

Abstract
T cells, which are involved in adaptive immunity, are essential in the elimination of tumor cells. Mature T cells can specifically recognize the antigen on the major histocompatibility complex (MHC) molecule through T‐cell receptors (TCR). The unique rearrangement mechanisms during T‐cell maturation provide great diversity to TCR, ensuring specific recognition between T cells and antigens. Thus, TCR repertoire analysis occupied an important position in T‐cell regarding research. Nowadays, next‐generation sequencing technology allows the simultaneous detection of TCR sequences with high throughput, and several evaluation indexes facilitate the measure of TCR repertoire. Based on this new methodology, discoveries are made across a range of tumor types. Results have shed light on the TCR repertoire differences between cancer patients and healthy control as well as between individual's lesions, paracancer, and peripheral blood samples. The potential of TCR repertoire as a biomarker for immunotherapy efficacy is also widely studied as TCR repertoire represents different baseline within individuals and shows dynamic change during treatment. Accurate delineation of the T‐cell repertoire can further the understanding of the immune system response to tumorigenesis. Still, existing researches are insufficient to clarify the specific clinical implications of TCR dynamic change and the definite role of TCR repertoire diversity during the treatment process. The results of some studies are even contrary. In this article, we reviewed TCR rearrangement mechanisms and analysis methods. Recent progress of TCR sequencing technology in tumor research is also discussed. In conclusion, intensive studies over an extended range of cancer types and a broadened group of subjects should be carried to solidify the TCR repertoire's position as an immunotherapy biomarker.
Keywords: diagnosis, diversity, immunotherapy, rearrangement, repertoire, sequencing, Solid tumor, T‐cell receptors
Abbreviations
- 5′ RACE
rapid amplification of 5′ complementary DNA ends
- C
constant
- CAR‐T
chimeric antigen receptor‐T
- CDR
complementary decision region
- CRC
colorectal cancer
- D
diversity
- dPCR
digital PCR
- gDNA
genomic DNA
- HEC
highly expanded clones
- HLA
human leukocyte antigen
- ICB
immunocheckpoint blocking therapy
- ICIs
immunocheckpoint inhibitors
- J
joining
- MHC
major histocompatibility complex
- MID
molecular identifiers
- MPR
major pathological response
- NGS
Next‐generation sequencing
- PBMC
peripheral blood mononuclear cell
- PD‐L1
programmed death‐ligand 1
- PFS
progression‐free survival
- TCR
T‐cell receptor
- TDLN
Tumor draining lymph nodes
- TIL
tumor‐infiltrating lymphocyte
- V
variable
1. INTRODUCTION
Since the year 2011, there have been rapid developments in the field of immunotherapy, and a range of immunocheckpoint inhibitors (ICIs) have been approved for a range of metastatic cancers, including malignant melanoma, lung, kidney, and bladder cancers [1, 2, 3, 4, 5]. In recent years, the clinical trial results of genetically engineered immune cells, including chimeric antigen receptor T‐cell (CAR‐T), TCR‐engineered T‐cell (TCR‐T), chimeric antigen receptor natural killer cell (CAR‐NK) and more, in the treatment of hematological tumors have also been encouraging [6, 7, 8]. Immunotherapy has nowadays developed into a new and promising therapy in addition to surgery, radiotherapy, chemotherapy, and molecular targeted therapy [9]. Havel et al.[10], however, found that the efficacy of ICIs might be influenced by certain factors such as tumor genomics, host germline genetics, programmed death‐ligand 1 (PD‐L1) levels, tumor microenvironment, and intestinal microbiome. Nevertheless, most of the existing studies predicting the clinical efficacy of immunocheckpoint blocking therapy (ICB) focused solely on single factors, possibly because the analyses are much easier to be conducted on single variate [11, 12, 13]. This may be a practical choice under current technical conditions but is still insufficient for comprehensively identifying patients who would benefit from immunotherapy. Unified predictive model for ICI efficacy should be developed, and different components affecting the tumor‐host interactions should be considered in the development of such a model [10].
T cells are the active cell population that mediates cellular immune response and function as an essential component in humoral immune system activation response. Regarding T cells, it has been found that the T‐cell receptor (TCR) repertoire has an important impact on a wide range of disease, including malignancy, autoimmune disorders, and infectious diseases, and is involved with the immune system in almost all human health conditions and diseases [14]. The majority of T cells in the human peripheral blood are made up of TCR α and β chains, while a small portion (1%‐5%) contains TCR δ and γ chains [15]. In recent years, enormous progress has been made in understanding TCR diversity in immunotherapy. ICIs, which target T cells to suppress their programmed death‐1 (PD‐1) receptor and activate an anti‐tumor immune response, have brought substantial transformations in the treatment of various cancer types. The α/β T cells are essential agents in cellular immunity due to their ability to recognize antigen‐mediated adaptive immunity presented on class I and II proteins of major histocompatibility complex (MHC), which encode antigen participating in the body's immune response and immune regulation, and determine tissue compatibility [16]. A complete TCR chain is composed of constant (C) regions and variable (V) regions. Those V regions mediate the specific recognition of different antigens. The V regions of TCR α/δ chains are encoded by the V gene and joining (J) genes, while the coding of β and γ chains require additional diversity (D) genes [17, 18, 19]. During T‐cell maturation, V/D/J gene fragment is rearranged to form a continuous sequence containing a single V, D, and J gene, and a random insertion or deletion is introduced at the junction sites of both V/D genes and D/J genes to produce a functional TCR sequence (Figure 1). So far, 1013 different types of TCR sequences have been found [20, 21]. The V region encoded by V/D/J gene rearrangement is also known as a complementary determining region 1‐3 (CDR1‐3). CDR1 and CDR2 are encoded by the V gene, while CDR3 is encoded by the junction region V/J or V/D/J, which varies greatly and is in direct contact with antigens [22]. Recently, researches have been focusing on investigating the state of the immune spectrum in different disease conditions such as cancer [23, 24], autoimmunity [25], inflammation [26], and infectious diseases [27]. The total TCR of all T cells, also known as TCR profile, can change dramatically as these diseases progress [24, 25, 26, 27]. Some studies have attempted to identify specific types of T‐cell clones involved in the pathogenesis of cancer by analyzing the tumor‐infiltrating lymphocyte (TIL) spectrum [15, 28]. At the same time, the great diversity of TCR sequences within and between individuals represents the biggest challenge in TCR research as there is still technical limitations in obtaining unbiased absolute count data of TCR clonotypes [21].
FIGURE 1.
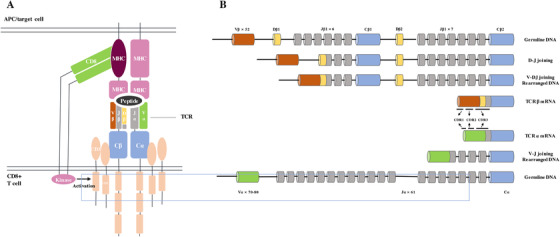
Structure and rearrangement mechanisms of TCR. (A) Mature TCR recognizes antigens presented on MHC molecules. (B) TCR alleles on the genome are rearranged to form a mature TCR sequence. Rearrangement and recombination of V, D, and J segments for TCR β chains and V and J segments for TCR α chains guarantee antigen repertoire diversity. This variability (combinatorial diversity) is further increased by the addition or deletion of nucleotides at the junction sites (junctional diversity)
Abbreviations: TCR: T‐cell receptor(s); MHC: major histocompatibility complex.
Next‐generation sequencing (NGS), a method that allows large‐scale parallel sequencing of millions of DNA molecules, has revolutionized genomic research [29]. NGS can be used for comprehensive mapping of complex TCR repertoire, including low‐frequency clonal sequences, with its advantages in greater sequencing depth and significantly more accurate quantification of TCR clonotype abundance than spectratyping, where the number of clonotypes is indicated by the number of different CDR3 lengths [21]. Although there are many TCR sequencing methods at the single‐cell level, this paper mainly focused on discussing bulk sequencing for a cell population, compared with the former, which is more commonly used to study TCR diversity and to compare different profiling methods in large groups of patients with solid tumor. In this article, we reviewed the TCR library construction method and a series of evaluation parameters. We also summarized the real‐world application of TCR library analysis technology in the diagnosis and treatment of solid tumors. Further, we comprehensively discussed the feasibility and practicality of using TCR as an immunotherapy biomarker for predicting the treatment and prognosis of cancer patients, thus providing valuable information for subsequent research.
2. TCR REPERTOIRE ANALYSIS VIA NGS
2.1. TCR sequencing materials
Both genomic DNA (gDNA) and RNA can be used for TCR repertoire analysis [14, 30]. Yet, each has its own advantages and limitations. gDNA has higher stability and is more adaptable as sequencing material when samples are under complex real‐world conditions where RNA is more vulnerable to degradation. At the same time, the TCR sequence on gDNA has a constant copy number in each cell, so that the quantitative analysis of TCR with gDNA as the sequencing material can better reflect the characteristics of the T‐cell population [30]. However, gDNA cannot reflect the differences in TCR expression. Further, introns located upstream and downstream of the coding region may interfere with primer binding and affect sequencing accuracy [21].
By using RNA as sequencing materials, it is possible to avoid the above‐mentioned problems. The minimum requirement of volume for starting the RNA template is lower than the DNA template. However, since each cell contains more than one transcript template and the number of transcripts can significantly differ between cells, a quantitative analysis of the results obtained by the RNA template cannot accurately reduce cell clone of amplification, posing challenges on single‐cell analysis at the meantime [31]. Considering this, we mainly discussed the sequencing approach targeting cell clones rather than single‐cell here as mentioned before. To quantitatively study TCR RNA molecule copy number and clonality in T cells, Ma et al. published a study on TCR sequencing based on RNA library construction [32]. Using molecular identifier (MID) cluster analysis data, the authors evaluated the sequencing results via TCR amplification using microdroplet digital PCR (dPCR). Their data suggested a high accuracy and sensitivity when using MID to correct errors caused by PCR bias and the use of the sequencing process to calculate the number and diversity of TCR with RNA as raw material.
2.2. TCR library construction
The most common library construction methods include multiplex PCR and rapid amplification of 5′ complementary DNA ends (5′ RACE) (Figure 2).
FIGURE 2.
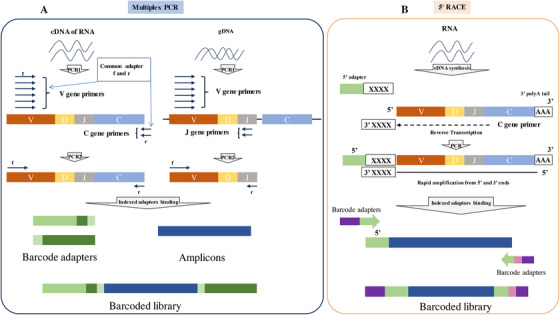
Process of TCR library construction. (A) The process of multiplex PCR. (B) The process of 5′ RACE
Abbreviations: TCR: T‐cell receptor(s); PCR: polymerase chain reaction; 5′ RACE: 5′ rapid amplification of cDNA ends.
In multiplex PCR (Figure 2A) for which both gDNA and RNA can be used as starting materials, the CDR sequence of TCR is specifically amplified with complementary pairs of primers at both ends of the CDR sequence. Commonly, a series of mixed primers located on the known V allele are designed and paired with primers located in the J or C region of the TCR α/δ chain. [30, 33]. However, the library construction method using multiplex PCR represents a big shortcoming due to its lack of sequence diversity. The reasons behind this shortage are as follows: (1) primer designed on the basis of a known sequence results in only the known V alleles can be amplified in TCR sequences; (2) sequence abundance may also be biased due to the amplification preference of different primer pairs. Technologies are developed to solve this problem, one of which is using mRNA as raw material and adding a cap during mRNA reverse transcription to replace the uncertain V region. The addition of molecular barcodes and adjustment of primer concentrations can also be used to avoid inaccuracies in abundance caused by amplification bias and thus optimize sequence diversity [34, 35].
Using only RNA samples, 5′ RACE (Figure 2B) represents another effective way for TCR library construction [36]. This technique uses reverse transcriptase with terminal transferase activity for reverse transcription and generates complementary DNA (cDNA) which is added with an additional non‐template sequence, usually deoxycytidine triphosphate (dCTP), at the 3′ end. Subsequently, a poly‐G template‐switch‐oligonucleotide binds to the 3′‐terminal sequence of the first strand and initiates the polymerization reaction [37]. The amplicons obtained via this method have a complete 5′ terminal of TCR, which means that the complete V gene sequence can be retained. Meanwhile, since the oligonucleotide introduced in the chain replacement step can serve as a common forward primer‐binding site, subsequent template amplification links only need one primer set to achieve synchronous amplification of different V genes, including known and unknown V genes. A single primer set can also be used to effectively avoid bias caused by amplification preference. This method is currently considered as the gold standard for TCR repertoire sequencing because it can detect all types of V genes accurately as well as avoid the impact of sequencing bias on the results effectively [38, 39, 40]. Table 1 shows the basic differences between multiplex PCR and 5′ RACE.
TABLE 1.
Pros and Cons when using multiplex PCR and 5′ RACE for TCR library construction
| Items | Multiplex PCR | 5′ RACE |
|---|---|---|
| Core principles | A series of mixed primers which are located on the known V allele are designed and paired with primers located in the J or C region. | This method relies on reverse transcriptase activity. Next, dCTP is added to the cDNA 3′ end as a primer binding domain during the first strand synthesis reaction. |
| Advantages | The method is compatible with gDNA and RNA. | (1) Can cover the entire V gene and retain the whole TCR and V/D/J region. (2) Avoid amplification bias. |
| Disadvantages | (1) Unable to detect mutation information for the V region. (2) Primer amplification bias | This method can only start with RNA, so it is more demanding than other techniques. Besides, the repeatability may be affected. |
| Correction | Adjust primer concentration or use a unique molecular identifier | ‐ |
Abbreviations: TCR: T‐cell receptor(s); PCR:Polymerase chain reaction; 5′ RACE: 5′ Rapid Amplification of cDNA Ends
2.3. Description and evaluation of TCR repertoire
Analysis of the immune repertoire usually involves one or more diversity indexes, such as Shannon entropy, clonality, and high‐expanded clone (HEC) ratio, which are used to evaluate the amplification status of different TCR sequences and determine whether there is a high expansion of a few T‐cell clones. The different indexes are explained one by one in the following paragraphs.
As shown in the formula below, the Shannon entropy index takes into account both the number of T‐cell clone types “n” and the frequency “pi” of each clone, where “pi” is the proportion of the i‐th clone in the TCR library with n clones. Shannon entropy can measure the diversity of T‐cell clones as it reflects the CDR variability. The higher the index, the higher the diversity of T‐cell clones in the sample [41].
Shannon entropy:
Clonality is based on the normalized Shannon entropy [42], which is inversely related to the diversity of T‐cell clones. The clonality value is always between 0‐1, which facilitates the comparison of the difference between two TCR repertoires with a different number of clones. Unlike the Shannon entropy, the clonality index is used to measure T‐cell expansion and indicates the frequency of clone expansion in some clones. The larger the value, the smaller the sample diversity.
Clonality:
In addition to the Shannon entropy and clonality, the HEC ratio is also used for the description of the immune group library state. The HEC ratio is calculated as the sum of the abundance of all sequences with the abundance higher than the threshold. The common threshold is 0.01% or 0.1%, and its threshold setting can be adjusted according to research needs [3].
High expanded clone (HEC):
Figure 3 shows the association between different statistical parameters and the status of the immune repertoire. It can be seen that some TCR sequences have high expanded clones in the left circle (Figure 3A), illustrating that the TCR repertoire has higher clonality, lower Shannon entropy, and thus lower diversity than that represented by the circle on the right (Figure 3B), which shows a typical immune repertoire without high expanded clone. The right TCR repertoire has lower clonality but a higher Shannon index and repertoire diversity. Generally, it is considered that the TCR repertoire of a healthy status has higher diversity than that of disease status, making it a potential novel biomarker for immunotherapy and a predictor for clinical prognosis [43, 44]. Shannon entropy, clonality, and HEC have been widely used as evaluation parameters in assessing TCR repertoire diversity in recent studies [1, 45, 46], through which it quantifies and facilitates the assessment of a patient's physical condition and his/her prognosis.
FIGURE 3.
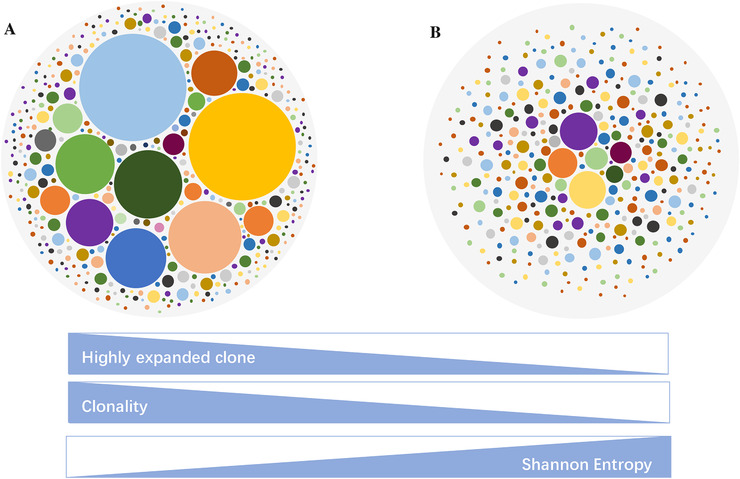
Examples of different immune repertoire expansion status. Each dot represents a unique TCR sequence, and the area of the dot represents the abundance of the TCR sequence. (A) Circle represents a typical TCR repertoire with significant clonal expansion. (B) Circle represents a typical TCR repertoire without clonal expansion. The bar below represents the association between different statistical parameters and the status of immune repertoire
Abbreviations: TCR: T‐cell receptor(s).
3. APPLICATION OF TCR SEQUENCING IN SOLID TUMORS
3.1. Differences in TCR repertoire between solid tumor, paracancer, and peripheral blood samples
The tumor microenvironment indicates the cellular environment in which the tumor exists. It includes tumor cells, different types of normal cells, surrounding blood vessels, and signaling molecules [47]. Due to the influence of tumor microenvironment complexity, the distribution of T‐cell repertoire in tumors, normal tissues, and peripheral blood is heterogeneous [47]. Real‐world studies have shown that the characteristics of TCR during the malignancy development or from various sample types may be different [43, 44, 48]. Therefore, it is worth exploring the TCR repertoire differences between tumoral and adjacent normal tissues, as well as the consistency of TCRs between tumoral tissues and peripheral blood.
Using the 5′ RACE method, one study constructed and sequenced TCR libraries for peripheral blood mononuclear cells from patients with cervical cancer (CC), cervical intraepithelial neoplasia (CIN), and healthy women. CC patient's sentinel lymph node and tumor tissue samples were also paired and analyzed [43]. The results showed that TCR diversity in the peripheral blood decreased with tumor progression, where the lowest number of unique sequences of TCR were identified in CC patients, followed by CIN patients while the highest number was within the healthy population. In other words, the highest diversity was found in peripheral blood samples of the healthy population according to the calculation results of Shannon entropy. Moreover, TCR repertoire diversity in sentinel lymph nodes from CC patients was higher than in tumor tissues. The study suggested fewer clonotypes in the TCR repertoire of sentinel lymph nodes indicated poorer prognosis of the patients. Based on the discovery, the TCR repertoire may have the potential to be an immune monitoring indicator and a prognosis prediction biomarker for CC patients [43].
Tumor draining lymph nodes (TDLN) are located in the lymphatic drainage pathway of the primary tumor and are the most likely sites to have metastatic cancer cells in various solid tumors. Matsuda et al. [44] showed that compared with non‐metastatic TDLN, the TCR diversity of metastatic TDLN was significantly reduced for colorectal cancer. Moreover, principal component analysis (PCA) showed that the transferred TDLNs shared similar TCR sequences. These results suggest that cancer‐reactive T‐cell clones can be enriched in precancerous lesions, tumors, and metastatic lymph nodes, which indicate a lower Shannon entropy and diversity.
Contrarily, Wang et al. [48] analyzed TCR β repertoire characteristics of the tumor and paired paracancerous tissues in 15 patients with lung cancer. Their results showed that the proportion of HEC in normal lung tissues was significantly higher than that in the tumor. Besides, TCR diversity of cancerous tissues was also considerably higher than that in healthy lung tissues. Moreover, Wang et al. [48] found that younger patients had a significantly higher TCR diversity than older patients, and higher TCR diversity in tumors was related to worse cancer outcomes. Compared with the above cancer types, their data indicated that patients with lung cancer might have distinct T‐cell immune microenvironments.
Based on the studies mentioned above, we observed that some conclusions in different tumors seem to be opposite, for instance, CC patients tend to have lower TCR repertoire diversity of peripheral blood samples compared to the healthy population [43] while a significantly higher TCR diversity was observed in lung cancer tissues than in normal lung tissues [48]. Moreover, as for the association between TCR repertoire diversity and prognosis, the results are still ambiguous. Inconsistent with the findings of Wang et al. [48] in their lung cancer group, diversity in tumor tissues or sentinel lymph nodes showed no significant differences between the progressive CC patients group and the non‐progressive CC patients group. It has also been reported that the TCR repertoire has great prognostic value in prostate cancer [49] and metastatic melanoma [2]. To further understand the availability of TCR repertoire in predicting cancer outcomes, more researches are required. Moreover, there is a clinical need to identify patients who would have better immunotherapeutic response and to understand therapeutic resistance mechanisms [2]. Due to this, a lot of works have been done on host immune infiltrates [50, 51, 52, 53], and has attracted a lot of attentions for investigations on TCR repertoire.
3.2. Dynamic changes of TCR repertoire during tumor treatment
For patients with advanced cancer who fail to undergo surgery or biopsy due to a range of reasons, tumor tissue may be unavailable through the treatment period. For these patients, blood samples are commonly analyzed. During tumor treatment, the peripheral blood TCR repertoire may significantly change.
In 2019, Liu et al. [45] examined the association between TCR similarity/diversity in peripheral blood and survival benefit for advanced lung cancer patients, before and after several lines of therapy, including chemotherapy, radiotherapy, tyrosine kinase inhibitor therapy (also known as targeted therapy), surgery, and anti‐angiogenic therapy. Most patients in this study received these therapies as combination treatment. Their results showed that all patients with a significant increase (> 10%) in Shannon diversity after treatment exhibited durable clinical benefit (DCB) and longer progression‐free survival (PFS) than those with stable or reduced immune repertoire diversity. Another TCR analysis method, similarity analysis, was also used in this study [46]. Similar findings were observed by this method, compared with Shannon diversity analysis. Combining the diversity and similarity analysis results, it was shown that all patients (11/11) with either increased diversity (> 10%) or a stable diversity (increased or decreased by < 10%) but high similarity with paired samples in the immune repertoire exhibited DCB and longer PFS. Therefore, the dynamic changes of peripheral blood immune pools before and after treatment may be used as a useful prognostic indicator in lung cancer treatment.
Liu et al. [3] investigated the changes in peripheral blood TCR repertoire before and after radical surgery in 19 colorectal cancer patients (T1M0N0, stage I). Five healthy subjects were included as controls. They defined TCR clones with a frequency > 0.5% as HECs. The preoperative patient group showed a significantly higher HEC ratio when compared with healthy control while after surgery, the patient group's HECs fluctuated and decreased generally. This suggested that the immune repertoire could change over surgery and might be used as a powerful tool for predicting colorectal cancer surgery prognosis.
Furthermore, TCR was dynamically associated with the patient's physical state during treatment. After PD‐1 blockade therapy, the number of T‐cell clones found in both the tumor and peripheral blood systemically increased in eight patients with untreated, surgically resectable, early‐stage (I, II, or IIIA) non‐small‐cell lung cancer (NSCLC) [1]. Besides, mutation‐associated, neoantigen‐specific T‐cell clones from a primary tumor with a complete response on pathological assessment rapidly expanded in peripheral blood at 2 to 4 weeks after PD‐1 blockade with nivolumab [1]. Moreover, some new clones appeared, which indicated that TCR repertoire analysis could be applied in the dynamic monitoring of tumor immunotherapy. Detecting the types of specific clones that change significantly during treatment will be an interesting subject.
As mentioned above, peripheral blood is easily obtainable throughout the whole course of cancer. Based on current study results, peripheral blood TCR repertoire analysis may have an indication for treatment efficacy in a range of cancer types, while more evidence is still required to validate the association between dynamic TCR repertoire alteration and tumor outcome.
3.3. TCR repertoire analysis in immune therapy
Several studies have shown that deep sequencing‐based T‐cell libraries can serve as biomarkers for immune responses in cancer patients [54]. In 2014, Tumeh et al. [51] first reported the correlation between TIL TCR diversity and the PD‐1 inhibitor efficacy in melanoma. The proliferation of T cells and higher TCR clonality were found in patients with metastatic melanoma treated with pembrolizumab compared to patients with disease progression. Furthermore, Roh et al. [2] explored the relationship between tumor TCR clonality and immune therapy efficacy in advanced melanoma patients to clarify the mechanism underlying immune checkpoint inhibitor resistance. They found higher TCR clonality in patients who responded to PD‐L1 blockade after cytotoxic T‐lymphocyte‐associated protein 4 (CTLA‐4) treatment failure compared to non‐responders, confirming previous reports that TCR clonality was associated with response to PD‐1 blockade.
Hopkins et al. [54] demonstrated that TCR diversity and clonal changes in T cells were associated with immunotherapy efficacy for metastatic or locally advanced pancreatic cancer. In the CTLA‐4 treatment group, patients with higher pretreatment TCR diversity or more posttreatment expanded clones had longer survival (mPFS, 8.66 months vs. 4.28 months, and 13.23 months vs. 4.55 months, respectively) while similar results were not observed in the PD‐1 treatment group. In 2018, Forde et al. [1] evaluated the safety and efficacy of immune checkpoint inhibitors in patients with untreated, surgically resectable, early‐stage (I, II, or IIIA) NSCLC. The patients underwent neoadjuvant therapy and those who achieved significant pathological response after neoadjuvant treatment had higher TCR clonality in TILs, thus suggesting that TCR repertoire sequencing could be applied in the evaluation of immunotherapy efficacy. This suggests that TCR repertoire analysis has the potential to be a biomarker for selecting patients who may respond to immunotherapy. Still, studies on more cancer types and related clinical trials should be conducted before acclaiming its clinical reliability.
The functional study of the T‐cell population in real‐world clinical applications not only expands our understanding on T‐cell immunity during tumor development but also provides an experimental basis for further research on tumor pathogenesis and tumor immunotherapy. For example, chimeric antigen receptor‐T (CAR‐T) has changed the treatment landscape of B‐cell non‐Hodgkin's lymphoma and acute lymphoblastic leukemia [55, 56]. Several CAR‐T cell platforms targeting B cell maturation antigen are also under active clinical trials for refractory and/or relapsed multiple myeloma, and more targets such as C‐type lectin‐like molecule‐1 (CLL‐1), epidermal growth factor receptor (EGFR), NKG2D and mesothelin are being studied in CAR‐T cell trials for leukemia and solid tumors [57, 58]. Besides, TCR‐T and other TCR‐modified therapy based on research have been evolving rapidly and has become a promising strategy against various types of cancer, especially solid tumors [6]. Due to these developments, therapies based on engineered T cells have become a topic of interest in clinical practice.
4. FUTURE DIRECTIONS
TCR is an essential molecule for T cells to recognize neoantigens in tumors. Monitoring the high amplification of TCR clones in tumors or peripheral blood may help assessing the status for an immune response. NGS technology provides a feasible method to simultaneously identify a large number of TCR clones and is widely used in the clinic for TCR repertoire analysis. This article summarizes the studies which have reported the characteristics of immune banks based on NGS technology over the past three years. Different TCR library amplification states have been found between tumor and healthy tissues, tumor and precancerous lesions, and tumor tissues and peripheral blood. Therefore, to some extent, the TCR clonality of a tumor can be used as a useful biomarker for immunotherapy response and efficacy. Moreover, based on TCR sequencing, the dynamic changes of TCR cloning during treatment can be analyzed to understand the clinical benefits of tumor treatment.
However, due to the diversity of tumor antigens and the complexity of TCR and antigen recognition mechanisms, the significance of TCR tracking specific sequences is limited [35]. Compared with PD‐L1 immunohistochemical detection and targeted sequencing of liquid biopsy, TCR group library analysis is a complex and costly procedure. Although TCR analysis indicators, such as Shannon entropy, clonality, and HEC, can help to understand the relationship between TCR clonality and solid tumors, the association between TCR repertoire and clinical benefit needs to be supported by a larger scale of data. As mentioned earlier, human leukocyte antigen, MHC, neoantigen, intestinal microorganisms, and TIL are contributing factors to the efficacy of immunocheckpoint inhibitors. Careful consideration in these fields may be the direction for future efforts. However, the TCR immunorepository could still be used as a tumor immune marker with potential clinical significance in predicting prognosis and monitoring treatment efficacy. In the future, more advanced sequencing, library construction technologies, and comprehensive analytic algorithms could further elucidate the evaluation and comparison of TCR repertoire.
DECLARATIONS
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
COMPETING INTERESTS
The authors declare that they have no competing interests.
FUNDING
This research was supported in part by “Model establishment of early lung cancer diagnosis and treatment and early lung cancer driver gene research project of Liaoning province” (2019JH8/10300089).
AUTHORS' CONTRIBUTIONS
YYL contributed substantially to the conception and design of this study. NL collected material and wrote the manuscript. JNY, WJT, and LM contributed to the revision of the article. All authors have read and approved the final manuscript.
ACKNOWLEDGMENTS
Not applicable.
Li N, Yuan J, Tian W, Meng L, Liu Y. T‐cell receptor repertoire analysis for the diagnosis and treatment of solid tumor: A methodology and clinical applications. Cancer Commun. 2020;40:473–483. 10.1002/cac2.12074
REFERENCES
- 1. Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR, Hellmann MD, et al. Neoadjuvant PD‐1 blockade in resectable lung cancer . New New England Journal of Medicine. 2018;378(21):1976‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roh W, Chen P‐L, Reuben A, Spencer CN, Prieto PA, Miller JP, et al. Integrated molecular analysis of tumor biopsies on sequential CTLA‐4 and PD‐1 blockade reveals markers of response and resistance. Sci Transl Med. 2017;9(379):eaah3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu X, et al. A comprehensive study of immunology repertoires in both preoperative stage and postoperative stage in patients with colorectal cancer. Molecular Genetics & Genomic Medicine. 2019;7(3):e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Unverzagt S, et al. Immunotherapy for metastatic renal cell carcinoma. Cochrane Database of Systematic Reviews. 2017(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lerner SP, Bajorin DF, Dinney CP, Efstathiou JA, Groshen S, Hahn NM, et al. Summary and Recommendations from the National Cancer Institute's Clinical Trials Planning Meeting on Novel Therapeutics for Non‐Muscle Invasive Bladder Cancer. Bladder Cancer. 2016;2(2):165‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang J, Wang L. The Emerging World of TCR‐T Cell Trials Against Cancer: A Systematic Review. Technol Cancer Res Treat. 2019;18:153303381983106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang W, Jiang J, Wu C. CAR‐NK for tumor immunotherapy: Clinical transformation and future prospects. Cancer Lett. 2020;472:175‐80. [DOI] [PubMed] [Google Scholar]
- 8. Singh AP, Zheng X, Lin‐Schmidt X, Chen W, Carpenter TJ, Zong A, et al. Development of a quantitative relationship between CAR‐affinity, antigen abundance, tumor cell depletion and CAR‐T cell expansion using a multiscale systems PK‐PD model. mAbs. 2020;12(1):1688616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Qiu H. Safety and efficacy of toripalimab in advanced gastric cancer: A new clinical trial bringing hope for immunotherapy in gastric cancer. Cancer Commun. 2020;40(4):194‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19(3):133‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti‐PD‐1 immunotherapy in melanoma patients. Science 2018;359(6371):97‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Larkin J, Chiarion‐Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373(1):23‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T‐cell invigoration to tumour burden ratio associated with anti‐PD‐1 response. Nature. 2017;545(7652):60‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Woodsworth DJ, Castellarin M, Holt RA. Sequence analysis of T‐cell repertoires in health and disease. Genome Medicine. 2013;5(10):98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lo Presti E, Dieli F, Meraviglia S. Tumor‐infiltrating γδ T lymphocytes: pathogenic role, clinical significance and differential programming in the tumor microenvironment. Front Immunol. 2014;5:607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xiong Na, Raulet DH. Development and selection of γδ T cells. Immunol Rev. 2007;215(1):15‐31. [DOI] [PubMed] [Google Scholar]
- 17. Schatz DG, Ji Y. Recombination centres and the orchestration of V (D) J recombination. Nat Rev Immunol. 2011;11(4):251‐63. [DOI] [PubMed] [Google Scholar]
- 18. Miles, JJ, Douek, DC, Price, DA. Bias in the αβ T‐cell repertoire: implications for disease pathogenesis and vaccination. Immunology & Cell Biology. 2011;89(3):375‐87. [DOI] [PubMed] [Google Scholar]
- 19. Burtrum DB, et al., TCR gene recombination and alpha beta‐gamma delta lineage divergence: productive TCR‐beta rearrangement is neither exclusive nor preclusive of gamma delta cell development. J Immunol. 1996;157(10):4293‐96. [PubMed] [Google Scholar]
- 20. Turner, SJ, Doherty PC, Mccluskey J, Rossjohn J. Structural determinants of T‐cell receptor bias in immunity. Nat Rev Immunol. 2006;6(12):883‐94. [DOI] [PubMed] [Google Scholar]
- 21. Laydon DJ, Bangham CRM, Asquith B. Estimating T‐cell repertoire diversity: limitations of classical estimators and a new approach. Philosophical Transactions of the Royal Society B: Biological Sciences. 2015;370(1675):20140291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Davis MM, Boniface JJ, Reich Z, Lyons D, Hampl J, Arden B, Chien Y‐H. Ligand recognition by αβ T cell receptors. Annu Rev Immunol. 1998;16(1):523‐44 [DOI] [PubMed] [Google Scholar]
- 23. Jones SJm, Laskin J, Li YY, Griffith OL, An J, Bilenky M, et al. Evolution of an adenocarcinoma in response to selection by targeted kinase inhibitors. Genome Biol. 2010;11(8):R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu D, Sherwood A, Fromm JR, Winter SS, Dunsmore KP, Loh ML, et al. High‐throughput sequencing detects minimal residual disease in acute T lymphoblastic leukemia. Sci Transl Med. 2012;4(134):134ra63‐134ra63. [DOI] [PubMed] [Google Scholar]
- 25. Backes C, Ludwig N, Leidinger P, Harz C, Hoffmann J, Keller A, Meese E, et al. Immunogenicity of autoantigens. BMC Genomics. 2011;12(1):340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klarenbeek PL, De Hair MJH, Doorenspleet ME, Van Schaik BDC, Esveldt REE, Van De Sande MGH, et al. Inflamed target tissue provides a specific niche for highly expanded T‐cell clones in early human autoimmune disease. Ann Rheum Dis. 2012;71(6):1088‐93. [DOI] [PubMed] [Google Scholar]
- 27. Chen H, Ndhlovu ZM, Liu D, Porter LC, Fang JW, Darko S, et al. TCR clonotypes modulate the protective effect of HLA class I molecules in HIV‐1 infection. Nat Immunol. 2012;13(7):691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao Y‐J, Zhang J, Shi F, Hu Z‐P, Wu J‐P, Wu G‐J, et al. Expression of PD‐1 on CD4+ tumor‐infiltrating lymphocytes in tumor microenvironment associated with pathological characteristics of breast cancer. Journal of Immunology Research. 2018;2018:1‐8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brown SD, Raeburn LA, Holt RA. Profiling tissue‐resident T cell repertoires by RNA sequencing. Genome Medicine. 2015;7(1):125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dziubianau M, Hecht J, Kuchenbecker L, Sattler A, Stervbo U, Rödelsperger C, et al. TCR Repertoire Analysis by Next Generation Sequencing Allows Complex Differential Diagnosis of T Cell‐Related Pathology. Am J Transplant. 2013;13(11):2842‐54. [DOI] [PubMed] [Google Scholar]
- 31. Wu T, Wu X, Wang H‐Y, Chen L. Immune contexture defined by single cell technology for prognosis prediction and immunotherapy guidance in cancer. Cancer Commun. 2019;39(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ma Ke‐Y, He C, Wendel BS, Williams CM, Xiao J, Yang H, et al. Immune repertoire sequencing using molecular identifiers enables accurate clonality discovery and clone size quantification. Front Immunol. 2018;9:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Correia‐Neves M, Waltzinger C, Mathis D, Benoist C. The shaping of the T cell repertoire. Immunity 2001;14(1):21‐32. [DOI] [PubMed] [Google Scholar]
- 34. Carlson CS, Emerson RO, Sherwood AM, Desmarais C, Chung M‐W, Parsons JM, et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nat Commun. 2013;4(1):1‐9. [DOI] [PubMed] [Google Scholar]
- 35. Peng Q, Vijaya Satya R, Lewis M, Randad P, Wang Y. Reducing amplification artifacts in high multiplex amplicon sequencing by using molecular barcodes. BMC Genomics. 2015;16(1):589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Anonymous . Rapid amplification of 5′ complementary DNA ends (5′ RACE). Nat Methods. 2005;2(8):629‐30. [DOI] [PubMed] [Google Scholar]
- 37. Zhu YY, Machleder EM, Chenchik A, Li R, Siebert PD. Reverse transcriptase template switching: A SMART™ approach for full‐length cDNA library construction. BioTechniques. 2001;30(4):892‐7. [DOI] [PubMed] [Google Scholar]
- 38. O'Connell AE, et al., Next generation sequencing reveals skewing of the T and B cell receptor repertoires in patients with Wiskott–Aldrich syndrome. Front Immunol. 2014;5:340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mamedov IZ, Britanova OV, Zvyagin IV, Turchaninova MA, Bolotin DA, Putintseva EV, et al. Preparing unbiased T‐cell receptor and antibody cDNA libraries for the deep next generation sequencing profiling. Front Immunol. 2013;4:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shugay M, Britanova OV, Merzlyak EM, Turchaninova MA, Mamedov IZ, Tuganbaev TR, et al. Towards error‐free profiling of immune repertoires. Nat Methods. 2014;11(6):653. [DOI] [PubMed] [Google Scholar]
- 41. Stewart JJ, Lee CY, Ibrahim S, Watts P, Shlomchik M, Weigert M, et al. A Shannon entropy analysis of immunoglobulin and T cell receptor. Mol Immunol. 1997;34(15):1067‐82. [DOI] [PubMed] [Google Scholar]
- 42. Dewitt WS, Emerson RO, Lindau P, Vignali M, Snyder TM, Desmarais C, et al. Dynamics of the cytotoxic T cell response to a model of acute viral infection. J Virol. 2015;89(8):4517‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cui J‐H, Lin K‐R, Yuan S‐H, Jin Ya‐B, Chen X‐P, Su Xi‐K, et al. TCR repertoire as a novel indicator for immune monitoring and prognosis assessment of patients with cervical cancer. Front Immunol. 2018;9:2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Matsuda T, Miyauchi E, Hsu Yu‐W, Nagayama S, Kiyotani K, Zewde M, et al. TCR sequencing analysis of cancer tissues and tumor draining lymph nodes in colorectal cancer patients. OncoImmunology. 2019;8(6):e1588085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu YY, Yang QF, Yang JS, Cao RB, Liang JY, Liu YT, et al. Characteristics and prognostic significance of profiling the peripheral blood T‐cell receptor repertoire in patients with advanced lung cancer. Int J Cancer. 2019;145(5):1423‐1431. [DOI] [PubMed] [Google Scholar]
- 46. Rempala GA, Seweryn M. Methods for diversity and overlap analysis in T‐cell receptor populations. J Math Biol. 2013;67(6‐7):1339‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hui L, Chen Ye. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015;368(1):7‐13. [DOI] [PubMed] [Google Scholar]
- 48. Wang X, et al., Characterization of Distinct T Cell Receptor Repertoires in Tumor and Distant Non‐tumor Tissues from Lung Cancer Patients. Genomics Proteomics Bioinformatics. 2019;17(3):287‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sheikh N, Cham J, Zhang Li, Devries T, Letarte S, Pufnock J, et al. Clonotypic diversification of intratumoral T cells following sipuleucel‐T treatment in prostate cancer subjects. Cancer Res. 2016;76(13):3711‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hamid O, et al. Safety and tumor responses with lambrolizumab (anti–PD‐1) in melanoma. N Engl J Med. 2013;369(2):134‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515(7528):568‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160(1‐2):48‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Angelova M, Charoentong P, Hackl H, Fischer ML, Snajder R, Krogsdam AM, et al. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015;16(1):64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hopkins AC, Yarchoan M, Durham JN, Yusko EC, Rytlewski JA, Robins HS, et al. T cell receptor repertoire features associated with survival in immunotherapy‐treated pancreatic ductal adenocarcinoma. JCI Insight. 2018;3(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chavez JC, Bachmeier C, Kharfan‐Dabaja MA. CAR T‐cell therapy for B‐cell lymphomas: clinical trial results of available products. Therapeutic Advances in Hematology. 2019;10:204062071984158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19‐28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25‐224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liu D, CAR‐T “the living drugs”, immune checkpoint inhibitors, and precision medicine: a new era of cancer therapy. 2019, Springer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. D'Angelo S, et al. Antitumor activity associated with prolonged persistence of adoptively transferred NY‐ESO‐1 (c259) T cells in synovial sarcoma. Cancer Discov. 2018;8(8):944‐57. 10.1158/2159-8290. CD‐17‐1417.[PubMed][Google Scholar]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.