

Abstract
Rationale:
Zonula occludens-1 (ZO-1), a plasma membrane-associated scaffolding protein regulates signal transduction, transcription and cellular communication. Global deletion of ZO-1 in the mouse is lethal by embryonic day 11.5. The function of ZO-1 in cardiac myocytes (CM) is largely unknown.
Objective:
To determine the function of CM ZO-1 in the intact heart, given its binding to other CM proteins that have been shown instrumental in normal cardiac conduction and function.
Methods and Results:
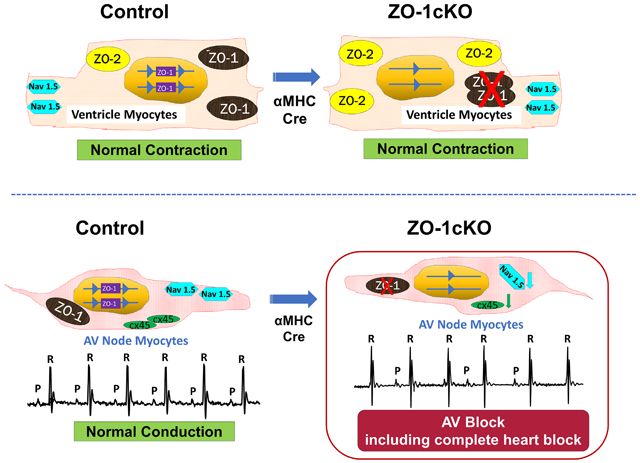
We generated ZO-1 CM-specific knockout (KO) mice using α-Myosin Heavy Chain-nuclear Cre, (ZO-1cKO), and investigated physiological and electrophysiological function by echocardiography, surface ECG and conscious telemetry, intracardiac electrograms and pacing, and optical mapping studies. ZO-1cKO mice were viable, had normal Mendelian ratios, and had a normal lifespan. Ventricular morphometry and function were not significantly different between the ZO-1cKO vs control (CTL) mice, basally in young or aged mice, or even when hearts were subjected to hemodynamic loading. Atrial mass was increased in ZO-1cKO. Electrophysiological and optical mapping studies indicated high-grade atrioventricular (A-V) block in ZO-1cKO comparing to CTL hearts. While ZO-1 associated proteins such as vinculin, connexin 43, N-cadherin, and α-catenin showed no significant change with the loss of ZO-1, Connexin-45 and Coxsackie-adenovirus (CAR) proteins were reduced in atria of ZO-1cKO. Further, with loss of ZO-1, ZO-2 protein was increased significantly in ventricular CMs in a presumed compensatory manner, but was still not detected in the AV nodal myocytes. Importantly, the expression of the sodium channel protein NaV1.5 was altered in AV nodal cells of the ZO-1cKO vs. CTL.
Conclusion:
ZO-1 protein has a unique physiological role in cardiac nodal tissue. This is in alignment with its known interaction with CAR and Cx45, and a new function in regulating the expression of NaV1.5 in AV node. Uniquely, ZO-1 is dispensable for function of the working myocardium.
Keywords: Zonula occludens, conduction system, atrioventricular block, coxsackie adenovirus receptor, vinculin, arrhythmia (heart rhythm disorders), cardiac gap junction connexins, gene targeting
Subject Terms: Animal Models of Human Disease, Arrhythmias, Basic Science Research, Electrophysiology
Graphical Abstract
INTRODUCTION
The zonula occludens (ZO) family consists of ZO-1, −2 and −3 proteins (also known as tight junction protein (TJP)1, TJP2 and TJP3, respectively). ZO proteins are members of the membrane-associated guanylate kinase (MAGUK) family of proteins that were first identified in Drosophila Melanogaster1, that assemble multiprotein complexes adjacent to the cell membrane. ZO proteins were initially found to be components of tight junctions2 and later also found in adherens junctions (AJs)3-6, and gap junctions (GJ)7, 8. In cardiac myocytes (CMs) that do not have tight junctions, ZO-1 colocalizes with cadherins in AJs as they form components of the intercalated discs (ICD)9, 10, and helps to maintain normal function of various ICD junctions. Furthermore, ZO proteins are also recognized to be important for signal transduction, modulation of transcription, and cellular communication. Recently, De Bortoli et al. identified pathogenic variants in ZO-1 that were associated with human arrhythmogenic cardiomyopathy11.
ZO proteins carry three PDZ domains, named for the three proteins: Post-synaptic density protein (PSD5/SAP90), Drosophila disc large tumor suppressor, and ZO-1. In addition, they have SH3 and GUK domains. ZO-1 may complex with ZO-2 and ZO-312, 13. During development, ZO-1 and ZO-2 are expressed ubiquitously, while ZO-3 is restricted to epithelial cells14.
All three ZO proteins have been genetically ablated in the mouse. Global ZO-1–deficient mice showed 100% embryonic lethality by embryonic day (E) 11.5 with extensive apoptosis14, neural tube / notochord defects, with both embryonic and extraembryonic defects including defective chorioallantoic fusion. ZO-1 loss did not alter ZO-2 or ZO-3 expression in these mice. ZO-2 ablated mice showed abnormal gastrulation, cell proliferation and apoptosis with lethality around the time of implantation.15 Why ZO-2 loss led to these abnormalities led to proposals that leaky TJs, abnormal GJ and perhaps even perturbation of gene expression, given ZO-2’s interaction with transcription factors, was causal. In contrast, ZO-3 global deletion did not cause any changes in normal development, postnatal growth or function15, 16.
ZO-1 binds to many proteins in multiple cell-types. These include connexins (Cx), N-cadherin, α-actinin, α-catenin, occludin, claudin, shroom-2, nephrin, F-actin and afadin, highlighting its role as an important cytosolic scaffolding protein17, 18. In TJ, ZO-1 directly binds to adhesion molecules such as claudins and occludin, via its PDZ-1 and GUK domains, respectively19, 20. In CM, the second PDZ domain of ZO-1 directly interacts with the GJ protein, Cx4321-23 and Cx4524.
Previously, we reported that the Coxsackievirus and adenovirus receptor (CAR), a transmembrane adhesion molecule localized at GJ, mediates atrioventricular-node (AVN) function24. Disruption of CAR expression in mouse CM resulted in AVN block, with late onset of cardiomyopathy. This was associated with decreases in ZO-1 and Cx-45 expression levels, and loss of their localization at the AV-node24. In that study, we found that CAR interacted with ZO-1 as had been previously demonstrated 25, and that CAR binding to ZO-1, as well as Cx45 binding to ZO-1, was required for complex formation between CAR, ZO-1 and Cx45.
In separate work, we found also that the AJ / ICD-expressed protein Vinculin (Vcl) also colocalized at the ICD with Cx43 and ZO-1. When Vcl was ablated from CMs, ZO-1 expression was lost from the ICD and Cx43 became maldistributed and mice showed ventricular tachycardia and sudden death26. We also showed that Vcl directly binds ZO-127.
We therefore hypothesized that ZO-1 would be required for AVN function by facilitating molecular interactions between CAR and Cx45, and perhaps other scaffolding or channel proteins, and that it would also be essential for maintaining normal working CM (e.g. ventricular CM) function, perhaps by maintaining localization of Cx43 and vinculin.
Somewhat surprisingly, the data demonstrates that absence of ZO-1 from CM did not affect ventricular structure or function, but led to varied degrees of AV block, including complete heart block. ZO-2 was seen to increase in the isolated ventricular CM suggesting that ZO-2 could substitute for loss of ZO-1, and preserve CM function, except in nodal tissue. Further, the qualitative expression of the sodium channel NaV1.5, that has been linked to conduction system disease, was altered in AV nodal cells. Thus, this data indicates that ZO-1 protein plays a critical role in the function of the cardiac conduction system but is dispensable for mechanical function of the intact heart.
METHODS
An expanded Methods section is available in the Online Supplement.
The authors declare that all supporting data are available within the article, and its Online Supplement.
Within specific genotypes, animals, or samples from them, were randomly assigned to experiments. Studies were performed in a blinded manner. Unless specified in the figure legends, male mice were used.
Animal care.
All animal procedures were performed and approved by the University of California–San Diego Institutional Animal Care and Use Committee.
Gene targeting and generation of ZO-1cKO.
Techniques for generation of ZO-1cKO mice were as previously described28, 29 and detailed in Online Supplement.
Surface electrocardiography, conscious telemetry, transverse aortic constriction, echocardiography, intracardiac electrophysiology and optical mapping.
Procedures were as performed previously26, 30, 31 with EP study detailed in the Online Supplement.
Statistics.
Data were analyzed using GraphPad Prism 7 software (GraphPad Software, San Diego, CA, USA). All data are represented as mean ± SD. The data obtained from all analyses were evaluated using either an un-paired parametric t-test or 2-way ANOVA, as outlined, with P≤0.05 regarded as significant for all studies. Further details are provided in the Online Supplement.
RESULTS
Generation of ZO-1 cardiac myocyte specific knockout mice.
Given the embryonic lethality found in the global ZO-1 KO mice14, we generated mice with a ZO-1 floxed allele that targeted exon 3 of the ZO-1 gene (Online Figure IA and IB). Appropriately targeted R1 embryonic stem (ES) cells were used to produce mice which germline transmitted the targeted allele, and then bred into the Black Swiss background to generate ZO-1f/+ mice. These mice were mated with α-Myosin Heavy Chain (MHC)-nuclear Cre mice32,26 to generate CM-specific ZO-1 KO mice (ZO-1f/f x αMHC-Cre, ZO-1cKO). Control mice utilized throughout the study were ZO-1f/+ x αMHC-Cre (CTL).
To determine whether deletion of the ZO-1 allele caused protein reduction using this scheme, samples from the four cardiac chambers were dissected and Western blotting was performed on whole tissue protein lysates from the chambers, to detect ZO-1. Expression of ZO-1 was decreased 60–97% in all the chambers (Figure 1A, Online Figure IC). Reasoning that the residual ZO-1 in these samples could come from non-myocytes we isolated CM and prepared protein from ZO-1cKO and CTL hearts. Western blotting of these samples showed virtually complete loss of ZO-1 expression in the ZO-1cKO vs. CTL myocytes (Figure 1B, Online Figure IC). These biochemical results were confirmed by immunomicroscopy showing loss of ZO-1 protein from the ICD in the ZO-1cKO compared to CTL. Residual ZO-1 co-localized with endothelial cells that were present between myocytes (Figure 1C, D).
Figure 1. ZO-1 protein expression is virtually absent from CM in the ZO-1cKO, with only residual expression seen in non-CM cells in heart.

(A, B) ZO-1 expression was evaluated by Western blotting of protein lysates with an anti-ZO-1 antibody, using samples from (A) tissue samples discretely obtained from the four cardiac chambers and (B) Isolated adult CM lysates. GAPDH was used as a loading control. Only minimal ZO-1 expression was evident in CM samples of ZO-1cKO compared to CTL, indicating that residual expression in tissue was related to expression of ZO-1 in non-myocytes within the heart. WT samples of CM protein were also analyzed here as reference. Densitometric quantification of tissue samples showed protein expression in ZO-1cKO, relative to CTL to be: RA – 37%, LA – 37%, RV – 13% and LV – 39%, yet densitometric evaluation of Western blots of isolated CM protein showed only 3.3% residual expression in cKO vs. CTL (n=4 for CTL and ZO-1cKO). (See details of densitometry in Online Figure IC.) (C, D) Sections of left ventricle were evaluated using immunomicroscopy with antibodies to discretely detect (C) ZO-1 (red) and Desmoplakin (green), used as a marker of the cell-cell junction, which showed typical intercalated disc staining for ZO-1 in the CTL mice (arrows) but not in the ZO-1cKO mice, and (D) Residual ZO-1 (Green) localized with endothelial cells (marked by PECAM, Red) in ZO-1cKO heart, again supporting the concept that residual ZO-1 protein detected in the cKO heart was related to expression in non-CM. DAPI was used to mark nuclei blue. (Scale bar: 40 μm.)
ZO-1-cKO mice live a full life span, and show no significant basal histological or contractile abnormalities in the ventricles, nor abnormal responses to hemodynamic challenge with aortic constriction.
The ZO-1cKO mice were born, had a normal Mendelian distribution of genotypes, and were found to have a similar life span to littermate controls, to the age of 22-23 months. No obvious morphological differences were noted in the ventricles of ZO-1cKO vs. CTL, and histological evaluation of the ventricles by hematoxylin and eosin staining (Figure 2A) and using Mason’s trichrome (to detect fibrosis – not shown) did not detect any abnormalities in the ZO-1cKO ventricles.
Figure 2. Changes in atrial normalized weights were noted in ZO-1cKO vs. CTL mouse hearts.

(A) Representative H&E sections did not show alteration in histology of ZO-1cKO hearts vs. CTL. (Scale Bar: Ab and Ae: 2.5mm; Aa,c,d,f: 500uM.) (B) Normalized weights of RA and LA were significantly increased in ZO-1cKO vs. CTL Normality was evaluated using the Shapiro-Wilk test. Data are represented as mean ± SD. Unpaired two-tailed Student’s t-test with Welch correction was used to assess the p value by groups. (CTL n=11, M=6, F=5; ZO-1cKO n=10, M=5, F=5). No differences were seen in combined ventricular weights between groups (data not shown).
However, histological examination of the ZO-1cKO mice showed thicker atrial walls than CTL. Analysis was performed and showed both right atria (RA) and left atria (LA) had greater normalized weight in the ZO-1cKO vs. CTL (RA / Tibia Length and LA / Tibia Length) at 4–6 months of age (Figure 2B). Remarkably, assessment of the ventricles not only showed no histological differences between ZO-1cKO and CTL, but also showed no variance of ventricular normalized weights up to 9 months of age (Data not shown). Myocardial tissues from the various chambers were also examined using transmission electron microscopy (TEM) and did not show significant differences in the ultrastructure of specimens from the two groups. (Online Figure II) While minimal increase in interstitial matrix was noted between myocytes of the cKO atria as compared to CTL, there was no alterations of the CMs or ICDs in ZO-1cKO ventricles or atria.
To investigate cardiac function, we next evaluated the ZO-1cKO and CTL groups by echocardiography (Echo) at 4, 10, and 88 weeks of age. Echo parameters were assessed at each time point and did not show any significant differences (Online Figure IIIA).
Given the critical localization of ZO-1 expression in the ICD, we reasoned that the ZO-1cKO mice might not be tolerant of stress compared to CTL. Therefore, transverse aortic constriction (TAC) was performed to hemodynamically challenge the heart. Mice (11–12w age) were challenged with TAC and followed for 4w following surgery. No significant differences in echocardiographic parameters were detected in ZO-1cKO vs. CTL at 2, 3, or 4w post TAC (Online Figure IIIB). At study termination, pressure gradients (left and right carotid pressures) were measured and showed no differences between groups. (ZO-1cKO gradient = 66.8 +/− 21.7mmHg; CTL gradient = 64.6+/− 20.9mmHg (mean +/− SD) ) Likewise, at study termination, analysis was not significantly different between groups, except for higher atrial normalized weights in ZO-1cKO vs. CTL (Online Figure IIIC), in line with basal evaluation (Figure 2).
Abnormal rhythm in the ZO-1cKO mice.
Given the potential of a rhythm disturbance in the ZO-1cKO, we performed surface ECGs and conscious telemetry. ECGs in lightly anesthetized 11 to 12w mice demonstrated abnormal conduction in the ZO-1cKO mice, with indications of varied degrees of high-grade AV block (Figure 3A). Similar findings were detected in mice as young as 5w (data not shown). High-resolution signal averaging of these ECGs demonstrated no significant difference in the QRS duration between the ZO-1cKO and CTL mice (Figure 3B), but showed evidence of high-grade AV block, including complete heart block, in the ZO-1cKO. In the ZO-1cKO mice P-waves did not appear in the signal averaged ECG because of variability in the PR interval and the atrial rate was frequently decreased (Figure 3B), indicating the presence of sinoatrial (SA) abnormalities in addition to AV conduction delay. Next we performed conscious telemetry and saw that all CTL mice had normal sinus rhythm and normal AV conduction, however all ZO-1cKO mice had abnormal AV conduction. In addition, surface ECG and telemetric monitoring showed that P-waves were present prior to all QRS complexes in CTL mice, but were not always detected in the ZO-1cKO, suggestive that SA nodal dysfunction might also be present in the ZO-1cKO (Figure 3C, average view 3D).
Figure 3. AV–nodal block is detected in ZO-1cKO mice vs. CTL mice.

(A) Surface electrocardiograms obtained from lightly anesthetized 4-6m ZO-1cKO and CTL demonstrated complete AV dissociation in ZO-1cKO mice, with no abnormalities in CTL. Arrow indicates P-wave. (B) Signal-averaged beat representative of 50 high-resolution beats obtained from ZO-1cKO and CTL mice. P-wave (arrow) and QRS complexes are clearly shown in CTL mouse, but signal-averaged P-waves could not be displayed for the ZO-1cKO tracing because P-waves were not associated in a regular interval related to the QRS complex in the cKO mice. (C) Telemetric ECG analysis in conscious mice. All CTL mice had normal AV conduction. ZO-1cKO mice had high-grade AV block with none of the ZO-1cKO mice showing normal AV conduction. (D) Like the surface ECG, representative signal averaged beats (from 50 beats) is displayed with arrow = P wave. No P-Wave could be identified again for the ZO-1cKO samples. (n=8; M=4, F=4, for both CTL and ZO-1cKO)
Detailed electrophysiological study and optical mapping shows clearly abnormal AV conduction in the ZO-1-cKO mice.
Next we turned to more detailed analyses, first with intracardiac electrocardiograms obtained using an octapolar lead (EPA-800, Millar, Houston, Texas) via the right internal jugular vein. Lightly sedated CTL mice showed normal sinus rhythm, and no evidence of AV conduction abnormalities. In contrast, in ZO-1cKO, P waves occurred irregularly, at a slower rate, and P-waves were dissociated from the QRS complexes. (Figure 4A). At times in these recordings, atrial rates appeared slower than the ventricular rate. Averaged measurements of 20 continuous beats were used to measure P-P and R-R intervals from the various groups, and showed P-P intervals that were significantly increased in ZO-1cKO vs. CTL, but no significant differences in R-R intervals between groups (Figure 4B). To more accurately evaluate for AV block, we performed atrial overdrive pacing to increase the atrial rate and evaluate the subsequent ventricular conduction. CTL mice routinely showed that each atrial paced beat was associated with a corresponding ventricular depolarization. In contrast, atrial pacing in the ZO-1cKO was associated with AV block (Figure 4C).
Figure 4. Intracardiac electrophysiology shows abnormal AV conduction in the ZO-1-cKO mice.

(A) Native intracardiac electrograms were recorded from ZO-1cKO and CTL mice. Shown are typical baseline of CTL and ZO-1cKO mice. Upper panels in each display show native intracardiac recordings of atria (A), ventricle (V) and surface ECG (S) with ladder diagrams below for atrial (A), atrioventricular (AV) and ventricular (V) conduction. Normal conduction was evident in the control mice, but abnormal AV conduction was found in all the cKO mice. (B) P-P and R-R intervals were measured in the mice. P-P intervals showed significant differences in the ZO-1cKO vs. CTL mice. No significant differences were detected in R-R intervals between groups. Normality was evaluated with the Shapiro-Wilk test. Data are represented as mean ± SD. Unpaired two-tailed Student’s t-test with Welch correction was used to assess the p value by groups. Left - Averaged P-P and R-R intervals, Right - Representative Individual P-P intervals from three CTL and three cKO mice show consistent values in CTL and great variance in cKO mice.
(C) In CTL, atrial pacing (arrow=pacing spike) showed atrial capture (triangle) and subsequent ventricular depolarization. However, in ZO-1cKO mice atrial pacing was not associated with a ventricular depolarization. Corresponding intracardiac recordings (Upper panels) and ladder diagrams (lower panels) are displayed from representative animals. (n=11, M=6, F=5 for both groups)Figure 5
Given the telemetry and intracardiac recordings, we next used optical mapping to evaluate heart rhythm and action potential propagation in isolated hearts from 6–8m mice (n=4, each group). CTL hearts demonstrated clear sinus rhythm and normal conduction while ZO-1cKO hearts displayed a slow atrial rate and periodic escape beats. Optical mapping of the epicardial membrane potential revealed that escape beats had normal ventricular activation patterns suggesting appropriate Purkinje activation in both groups (Online Figure IV). ZO-1cKO beats that showed no evidence of atrial activation, showed a ventricular activation pattern consistent with a high junctional origin of the escape beat (Figure 5A). Atrial pacing at 150ms cycle lengths, uncovered AV nodal conduction abnormalities in the hearts from all ZO-1cKO hearts, while all CTL hearts maintained normal AV conduction (n=4 each). All ZO-1cKO demonstrated complete heart block at 100 ms cycle length atrial pacing. (Video demonstrations of the electrical propagation during intrinsic and atrial pacing are in Online. videos 1-4.) No difference was seen in total ventricular activation time between groups, but a trend towards significantly longer atrial activation was seen in the cKO hearts (Figure 5B). Similarly, there were no differences in ventricular conduction velocity (Figure 5C) measured during pacing on the anterior LV surface (BCL=150ms). Atrial conduction velocities measured during intrinsic rhythm showed significantly slowed LA conduction (Figure 5C). Finally, ventricular action potential duration was not different between groups (Figure 5D).
Figure 5. Optical mapping shows a slow atrial rate and periodic ventricular escape beats in ZO-1-cKO mice.

(A) Activation maps of isolated hearts showed anterior activation during intrinsic rhythm. CTL hearts demonstrated clear evidence of sinus rhythm and normal activation patterns. Isolated ZO-1cKO hearts demonstrated AV conduction abnormalities consistent with previously described ECG results. Beats with apparent AV conduction (ZO-1cKO, beat 1) had slow atrial conduction, long AV delays, and a ventricular activation pattern with dual breakthrough sites, suggesting Purkinje activation. Ventricular beats with no apparent atrial activation (ZO-1cKO, beat 2) had similar ventricular activation patterns suggesting a high junctional origin of the escape beat.(B) There was no difference in ventricular total activation time between the two groups, but a trend towards longer atrial activation time in the ZO-1cKO vs. the CTL hearts. (C) The ventricular conduction velocity (LV epicardial pacing; BCL = 150 ms) was not different between the two groups, but left atrial conduction was significantly slower for ZO-1cKO hearts during the intrinsic rhythm (p=0.023).(D) Action potential duration at 80% of repolarization (APD80) was not significantly different between the two groups (LV epicardial pacing; BCL = 150 ms).
Data presented as mean +/− SD. Statistical analysis was performed using the Shapiro-Wilk test to assess normality and an unpaired Student’s t-test to compare. (n=4 for both groups.) BCL, basic cycle length.
No morphological changes were detected in conduction system tissue.
To determine whether absence of ZO-1 causes morphological changes in the SA or AV nodes and associated conduction pathways, we crossed HCN4-GFP-ERT2 mice to the ZO-1cKO, to allow for GFP labeling of conduction system cells. These mice were termed ZO-1cKO-G and ZO-1CTL-G. SA and AV nodal tissue, and conduction system components, labeled by HCN4 nuGFP expression, did not appear varied in the ZO-1cKO vs. CTL (Figure 6).
Figure 6. General morphology of SAN and AV nodes were not changed in the ZO-1cKO hearts vs. CTL.

HCN4-ER-GFP was used to label conduction tissue in isolated and dissected hearts. (A - D) representative CTL heart; E - H representative ZO-1cKO heart, both mated to HCN4-ER-GFP. Dorsal anterior view (A, E), dorsal view (B, F), and right dorsolateral view (C, G) of the intact heart. After dissecting the junction of the right atria and right ventricle, the SA and AV nodes were seen from the dorsal view (D, H). SA and AV nodal tissue was viewed as GFP (green) labeled tissue (SAN, sinoatrial node; AVN, Atrioventricular node; RA, right atria; SVC, superior vena cava; LA, left atria; PA, Pulmonary artery; PV, Pulmonary Vein). (Scale bar: 2mm.)
Expression of ZO-1 associated proteins occurs in a differential manner in atrial vs ventricular tissue when ZO-1 is deleted from cardiomyocytes.
To assess how loss of ZO-1 might impact the localization and the expression of ZO-bound or complexed ICD or ICD-associated proteins in the CM we evaluated expression of a panel of proteins from tissue dissected from the four cardiac chambers of mice at 6m. Western blotting showed no difference in expression levels of vinculin, N-cadherin, Cx43, phospho-Cx43 (368), β & γ-catenin, Cx40 and Cx30.2 (Online Figure V). Immunomicroscopic analyses of Cx43 and desmoplakin (DSP) (Online Figure VI), N-cadherin, β & γ-catenin (Online Figures VII and VIII) qualitatively agreed with the Western blot result. In particular, recognizing that the Cx43 carboxyl-terminus appears to regulate GJ organization along with ZO-133, we noted that localization of Cx43 was essentially the same in the ZO-1cKO vs. CTL mice, in support of our finding preserved ventricular function in the cKO hearts.
Cx45 expression in whole atrial tissue was reduced by Western blotting in ZO-1cKO. (Figure 7 A, B), and immunomicroscopy showed alterations in Cx45 in AV nodal cells of ZO-1cKO vs. CTL (Figure 7 C). While there was no clear difference in ZO-2 expression level at the tissue level (Online Figure V), when ventricular CM were isolated, ZO-2 expression was found to be significantly increased in ZO-1cKO as compared to CTL (Figure 7D & E). ZO-2 protein is detected in working myocytes but not in AV node myocytes, and was still not detected in AV node when ZO-1 was deleted (Figure 7F, G and H). CAR protein was also reduced in atrial specimens from cKO vs. CTL detected by Western blot and immunostaining, without changes in ventricular tissue or cells (Online Figure IX, A, B, C). No significant qualitative difference in CAR expression was seen in AV nodal tissue. (Online Figure IXC)
Figure 7. Assessment of ZO-1 associated proteins shows alteration of expression of Cx45, ZO-2 and Nav1.5 in ZO-1cKO vs. CTL samples. (A, B, C) Cx45 was reduced in atrial tissue and AV nodal cells from ZO-1cKO vs. CTL hearts.

(A,B) Western blot (A) shows expression of Cx45 was downregulated in atrial tissue samples of ZO-1cKO vs. CTL mice. (B) Densitometric quantitation of Western blot of CX45, normalized to GAPDH (loading control) shows a significant reduction in ZO-1cKO vs CTL heart samples. Normality was assessed with the Shapiro-Wilk test. Data are represented as mean ± SD. Unpaired two-tailed Student’s t-test with Welch correction was used to assess the p value by groups. (n = 3 for both groups). (C) Cx45 protein was visibly reduced in AV node and working myocytes by immunomicroscopy. Yellow lines show separation of ventricle from AV nodal tissue in c and c’. HCN4 nuclear GFP was used to mark AV nodal cells. (CX45=gray, HCN4 nuclear GFP=green) (Scale bar: 20uM.) (D, E, F, G) ZO-2 was increased in isolated cardiac myocyte samples from ZO-1cKO vs. CTL hearts. (D) ZO-1 was virtually absent while ZO-2 expression was upregulated in isolated ventricular CM samples of ZO-1cKO vs. CTL evaluated by Western blot. (E) Densitometric expression level was determined for ZO-1 and ZO-2 normalized to GAPDH as a loading control. Densitometric quantitation of Western blot of CX45, normalized to GAPDH (loading control) showed a significant reduction in ZO-1cKO vs CTL heart samples. (n=3 for both groups). (F, G, H) ZO-2 protein localizes in the working CM (ventricle) (F) but was not detected in the AV node (G, H), and was still not detected in the AV node when ZO-1 was deleted from CM in ZO-1cKO hearts. Box in panels d and d’ of panel G is shown as a magnified view in all panels of H. (In ‘Merge’ panel of F, G and H: N-Cadherin=green, ZO-2=red, Nuclei DAPI=blue; then in panel G and H - HCN4 nuGFP=Pink and marks AV nodal cells;) (Scale Bar: 20uM in F and G, 5uM in H.)
Finally, we reasoned that a key interacting partner of ZO-1 could be the cardiac sodium channel Nav1.5, given that ZO-1 has a PDZ domain that could interact with Nav1.5, and since Nav1.5 interacts with other proteins also found in the ICD. As shown in Figure 7I, J and Online Figure X, loss of ZO-1 from AV nodal cells (marked by HCN4 nuGFP as well as contactin-234, 35), caused qualitative alteration in the expression of Nav1.5 is seen, without significant alteration in ventricular cells, as amplified by data in Figure 7K.
DISCUSSION
In this study we focused on the function of ZO-1 specifically in the cardiac myocyte by generating a ZO-1 CM-specific KO mouse model, using a floxed ZO-1 allele mated to a well-characterized αMHC-Cre transgenic mouse32, 36 that expresses Cre specifically in CM by E9.5. We found that mice with homozygous CM-specific deletion of ZO-1 are viable, have normal lifespans, and have no evidence of histological or morphological changes in the myocardium except for increased atrial weights. Further, normal contractile function was evident basally, and responses to hemodynamic loading provoked by TAC were similar to that observed in CTL mice. Most strikingly, we found electrophysiologic abnormalities in the ZO-1cKO when evaluated by surface ECGs, conscious telemetry, invasive electrophysiological study and optical mapping. Specifically, we noted defective nodal function and no indication of abnormal Purkinje system activation in ZO-1cKO. Together, these findings suggest unique physiological roles for ZO-1 in the conduction system, as distinct from its role in working cardiac myocytes. A model illustrating this unique role of ZO-1 in conduction system vs. contractile myocardium is shown in Figure 8.
Figure 8. Proposed model illustrating concepts of AV node dysfunction with preserved ventricular function in ZO-1cKO mice.

(A, C) CTL hearts have normal intercellular communication in AV node gap junctions (A) as well as transmission across ventricular intercalated disks (C). (B, D) When ZO-1 expression was deleted from CM in ZO-1cKO AV nodal tissue (B), Cx45 expression is reduced as ZO-1 is lost, though no increase in ZO-2 expression was seen. NaV1.5 expression was also altered in these AV nodal cells when ZO-1 was lost. Together, loss of ZO-1 leads to abnormal AV conduction. (D) In contrast, in the intercalated disk of ZO-1cKO ventricle, ZO-2 expression is increased allowing for ZO-2 function to replace ZO-1, and thus continued normal communication and function of KO ventricular tissue.
ZO proteins were initially detected in TJ2 and AJs3-6 in endothelial and epithelial cells, but have also been identified in CM gap junctions (GJ)7, 8. Though the ZO protein family has three members - ZO-1, −2 and −3, only ZO-1 and ZO-2 are expressed in CM9, 37-39. Data have shown that ZO proteins may regulate GJ size and distribution 40 and connect AJs to GJs. While TJ are not present in CM, the area bridging two CM, the ICD, contains AJs and GJs, making the ICD essential for both strengthening the connection and intracellular communication between CMs41. The Cx-containing GJ allow for rapid conductance of action potentials and thus electrical coupling of CM, so that the working myocardium can contract as a syncytium42-44. GJ also have an important role in transmission of electrical signaling in the SA and AV nodes45. Of note is recent work which identified pathogenic variants in the ZO-1 gene associated with arrhythmogenic cardiomyopathy11.
We confirmed ZO-1 protein localized in the ICDs (Figure 1C) and that it colocalized with Cx43 (Online Figure VI) in the ventricle of the mouse heart. Despite this, given the essential function of the ICD as described above, we were surprised to find that with the loss of ZO-1 expression from the CM ICD in the ZO-1cKO, ventricular contractile function was not altered, either basally or with hemodynamic stress (Online Figure III).
Prior work in our lab had studied the role of ICD and costameric proteins, including CAR and Vcl, a component of the integrin-talin-vinculin complex24, 26, 27. Use of CM-specific CAR KO mice (CARcKO) illustrated that normal expression of this adhesion protein was essential for normal AV node conduction and cardiac function. Likewise, ablation of VCL expression from the CM in VCL-cKO, showed that VCL was necessary for preservation of normal ventricular function, and also that loss of CM VCL led to ventricular arrythmias. CAR was found to form a complex with ZO-1 and Cx45 via its PDZ binding motif, and also when reduced in the CM, led to reduction of both ZO-1 expression in the ICD and Cx45 expression, in the AV node. Similarly, loss of VCL from the CM in the VCLcKO caused loss of ZO-1 from ICD. VCL and ZO-1 were also shown by us to be direct binding partners27. Based on these findings, we hypothesized that normal CM ZO-1 expression would be essential for both cardiac conduction and contractile function.
Surprisingly, we found no alteration in ventricular function here, nor did we find alterations in the expression of either CAR or Vcl expression in the ZO-1cKO ventricle (Online Figure V, IX). We posit that ventricular function was preserved in the absence of ZO-1, due to the increased expression of ZO-2 that occurred in ventricular myocytes.
While no VCL changes were noted in atrial tissue, CAR expression was reduced there, along with our findings of AV and SA nodal abnormalities. Further, expression of the CAR-linked connexin, Cx45, a key conduction system connexin46, 47, was also reduced in cKO atrial tissue compared to control. It is important to note that nodal tissue is typically included in right atrial samples, as we evaluated the expression of these proteins.
Intriguingly, while ZO-2 expression increased in the working ventricular myocardium with ZO-1 loss, this was not evident in the AV node (Figure 7D - 7H). Therefore, the current data demonstrates that the decrease in AV node ZO-1 that was observed in CAR deficient hearts, and in the current ZO-1cKO, leads to AV conduction delay.
In addition to Cx45, we evaluated the expression of multiple connexins that have been associated with cardiovascular function and conduction, as well as other proteins expressed in the ICD. Expression of adherens junction proteins (N-cadherin, α-catenin, Y-Catenin as well as Vcl), and gap junction proteins (Cx40, Cx43 and Cx30.2) proteins were not significantly different in the ZO-1cKO vs. CTL (Online Figure V). While immunolocalization of all these Cx proteins could be instructive, it is worthwhile to note that combined knockout of Cx30.2 and Cx40 in mice did not alter AV impulse propagation48.
Cx45 is of particular interest in conduction abnormalities since it is one of the main connexins in the SA node and the AV node, with limited expression in the atria and ventricles49. Cx45 is strongly expressed in the early embryonic myocardium and is required for cardiac development47, 50. Homozygous Cx45-deficient mice are embryonic lethal at around E10 due to heart failure with conduction block47, heterozygous Cx45 KO mice display AV-septal defects and slowed AV-conduction51. In the adult heart, Cx45 is expressed mainly in the SA and AV nodes and His-Purkinje system, but has also been shown to be expressed in atrial and ventricular myocytes49. It is not essential for survival of adult mice as inducible, CM-specific deletion of Cx45 leads to delayed AV conduction, but normal ventricular function52.
Cx45 has also been linked to nonimmune familial AV-block in man46 and a disease-causing Cx45 mutant (p.R75H) was found in 2 unrelated families who presented with progressive AV block, and ultimately atrial standstill, without ventricular conduction abnormalities46. Further, two groups have been shown that Cx45 interacts directly with ZO-1, but these studies were not carried out in CM53, 54. Still, these results are in line with our findings since ZO-1 loss resulted in reduced expression of Cx45.
Importantly, we found that with loss of ZO-1 from CMs, qualitative expression of the sodium channel NaV1.5 was reduced in AV nodal cells, but not ventricular CMs. (Figure 7I-K and Online Figure X). These results suggest that ZO-1 is necessary for proper expression of Nav1.5 in the AV nodal cells. While variants / mutations in NaV1.5 has been linked to a variety of cardiac diseases, included amongst these are correlation with SA nodal abnormalities (sick sinus syndrome) and also other progressive cardiac conduction system disorders, suggesting that perhaps ZO-1 may be a component of these disease states55, 56.
We respect that there are several limitations to our study. Experiments are required to further elucidate the basis for the unique findings in our model. Since we propose that ZO-2 is be able to replace ZO-1 function in the working CM, future studies where both ZO-1 and ZO-2 protein expression is deleted from all CMs will be necessary to discern the general function of ZO proteins in the ventricle. We are also aware that studies have begun to show how abnormalities of ICD proteins, particularly those that contain PDZ domains, can alter sodium and potassium channel function and predispose to arrhythmias and conduction disturbances57-61. Though we have shown here that NaV1.5 expression is changed in AV nodal cells deficient in ZO-1, future functional studies are necessary. Also, since ZO-1 complexes with CAR and binds VCL, experiments are needed to fully explain how these three proteins function individually and together, to affect cardiac conduction and ventricular function.
In sum, we have identified a unique role for ZO-1 in cardiac conduction that has not been previously recognized. Future studies, currently underway, are necessary to fully define the role of ZO proteins in all CM in various regions of the heart.
Supplementary Material
Major Resources Table.
In order to allow validation and replication of experiments, all essential research materials listed in the Methods should be included in the Major Resources Table below. Authors are encouraged to use public repositories for protocols, data, code, and other materials and provide persistent identifiers and/or links to repositories when available. Authors may add or delete rows as needed.
| Animals (in vivo studies) Genetically Modified Animals | ||||||
|---|---|---|---|---|---|---|
| Species | Vendor or Source |
Background Strain | Other Information |
Persistent ID / URL | ||
| Parent - Male | Mouse | N/A | ZO-1 f/+, Cre | |||
| Parent - Female | Mouse | N/A | ZO-1 f/f or ZO-1 f/+ | |||
| Parent - Male | Mouse | N/A | HCN4-GFP-ERT2 | |||
| Antibodies | ||||||
| Target antigen | Vendor or Source |
Catalog # | Working concentration |
Lot # (preferred but not required) |
Persistent ID / URL | |
| For immunostaining | ||||||
| ZO-1 | Millipore-Sigma | MATB11 | 1:50 | |||
| ZO-2 | Abgent | ABO11279 | 1:25 | |||
| Desmoplakin | AbD Serotec | 2722-5204 | 1:200 | |||
| PECAM/CD31 | R & D Systems | AF3628 | 1:100 | |||
| Connexin 43 | Invitrogen | 71-0700 | 1:500 | |||
| β-catenin | Abcam | ab16051 | 1:100 | |||
| Y-catenin | ThermoFisher | PA5-17320 | 1:50 | |||
| N-cad | BD Biosciences | 610920 | 1:100 | |||
| contactin-2 | R & D Systems | AF439 | 1:50 | |||
| donkey anti-Rabbit Alex-488 | Jackson Lab | 711-545-152 | 1:100 | |||
| anti-Rabbit Alex-647 | Jackson Lab | 711-605-152 | 1:200 | |||
| cy3 donkey anti-Rat | Jackson Lab | 712-165-153 | 1:200 | |||
| cy5 donkey anti-Mouse | Jackson Lab | 715-175-151 | 1:200 | |||
| cy3 donkey anti-goat | Jackson Lab | 705-165-147 | 1:200 | |||
| DAPI | ThermoFisher | D1306 | 10ug/ml | |||
| Fluoromount-G™ | eBioscience | 00-4958-02 | ||||
| For western-Blot | ||||||
| ZO-1 | Millipore-Sigma | MATB11 | 1:1000 | |||
| Desmoplakin | AbD Serotec | 2722-5204 | 1:1000 | |||
| Connexin 43 | Invitrogen | 71-0700 | 1:5000 | |||
| p-Connexin 43 | Cell Signaling Technology | 3511 | 1:1000 | |||
| Vinculin | Sigma | V9131 | 1;10000 | |||
| N-cad | UIHB | MNCD2 | 1:1000 | |||
| CAR | Santa Cruz Biotechnology Inc. |
SC-15405 | 1:1000 | |||
| β-catenin | Abcam | AB16051 | 1:1000 | |||
| Υ-catenin | Santa Cruz Biotechnology Inc. | SC-7900 | 1:1000 | |||
| ZO-2 | Abgent | ABO11279 | 1:1000 | |||
| Connexin 30.2 | Invitrogen | 40-7400 | 1:1000 | |||
| Connexin 40 | Invitrogen | 36-5000 | 1:1000 | |||
| Connexin 45 | Invitrogen | 41-4700 | 1:1000 | |||
| GAPDH | Millipore-Sigma | MAB374 | 1:10000 | |||
| donkey anti-Mouse | Jackson Lab | 615-035-214 | 1:2500 | |||
| donkey anti-rabbit | Jackson Lab | 711-035-152 | 1:2500 | |||
| donkey anti-rat | Jackson Lab | 712-035-153 | 1:2500 | |||
| donkey anti-goat | Jackson | Lab | 705-035-003 | 1:2500 | ||
NOVELTY AND SIGNIFICANCE.
What Is Known?
Intercalated disks (ID) that interconnect cardiac myocytes (CMs), contain three types of junctions, with two providing a means for strong cellular attachment (adherens junctions (AJ) and desmosomes) and a third (gap junctions (GJ)) directing essential electrical coupling and transmission of small metabolites between cells.
There are three Zonula Occludens (ZO) proteins – ZO-1,2 and 3, with only ZO-1 and 2 expressed in AJ and GJ of the CM IDs.
CM ZO proteins are important for GJ remodeling and control of GJ size.
What New Information Does This Article Contribute?
CM-specific deletion of the mouse ZO-1 gene was performed and produced mice termed ZO-1 CM-specific knockout (ZO-1cKO) that are viable, have normal lifespan and normal ventricular contractile function.
The ZO-1cKO mice showed varied degrees of atrioventricular (AV) block, including complete heart block.
With loss of ZO-1 from CMs, ZO-2 was seen to increase in ventricular myocytes but not in atrioventricular nodal cells.
ZO-1 loss from AV node tissue lead to altered microscopic expression of the sodium channel NaV1.5 in AV node tissue.
Interconnections of CMs via IDs is essential to maintain integrity of the myocardium and allow synchronous electrical activity and muscular contraction. Studies show that defective ID proteins can lead to cardiac contractile dysfunction or rhythm disturbances. ZO proteins are found in the ID, but their function in CMs is not fully understood. Given the known binding of ZO-1 to other CM proteins, we produced ZO-1cKO mice to explore how loss of CM ZO-1 would alter heart function. ZO-1cKO mice were viable, had normal lifespan and showed normal contractile function, even with hemodynamic challenge. Yet, electrocardiograms, intracardiac electrophysiology and optical mapping studies showed evidence of AV block, including complete heart block, in these mice. With loss of ZO-1 from CMs, ZO-2 expression increased significantly in ventricular CMs but not in AV node tissue, suggesting it could substitute for ZO-1 in working myocardium, but not in AV node. Further, in ZO-1cKO, qualitative alteration of the sodium channel NaV1.5, was seen in AV node. Thus ZO-1 protein appears dispensable for function of the working CM and has a unique physiological role in cardiac nodal tissue. This increased understanding of CM ZO-1 provides novel information relevant to cardiac conduction system function and disease.
ACKNOWLEDGEMENT
Dr. Robert Gourdie (Virginia Tech Carilion Research Institute) kindly provided a NaV1.5 antibody and Dr. Ulrike Janssen-Bienhold (University of Oldenburg, Germany) provided an antibody for Cx 45 antibody.
SOURCE OF FUNDING
This project was supported by the NIH to KUK and ADM (U01 HL126273 and R01 HL137100) and also by NINDS NS047101 to the UCSD Microscopy Core.
Nonstandard Abbreviations and Acronyms:
- AJ
adherens junction
- AV
atrioventricular
- AVN
atrioventricular node
- BW
body weight.
- CAR
coxsackievirus and adenovirus receptor
- CARcKO
CAR cardiomyocyte-specific KO mice
- CM
cardiac myocytes
- CX
connexin
- DSP
desmoplakin
- E
embryonic day
- ECG
electrocardiography
- ECHO
echocardiography
- EP
electrophysiology
- GJ
gap junction
- HR
heart rate
- ICD
intercalated discs
- PSD5/90
post-synaptic density protein
- TJ
tight junction
- VCL
vinculin
- ZO
zonula occludens
Footnotes
DISCLOSURES
ADM is a co-founder of Insilicomed and Vektor Medical and serves on the scientific advisory boards. He has an equity interest in Insilicomed. His research grants, including those acknowledged here, have been identified for conflict of interest management by the University of California San Diego based on the overall scope of the research and its potential benefit to these entities. The author is mandated to include this disclosure in all publications that acknowledge this grant support. However, the specific research subject and findings reported here did not involve either company in any way and have no relationship whatsoever to the business activities or scientific interests of either company. The terms of this arrangement have been reviewed and approved by the University of California San Diego in accordance with its conflict of interest policies
REFERENCES
- 1.Woods DF, Bryant PJ. The discs-large tumor suppressor gene of drosophila encodes a guanylate kinase homolog localized at septate junctions. Cell. 1991;66:451–464 [DOI] [PubMed] [Google Scholar]
- 2.Stevenson BR, Siliciano JD, Mooseker MS, Goodenough DA. Identification of zo-1: A high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J Cell Biol. 1986;103:755–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Itoh M, Nagafuchi A, Yonemura S, Kitani-Yasuda T, Tsukita S, Tsukita S. The 220-kd protein colocalizing with cadherins in non-epithelial cells is identical to zo-1, a tight junction-associated protein in epithelial cells: Cdna cloning and immunoelectron microscopy. J Cell Biol. 1993;121:491–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furuse M, Itoh M, Hirase T, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S. Direct association of occludin with zo-1 and its possible involvement in the localization of occludin at tight junctions. J Cell Biol. 1994;127:1617–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fanning AS, Jameson BJ, Jesaitis LA, Anderson JM. The tight junction protein zo-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem. 1998;273:29745–29753 [DOI] [PubMed] [Google Scholar]
- 6.Bazzoni G, Martinez-Estrada OM, Orsenigo F, Cordenonsi M, Citi S, Dejana E. Interaction of junctional adhesion molecule with the tight junction components zo-1, cingulin, and occludin. J Biol Chem. 2000;275:20520–20526 [DOI] [PubMed] [Google Scholar]
- 7.Jesaitis LA, Goodenough DA. Molecular characterization and tissue distribution of zo-2, a tight junction protein homologous to zo-1 and the drosophila discs-large tumor suppressor protein. J Cell Biol. 1994;124:949–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inoko A, Itoh M, Tamura A, Matsuda M, Furuse M, Tsukita S. Expression and distribution of zo-3, a tight junction maguk protein, in mouse tissues. Genes Cells. 2003;8:837–845 [DOI] [PubMed] [Google Scholar]
- 9.Itoh M, Morita K, Tsukita S. Characterization of zo-2 as a maguk family member associated with tight as well as adherens junctions with a binding affinity to occludin and alpha catenin. J Biol Chem. 1999;274:5981–5986 [DOI] [PubMed] [Google Scholar]
- 10.Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S. Direct binding of three tight junction-associated maguks, zo-1, zo-2, and zo-3, with the cooh termini of claudins. J Cell Biol. 1999;147:1351–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Bortoli M, Postma AV, Poloni G, Calore M, Minervini G, Mazzotti E, Rigato I, Ebert M, Lorenzon A, Vazza G, Cipriani A, Bariani R, Perazzolo Marra M, Husser D, Thiene G, Daliento L, Corrado D, Basso C, Tosatto SCE, Bauce B, van Tintelen JP, Rampazzo A. Whole-exome sequencing identifies pathogenic variants in tjp1 gene associated with arrhythmogenic cardiomyopathy. Circ Genom Precis Med. 2018;11:e002123. [DOI] [PubMed] [Google Scholar]
- 12.Gumbiner B, Lowenkopf T, Apatira D. Identification of a 160-kda polypeptide that binds to the tight junction protein zo-1. Proc Natl Acad Sci U S A. 1991;88:3460–3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balda MS, Anderson JM. Two classes of tight junctions are revealed by zo-1 isoforms. Am J Physiol. 1993;264:C918–924 [DOI] [PubMed] [Google Scholar]
- 14.Katsuno T, Umeda K, Matsui T, Hata M, Tamura A, Itoh M, Takeuchi K, Fujimori T, Nabeshima Y, Noda T, Tsukita S, Tsukita S. Deficiency of zonula occludens-1 causes embryonic lethal phenotype associated with defected yolk sac angiogenesis and apoptosis of embryonic cells. Mol Biol Cell. 2008;19:2465–2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu J, Kausalya PJ, Phua DC, Ali SM, Hossain Z, Hunziker W. Early embryonic lethality of mice lacking zo-2, but not zo-3, reveals critical and nonredundant roles for individual zonula occludens proteins in mammalian development. Mol Cell Biol. 2008;28:1669–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adachi M, Inoko A, Hata M, Furuse K, Umeda K, Itoh M, Tsukita S. Normal establishment of epithelial tight junctions in mice and cultured cells lacking expression of zo-3, a tight-junction maguk protein. Mol Cell Biol. 2006;26:9003–9015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fanning AS, Anderson JM. Zonula occludens-1 and −2 are cytosolic scaffolds that regulate the assembly of cellular junctions. Ann N Y Acad Sci. 2009;1165:113–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fanning AS, Lye MF, Anderson JM, Lavie A. Domain swapping within pdz2 is responsible for dimerization of zo proteins. J Biol Chem. 2007;282:37710–37716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitic LL, Anderson JM. Molecular architecture of tight junctions. Annu Rev Physiol. 1998;60:121–142 [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez-Mariscal L, Betanzos A, Avila-Flores A. Maguk proteins: Structure and role in the tight junction. Semin Cell Dev Biol. 2000;11:315–324 [DOI] [PubMed] [Google Scholar]
- 21.Giepmans BN, Moolenaar WH. The gap junction protein connexin43 interacts with the second pdz domain of the zona occludens-1 protein. Curr Biol. 1998;8:931–934 [DOI] [PubMed] [Google Scholar]
- 22.Giepmans BN, Verlaan I, Moolenaar WH. Connexin-43 interactions with zo-1 and alpha- and beta-tubulin. Cell Commun Adhes. 2001;8:219–223 [DOI] [PubMed] [Google Scholar]
- 23.Toyofuku T, Yabuki M, Otsu K, Kuzuya T, Hori M, Tada M. Direct association of the gap junction protein connexin-43 with zo-1 in cardiac myocytes. J Biol Chem. 1998;273:12725–12731 [DOI] [PubMed] [Google Scholar]
- 24.Lim BK, Xiong D, Dorner A, Youn TJ, Yung A, Liu TI, Gu Y, Dalton ND, Wright AT, Evans SM, Chen J, Peterson KL, McCulloch AD, Yajima T, Knowlton KU. Coxsackievirus and adenovirus receptor (car) mediates atrioventricular-node function and connexin 45 localization in the murine heart. J Clin Invest. 2008;118:2758–2770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cohen CJ, Shieh JTC, Pickles RJ, Okegawa T, Hsieh JT, Bergelson JM. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. P Natl Acad Sci USA. 2001;98:15191–15196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zemljic-Harpf AE, Miller JC, Henderson SA, Wright AT, Manso AM, Elsherif L, Dalton ND, Thor AK, Perkins GA, McCulloch AD, Ross RS. Cardiac-myocyte-specific excision of the vinculin gene disrupts cellular junctions, causing sudden death or dilated cardiomyopathy. Mol Cell Biol. 2007;27:7522–7537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zemljic-Harpf AE, Godoy JC, Platoshyn O, Asfaw EK, Busija AR, Domenighetti AA, Ross RS. Vinculin directly binds zonula occludens-1 and is essential for stabilizing connexin-43-containing gap junctions in cardiac myocytes. J Cell Sci. 2014;127:1104–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J, Kubalak SW, Minamisawa S, Price RL, Becker KD, Hickey R, Ross J Jr., Chien KR. Selective requirement of myosin light chain 2v in embryonic heart function. J Biol Chem. 1998;273:1252–1256 [DOI] [PubMed] [Google Scholar]
- 29.Liang X, Wang G, Lin L, Lowe J, Zhang Q, Bu L, Chen Y, Chen J, Sun Y, Evans SM. Hcn4 dynamically marks the first heart field and conduction system precursors. Circ Res. 2013;113:399–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rockman HA, Ono S, Ross RS, Jones LR, Karimi M, Bhargava V, Ross J Jr., Chien KR. Molecular and physiological alterations in murine ventricular dysfunction. Proc Natl Acad Sci U S A. 1994;91:2694–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zemljic-Harpf AE, Miller JC, Henderson SA, Wright AT, Manso AM, Elsherif L, Dalton ND, Thor AK, Perkins GA, McCulloch AD, Ross RS. Cardiac-myocyte-specific excision of the vinculin gene disrupts cellular junctions, causing sudden death or dilated cardiomyopathy. Mol.Cell Biol. 2007;27:7522–7537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abel ED, Kaulbach HC, Tian R, Hopkins JC, Duffy J, Doetschman T, Minnemann T, Boers ME, Hadro E, Oberste-Berghaus C, Quist W, Lowell BB, Ingwall JS, Kahn BB. Cardiac hypertrophy with preserved contractile function after selective deletion of glut4 from the heart. J Clin Invest. 1999;104:1703–1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palatinus JA, Rhett JM, Gourdie RG. The connexin43 carboxyl terminus and cardiac gap junction organization. Biochim Biophys Acta. 2012;1818:1831–1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pallante BA, Giovannone S, Fang-Yu L, Zhang J, Liu N, Kang G, Dun W, Boyden PA, Fishman GI. Contactin-2 expression in the cardiac purkinje fiber network. Circ Arrhythm Electrophysiol. 2010;3:186–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhattacharyya S, Bhakta M, Munshi NV. Phenotypically silent cre recombination within the postnatal ventricular conduction system. Plos One. 2017;12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manso AM, Li RX, Monkley SJ, Cruz NM, Ong S, Lao DH, Koshman YE, Gu YS, Peterson KL, Chen J, Abel ED, Samarel AM, Critchley DR, Ross RS. Talin1 has unique expression versus talin 2 in the heart and modifies the hypertrophic response to pressure overload. Journal of Biological Chemistry. 2013;288:4252–4264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stevenson BR, Goodenough DA. Zonulae occludentes in junctional complex-enriched fractions from mouse liver: Preliminary morphological and biochemical characterization. J Cell Biol. 1984;98:1209–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haskins J, Gu L, Wittchen ES, Hibbard J, Stevenson BR. Zo-3, a novel member of the maguk protein family found at the tight junction, interacts with zo-1 and occludin. J Cell Biol. 1998;141:199–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsukita S, Furuse M, Itoh M. Molecular architecture of tight junctions: Occludin and zo-1. Soc Gen Physiol Ser. 1997;52:69–76 [PubMed] [Google Scholar]
- 40.Hunter AW, Barker RJ, Zhu C, Gourdie RG. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell. 2005;16:5686–5698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vermij SH, Abriel H, van Veen TA. Refining the molecular organization of the cardiac intercalated disc. Cardiovasc Res. 2017 [DOI] [PubMed] [Google Scholar]
- 42.Delmar M, Makita N. Cardiac connexins, mutations and arrhythmias. Curr Opin Cardiol. 2012;27:236–241 [DOI] [PubMed] [Google Scholar]
- 43.Rhett JM, Jourdan J, Gourdie RG. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol Biol Cell. 2011;22:1516–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saffitz JE, Laing JG, Yamada KA. Connexin expression and turnover : Implications for cardiac excitability. Circ Res. 2000;86:723–728 [DOI] [PubMed] [Google Scholar]
- 45.Jansen JA, van Veen TA, de Bakker JM, van Rijen HV. Cardiac connexins and impulse propagation. J Mol Cell Cardiol. 2010;48:76–82 [DOI] [PubMed] [Google Scholar]
- 46.Seki A, Ishikawa T, Daumy X, Mishima H, Barc J, Sasaki R, Nishii K, Saito K, Urano M, Ohno S, Otsuki S, Kimoto H, Baruteau AE, Thollet A, Fouchard S, Bonnaud S, Parent P, Shibata Y, Perrin JP, Le Marec H, Hagiwara N, Mercier S, Horie M, Probst V, Yoshiura KI, Redon R, Schott JJ, Makita N. Progressive atrial conduction defects associated with bone malformation caused by a connexin-45 mutation. J Am Coll Cardiol. 2017;70:358–370 [DOI] [PubMed] [Google Scholar]
- 47.Nishii K, Kumai M, Egashira K, Miwa T, Hashizume K, Miyano Y, Shibata Y. Mice lacking connexin45 conditionally in cardiac myocytes display embryonic lethality similar to that of germline knockout mice without endocardial cushion defect. Cell Commun Adhes. 2003;10:365–369 [DOI] [PubMed] [Google Scholar]
- 48.Schrickel JW, Kreuzberg MM, Ghanem A, Kim JS, Linhart M, Andrie R, Tiemann K, Nickenig G, Lewalter T, Willecke K. Normal impulse propagation in the atrioventricular conduction system of cx30.2/cx40 double deficient mice. J Mol Cell Cardiol. 2009;46:644–652 [DOI] [PubMed] [Google Scholar]
- 49.Baruteau AE, Probst V, Abriel H. Inherited progressive cardiac conduction disorders. Curr Opin Cardiol. 2015;30:33–39 [DOI] [PubMed] [Google Scholar]
- 50.Kumai M, Nishii K, Nakamura K, Takeda N, Suzuki M, Shibata Y. Loss of connexin45 causes a cushion defect in early cardiogenesis. Development. 2000;127:3501–3512 [DOI] [PubMed] [Google Scholar]
- 51.Kruger O, Maxeiner S, Kim JS, van Rijen HV, de Bakker JM, Eckardt D, Tiemann K, Lewalter T, Ghanem A, Luderitz B, Willecke K. Cardiac morphogenetic defects and conduction abnormalities in mice homozygously deficient for connexin40 and heterozygously deficient for connexin45. J Mol Cell Cardiol. 2006;41:787–797 [DOI] [PubMed] [Google Scholar]
- 52.Frank M, Wirth A, Andrie RP, Kreuzberg MM, Dobrowolski R, Seifert G, Offermanns S, Nickenig G, Willecke K, Schrickel JW. Connexin45 provides optimal atrioventricular nodal conduction in the adult mouse heart. Circulation Research. 2012;111:1528–1538 [DOI] [PubMed] [Google Scholar]
- 53.Laing JG, Manley-Markowski RN, Koval M, Civitelli R, Steinberg TH. Connexin45 interacts with zonula occludens-1 and connexin43 in osteoblastic cells. J Biol Chem. 2001;276:23051–23055 [DOI] [PubMed] [Google Scholar]
- 54.Kausalya PJ, Reichert M, Hunziker W. Connexin45 directly binds to zo-1 and localizes to the tight junction region in epithelial mdck cells. FEBS Lett. 2001;505:92–96 [DOI] [PubMed] [Google Scholar]
- 55.Veerman CC, Wilde AA, Lodder EM. The cardiac sodium channel gene scn5a and its gene product nav1.5: Role in physiology and pathophysiology. Gene. 2015;573:177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Detta N, Frisso G, Salvatore F. The multi-faceted aspects of the complex cardiac nav1.5 protein in membrane function and pathophysiology. Biochim Biophys Acta. 2015;1854:1502–1509 [DOI] [PubMed] [Google Scholar]
- 57.Rizzo S, Lodder EM, Verkerk AO, Wolswinkel R, Beekman L, Pilichou K, Basso C, Remme CA, Thiene G, Bezzina CR. Intercalated disc abnormalities, reduced na(+) current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc Res. 2012;95:409–418 [DOI] [PubMed] [Google Scholar]
- 58.Willis BC, Ponce-Balbuena D, Jalife J. Protein assemblies of sodium and inward rectifier potassium channels control cardiac excitability and arrhythmogenesis. Am J Physiol Heart Circ Physiol. 2015;308:H1463–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheng L, Yung A, Covarrubias M, Radice GL. Cortactin is required for n-cadherin regulation of kv1.5 channel function. J Biol Chem. 2011;286:20478–20489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vite A, Radice GL. N-cadherin/catenin complex as a master regulator of intercalated disc function. Cell Commun Adhes. 2014;21:169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Agullo-Pascual E, Lin X, Leo-Macias A, Zhang M, Liang FX, Li Z, Pfenniger A, Lubkemeier I, Keegan S, Fenyo D, Willecke K, Rothenberg E, Delmar M. Super-resolution imaging reveals that loss of the c-terminus of connexin43 limits microtubule plus-end capture and nav1.5 localization at the intercalated disc. Cardiovasc Res. 2014;104:371–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kanki H, Suzuki H, Itohara S. High-efficiency cag-flpe deleter mice in c57bl/6j background. Exp Anim. 2006;55:137–141 [DOI] [PubMed] [Google Scholar]
- 63.Zhang J, Felder A, Liu Y, Guo LT, Lange S, Dalton ND, Gu Y, Peterson KL, Mizisin AP, Shelton GD, Lieber RL, Chen J. Nesprin 1 is critical for nuclear positioning and anchorage. Hum Mol Genet. 2010;19:329–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kreuzberg MM, Schrickel JW, Ghanem A, Kim JS, Degen J, Janssen-Bienhold U, Lewalter T, Tiemann K, Willecke K. Connexin30.2 containing gap junction channels decelerate impulse propagation through the atrioventricular node. Proc Natl Acad Sci U S A. 2006;103:5959–5964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Veeraraghavan R, Hoeker GS, Alvarez-Laviada A, Hoagland D, Wan X, King DR, Sanchez-Alonso J, Chen C, Jourdan J, Isom LL, Deschenes I, Smyth JW, Gorelik J, Poelzing S, Gourdie RG. The adhesion function of the sodium channel beta subunit (beta1) contributes to cardiac action potential propagation. Elife. 2018;7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Z, Stroud MJ, Zhang J, Fang X, Ouyang K, Kimura K, Mu Y, Dalton ND, Gu Y, Bradford WH, Peterson KL, Cheng H, Zhou X, Chen J. Normalization of naxos plakoglobin levels restores cardiac function in mice. J Clin Invest. 2015;125:1708–1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bayly PV, KenKnight BH, Rogers JM, Hillsley RE, Ideker RE, Smith WM. Estimation of conduction velocity vector fields from epicardial mapping data. IEEE Trans Biomed Eng. 1998;45:563–571 [DOI] [PubMed] [Google Scholar]
- 68.Zhang J, Bang ML, Gokhin DS, Lu Y, Cui L, Li X, Gu Y, Dalton ND, Scimia MC, Peterson KL, Lieber RL, Chen J. Syncoilin is required for generating maximum isometric stress in skeletal muscle but dispensable for muscle cytoarchitecture. Am J Physiol Cell Physiol. 2008;294:C1175–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H, Fan J, Zhao Y, Zhang X, Dai B, Zhan J, Yin Z, Nie X, Fu XD, Chen C, Wang DW. Nuclear mir-320 mediates diabetes-induced cardiac dysfunction by activating transcription of fatty acid metabolic genes to cause lipotoxicity in the heart. Circ Res. 2019;125:1106–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.