

SUMMARY
Diverse accessory subunits are involved in the recruitment of Polycomb Repressive Complex 2 (PRC2) to CpG island (CGI) chromatin. Here we report the crystal structure of a SUZ12-RBBP4 complex bound to fragments of the accessory subunits PHF19 and JARID2. Unexpectedly, this complex adopts a dimeric structural architecture, accounting for PRC2 self-association that has long been implicated. The intrinsic PRC2 dimer is formed via domain swapping, involving RBBP4 and the unique C2 domain of SUZ12. MTF2 and PHF19 associate with PRC2 at around the dimer interface and stabilize the dimer. Conversely, AEBP2 binding results in a drastic movement of the C2 domain, disrupting the intrinsic PRC2 dimer. PRC2 dimerization enhances CGI DNA binding by PCLs in pairs in vitro, reminiscent of the widespread phenomenon of transcription factor dimerization in active transcription. Loss of PRC2 dimerization impairs histone H3K27 trimethylation (H3K27me3) on chromatin at developmental gene loci in mouse embryonic stem cells.
eTOC Blurb
PRC2 dimerization has long been noted, with unclear functions. Chen S. et al. revealed the structural basis of PRC2 dimerization, in the context of PRC2-PCL (PRC2.1), which is drastically different from PRC2-AEBP2 (PRC2.2) in structural architecture. PRC2 dimerization controls CGI DNA binding in vitro and H3K27me3 deposition in vivo, possibly through an avidity effect.
Graphical Abstarct
INTRODUCTION
Polycomb group (PcG) proteins maintain epigenetic memory of cell identity, a fundamental biological phenomenon in which the cell type-specific gene expression pattern is stably inherited through cell division (Steffen and Ringrose, 2014). PcG proteins assemble into diverse repressive chromatin complexes with distinct enzyme activities for histone modification. Polycomb Repressive Complex 2 (PRC2) mediates trimethylation of histone H3 lysine 27 (H3K27me3), hallmark of gene repression (Schuettengruber et al., 2017).
PRC2 function is subjected to intricate cellular regulation, which correlates with the tremendous compositional complexity of PRC2 (Schuettengruber et al., 2017). The mammalian paralogs EZH1 and EZH2 contain a SET (Su(var)3–9, Enhancer-of-zeste and Trithorax) domain for lysine methylation and serve as the catalytic subunit of PRC2. Besides EZH1/2, other core subunits include EED, SUZ12 and RBBP4/7. Notably, H3K27me3 is recognized by both the aromatic cage of EED and the Stimulation-Responsive Motif (SRM) of EZH2 to confer allosteric stimulation of PRC2 (Jiao and Liu, 2015; Justin et al., 2016; Margueron et al., 2009), which is partially responsible for heritable gene repression in addition to other mechanisms (Coleman and Struhl, 2017; Hansen et al., 2008; Hojfeldt et al., 2018; Laprell et al., 2017; Margueron et al., 2009). An increasing number of the accessory subunits of PRC2 are being identified (Vizan et al., 2015); many of them are members of distinct PRC2 holo complexes. At least two classes of PRC2 holo complexes exist in cells depending on the mutually exclusive binding of the accessory subunits to the PRC2 core complex: EPOP, PALI1/2 and polycomb-like (PCL) proteins, including PHF1 (a.k.a. PCL1), MTF2 (a.k.a. PCL2) and PHF19 (a.k.a. PCL3), belong to one class (PRC2.1); JARID2 and AEBP2 are present in the other (PRC2.2) (Conway et al., 2018; Grijzenhout et al., 2016). Atypical hybrid PRC2 holo complexes containing both PCLs and JARID2 may also form in the absence of AEBP2 (Grijzenhout et al., 2016). While loss of PCLs or JARID2 impairs chromatin targeting of PRC2 in vivo (Schuettengruber et al., 2017), JARID2 and AEBP2 facilitate PRC2 binding to nucleosomes in vitro (Chen et al., 2018; Lee et al., 2018; Son et al., 2013; Wang et al., 2017). Many of the accessory subunits are known to play a critical, pleiotropic role in embryonic development (Vizan et al., 2015).
PRC2 binding to cis-acting DNA elements is indispensible for epigenetic inheritance (Coleman and Struhl, 2017; Laprell et al., 2017). PRC2 binding sites correlate with hypomethylated CpG islands (CGIs) in mammalian cells (Deaton and Bird, 2011). Histone methylation by PRC2 and CGI methylation by DNA methyltransferases (DNMTs) represent two different, yet interconnected cellular mechanisms for maintaining gene repression (Deaton and Bird, 2011). Inhibition of transcription triggers the de novo recruitment of PRC2 to CGIs that are intrinsically depleted of nucleosomes (Riising et al., 2014). GC-rich content in CGI promoters is also recognized by numerous transcription factors, a genomic feature possibly underlying the competition between PRC2 targeting and active transcription (Deaton and Bird, 2011; Wang et al., 2012). Notably, both endogenous and synthetic CGIs that lack DNA methylation and transcription factor binding are sufficient for PRC2 targeting in vivo (Jermann et al., 2014; Mendenhall et al., 2010; Wachter et al., 2014). Nonetheless, how PRC2 is recruited to specific CGIs in mammalian cells remains a long-standing question in the field.
Recent progress in structural biology has greatly advanced the mechanistic understanding of function and regulation of PRC2. In particular, the structures of the PRC2 core and subcomplexes bound to fragments of JARID2 and AEBP2 reveal a central role of SUZ12 in organizing the structural architecture of the holo complexes: the C-terminal VEFS (VRN2-EMF2-FIS2-Su(z)12) domain of SUZ12 (SUZ12(VEFS)) acts as an integral component of the minimal catalytic module, EZH2-EED-SUZ12(VEFS) (Jiao and Liu, 2015; Justin et al., 2016); the N-terminal region of SUZ12 (SUZ12(N)) together with RBBP4 provides binding surfaces for JARID2 and AEBP2 (Chen et al., 2018; Kasinath et al., 2018), which overlap with those for EPOP and PCLs, respectively (Chen et al., 2018). The structure of PCL-containing PRC2 is unknown, although functional insights have started to emerge from the structures of the Tudor and Extended Homologous (EH) domains of PCLs. While the Tudor domain can link PRC2 to chromatin by recognizing the active H3K36me3 histone mark (Ballare et al., 2012; Cai et al., 2013; Musselman et al., 2012), the EH domain enhances PRC2 binding to chromatin through a direct interaction with the linker DNA (Choi et al., 2017; Li et al., 2017; Perino et al., 2018).
Here we report that the PRC2-PCL holo complex adopts an intrinsic dimeric structural architecture mediated by SUZ12 and RBBP4. SUZ12 undergoes a dramatic conformational change in switching between PCL-bound and AEBP2-bound states in the two distinct classes of the holo complexes. Disruption of the PRC2 dimer weakens CGI DNA binding in vitro and impairs H3K27me3 deposition on chromatin in vivo in mouse embryonic stem cells (mESCs). Our results provide insights into the chromatin recruitment of PRC2-PCL achieved based on this dimeric structural framework: PCLs stabilize the dimeric state of the PRC2 core complex, which reciprocally enhances the binding of PCLs to CGI DNAs in pairs for gene repression, reminiscent of DNA binding by many dimeric transcription factors in active transcription.
RESULTS
Overall Crystal Structure of a PRC2-PHF19 Complex
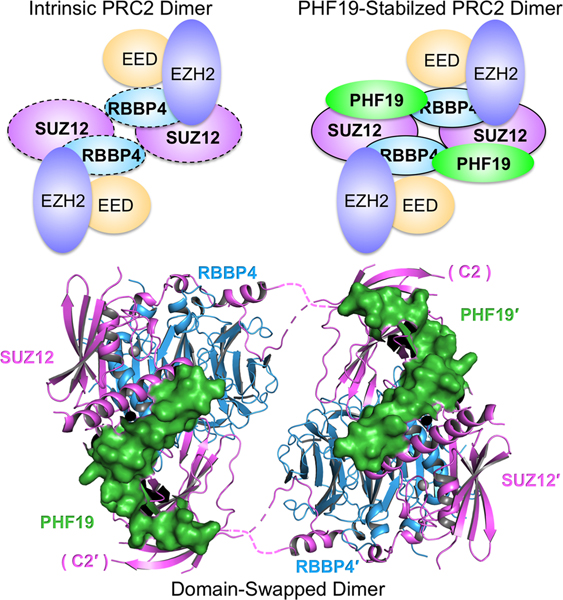
We have previously determined the crystal structure of a SUZ12(N)-RBBP4 binary complex bound to fragments of AEBP2 and JARID2, which adopts a monomeric structural architecture (Chen et al., 2018). We now present the crystal structure of a related complex containing PHF19 in place of AEBP2, which unexpectedly assembles into a dimer (Fig. 1A, 1B and 1C and Table 1). The existence of a PRC2 dimer in Drosophila, mice and humans has long been implicated in literatures primarily based on size-exclusion chromatography (SEC) in the context of nuclear extracts (Ballare et al., 2012; Casanova et al., 2011; Grijzenhout et al., 2016; Margueron et al., 2008; O’Connell et al., 2001; Tie et al., 2003). In addition, EZH2 was found to be present in EZH1-containing PRC2 when both proteins were coexpressed with other core subunits in Sf9 insect cells, suggesting self-association of PRC2 (Son et al., 2013). Fully purified PRC2 also behaved as a dimer in the previous SEC, co-immunoprecipitation (Co-IP) and SEC-MALS (Multi-Angle Light Scattering) experiments (Davidovich et al., 2014). The current study provided structural evidence for dimerization of the PRC2 core complex and the PRC2-PHF19 holo complex.
Fig. 1. Overall structure of the SUZ12(N)-RBBP4-PHF19(RC)-JARID2(TR) heterotetrameric complex in the dimeric state.

(A) Domain structures of the proteins in the crystal. ZnB, Zinc-finger Binding; WDB1, WD40-Binding 1; Zn, Zinc-finger; WDB2, WD40-Binding 2; NT, N-Terminal; DS, Dimer Stabilization; SC, Short Connecting; C2B, C2-Binding; CT, C-Terminal; TR, Transrepression. C2 and βL7 are not abbreviated names.
(B) Cartoon representation of the dimeric complex in Top View. Disordered protein loops are displayed as dotted lines. Hinge loops that connect to the C2 domains are indicated by black arrows. The second protomer of the dimer is labeled with a prime symbol.
(C) Surface representation of the dimeric complex in Side View. The rotational matrix that relates the Top View and Side View is indicated.
See also Fig. S1.
Table 1.
Diffraction data collection and structure refinement statistics
| Crystal | SUZ12-RBBP4-PHF19-JARID2 |
|---|---|
| Diffraction data | |
| Wavelength (Å) | 0.97946 |
| Space group | C2221 |
| a, b, c (Å) | 127.7, 139.6, 268.1 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| Resolution range (Å) | 50.00 – 2.90 (2.95 – 2.90) |
| Rsym or Rmerge | 0.138 (1.515) |
| Mean I/σ (I) | 17.3 (1.0) |
| Rpim (%) | 4.9 (64.7) |
| CC1/2 (%) | 85.4(57.1) |
| Completeness (%) | 98.9(89.0) |
| Redundancy | 8.0 (5.4) |
| Refinement | |
| Bumber of reflections | 44020 |
| Rwork/Rfree | 0.18/0.23 |
| Number of non-hydrogen atoms | 11807 |
| Suz12 | 4645 |
| Rbbp4 | 6083 |
| Phf19 | 803 |
| Jarid2 | 282 |
| Ligand (zinc ion) | 2 |
| Protein residues | 1454 |
| Average B-factor (Å2) | 67.57 |
| Suz12 | 70.1 |
| Rbbp4 | 47.6 |
| Phf19 | 85.5 |
| Jarid2 | 57.5 |
| Ligand (zinc ion) | 41.7 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.009 |
| Bond angles (º) | 1.12 |
| Ramachandran | |
| Favored (%) | 95.90 |
| Allowed (%) | 4.10 |
| Outliers (%) | 0.00 |
Statistics for the highest-resolution shell are shown in parentheses.
We will introduce the overall structure of the dimeric complex first and provide the detailed structure-function relationship in the sections that follow. The “reversed chromodomain” (RC) of PHF19 (PHF19(RC)) is included in the current structure, and it was previously shown to compete with AEBP2 for SUZ12 binding in the different classes of the PRC2 holo complexes (Fig. 1A) (Chen et al., 2018). We also used a peptide corresponding to the transrepression (TR) domain of JARID2 (JARID2(TR); residues 147–165) that appeared to improve the crystal quality for structure determination (Fig. 1A). Although a PRC2 holo complex with both PCLs and JARID2 has so far only been identified in cells that lack AEBP2 (Grijzenhout et al., 2016), the JARID2(TR) was unlikely to change the structure of SUZ12(N)-RBBP4-PHF19(RC). Indeed, the JARID2(TR) occupies essentially the same surface of SUZ12 as shown in the previous AEBP2 and JARID2-containing crystal structure (Chen et al., 2018), with little crosstalk with the PHF19(RC) (Fig. S1A). In this paper, we will not discuss JARID2 binding any further. PCLs and EPOP belong to the same class of PRC2 holo complex, PRC2.1. We showed previously that the C-Terminal domain of EPOP (residues 285–379) (EPOP(CT)) competed with the JARID2(TR) for SUZ12(N)-RBBP4 binding (Chen et al., 2018; Liefke and Shi, 2015). We also reconstituted SUZ12(N)-RBBP4-PHF19(RC)-EPOP(CT) (Fig. S1B), but this specific complex did not produce crystals.
Single-wavelength anomalous dispersion (SAD) signals of sulfur atoms were extensively used to facilitate model building and were critical for confirming the registers. Extensive efforts were invested to collect a variety of sulfur SAD datasets, which clearly marked the locations of eleven endogenous cysteine and methionine residues on SUZ12, four designed methionine mutants on SUZ12 and one endogenous cysteine residue on PHF19 (Fig. S1C).
Overall, the dimer is formed by two symmetry-related protomers correlated by a 2-fold rotational axis (Fig. 1B and 1C). Each protomer contains a pair of flexible polypeptide hinge loops (Fig. 1B). The dimer interface is located between RBBP4 and the C2 domain of SUZ12 (SUZ12(C2)), which was previously named based on the structural homology to the canonical phospholipid-binding C2 domain (Chen et al., 2018). The PHF19(RC) is not involved in dimerization directly, but rather appears to stabilize the PRC2 dimer by simultaneously binding to and thus “gluing” together the SUZ12(C2) from one protomer and the main SUZ12(N)-RBBP4 protein body from the other (Fig. 1B and 1C).
The PRC2 core complex forms an intrinsic dimer through domain swapping
Domain swapping is a widespread mechanism for oligomer assembly (Liu and Eisenberg, 2002). A prominent feature of the PHF19(RC)-bound SUZ12(N)-RBBP4 complex is formation of a domain-swapped dimer between the core subunits (Fig. 1B). Each protomer exchanges the C2 domain of SUZ12 to interact with RBBP4 from the reciprocal protomer (Fig. 1B). The hinge loops that connect to the C2 domain appear to be too short to allow formation of a closed monomer with the same set of interactions with RBBP4 as observed in the dimer (Fig. 2A). On the other hand, the working length of the hinge loops can be extended due to melting of local secondary structures, which may lead to greater flexibility between the two protomers than observed in the crystal structure.
Fig. 2. Dimerization of the PRC2 core complex.
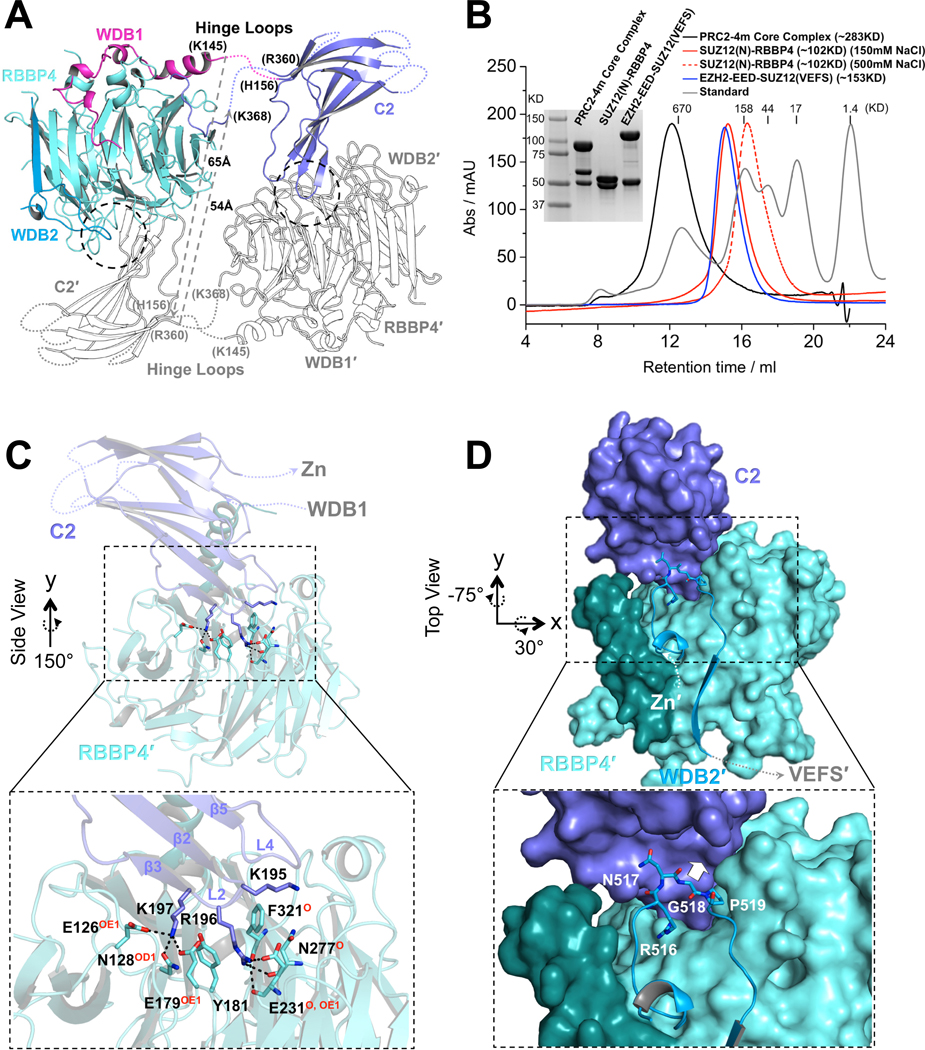
(A) Structure of one protomer of the PRC2 core complex in cartoon representation. Only SUZ12(N) and RBBP4 are shown. Domains within only one protomer are colored. The SUZ12(ZnB) and SUZ12(Zn) domains are removed for clarity. SUZ12 residues at the junction of the disordered hinge loops are labeled. The minimal lengths of a pair of hypothetical hinge loops between the WDB1 domain from one protomer and the C2 domain from the other protomer that would exist in a closed complex are indicated by dotted gray lines.
(B) SEC elution profiles of the four-member PRC2 core complex (PRC2–4m), SUZ12(N)-RBBP4 (at both 150mM and 500mM salt), and EZH2-EED-SUZ12(VEFS). SDS-PAGE gel image is provided. In the last complex, EZH2 and SUZ12(VEFS) were expressed as a fusion protein.
(C) Zoom-in view of the dimer interface between the SUZ12(C2) domain and RBBP4. SUZ12(C2) residues on the dimer interface are shown as sticks. RBBP4 residues involved in SUZ12(C2) binding are indicated, with hydrogen bonding atoms highlighted in red.
(D) Zoom in view of the dimer interface between the SUZ12(C2) and SUZ12(WDB2) domains. Steric clash to the SUZ12(C2) would be imposed by bulky amino acids at residue G518 and is indicated by a white arrow.
See also Fig. S2.
In line with the crystal structure, we noted that the purified full-length four-member PRC2 core complex (EZH2-EED-SUZ12-RBBP4; also referred to as ‘PRC2–4m’ below) ran like a dimer on a SEC column as reported previously (Davidovich et al., 2014). The EZH2-EED-SUZ12(VEFS)) moiety and the SUZ12(N)-RBBP4 moiety that together compose the entire PRC2 core complex behaved as a monomer and a dimer, respectively. Moreover, the dimeric SUZ12(N)-RBBP4 moiety was converted into a monomer in the presence of 500mM NaCl (Fig. 2B). Together, these data indicated that SUZ12(N)-RBBP4 might contribute to dimerization of the PRC2 core complex in solution (Fig. 2B).
Both SUZ12 and RBBP4 appear to be indispensible for PRC2 dimerization. A main dimer interface is located around the overall electronegative central cavity of the WD40 domain of RBBP4 and involves three positively charged residues, K195, R196 and K197, at the tip of the L2 loop of the SUZ12(C2) domain (Fig. 2C and S2A). In particular, residue R196 is sandwiched by two aromatic residues, Y181 and F321, from inside the central cavity of RBBP4 and is stabilized by combined hydrophobic, cation-π, charge-charge and hydrogen-bonding interactions (Fig. 2C). Residue K197 is captured by additional interactions on the top surface of RBBP4 (Fig. 2C). Furthermore, a well-structured loop within the second WD40-Binding domain of SUZ12 (SUZ12(WDB2)) fits into a concave surface formed by the C2 domain, likely contributing to the dimer formation as well (Fig. 2D). In particular, residue G518 at the tip of the structured loop occupies the binding interface, such that bulky residues at this position would clash with the C2 domain (Fig. 2D). Key residues for dimerization revealed by the structure will be examined by mutagenesis in the next section in the contexts of both the core and holo complexes.
Dimerization is an intrinsic property of the PRC2 core complex. The dimer interface on SUZ12 and RBBP4 is highly conserved between humans and Drosophila (Fig. S2B), indicating that the Drosophila PRC2 core complex may adopt a dimeric structure as well through a similar set of molecular interactions. In addition, the dimer interface on RBBP4 largely overlaps with the binding sites for other cellular factors, including FOG1, PHF6, AEBP2 and the unmethylated histone H3K4 tail (Chen et al., 2018; Kasinath et al., 2018; Lejon et al., 2011; Liu et al., 2015; Schmitges et al., 2011), raising the possibility that the PRC2 dimer may be subjected to disruption by competitive binding in cells.
PHF19 stabilizes the intrinsic PRC2 dimer
The PHF19(RC) domain contains four structural regions, including a Dimer Stabilization (DS) helix, a Short Connecting (SC) helix, a C2-Binding (C2B) domain and a C-Terminal (CT) tail. Notably, the PHF19(DS) helix is conserved in MTF2 and Drosophila PCL but not in PHF1 (Fig. 3A), suggestive of functional divergence of PHF1.
Fig. 3. Stabilization of the intrinsic PRC2 dimer by PHF19.

(A) Sequence alignment of the RC domains of PHF19, MTF2, PHF1 and Drosophila PCL. Functional domains are indicated. The arginine and leucine residues that form the R-W-L triad with residue W334 of the SUZ12(C2) are indicated by filled circles, and the tryptophan residue that forms a hydrogen bond with residue T333 of the SUZ12(C2) is indicated by a filled triangle.
(B) Interactions of the DS helix of PHF19 with the ZnB helix of SUZ12 and the NT helix of RBBP4. Residues mediating hydrophobic and hydrogen bonding interactions are displayed as sticks.
(C) Interactions of the SUZ12(C2), PHF19(C2B) and PHF19(CT) domains. The DS and SC domains of PHF19 are removed from the view for clarity.
(D) Stabilization of the SUZ12(N)-RBBP4 dimer by the PHF19(RC). Co-IP was used to assess the dimer formation. In (D) and (F), equal amounts of the SUZ12(N)-RBBP4 binary complex containing both FLAG-SUZ12(N) and HA-SUZ12(N) were bound to anti-FLAG resin. Anti-FLAG signals served as input control. HA-SUZ12 bound via protein dimerization was assessed by anti-HA antibody. Formation of the intrinsic dimer is indicated by the control lane.
(E) Dimer stabilization and disruption of the four-member PRC2 core complex (PRC2–4m). Different from (D) and (F), equal amounts of PRC2–4m containing both FLAG-EZH2 and Myc-EZH2 were used in (E) and (H). While anti-FLAG signals served as input control, anti-Myc signals indicated the extent of PRC2 dimerization.
(F) Critical role of the DS helix of PHF19 in dimer stabilization. PHF19(RC) corresponding to residues 500–580 was used for crystallization. PHF19 (residues 531–580) is visible in the crystal structure. PHF19(residues 544–580) lacks the DS helix.
(G) Differential dimer stabilization activities of the RC domains of PHF19, MTF2 and PHF1.
(H) Effect of SUZ12 mutations, SUZ123D, SUZ12R196A, SUZ12W334A and SUZ12G518W, on the intrinsic and PHF19-stabilzied dimers of PRC2–4m. Anti-FLAG signals served as input control and anti-Myc signals represented the extent of PRC2 dimerization.
(I) Summary of the structural mechanism of the defect in PRC2 dimerization caused by SUZ12 mutations.
See also Fig. S3.
Extensive interactions between SUZ12(N)-RBBP4 and the PHF19(RC) underlie the regulation of PRC2 function by PHF19. First, the PHF19(DS) helix packs against two other helices, including the Zinc-finger Binding (ZnB) helix of SUZ12 (SUZ12(ZnB)) and N-Terminal (NT) helix of RBBP4 (RBBP4(NT)) (Fig. 1A and 3B). Second, the PHF19(C2B) domain that consists of the antiparallel β1 and β2 strands is laterally added to the β7 strand of the C2 domain of SUZ12 (Fig. 3C). Notably, the conserved L7 loop of the C2 domain that is disordered in the previous AEBP2-bound structure is now partially folded into a β strand, designated as βL7, which structurally locks the PHF19(C2B) onto the SUZ12(C2) (Fig. 3C and S2A). Residue W334 on βL7 is at the center of the “lock”: it forms an R-W-L triad with residues R561 and L571 from the β1 and β2 strands of the PHF19(C2B), respectively, mediating cation-π and hydrophobic interactions (Fig. 3C). Finally, the PHF19(CT) tail fits into a binding cleft formed by the structured loop of the SUZ12(WDB2) (Fig. 3C).
Overall, the PRC2-PHF19 complex displays a 2:2 binding stoichiometry. While the PHF19(DS) and PHF19(CT) are bound to the SUZ12(N)-RBBP4 protein body of one protomer, the PHF19(C2B) is associated with the SUZ12(C2) of the other protomer (Fig. 1B). In this way, PHF19 ties together the two protomers and stabilizes the dimeric structure of SUZ12(N)-RBBP4 (Fig. 1B).
To study dimer stabilization in solution, we first confirmed that the dimer of SUZ12(N)-RBBP4 was stabilized by the PHF19(RC) by Co-IP (Fig. 3D), as observed in the crystal structure. In the Co-IP assay, equal amounts of the SUZ12(N)-RBBP4 binary complex containing both FLAG-SUZ12(N) and HA-SUZ12(N) were bound to anti-FLAG resin. HA-SUZ12 bound via protein dimerization was assessed by anti-HA antibody, while anti-FLAG signals served as input control. In contrast to the PHF19(RC), a polypeptide containing the C2-Binding domain and the H3K4 Displacement domain of AEBP2 (AEBP2(C2B-H3K4D)) disrupted the dimer (Fig. 3D and S1A), agreeing with the monomeric structure of the AEBP2-containing PRC2 complexes (Chen et al., 2018; Kasinath et al., 2018). In line with these results, SUZ12(N)-RBBP4 behaved as a dimer and a monomer on a SEC column, when bound to the PHF19(RC) and the AEBP2(C2B-H3K4D), respectively (Fig. S3A). The PHF19(RC) was indeed sufficient to stabilize the dimer of PRC2–4m (Fig. 3E). Additionally, the dimer stabilization activity of the PHF19(RC) was lost upon deletion of the PHF19(DS) helix (Fig. 3F), while SUZ12(N)-RBBP4 binding remained unaffected (Fig. S3B). Similarly, the PHF1(RC) only marginally stabilized the dimer, likely due to the lack of the dimer stabilization helix found in other PCLs (Fig. 3A and 3G). MTF2-mediated stabilization and AEBP2-mediated disruption of the intrinsic PRC2 dimer were also validated by Co-IP in the context of the full complexes, which were transiently expressed in HEK293T cells (Fig. S3C). Consistently, the purified PRC2-MTF2 (~360 kDa) and PRC2-AEBP2 (~320 kDa) holo complexes behaved like a dimer and a monomer on a SEC column, respectively (Fig. S3D).
PRC2 dimerization is directly mediated by SUZ12 residues; the current structure allowed us to design a panel of SUZ12 mutations that specifically disrupt the dimer in solution. Residues K195, R196 and K197 of the SUZ12(C2) are located on the dimer interface (Fig. 2C). PRC2–4m harboring the K195D/R196D/K197D triple mutation of SUZ12 (SUZ123D) did not form the dimer in the Co-IP assay; furthermore, the dimer stabilization activity of the PHF19(RC) was unable to rescue the dimerization defect (Fig. 3H, 3I, S3E). In fact, the less dramatic R196A single mutation of SUZ12 (SUZ12R196A) was sufficient to disrupt the PRC2 dimer (Fig. 3H, 3I and S3E). Different from these two mutants, the intrinsic PRC2 dimer stayed intact in the presence of the G518W mutation of SUZ12 (SUZ12G518W) (Fig. 2D); however, PHF19(RC)-mediated dimer stabilization was diminished, likely owing to the steric hindrance on the dimer interface in the PHF19-bound state (Fig. 3H and 3I and S3E). Finally, residue W334 on the βL7 strand of the SUZ12(C2) locks the PHF19(RC) (Fig. 3C), and we found that the W334A mutation of SUZ12 (SUZ12W334A) disrupted PHF19 binding to PRC2 (Fig. S3F). In this case, although the intrinsic PRC2 dimer was not disturbed, the PHF19(RC) could not stabilize the dimer any more due to the lack of physical binding (Fig. 3H, 3I, S3E and S3F).
Together, we were able to categorize these SUZ12 mutants into three classes of separation-of-function mutants, each corresponding to a distinct structural mechanism of PRC2 dimerization defect: SUZ123D and SUZ12R196A disrupted both the intrinsic and PHF19-stabilized PRC2 dimers, without compromising PHF19 binding (Fig. 3I); SUZ12G518W was incompatible with the PHF19-bound dimer, leaving the intrinsic dimer intact and PHF19 binding unaffected (Fig. 3I); SUZ12W334A did not affect the intrinsic dimer, but it lost PHF19 binding and abolished dimer stabilization (Fig. 3I). We expect that these mechanistically distinct SUZ12 mutations can serve as a useful tool for dissecting the role of PRC2 dimerization in gene repression and epigenetic inheritance in vivo.
PRC2-PHF19 and PRC2-AEBP2 are drastically different in structural organization
PRC2-PHF19 and PRC2-AEBP2 represent two classes of the PRC2 holo complexes that are mutually exclusive. Different structural architectures can dictate how the holo complexes engage chromatin. The structural models of AEBP2 largely differ between the recent crystal structure and cryo-EM structure (Chen et al., 2018; Kasinath et al., 2018). Towards a more complete understanding of the structural architecture of PRC2-AEBP2, we fitted the crystal structure into the cryo-EM electron density map, which allowed us to extend the crystal structure with an α helix located in the center of the holo complex and connecting the N-terminal zinc finger to the C2B domain of AEBP2 (Fig. S4A). The exact register of the central connecting helix of AEBP2 (AEBP2(CC)) could not be determined unambiguously based on the published data, although we estimated that it spans residues 400–415 of AEBP2 (AEBP2isoform 1, Q6ZN18–1) (Fig. S4A).
Aligning the latest structural model of PRC2-AEBP2 constructed above to the current dimeric structure of SUZ12(N)-RBBP4-PHF19(RC) generated a structural model of PRC2-PHF19 in the dimeric state (Fig. 4A). Our previous biochemical data suggested that both PHF19 and AEBP2 bound to the SUZ12(C2) domain, which might at least in part account for the mutual exclusivity of these two accessory subunits (Chen et al., 2018). The current structure confirmed this model directly. In addition, structural alignment between SUZ12(N)-RBBP4-PHF19(RC) and PRC2-AEBP2 indicated that the PHF19(DS) and AEBP2(CC) helices occupy overlapped PRC2 surfaces, which may also contribute to the mutual exclusivity (Fig. 4A and 4B).
Fig. 4. Distinct structural architectures of the PRC2-PCL and PRC2-AEBP2 complexes.

(A) Structure of PRC2-AEBP2 was constructed by fitting the crystal structures of EZH2-EED-SUZ12(VEFS) (PDB: 5HYN) and SUZ12(N)-RBBP4-AEBP2(C2B-H3K4D) (PDB: 5WAI) into a cryo-EM density map of the corresponding holo complex (EMD-7334). To model the PRC2-PHF19 dimer, PRC2-AEBP2 was structurally aligned to the dimeric SUZ12(N)-RBBP4-PHF19(RC). The two C2 domains in PRC2-PHF19 are colored in purple and labeled as C2PHF19 and C2′ PHF19 (other parts of this protomer are shown as outlines for clarity). The C2 domain PRC2-AEBP2, C2AEBP2, is colored in orange, the AEBP2(C2B-H3K4D) in magenta, and the PHF19(RC) in green.
The black arrow pointing from C2PHF19 to C2AEBP2 indicates the movement of the C2 domain induced by AEBP2 binding that disrupts the intrinsic PRC2 dimer, along the hinge loops indicated by a black arrowhead. The dotted red oval indicates that the C2 domain in PRC2-PHF19 would clash with the EZH2-EED-SUZ12(VEFS) moiety from PRC2-AEBP2. To avoid the steric clash, the EZH2-EED-SUZ12(VEFS) moiety may move along the linker (red arrowhead) between the SUZ12(N) and SUZ12(VEFS) as indicated by the dotted red arrow (and the dotted gray arrow in the other protomer). The dotted black rectangle indicates the overlapped binding surface for the PHF19(DS) and AEBP2(CC) helices.
(B) Zoom-in view of the structural alignment of the SUZ12(C2)-PHF19(RC) and SUZ12(C2)-AEBP2(C2B-H3K4D) interactions. The polarities of the PHF19(DS) and AEBP2(CC) helices are opposite. The black double-headed arrow indicates that the position of the C2 domain of SUZ12 is completely different in PRC2-PHF19 and PRC2-AEBP2.
See also Fig. S4.
Further analysis highlighted several structural features that explained how the drastically different architectures of the PRC2-PHF19 and PRC2-AEBP2 holo complexes arise and might result in functional consequences (Fig. 4A and 4B). First, the PHF19(DS) and AEBP2(CC) helices point to opposite directions on the SUZ12(N)-RBBP4 protein body, placing their respective C2-Binding domains and the bound SUZ12(C2) domains in completely different locations (Fig. 4B). Second, the SUZ12(C2) in PRC2-PHF19 would clash with the EZH2-EED-SUZ12(VEFS) moiety from PRC2-AEBP2 (Fig. 4A), suggesting this moiety might be relocated relative to the SUZ12(N)-RBBP4 moiety to accommodate the SUZ12(C2) in PRC2-PHF19 (Fig. 4A). Finally, the remarkable structural difference between PRC2-AEBP2 and PRC2-PHF19 is also accompanied by disruption and stabilization of the intrinsic PRC2 dimer, respectively, which we speculate to dictate distinct structural modes of CGI chromatin binding.
Different versions of AEBP2 were used and correlated with different oligomerization states of PRC2-AEBP2 observed in some previous studies (Cao and Zhang, 2004; Chen et al., 2018; Davidovich et al., 2014; Grijzenhout et al., 2016; Kasinath et al., 2018). For a direct comparison, we purified three representative PRC2-AEBP2 holo complexes, containing AEBP2isoform 1 (Q6ZN18–1, residues 1–517), AEBP2isoform 3 (Q6ZN18–3, residues 1–301), or AEBP2partial (NP_694939.1, residues 1–295). AEBP2partial closely resembles AEBP2isoform 3 in protein length and sequence (Fig. S4B); in comparison, AEBP2isoform 1 also contains a unique, intrinsically disordered N-terminal region of over 200 amino acids with several glutamate/aspartate-rich, serine-rich and glycine-rich patches (Fig. S4B). All the three versions of AEBP2 contain an identical C2B-H3K4D region that was sufficient to disrupt the intrinsic PRC2 dimer (Fig. 3H and S4B).
While PRC2-AEBP2isoform 1 behaved like a dimer in SEC as reported (Fig. S4C) (Davidovich et al., 2014; Grijzenhout et al., 2016), PRC2-AEBP2partial and PRC2-AEBP2 isoform 3 were eluted in later fractions probably as monomers (Fig. S4C) (Cao and Zhang, 2004; Chen et al., 2018; Kasinath et al., 2018). We noted that excessive negative charges within the unique N-terminal region of AEBP2isoform 1 might abnormally increase the apparent hydrodynamic radius of PRC2-AEBP2isoform 1 on the SEC column (Fig. S4B and S4C) (Marsh and Forman-Kay, 2010). Indeed, similar to AEBP2partial, AEBP2isoform 1 abolished PRC2 dimerization under our assay condition, when the holo complex was transiently expressed in HEK293T cells (Fig. S4D), likely through the structural rearrangement of the intrinsic PRC2 dimer induced by the AEBP2(C2B-H3K4D) region as discussed above (Fig. 4A and 4B). Future structural study of PRC2-AEBP2isoform 1 resolving the unique N-terminal region of AEBP2isoform 1 is needed to provide a clearer picture of this holo complex.
Dimerization is required for the stable binding of PRC2-PCL to CGI DNAs
Although the EZH1-containing PRC2 core complex exhibited nucleosome-binding activity, the EZH2-containing counterpart in the absence of the accessory subunits displayed weak affinity for nucleosomes (Son et al., 2013). The EH domains of PCLs were previously shown to interact with the linker DNA and facilitate PRC2 binding to CGI chromatin (Choi et al., 2017; Li et al., 2017). An efficient mechanism to enhance DNA/chromatin binding is to increase binding avidity through protein dimerization. In this regard, CGI DNA binding by PCLs may be promoted due to the dimeric structural platform provided by the PRC2 core complex. We next studied the binding of PRC2-MTF2 to a natural mouse CGI DNA from the LHX6 gene (referred as CGILHX6 below) that was sufficient to recruit PRC2 at both endogenous and ectopic loci in mESCs (Jermann et al., 2014).
MTF2 is highly expressed and involved in PRC2 targeting in mESCs (Casanova et al., 2011; Zhang et al., 2011). We used the reconstituted PRC2-MTF2 holo complex for the electrophoretic mobility shift assay (EMSA) (Fig. 5A). PRC2-MTF2 bound tightly to a piece of 100 bp CGILHX6 DNA with a binding affinity of 6.3 nM. Notably, the binding affinity was decreased by only about 2 folds to 15.0 nM in the presence of 200 ng/μl competitor yeast tRNA, which translates into an at least 10,000-fold molar concentration compared to the 32P-labeled CGILHX6 DNA probe (Fig. 5B, 5C and 5D). PRC2-MTF2 bound poorly to an AT-rich DNA probe and to single-stranded CGILHX6 DNA probes, confirming the specificity of the EMSA (Fig. S5A and S5B).
Fig. 5. Determining role of PRC2 dimerization in DNA binding by PRC2-PCL.

(A) SDS-PAGE gel of the reconstituted PRC2-MTF2 holo complex
(B) and (C) EMSA of the binding of the PRC2-MTF2 holo complex to the 32P-labeled CGILHX6 DNA probe in the absence and presence of yeast tRNA.
(D) Quantification of the binding affinities measured in (B) and (C). (D) and (G) were based on three independently performed EMSAs. Graphs display mean ± SEM. GraphPad Prism 8.0 was used for data analysis.
(E) and (F) EMSA of the binding of the SUZ12(N)-RBBP4-MTF2 ternary complex to the CGILHX6 DNA probe in the presence of yeast tRNA. The binding condition is identical to (C). SUZ12WT and SUZ123D were assayed.
(G) Quantification of the binding affinities measured in (E) and (F).
(H) Schematic of the biotinylated DNA pull-down assay.
(I) and (J) Role of PRC2 dimerization in DNA binding. EZH2, EED, MTF2 and PHF19 all contained an N-terminal FLAG tag. Input controls were indicated by anti-FLAG signals (holo complexes, the lower two panels) and SYBR Gold signals (DNA probe, the middle panel). PRC2-MTF2 or PRC2-PHF19 released by the restriction enzyme was detected as anti-FLAG signals (the top panel).
See also Fig. S5.
Both PRC2–4m and PRC2–4m bound to the MTF2(RC) domain exhibited marginal binding to the CGILHX6 DNA probe, which was eliminated upon addition of the yeast tRNA (Fig. S5C and S5D). This data agreed with the previous finding that the EH domain of MTF2 N-terminal to the RC domain directly mediated DNA binding (Li et al., 2017). However, when PRC2 was omitted, a corresponding N-terminal fragment of MTF2 (residues 1–539) alone either formed aggregates with the DNA probe in the absence of the yeast tRNA competitor or displayed a largely weakened binding in its presence (Fig. S5E and S5F). As a control, MTF2 (residues 1–539) was purified as a monomer with low polydispersity as indicated by the SEC-MALS result (Fig. S5G). Collectively, PRC2 appeared to be necessary for the specific and tight binding of MTF2 to the CGILHX6 DNA. A similar observation was made in a recent genome-wide ChIP-seq study (Hojfeldt et al., 2019).
To dissect the contribution of protein dimerization in PRC2-MTF2 binding to DNA, we compared the DNA-binding affinity of full-length MTF2 in the context of WT and a dimer-disrupting mutant of the SUZ12(N)-RBBP4 binary complex (Fig. 5E, 5F, 5G and S5H). The SUZ12(N) fragment was formerly shown to be sufficient to occupy normal PRC2 binding loci in vivo, when bound to the accessory subunits (Hojfeldt et al., 2018). The SUZ12(N)-RBBP4-MTF2 ternary complex bound to the CGILHX6 DNA with a binding affinity of 17.6 nM (Fig. 5E and 5G), comparable to that for the PRC2-MTF2 holo complex under the same binding condition in the presence of tRNA (Fig. 5C and 5D). In stark contrast, the same complex harboring the SUZ123D dimer-disrupting mutation of SUZ12 bound DNA with an affinity reduced by at least 25 folds (Fig. 5F and 5G). This data supported a dimer-mediated avidity effect on DNA binding by MTF2, although the dimerized SUZ12(N)-RBBP4 scaffold might also contribute to interactions with the DNA probe. As a control, SUZ12 containing a comparable charge reversal triple mutation outside of the dimer interface, K534D/R535D/K537D, displayed minimal disturbance to DNA binding (Fig. S5I).
In addition to the quantitative EMSA with fully purified components, we also assessed CGI DNA binding by PRC2-MTF2 and PRC2-PHF19 in nuclear extracts, using an orthogonal biotinylated DNA pull-down assay. The synthetic biotinylated CGILHX6 DNA probe (biotin-CGILHX6) pre-bound to avidin beads was added to nuclear extracts of HEK293T cells expressing PRC2-MTF2 or PRC2-PHF19 (Fig. 5H). The WT or mutant holo complex specifically bound to the DNA probe was released by restriction enzyme digestion (Fig. 5H). Specificity of the pull-down assay was supported by two proof-of-principle experiments. First, a double alanine mutation of two lysine residues in the EH domain of MTF2 on the DNA binding interface (MTF2K338A/K339A) and the corresponding mutation in PHF19 (PHF19K331A/K332A) abolished DNA binding in the respective pull-down assays (Fig. S5J) (Li et al., 2017). Second, an inactivating mutation of EZH2 (EZH2H689A) did not cause any defect in CGI DNA binding (Fig. S5J). Endogenous PCLs were unlikely to influence the result since overexpressed proteins were dominating (Fig. S5K).
Compared to WT PRC2-MTF2, CGI DNA binding in nuclear extracts was largely diminished for the holo complex containing the SUZ123D or SUZ12R196A dimer-disrupting mutation and for the holo complex lacking the dimer stabilization helix of MTF2 (MTF2ΔDS) (Fig. 5I). Comparable results were obtained for the corresponding WT and mutant PRC2-PHF19 holo complexes (Fig. 5J), underscoring the critical role of PRC2 dimerization in CGI DNA binding by PRC2-MTF2 and PRC2-PHF19.
Loss of PRC2 dimerization impairs H3K27 trimethylation on chromatin in mESCs
PRC2 maintains mESC pluripotency and regulates cell fate determination during mESC differentiation. To study how PRC2 dimerization impacts chromatin structure and gene regulation in vivo, we generated human SUZ12WT and SUZ123D-expressing mESC lines based on a SUZ12 knockout (KO) mESC line using lentiviral vectors (Fig. 6A) (Hojfeldt et al., 2018). We selected single mESC clones that expressed comparable amounts of SUZ12WT and SUZ123D for further investigation (Fig. 6A). mESCs were normally grown in the 2i condition unless otherwise stated (Ying et al., 2008).
Fig. 6. Contribution of PRC2 dimerization to H3K27me3 on chromatin in vivo.

(A) Western blot confirming SUZ12 expression levels in SUZ12WT and SUZ123D mESCs grown in the 2i condition, using an anti-SUZ12 antibody. Global H3K27me3 levels were detected. Anti-GAPDH signals were used as loading controls.
(B) SEC profiles for endogenous EZH2 (upper panel) and MTF2 (lower panel) in SUZ12WT and SUZ123D mESCs. Elution profile was depicted according to Western blot signals. Positions of a 670KD (Bio-Rad SEC standard) and a 200KD (molecular weight of the SUZ12(N)-RBBP4 dimer) marker protein are indicated by arrows.
(C) The same as (A), for SUZ12WT and SUZ123D HEK293T cells.
(D) H3K27me3 enrichment by ChIP-qPCR on gene loci not targeted by PRC2. In (D), (E), (F) and (G), SUZ12WT and SUZ123D mESCs grown in three different days were used. Graphs display mean ± SEM from a total of six ChIP-qPCR reactions. GraphPad Prism 8.0 was used for data analysis. P values were derived from two-tailed Student’s t-test: ns (p>0.05); * (p≤0.05); ** (p≤0.01); *** (p≤0.001); **** (p≤0.0001).
(E) H3K27me3 enrichment on PRC2 targets in mESCs grown under the 2i condition.
(F) and (G) The same as (D) and (E), except that mESCs were grown under the serum condition.
Endogenous mouse EZH2 from nuclear extracts of SUZ12WT mESCs was eluted from a Superpose 6 SEC column as a broad peak with apparent molecular weights roughly ranging from a monomeric to dimeric holo complex (Fig. 6B). In stark contrast, EZH2 from SUZ123D mESCs was mostly eluted in a relatively narrow peak corresponding to a monomeric holo complex (Fig. 6B), suggesting a PRC2 dimer was converted into a monomer. A similar change of the SEC elution profile was observed for MTF2 from SUZ123D mESCs, confirming that MTF2 is a part of a dimeric holo complex (Fig. 6B). No difference was observed for either the longer isoform (e.g. isoform 1) or the shorter isoform (e.g. isoform 3) of endogenous mouse AEBP2 from the SUZ12WT and SUZ123D mESCs in the same experiment (Fig. S6).
SUZ12WT and SUZ123D rescued the global H3K27me3 level, to a comparable extent (Fig. 6A). Indeed, the SUZ12(VEFS) domain that lacks the entire SUZ12(N) was shown to be sufficient to restore the global H3K27me3 level in SUZ12 KO mESCs, through non-specific H3K27 methylation on chromatin (Hojfeldt et al., 2018). We also generated a SUZ12 CRISPR KO HEK293T cell line and expressed SUZ12WT and SUZ123D using lentiviral vectors. Similar to mESCs, the global H3K27me3 levels differed marginally between the two engineered HEK293T cell lines (Fig. 6C).
To test whether PRC2 dimerization regulates H3K27me3 at specific gene loci, we analyzed H3K27me3 enrichment on a series of developmental genes in SUZ12WT and SUZ123D mESCs by ChIP-qPCR. These genes were known to be PRC2 targets in mESCs (Boyer et al., 2006; Pasini et al., 2007; Walker et al., 2010). As a control, H3K27me3 was not affected by SUZ123D on OCT4 or NANOG genes (Fig. 6D), known not to be targeted by PRC2 (Pasini et al., 2007). SUZ123D impaired H3K27me3 enrichment on the TBX3, SATB2 and GATA4 gene loci, as compared to SUZ12WT (Fig. 6E). Defect was less profound for other loci, including PAX3, FGF5 and HOXA7 (Fig. 6E), suggesting that PRC2 dimerization was differentially required for H3K27me3 on distinct genes under the 2i condition. In comparison, H3K27me3 enrichment on all the six PRC2 target gene loci was robustly affected by the dimer-disrupting mutation, when SUZ12WT and SUZ123D mESCs were grown in the serum condition (Fig. 6F and 6G). Our ChIP-qPCR analysis of H3K27me3 enrichment in SUZ12WT and SUZ123D mESCs provided only a glimpse of the importance of PRC2 dimerization on a set of known PRC2 targets; genome-wide ChIP-seq analysis in these cell lines as well as mESCs harboring other separation-of-function SUZ12 mutations will allow us to gain a more quantitative and complete view on this subject (Fig. 3I).
DISCUSSION
How mammalian PRC2 engages CGI chromatin is a big mystery in the field. Emerging evidence suggests that the accessory subunits, including PCLs and AEBP2, are important for PRC2 targeting by directly binding to the linker DNA (Choi et al., 2017; Li et al., 2017; Perino et al., 2018; Wang et al., 2017). In the current study, we show that the PRC2-PCL holo complexes display a dimeric structural architecture and that dimerization plays a determining role in PRC2-PCL binding to an endogenous CGI DNA in vitro and PRC2 targeting to developmental genes in vivo in mESCs, possibly through an avidity effect. Our results provide a distinct structural framework for thinking of chromatin binding and gene regulation by PRC2-PCL.
PRC2-PCL dimerization is mediated by the PRC2 core complex: the C2 domain of SUZ12 in one protomer is swapped to interact with RBBP4 in the reciprocal protomer (Fig. 7). PCLs are not involved in dimerization per se. However, the RC domains of PHF19 and MTF2 stabilize the PRC2 dimer, while the RC domain of PHF1 displays a greatly reduced dimer stabilization activity due to divergence in protein sequence (Fig. 7). In stark contrast, AEBP2 disrupts the intrinsic PRC2 dimer by inducing a large movement of the C2 domain of SUZ12, indicative of dissimilar modes of DNA binding by these two classes of the holo complexes (Fig. 7) (Chen et al., 2018; Kasinath et al., 2018).
Fig. 7. Schematic model of the structural rearrangement between PRC2-PCL and PRC2-AEBP2 and of chromatin binding by PRC2-PCL.

The dotted curved lines represent the hinge loops that display conformational flexibility (black arrows). The DNA-binding domains of PCLs and AEBP2 are simplified as DBD. The H3K27 histone tail is bound by the SET domain of EZH2. Although the two EZH2-EED-SUZ12(VEFS) moieties in the PRC2-PCL dimer are drawn distal to each other in 2D, they may become proximal to each other in 3D, as they will move to avoid steric clash with the C2 domain. Black star symbolizes PRC2 dimer-disrupting mutations, which impair H3K27me3 on chromatin. The cartoons are not drawn to scale.
Homo- and heterodimerization of transcription factors are a widespread phenomenon in active transcription and are believed to account for the specificity and complexity of gene regulation (Amoutzias et al., 2008). In a similar but more complicated manner, PRC2 dimerization may also contribute to DNA binding specificity, by tethering two PCL molecules in one holo complex and thus imposing a spatial constraint between the two PCL-bound DNA motifs on the three-dimensional chromatin. On the other hand, the diversity of CGI chromatin can be accommodated by the conformational plasticity of PRC2-PCL conferred by the flexible hinge loops, such as the ones connecting to the C2 domain of SUZ12 (Fig. 7). Different paralogs and isoforms of the core and accessory subunits, like EZH1/2, Rbbp4/7, PCLs and so on, may be combined within the dimer platform to generate enormous complexity in composition and fine-tune PRC2 function. Furthermore, PRC2 dimerization is dynamic and subjected to differential regulation by AEBP2, PHF1, MTF2, PHF19 and possibly other accessory subunits, which may add an additional layer of complexity in gene regulation.
Although we only observed dimerization in our reconstituted system in vitro, formation of a PRC2 oligomer of a higher valency is not impossible under certain conditions in vivo, given the domain-swapping nature of the PRC2 dimer and the apparent flexibility of the hinge loops that connect to the swapped domains. Alternatively, some of the accessory subunits may also oligomerize, in which case both the core and accessory subunits may act together to promote oligomerization of the PRC2 holo complexes. It was previously shown that oligomerization of PRC1 through the sterile alpha motif (SAM) domain is required for PRC1 clustering and stable chromatin binding during gene silencing (Isono et al., 2013). PRC2 function may be regulated in a similar manner. In this regard, the specific dimer-disrupting mutations of SUZ12 revealed in our study provide a clean system for directly testing this hypothesis in vivo.
LEAD CONTACT AND MATERIALS AVAILABILITY
Further inquiries and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xin Liu (xin.liu@utsouthwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse embryonic stem cells
SUZ12 KO mESC line was kindly provided by Dr. Kristian Helin (Memorial Sloan Kettering Cancer Center) (Hojfeldt et al., 2018). mESCs were grown on gelatin-coated plates in Gibco™ KnockOut™ DMEM media (GIBCO) supplemented with 15% FBS (HyClone) (v/v), 1×Glutamax (Gibco), 1×non-essential amino acids (Sigma), 1×penicillin/streptomycin (Sigma), 0.1mM β-mercaptoethanol (Sigma) and leukemia inhibitory factor (Lab prep). For growth in 2i medium condition, mESCs were cultured in 1:1 mix of DMEM/F12 and Neurobasal media (Gibco) supplemented with 1×penicillin/streptomycin (Sigma), 0.3% BSA, 0.5×Glutamax (Gibco), 0.5% N-2 supplement (Gibco), 1% B27 supplement (Gibco) and leukemia inhibitory factor (produced in the lab). The GSK inhibitor (CHIR99021) (Cayman Chemical) and MEK inhibitor (PD0325901) (Cayman Chemical) were also added in 2i medium at final concentrations of 3 mM and 1 mM, respectively. SUZ12WT and SUZ123D knock-in mESCs were generated by infecting SUZ12 KO mESCs with 3μg /mL polybrene and lentivirus harboring WT or mutant SUZ12 for 24 hr, followed by puromycin selection. mESCs were passaged every two days.
HEK293T cells
HEK293T cells (ATCC) were cultured in DMEM medium supplemented with 10% FBS (Sigma), 1× penicillin/streptomycin (Sigma).
METHOD DETAILS
Protein Expression and Purification
Human PRC2 Complexes
Human SUZ12(N) (residues 76–545)-RBBP4 binary complex, four-member PRC2 core complex (EZH2-EED-SUZ12-RBBP4), PRC2 holo complexes containing different AEBP2 isoforms (PRC2-AEBP2isoform 1, PRC2-AEBP2isoform 3, and PRC2-AEBP2old) were co-expressed in Sf9 insect cells and were purified essentially the same as previously described (see Key Resources Table) (Chen et al., 2018). Human EZH2-EED-SUZ12(VEFS) ternary complex was expressed and purified from S. cerevisiae BY4741 strain the same as previously described (Chen et al., 2018). HEK293T cells (ATCC) were also used for WT and mutant PRC2 core complex expression and purification for functional studies. Briefly, Myc-EZH2, His6-EED and HA-RBBP4 cDNA were inserted into a pCS2+ vector. WT and mutant SUZ12 cDNAs were tagged at the 5′ end with a TAP tag cassette consisting of a Protein A and an HA separated by a TEV protease site. All mutant forms were generated by Quick-change mutagenesis using PfuUltra High-Fidelity DNA Polymerase and verified by sequencing. To generate WT or mutant SUZ12-containing PRC2 complexes, HEK293T cells were transiently co-transfected with the desired combination of plasmids. Cells were harvested 48 hours post transfection. The complex was captured by IgG resin and proteins were released from IgG resin by TEV cleavage overnight at 4°C. The purity of the protein complexes was assessed by SDS-PAGE.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GAPDH Loading Control Monoclonal Antibody (GA1R) | Invitrogen™ | Cat# MA5–15738 |
| Monoclonal ANTI-FLAG® M2 antibody | Sigma-Aldrich | Cat# F1804 |
| AEBP2 (D7C6X) Rabbit mAb | Cell Signaling | Cat# 14129S |
| Tri-Methyl-Histone H3 (Lys27) (C36B11) Rabbit mAb | Cell Signaling | Cat# 9733 |
| EZH2 (D2C9) XP® Rabbit mAb | Cell Signaling | Cat# 5246S |
| SUZ12 (D39F6) XP® Rabbit mAb | Cell Signaling | Cat# 3737S |
| Monoclonal Anti-HA antibody produced in mouse | Sigma-Aldrich | Cat# H9658 |
| Myc-Tag antibody (mAb) | Active motif | Cat# 39279 |
| MTF2 Antibody | Protein tech | Cat# 16208–1-AP |
| PHF19 Antibody | Cell Signaling | Cat# 77271S |
| HA-Tag (C29F4) Rabbit mAb | Cell Signaling | Cat# 3724S |
| RBBP4 Antibody | Bethyl Laboratories | Cat# A301–206A |
| Anti-EED Antibody, clone AA19 | EMD Millipore | Cat# 05–1320 |
| Bacterial and Yeast Strains | ||
| Rosetta 2(DE3) competent bacterial cells | Novagen | Cat# 71402 |
| S. cerevisiae BY4741 strain | Euroscarf | Cat# Y00000 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dynabeads™ Protein A for Immunoprecipitation | Invitrogen™ | Cat# 10002D |
| Dynabeads™ Protein G for Immunoprecipitation | Invitrogen™ | Cat# 10004D |
| Pierce™ 16% Formaldehyde (w/v), Methanol-free | Thermo Scientific™ | Cat# 28908 |
| cOmplete, EDTA-free proteinase inhibitor cocktail | Sigma-Aldrich | Cat# 11873580001 |
| RNase A | Thermo Scientific™ | Cat# EN0531 |
| Proteinase K, recombinant, PCR Grade | Sigma-Aldrich | Cat# 3115836001 |
| Glycogen | Invitrogen™ | Cat# 10814010 |
| SYBR™ Green PCR Master Mix | Applied Biosystems™ | Cat# 4309155 |
| Gelatin solution-100 mL Type B, 2% in H2O | Sigma-Aldrich | Cat# G-1393 |
| DMEM/F12 (1:1) | Gibco™ | Cat# 11320033 |
| Neurobasal Medium | Gibco™ | Cat# 21103049 |
| Gibco™ KnockOut™ DMEM | Gibco™ | Cat# 10829018 |
| GlutaMAX™ Supplement | Gibco™ | Cat# 35050061 |
| BSA Fraction V 7.5% | Gibco™ | Cat# 15260037 |
| MEM Non-essential Amino Acid Solution (100×) | Sigma-Aldrich | Cat# M7145–100ML |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat# M3148–25ML |
| N-2 supplement | Gibco™ | Cat# 17502048 |
| B27 supplement | Gibco™ | Cat# 17504044 |
| PD0325901-MEK inhibitor | Cayman | Cat# 391210109 |
| CHIR99021 (GSK3 inhibitor) | Cayman | Cat# NC0226336 |
| TrypLE™ Express Enzyme (1X), phenol red | Gibco™ | Cat# 12605028 |
| Penicillin-Streptomycin | Sigma-Aldrich | Cat# P0781–100ML |
| HyClone™ Fetal Bovine Serum, USDA Tested | GE Healthcare | Cat# SH3091003 |
| Dulbecco’s Modified Eagle’s Medium - high glucose | Sigma-Aldrich | Cat# D6429–500ML |
| Opti-MEM-Reduced Serum Medium | Gibco™ | Cat# 31985062 |
| Xtreme Gene 9 DNA transfection | Roche | Cat# 6365787001 |
| Lenti-X™ Concentrator | Clontech | Cat# 631232 |
| Gibco™ Sterile Puromycin Dihydrochloride | Gibco™ | Cat# A1113803 |
| IgG Sepharose™ 6 Fast Flow Agarose | GE Healthcare | Cat# 17096901 |
| Strep-Tactin® Superflow® high capacity 50% suspension | IBA-Lifesciences | Cat# 21208010 |
| Pierce™ Anti-DYKDDDDK Magnetic Agarose | Thermo Scientific™ | Cat# A36797 |
| Pierce™ Avidin Agarose | Thermo Scientific™ | Cat# 20219 |
| Pierce™ Glutathione Agarose | Thermo Scientific™ | Cat# 16101 |
| HIS-Select® Nickel Affinity Gel | Sigma-Aldrich | Cat# P6611–25ML |
| d-Desthiobiotin | Sigma-Aldrich | Cat# D1411–1G |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | Thermo Scientific™ | Cat# 34580 |
| SYBR Gold | ThermoFisher | Cat# S11494 |
| IPTG | Gold Biotechnology | Cat# I2481C |
| PfuUltra High-Fidelity DNA Polymerase | Agilent Technologies | Cat# 600385 |
| ProtoScript® II First Strand cDNA Synthesis Kit | NEB | Cat# E6560S |
| Bio-Rad gel filtration standard | BioRad | cat # 1511901 |
| Yeast tRNA (10 mg/mL) | Invitrogen | cat # AM7119 |
| Amersham Hybond-N+ | GE Healthcare | cat # RPN203B |
| EMD Millipore™ Novagen™ Mouse Genomic DNA | EMD Millipore | Cat# 69239–3 |
| Deposited Data | ||
| Crystal structure of SUZ12(N)-RBBP4-PHF19(RC)-JARID2(TR) | This study | PDB: 6NQ3 |
| Uncropped gel images | This study | http://dx.doi.org/10.17632/j8mddywf7j.1 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat# CRL-3216 |
| SUZ12-KO HEK293T | This study | N/A |
| SUZ12-KO + SUZ12WT HEK293T | This study | N/A |
| SUZ12-KO + SUZ123D HEK293T | This study | N/A |
| SUZ12-KO mESCs | Kristian Helin Laboratory | N/A |
| SUZ12-KO + SUZ12WT mESCs | This study | N/A |
| SUZ12-KO + SUZ123D mESCs | This study | N/A |
| Recombinant DNA | ||
| pCS2-Myc-EZH2 | This paper | N/A |
| pCS2-FLAG-EZH2 | This paper | N/A |
| pCS2-FLAG-EZH2(H689A) | This paper | N/A |
| pCS2-His6-EED | This paper | N/A |
| pCS2-HA-RBBP4 | This paper | N/A |
| pCS2-ProteinA-TEV-HA-SUZ12WT | This paper | N/A |
| pCS2-ProteinA-TEV-HA-SUZ123D | This paper | N/A |
| pCS2-ProteinA-TEV-HA-Suz12R196A | This paper | N/A |
| pCS2-ProteinA-TEV-HA-Suz12W334A | This paper | N/A |
| pCS2-ProteinA-TEV-HA-Suz12G518W | This paper | N/A |
| pCS2-Flag-PHF19WT | This paper | N/A |
| pCS2-Flag-PHF19ΔDS(531−544) | This paper | N/A |
| pCS2-Flag-PHF19K331A/K332A | This paper | N/A |
| pCS2-Flag-MTF2WT | This paper | N/A |
| pCS2-Flag-MTF2ΔDS(544−557) | This paper | N/A |
| pCS2-Flag-MTF2K338A/K339A | This paper | N/A |
| pET-28a-His6-SUMO-StrepII-Flag-MTF2(1–593) | This paper | N/A |
| pGEX-4T1-TEV-PHF19(500–580) | This paper | N/A |
| pGEX-4T1-TEV-MTF2(507–593) | This paper | N/A |
| pET-28a-His6-SUMO-PHF1(493–567) | This paper | N/A |
| pGEX-4T1-TEV-AEBP2(407–503) | This paper | N/A |
| pFastBac-Flag-Ezh2(1–746) | This paper | N/A |
| pFastBac-His6-Thrombin-RBBP4(1–425) | This paper | N/A |
| pACEBac1-His6-Flag-EED(1–441)-3C-StrepII | This paper | N/A |
| pACEBac1-His6-Flag-SUZ12(1–739)-TEV-StrepII | This paper | N/A |
| pACEBac1-StrepII-3C-AEBP2(1–295)-His6 | This paper | N/A |
| pACEBac1-StrepII-AEBP2(1–301) | This paper | N/A |
| Software and Algorithms | ||
| Pymol | The PyMOL Molecular Graphics System, Schrö dinger, LLC. | N/A |
| PHENIX | https://www.phenix-online.org | N/A |
| Coot | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ | N/A |
PHF19(RC), MTF2(RC) and PHF1(RC)
The cDNA sequences encoding human PHF19(RC) (residues 500–580) and MTF2(RC) (residues 507–593) were subcloned into pGEX-4T1 vector with a TEV cleavage site inserted. The cDNA of PHF1(RC) (residues 493–567) was inserted into the pET-28a-Sumo vector. E.coli Rosetta 2(DE3) cells transformed with the expression plasmids were induced with 0.5 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG) at OD600 of 0.8 for 15 h at 20°C. For purification of PHF19(RC) and MTF2(RC), cells were harvested and lysed by sonication in GST binding buffer (50mM Tris-HCl, pH 8.0, 150mM NaCl, 10% glycerol and 5mM DTT). After centrifugation, the supernatant was incubated with GST resin for 2 hours at 4°C with mixing. Bound proteins were then eluted by incubating GST resin with TEV protease overnight at 4°C. Proteins eluted from the GST resin were pooled, concentrated, and loaded onto a preparative Superdex S200 size exclusion chromatography (SEC) column (GE Healthcare) in the SEC buffer (20mM Tris-HCl, pH 8.0, 100mM NaCl and 2mM DTT). For PHF1(RC) purification, the harvested cells were lysed by sonication in Ni-NTA binding buffer (50mM HEPES, pH 7.4, 150mM NaCl, 10% glycerol and 5mM 2-mercaptoethanol supplemented with 1mM PMSF). The cell lysate was clarified by ultracentrifugation and the supernatant was applied to Ni-NTA resin at 4°C. The beads were washed using Ni-NTA wash buffer (50mM Hepes, pH 7.4, 300mM NaCl, 20mM imidazole, 10% glycerol and 5mM 2-mercaptoethanol). His6-Sumo-PHF1(RC) was then eluted and incubated with SUMO protease overnight at 4°C to remove the His6-Sumo tag. PHF1(RC) was further purified by Mono S ion exchange chromatography followed by SEC. PHF19(RC)Δ500−530 (corresponding to residues 531–580) and PHF19(RC)Δ500−543 (corresponding to residues 544–580) were expressed and purified essentially the same as PHF19(RC).
Full-length MTF2
Full-length MTF2 (residues 1–593) was inserted into the pET-28a-Sumo vector with an N-terminal His6-Sumo-FLAG-StrepII tag. E.coli Rosetta 2(DE3) cells transformed with the expression plasmid were grown in LB medium contains 10μM ZnCl2 at 37°C to an OD600 of 0.6 and induced with 0.3mM IPTG at 20°C for 6 hrs. The cells were harvested and lysed by sonication in lysis buffer (50mM HEPES pH 7.4, 500mM NaCl, 10μM ZnCl2, 5% Glycerol, 2mM 2-mercaptoethanol and 1mM PMSF). After centrifugation, the supernatant was applied to Ni-NTA resin at 4°C. The protein bound to Ni-NTA resin was extensively washed using Ni-NTA wash buffer (50mM HEPES pH 7.4, 500mM NaCl, 10μM ZnCl2, 5% Glycerol and 2mM 2-mercaptoethanol) and then eluted with the wash buffer supplemented with 250 mM imidazole. The Strep-Tactin column (IBA) was used for further purification after the His6-Sumo tag was removed from MTF2 (residues 1–593) by SUMO protease. Protein eluted from the Strep-Tactin column (IBA) was pooled, flash frozen in liquid nitrogen and stored at −80°C.
Reconstitution of PRC2-MTF2 and SUZ12(N)-RBBP4-MTF2
The purified PRC2 core complex (EZH2-EED-SUZ12-RBBP4) was incubated with the full-length MTF2 (residues 1–593) in a buffer containing 50mM HEPES pH 7.4, 1M NaCl, 10μM ZnCl2, 5% Glycerol and 2mM 2-mercaptoethanol for 4 hours at 4°C with mixing. The reconstituted PRC2-MTF2 holo complex was then dialyzed into low salt buffer (50mM Tris-HCl, pH 8.0, 150mM NaCl, 10% glycerol, 10μM ZnCl2 and 2mM DTT) overnight at 4°C. The reconstituted PRC2-MTF2 holo complex was clarified by ultracentrifugation and the supernatant was further purified via Mono S ion exchange chromatography in Buffer A (20mM Tris-HCl pH8.0, 300mM NaCl, 5mM 2-mercaptoethanol) and Buffer B (20mM Tris-HCl pH8.0, 1M NaCl, 5mM 2-mercaptoethanol). The complex was eluted by a linear gradient from 300 mM to 1 M NaCl and the eluted fractions were analyzed by SDS-PAGE. The peak fractions were pooled, concentrated and then loaded onto a Superose 6 10/300 GL column (GE Healthcare) in SEC buffer (20mM Tris pH 8.0, 150mM NaCl and 2mM DTT). The SUZ12(N)-RBBP4-MTF2 (residues 1–593) ternary complex was reconstituted essentially the same as the PRC2-MTF2 holo complex, except that the PRC2 core complex was replaced by SUZ12(N)-RBBP4 binary complex in complex assembly.
Crystallization and Structure Determination
The SUZ12(N)-RBBP4 binary complex was incubated with about 3 molar excess of PHF19(RC) for 1 hour on ice, followed by size exclusion chromatography to remove the extra PHF19(RC). We initially crystallized the SUZ12(N)-RBBP4-PHF19(RC) ternary complex. In order to improve the crystal quality, a peptide corresponding to the JARID2(TR) (residues 147–165) was added to the ternary complex in a 3:1 molar ratio prior to crystallization. Crystals of the heterotetrameric complex were grown by mixing 1μl protein solution at 5 to 10 mg/ml with 1μl of the reservoir solution containing 12% PEG3350, 0.1M Sodium malonate. Crystals were cryoprotected in a reservoir solution containing 25% PEG400 and subsequently flash frozen in liquid nitrogen.
Diffraction data were collected at Advanced Light Source (ALS) beamline 5.0.2, Stanford Synchrotron Radiation Lightsource (SSRL) beamlines 9–2 and 12–2, and Advanced Photo Source (APS) beamline 19ID. Images were indexed, integrated, and scaled with HKL2000 package and further processed with the CCP4 suite of programs (Otwinowski and Minor, 1997; Winn et al., 2011). The structures were solved by molecular replacement with Phaser using a previously determined crystal structure of SUZ12-RBBP4 binary as a search model (McCoy et al., 2007). Refinement and model building were carried out by using Coot, Phenix and autoBUSTER (Adams et al., 2010; Bricogne G, 2010; Emsley et al., 2010). Statistics for data collection, phase calculation, and refinement are summarized in Table 1. Structure images were prepared using PyMOL software (The PyMOL Molecular Graphics System).
Analytical Gel Filtration
The SEC analysis of the human SUZ12(N)-RBBP4 binary complex, the EZH2-EED-SUZ12(VEFS) ternary complex, the PRC2 core complex (EZH2-EEDSUZ12-RBBP4), the PRC2-holo complex, and the PRC2-AEBP2 holo complexes was performed on Superose 6 10/300 GL column (GE Healthcare) in SEC buffer (20mM Tris pH 8.0, 150mM NaCl and 2mM DTT). Thyroglobulin (670 kDa), gamma globulin (158 kDa), ovalbumin (44 kDa), myoglobin (17 kDa) and vitamin B12 (1.35 kDa) from Bio-Rad gel filtration standard (Bio-Rad) were used as calibrants. Protein samples were injected at a flow rate of 0.4 ml/min. Elution profile was monitored at 280 nm.
PRC2 Dimerization Co-IP Assay
HEK293T cells (ATCC) were used in PRC2 dimerization assay. EZH2 was subcloned into the pCS2+ vector with an N-terminal FLAG tag or Myc tag to generate FLAG-EZH2 or Myc-EZH2, respectively. The other subunits of PRC2 complex were subcloned in to the pCS2+ vector with the following modifications: the EED cDNA was fused to an N-terminal His6-tag, RBBP4 to an N-terminal HA tag, and SUZ12 cDNA (WT or mutants) to an N-terminal ProteinA-TEV-HA tag. Constructs for the expression of FLAG-EZH2, Myc-EZH2, His6-EED, HA-RBBP4 and WT or mutant ProteinA-TEV-HA-SUZ12 were co-transfected into HEK293T cells. After 48 hours, cells were harvested and the nuclear extracts were prepared followed by standard protocol. 15μg of purified AEBP2(C2B-H3K4D) or PHF19(RC) fragment was added to the nuclear extracts before SUZ12 was captured by IgG resin. Non-specifically bound species were removed by extensive wash, and bound proteins were eluted by incubating IgG resin with TEV protease overnight at 4°C. Around 1.5μg of PRC2 (core or AEBP2/PHF19 fragment-bound complexes) eluted from IgG resin was bound to anti-FLAG resin. After extensive wash with pull-down buffer (50mM Tris-HCl, pH 8.0, 150mM NaCl, 10% glycerol, 0.1% NP40 and 2mM DTT), the bound proteins were eluted from anti-FLAG resin by FLAG peptide. Anti-FLAG and anti-Myc signals were examined by Western blot with respective anti-epitope antibodies. For the dimerization assay using full-length AEBP2, FLAG-EZH2, MYC-EZH2, HA-SUZ12, HA-RRBP4 and ProteinA-3C-AEBP21−295 or ProteinA-TEV-AEBP21−517 were cotransfected into HEK293T cells. Respective complexes were purified by IgG resin and released from the resin by incubation with TEV or 3C proteases overnight. The quality of the purified PRC2-AEBP2 complexes was first assessed by both SDS-PAGE Coomassie blue staining and Western blot, and the dimerization assay was then performed following the same protocol as described above for the AEBP2(C2B-H3K4D) and PHF19(RC) fragments.
Eletrophoretic Mobility Shift Assay (EMSA)
A 100 bp CGILhx6 DNA were amplified from mouse genomic DNA by PCR and end-labeled with gamma-32P-ATP (PerkinElmer). 200 pM of 32P labeled DNA was incubated with an increasing amount of protein complexes in 10 μl binding buffer (25mM Tris, pH 7.5, 100mM KCl, 0.05% NP40, 2.5mM MgCl2, 5% Glycerol, and 2mM 2-mercaptoethanol) for 1 hour on ice. 200ng/μl competitor yeast tRNA (Invitorgen) was added into the binding buffer wherever indicated. Sample was then loaded onto 0.8% agarose gel in 1XTBE at 4°C and resolved by electrophoresis at 100 V for 80 min. Gels were vacuum dried for 120 min at 80°C on an Amersham Hybond-N+ membrane (GE Healthcare) and Whatman 3 mm chromatography paper. Dried gels were exposed to phosphorimaging plates (GE Healthcare) and signal acquisition was carried out by using a Molecular Imager PharosFX phosphorimager Systems (Bio-Rad). The sequence of the CGILhx6 DNA probe is provided below with CpG dinucleotides highlighted in red):
GTGGTGGTGGTGGTTGAGAAAATAAAACCCAGAGCGCTAGGAGCATCCGCCCGCTCGACGCGCGCCGGGAAATTGAAGCGGGGATATTGACACCGATTCA
An AT-rich DNA probe was used as a negative control and the sequence is provided below:
TATATATATTTTATATATATATAAATTAATATATAATTATATTATATTAAAATTAATATTATATTTAAATTATTATATATATATAAATAATTTAATTATA
DNA Pull-down Assay
HEK293T cells (ATCC) were used for preparation of nuclear extracts overexpressing the WT or mutant PRC2 holo complexes containing MTF2 or PHF19. Briefly, EZH2(WT/Mutant), PHF19(WT/Mutant) and MTF2(WT/Mutant) were subcloned into the pCS2+ vector with an N-terminal FLAG tag. The other subunits of PRC2 were the same as the ones used in PRC2 Dimerization Co-Immunoprecipitation (Co-IP) Assay described above, including: pCS2-His6-EED, pCS2-HA-RBBP4 and pCS2-ProteinA-TEV-HA-SUZ12(WT/Mutant). To generate nuclear extracts containing the WT or mutant PRC2-PHF19 or PRC2-MTF2 holo complexes, HEK293T cells were transiently transfected with the desired combination of plasmids. 48 hours post transfection, cells were harvested and the nuclear extracts were prepared following a standard protocol. The nuclear extracts were dialyzed into the DNA binding buffer (25mM Tris, pH 7.5, 100mM KCl, 0.05% NP40, 2.5mM MgCl2, 5% Glycerol and 2mM 2-mercaptoethanol) O/N and clarified by centrifugation at top speed to remove the precipitates. The biotinylated CGILhx6 DNA was prepared by PCR by using primers labeled with biotin. The Bstz17I cleave site was inserted between the 5’-biotin label and the actual CpG DNA sequence. 2μg biotinylated CGILhx6 was pre-bound to avidin beads for 2 hours and the excessive unbound DNA were removed. The DNA-bound beads were then incubated with a total of 400μl nuclear extracts for 1hr and subjected to wash for 5 times using the same DNA binding buffer, followed by washing 2 times using Bstz17I digestion buffer (NEBuffer 3.1). The bound proteins were released by incubating avidin resin with Bstz17I overnight at 16°C and detected by Western blot. The sequence of the biotinylated CGILhx6 DNA probe is provided below (CpG dinucleotides in red and Bstz17I site underlined):
Biotin-GGAATTCCATATGAAGTATACAAGTGGTGGTGGTGGTTGAGAAAATAAAACCCAGAGCGCTAGGAGCATCCGCCCGCTCGACGCGCGCCGGGAAATTGAAGCGGGGATATTGACACCGATTCA
GST Pull-down Assay for PRC2 Core Complexes
Briefly, GST-tagged PHF19(RC) or GST alone control were pre-incubated with GST beads and then mixed with the WT and mutant PRC2 core complexes purified from HEK293T cells in the binding buffer (50mM Tris-HCl, pH 8.0, 150mM NaCl, 2mM DTT, 10% Glycerol and 0.1% NP40) for 1 hour at 4°C. After extensive wash with the same binding buffer, the supernatants were eluted by GSH and subjected to Western blot analysis.
SEC-MALS
Size Exclusion Chromatography–MultiAngle Light Scattering (SEC-MALS) experiment was performed at 25°C. 2 mg/mL Protein samples were filtered through a 0.22-μm centrifugal filter and then loaded on to a Superdex 200 10/300 Increase column equilibrated with Buffer (50mM Tris, pH 8.0, 500mM NaCl and 0.5mM TCEP at room temperature) at a flow rate of 0.5 mL/min. The mobile phase was pumped through a Shimadzu LC20-AD unit with inline degassing and was subjected to MALS using a Wyatt TREOS II light-scattering detector and a Wyatt Optilab tREX differential-refractive-index detector. Data were analyzed with Wyatt’s ASTRA software version 7.3.0.11.
Lentivirus Production and Delivery
Human SUZ12WT or SUZ123D cDNAs were subcloned into the pCDH-EF1 vector with an N-terminal 3×FLAG tag for lentivirus production. The pCDH-EF1–3XFLAG-SUZ12-Puro WT or mutant plasmid (5μg) was co-transfected with psPAX2 (5μg) and pVSV-G (0.5 μg) packaging vectors into HEK293T cells. After 48 hr transfection, the virus-containing medium was collected and centrifuged at 200g for 10min. The supernatant was filtered by 0.45μm and concentrated by adding 1/3 volume Lenti-X™ concentrator (Clontech) at 4°C for overnight. The viral pellet was collected by centrifuged at 1500g for 45min and re-suspended in 2i medium for mouse embryonic stem cell infection or DMEM medium for HEK293T cell infection.
CRISPR-Mediated SUZ12 Knockout in HEK293T Cells
SUZ12 KO HEK293T cell line was constructed by using the CRISPR/Cas9 gene-editing system. Single-guide RNA (sgRNA) targeting sequences (5’-AAATCCGGCGGCGGGAGCTG −3’) was synthesized and inserted into the pSpCas9(BB)-2A-GFP (PX458) expression vector (Addgene). HEK293T cells were transfected with px458-Suz12(gRNA) by PEI reagent and sorted by FACS. Single-cell colonies were picked and the knockout efficiency was determined by Western blot at protein expression levels. SUZ12WT and SUZ123D knock-in HEK293T cell lines were generated by infecting SUZ12 KO HEK293T cells with lentiviruses harboring the corresponding constructs, followed by selection by puromycin.
Preparation of mESC Nuclear Extracts
To prepare mESC nuclear extracts for gel filtration elution profile analysis, mESCs were harvested from 15-cm dishes at 80% confluency and washed two times in cold PBS. Intact nuclei were prepared by resuspending cell pellet in 5 volumes of hypotonic buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.5%NP40, 1mM DTT, 1×protease inhibitors). Nuclei were collected by centrifugation at 1000 x g at 4°C for 10 min. Nuclei pellet was resuspended in 5 volumes of ice cold high salt buffer (20 mM HEPES pH 7.9, 420mM KCl, 1.5mM MgCl2, 0.2mM EDTA, 25% Glycerol) supplemented with protease inhibitors, and incubated on rotator at 4°C for 1 hr. The mESC nuclear extracts were collected by centrifugation at 18,000 x g at 4°C for 20 min, before being dialyzed into the SEC buffer (20mM Tris pH 8.0, 100mM NaCl and 2mM DTT).
Chromatin Immunoprecipitation Assays
ChIP experiments for H3K27me3 were carried out according to standard protocols. Briefly, mESCs were cross-linked in PBS by addition of 1% formaldehyde (Sigma) in single cell suspension at room temperature for 10 min with rotation, and quenched with 125mM glycine for 5 min. Cross-linked cells were washed once with cold PBS, pelleted, and then lysed in Farnham Lysis Buffer (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% NP-40, 1 mM DTT and 1x protease inhibitor cocktail). A crude nuclear pellet was collected by gentle centrifugation (2000rpm, 5min at 4°C) and further resuspended in Sonication Buffer (50 mM Tris-HCl pH 7.9, 1% SDS, 10 mM EDTA, 1 mM DTT, and 1× protease inhibitor cocktail). The chromatin DNA were sonicated at 4°C by using a Biorupter (Diagenode) on the high setting for four cycles of 5 min (30s on/30s off) to generate genomic DNA fragments of 200–500 bp size distribution. The soluble chromatin was diluted 1:10 with ChIP Dilution Buffer (20 mM Tris-HCl pH 7.9, 0.5% Triton X-100, 2 mM EDTA, 150 mM NaCl, 1 mM DTT, and 1× protease inhibitor cocktail) and then clarified by centrifugation at max speed at 4°C for 15 min. After saving 10% as input sample, each ChIP was carried out using 50–200 μg of sonicated genomic DNA from mESCs to incubate with 5 μg of anti-H3K27me3 antibody overnight at 4°C. 50μl Dynabeads Protein A and 50μl Dynabeads Protein G (Invitrogen) mixture was pre-blocked with 0.5% BSA in ChIP Dilution Buffer overnight and used to capture the antibody-chromatin complex. Beads were washed with low salt (20 mM Tris-HCl pH 8.0, 2 mM EDTA, 1% Triton X-100, 0.1% SDS, 150 mM NaCl), high salt (20 mM Tris-HCl pH 8.0, 2 mM EDTA, 1% Triton X-100, 0.1% SDS, 500 mM NaCl), LiCl (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 1% NP40, 1% NaDOC, 250 mM LiCl), and TE (pH 8.0) wash buffers. Each wash was performed on the rotator at RT for 5min. The immunoprecipitated genomic DNA was eluted in Elution Buffer (50 mM Tris pH8.0, 10mM EDTA, 1% SDS) and the crosslinks were reversed by incubation in elution buffer (50 mM Tris pH8.0, 10mM EDTA, 1% SDS) at 65°C overnight. The genomic DNA was further digested with proteinase K and RNase A, and extracted with phenol:chloroform:isoamyl alcohol. H3K27me3 enrichment was analyzed by qPCR by using gene-specific primers listed in Table S1. All ChIP-qPCR assays were performed for at least three times with independent biological replicates.
QUANTIFICATION AND STATISTICAL ANALYSIS
Graphs display the mean ± SEM from at least three independently performed assays. GraphPad Prism 8.0 was used for statistical analysis.
DATA AND CODE AVAILABILITY
The crystal structure and structure factors described in this study have been deposited in the Protein Data Bank under accession numbers PDB: 6NQ3. Original gel images have been uploaded to Mendeley Data: http://dx.doi.org/10.17632/j8mddywf7j.1
Supplementary Material
Highlights.
Crystal structure reveals the molecular mechanism of PRC2 dimerization
Polycomb-like (PCL) proteins MTF2 and PHF19 stabilize the intrinsic PRC2 dimer
Dimerization enhances PRC2-PCL binding to an endogenous CGI DNA in vitro
Loss of PRC2 dimerization impairs H3K27me3 deposition on chromatin in mESCs
Acknowledgments
The SUZ12 KO mESC line was a gift from Dr. Kristian Helin at Memorial Sloan Kettering Cancer Center. The cDNA of EPOP was kindly provided by Dr. Yang Shi at Harvard Medical School and Children’s Hospital Boston. The SEC-MALS was performed with the help of Dr. Chad Brautigam from Macromolecular Biophysics Resource at UT Southwestern Medical Center. This research was supported by Welch Foundation research grant I-1790, CPRIT research grant R1119, Rita Allen Foundation research grant, and NIH grants GM114576 and GM121662 to X.L. X.L. is a W.W. Caruth, Jr., Scholar in Biomedical Research. This research also received support from the Cecil H. and Ida Green Center Training Program in Reproductive Biology Sciences Research. This research used the Protein and Monoclonal Antibody Production Shared Resource of Baylor College of Medicine. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02–06CH11357. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. DOE under contract no. DE-AC02–05CH11231. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02–76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- Amoutzias GD, Robertson DL, Van de Peer Y, and Oliver SG (2008). Choose your partners: dimerization in eukaryotic transcription factors. Trends in biochemical sciences 33, 220–229. [DOI] [PubMed] [Google Scholar]
- Ballare C, Lange M, Lapinaite A, Martin GM, Morey L, Pascual G, Liefke R, Simon B, Shi Y, Gozani O, et al. (2012). Phf19 links methylated Lys36 of histone H3 to regulation of Polycomb activity. Nature structural & molecular biology 19, 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. (2006). Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441, 349–353. [DOI] [PubMed] [Google Scholar]
- Cai L, Rothbart SB, Lu R, Xu B, Chen WY, Tripathy A, Rockowitz S, Zheng D, Patel DJ, Allis CD, et al. (2013). An H3K36 methylation-engaging Tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Molecular cell 49, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, and Zhang Y (2004). SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Molecular cell 15, 57–67. [DOI] [PubMed] [Google Scholar]
- Casanova M, Preissner T, Cerase A, Poot R, Yamada D, Li X, Appanah R, Bezstarosti K, Demmers J, Koseki H, et al. (2011). Polycomblike 2 facilitates the recruitment of PRC2 Polycomb group complexes to the inactive X chromosome and to target loci in embryonic stem cells. Development 138, 1471–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Jiao L, Shubbar M, Yang X, and Liu X (2018). Unique Structural Platforms of Suz12 Dictate Distinct Classes of PRC2 for Chromatin Binding. Molecular cell 69, 840–852 e845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Bachmann AL, Tauscher K, Benda C, Fierz B, and Muller J (2017). DNA binding by PHF1 prolongs PRC2 residence time on chromatin and thereby promotes H3K27 methylation. Nature structural & molecular biology. [DOI] [PubMed] [Google Scholar]
- Coleman RT, and Struhl G (2017). Causal role for inheritance of H3K27me3 in maintaining the OFF state of a Drosophila HOX gene. Science 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway E, Jerman E, Healy E, Ito S, Holoch D, Oliviero G, Deevy O, Glancy E, Fitzpatrick DJ, Mucha M, et al. (2018). A Family of Vertebrate-Specific Polycombs Encoded by the LCOR/LCORL Genes Balance PRC2 Subtype Activities. Molecular cell 70, 408–421 e408. [DOI] [PubMed] [Google Scholar]
- Davidovich C, Goodrich KJ, Gooding AR, and Cech TR (2014). A dimeric state for PRC2. Nucleic acids research 42, 9236–9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton AM, and Bird A (2011). CpG islands and the regulation of transcription. Genes Dev 25, 1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grijzenhout A, Godwin J, Koseki H, Gdula MR, Szumska D, McGouran JF, Bhattacharya S, Kessler BM, Brockdorff N, and Cooper S (2016). Functional analysis of AEBP2, a PRC2 Polycomb protein, reveals a Trithorax phenotype in embryonic development and in ESCs. Development 143, 2716–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KH, Bracken AP, Pasini D, Dietrich N, Gehani SS, Monrad A, Rappsilber J, Lerdrup M, and Helin K (2008). A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol 10, 1291–1300. [DOI] [PubMed] [Google Scholar]
- Hojfeldt JW, Hedehus L, Laugesen A, Tatar T, Wiehle L, and Helin K (2019). Non-core Subunits of the PRC2 Complex Are Collectively Required for Its Target-Site Specificity. Molecular cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojfeldt JW, Laugesen A, Willumsen BM, Damhofer H, Hedehus L, Tvardovskiy A, Mohammad F, Jensen ON, and Helin K (2018). Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nature structural & molecular biology 25, 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isono K, Endo TA, Ku M, Yamada D, Suzuki R, Sharif J, Ishikura T, Toyoda T, Bernstein BE, and Koseki H (2013). SAM domain polymerization links subnuclear clustering of PRC1 to gene silencing. Dev Cell 26, 565–577. [DOI] [PubMed] [Google Scholar]
- Jermann P, Hoerner L, Burger L, and Schubeler D (2014). Short sequences can efficiently recruit histone H3 lysine 27 trimethylation in the absence of enhancer activity and DNA methylation. Proceedings of the National Academy of Sciences of the United States of America 111, E3415–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao L, and Liu X (2015). Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 350, aac4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E, De Marco V, Haire LF, Walker PA, Reinberg D, et al. (2016). Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nature communications 7, 11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasinath V, Faini M, Poepsel S, Reif D, Feng XA, Stjepanovic G, Aebersold R, and Nogales E (2018). Structures of human PRC2 with its cofactors AEBP2 and JARID2. Science 359, 940–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprell F, Finkl K, and Muller J (2017). Propagation of Polycomb-repressed chromatin requires sequence-specific recruitment to DNA. Science 356, 85–88. [DOI] [PubMed] [Google Scholar]
- Lee CH, Holder M, Grau D, Saldana-Meyer R, Yu JR, Ganai RA, Zhang J, Wang M, LeRoy G, Dobenecker MW, et al. (2018). Distinct Stimulatory Mechanisms Regulate the Catalytic Activity of Polycomb Repressive Complex 2. Molecular cell 70, 435–448 e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejon S, Thong SY, Murthy A, AlQarni S, Murzina NV, Blobel GA, Laue ED, and Mackay JP (2011). Insights into association of the NuRD complex with FOG-1 from the crystal structure of an RbAp48.FOG-1 complex. The Journal of biological chemistry 286, 1196–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Liefke R, Jiang J, Kurland JV, Tian W, Deng P, Zhang W, He Q, Patel DJ, Bulyk ML, et al. (2017). Polycomb-like proteins link the PRC2 complex to CpG islands. Nature 549, 287–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liefke R, and Shi Y (2015). The PRC2-associated factor C17orf96 is a novel CpG island regulator in mouse ES cells. Cell Discov 1, 15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, and Eisenberg D (2002). 3D domain swapping: as domains continue to swap. Protein science : a publication of the Protein Society 11, 1285–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Li F, Zhang B, Li S, Wu J, and Shi Y (2015). Structural basis of plant homeodomain finger 6 (PHF6) recognition by the retinoblastoma binding protein 4 (RBBP4) component of the nucleosome remodeling and deacetylase (NuRD) complex. The Journal of biological chemistry 290, 6630–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ 3rd, Voigt P, Martin SR, Taylor WR, De Marco V, et al. (2009). Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461, 762–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, and Reinberg D (2008). Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Molecular cell 32, 503–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh JA, and Forman-Kay JD (2010). Sequence determinants of compaction in intrinsically disordered proteins. Biophysical journal 98, 2383–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendenhall EM, Koche RP, Truong T, Zhou VW, Issac B, Chi AS, Ku M, and Bernstein BE (2010). GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet 6, e1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman CA, Avvakumov N, Watanabe R, Abraham CG, Lalonde ME, Hong Z, Allen C, Roy S, Nunez JK, Nickoloff J, et al. (2012). Molecular basis for H3K36me3 recognition by the Tudor domain of PHF1. Nature structural & molecular biology 19, 1266–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell S, Wang L, Robert S, Jones CA, Saint R, and Jones RS (2001). Polycomblike PHD fingers mediate conserved interaction with enhancer of zeste protein. The Journal of biological chemistry 276, 43065–43073. [DOI] [PubMed] [Google Scholar]
- Pasini D, Bracken AP, Hansen JB, Capillo M, and Helin K (2007). The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Molecular and cellular biology 27, 3769–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perino M, van Mierlo G, Karemaker ID, van Genesen S, Vermeulen M, Marks H, van Heeringen SJ, and Veenstra GJC (2018). MTF2 recruits Polycomb Repressive Complex 2 by helical-shape-selective DNA binding. Nature genetics 50, 1002–1010. [DOI] [PubMed] [Google Scholar]
- Riising EM, Comet I, Leblanc B, Wu X, Johansen JV, and Helin K (2014). Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Molecular cell 55, 347–360. [DOI] [PubMed] [Google Scholar]
- Schmitges FW, Prusty AB, Faty M, Stutzer A, Lingaraju GM, Aiwazian J, Sack R, Hess D, Li L, Zhou S, et al. (2011). Histone methylation by PRC2 is inhibited by active chromatin marks. Molecular cell 42, 330–341. [DOI] [PubMed] [Google Scholar]
- Schuettengruber B, Bourbon HM, Di Croce L, and Cavalli G (2017). Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 171, 34–57. [DOI] [PubMed] [Google Scholar]
- Son J, Shen SS, Margueron R, and Reinberg D (2013). Nucleosome-binding activities within JARID2 and EZH1 regulate the function of PRC2 on chromatin. Genes Dev 27, 2663–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen PA, and Ringrose L (2014). What are memories made of? How Polycomb and Trithorax proteins mediate epigenetic memory. Nature reviews. Molecular cell biology 15, 340–356. [DOI] [PubMed] [Google Scholar]
- Tie F, Prasad-Sinha J, Birve A, Rasmuson-Lestander A, and Harte PJ (2003). A 1-megadalton ESC/E(Z) complex from Drosophila that contains polycomblike and RPD3. Molecular and cellular biology 23, 3352–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizan P, Beringer M, Ballare C, and Di Croce L (2015). Role of PRC2-associated factors in stem cells and disease. FEBS J 282, 1723–1735. [DOI] [PubMed] [Google Scholar]
- Wachter E, Quante T, Merusi C, Arczewska A, Stewart F, Webb S, and Bird A (2014). Synthetic CpG islands reveal DNA sequence determinants of chromatin structure. eLife 3, e03397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker E, Chang WY, Hunkapiller J, Cagney G, Garcha K, Torchia J, Krogan NJ, Reiter JF, and Stanford WL (2010). Polycomb-like 2 associates with PRC2 and regulates transcriptional networks during mouse embryonic stem cell self-renewal and differentiation. Cell Stem Cell 6, 153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Y, et al. (2012). Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome research 22, 1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Paucek RD, Gooding AR, Brown ZZ, Ge EJ, Muir TW, and Cech TR (2017). Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA. Nature structural & molecular biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, and Smith A (2008). The ground state of embryonic stem cell self-renewal. Nature 453, 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Jones A, Sun CW, Li C, Chang CW, Joo HY, Dai Q, Mysliwiec MR, Wu LC, Guo Y, et al. (2011). PRC2 complexes with JARID2, MTF2, and esPRC2p48 in ES cells to modulate ES cell pluripotency and somatic cell reprogramming. Stem Cells 29, 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The crystal structure and structure factors described in this study have been deposited in the Protein Data Bank under accession numbers PDB: 6NQ3. Original gel images have been uploaded to Mendeley Data: http://dx.doi.org/10.17632/j8mddywf7j.1