

The fight against SARS-CoV-2 coronavirus is the centerpiece of the current global pandemic that has stormed every corner of the world, claiming hundreds of thousands of lives. The scientific community has come together in an unprecedented effort to find a solution to the COVID-19 pandemic and to develop an efficient vaccine against the SARS-CoV-2 virus. In this scenario, the Amaro lab has seized the day by performing massive molecular dynamics (MD) simulations and making a leap in strategizing a model for vaccine development (ref (1)).
Coronaviruses such as SARS-CoV and SARS-CoV-2 enter the host cell by interacting with the heavily glycosylated spike protein, where each monomer in the homotrimer contains two subunits (S1 and S2) that can be divided into three domains—head, stalk, and cytoplasmic tail (Figure 1A).2 S1 is composed of an N-terminal domain (NTD) and receptor-binding domain (RBD) that contains the receptor-binding motif (RBM) responsible for tissue tropism as it interacts with angiotensin-converting enzyme 2 (ACE2). This interaction between the RBM and ACE2 is the key step in the pathogenesis of the infection.3 Experimental studies have shown that the D614G mutation in the SARS-CoV-2 spike protein reduced shedding of the S1 protein prompted by the proteolytic cleavage of S1 and S2, thus, increasing infectivity.4 This indicated S1 as the kernel in the route of infection and thereby its surfaces as a potential target for developing vaccine and therapeutic agents. Similar to other viral fusion proteins, SARS-CoV-2 spikes are covered with a sugar coat—referred to as “glycan shield”—presenting 22 N-glycans per monomer (Figure 1B).5 Though the glycan shield of the SARS-CoV-2 spike protein is not as effective when compared to the one on HIV-1 Env protein, it still deploys a similar camouflaging stratagem to evade the host cell immune response.
Figure 1.
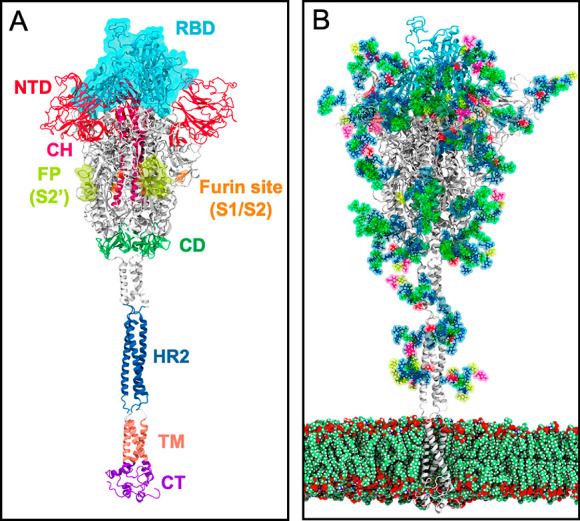
(a) Three-dimensional structure of the full-length model of the spike protein in open conformation. The model (based on the PDB ID: 6VSB)2 was developed by the Amaro lab.1 The protein is shown as ribbons, highlighting individual protein domains using different colors. The critical receptor-binding domain (RBD) is shown in the molecular surface. (b) Glycosylated spike protein embedded in a realistic membrane environment. All structural models were downloaded from https://amarolab.ucsd.edu/covid19.php.
In this issue of ACS Central Science, the Amaro lab presents massive all-atom MD simulations of the glycosylated full-length model of the SARS-CoV-2 spike protein in a realistic membrane/aqueous environment.1 This system encompassing ∼1.7 million atoms has been simulated over multiple microseconds by pushing the boundaries of the state-of-the-art Frontera computer cluster at the Texas Advanced Computing Center (TACC) established by the National Science Foundation. Molecular simulations have been carried out on different conformations of the spike protein, viz., open (displaying one RBD in the “up” conformation) and closed (showing three RBDs “down”). This revealed that the two N-linked glycans (N165 and N234) also hold a structural role in modulating the conformational dynamics of the RBD, beyond solely shielding the spike protein (Figure 2A). These glycans, located at strategic positions on the NTD, are found to “lock-and-load” the adjacent RBD in the “up” state for infection. To probe the importance of these N-glycans, a double mutant of the open spike (N165A and N243A) was generated and subjected to MD simulations. As a result, the stability of the RBD in the “up” state is markedly disrupted by alanine mutation of N165 and N234 owing to the loss of the respective N-linked glycans. These observations inspired the McLellan lab to implement biolayer interferometry experiments and reveal an ∼10 and ∼40% reduction of ACE2 binding to the N165A and N234A spike variants, respectively (Figure 2B). These critical insights bolster a viable approach for modulating the conformational plasticity of the RBD, thereby harnessing the alteration of the SARS-CoV-2 spike glycosylation as a strategy for vaccine development.
Figure 2.

(a) Structural role of the N-glycans at N165 and N234 (van der Waals representation) in stabilizing the RBD of chain A (RBD-A, cyan) in the “up” conformation. (b) Binding responses for biolayer interferometry measurements of ACE2 binding to spike variants. Reproduced with permission from ref (1). Copyright 2020 American Chemical Society.
A model of the fully glycosylated spike protein was earlier proposed by Woo and coauthors,6 yet the dynamics and the role of glycans were not fully uncovered. Amaro and co-workers, in their recent article, provide a complete overview of the glycan shield around the spike protein in the time scale of nanoseconds to microseconds.1 Interestingly, the region of the spike protein that is most effectively camouflaged by glycans is the stalk, with ∼90% overall coverage. However, the head of the spike protein is considerably less shielded (∼62%) by glycans as well as the RBD in the “up” state when compared to the “down” state. Hence, by revealing the vulnerabilities of the spike’s glycan shield and identifying the weak points of the main molecular machine of SARS-CoV-2, the study highlighted in this First Reaction provides unprecedented insights that can smoothen the way for the development of new drugs against this virus.1
In this critical time, where the need of a vaccine is of the utmost urgency, molecular simulations have the opportunity to unravel the mechanistic intricacies of the SARS-CoV-2 virus detailed at the atomic level. Yet, as also shown by Amaro and team, experimental corroboration with computational findings is critical to advance the development of therapeutic agents and a promising vaccine. Recent investigations, integrating cryo-electron microscopy (cryo-EM) and MD simulations, have shown that the stalk domain of the spike contains three hinges that confer remarkable orientational freedom.7 Multimicrosecond MD simulations of a patch of viral membrane with full-length glycosylated spike proteins have also shown that an intrinsically flexible glycan coat that can shield and/or dynamically expose a larger structure than what is expected from static structures.8 Further molecular simulation efforts focused on the intricacies of the binding of the SARS-CoV-2 RBD to ACE2 revealed critical hydrophobic regions and hydrogen bonding networks.9 Recently, using cryo-EM and tomography, Ke and co-workers released a model of the intact SARS-CoV-2 virion.10 In light of this extraordinary wealth of information, the synergy between experiments and atomic-scale computer simulations opens the possibility of performing mesoscale all-atom MD simulations of the complete virion. This would not only help in fighting the current global pandemic by providing more realistic data but also be a paramount step toward the dream of “simulating a cell”.
In summary, the Amaro group offers deep insights into the molecular determinants playing a pivotal role in the virulence of the SARS-CoV-2 coronavirus, which is not only fatal to human lives but also destructive to their psychological state and the economy at large. With billions of lives at stake, the world is looking upon the scientific community for a vaccine that could make them immune to this deadly virus and help them fall back to their former normalcy. In the current situation, the findings from extensive MD simulations provide novel mechanistic insights into the SARS-CoV-2 spike protein and its glycan coat that can be exploited for the development of a potent vaccine and therapeutic agents against this lethal virus. Conclusively, the synergistic efforts from different scientific fields provide hope for a society free of SARS-CoV-2 infection in the near future.
Acknowledgments
The Palermo lab acknowledges funding from the National Science Foundation (Grant No. CHE-1905374) and the National Institute of Health (Grant R01 EY027440). The graphical abstract was prepared using CellPAINT (https://ccsb.scripps.edu/cellpaint/).
References
- Casalino L.; Gaieb Z.; Goldsmith J. A.; Hjorth C. K.; Dommer A. C.; Harbison A. M.; Fogarty C. A.; Barros E. P.; Taylor B. C.; McLellan J. S.; Fadda E.; Amaro R. E.. Beyond Shielding: The Roles of Glycans in SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, ASAP. 10.1021/acscentsci.0c01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D.; Wang N.; Corbett K. S.; Goldsmith J. A.; Hsieh C.-L.; Abiona O.; Graham B. S.; McLellan J. S. Cryo-EM Structure of the 2019-NCoV Spike in the Prefusion Conformation. Science 2020, 367, 1260–1263. 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Zhang Y.; Wu L.; Niu S.; Song C.; Zhang Z.; Lu G.; Qiao C.; Hu Y.; Yuen K.-Y.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904.e9. 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B.; Fischer W. M.; Gnanakaran S.; Yoon H.; Theiler J.; Abfalterer W.; Hengartner N.; Giorgi E. E.; Bhattacharya T.; Foley B.; et al. Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827.e19. 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y.; Allen J. D.; Wrapp D.; McLellan J. S.; Crispin M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo H.; Park S.; Choi Y. K.; Park T.; Tanveer M.; Cao Y.; Kern N. R.; Lee J.; Yeom M. S.; Croll T. I.; Seok C.; Im W. Developing a fully-glycosylated full-length SARS-CoV-2 spike protein model in a viral membrane. J. Phys. Chem. B 2020, 124, 7128–7137. 10.1021/acs.jpcb.0c04553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turoňová B.; Sikora M.; Schürmann C.; Hagen W. J.; Welsch S.; Blanc F. E.; von Bülow S.; Gecht M.; Bagola K.; Hörner C.; et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, eabd5223 10.1126/science.abd5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikora M.; von Bülow S.; Blanc F. E. C.; Gecht M.; Covino R.; Hummer G.. Map of SARS-CoV-2 spike epitopes not shielded by glycans. bioRxiv, 2020. 10.1101/2020.07.03.186825. [DOI] [PMC free article] [PubMed]
- Wang Y.; Liu M.; Gao J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 13967–13974. 10.1073/pnas.2008209117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Z.; Oton J.; Qu K.; Cortese M.; Zila V.; McKeane L.; Nakane T.; Zivanov J.; Neufeldt C. J.; Cerikan B.. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020. 10.1038/s41586-020-2665-2. [DOI] [PMC free article] [PubMed] [Google Scholar]