

Abstract
BACKGROUND:
The mechanisms of aberrant circulating platelet behavior following injury remain unclear. Platelets retain megakaryocyte immature ribonucleic acid (RNA) splicing and protein synthesis machinery to alter their functions based on physiologic signals. We sought to identify fluctuating platelet-specific RNA transcripts in cell-free plasma (CFP) from traumatic brain injury (TBI) patients as proof-of-concept for using RNA sequencing to improve our understanding of postinjury platelet behavior. We hypothesized that we could identify differential expression of activated platelet-specific spliced RNA transcripts from CFP of patients with isolated severe fatal TBI (fTBI) compared with minimally injured trauma controls (t-controls), filtered by healthy control (h-control) data sets.
METHODS:
High-read depth RNA sequencing was applied to CFP from 10 patients with fTBI (Abbreviated Injury Scale [AIS] for head ≥3, AIS for all other categories <3, and expired) and five t-controls (Injury Severity Score ≤1, and survived). A publicly available CFP RNA sequencing data set from 23 h-controls was used to determine the relative steady state of splice-form RNA transcripts discoverable in CFP. Activated platelet-specific spliced RNA transcripts were derived from studies of ex vivo platelet activation and identified by splice junction presence greater than 1.5-fold or less than 0.67-fold ex vivo nonactivated platelet-specific RNA transcripts.
RESULTS:
Forty-two differentially spliced activated platelet-specific RNA transcripts in 34 genes were altered in CFP from fTBI patients (both upregulated and downregulated).
CONCLUSION:
We have discovered differentially expressed activated platelet-specific spliced RNA transcripts present in CFP from isolated severe fTBI patients that are upregulated or downregulated compared with minimally injured trauma controls. This proof-of-concept suggests that a pool of immature platelet RNAs undergo splicing events after injury for presumed modulation of platelet protein products involved in platelet function. This validates our exploration of injury-induced platelet RNA transcript modulation as an upstream “liquid biopsy” to identify novel postinjury platelet biology and treatment targets for aberrant platelet behavior.
Keywords: Platelet activation, next generation sequencing, ribonucleic acid sequencing, cell-free plasma, prespliced RNA complexes, dynamically regulated RNA splicing
Platelets are cardinal in vascular homeostasis via formation and breakdown of clot, endothelial health, and immunoregulation.1–3 Given hemorrhage still accounts for the majority of preventable deaths after injury,4,5 it is alarming that nearly half of trauma patients exhibit ex vivo evidence of aberrant functional behavior in the activation and aggregation of circulating platelets following injury.6,7 Specifically, despite increases in the translocation of platelet activation surface markers following injury, platelet aggregatory function has been shown to be impaired in both static aggregometry assays as well as shear stress and flow enriched microfluidic environments.6–9 Clinically, injury patterns, such as traumatic brain injury (TBI), characterized by a significant burden of damage to endothelial rich vascular beds, have been found to have the strongest associations with increased platelet activation yet impaired platelet aggregation.6,7,10,11 However, these phenotypes of altered postinjury platelet function have also been demonstrated in mildly injured cohorts,12 questioning whether these findings are physiologic or maladaptive.
To date, our understanding of postinjury platelet behavior has primarily relied upon ex vivo identifications of downstream functional changes in platelet activation and aggregation following tissue damage. Recent studies have exposed the possibility of multiple pathways responsible for these downstream functional changes, including histone driven platelet structural changes,13 impaired calcium signaling,13,14 and changes in the von-Willebrand factor-platelet axis.15 However, the upstream molecular regulators of these altered pathways remain unknown. Importantly, platelets are known to retain immature ribonucleic acid (RNA) transcripts and active RNA editing machinery from megakaryocyte progenitor cells.16 Furthermore, physiologic stimuli and platelet activation lead to splicing of these immature RNA transcripts to create functionally relevant proteins involved in platelet physiology.17–22 Accordingly, molecular technologies including RNA sequencing have been recently shown to dynamically fine-tune measurements of platelet function in both health and disease.22–24 Consequently, elucidating changes in platelet-specific RNA transcripts may present a promising avenue for exposing upstream regulators of downstream functional changes in postinjury platelet behavior.
Therefore, we sought to identify fluctuating platelet-specific RNA transcripts present in cell-free plasma (CFP) from TBI patients as proof-of-concept for using RNA sequencing to improve our understanding of postinjury platelet behavior. We hypothesized that by using RNA sequencing of CFP, we could identify differential expression of activated platelet-specific spliced RNA transcripts in patients with isolated severe fatal TBI (fTBI) compared with minimally injured trauma controls (t-controls), filtered by healthy control (h-control) data sets.
METHODS
Patient Enrollment, Blood Specimen Collection, Sample Selection, Cohorts, CFP Processing
Whole blood samples were prospectively collected on patients on arrival to the Emergency Department at a Level I urban trauma center from 2005 to 2011 as part of a longitudinal study (Activation of Coagulation and Inflammation after Trauma [ACIT]) evaluating abnormalities of inflammation and coagulation after injury.6,25–27 Blood samples were collected via initial placement of a 16G or larger peripheral IV in standard laboratory vacuum-sealed tubes containing 3.2% (0.109 M) sodium citrate. The samples were centrifuged for 10 minutes at 2960 relative centrifugal field at room temperature into platelet-poor plasma. Platelet-poor plasma was aliquoted, and immediately placed into a −80°C freezer. Samples were collected prior to the patient receiving any blood product transfusions; therefore, none were subject to any variation from transferred RNA. Demographic, injury, clinical, and outcome data were prospectively collected. Patients were retrospectively excluded if they were found to be on anticoagulant or antiplatelet medications, or possessed a preexisting bleeding diathesis at the time of injury. The study was approved by the University of California Committee on Human Research.
Sample selection from the described banked plasma samples was carefully performed prior to RNA sequencing. Specifically, the 15 plasma samples sequenced were selected based on: (1) confirmation of stable processing and storage protocols, (2) single freeze-thaw cycle, and (3) an attempt to produce clinical homogeneity in an effort to limit as much injury confounding as possible. The patients from which plasmas were selected are outlined in Table 1, and their aggregate demographics and outcomes are outlined in Table 2. The cohorts were determined based on the following:
Isolated Severe fTBI Cohort. Ten fTBI patients had CFP sequenced. Traumatic brain injury was defined by clinical notes and chart review for imaging-identified intracranial hemorrhage and confirmed with Abbreviated Injury Scale (AIS) head score of 3 or greater. From this cohort, isolated severe fTBI was defined as AIS all other categories score less than 3 and expired. Further, to be as confident as possible that there was a breakdown of the blood-brain barrier for presumed discharge of activated platelet-specific RNA into peripheral circulation, patients with penetrating mechanisms of injury were preferentially chosen.
Minimally Injured t-control Cohort. Five t-control patients had CFP sequenced. t-Control was defined as Injury Severity Scores (ISS) of 1 or less, and survived. Further, to be as confident as possible that these patients did not have significant burdens of tissue injury, patients with little to no injury burden were chosen.
h-control Cohort. A publicly available data set of CFP RNA sequencing from 23 independent h-control patient samples was used for classification of organospecific transcriptome enrichment to model transcript stability for identifying platelet-specific genes in fTBI.28
TABLE 1.
Clinical Notes for the Isolated Severe fTBI vs. t-Control Patients
| Cohort | Clinical Synopsis | Autopsy Cause of Death |
|---|---|---|
| fTBI | A 20-year-old man with isolated GSW to head who presented in PEA arrest, but regained pulses with resuscitation and underwent decompressive craniectomy—declared brain dead | GSW head Brain dead |
| fTBI | An 18-year-old man with multiple GSWs to head complicated by intracranial midline shift, herniation, and loss of brainstem reflexes—declared brain dead | GSW head Brain dead |
| fTBI | An 18-year-old man who was bluntly assaulted to head found to have diffuse SAH—declared brain dead | BFI to head Brain dead |
| fTBI | A 53-year-old man who was found down with head trauma who underwent decompressive hemicraniectomy for a large SDH—declared brain dead | BFI to head Brain dead |
| fTBI | A 36-year-old man who was found down with head trauma and without a pulse and received resuscitation with return of circulation. He was found to have a large SDH with uncal herniation—declared brain dead | BFI to head Brain dead |
| fTBI | A 65-year-old man who fell down 12 stairs with a large SDH and parenchymal contusions—declared brain dead | BFI to head Brain dead |
| fTBI | A 28-year-old woman with isolated GSW to head complicated by SDH, diffuse SAH, and uncal herniation—declared brain dead | GSW head Brain dead |
| fTBI | A 20-year-old man with isolated GSW to the head found to have SDH and intraparenchymal hemorrhage—declared brain dead | GSW head Brain dead |
| fTBI | A 30-year-old man with isolated GSW to head who presented in PEA arrest, but regained pulses with resuscitation, found to have large SDH, IVH, and loss of brainstem reflexes—declared brain dead | GSW head Brain dead |
| fTBI | A 23-year-old man with isolated GSW to head with significant blood loss and associated shock who underwent decompressive hemicraniectomy—declared brain dead | GSW head Brain dead |
| t-control | A 34-year-old man who was in a motor vehicle collision—no injuries identified | n/a |
| t-control | A 50-year-old found down with occipital hematoma—no injuries identified | n/a |
| t-control | A 21-year-old man with GSWs to left and right thigh—minor soft tissue injuries only | n/a |
| t-control | A 24-year-old man with stab wound to the left wrist—minor soft tissue injuries only | n/a |
| t-control | A 30-year-old man who was in a motor vehicle collision—no injuries identified | n/a |
Cause of death determined by autopsy.
GSW, gunshot wound; BFI, blunt force injury; SDH, subdural hematoma; SAH, subarachnoid hemorrhage; IVH, intraventricular hemorrhage; GCS, Glasgow Coma Scale; ICU, intensive care unit; PEA, pulseless electrical activity.
TABLE 2.
Aggregate Demographics/Outcomes for Minimally Injured t-Controls and Isolated Severe fTBI Patients
| t-Control (n = 5) | fTBI (n = 10) | SMD (95% CI) | |
|---|---|---|---|
| Age, y | 26 (24–27) | 25 (20–36) | >0.1 (−1.1–1.1) |
| Male | 100% | 90% | N/A |
| Body mass index, kg/m2 | 24 +/− 1 | 26 +/− 3 | 0.4 (−1.2–1.9) |
| Blunt mechanism | 60% | 40% | N/A |
| ISS | 1 (1–1) | 26 (26–30) | 17.5 (10.8–24.2) |
| Presentation Glasgow Coma Scale | 14 (13–15) | 3 (3–3) | −5.8 (−8.3–3.3) |
| Isolated Severe fTBI | 0% | 100% | N/A |
| Prehospital crystalloid volume (mL) | 100 (100–400) | 200 (50–400) | −0.3 (−1.6–1.1) |
| Presentation temperature (°) | 36.8 ± 0.4 | 35.1 ± 1.1 | −1.6 (−3.0–−0.1) |
| Presentation pH | 7.2 ± 0.2 | 7.2 ± 0.2 | −0.1 (−1.4–1.2) |
| Presentation international normalized ratio | 1.1 (1–1.1) | 1.2 (1.1–1.3) | 0.8 (−0.3–1.9) |
| Presentation prothrombin time (sec) | 28 ± 6 | 39 ± 16 | 0.9 (−0.25–2.0) |
| Presentation platelet count (× 109/L) | 274 (202–347) | 257 (218–286) | −0.5 (−1.6–0.6) |
| Presentation blood alcohol level (mg/dL) | 241 (10–471) | 212 (0–293) | <−0.1 (−1.8–1.8) |
| Transfused in 24 h | 0% | 80% | N/A |
| Total hospital days | 1 (1–2) | 2 (2–2) | 0.88 (−0.25–2.0) |
| Total ICU days (to 28 days) | 0 (0–0) | 2 (2–2) | 2.9 (1.3–4.4) |
| Ventilator-free days (to 28 days) | 28 (28–28) | 0 (0–0) | N/A |
| Acute respiratory distress syndrome | 0% | 40% | N/A |
| Multi-organ failure | 0% | 0% | N/A |
| Mortality at discharge | 0% | 100% | N/A |
| Hours to death | N/A | 26 (17–38) | N/A |
Patient demographics for the 15 patients. Data are mean ± SD, median (interquartile range), or percentage. The SMD and 95% CI between t-control and fTBI patients were calculated for continuous variables. Data for skewed variables reported as median with inter-quartile ranges. Ventilator-free days are counted for the first 28 days of hospitalization. Patients who expired received 0 ventilator-free days.
SMD, standardized mean difference; 95% confidence intervals, 95% CI.
RNA Sequencing of CFP in Isolated Severe fTBI and Minimally Injured t-Control Cohorts
The RNA sequencing of banked CFP was performed. Total RNA was isolated from 400 microliters of CFP using RNAeasy Qiagen kits. Two hundred nanograms of total RNA per sample was sent for random primer based complementary DNA amplification and sequencing at high-read depth (~20 million reads per sample).28
RNA Sequencing Analyses
Pertinent FASTQ files were downloaded to a local server, aligned, and mapped using Star2 software at standard default settings, the Hg19 version of the human genome from UCSC databases, and the Hg19 annotation format from the Illumina iGenomes database to produce differential expression (raw counts) or sjout (splice-forms) outputs. To avoid sequence mapping batch effects, all RNA sequencing data (including the publicly available data sets22,28) were mapped in an identical manner: mapped count normalized where appropriate, and represented as fractional counts (Fragments Per Kilobase of transcript per Million mapped reads [FPKM]) for gene filtering analysis. To determine if fluctuating platelet-specific genes were detectible in the CFP of the fTBI patients that we sequenced from ACIT, three publicly available RNA sequencing data sets22,28,29 were used to create a biologically plausible enriched data set of activated platelet-specific spliced RNA transcripts discoverable in CFP from fTBI patients (Fig. 1A–D).
Figure 1.
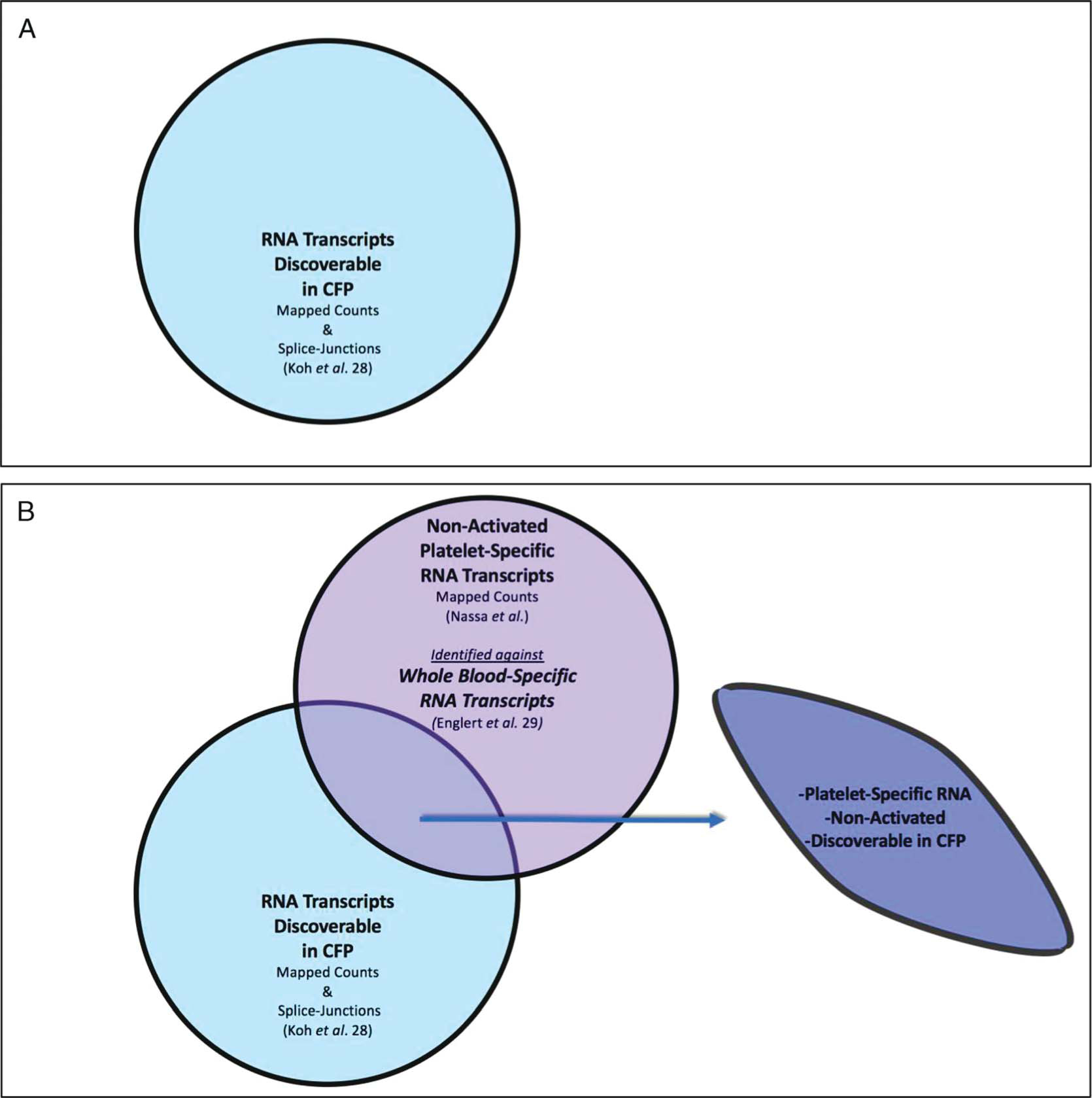
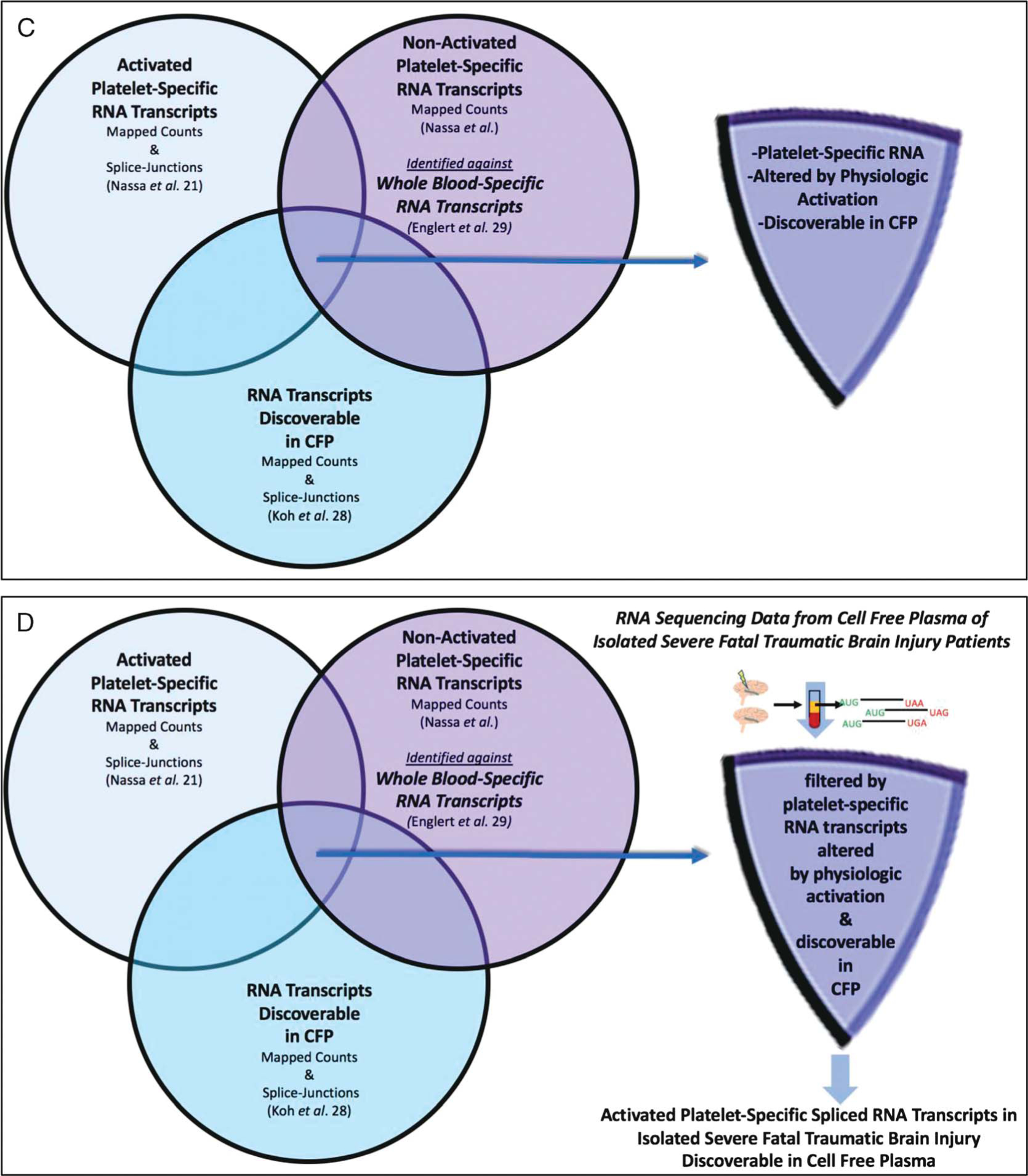
(Panels A-D). Filtering and validating methodology to identify activated platelet-specific spliced RNA transcripts discovered in CFP from isolated severe fatal traumatic brain injury patients. Publicly available RNA sequencing data sets22,28,29 were used to generate lists of activated platelet-specific spliced RNA transcripts discoverable in CFP and applied to isolated severe fTBI patient CFP sequencing data. Panel A: RNA transcripts discoverable in CFP (1427 RNA splice-form variant transcripts). Panel B: nonactivated platelet-specific RNA transcripts discoverable in CFP (685 platelet-specific RNA transcripts). Panel C: activated platelet-specific RNA transcripts discoverable in CFP (780 activated platelet-specific RNA splice-form variant transcripts). Panel D: activated platelet-specific spliced RNA transcripts discoverable in CFP from isolated severe fTBI patients (42 spliced RNA transcripts in 34 genes).
RNA Transcripts Discoverable in CFP (1427 RNA splice-form variant transcripts, Fig. 1A):
A publicly available data set from the CFP (conserved collection and processing methodology to ACIT) of 23 h-control patient samples was used to determine the relative steady state of splice-form RNA transcripts discoverable in CFP (identified by presence of splice junctions, >25 counts/sample and read count stability [average/SD > 1]).28
Nonactivated Platelet-Specific RNA Transcripts Discoverable in CFP (685 platelet-specific RNA transcripts, Fig. 1B):
Platelet-specific RNA transcript lists were derived from ex vivo nonactivated platelet RNA sequencing,21 and identified against whole blood-specific genes29 to create a ranked list of nonactivated platelet-specific (i.e., organospecific) RNA transcripts discoverable in CFP.
Activated Platelet-Specific RNA Transcripts Discoverable in CFP (780 activated platelet-specific RNA splice-form variant transcripts, Fig. 1C):
Activated platelet-specific spliced RNA transcript lists were derived from studies of ex vivo platelet activation with collagen or a thrombin analog (thrombin receptor activating peptide).22 These were identified if they demonstrated an altered steady state in CFP, defined by splice junction presence greater than 1.5 or less than 0.67 fold the ex vivo nonactivated platelet-specific RNA transcripts.
Activated Platelet-Specific Spliced RNA Transcripts Discoverable in CFP from Isolated Severe fTBI Patients (42 spliced RNA transcripts in 34 genes, Fig. 1D; Table 3):
TABLE 3.
Differential Expression of Activated Platelet-Specific Spliced RNA Transcripts in CFP of Isolated Severe fTBI vs. t-Control Patients
| Gene Name | Gene Name/Function | fTBI/t-Control (Fold Change) | Platelet Gene Rank |
|---|---|---|---|
| AKT3 | RAC-γ serine/threonine protein kinase | ↓ >4× | 2,581 |
| ANKRD12 | Ankyrin-repeat containing transcriptional regulator | ↓ > 10× | 968 |
| ARHGAP18 | Rho GTPase activating protein | ↓ > 5× | 580 |
| ARHGAP21 | CDC42 GTPase activating protein, actin regulation | ↓ >8× | 1,603 |
| ARHGAP21 | CDC42 GTPase activating protein, actin regulation | ↓ > 1,000× | 1,603 |
| ARHGEF12 | Rho guanine nucleotide exchange factor | ↓ >2× | 1,271 |
| ARHGEF12 | Rho guanine nucleotide exchange factor | ↑3× | 1,271 |
| CALD1 | Caldesmon 1, cytoskeletal regulation | ↑30× | 1,627 |
| CCL5 | Histamine and eosinophil activation | ↓ >8× | 72 |
| CDK2AP1 | Cyclin-dependent kinase 2-associated protein 1 | ↓ > 25× | 451 |
| CHD9 | Chromodomain helicase DNA binding protein 9 | ↓ >8× | 2,251 |
| CSNK1G3 | Casein kinase1 γ 3, intracellular signaling | ↓ >8× | 1,850 |
| DNM3 | Microtubules and vesicular transport | ↑14× | 602 |
| DNM3 | Microtubules and vesicular transport | ↑2× | 602 |
| EIF4G3 | Eukaryotic translation initiation factor 4 γ 3 | ↓ > 250× | 877 |
| F13A1 | Factor XIII A Subunit | ↓ > 25× | 47 |
| FIP1L1 | Factor interacting with PAPOLA and CPSF1, pre-mRNA 3′ end processing | ↓ > 207× | 1,668 |
| FNBP1L | Formin binding protein 1-like, cytoskeletal regulation | ↓ > 150× | 4,067 |
| GRB14 | Growth factor receptor bound protein 14 | ↓ > 336× | 3,729 |
| HBA1 | Hemoglobin subunit α 1 | ↓ > 348× | 25 |
| IQGAP2 | IQ motif containing GTPase activating protein, cytoskeletal regulator | ↓ > 22× | 1,720 |
| IQGAP2 | IQ motif containing GTPase activating protein, cytoskeletal regulator | 1× | 1,720 |
| IQGAP2 | IQ motif containing GTPase activating protein, cytoskeletal regulator | ↓ > 17× | 1,720 |
| KIF2A | Kinesin family member, microtubule motor | ↑6× | 164 |
| LIMS1 | LIM Zn finger domain containing Integrin signaling adaptor | 1× | 209 |
| LTBP1 | Latent transforming growth factor binding protein 1 | ↓ > 46× | 824 |
| MAP4K5 | MAP kinase kinase kinase kinase 5, JNK signaling | ↓ > 6× | 5,254 |
| MBNL3 | Muscleblind-like splicing regulator 3 | ↓ > 176× | 1,394 |
| MLLT3 | Super elongation complex subunit | ↓ > 51× | 4,178 |
| NAP1L1 | Nucleosome assembly protein 1-like 1 | ↑19× | 168 |
| NAP1L1 | Nucleosome assembly protein 1-like 1 | 1× | 168 |
| NLK | Nemo-like kinase | ↓ > 648 × | 2,165 |
| PF4 | Chemokine, released from alpha granules | ↑3× | 13 |
| PF4 | Chemokine, released from alpha granules | ↑1.5× | 13 |
| PLEK | Pleckstrin, platelet activation, and vesicle secretion | ↑4× | 431 |
| PTGS1 | Cyclooxygenase 1, prostaglandin biosynthesis | ↓ > 357× | 237 |
| PTGS1 | Cyclooxygenase 1, prostaglandin biosynthesis | ↓ > 406× | 237 |
| RAP1B | Regulation of integrin-mediated cell signaling, and outside in signaling in platelets | ↓ > 117× | 261 |
| RBPMS2 | RNA-binding protein with multiple splicing 2 | ↓ > 118× | 8,079 |
| SH3BGRL2 | SH3 domain binding glutamate-rich protein-like 2 | ↓ > 10× | 189 |
| SPARC | Osteonectin, activation-dependent platelet secretion, bone, and ECM metabolism | ↑1.5× | 85 |
| TMSB4X | Thymosin β 4, actin regulation, tissue regeneration | ↓ > 1000× | 24 |
Platelet Gene Rank, absolute gene count from normalized ex vivo platelet control22; GTP, guanosine triphosphate; CDC42, Cell Division Cycle 42; PAPOLA, polynucleotide adenylyltransferase alpha; CPSF1, cleavage and polyadenylation specific factor 1; IQ, isoleucine-glutamine; LIM, Lin11, Isl-1 and Mec-3; MAP, mitogen-activated protein; JNK, c-Jun N-terminal kinase; ECM, extracellular matrix; SH3, Src (Sarcoma) homology 3.
RNA sequencing data from CFP of fTBI patients was filtered by activated platelet-specific RNA transcripts discoverable in CFP. These genes were accepted if there is greater than 1.5-fold change in transcript levels.
Clinical Data Analyses
Data are presented as mean (± standard deviation), median (interquartile range), or percentage. The standardized mean differences and 95% confidence intervals between t-control and fTBI patients were calculated for continuous variables.
RESULTS
Due to the 60% rate of penetrating mechanisms in the fTBI cohort, 80% received transfusions. All of the patients with fTBI were declared brain dead, and they expired early after injury (26 median hours to death; Table 1 and Table 2). None of the t-control patients received transfusions, developed organ failure, or required the ICU. All of the t-control patients survived and were discharged with short hospital stays (Table 1 and Table 2).
A total of 42 differentially spliced activated platelet-specific RNA transcripts in 34 genes were identified as altered in fTBI (Table 3). Fatal TBI patients had both upregulated and downregulated activated platelet-specific RNA transcripts discovered in CFP (Table 3). Figure 2 is a representation of activated platelet-specific RNA splicing events in fTBI using F13A1 gene (encodes factor XIII, subunit “A”) as an example.
Figure 2.
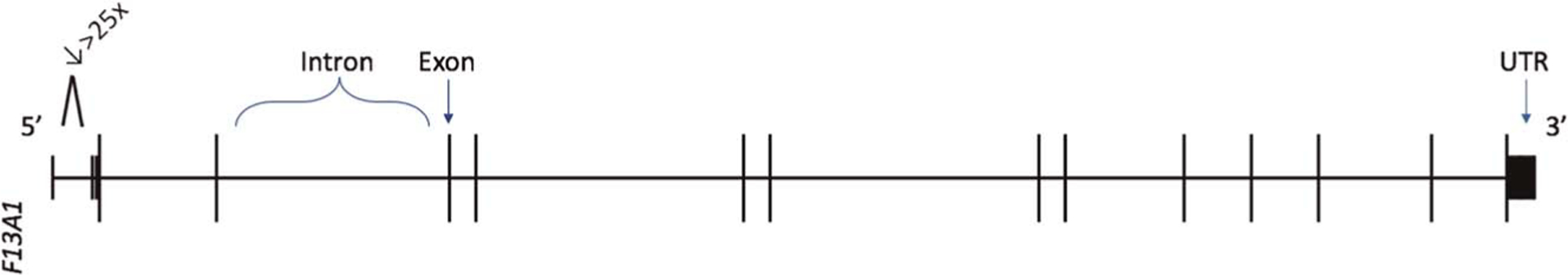
Activated platelet-specific RNA splicing events in isolated severe fTBI visualized using F13A1 gene (encodes Factor XIII Subunit “A”) as a representative example. Differentially discovered splice junctions were used to query the hg19 genome assembly in the University of California at Santa Cruz Genome Browser. Horizontal bars are representing introns and vertical bars are representing exon junctions (not to scale). Numeric decrease (↓) represents the fold change of F13A1 spliced RNA transcript discovered in isolated severe fTBI vs. minimally injured t-control. UTR, untranslated region. 5′ and 3′ ends noted accordingly.
DISCUSSION
We have presented proof-of-concept that it is possible to leverage molecular gene sequencing technologies to identify differential expression of activated platelet-specific spliced RNA transcripts present in the CFP of patients with isolated severe fTBI. This finding accelerates our trajectory toward identifying upstream molecular regulators of aberrant postinjury platelet behavior using RNA sequencing.
The molecular technique of high-read depth RNA sequencing is in fact optimal for addressing unsolved biologic dilemmas in the setting of injury because it is based on the tenant that innate tissue damage leads to changes in the nucleic acids signatures of circulating cellular components.30 Accordingly, there is tremendous equipoise to leverage molecular technologies toward improving our understanding of complex biologic processes in the setting of injury that are evidenced to be altered in ex vivo settings, representative of the in vivo biology. As such, the biology of platelets in the setting of tissue damage is ideal for applying this methodology. Although ex vivo aberrancy in postinjury platelet behavior remains a consistent finding in trauma patients,6–8,31 the mechanisms and pathways responsible for these findings remain largely unresolved, despite recent advances exposing changes that occur in platelet structure, signaling, and immunoregulation.13–15,32–34 In addition, a role for identifying differential platelet-specific genes has emerged in multiple disease conditions outside the field of trauma, including for determining the mechanisms of endothelial cell regulation in myocardial infarction,35 for understanding the pathogenesis of essential thrombocytopenia,36 and for early diagnosis of malignancy.37
This is because platelet protein synthesis is under transcriptional control and has been shown to be responsive to external signaling and platelet activation. In fact, hemostatic protein factor production by platelets increases in response to ex vivo activation.17 Though platelets are subcellular in size (approximately 2–3 microns in diameter), they are extremely active biologically. Furthermore, due to their size constraints combined with a need for multiple protein products to perform tasks critical to vascular homeostasis and immune function, platelets carry a transcriptome that can undergo maturation by splicing events during activation, thereby resulting in accumulation of corresponding functional protein products as part of an activation cascade.22–24 This is of notable importance in the behavior of platelets after injury, because multiple investigators have identified that platelets translocate and expose platelet activation surface markers, consistent with morphologic evidence of platelet activation via multiple surface receptor pathways.7,14 Therefore, the ability to identify differential gene expression upon platelet activation presents a promising avenue for understanding platelet biology in a disease state, such as trauma, characterized by substantial activation of circulating platelets.22
The identification of RNA splicing events in circulating platelets induced by tissue damage driven signaling offers a way to decode postinjury platelet biologic patterns. This is feasible given the vast array of transcriptome data obtained through sequencing, that is highly specific to physiology, and can be obtained at a low cost relative to the number of identified genetic features.28 However, despite this benefit, our model is dependent on assumptions concerning the discovery of splicing events of platelet-specific RNA transcripts in a cell-free space. Specifically, to elucidate unique platelet-specific RNA splicing events, we created three gene list filters: (i) RNA transcripts discoverable in CFP,27 (ii) platelet-specific RNA transcripts from ex vivo nonactivated platelets,22 and (iii) platelet-specific RNA transcripts from ex vivo activated platelets.21 From this, we were able to create a proof-of-concept model of the intersection of physiologically driven platelet-specific RNA transcript splicing events discoverable in a cell-free space. The vast majority of identified RNA transcripts were highly ranked platelet-specific genes (top 10%), thus providing reassurance that platelet gene uniqueness mostly correlated with high expression and discovery in CFP. Interestingly, platelet-specific RNA splicing events were identified in the CFP of minimally injured trauma control patients as well (data not shown), suggesting intriguingly that high degrees of platelet activation may occur despite lesser burden of innate tissue damage than would be expected with isolated severe fTBI.
We recognize several limitations of this study including that the sample size is small and the data lacks power to calculate a false discovery rate. However, the intent of the study was not to focus or make claim to discovery of specific genes, but rather to test a methodology that could be useful to identify physiologically driven differential gene expression in response to tissue damage induced biologic changes. As in all blood-based studies of populations of injured patients, the samples are derived from patients with different genetic backgrounds, varying mechanisms of injury, and have undergone processing and storage, thereby allowing for many unmeasured confounding factors.
However, we chose to only sequence samples that could be confirmed to have standardized spin methods and single freeze thaw cycles. We then used clinical criteria to attempt to produce homogenous cohorts. Due to these methodologic criteria, the samples chosen were a minimum of 8 years old and a maximum of 14 years old and had been banked for other purposes. These characteristics are likely to contribute to the potential for RNA degradation, particularly because the samples were not obtained with the intent of studying RNA expression, and an RNAse-free environment could not be ensured. Therefore, the potential for RNA degradation must be taken into account when interpreting these data; however, the large fold increases in expression identified are not likely to be explained by degradation.
The isolated severe fTBI patients were chosen in an effort to ensure isolated injury to the brain and to be as confident as possible that there was breakdown of the blood-brain barrier for presumed discharge of activated platelet-specific RNA into peripheral circulation. This was done by preferentially selecting patients with isolated severe fTBI from penetrating mechanisms (60% of samples). Conversely, the minimally injured trauma control patients were activated as trauma patients, but were found have no significant injuries (ISS scores of 0 or 1). We consider these patients “sick” controls because although they had alterations in their physiology and potentially biology, they did not have clinical findings of significant tissue injury and can be presumed to not have had significant breakdown of their blood brain barrier. Furthermore, we used true “healthy” control data sets to filter the genomics data in the model we created.
It should also be noted that there were intoxicated patients in both the fTBI cohort and the t-control cohort. Although intoxication has effects on coagulation biology, it is also a risk factor for general trauma and brain injury. In addition, the t-control and fTBI cohorts were relatively matched in blood alcohol levels (median levels, 241 mg/dL vs. 212 mg/dL, respectively). Finally, there is conflicting data regarding the in vivo and ex vivo effects of alcohol on platelet function, but some studies suggest that alcohol may reduce the circulating number of platelets and impair platelet hemostatic functions.38–40
Ultimately, we make no conclusions regarding clinical interpretations of our results, as the sample sizes are small and the sample selection was solely to attempt to tightly control an in vivo biology in a human model from samples previously collected. Ultimately, these cohorts are likely not the “interest” populations to use this methodology on, and for this reason, we made no hypotheses about associations of clinical characteristics or patient outcomes with gene signatures, solely that platelet-specific gene signatures could be identified between an injured and a noninjured population, in the plasma space.
Finally, the CFP genomic space is complex, containing highly degraded “free” RNA from many cells, along with exosomes, bound RNA, and a large fraction of platelet-specific RNA.28 In fact, by our estimation 2% to 40% of RNA in CFP is platelet-derived, thus, there is abundant source material that could represent various states of platelet activation, renewal, and degradation (data not shown). Our results suggest that there are species of RNA likely derived from platelets, and following injury that they undergo extreme perturbations in content and kind. However, it remains unknown what the origin of the platelet-specific RNA transcripts in the CFP space are. Although the origin may be fragments of platelets or discharged procoagulant and proinflammatory platelet contents, megakaryocytes and leukocytes also have overlapping signatures with platelets. This biologic space is not only unlikely to contain megakaryocytes or leukocytes in abundance but also cannot be presumed to contain significant numbers of whole platelets and is, therefore, not the ideal sampling space for elucidating the exact molecular basis of aberrant postinjury platelet biology. However, we believe CFP is ideal for an initial proof-of-concept model because it is a logistically feasible space to sample in patients.
In addition, our filtering methodology is supported by identification of biologically relevant platelet-specific spliced gene variants in the cell-free space. As an example of the ability to identify potentially biologically relevant changes, the identification of fold changes in the RNA transcript that encodes factor XIII subunit A (F13A1, Table 3, Fig. 2) in CFP in the setting of isolated severe fTBI is likely biologically relevant and may prove itself to be an important finding and a potential molecular target.41 It is known that factor XIII is a heterotetramer that consists of two catalytic “A” subunits and two noncatalytic “B” subunits, and the interaction of these subunits is critical for hemostasis via stabilizing crosslinked fibrin from degradation by fibrinolytic machinery.42 Notably, the “A” subunits are encoded by the F13A1 gene are synthesized in megakaryocytes, exist in the cytoplasm of platelets, and factor XIII “A” subunit genetic deficiencies cause bleeding diatheses. Moreover, recombinant factor XIII “A2” is used as a substitution therapy in factor XIII-A deficiencies.42–44 Therefore, the discovery of a 25-fold decrease of the RNA transcript variant of this critical platelet-specific (47th highest ranked platelet gene out of 26,364 annotated genes) hemostatic protein in the CFP of isolated severe fTBI compared with minimally injured trauma controls could prove itself to be relevant in the future.
We can now apply this methodology to isolated platelets from injured patients, prospectively, with control over enrollment, collection, and processing, and in combination with assays of platelet physiology. With this, we will be able to identify if the signature patterns in the CFP are truly from platelets/platelet-derived contents versus from megakaryocytes or leukocyte lineages. Ultimately, if platelet-specific genes associated with functional changes in postinjury platelet behavior successfully overlap to what can be identified in the cell-free space, it could support development of innovative and precise point-of-care diagnostics. This type of “liquid biopsy” could be useful for the purpose of identifying postinjury biologic alterations, guiding molecularly targeted treatments, and improving overall outcomes in injured patients. Overall, we recognize that to further interpret platelet-specific gene patterns in injury, we will require larger and more varied RNA sequencing from CFP samples, and importantly from isolated platelets paralleled with simultaneously measured platelet physiology.
CONCLUSION AND FUTURE DIRECTIONS
Until now, the fluctuating platelet-specific RNA transcriptome has not been characterized in the setting of injury. Using high-read depth RNA sequencing, we have identified a list of 42 differentially spliced platelet-specific RNA transcripts in the CFP from patients with isolated severe fTBI. This novel proof-of-concept finding may indicate that a pool of resident platelet immature RNAs undergo modifications, including splicing events for presumed modulation of platelet protein expression involved in platelet function after injury. Much work remains as we learn about the discovery of platelet-specific genes in the CFP space and its relevance to postinjury platelet behavior. Yet, the finding that activated platelet-specific spliced RNA transcripts are present supports our focus on injury-induced platelet RNA transcript modulation and may ultimately form the foundation of “liquid biopsy” to identify treatment targets for aberrant postinjury platelet behavior. This is a first step in establishing a genomic road map for more precise gene sequencing experiments on isolated platelets. Ultimately, applying sequencing techniques to improve our understanding of the molecular basis of altered postinjury platelet physiology may allow for improved targeted therapies for trauma-induced coagulopathy and associated hemorrhage, organ failure, and death.
DISCUSSION.
MARTIN SCHREIBER, M.D. (Portland, Oregon): Karen Brasel told me not to thank anyone or compliment anyone, because it takes too much time, so I’ll just get right into it.
The authors compared specific RNA expression on admission in cell free plasma between ten patients with isolated head injury who died and five control patients with an ISS less than or equal to one.
Twenty-three human controls defined platelet specific RNA. The authors show substantial changes in RNA expression in TBI patients compared to the controls.
The samples in this study are a minimum of eight years old and a maximum of 14 years old. How are the samples managed from the time of blood draw, and how long until processing?
Was this consistently the same? Have they been previously thawed and frozen? Could the age of the samples and thawing and refreezing affect the results of the study?
RNA is very unstable and begins degrading immediately, so management of the samples is critical, and these samples were not obtained with the intention of studying RNA expression, so an RNaAse-free environment was not insured, meaning a significant degradation of samples could have occurred as samples were procured and processed. Please comment.
All the isolated TBI patients died. Did they die from TBI, or did they die from comorbidities that might affect the results of the study?
Sixty percent of the isolated TBI patients suffered penetrating trauma, which is not generally representative of civilian TBI.
If the causes of death were not modifiable, was this the best patient population to study because examination of RNA expression would presumably result in a change in management that would improve outcomes?
How were these 15 patients chosen of the many possible patients eligible over the six years where blood was obtained?
Why did 80 percent of isolated TBI patients require a blood transfusion? Substantial bleeding is not generally a major characteristic of isolated TBI.
Why did the control group with ISS less than or equal to one have a mean PH of 7.2? These are essentially non-injured patients. It is not clear to me how this information will affect the clinical care of TBI patients. What will we do with this information, and how will it affect management?
Fundamentally, the authors show that RNA expression of patient-specific RNA changes in moribund TBI patients, but are the data accurate, and are the data actionable? Thank you.
SCOTT C. BRAKENRIDGE, M.D. (Gainesville, Florida): Super exciting work, and I understand Dr. Schreiber’s sampling concerns, but I want to ask you a theoretical question instead, which is what exactly are you going to be measuring? The traditional thought is that platelets are not transcriptionally active, are these differences in expression within megakaryocytes before the platelets are produced and that’s what you’re measuring, or are these splice variants in the platelets afterwards? What is this going to assess? Exciting and important work.
MATTHEW KUTCHER, M.D. (Jackson, Mississippi): Wonderful work, again, from some leaders in the field in this. I wonder, just in dealing with the activated platelet transcripts that you used to select your RNA transcription, kind of splice variants of interest, what did you use, what was used to activate platelets in that circumstance? Was it a specific agonist; were there multiple agonists tried? Thank you.
LUCY Z. KORNBLITH, M.D. (San Francisco, California): Thank you. Thank you, Dr. Schreiber, for your thoughtful comments, and for sharing the discussion with me ahead of time.
I’m going to address your questions on how the samples were managed, specifics of isolation and storage, and about patient selection altogether.
So, the samples were immediately centrifuged at 2960 RCF for ten minutes. This is consistent across all the samples, because they all came from this longitudinal study with a stable processing and storage protocol; and it’s consistent with how the plasma was processed for the healthy control dataset as well.
Now, the 15 samples included were chosen very purposefully. First, I chose the ten TBI patients and the five controls over such a lengthy time period that you pointed out, because I was choosing from plasma that had been banked for prior purposes; therefore, in order to be strict with criteria about which plasma was appropriate I chose only those that I could confirm correct collection and spin methodology, single freeze-thaw cycles, and fit the strict clinical criteria I was looking for to produce as homogenous a population as I could in a human study, which brings me to your question about clinical characteristics.
So, for the TBI patients, I did not choose them to mimic civilian injured populations, but rather so that I could produce homogeneity, which was important in an effort to try and limit as much injury confounding as possible.
So, who were these patients? They were patients in which I thought that there was discharge of activated platelet specific RNA into peripheral circulation-isolated fatal brain injury primarily from penetrating trauma to the head, to be as sure as possible that there was breakdown of the blood-brain barrier.
These were fatal injuries, and 80 percent of them received transfusion due to the predominance of penetrating mechanism.
And then for the uninjured population with the PH of 7.2, based on my chart review I think most of this was due to intoxication and concomitant dehydration. To ensure homogeneity in this group as well, these patients all had an ISS score of 0 or 1 and no significant injuries.
Ultimately, these aren’t the cohorts that are going to necessarily matter as the interest populations. They were chosen for those characteristics that I mentioned.
And for this reason, we really made no hypothesis about associations with the gene signatures and anything clinically.
And in the future, to address Dr. Brakenridge’s and Dr. Kutcher’s questions together, what is going to be important is doing this in isolated platelets, which is underway currently. The activators we use are ADP, collagen, and thrombin, as well as trauma patient plasma.
And finally, to answer the question about transcription, we are currently focused on post-transcriptional changes in circulating platelets via splicing. They do not have a nucleus, but they have RNA machinery and prior to mRNA translation, there is a process of differential inclusion or exclusion of regions of pre-mRNA, allowing them to alter their translational products to produce various proteins associated with their needed physiology.
Thank you.
ACKNOWLEDGMENTS
Erin Ross, BS, and John Parks, BA are acknowledged for their assistance with data collection.
Funding: L.Z.K. is supported by NIH 1K23GM130892-01, Eastern Association for the Surgery of Trauma Research Scholarship, and University of California San Francisco Irene Perstein Award. R.A.C. is supported by NIH K01ES026834 and DoD W911QY-15-C-0044. M.J.C. is supported by DoD W911QY-15-C-0044 and NIH UM1HL120877. C.M.V.B. is supported by a University of California San Francisco Program for Breakthrough Biomedical Research Award. A.P. is supported by NIH K08GM110497. Z.A.M. is supported by The National Center for Advancing Translational Sciences of the NIH award 5TL1TR001871-04.
Footnotes
Meeting presentation: To be presented at the 78th annual meeting of the American Association for the Surgery of Trauma; September 18–21, 2019; in Dallas, Texas.
DISCLOSURE
The authors declare no conflicts of interest.
REFERENCES
- 1.Nachman RL, Rafii S. Platelets, petechiae, and preservation of the vascular wall. N Engl J Med. 2008;359(12):1261–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoffman M, Monroe DM 3rd. A cell-based model of hemostasis. J Thromb Haemost. 2001;85(6):958–965. [PubMed] [Google Scholar]
- 3.Vieira-de-Abreu A, Campbell RA, Weyrich AS, Zimmerman GA. Platelets: versatile effector cells in hemostasis, inflammation, and the immune continuum. Semin Immunopathol. 2012;34(1):5–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cannon JW. Hemorrhagic shock. N Engl J Med. 2018;378(19):1852–1853. [DOI] [PubMed] [Google Scholar]
- 5.Callcut RA, Kornblith LZ, Conroy AS, et al. The why and how our trauma patients die: a prospective Multicenter Western Trauma Association study. J Trauma Acute Care Surg. 2019;86(5):864–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kutcher ME, Redick BJ, McCreery RC, Crane IM, Greenberg MD, Cachola LM, Nelson MF, Cohen MJ. Characterization of platelet dysfunction after trauma. J Trauma Acute Care Surg. 2012;73(1):13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacoby RC, Owings JT, Holmes J, Battistella FD, Gosselin RC, Paglieroni TG. Platelet activation and function after trauma. J Trauma Acute Care Surg. 2001; 51(4):639–647. [DOI] [PubMed] [Google Scholar]
- 8.Li R, Elmongy H, Sims C, Diamond SL. Ex vivo recapitulation of trauma-induced coagulopathy and preliminary assessment of trauma patient platelet function under flow using microfluidic technology. J Trauma Acute Care Surg. 2016;80(3):440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solomon C, Traintinger S, Ziegler B, Hanke A, Rahe-Meyer N, Voelckel W, Schöchl H. Platelet function following trauma. J Thromb Haemost. 2011; 106(8):322–330. [DOI] [PubMed] [Google Scholar]
- 10.Davis PK, Musunuru H, Walsh M, et al. Platelet dysfunction is an early marker for traumatic brain injury-induced coagulopathy. Neurocrit Care. 2013;18(2):201–208. [DOI] [PubMed] [Google Scholar]
- 11.Donahue DL, Beck J, Fritz B, Davis P, Sandoval-Cooper MJ, Thomas SG, Yount RA, Walsh M, Ploplis VA, Castellino FJ. Early platelet dysfunction in a rodent model of blunt traumatic brain injury reflects the acute traumatic coagulopathy found in humans. J Neurotrauma. 2014;31(4):404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sirajuddin S, Valdez C, DePalma L, Maluso P, Singhal R, Schroeder M, Sarani B. Inhibition of platelet function is common following even minor injury. J Trauma Acute Care Surg. 2016;81(2):328–332. [DOI] [PubMed] [Google Scholar]
- 13.Vulliamy P, Gillespie S, Armstrong PC, Allan HE, Warner TD, Brohi K. His-tone H4 induces platelet ballooning and microparticle release during trauma hemorrhage. Proc Natl Acad Sci U S A. 2019;116(35):17444–17449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verni CC, Davila AJr., Balian S, Sims CA, Diamond SL. Platelet dysfunction during trauma involves diverse signaling pathways and an inhibitory activity in patient-derived plasma. J Trauma Acute Care Surg. 2019; 86(2):250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kornblith LZ, Robles AJ, Conroy AS, Hendrickson CM, Calfee CS, Fields AT, Callcut RA, Cohen MJ. Perhaps it’s not the platelet: ristocetin uncovers the potential role of von Willebrand factor in impaired platelet aggregation following traumatic brain injury. J Trauma Acute Care Surg. 2018; 85(5):873–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denis MM, Tolley ND, Bunting M, et al. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell. 2005; 122(3):379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwertz H, Tolley ND, Foulks JM, et al. Signal-dependent splicing of tissue factor pre-mRNA modulates the thrombogenicity of human platelets. J Exp Med. 2006;203(11):2433–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weyrich AS, Lindemann S, Tolley ND, Kraiss LW, Dixon DA, Mahoney TM, Prescott SP, McIntyre TM, Zimmerman GA. Change in protein phenotype without a nucleus: translational control in platelets. Semin Thromb Hemost. 2004;30(4):491–498. [DOI] [PubMed] [Google Scholar]
- 19.Amisten S, Braun OO, Bengtsson A, Erlinge D. Gene expression profiling for the identification of G-protein coupled receptors in human platelets. Thromb Res. 2008;122(1):47–57. [DOI] [PubMed] [Google Scholar]
- 20.Brogren H, Karlsson L, Andersson M, Wang L, Erlinge D, Jern S. Platelets synthesize large amounts of active plasminogen activator inhibitor 1. Blood. 2004;104(13):3943–3948. [DOI] [PubMed] [Google Scholar]
- 21.Warshaw AL, Laster L, Shulman NR. Protein synthesis by human platelets. J Biol Chem. 1967;242(9):2094–2097. [PubMed] [Google Scholar]
- 22.Nassa G, Giurato G, Cimmino G, et al. Splicing of platelet resident premRNAs upon activation by physiological stimuli results in functionally relevant proteome modifications. Sci Rep. 2018;8(1):498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bray PF, McKenzie SE, Edelstein LC, et al. The complex transcriptional landscape of the anucleate human platelet. BMC Genomics. 2013;14:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rondina MT, Weyrich AS. Regulation of the genetic code in megakaryocytes and platelets. J Thromb Haemost. 2015;13(Suppl 1):S26–S32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kutcher ME, Kornblith LZ, Vilardi RF, Redick BJ, Nelson MF, Cohen MJ. The natural history and effect of resuscitation ratio on coagulation after trauma: a prospective cohort study. Ann Surg. 2014;260(6):1103–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohen MJ, Call M, Nelson M, Calfee CS, Esmon CT, Brohi K, Pittet JF. Critical role of activated protein C in early coagulopathy and later organ failure, infection and death in trauma patients. Ann Surg. 2012;255(2):379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kornblith LZ, Kutcher ME, Redick BJ, Calfee CS, Vilardi RF, Cohen MJ. Fibrinogen and platelet contributions to clot formation: implications for trauma resuscitation and thromboprophylaxis. J Trauma Acute Care Surg. 2014; 76(2):255–256; discussion 262–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koh W, Pan W, Gawad C, Fan HC, Kerchner GA, Wyss-Coray T, Blumenfeld YJ, El-Sayed YY, Quake SR. Noninvasive in vivo monitoring of tissue-specific global gene expression in humans. Proc Natl Acad Sci U S A. 2014;111(20):7361–7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Englert JA, Cho MH, Lamb AE, et al. Whole blood RNA sequencing reveals a unique transcriptomic profile in patients with ARDS following hematopoietic stem cell transplantation. Respir Res. 2019;20(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zetterberg H, Smith DH, Blennow K. Biomarkers of mild traumatic brain injury in cerebrospinal fluid and blood. Nat Rev Neurol. 2013;9(4):201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Solomon C, Traintinger S, Ziegler B, Hanke A, Rahe-Meyer N, Voelckel W, Schöchl H. Platelet function following trauma. A multiple electrode aggregometry study. Thromb Haemost. 2011;106(2):322–330. [DOI] [PubMed] [Google Scholar]
- 32.Ding N, Chen G, Hoffman R, Loughran PA, Sodhi CP, Hackam DJ, Billiar TR, Neal MD. Toll-like receptor 4 regulates platelet function and contributes to coagulation abnormality and organ injury in hemorrhagic shock and resuscitation. Circ Cardiovasc Genet. 2014;7(5):615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGhan LJ, Jaroszewski DE. The role of toll-like receptor-4 in the development of multi-organ failure following traumatic haemorrhagic shock and resuscitation. Injury. 2012;43(2):129–136. [DOI] [PubMed] [Google Scholar]
- 34.Kral JB, Schrottmaier WC, Salzmann M, Assinger A. Platelet interaction with innate immune cells. Transfus Med Hemother. 2016;43(2):78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gidlof O, van der Brug M, Ohman J, Gilje P, Olde B, Wahlestedt C, Erlinge D. Platelets activated during myocardial infarction release functional miRNA, which can be taken up by endothelial cells and regulate ICAM1 expression. Blood. 2013;121(19):3908–3917, S1–26. [DOI] [PubMed] [Google Scholar]
- 36.Tran JQD, Pedersen OH, Larsen ML, Grove EL, Kristensen SD, Hvas AM, Nissen PH. Platelet microRNA expression and association with platelet maturity and function in patients with essential thrombocythemia. Platelets. 2020;31(3):365–372. [DOI] [PubMed] [Google Scholar]
- 37.Sol N, Wurdinger T. Platelet RNA signatures for the detection of cancer. Cancer Metastasis Rev. 2017;36(2):263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu L, Chen M, Zhao L, Zhao Q, Hu R, Zhu J, Yan R, Dai K. Ethanol induces platelet apoptosis. Alcohol Clin Exp Res. 2017;41(2):291–298. [DOI] [PubMed] [Google Scholar]
- 39.Fehr M, Galliard-Grigioni KS, Reinhart WH. Influence of acute alcohol exposure on hemorheological parameters and platelet function in vivo and in vitro. Clin Hemorheol Microcirc. 2008;39(1–4):351–358. [PubMed] [Google Scholar]
- 40.Dunn EL, Cohen RG, Moore EE, Hamstra RD. Acute alcohol ingestion and platelet function. Arch Surg. 1981;116(8):1082–1083. [DOI] [PubMed] [Google Scholar]
- 41.Ribizzi G, Farinini D, Gentile R, Rizzi D, Serrati C. Factor XIII deficiency and head trauma: management and therapy. Neurol Sci. 2015;36(10):1933–1934. [DOI] [PubMed] [Google Scholar]
- 42.Poon MC, Russell JA, Low S, Sinclair GD, Jones AR, Blahey W, Ruether BA, Hoar DI. Hemopoietic origin of factor XIII a subunits in platelets, monocytes, and plasma. Evidence from bone marrow transplantation studies. J Clin Invest. 1989;84(3):787–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katona E, Penzes K, Csapo A, Fazakas F, Udvardy ML, Bagoly Z, Orosz ZZ, Muszbek L. Interaction of factor XIII subunits. Blood. 2014;123(11):1757–1763. [DOI] [PubMed] [Google Scholar]
- 44.Radek JT, Jeong JM, Wilson J, Lorand L. Association of the a subunits of recombinant placental factor XIII with the native carrier B subunits from human plasma. Biochemistry. 1993;32(14):3527–3534. [DOI] [PubMed] [Google Scholar]