of VEGFR-2 proved that molecule 57 shows a binding mode similar to that of other VEGFR-2 inhibitors as lenvatinib: both displayed H-bonds between quinoline nitrogen and Cys919, and between the carbonyl group (in the aminoacyl and in urea moieties in derivative 57 and tivozanib, respectively) and Asp1046. However, 57 showed some additional π-interaction with Ile888 and Phe918 [134].
Figure 31.

Structure of compound 57, a 7-Chloro-4-(piperazin-1-yl)quinoline derivative with VEGFR-2 inhibition activity [134].
In 2008, Westman et al. published a series of quinoline-3-carboxylic acid derivatives (Figure 32), acting as VEGFR-2 and -3 inhibitors and with good potency and high selectivity for the treatment and prevention of cell proliferative and/or differentiation disorders. Compounds 58 and 59 showed IC50 values against VEGFR-2 of 1.2 μM and 2.5 μM, and against VEGFR-3 of 0.53 μM and 0.35 μM respectively [135].
Figure 32.

Quinoline-3-carboxylic acid derivatives with VEGFR-2 and VEGFR-3 inhibition activity [135].
Different quinoline derivatives are reported to act as dual inhibitors of VEGFR and other targets involved in carcinogenic processes. For example, Li et al. discovered some 3-aryl-quinoline derivatives 60 and 61 as dual inhibitors of both VEGFR-2 and ERα. Two molecules (Figure 33) showed interesting IC50 against ERα and VEGFR compared to the reference compounds, tamoxifen and sunitinib, respectively (for compound 60, ERα IC50 = 2.33 μM and VEGFR-2 IC50 = 104 nM; for 61, ERα IC50 = 1.78 μM and VEGFR-2 IC50 = 86 nM) [136].
Figure 33.

Structure of 3-aryl-quinoline derivatives 60 and 61, dual VEGFR-2/ERα inhibitors [136].
2.5. Quinoline Derivatives as Inhibitors of Ras/Raf/MEK Pathway
EGFR and VEGFR extracellular activations trigger the intracellular cascade of signalling connected to Ras/Raf/MEK/ERK. This deregulated pathway, in neoplastic cells, improve the tumour genesis, acting on the survival processes of cellular proliferation; such as differentiation, apoptosis, angiogenesis.
Several quinoline derivatives, tested as inhibitors of Ras/Raf/MEK cascade, demonstrate promising results both in antiproliferative and enzymatic inhibition assays. In detail, Feng and co-workers developed quinoline KAL-21404358 (Figure 34), the first ligand of the K-Ras P110 allosteric pocket able to disrupt downstream pathways (Raf/MEK/ERK and PI3K/AkT/mTOR). Through a combination of computational methods and with the validation of biochemical assays, the authors proposed a mechanism of action for KAL-21404358. The quinoline compound could interfere with the protein-protein interactions, binding and stabilizing K-Ras in its inactive GDP-bound state, then, it halts the nucleotide exchange process and the subsequent activation of Ras [137].
Figure 34.

Quinoline-based molecules with inhibition activity on Ras.
In 2016, Li et al. filled a patent of a new set of fused-tricyclic compounds containing quinoline scaffold with a promising inhibition activity on the mutated form of Gly12Cys K-Ras (Figure 34). The analysis of the catalytic binding site, highlights that Cys12 forms a covalent bond with the electrophilic acryloyl moiety of quinolines 62 and 63 [138].
El-Gamal et al. synthesized two class of Raf inhibitors possessing quinoline scaffold. In the first one, a diarylamide moiety, through a S or an O atom, was linked at the C-3 of a dimethoxy/dihydroxy-quinoline scaffold. Biological assays realized by National Cancer Institute demonstrated that the most active compounds were dimethoxyquinolines with an oxygen as linker and with electron-withdrawing groups (-Cl or CF3) on the terminal ring. One of the most active compounds, 64 (Figure 35a), exhibited GI50 values in the micromolar range against the full panel of NCI cancer cell lines and inhibited C-Raf kinase activity by 76.65% at 10 μM [139].
Figure 35.

(a) Structures of diarylurea derivative sorafenib and of quinoline inhibitors 64,65 as selective inhibitors of Raf.; (b) The best docked pose of 64 within the C-Raf active site (two H-bond stabilize the ligand-protein complex: amidic carbonyl oxygen-Thr421, quinolinyl nitrogen-Asn472); (c) best docked pose for 65 in C-Raf active site (three H-bond stabilize the interaction: urea carbonyl oxygen-catalytic Lys470, urea terminal nitrogen-Asp486, quinolinyl nitrogen-Lys431) [139,140].
In the second class of quinoline compounds, the same authors substituted the amide linker with urea one to obtain diarylurea derivatives. In NCI five-dose screening protocol, molecule 65, with the insertion of 4-chloro-3-(trifluoromethyl)phenylurea moiety, was the most promising compound, with a relevant antiproliferative effect compared to the reference diarylurea drug sorafenib (Figure 35a). Inhibition assays of C-Raf kinase evidenced quinoline 65 as more active than the lead compound 64 (for 65 %inhibition at 10 μM = 99.67% and IC50 = 0.10 μM). Docking studies of the active site of C-Raf kinase (PDB ID: 3OMV) further explained the high potency of 65 with respect to 64. Indeed, compound 65, due to the greater flexibility of the urea linker, formed additional H-bonds with Asp486 and Lys470 (Figure 35b,c) [140].
El-Damasy et al. designed and synthetized several quinoline analogues of sorafenib, substituting the central phenoxy nucleus with a quinoline one and inserting the two recurrent moieties of the lead compound, the arylurea/arylamide and the N-methyl picolinamide, respectively at the C-2 and C-5 positions of the quinoline ring system.
NCI antiproliferative assays of urea derivatives with fluorinated phenyl ring, as 66 and 67 (Figure 36a), showed GI50 values in the low-submicromolar range for the majority of the tested cell lines. Furthermore, compound 67 obtained remarkable results in the kinase inhibition assays, with high selectivity against Raf family and IC50 of 316 nM and 61 nM against BRAFV600E and C-Raf, respectively (for the reference sorafenib BRAFV600E IC50 = 38 nM and C-Raf IC50 = 6 nM); instead of quinoline derivative 66, that exerted its inhibitory effect only against C-Raf. Docking studies showed a similar binding mode between 67 and sorafenib in the catalytic kinase domain of BRAFV600E, indeed both molecules formed interactions through the picolinamide, urea, and trifluoromethylphenyl moieties (Figure 36b); on the other hand, the absence of activity of 66 against BRAFV600E was justified by the unfit orientation of the 2,4-difluorophenyl group, far from the hydrophobic pocket of the allosteric site (Figure 36c) [141].
Figure 36.

(a) Quinoline inhibitors of Raf compared with the reference compound sorafenib; (b) overlay of the docked pose of compound 67 (magenta) and co-crystalized sorafenib (black) in the catalytic kinase domain of BRAFV600E (PDB id: 1UVJ); (c) superimposition of the docked pose of compound 67 (magenta) and 66 (orange) in the catalytic kinase domain of BRAFV600E (PDB id: 1UVJ) [141,142].
Considering 67 as lead compound, the same authors, by the substitution of the urea linker with an amine one, developed a new series of 2-anilinoquinolines bearing the N-methylpicolinamide group at the C-5 position of the quinoline scaffold. The derivative 4-chloro-3-(trifluoromethyl)aniline 68 (Figure 36a) manifested more remarkable antiproliferative effects than compound 67 and sorafenib, especially against melanoma and breast cancer panels. However, the replacement of the urea linker with an amine in compound 68 has a negative effect on Raf kinase inhibition, that resulted modestly. Docking studies in the catalytic kinase domain of BRAFV600E, in fact, underline the capability of quinoline 67 to form additional interactions right through its urea moiety; furthermore, the short amine spacer inhibited the insertion of the 4-chloro-3-(trifluoromethyl)phenyl group within the hydrophobic allosteric site adjacent the ATP binding site, precluding some stabilizing hydrophobic interactions [142].
Li et al. developed quinoline derivative 69 (Figure 37) with urea and a 4-fluoro-3-(trifluoromethyl)phenyl moieties, as sorafenib. It exhibited higher antiproliferative activity than the reference compound, against Hep G2, A549 and KCC-853 cancer cell lines and also a higher inhibitory activity against C-Raf (for 69 C-Raf IC50 = 8.7 nM; for sorafenib C-Raf IC50 = 28.5 nM) [143].
Figure 37.

3,3-dimethyl-1H-pyrrolo[3,2-g]quinolin-2(3H)-one derivative as potent C-Raf inhibitor [143].
In the Ras/Raf/MEK signalling cascade, MEK has been also identified as an interesting target for quinoline-based agents able to inhibit its catalytic activity.
Zhang et al. developed 4-(phenoxyanilino)quinoline 70 (Figure 38), that showed high selectivity against MEK enzyme with an IC50 value of 25 nM. SAR studies demonstrated that the cyanoquinoline core is crucial in the inhibition of the enzyme if compared with the less active quinazoline analogues [144]. The same authors also explored the effects of different substitutions on the 4-anilino group in some 4-anilino-3-cyano-6-methoxy-7-(3-morpholino-4-yl-propoxy)quinolines; compounds 71 and 72 (Figure 38), with a phenoxy and phenylsulfonyl substitution respectively, were the most potent (MEK IC50 of 7 and 5,7 nM, respectively) [145].
Figure 38.

Structures of some MEK inhibitors with 4-anilino-quinoline-3-carbonitrile core [144,145,146,147].
Berger and coworkers discovered some ATP competitive inhibitors of MEK1 kinase with a 6,7-dialkoaxy-3-quinolinecarbonitrile core, as 73 and 74 (Figure 38), in which the 4-anilino moiety is substituted with the 1-methyl-1H-imidazol-2-ylsulfonyl one. Compound 73 exhibited a MEK IC50 = 3 nM, exceptional in vitro, (IC50 = 7 nM), and in vivo activity against LoVo xenografts in nude mice [146].
In order to improve the solubility and the oral bioavailability, the same authors further developed molecule 74, with an alkenyl group at C-7 of the quinoline core; this compound demonstrated good in vitro potency (MEK1 IC50 = 12 nM) and high plasma levels after oral dosing in H358 xenograft models [147].
Polycondensed quinoline systems, as the 1H-imidazo[4,5-c]quinoline 75 (Figure 39) show interesting results in MEK kinase inhibition assays, with IC50 values lower than 100 μM; it was demonstrated that the high selectivity for MEK over other kinases depends on the –F substituent at the C-7 position of the quinoline nucleus [148].
Figure 39.

Structure of MEK inhibitor 1H-imidazo[4,5-c]quinoline 75.
An interesting derivative of ursolic acid possessing a condensed chloro-substituted quinoline nucleus and a hydrazide moiety was developed recently by Jin et al. (76 in Figure 40). Compound 76 showed remarkable results as antiproliferative agent, capability to inhibit MEK1 kinase activity (MEK1 IC50 = 64 nM) and activation of Ras/Raf/MEK/ERK pathway. Molecular docking analysis into the MEK1 binding site proved that the ursolic acid skeleton ensured the correct orientation of the hydrazide side chain and of the quinoline ring into the pocket, while the latter were involved in stabilizing interactions with the amino acids [149].
Figure 40.

(a) structure of the quinoline derivative of ursolic acid 76; (b) binding pose of 76 within MEK1 kinase domain (PDB id: 3EQF); (c) ligand interactions of 76 docked into MEK1 active site [149].
3. Conclusions
The quinoline ring system is a scaffold highly frequent in druggable molecules. Currently, a great amount of quinoline derivatives exhibit pharmaceutical activity as antibacterial, antifungal, antimalarial, anthelmintic, local anaesthetic, antipsychotic, and anticancer drugs.
In the anticancer field, the quinoline nucleus has been observed as crucial molecular moiety recurrent in several inhibitors of kinases, for the treatments of a wide range of tumours in the targeted therapeutic approach.
In particular, this review summarizes the biological data of several quinoline compounds, which are active on c-Met, VEGF, and EGF receptors, and on the related proteins involved in the intracellular signalling cascades. Overexpressed in tumour cells, these three protein kinases receptors trigger carcinogenic pathways closely connected with each other, regulating the survival processes in the cell, such as proliferation, apoptosis, differentiation, and angiogenesis.
The druggable attitude of the quinoline scaffold probably lies on its proved biocompatibility, versatility, and chemical accessibility, from which new derivatives can be easily designed and synthesized.
The quinoline nucleus is an electron-deficient ring system with tertiary base properties. The presence of nitrogen withdraws electrons by resonance, interfering with the equal distribution of the π-electron density and suggesting a chemical behaviour similar to the pyridine.
In view of an SAR study, the analysis of the interactions of the quinoline molecules in the protein binding sites, under investigation, highlights recurrent hydrogen bonds with the nitrogen of the quinoline ring and π-π stacking complexes with complementary amino acid residues. The presence of flexible moiety, especially at C-4, C-6, and C-7 positions, frequently consolidates the force of the ligand–protein complexes.
All the biological data of the quinoline compounds, analysed in the present review, are reported in detail, confirming the antiproliferative activity and the pivotal importance of this ring system in the efficacy of several approved drugs. In drug discovery, the information recovered in this manuscript could help in the design and rationale optimization studies for the development of new quinoline multitargets molecules.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Marella A., Tanwar O.P., Saha R., Ali M.R., Srivastava S., Akhter M., Shaquiquzzaman M., Alam M.M. Quinoline: A versatile heterocyclic. Saudi. Pharm. J. 2013;21:1–12. doi: 10.1016/j.jsps.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Afzal O., Kumar S., Haider M.R., Ali M.R., Kumar R., Jaggi M., Bawa S. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2015;97:871–910. doi: 10.1016/j.ejmech.2014.07.044. [DOI] [PubMed] [Google Scholar]
- 3.Musiol R. An overview of quinoline as a privileged scaffold in cancer drug discovery. Expert Opin. Drug Discov. 2017;12:583–597. doi: 10.1080/17460441.2017.1319357. [DOI] [PubMed] [Google Scholar]
- 4.Roskoski R. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019;144:19–50. doi: 10.1016/j.phrs.2019.03.006. [DOI] [PubMed] [Google Scholar]
- 5.Roskoski R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol. Res. 2019;152:104609. doi: 10.1016/j.phrs.2019.104609. [DOI] [PubMed] [Google Scholar]
- 6.Cortes J.E., Gambacorti-Passerini C., Deininger M.W., Mauro M.J., Chuah C., Kim D.W., Dyagil I., Glushko N., Milojkovic D., le Coutre P., et al. Bosutinib Versus Imatinib for Newly Diagnosed Chronic Myeloid Leukemia: Results From the Randomized BFORE Trial. J. Clin. Oncol. 2018;36:231–237. doi: 10.1200/JCO.2017.74.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zschäbitz S., Grüllich C. Lenvantinib: A Tyrosine Kinase Inhibitor of VEGFR 1-3, FGFR 1-4, PDGFRα, KIT and RET. Recent Results Cancer Res. 2018;211:187–198. doi: 10.1007/978-3-319-91442-8_13. [DOI] [PubMed] [Google Scholar]
- 8.Markowitz J.N., Fancher K.M. Cabozantinib: A Multitargeted Oral Tyrosine Kinase Inhibitor. Pharmacotherapy. 2018;38:357–369. doi: 10.1002/phar.2076. [DOI] [PubMed] [Google Scholar]
- 9.Deeks E.D. Neratinib: First Global Approval. Drugs. 2017;77:1695–1704. doi: 10.1007/s40265-017-0811-4. [DOI] [PubMed] [Google Scholar]
- 10.Yamaoka T., Kusumoto S., Ando K., Ohba M., Ohmori T. Receptor Tyrosine Kinase-Targeted Cancer Therapy. Int. J. Mol. Sci. 2018;19:3491. doi: 10.3390/ijms19113491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y., Xia M., Jin K., Wang S., Wei H., Fan C., Wu Y., Li X., Li G., Zeng Z., et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer. 2018;17:45. doi: 10.1186/s12943-018-0796-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Birchmeier C., Birchmeier W., Gherardi E., Vande Woude G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 13.Organ S.L., Tsao M.S. An overview of the c-MET signaling pathway. Ther. Adv. Med. Oncol. 2011;3:S7–S19. doi: 10.1177/1758834011422556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson M., Kochhar K., Nakamura T., Iyer A. Hepatocyte growth factor-induced signal transduction in two normal mouse epithelial cell lines. Biochem. Mol. Biol. Int. 1995;36:465–474. [PubMed] [Google Scholar]
- 15.Goyal L., Muzumdar M.D., Zhu A.X. Targeting the HGF/c-MET pathway in hepatocellular carcinoma. Clin. Cancer Res. 2013;19:2310–2318. doi: 10.1158/1078-0432.CCR-12-2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maroun C.R., Rowlands T. The Met receptor tyrosine kinase: A key player in oncogenesis and drug resistance. Pharmacol. Ther. 2014;142:316–338. doi: 10.1016/j.pharmthera.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 17.Emerling B.M., Akcakanat A. Targeting PI3K/mTOR signaling in cancer. Cancer Res. 2011;71:7351–7359. doi: 10.1158/0008-5472.CAN-11-1699. [DOI] [PubMed] [Google Scholar]
- 18.Liu P., Cheng H., Roberts T.M., Zhao J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown K.K., Toker A. The phosphoinositide 3-kinase pathway and therapy resistance in cancer. F1000Prime Rep. 2015;7:13. doi: 10.12703/P7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang J., Jiang X., Jiang Y., Guo M., Zhang S., Li J., He J., Liu J., Wang J., Ouyang L. Recent advances in the development of dual VEGFR and c-Met small molecule inhibitors as anticancer drugs. Eur. J. Med. Chem. 2016;108:495–504. doi: 10.1016/j.ejmech.2015.12.016. [DOI] [PubMed] [Google Scholar]
- 21.Guardiola S., Varese M., Sanchez-Navarro M., Giralt E. A Third Shot at EGFR: New Opportunities in Cancer Therapy. Trends Pharmacol. Sci. 2019;40:941–955. doi: 10.1016/j.tips.2019.10.004. [DOI] [PubMed] [Google Scholar]
- 22.Lemmon M.A., Schlessinger J., Ferguson K.M. The EGFR family: Not so prototypical receptor tyrosine kinases. Cold Spring Harb. Perspect Biol. 2014;6:a020768. doi: 10.1101/cshperspect.a020768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Citri A., Yarden Y. EGF-ERBB signalling: Towards the systems level. Nat. Rev. Mol. Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 24.Shibuya M. VEGF-VEGFR Signals in Health and Disease. Biomol. Ther. (Seoul) 2014;22:1–9. doi: 10.4062/biomolther.2013.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrara N., Gerber H.P., LeCouter J. The biology of VEGF and its receptors. Nat. Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 26.De Luca A., Maiello M.R., D’Alessio A., Pergameno M., Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: Role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets. 2012;16(Suppl. 2):S17–S27. doi: 10.1517/14728222.2011.639361. [DOI] [PubMed] [Google Scholar]
- 27.Asati V., Mahapatra D.K., Bharti S.K. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2016;109:314–341. doi: 10.1016/j.ejmech.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 28.Yarden Y. The EGFR family and its ligands in human cancer: Signalling mechanisms and therapeutic opportunities. Eur. J. Cancer. 2001;37(Suppl. 4):S3–S8. doi: 10.1016/S0959-8049(01)00230-1. [DOI] [PubMed] [Google Scholar]
- 29.Yakes F.M., Chen J., Tan J., Yamaguchi K., Shi Y., Yu P., Qian F., Chu F., Bentzien F., Cancilla B., et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011;10:2298–2308. doi: 10.1158/1535-7163.MCT-11-0264. [DOI] [PubMed] [Google Scholar]
- 30.Underiner T.L., Herbertz T., Miknyoczki S.J. Discovery of small molecule c-Met inhibitors: Evolution and profiles of clinical candidates. Anticancer Agents Med. Chem. 2010;10:7–27. doi: 10.2174/1871520611009010007. [DOI] [PubMed] [Google Scholar]
- 31.Qian F., Engst S., Yamaguchi K., Yu P., Won K.A., Mock L., Lou T., Tan J., Li C., Tam D., et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 2009;69:8009–8016. doi: 10.1158/0008-5472.CAN-08-4889. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Q.W., Ye Z.D., Shen C., Tie H.X., Wang L., Shi L. Synthesis of novel 6,7-dimethoxy-4-anilinoquinolines as potent c-Met inhibitors. J. Enzyme Inhib. Med. Chem. 2019;34:124–133. doi: 10.1080/14756366.2018.1533822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li S., Huang Q., Liu Y., Zhang X., Liu S., He C., Gong P. Design, synthesis and antitumour activity of bisquinoline derivatives connected by 4-oxy-3-fluoroaniline moiety. Eur. J. Med. Chem. 2013;64:62–73. doi: 10.1016/j.ejmech.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 34.Parikh P.K., Ghate M.D. Recent advances in the discovery of small molecule c-Met Kinase inhibitors. Eur. J. Med. Chem. 2018;143:1103–1138. doi: 10.1016/j.ejmech.2017.08.044. [DOI] [PubMed] [Google Scholar]
- 35.Liu X., Kou J., Xiao Z., Tian F., Hu J., Zheng P., Zhu W. Design, Synthesis and Biological Evaluation of 6,7-Disubstituted-4-phenoxyquinoline Derivatives Bearing Pyridazinone Moiety as c-Met Inhibitors. Molecules. 2018;23:1543. doi: 10.3390/molecules23071543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu J., Yang D., Yang X., Nie M., Wu G., Wang Z., Li W., Liu Y., Gong P. Design, synthesis and biological evaluation of novel 4-phenoxyquinoline derivatives containing 3-oxo-3,4-dihydroquinoxaline moiety as c-Met kinase inhibitors. Bioorg. Med. Chem. 2017;25:4475–4486. doi: 10.1016/j.bmc.2017.06.037. [DOI] [PubMed] [Google Scholar]
- 37.Wang X., Jiang N., Zhao S., Xi S., Wang J., Jing T., Zhang W., Guo M., Gong P., Zhai X. Design, synthesis and biological evaluation of novel 4-(2-fluorophenoxy)quinoline derivatives as selective c-Met inhibitors. Bioorg. Med. Chem. 2017;25:886–896. doi: 10.1016/j.bmc.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 38.Liu M., Hou Y., Yin W., Zhou S., Qian P., Guo Z., Xu L., Zhao Y. Discovery of a novel 6,7-disubstituted-4-(2-fluorophenoxy)quinolines bearing 1,2,3-triazole-4-carboxamide moiety as potent c-Met kinase inhibitors. Eur. J. Med. Chem. 2016;119:96–108. doi: 10.1016/j.ejmech.2016.04.035. [DOI] [PubMed] [Google Scholar]
- 39.Zhou S., Liao H., Liu M., Feng G., Fu B., Li R., Cheng M., Zhao Y., Gong P. Discovery andw biological evaluation of novel 6,7-disubstituted-4-(2-fluorophenoxy)quinoline derivatives possessing 1,2,3-triazole-4-carboxamide moiety as c-Met kinase inhibitors. Bioorg. Med. Chem. 2014;22:6438–6452. doi: 10.1016/j.bmc.2014.09.037. [DOI] [PubMed] [Google Scholar]
- 40.Liao W., Hu G., Guo Z., Sun D., Zhang L., Bu Y., Li Y., Liu Y., Gong P. Design and biological evaluation of novel 4-(2-fluorophenoxy)quinoline derivatives bearing an imidazolone moiety as c-Met kinase inhibitors. Bioorg. Med. Chem. 2015;23:4410–4422. doi: 10.1016/j.bmc.2015.06.026. [DOI] [PubMed] [Google Scholar]
- 41.Liao W., Xu C., Ji X., Hu G., Ren L., Liu Y., Li R., Gong P., Sun T. Design and optimization of novel 4-(2-fluorophenoxy)quinoline derivatives bearing a hydrazone moiety as c-Met kinase inhibitors. Eur. J. Med. Chem. 2014;87:508–518. doi: 10.1016/j.ejmech.2014.09.095. [DOI] [PubMed] [Google Scholar]
- 42.Tang Q., Zhao Y., Du X., Chong L., Gong P., Guo C. Design, synthesis, and structure-activity relationships of novel 6,7-disubstituted-4-phenoxyquinoline derivatives as potential antitumor agents. Eur. J. Med. Chem. 2013;69:77–89. doi: 10.1016/j.ejmech.2013.08.019. [DOI] [PubMed] [Google Scholar]
- 43.Li S., Zhao Y., Wang K., Gao Y., Han J., Cui B., Gong P. Discovery of novel 4-(2-fluorophenoxy)quinoline derivatives bearing 4-oxo-1,4-dihydrocinnoline-3-carboxamide moiety as c-Met kinase inhibitors. Bioorg. Med. Chem. 2013;21:2843–2855. doi: 10.1016/j.bmc.2013.04.013. [DOI] [PubMed] [Google Scholar]
- 44.Qi B., Tao H., Wu D., Bai J., Shi Y., Gong P. Synthesis and biological evaluation of 4-phenoxy-6,7-disubstituted quinolines possessing semicarbazone scaffolds as selective c-Met inhibitors. Arch. Pharm. (Weinheim) 2013;346:596–609. doi: 10.1002/ardp.201300087. [DOI] [PubMed] [Google Scholar]
- 45.Gong P., Zhao Y., Liu Y., Zhai X. Novel quinoline derivatives and their applications. 9783499 B2. U.S. Patent. 2017 Oct 10;
- 46.Kung P.P., Funk L., Meng J., Alton G., Padrique E., Mroczkowski B. Structure activity relationships of quinoline-containing c-Met inhibitors. Eur. J. Med. Chem. 2008;43:1321–1329. doi: 10.1016/j.ejmech.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 47.Xu H. Preparation of Quinoline Compounds Containing 1,2,4-Triazine-Dione and Use as c-Met Kinase Inhibitors for Treating Proliferative Diseases. 20130252958 A1. U.S. Patent. 2013 Sep 26;
- 48.Tang Q., Duan Y., Xiong H., Chen T., Xiao Z., Wang L., Xiao Y., Huang S., Xiong Y., Zhu W., et al. Synthesis and antiproliferative activity of 6,7-disubstituted-4-phenoxyquinoline derivatives bearing the 1,8-naphthyridin-2-one moiety. Eur. J. Med. Chem. 2018;158:201–213. doi: 10.1016/j.ejmech.2018.08.066. [DOI] [PubMed] [Google Scholar]
- 49.Zhang W., Ai J., Shi D., Peng X., Ji Y., Liu J., Geng M., Li Y. Discovery of novel type II c-Met inhibitors based on BMS-777607. Eur. J. Med. Chem. 2014;80:254–266. doi: 10.1016/j.ejmech.2014.04.056. [DOI] [PubMed] [Google Scholar]
- 50.Li S., Jiang R., Qin M., Liu H., Zhang G., Gong P. Synthesis and antitumor activity of novel 4-(2-fluorophenoxy)quinoline derivatives bearing the 4-oxo-1,4-dihydroquinoline-3-carboxamide moiety. Arch. Pharm. (Weinheim) 2013;346:521–533. doi: 10.1002/ardp.201300029. [DOI] [PubMed] [Google Scholar]
- 51.Tang Q., Zhang G., Du X., Zhu W., Li R., Lin H., Li P., Cheng M., Gong P., Zhao Y. Discovery of novel 6,7-disubstituted-4-phenoxyquinoline derivatives bearing 5-(aminomethylene)pyrimidine-2,4,6-trione moiety as c-Met kinase inhibitors. Bioorg. Med. Chem. 2014;22:1236–1249. doi: 10.1016/j.bmc.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 52.Liu Z., Wang R., Guo R., Hu J., Li R., Zhao Y., Gong P. Design, synthesis and biological evaluation of novel 6,7-disubstituted-4-phenoxyquinoline derivatives bearing 4-oxo-3,4-dihydrophthalazine-1-carboxamide moieties as c-Met kinase inhibitors. Bioorg. Med. Chem. 2014;22:3642–3653. doi: 10.1016/j.bmc.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 53.Zhou S., Ren J., Liu M., Ren L., Liu Y., Gong P. Design, synthesis and pharmacological evaluation of 6,7-disubstituted-4-phenoxyquinoline derivatives as potential antitumor agents. Bioorg. Chem. 2014;57:30–42. doi: 10.1016/j.bioorg.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 54.Zhai X., Bao G., Wang L., Cheng M., Zhao M., Zhao S., Zhou H., Gong P. Design, synthesis and biological evaluation of novel 4-phenoxy-6,7-disubstituted quinolines possessing (thio)semicarbazones as c-Met kinase inhibitors. Bioorg. Med. Chem. 2016;24:1331–1345. doi: 10.1016/j.bmc.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 55.Wang Y., Ai J., Chen Y., Wang L., Liu G., Geng M., Zhang A. Synthesis and c-Met kinase inhibition of 3,5-disubstituted and 3,5,7-trisubstituted quinolines: Identification of 3-(4-acetylpiperazin-1-yl)-5-(3-nitrobenzylamino)-7- (trifluoromethyl)quinoline as a novel anticancer agent. J. Med. Chem. 2011;54:2127–2142. doi: 10.1021/jm101340q. [DOI] [PubMed] [Google Scholar]
- 56.Wu K., Ai J., Liu Q., Chen T., Zhao A., Peng X., Wang Y., Ji Y., Yao Q., Xu Y., et al. Multisubstituted quinoxalines and pyrido[2,3-d]pyrimidines: Synthesis and SAR study as tyrosine kinase c-Met inhibitors. Bioorg. Med. Chem. Lett. 2012;22:6368–6372. doi: 10.1016/j.bmcl.2012.08.075. [DOI] [PubMed] [Google Scholar]
- 57.Wang Y., Ai J., Yue J., Peng X., Ji Y., Zhao A., Gao X., Wang Y., Chen Y., Liu G., et al. Further SAR studies on 3,5-diamino-7-trifluoromethylquinolines as highly potent tyrosine kinase c-Met inhibitors: Efforts to correct hERG inhibition. MedChemComm. 2012;3:1423–1427. doi: 10.1039/c2md20192e. [DOI] [Google Scholar]
- 58.Nishii H., Chiba T., Morikami K., Fukami T.A., Sakamoto H., Ko K., Koyano H. Discovery of 6-benzyloxyquinolines as c-Met selective kinase inhibitors. Bioorg. Med. Chem. Lett. 2010;20:1405–1409. doi: 10.1016/j.bmcl.2009.12.109. [DOI] [PubMed] [Google Scholar]
- 59.Lien V.T., Pettersen S., Haugen M.H., Olberg D.E., Maelandsmo G.M., Klaveness J. Design, synthesis and biological evaluation of 6-substituted quinolines derived from cabozantinib as c-Met inhibitors. Arch. Pharm. (Weinheim) 2019;352:e1900101. doi: 10.1002/ardp.201900101. [DOI] [PubMed] [Google Scholar]
- 60.Cui J.J., McTigue M., Nambu M., Tran-Dube M., Pairish M., Shen H., Jia L., Cheng H., Hoffman J., Le P., et al. Discovery of a novel class of exquisitely selective mesenchymal-epithelial transition factor (c-MET) protein kinase inhibitors and identification of the clinical candidate 2-(4-(1-(quinolin-6-ylmethyl)-1H-[1,2,3]triazolo[4,5-b]pyrazin-6-yl)-1H-pyrazol-1 -yl)ethanol (PF-04217903) for the treatment of cancer. J. Med. Chem. 2012;55:8091–8109. doi: 10.1021/jm300967g. [DOI] [PubMed] [Google Scholar]
- 61.Diamond S., Boer J., Maduskuie T.P., Falahatpisheh N., Li Y., Yeleswaram S. Species-specific metabolism of SGX523 by aldehyde oxidase and the toxicological implications. Drug Metab. Dispos. 2010;38:1277–1285. doi: 10.1124/dmd.110.032375. [DOI] [PubMed] [Google Scholar]
- 62.Infante J.R., Rugg T., Gordon M., Rooney I., Rosen L., Zeh K., Liu R., Burris H.A., Ramanathan R.K. Unexpected renal toxicity associated with SGX523, a small molecule inhibitor of MET. Invest. New Drugs. 2013;31:363–369. doi: 10.1007/s10637-012-9823-9. [DOI] [PubMed] [Google Scholar]
- 63.Lolkema M.P., Bohets H.H., Arkenau H.T., Lampo A., Barale E., de Jonge M.J.A., van Doorn L., Hellemans P., de Bono J.S., Eskens F.A.L.M. The c-Met Tyrosine Kinase Inhibitor JNJ-38877605 Causes Renal Toxicity through Species-Specific Insoluble Metabolite Formation. Clin. Cancer Res. 2015;21:2297–2304. doi: 10.1158/1078-0432.CCR-14-3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang J.W., Xiao W., Gao Z.T., Yu Z.T., Zhang J.Y.J. Metabolism of c-Met Kinase Inhibitors Containing Quinoline by Aldehyde Oxidase, Electron Donating, and Steric Hindrance Effect. Drug Metab. Dispos. 2018;46:1847–1855. doi: 10.1124/dmd.118.081919. [DOI] [PubMed] [Google Scholar]
- 65.Zhao F., Zhang J., Zhang L., Hao Y., Shi C., Xia G., Yu J., Liu Y. Discovery and optimization of a series of imidazo[4,5-b]pyrazine derivatives as highly potent and exquisitely selective inhibitors of the mesenchymal-epithelial transition factor (c-Met) protein kinase. Bioorg. Med. Chem. 2016;24:4281–4290. doi: 10.1016/j.bmc.2016.07.019. [DOI] [PubMed] [Google Scholar]
- 66.Zhao F., Zhang L.D., Hao Y., Chen N., Bai R., Wang Y.J., Zhang C.C., Li G.S., Hao L.J., Shi C., et al. Identification of 3-substituted-6-(1-(1H-[1,2,3]triazolo[4,5-b]pyrazin-1-yl)ethyl)quinoline derivatives as highly potent and selective mesenchymal-epithelial transition factor (c-Met) inhibitors via metabolite profiling-based structural optimization. Eur. J. Med. Chem. 2017;134:147–158. doi: 10.1016/j.ejmech.2017.03.085. [DOI] [PubMed] [Google Scholar]
- 67.Knight S.D., Adams N.D., Burgess J.L., Chaudhari A.M., Darcy M.G., Donatelli C.A., Luengo J.I., Newlander K.A., Parrish C.A., Ridgers L.H., et al. Discovery of GSK2126458, a Highly Potent Inhibitor of PI3K and the Mammalian Target of Rapamycin. ACS Med. Chem. Lett. 2010;1:39–43. doi: 10.1021/ml900028r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Munster P., Aggarwal R., Hong D., Schellens J.H., van der Noll R., Specht J., Witteveen P.O., Werner T.L., Dees E.C., Bergsland E., et al. First-in-Human Phase I Study of GSK2126458, an Oral Pan-Class I Phosphatidylinositol-3-Kinase Inhibitor, in Patients with Advanced Solid Tumor Malignancies. Clin. Cancer Res. 2016;22:1932–1939. doi: 10.1158/1078-0432.CCR-15-1665. [DOI] [PubMed] [Google Scholar]
- 69.Nishimura N., Siegmund A., Liu L., Yang K., Bryan M.C., Andrews K.L., Bo Y., Booker S.K., Caenepeel S., Freeman D., et al. Phospshoinositide 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) dual inhibitors: Discovery and structure-activity relationships of a series of quinoline and quinoxaline derivatives. J. Med. Chem. 2011;54:4735–4751. doi: 10.1021/jm200386s. [DOI] [PubMed] [Google Scholar]
- 70.Lv X., Ying H., Ma X., Qiu N., Wu P., Yang B., Hu Y. Design, synthesis and biological evaluation of novel 4-alkynyl-quinoline derivatives as PI3K/mTOR dual inhibitors. Eur. J. Med. Chem. 2015;99:36–50. doi: 10.1016/j.ejmech.2015.05.025. [DOI] [PubMed] [Google Scholar]
- 71.Zhang J., Lv X., Ma X., Hu Y. Discovery of a series of N-(5-(quinolin-6-yl)pyridin-3-yl)benzenesulfonamides as PI3K/mTOR dual inhibitors. Eur. J. Med. Chem. 2017;127:509–520. doi: 10.1016/j.ejmech.2017.01.016. [DOI] [PubMed] [Google Scholar]
- 72.Ma X., Shen L., Zhang J., Liu G., Zhan S., Ding B., Lv X. Novel 4-Acrylamido-Quinoline Derivatives as Potent PI3K/mTOR Dual Inhibitors: The Design, Synthesis, and. Front. Chem. 2019;7:236. doi: 10.3389/fchem.2019.00236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu Y., Wan W.Z., Li Y., Zhou G.L., Liu X.G. Recent development of ATP-competitive small molecule phosphatidylinostitol-3-kinase inhibitors as anticancer agents. Oncotarget. 2017;8:7181–7200. doi: 10.18632/oncotarget.12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ma X., Lv X., Qiu N., Yang B., He Q., Hu Y. Discovery of novel quinoline-based mTOR inhibitors via introducing intra-molecular hydrogen bonding scaffold (iMHBS): The design, synthesis and biological evaluation. Bioorg. Med. Chem. 2015;23:7585–7596. doi: 10.1016/j.bmc.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 75.Ma X., Qiu N., Yang B., He Q., Hu Y. Novel quinoline-derived mTOR inhibitors with remarkable enzymatic and cellular activities: The design, synthesis and biological evaluation. Med. Chem. Commun. 2016;7:297–310. doi: 10.1039/C5MD00401B. [DOI] [Google Scholar]
- 76.Zhao B., Lei F., Wang C., Zhang B., Yang Z., Li W., Zhu W., Xu S. Design, Synthesis and Biological Evaluation of Novel Phenylsulfonylurea Derivatives as PI3K/mTOR Dual Inhibitors. Molecules. 2018;23:1553. doi: 10.3390/molecules23071553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Garcia-Echeverria C., Stauffer F., Furet P. 1,3-Dihydro-Imidazo[4,5-c]Quinolin-2-Ones as Lipid Kinase Inhibitors. PCT/EP2006/004725. U.S. Patent. 2006 Nov 23;
- 78.Maira S.M., Stauffer F., Brueggen J., Furet P., Schnell C., Fritsch C., Brachmann S., Chène P., De Pover A., Schoemaker K., et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 79.Serra V., Markman B., Scaltriti M., Eichhorn P.J., Valero V., Guzman M., Botero M.L., Llonch E., Atzori F., Di Cosimo S., et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68:8022–8030. doi: 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 80.Chiarini F., Grimaldi C., Ricci F., Tazzari P.L., Evangelisti C., Ognibene A., Battistelli M., Falcieri E., Melchionda F., Pession A., et al. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblastic leukemia. Cancer Res. 2010;70:8097–8107. doi: 10.1158/0008-5472.CAN-10-1814. [DOI] [PubMed] [Google Scholar]
- 81.Bendell J.C., Kurkjian C., Infante J.R., Bauer T.M., Burris H.A., Greco F.A., Shih K.C., Thompson D.S., Lane C.M., Finney L.H., et al. A phase 1 study of the sachet formulation of the oral dual PI3K/mTOR inhibitor BEZ235 given twice daily (BID) in patients with advanced solid tumors. Investig. New Drugs. 2015;33:463–471. doi: 10.1007/s10637-015-0218-6. [DOI] [PubMed] [Google Scholar]
- 82.Yang F., Qian X.J., Qin W., Deng R., Wu X.Q., Qin J., Feng G.K., Zhu X.F. Dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 has a therapeutic potential and sensitizes cisplatin in nasopharyngeal carcinoma. PLoS ONE. 2013;8:e59879. doi: 10.1371/journal.pone.0059879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fazio N., Buzzoni R., Baudin E., Antonuzzo L., Hubner R.A., Lahner H., WW D.E.H., Raderer M., Teule A., Capdevila J., et al. A Phase II Study of BEZ235 in Patients with Everolimus-resistant, Advanced Pancreatic Neuroendocrine Tumours. Anticancer Res. 2016;36:713–719. [PMC free article] [PubMed] [Google Scholar]
- 84.Seixas J.D., Luengo-Arratta S.A., Diaz R., Saldivia M., Rojas-Barros D.I., Manzano P., Gonzalez S., Berlanga M., Smith T.K., Navarro M., et al. Establishment of a structure-activity relationship of 1H-imidazo[4,5-c]quinoline-based kinase inhibitor NVP-BEZ235 as a lead for African sleeping sickness. J. Med. Chem. 2014;57:4834–4848. doi: 10.1021/jm500361r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kumar S., Sharma R., Zahler R., Sahu B., Agarwal V.R., Naik N. Substituted Imidazoquinoline Derivatives as Kinase Inhibitors. PCT/IB2011/053161. U.S. Patent. 2012 Jan 19;
- 86.Venkatesha V.A., Joshi A., Venkataraman M., Sonawane V., Bhatia D., Tannu P., Bose J., Choudhari S., Srivastava A., Pandey P.K., et al. P7170, a novel inhibitor of mTORC1/mTORC2 and Activin receptor-like Kinase 1 (ALK1) inhibits the growth of non small cell lung cancer. Mol. Cancer. 2014;13:259. doi: 10.1186/1476-4598-13-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jalota-Badhwar A., Bhatia D.R., Boreddy S., Joshi A., Venkatraman M., Desai N., Chaudhari S., Bose J., Kolla L.S., Deore V., et al. P7170: A Novel Molecule with Unique Profile of mTORC1/C2 and Activin Receptor-like Kinase 1 Inhibition Leading to Antitumor and Antiangiogenic Activity. Mol. Cancer Ther. 2015;14:1095–1106. doi: 10.1158/1535-7163.MCT-14-0486. [DOI] [PubMed] [Google Scholar]
- 88.Bean J.R., Hosford S.R., Symonds L.K., Owens P., Dillon L.M., Yang W., Shee K., Schwartz G.N., Marotti J.D., Muller K.E., et al. The PI3K/mTOR dual inhibitor P7170 demonstrates potent activity against endocrine-sensitive and endocrine-resistant ER+ breast cancer. Breast Cancer Res. Treat. 2015;149:69–79. doi: 10.1007/s10549-014-3201-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vicier C., Dieci M.V., Arnedos M., Delaloge S., Viens P., Andre F. Clinical development of mTOR inhibitors in breast cancer. Breast Cancer Res. 2014;16:203. doi: 10.1186/bcr3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Barda D.A., Mader M.M. Imidazo[4,5-c]Quinolin-2-one Compound and its Use as PI3 Kinase/mTor Dual Inhibitor. PCT/US2012/020897. U.S. Patent. 2012 Jul 19;
- 91.Smith M.C., Mader M.M., Cook J.A., Iversen P., Ajamie R., Perkins E., Bloem L., Yip Y.Y., Barda D.A., Waid P.P., et al. Characterization of LY3023414, a Novel PI3K/mTOR Dual Inhibitor Eliciting Transient Target Modulation to Impede Tumor Growth. Mol. Cancer Ther. 2016;15:2344–2356. doi: 10.1158/1535-7163.MCT-15-0996. [DOI] [PubMed] [Google Scholar]
- 92.Bendell J.C., Varghese A.M., Hyman D.M., Bauer T.M., Pant S., Callies S., Lin J., Martinez R., Wickremsinhe E., Fink A., et al. A First-in-Human Phase 1 Study of LY3023414, an Oral PI3K/mTOR Dual Inhibitor, in Patients with Advanced Cancer. Clin. Cancer Res. 2018;24:3253–3262. doi: 10.1158/1078-0432.CCR-17-3421. [DOI] [PubMed] [Google Scholar]
- 93.Li X., Dong Q., Chen Y., Wang B., Bai D. Imidazo Quinoline Derivative and Medicinal Salt Thereof, Preparation Method Thereof and Use in Medicine Thereof. PCT/CN2012/081330. U.S. Patent. 2013 Apr 18;
- 94.Xie C., Chen X., Zheng M., Liu X., Wang H., Lou L. Pharmacologic characterization of SHR8443, a novel dual inhibitor of phosphatidylinositol 3-kinase and mammalian target of rapamycin. Oncotarget. 2017;8:107977–107990. doi: 10.18632/oncotarget.22439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chang K.Y., Tsai S.Y., Wu C.M., Yen C.J., Chuang B.F., Chang J.Y. Novel phosphoinositide 3-kinase/mTOR dual inhibitor, NVP-BGT226, displays potent growth-inhibitory activity against human head and neck cancer cells in vitro and in vivo. Clin. Cancer Res. 2011;17:7116–7126. doi: 10.1158/1078-0432.CCR-11-0796. [DOI] [PubMed] [Google Scholar]
- 96.Glienke W., Maute L., Wicht J., Bergmann L. The dual PI3K/mTOR inhibitor NVP-BGT226 induces cell cycle arrest and regulates Survivin gene expression in human pancreatic cancer cell lines. Tumour Biol. 2012;33:757–765. doi: 10.1007/s13277-011-0290-2. [DOI] [PubMed] [Google Scholar]
- 97.Kampa-Schittenhelm K.M., Heinrich M.C., Akmut F., Rasp K.H., Illing B., Dohner H., Dohner K., Schittenhelm M.M. Cell cycle-dependent activity of the novel dual PI3K-MTORC1/2 inhibitor NVP-BGT226 in acute leukemia. Mol. Cancer. 2013;12:46. doi: 10.1186/1476-4598-12-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Baumann P., Schneider L., Mandl-Weber S., Oduncu F., Schmidmaier R. Simultaneous targeting of PI3K and mTOR with NVP-BGT226 is highly effective in multiple myeloma. Anticancer Drugs. 2012;23:131–138. doi: 10.1097/CAD.0b013e32834c8683. [DOI] [PubMed] [Google Scholar]
- 99.Katanasaka Y., Kodera Y., Yunokawa M., Kitamura Y., Tamura T., Koizumi F. Synergistic anti-tumor effects of a novel phosphatidyl inositol-3 kinase/mammalian target of rapamycin dual inhibitor BGT226 and gefitinib in non-small cell lung cancer cell lines. Cancer Lett. 2014;347:196–203. doi: 10.1016/j.canlet.2014.02.025. [DOI] [PubMed] [Google Scholar]
- 100.Simioni C., Cani A., Martelli A.M., Zauli G., Alameen A.A., Ultimo S., Tabellini G., McCubrey J.A., Capitani S., Neri L.M. The novel dual PI3K/mTOR inhibitor NVP-BGT226 displays cytotoxic activity in both normoxic and hypoxic hepatocarcinoma cells. Oncotarget. 2015;6:17147–17160. doi: 10.18632/oncotarget.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Minami H., Fujiwara Y., Muro K., Sato M., Moriya A. Phase I study of BGT226, a pan-PI3K and mTOR inhibitor, in Japanese patients with advanced solid cancers. Cancer Chemother Pharmacol. 2019;84:337–343. doi: 10.1007/s00280-019-03883-6. [DOI] [PubMed] [Google Scholar]
- 102.Xiao Z., Lei F., Chen X., Wang X., Cao L., Ye K., Zhu W., Xu S. Design, synthesis, and antitumor evaluation of quinoline-imidazole derivatives. Arch. Pharm. (Weinheim) 2018;351:e1700407. doi: 10.1002/ardp.201700407. [DOI] [PubMed] [Google Scholar]
- 103.Bahekar R., Dave B., Soman S., Patel D., Chopade R., Funde R., Kumar J., Sachchidanand S., Giri P., Chatterjee A., et al. Discovery of 1,3-dihydro-2H-imidazo[4,5-c]quinolin-2-ones based novel, potent and PI3Kdelta selective inhibitors. Bioorg. Med. Chem. Lett. 2019;29:1313–1319. doi: 10.1016/j.bmcl.2019.04.007. [DOI] [PubMed] [Google Scholar]
- 104.Reddy E., Reddy M.V. 3-Aminothieno[3,2-c]Quinoline Derivatives, Methods of Preparation and Uses. PCT/US2013/027778. U.S. Patent. 2013 Sep 26;
- 105.Guo Q., Yu C., Zhang C., Li Y., Wang T., Huang Z., Wang X., Zhou W., Qin Z., Wang C., et al. Highly Selective, Potent, and Oral mTOR Inhibitor for Treatment of Cancer as Autophagy Inducer. J. Med. Chem. 2018;61:881–904. doi: 10.1021/acs.jmedchem.7b01402. [DOI] [PubMed] [Google Scholar]
- 106.Liu Q., Chang J.W., Wang J., Kang S.A., Thoreen C.C., Markhard A., Hur W., Zhang J., Sim T., Sabatini D.M., et al. Discovery of 1-(4-(4-propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benzo[h][1,6]naphthyridin-2(1H)-one as a highly potent, selective mammalian target of rapamycin (mTOR) inhibitor for the treatment of cancer. J. Med. Chem. 2010;53:7146–7155. doi: 10.1021/jm101144f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liu Q., Wang J., Kang S.A., Thoreen C.C., Hur W., Ahmed T., Sabatini D.M., Gray N.S. Discovery of 9-(6-aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl)benzo[h][1,6]naphthyridin-2(1H)-one (Torin2) as a potent, selective, and orally available mammalian target of rapamycin (mTOR) inhibitor for treatment of cancer. J. Med. Chem. 2011;54:1473–1480. doi: 10.1021/jm101520v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wissner A., Berger D.M., Boschelli D.H., Floyd M.B., Jr., Greenberger L.M., Gruber B.C., Johnson B.D., Mamuya N., Nilakantan R., Reich M.F., et al. 4-Anilino-6,7-dialkoxyquinoline-3-carbonitrile inhibitors of epidermal growth factor receptor kinase and their bioisosteric relationship to the 4-anilino-6,7-dialkoxyquinazoline inhibitors. J. Med. Chem. 2000;43:3244–3256. doi: 10.1021/jm000206a. [DOI] [PubMed] [Google Scholar]
- 109.Wissner A., Mansour T.S. The development of HKI-272 and related compounds for the treatment of cancer. Arch. Pharm. (Weinheim) 2008;341:465–477. doi: 10.1002/ardp.200800009. [DOI] [PubMed] [Google Scholar]
- 110.Wissner A., Overbeek E., Reich M.F., Floyd M.B., Johnson B.D., Mamuya N., Rosfjord E.C., Discafani C., Davis R., Shi X., et al. Synthesis and structure-activity relationships of 6,7-disubstituted 4-anilinoquinoline-3-carbonitriles. The design of an orally active, irreversible inhibitor of the tyrosine kinase activity of the epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor-2 (HER-2) J. Med. Chem. 2003;46:49–63. doi: 10.1021/jm020241c. [DOI] [PubMed] [Google Scholar]
- 111.Tsou H.R., Overbeek-Klumpers E.G., Hallett W.A., Reich M.F., Floyd M.B., Johnson B.D., Michalak R.S., Nilakantan R., Discafani C., Golas J., et al. Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity. J. Med. Chem. 2005;48:1107–1131. doi: 10.1021/jm040159c. [DOI] [PubMed] [Google Scholar]
- 112.Rabindran S.K., Discafani C.M., Rosfjord E.C., Baxter M., Floyd M.B., Golas J., Hallett W.A., Johnson B.D., Nilakantan R., Overbeek E., et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004;64:3958–3965. doi: 10.1158/0008-5472.CAN-03-2868. [DOI] [PubMed] [Google Scholar]
- 113.Yun C.H., Mengwasser K.E., Toms A.V., Woo M.S., Greulich H., Wong K.K., Meyerson M., Eck M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li X., Yang C., Wan H., Zhang G., Feng J., Zhang L., Chen X., Zhong D., Lou L., Tao W. Discovery and development of pyrotinib: A novel irreversible EGFR/HER2 dual tyrosine kinase inhibitor with favorable safety profiles for the treatment of breast cancer. Eur. J. Pharm. Sci. 2017;110:51–61. doi: 10.1016/j.ejps.2017.01.021. [DOI] [PubMed] [Google Scholar]
- 115.Ma F., Li Q., Chen S., Zhu W., Fan Y., Wang J., Luo Y., Xing P., Lan B., Li M., et al. Phase I Study and Biomarker Analysis of Pyrotinib, a Novel Irreversible Pan-ErbB Receptor Tyrosine Kinase Inhibitor, in Patients With Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer. J. Clin. Oncol. 2017;35:3105–3112. doi: 10.1200/JCO.2016.69.6179. [DOI] [PubMed] [Google Scholar]
- 116.Ma F., Ouyang Q., Li W., Jiang Z., Tong Z., Liu Y., Li H., Yu S., Feng J., Wang S., et al. Pyrotinib or Lapatinib Combined With Capecitabine in HER2-Positive Metastatic Breast Cancer With Prior Taxanes, Anthracyclines, and/or Trastuzumab: A Randomized, Phase II Study. J. Clin. Oncol. 2019;37:2610–2619. doi: 10.1200/JCO.19.00108. [DOI] [PubMed] [Google Scholar]
- 117.Blair H.A. Pyrotinib: First Global Approval. Drugs. 2018;78:1751–1755. doi: 10.1007/s40265-018-0997-0. [DOI] [PubMed] [Google Scholar]
- 118.Pannala M., Kher S., Wilson N., Gaudette J., Sircar I., Zhang S.H., Bakhirev A., Yang G., Yuen P., Gorcsan F., et al. Synthesis and structure-activity relationship of 4-(2-aryl-cyclopropylamino)-quinoline-3-carbonitriles as EGFR tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2007;17:5978–5982. doi: 10.1016/j.bmcl.2007.07.071. [DOI] [PubMed] [Google Scholar]
- 119.Ibrahim D.A., Abou El Ella D.A., El-Motwally A.M., Aly R.M. Molecular design and synthesis of certain new quinoline derivatives having potential anticancer activity. Eur. J. Med. Chem. 2015;102:115–131. doi: 10.1016/j.ejmech.2015.07.030. [DOI] [PubMed] [Google Scholar]
- 120.Aly R.M., Serya R.A.T., El-Motwally A.M., Esmat A., Abbas S., Abou El Ella D.A. Novel quinoline-3-carboxamides (Part 2): Design, optimization and synthesis of quinoline based scaffold as EGFR inhibitors with potent anticancer activity. Bioorg. Chem. 2017;75:368–392. doi: 10.1016/j.bioorg.2017.10.018. [DOI] [PubMed] [Google Scholar]
- 121.El-Sayed M.A., El-Husseiny W.M., Abdel-Aziz N.I., El-Azab A.S., Abuelizz H.A., Abdel-Aziz A.A. Synthesis and biological evaluation of 2-styrylquinolines as antitumour agents and EGFR kinase inhibitors: Molecular docking study. J. Enzyme Inhib. Med. Chem. 2018;33:199–209. doi: 10.1080/14756366.2017.1407926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Makawana J.A., Sangani C.B., Lin L., Zhu H.L. Schiff’s base derivatives bearing nitroimidazole and quinoline nuclei: New class of anticancer agents and potential EGFR tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2014;24:1734–1736. doi: 10.1016/j.bmcl.2014.02.041. [DOI] [PubMed] [Google Scholar]
- 123.George R.F., Samir E.M., Abdelhamed M.N., Abdel-Aziz H.A., Abbas S.E. Synthesis and anti-proliferative activity of some new quinoline based 4,5-dihydropyrazoles and their thiazole hybrids as EGFR inhibitors. Bioorg. Chem. 2019;83:186–197. doi: 10.1016/j.bioorg.2018.10.038. [DOI] [PubMed] [Google Scholar]
- 124.Abdelbaset M.S., Abdel-Aziz M., Ramadan M., Abdelrahman M.H., Abbas Bukhari S.N., Ali T.F.S., Abuo-Rahma G.E.A. Discovery of novel thienoquinoline-2-carboxamide chalcone derivatives as antiproliferative EGFR tyrosine kinase inhibitors. Bioorg. Med. Chem. 2019;27:1076–1086. doi: 10.1016/j.bmc.2019.02.012. [DOI] [PubMed] [Google Scholar]
- 125.Abdelsalam E.A., Zaghary W.A., Amin K.M., Abou Taleb N.A., Mekawey A.A.I., Eldehna W.M., Abdel-Aziz H.A., Hammad S.F. Synthesis and in vitro anticancer evaluation of some fused indazoles, quinazolines and quinolines as potential EGFR inhibitors. Bioorg. Chem. 2019;89:102985. doi: 10.1016/j.bioorg.2019.102985. [DOI] [PubMed] [Google Scholar]
- 126.Kubo K., Shimizu T., Ohyama S., Murooka H., Iwai A., Nakamura K., Hasegawa K., Kobayashi Y., Takahashi N., Takahashi K., et al. Novel potent orally active selective VEGFR-2 tyrosine kinase inhibitors: Synthesis, structure-activity relationships, and antitumor activities of N-phenyl-N’-{4-(4-quinolyloxy)phenyl}ureas. J. Med. Chem. 2005;48:1359–1366. doi: 10.1021/jm030427r. [DOI] [PubMed] [Google Scholar]
- 127.Nakamura K., Taguchi E., Miura T., Yamamoto A., Takahashi K., Bichat F., Guilbaud N., Hasegawa K., Kubo K., Fujiwara Y., et al. KRN951, a highly potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, has antitumor activities and affects functional vascular properties. Cancer Res. 2006;66:9134–9142. doi: 10.1158/0008-5472.CAN-05-4290. [DOI] [PubMed] [Google Scholar]
- 128.Taguchi E., Nakamura K., Miura T., Shibuya M., Isoe T. Anti-tumor activity and tumor vessel normalization by the vascular endothelial growth factor receptor tyrosine kinase inhibitor KRN951 in a rat peritoneal disseminated tumor model. Cancer Sci. 2008;99:623–630. doi: 10.1111/j.1349-7006.2007.00724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.De Luca A., Normanno N. Tivozanib, a pan-VEGFR tyrosine kinase inhibitor for the potential treatment of solid tumors. IDrugs. 2010;13:636–645. [PubMed] [Google Scholar]
- 130.Okamoto K., Ikemori-Kawada M., Jestel A., von König K., Funahashi Y., Matsushima T., Tsuruoka A., Inoue A., Matsui J. Distinct binding mode of multikinase inhibitor lenvatinib revealed by biochemical characterization. ACS Med. Chem. Lett. 2015;6:89–94. doi: 10.1021/ml500394m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.McTigue M., Murray B.W., Chen J.H., Deng Y.L., Solowiej J., Kania R.S. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. USA. 2012;109:18281–18289. doi: 10.1073/pnas.1207759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kajal K., Panda A.K., Bhat J., Chakraborty D., Bose S., Bhattacharjee P., Sarkar T., Chatterjee S., Kar S.K., Sa G. Andrographolide binds to ATP-binding pocket of VEGFR2 to impede VEGFA-mediated tumor-angiogenesis. Sci. Rep. 2019;9:4073. doi: 10.1038/s41598-019-40626-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yang Y., Shi L., Zhou Y., Li H.Q., Zhu Z.W., Zhu H.L. Design, synthesis and biological evaluation of quinoline amide derivatives as novel VEGFR-2 inhibitors. Bioorg. Med. Chem. Lett. 2010;20:6653–6656. doi: 10.1016/j.bmcl.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 134.Aboul-Enein M.N., El-Azzouny A.M., Ragab F.A., Hamissa M.F. Design, Synthesis, and Cytotoxic Evaluation of Certain 7-Chloro-4-(piperazin-1-yl)quinoline Derivatives as VEGFR-II Inhibitors. Arch. Pharm. (Weinheim) 2017;350 doi: 10.1002/ardp.201600377. [DOI] [PubMed] [Google Scholar]
- 135.Westman J., Wannberg J., Nekhotiaeva N., Backman U. Quinoline-S-Carboxylic Acid Derivatives as Tyrosine Kinase Inhibitors. PCT/EP2008/053763. U.S. Patent. 2008 Oct 9;
- 136.Li X., Wu C., Lin X., Cai X., Liu L., Luo G., You Q., Xiang H. Synthesis and biological evaluation of 3-aryl-quinolin derivatives as anti-breast cancer agents targeting ERalpha and VEGFR-2. Eur. J. Med. Chem. 2019;161:445–455. doi: 10.1016/j.ejmech.2018.10.045. [DOI] [PubMed] [Google Scholar]
- 137.Feng H., Zhang Y., Bos P.H., Chambers J.M., Dupont M.M., Stockwell B.R. K-Ras(G12D) Has a Potential Allosteric Small Molecule Binding Site. Biochemistry. 2019;58:2542–2554. doi: 10.1021/acs.biochem.8b01300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li L., Feng J., Long Y.O., Liu Y., Wu T., Ren P., Liu Y. Fused-Tricyclic Inhibitors of KRAS and Methods of Use Thereof. PCT/US2016/027673. U.S. Patent. 2016 Oct 20;
- 139.El-Gamal M.I., Khan M.A., Abdel-Maksoud M.S., Gamal El-Din M.M., Oh C.H. A new series of diarylamides possessing quinoline nucleus: Synthesis, in vitro anticancer activities, and kinase inhibitory effect. Eur. J. Med. Chem. 2014;87:484–492. doi: 10.1016/j.ejmech.2014.09.068. [DOI] [PubMed] [Google Scholar]
- 140.El-Gamal M.I., Khan M.A., Tarazi H., Abdel-Maksoud M.S., Gamal El-Din M.M., Yoo K.H., Oh C.H. Design and synthesis of new RAF kinase-inhibiting antiproliferative quinoline derivatives. Part 2: Diarylurea derivatives. Eur. J. Med. Chem. 2017;127:413–423. doi: 10.1016/j.ejmech.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 141.El-Damasy A.K., Seo S.H., Cho N.C., Kang S.B., Pae A.N., Kim K.S., Keum G. Design, synthesis, in-vitro antiproliferative activity and kinase profile of new picolinamide based 2-amido and ureido quinoline derivatives. Eur. J. Med. Chem. 2015;101:754–768. doi: 10.1016/j.ejmech.2015.07.025. [DOI] [PubMed] [Google Scholar]
- 142.El-Damasy A.K., Seo S.H., Cho N.C., Pae A.N., Kim E.E., Keum G. Design and synthesis of new 2-anilinoquinolines bearing N-methylpicolinamide moiety as potential antiproliferative agents. Chem. Biol. Drug Des. 2017;89:98–113. doi: 10.1111/cbdd.12836. [DOI] [PubMed] [Google Scholar]
- 143.Li Y., Shi X., Xie N., Zhao Y., Li S. 3,3-Dimethyl-1H-pyrrolo[3,2-g]quinolin-2(3H)-one derivatives as novel Raf kinase inhibitors. MedChemComm. 2013;4:367–370. doi: 10.1039/C2MD20275A. [DOI] [Google Scholar]
- 144.Zhang N., Wu B., Powell D., Wissner A., Floyd M.B., Kovacs E.D., Toral-Barza L., Kohler C. Synthesis and structure-activity relationships of 3-cyano-4-(phenoxyanilino)quinolines as MEK (MAPKK) inhibitors. Bioorg. Med. Chem. Lett. 2000;10:2825–2828. doi: 10.1016/S0960-894X(00)00580-1. [DOI] [PubMed] [Google Scholar]
- 145.Zhang N., Wu B., Eudy N., Wang Y., Ye F., Powell D., Wissner A., Feldberg L.R., Kim S.C., Mallon R., et al. MEK (MAPKK) inhibitors. Part 2: Structure-activity relationships of 4-anilino-3-cyano-6,7-dialkoxyquinolines. Bioorg. Med. Chem. Lett. 2001;11:1407–1410. doi: 10.1016/S0960-894X(01)00238-4. [DOI] [PubMed] [Google Scholar]
- 146.Berger D., Dutia M., Powell D., Wu B., Wissner A., Boschelli D.H., Floyd M.B., Zhang N., Torres N., Levin J., et al. Synthesis and evaluation of 4-anilino-6,7-dialkoxy-3-quinolinecarbonitriles as inhibitors of kinases of the Ras-MAPK signaling cascade. Bioorg. Med. Chem. Lett. 2003;13:3031–3034. doi: 10.1016/S0960-894X(03)00640-1. [DOI] [PubMed] [Google Scholar]
- 147.Berger D.M., Dutia M., Powell D., Floyd M.B., Torres N., Mallon R., Wojciechowicz D., Kim S., Feldberg L., Collins K., et al. 4-Anilino-7-alkenylquinoline-3-carbonitriles as potent MEK1 kinase inhibitors. Bioorg. Med. Chem. 2008;16:9202–9211. doi: 10.1016/j.bmc.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 148.Bock M.G., Moebitz H., Panigrahi S.K., Poddutoori R., Samajdar S. Compounds and Compositions as Inhibitors of Mek. PCT/IB2014/063918. U.S. Patent. 2015 Feb 9;
- 149.Jin X.Y., Chen H., Li D.D., Li A.L., Wang W.Y., Gu W. Design, synthesis, and anticancer evaluation of novel quinoline derivatives of ursolic acid with hydrazide, oxadiazole, and thiadiazole moieties as potent MEK inhibitors. J. Enzyme Inhib. Med. Chem. 2019;34:955–972. doi: 10.1080/14756366.2019.1605364. [DOI] [PMC free article] [PubMed] [Google Scholar]