

Abstract
Background:
In cystic fibrosis (CF), the spectrum and frequency of CFTR variants differ by geography and race/ethnicity. CFTR variants in White patients are well-described compared with Latino patients. No studies of CFTR variants have been done in patients with CF in the Dominican Republic or Puerto Rico.
Methods:
CFTR was sequenced in 61 Dominican Republican patients and 21 Puerto Rican patients with CF and greater than 60 mmol/L sweat chloride. The spectrum of CFTR variants was identified and the proportion of patients with 0, 1, or 2 CFTR variants identified was determined. The functional effects of identified CFTR variants were investigated using clinical annotation databases and computational prediction tools.
Results:
Our study found 10% of Dominican patients had two CFTR variants identified compared with 81% of Puerto Rican patients. No CFTR variants were identified in 69% of Dominican patients and 10% of Puerto Rican patients. In Dominican patients, there were 19 identified CFTR variants, accounting for 25 out of 122 disease alleles (20%). In Puerto Rican patients, there were 16 identified CFTR variants, accounting for 36 out of 42 disease alleles (86%) in Puerto Rican patients. Thirty CFTR variants were identified overall. The most frequent variants for Dominican patients were p.Phe508del and p.Ala559Thr and for Puerto Rican patients were p.Phe508del, p.Arg1066Cys, p.Arg334Trp, and p.I507del.
Conclusions:
In this first description of the CFTR variants in patients with CF from the Dominican Republic and Puerto Rico, there was a low detection rate of two CFTR variants after full sequencing with the majority of patients from the Dominican Republic without identified variants.
Keywords: CFTR variants, health disparities, Latino, whole genome sequencing
1 |. INTRODUCTION
Although the majority of the 80 000 people worldwide with cystic fibrosis (CF) are White, an increasing proportion of patients are of other races and ethnicities. In the United States (U.S.), the percentage of patients with CF who are Latino increased from 5.6% to 8.7% over the past 15 years.1 The increase in the Latino CF population is important as these patients have increased morbidity and mortality compared with the White CF population.2,3
Latino patients not only have a different clinical course than White patients, but they also have different CFTR variants.4 Latino patients are also more likely to have CFTR Class IV to V or uncharacterized variants whereas White patients are more likely to have Class I to III variants. Latino patients are more likely to have one or no CFTR variants identified, in part due to CF genetic panels and newborn screens having lower sensitivity to variants that are more common in the Latino population.5
CFTR variants are population-specific and the spectrum of known CFTR variants is based largely on investigations of White populations.6 Even among investigations of Latino populations, there have been limited efforts to describe the genetic profile of CF in the Caribbean.7 There is considerable genetic heterogeneity between Latino populations and within the Caribbean.8
In this study, full genetic sequencing of CFTR was done in Dominican and Puerto Rican patients with CF to describe the spectrum of CFTR variants. The proportion of patients with 0, 1, or 2 CFTR variants identified was determined. The functional impact of each identified CFTR variant was classified based on clinical databases and deleteriousness prediction algorithms.
2 |. METHODS
2.1 |. Study population
This was a cross-sectional study of CFTR variants in patients with CF in the Dominican Republic and Puerto Rico. All patients had a diagnosis of CF made by their clinical doctor based on the presence of clinical CF symptoms and a positive sweat chloride concentration (≥60 mmol/L) based on Cystic Fibrosis Foundation guidelines.9 Patients with an intermediate sweat chloride concentration (<60 mmol/L) were not included in our study. Patients 6 years of age and older were recruited from CF clinics in the Dominican Republic and Puerto Rico in 2017. Consent and assent were obtained from patients and their guardians as appropriate. This study was approved by the Western Institutional Review Board.
At the time of recruitment, CFTR variants identified through prior genotyping, sweat chloride concentration, demographic data, pancreatic sufficiency status, and pulmonary function percent predicted based on Global Lung Initiative was recorded for each patient. Blood was drawn for genetic analysis.
2.2 |. Whole genome sequencing analysis
DNA was isolated from whole blood using the Wizard Genomic DNA Purification kits (Promega, Fitchburg, WI). DNA samples were quantified by fluorescence using the Quant-iT PicoGreen dsDNA assay (Thermo Fisher Scientific, Waltham, MA) on a Spectramax fluorometer (Molecular Devices, Sunnyvale, CA). DNA samples were sequenced as part of the Trans-Omics for Precision Medicine (TOPMed) whole genome sequencing (WGS) program.9 WGS was performed at the Northwest Genomics Center on a HiSeqX system (Illumina, San Diego, CA) using a paired-end read length of 150 base pair, to a minimum of 30× mean genome coverage. Details on DNA sample handling, quality control, library construction, clustering and sequencing, read processing, and sequence data quality control are previously described.10 Variant calls were obtained from TOPMed data freeze 8 variant call format files. The term “variant” is used in place of “mutation” or “polymorphism”.11 Variants with a minimal depth of coverage of 10 reads were included in our analyses.
Variants were annotated in TOPMed using the WGSA pipeline.12 CFTR variants were annotated with reference to the NM_000492.3 transcript. Genetic variants in CFTR were extracted (ENSG00000001626; ENST00000003084) from chr7:117,465,784–117,715,971, which included segments 15 kb upstream of the CFTR transcription start site and 47 kb downstream of the last exon. Sequences aligned to hs38DH 1000 Genomes GRCh38/hg38 reference assembly using BWA-MEM were received as CRAM files from TOPMed.13 Chromosome 7 sequence reads were extracted from the CRAM files using Samtools v1.9.14 Copy number variation was detected using a bin size of 500 with CNVnator v0.3.3.15 Structural variation in chromosome 7, including deletions, duplications, inversions, and translocations, were detected with LUMPY express v0.2.13.16 The sequencing quality of variants that did not have value “PASS” in the FILTER field from TOPMed was checked by manual inspection of the sequencing reads alignment using Integrative Genome Viewer. Other possible FILTER values include centromere (variant overlaps with centromeric region), SVM (variant failed SVM filter), and duplicate discordances (variant with high mendelian or duplicate genotype discordance [3/5% or more]).10
Phased genotypes from TOPMed data freeze 8 were used to determine whether two variants are in cis or trans (see section below).17 These were statistically phased by applying Eagle 2.4 (Dec 13, 2017) to the whole panel of 137 977 samples included in TOPMed freeze 8. Phasing was done in 1 Mb chunks with 0.1 Mb overlap. The entire CFTR locus (chr7:117,465,784–117,715,971) falls within a single chunk. Phasing was limited to variants which pass all filters and starts with minDP10 genotypes to restrict to high quality genotypes. Phasing imputes any missing genotypes. Statistical phasing has limited accuracy for very rare variants (those seen in fewer than five individuals in the panel).18
2.3 |. Clinical annotations of variants
To determine the clinical impact of CFTR variants identified by sequencing, variants were first compared with the Clinical and Functional Translation of CFTR (CFTR2) database.19 The CFTR2 database provides functional classifications for variants with clinical and laboratory evidence of phenotypic consequence. These classifications include “CF-causing,” “varying clinical consequence,” “un-known significance,” and “non-CF-causing.” CFTR variants identified by sequencing that were not listed in the CFTR2 database were analyzed to identify common variants, defined by an allele frequency greater than 3% on Genome Aggregation Database (gnomAD) in the general non-CF population.9 Variants that are common in a non-CF population are unlikely to be disease-causing. All identified variants that were not in the CFTR2 database or had an allele frequency less than 3% were cross-referenced with two variant databases, ClinVar and Invitae, to determine the functional impact of the variant. ClinVar is a publicly available repository of genotype-phenotype investigations.20 Invitae is a clinical genetic sequencing laboratory.21 Variants in the ClinVar and Invitae databases were annotated as “pathogenic,” “likely pathogenic,” “uncertain significance,” or “benign.”
Genetic variants without clinical annotations in the CFTR2, ClinVar, or Invitae databases were analyzed for deleteriousness using three computational prediction algorithms: Combined Annotation Dependent Depletion (CADD), FATHMM-XF, and Rare Exome Variant Ensemble Learner (REVEL).22–24 Variants predicted to be deleterious had a scaled C-score ranking from CADD >16 or were predicted to be likely damaging by FATHMM-XF or had a REVEL score greater than 0.5.
Variants were categorized into five functional classifications based on databases and computational predictions as follows: (a) CF-disease causing variant, (b) variant of varying clinical consequence, (c) variant of uncertain significance, (d) variant predicted to be deleterious, and (e) Non-CF-disease causing or likely benign variant (Figure 1).
FIGURE 1.
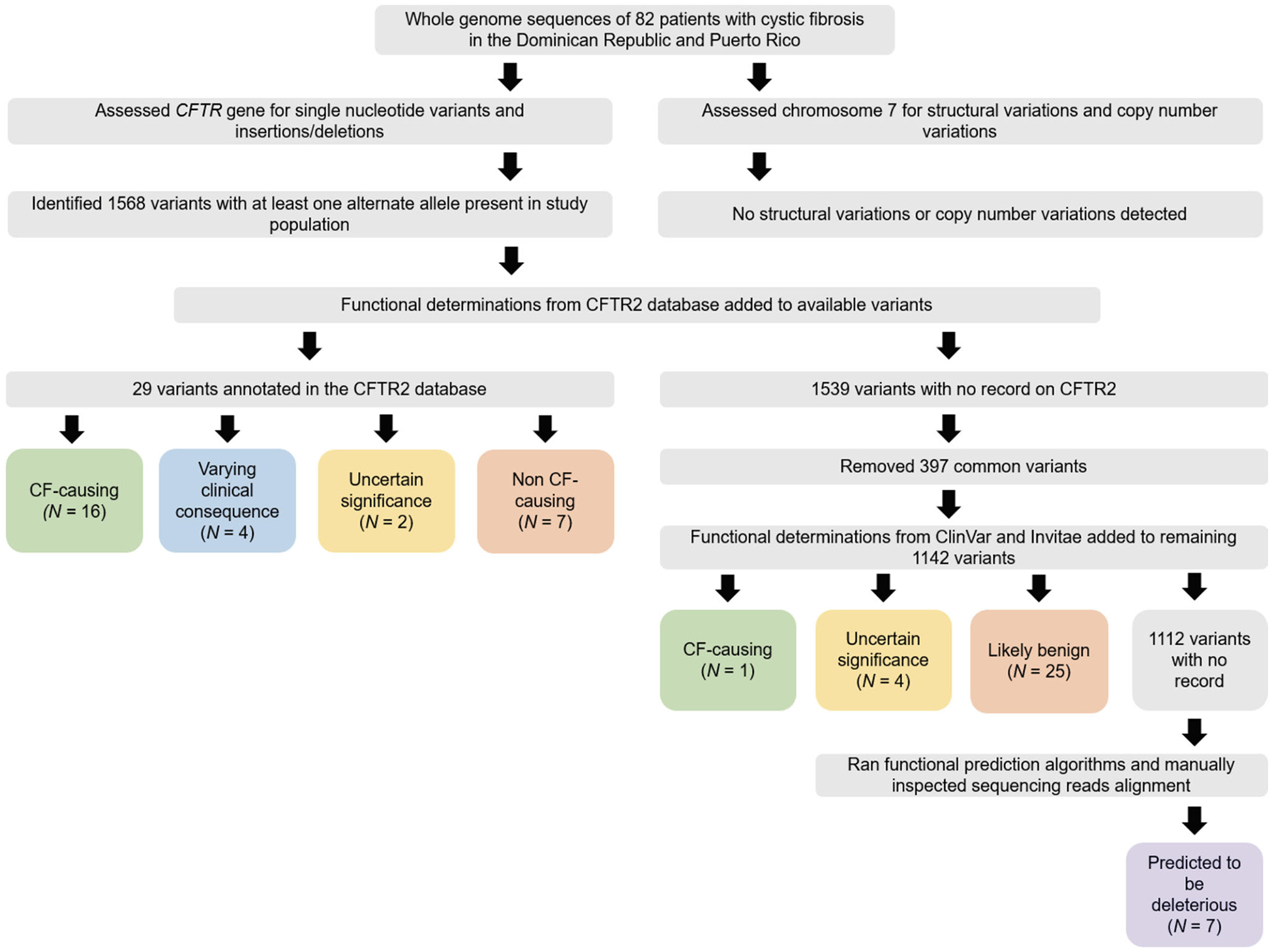
Flowchart for assignment of CFTR variant categories. CF, cystic fibrosis
The CFTR variants and genotype for each patient were determined. In patients with two variants, the phased genotype (variants in cis or trans) was assessed using BCFtools.25 Patients were categorized as fully identified CFTR genotype (two variants in trans) versus those who were not (with two variants in cis, one variant, or no variants).
3 |. RESULTS
3.1 |. Genotyping results before recruitment
Our study population consisted of 82 patients diagnosed with CF from the Dominican Republic (N = 61) and Puerto Rico (N = 21). At recruitment, 3% of Dominican patients had two identified CFTR variants, 3% had one variant, and 93% had not been genetically tested for CF. Among Puerto Rican patients, 57% of patients had two identified CFTR variants, 24% had one variant, 5% had no variants, and 14% had not been tested.
At the time of recruitment, Dominican patients were a median age of 10.6 years old and Puerto Rican patients were 15.4 years old. The majority of Dominican patients (86.9%) and Puerto Rican patients (81.0%) were pancreatic insufficient. The predicted percentage of average forced expiratory volume in 1 second was 91.7% in Dominican patients and was 83.4% in Puerto Rican patients.
3.2 |. WGS results
There were 1568 CFTR variants identified by WGS in our study population (Figure 1). No structural variation or copy number variation was detected in the CFTR region (ENSG00000001626; chr7:117,465,784–117,715,971). Of the 1568 variants identified, 29 variants were functionally classified in the CFTR2 database: 16 CF-disease causing variants, four varying clinical consequence variants, two variants of uncertain significance, and seven non-CF-causing variants. Of the 1539 CFTR variants not present in the CFTR2 database, 397 were identified as common variants in the general population, therefore were interpreted as benign and not analyzed further. Of the remaining 1142 variants, functional classification using the ClinVar and Invitae databases was determined in 30 variants: one CF-disease causing variant, four variants of uncertain significance, and 25 likely benign variants. There was no functional classification description for the 1112 remaining variants, so they were further annotated using three functional prediction algorithms. Seven variants were predicted to be deleterious by at least one computational prediction tool. Three of these seven variants were removed after manual inspection of the sequencing reads alignment suggested these were sequencing or alignment errors.
Overall, we identified 30 CFTR variants as follows: 16 CF-disease causing variants (Table 1), four variants of varying clinical consequence, six variants of uncertain significance, and four variants predicted to be deleterious (Table 2). The most frequent known disease-causing variants for Dominican patients were p.Phe508del (10%) and p.Ala559Thr (3%). The most frequent known disease-causing variants for Puerto Rican patients were p.Phe508del (33%), p.Arg1066Cys (33%), p.Arg334Trp (14%), and p.Ile507del l (14%).
TABLE 1.
Known disease-causing CFTR variants
| Occurrences, n (%) | |||||||
|---|---|---|---|---|---|---|---|
| # | Protein name | cDNA name | Position (GRCh38) | Reference allele | Alternate allele | D.R. | P.R. |
| 1 | p.Phe508del | c.1521_1523delCTT | 7:117559590 | ATCT | A | 6 (10%) | 7 (33%) |
| 2 | p.Arg1066Cys | c.3196C>T | 7:117611637 | C | T | --- | 7 (33%) |
| 3 | p.Arg334Trp | c.1000C>T | 7:117540230 | C | T | 1 (2%) | 3 (14%) |
| 4 | p.Ile507del | c.1519_1521delATC | 7:117559586 | TATC | T | --- | 3 (14%) |
| 5 | p.Gly542* | c.1624G>T | 7:117587778 | G | T | --- | 2 (10%) |
| 6 | --- | c.1680–886A>G | 7:117589467 | A | G | --- | 2 (10%) |
| 7 | p.Asn1303Lys | c.3909C>G | 7:117652877 | C | G | --- | 2 (10%) |
| 8 | p.Arg553* | c.1657C>T | 7:117587811 | C | T | 1 (2%) | 1 (5%) |
| 9 | --- | c.2988 + 1G>A | 7:117606754 | G | A | 1 (2%) | 1 (5%) |
| 10 | p.Ala559Thr | c.1675G>A | 7:117587829 | G | A | 2 (3%) | --- |
| 11 | --- | c.164 + 1G>A | 7:117504364 | G | A | --- | 1 (5%) |
| 12 | p.Tyr1092* | c.3276C>A | 7:117611717 | C | A | --- | 1 (5%) |
| 13 | --- | c.3368–2A>G | 7:117614611 | A | G | --- | 1 (5%) |
| 14 | p.Thr1220fs | c.3659delC | 7:117627711 | AC | A | --- | 1 (5%) |
| 15 | p.Ile148fs | c.442delA | 7:117531067 | CA | C | 1 (2%) | --- |
| 16 | p.Arg709* | c.2125C>T | 7:117592292 | C | T | 1 (2%) | --- |
Abbreviations: cDNA, complementary DNA; D.R., Dominican Republic; P.R., Puerto Rico.
TABLE 2.
Potentially disease-causing variants
| Occurrences, n (%) | |||||||
|---|---|---|---|---|---|---|---|
| # | Protein name | cDNA name | Position (GRCh38) | Reference allele | Alternate allele | D.R. | P.R. |
| Variants of varying clinical consequence | |||||||
| 1 | p.Arg74Trp | c.220C>T | 7:117509089 | C | T | 1 (2%) | 1 (5%) |
| 2 | --- | c.1210–34TG[11]T[5] | 7:117548628 | GTT | G | 2 (3%) | --- |
| 3 | --- | c.1210–34TG[12]T[5] | 7:117548630 | T | G | 1 (2%) | --- |
| 4 | p.Asp1270Asn | c.3808G>A | 7:117642528 | G | A | --- | 1 (5%) |
| Variants of uncertain significance | |||||||
| 1 | --- | c.−226G>T | 7:117479869 | G | T | 1 (2%) | --- |
| 2 | p.Val201Met | c.601G>A | 7:117535269 | G | A | --- | 1 (5%) |
| 3 | p.Ser589Asn | c.1766G>A | 7:117590439 | G | A | 1 (2%) | --- |
| 4 | p.Tyr1014Cys | c.3041A>G | 7:117610571 | A | G | 1 (2%) | --- |
| 5 | p.Arg1158Gln | c.3473G>A | 7:117627526 | G | A | 1 (2%) | --- |
| 6 | p.Asp1445Asn | c.4333G>A | 7:117666998 | G | A | 1 (2%) | --- |
| Variants predicted to be deleterious | |||||||
| 1 | --- | n.49–4832T>G | 7:117470859 | T | G | 3 (5%) | --- |
| 2 | --- | c.3674084T>G | 7:117711674 | T | G | 3 (5%) | --- |
| 3 | --- | c.1585–1361A>G | 7:117586378 | A | G | 1 (2%) | --- |
| 4 | --- | n.-2799A>G | 7:117710954 | A | G | 1 (2%) | --- |
Abbreviations: cDNA, complementary DNA; D.R., Dominican Republic; P.R., Puerto Rico.
Only 10% of Dominican patients had two CF-disease causing variants in trans compared with 81% of Puerto Rican patients (Table 3). Both Dominican patients (10%) and Puerto Rican patients (10%) had multiple CFTR variants in cis. Eleven percent of Dominican patients had only one CFTR variant identified; no Puerto Rican patients had only one CFTR variant identified. No variants were identified in 69% of Dominican patients and in 10% of Puerto Rican patients.
TABLE 3.
Type and frequency of CFTR genotypes
| CFTR Genotype | Dominican Republic, N (%) | Puerto Rico, N (%) |
|---|---|---|
| 2 CF-disease causing, trans | 6 (10%) | 17 (81%) |
| 2 CF-disease causing, cis | 1 (2%) | 1 (5%) |
| 2 VVCC, 1 VUS, cis | --- | 1 (5%) |
| 1 VUS, 1 predicted, cis | 1 (2%) | --- |
| 2 Predicted, cis | 3 (5%) | --- |
| 1 VVCC, 1 VUS, cis | 1 (2%) | --- |
| 1 CF-causing | 1 (2%) | --- |
| 1 VVCC | 3 (5%) | --- |
| 1 VUS | 2 (3%) | --- |
| 1 Predicted | 1 (2%) | --- |
| No variants | 42 (69%) | 2 (10%) |
Note: CFTR variants were categorized using three functional annotation databases as well as three computational tools for predicting deleteriousness. Cis and trans describe the relationship between two or more variants; cis refers to variants on the same gene copy while trans describes variants on different gene copies.
Abbreviations: CF, cystic fibrosis; VVCC, variant of varying clinical consequence; VUS, variant of uncertain significance.
The 30 identified CFTR variants accounted for 25 out of 122 disease alleles (20%) in Dominican patients and 36 out of 42 disease alleles (86%) in Puerto Rican patients.
4 |. DISCUSSION
In this first genetic description of CFTR variants in Dominican and Puerto Rican patients with CF, we found that there was a low rate of patients having two CFTR variants identified after full sequencing. The spectrum of CFTR variants differed between the two populations, which are of the same ethnicity in close geographic proximity. In the overall CF population in the U.S., 86% of patients have at least one copy of p.Phe508del. In contrast, in our study, only 9.8% of Dominican patients and 33% of Puerto Rican patients had at least one copy of p.Phe508del.1 The most frequent variants we found in Puerto Rican patients occurred at low rates in the general CF population in the U.S.: p.Ile507del is the 15th most common variant occurring in 0.8% of the general CF population but was observed in 14% of Puerto Rican patients in our study; p.Arg334Trp is the 25th most common variant occurring in 0.3% of the general CF population but was observed in 14% of Puerto Rican patients and 3% of Dominican patients in our study. p.Arg1066Cys was observed in a third of Puerto Rican patients in our study but is not in the top 25 most common variants in the general CF population. p.Ala559Thr was observed in 3% of Dominican patients in our study was not in the top 25 variants of the general CF population.1
The spectrum of CFTR variants varies between Latino populations across the world and also varied between the two specific Latino populations we studied: Dominicans and Puerto Ricans. In patients with CF in Spain, the most frequent CFTR variants were p.Phe508del (52%), p.Gly542x (8%), p.Asn1303Lys (3%), and 3849 + 10kbC→T (2%).26 In Latino patients with CF from across the U.S., the most frequent variants were p.Phe508del (37%), p.Gly542x (11%), and p.Arg334Trp (11%),27 but most frequent variants differed across the U.S. In the Southwestern U.S., the most frequent CFTR variants were p.Phe508del (47%), p.Gly542x (5%), and 3849 + 10kbC→T (3%),28 which was similar to the pattern observed in Southern California: p.Phe508del (52%), p.Gly542x (4%), 3849 + 10kbC→T (4%), and p.Ser549Asn (2%).29 In Illinois, the most frequent variants were p.Phe508del (52%), 3849 + 10kbC→T (7%), and p.Phe311del (7%).4 p.Phe508del was observed at a lower frequency in Dominican patients (10%) and Puerto Rican patients (33%) in this study than in the Latino populations described above. 3849 + 10kbC→T, a frequent variant in all referenced Latino populations, was not found in any patient in our study, which may be due to low frequencies in other Hispanic populations (2%–3%). p.Gly542X, one of the most frequent variants in Latino populations, was not present at all in Dominicans and observed only in 10% of Puerto Rican patients. The unique spectrum of CFTR variants in Dominican and Puerto Rican patients may be due to their heterogeneous genetic background, with a higher proportion of African ancestry than in Latino populations from the mainland U.S.8 Our findings highlight the need for investigating population-specific CFTR variants.
In this comprehensive genetic analysis of patients with clinically confirmed CF, 81% of Puerto Rican patients had disease-causing CFTR variants identified on both chromosomes compared with only 10% of the Dominican patients. Over two-thirds of Dominican patients had no identifiable variant in CFTR compared with 10% of Puerto Rican patients. We were surprised at the high proportion of Dominican patients without any identifiable CFTR variants. In contrast, sequencing analysis in other Latino populations with CF have reported much higher detection rate (approximately 95%) of CFTR variants.4,26 All the patients included in our study had clinical evidence of CFTR dysfunction with symptoms consistent with CF and a sweat chloride concentration of greater than 60 mmol/L. Analysis of nasal potential difference and functional analysis of the CFTR channel may increase our understanding of CFTR function in patients lacking CFTR variants. Other studies have described patients with symptoms of CF and elevated sweat chloride concentrations but without evidence of CFTR variants.30,31 Patients without 2 CFTR variants in trans may have variants in other genes such as the epithelial sodium channel (ENaC) or may have defective pathways that lead to CFTR dysfunction.32
We were also surprised that 10% of Dominican and 10% of Puerto Rican patients had multiple variants found on only one chromosome (ie, in cis). Genotype phase is not routinely analyzed in clinical sequencing of CFTR, so deleterious effects of different variants in cis may be more common in the general CF population than currently understood.
Understanding the spectrum and frequency of CFTR variants in diverse populations is important for improving CF genetic panels and newborn screening programs. Genetic panels and newborn screening programs are generally developed based on variant frequencies observed in the White population and have lower sensitivity (ie, higher false negative rate) when applied to a Latino population. The American College of Medical Genetics (ACMG)/American College of Obstetricians and Gynecologists (ACOG)-recommended CFTR 23 variant panel offered to pregnant women has a 76% detection rate in White patients with CF, while the detection rate is only 48% in Latino patients with CF.5,33 The ACOG panel would have detected only 7 of the 30 variants that we identified in our study (five variants in Dominicans, six variants in Puerto Ricans). Using the ACOG panel in our patients, 73% of patients would have no variants identified and 15% would have had only one variant identified.
Genetic screens and newborn screens should be sensitive to the target population and include the prevalent CFTR variants for all racial and ethnic groups to minimize false negative diagnoses. Detection via newborn screening is important as patients diagnosed via newborn screen demonstrate improved lung function and nutritional status compared with those not detected on a newborn screen.34 In the Illinois newborn screen, for example, Latino infants were more likely to have undefined variants and twice as likely to have only one variant identified compared to White infants.4 Latino patients have both more rare and novel CFTR variants so newborn or genetic screens will always be less effective for Latino patients if they do not include sequencing.35,36
CFTR genetic variant identification and functional classification have become increasingly valuable not only for CF phenotype prediction but also for identifying those patients who would benefit from CFTR modulator therapies.37 CFTR modulators target specific CFTR variants, which occur more frequently in White patients compared with minorities. As a result, only a third of Latino patients qualify compared with three-quarters of White patients.38 This is consistent with our studyʼs findings that only 5 of 82 patients (three Dominicans and two Puerto Ricans) were eligible for CFTR modulator pharmacotherapy. Only two of the 30 variants we identified were eligible CFTR modulator targets: p.Phe508del and p.Arg74Trp. The most common variant in Puerto Rican patients, p.Arg1066Cys, is not approved for CFTR modulators. To combat this disparity in access to life-altering pharmacogenetic therapies, the first step is to identify CFTR variants in CF populations, as we have done in this study, and then to describe the functional implications of the identified variants and investigate the protein response to CFTR modulators. The final step is to include Latino and other non-Latino non-White patients in clinical trials of CFTR modulators, as minorities are underrepresented in the majority of CF pharmacotherapy clinical trials.39
In silico prediction algorithms have been used to identify likely disease-contributing CFTR variants, but the utility of predictive algorithms is controversial as they cannot differentiate between variants that caused severe, moderate, or minimal reduction in CFTR function.40–42 Our study similarly found inconsistent predictions as the algorithms predicted five variants to be deleterious but were annotated by CFTR2 as “non-CF-causing” (Table S1). Additionally, we identified three variants that were predicted to be deleterious but were removed after manual inspection of the sequencing reads alignment.
Although we sequenced the majority of known patients with CF over 6 years old in both Puerto Rico and the Dominican Republic, our study was limited by a small number of patients. To fully understand CFTR variants in these populations, a genetic analysis of the general population of the Dominican Republic and Puerto Rico is needed. Our study identified multiple variants in cis, but our study was not designed to genotype parents to confirm genotype phase.
Our study results indicate that the spectrum of CFTR variants in an unstudied CF population cannot be inferred from another CF population, even if the racial and ethnic background is similar. Genetic panels and even genome sequencing have limitations in identifying CFTR variants in Latino patients with CF. Understanding the spectrum of CFTR variants in all populations with CF is the first step towards developing effective CF treatment for all patients. Studies of cystic fibrosis and pharmacotherapies need to include more racially diverse populations to make precision medicine socially precise.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the patients, families, recruiters, health care providers, and community clinics for their participation. In particular, the authors thank Yvelisse Mendez Arias and Fundación Dominicana de Apoyo a la Fibrosis Quística for their commitment to the cystic fibrosis community in the Dominican Republic. Whole genome sequencing (WGS) for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). WGS for “NHLBI TOPMed: Genes-environments and Admixture in Latino Asthmatics (GALA II) Study (phs000920)” was performed at University of Washington, Northwest Genomics Center (HHSN268201600032I). Centralized read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Phenotype harmonization, data management, sample-identity QC, and general study coordination were provided by the TOPMed Data Coordinating Center (3R01HL-120393-02S1; contract HHSN268201800001I).
Funding information
National Institute of Environmental Health Sciences, Grant/Award Number: R01ES015794; Cystic Fibrosis Foundation Therapeutics, Grant/Award Number: MCGARR16A0; National Institute on Minority Health and Health Disparities, Grant/Award Number: R01MD010443; National Heart, Lung, and Blood Institute, Grant/Award Numbers: 1K23HL133437-01A1, 1X01HL134589, 3R01HL-120393-02S1, R01HL117004; NHLBI/NIMHD, Grant/Award Number: R01HL135156
DISCLOSURES
MEM is supported by the NHLBI (1K23HL133437-01A1) and Cystic Fibrosis Foundation Therapeutics (MCGARR16A0). EGB is supported by the Sandler Family Foundation, the American Asthma Foundation, the RWJF Amos Medical Faculty Development Program, Harry Wm. and Diana V. Hind Distinguished Professor in Pharmaceutical Sciences II, National Institutes of Health (R01HL117004, 1 × 01HL134589), National Institute on Minority Health (R01MD010443), National Institute of Health and Environmental Health Sciences (R01ES015794). TOPMed Consortium is supported by the NHLBI (3R01HL-120393-02S1).
Data previously presented at the American Thoracic Society International Conference in May 2019 in Dallas, Texas.
Footnotes
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
TOPMed AUTHORSHIP LIST
The full TOPMed authorship list can be found at the TOPMed website (https://www.nhlbiwgs.org/topmed-banner-authorship.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
REFERENCES
- 1.Cystic Fibrosis Foundation. Cystic fibrosis Foundation Patient Registry, 2017 Annual Data Report to the Center Directors. Bethesda, MD: Cystic Fibrosis Foundation; 2017. [Google Scholar]
- 2.McGarry ME, Neuhaus JM, Nielson DW, Burchard E, Ly NP. Pulmonary function disparities exist and persist in Hispanic patients with cystic fibrosis: a longitudinal analysis. Pediatr Pulmonol. 2017;52(12):1550–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rho J, Ahn C, Gao A, Sawicki GS, Keller A, Jain R. Disparities in mortality of Hispanic patients with cystic fibrosis in the United States. A national and regional cohort study. Am J Respir Crit Care Med. 2018;198(8):1055–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watts KD, Layne B, Harris A, McColley SA. Hispanic infants with cystic fibrosis show low CFTR mutation detection rates in the Illinois newborn screening program. J Genet Couns. 2012;21(5):671–675. [DOI] [PubMed] [Google Scholar]
- 5.Schrijver I, Pique L, Graham S, Pearl M, Cherry A, Kharrazi M. The spectrum of CFTR variants in non-White cystic fibrosis patients. J Mol Diagn. 2016;18(1):39–50. [DOI] [PubMed] [Google Scholar]
- 6.Palomaki GE, FitzSimmons SC, Haddow JE. Clinical sensitivity of prenatal screening for cystic fibrosis via CFTR carrier testing in a United States panethnic population. Genet Med. 2004;6(5): 405–414. [DOI] [PubMed] [Google Scholar]
- 7.Stewart C, Pepper MS. Cystic fibrosis in the African diaspora. Ann Am Thorac Soc. 2017;14(1):1–7. [DOI] [PubMed] [Google Scholar]
- 8.Moreno-Estrada A, Gravel S, Zakharia F, et al. Reconstructing the population genetic history of the Caribbean. PLoS Genet. 2013;9(11): e1003925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farrell PM, White TB, Ren CL, et al. Diagnosis of cystic fibrosis: consensus guidelines from the Cystic Fibrosis Foundation. J Pediatr. 2017;181:S4–S15.e1. [DOI] [PubMed] [Google Scholar]
- 10.Taliun D, Harris DN, Kessler MD, et al. Sequencing of 53 831 diverse genomes from the NHLBI TOPMed Program. bioRxiv. 2019; Taliun, Daniel, et al. “Sequencing of 53 831 diverse genomes from the NHLBI TOPMed Program. BioRxiv. 2019:563866. [Google Scholar]
- 11.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu X, White S, Peng B, et al. WGSA: an annotation pipeline for human genome sequencing studies. J Med Genet. 2016;53(2):111–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arWiv preprint. 2013;arXiv:1303. 3997. [Google Scholar]
- 14.Li H, Handsaker B, Wysoker A, et al. The sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011; 21(6):974–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Layer RM, Chiang C, Quinlan AR, Hall IM. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15(6):R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loh P-R, Danecek P, Palamara PF, et al. Reference-based phasing using the haplotype reference consortium panel. Nat Genet. 2016;48(11):1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.CFTR2@Johns Hopkins. Home page [Internet]. http://cftr2.org/. Updated March 2019.
- 20.Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nykamp K, Anderson M, Powers M, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017;19(10):1105–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rogers MF, Shihab HA, Mort M, Cooper DN, Gaunt TR, Campbell C. FATHMM-XF: accurate prediction of pathogenic point mutations via extended features. Bioinformatics. 2018;34(3):511–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99(4):877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27(21):2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alonso MJ, Heine-Suñer D, Calvo M, et al. Spectrum of mutations in the CFTR gene in cystic fibrosis patients of Spanish ancestry. Ann Hum Genet. 2007;71(2):194–201. [DOI] [PubMed] [Google Scholar]
- 27.Sugarman EA, Rohlfs EM, Silverman LM, Allitto BA. CFTR mutation distribution among U.S. Hispanic and African American individuals: evaluation in cystic fibrosis patient and carrier screening populations. Genet Med. 2004;6(5):392–399. [DOI] [PubMed] [Google Scholar]
- 28.Grebe TA, Seltzer WK, DeMarchi J, et al. Genetic analysis of Hispanic individuals with cystic fibrosis. Am J Hum Genet. 1994;54(3):443–446. [PMC free article] [PubMed] [Google Scholar]
- 29.Wong LJC, Wang J, Zhang YH, et al. Improved detection of CFTR mutations in southern California Hispanic CF patients. Hum Mutat. 2001;18(4):296–307. [DOI] [PubMed] [Google Scholar]
- 30.Groman JD, Cutting GR. Variant cystic fibrosis phenotypes in the absence of CFTR mutations. N Engl J Med. 2002;347(6):401–407. [DOI] [PubMed] [Google Scholar]
- 31.Mekus F, Ballmann M, Bronsveld I, et al. Cystic-fibrosis-like disease unrelated to the cystic fibrosis transmembrane conductance regulator. Hum Genet. 1998;102(5):582–586. [DOI] [PubMed] [Google Scholar]
- 32.Agrawal PB, Wang R, Li HL, et al. The epithelial sodium channel is a modifier of the long-term nonprogressive phenotype associated with F508del CFTR mutations. Am J Respir Cell Mol Biol. 2017;57(6): 711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Committee on Genetics. Committee opinion no. 691: carrier screening for genetic conditions. Obstet Gynecol. 2017;129(3): e41–e55. [DOI] [PubMed] [Google Scholar]
- 34.Leung DH, Heltshe SL, Borowitz D, et al. Effects of diagnosis by newborn screening for cystic fibrosis on weight and length in the first year of life. JAMA Pediatrics. 2017;171(6):546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kharrazi M, Yang J, Bishop T, et al. Newborn screening for cystic fibrosis in California. Pediatrics. 2015;136(6):1062–1072. [DOI] [PubMed] [Google Scholar]
- 36.Alper M, Wong LJC, Young S, et al. Identification of novel and rare mutations in California Hispanic and African American cystic fibrosis patients. Hum Mutat. 2004;24(4):353. [DOI] [PubMed] [Google Scholar]
- 37.De Boeck K, Amaral MD. Progress in therapies for cystic fibrosis. Lancet Respir Med. 2016;4(8):662–674. [DOI] [PubMed] [Google Scholar]
- 38.McGarry ME, Ly NP. Unequal access to CFTR modulators in Hispanics. Pediatr Pulm. 2017;52:S396–297. [Google Scholar]
- 39.McGarry ME, McColley SA. Minorities are underrepresented in clinical trials of pharmaceutical agents for cystic fibrosis. Ann Am Thorac Soc. 2016;13(10):AnnalsATS.201603–192BC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raraigh KS, Han ST, Davis E, et al. Functional assays are essential for interpretation of missense variants associated with variable expressivity. Am J Hum Genet. 2018;102(6):1062–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lim RM, Silver AJ, Silver MJ, et al. Targeted mutation screening panels expose systematic population bias in detection of cystic fibrosis risk. Genet Med. 2016;18(2):174–179. [DOI] [PubMed] [Google Scholar]
- 42.Marson FAL, Bertuzzo CS, Ribeiro JD. Classification of CFTR mutation classes. Lancet Respir Med. 2016;4(8):37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.