

Abstract
Precision medicine based on cancer genomics is being applied in clinical practice. However, patients do not always derive benefits from genomic testing. Here, we performed targeted amplicon exome sequencing‐based panel tests, including 160 cancer‐related genes (PleSSision‐160), on 88 malignant ovarian tumors (high‐grade serous carcinoma, 27; endometrioid carcinoma, 15; clear cell carcinoma, 30; mucinous carcinoma, 6; undifferentiated carcinoma, 4; and others, 6 (immature teratoma, 1; carcinosarcoma, 3; squamous cell carcinoma, 1; and mixed, 1)), to assess treatment strategies and useful biomarkers for malignant ovarian tumors. Overall, actionable gene variants were found in 90.9%, and druggable gene variants were found in 40.9% of the cases. Actionable BRCA1 and BRCA2 variants were found in 4.5% of each of the cases. ERBB2 amplification was found in 33.3% of mucinous carcinoma cases. Druggable hypermutation/ultramutation (tumor mutation burden ≥ 10 SNVs/Mbp) was found in 7.4% of high‐grade serous carcinoma, 46.7% of endometrioid carcinoma, 10% of clear cell carcinoma, 0% of mucinous carcinoma, 25% of undifferentiated carcinoma, and 33.3% of the other cancer cases. Copy number alterations were significantly higher in high‐grade serous carcinoma (P < .005) than in other histologic subtypes; some clear cell carcinoma showed high copy number alterations that were correlated with advanced stage (P < .05) and worse survival (P < .01). A high count of copy number alteration was associated with worse survival in all malignant ovarian tumors (P < .05). Our study shows that targeted agents can be detected in approximately 40% of malignant ovarian tumors via multigene panel testing, and copy number alteration count can be a useful marker to help assess risks in malignant ovarian tumor patients.
Keywords: Actionable gene alteration, Clinical sequencing, Druggable gene alteration, Ovarian cancer, Precision medicine
Next‐generation sequencing‐based panel tests on 88 malignant ovarian tumors revealed that targeted agents can be detected in approximately 40% of malignant ovarian tumors. Copy number alteration count was found to be a useful marker to help assess risks in malignant ovarian tumor patients.

1. INTRODUCTION
Ovarian cancer is a devastating disease with a 5‐year survival rate of approximately 40%, and this survival rate has not improved in the past 30 years. 1 Approved molecular targeted drugs include antivascular endothelial growth factor monoclonal antibody, bevacizumab, and poly (ADP‐ribose) polymerase (PARP) inhibitors; olaparib, niraparib, and rucaparib. Currently, there is no established biomarker for bevacizumab, which can prolong progression‐free survival (PFS) for several months; however, cancer usually regrows after or during bevacizumab maintenance therapy. PARP inhibitors are effective in ovarian cancers with homologous recombination deficiency (HRD), 40% of which are attributed to BRCA1/BRCA2 germline or somatic variants. Novel biomarker‐based molecular target drugs are needed for high‐grade serous carcinoma (HGSC) without HRD, and other ovarian cancer histotypes, including endometrioid carcinoma (EC), clear cell carcinoma (CCC), and mucinous carcinoma (MC).
Next‐generation sequencing‐based genomic testing is currently used in clinical settings. MSK‐IMPACT and Foundation One CDx were approved by the US Food and Drug Administration. In Japan, Foundation One CDx and NCC OncoPanel received insurance coverage approval for advanced cancer patients who had progressive diseases after standard of care. However, only a low percentage of the patients undergo therapeutics recommended by genomic testing. It is reported that of the 59.4%‐85% of patients with actionable gene aberrations, only 13.3%‐24% received molecular targeted therapy. 2 , 3 We previously reported that only 10% of pancreatic cancer patients who underwent a targeted amplicon exome sequencing for 160 cancer‐related genes (PleSSision‐160) could be treated with therapeutic agents based on the results of genomic testing. 4 Clinical sequencing for ovarian cancer is necessary due to its poor prognosis; however, reports regarding ovarian cancer are scarce. 2 , 5 , 6 Currently, there is no comprehensive report on genomic testing of malignant ovarian tumors. Here, we report the results of PleSSision‐160 in malignant ovarian tumors, including four major histologic subtypes and rare tumor histologies, such as immature teratoma and carcinosarcoma.
2. MATERIALS AND METHODS
2.1. Patient population
This study included patients with malignant ovarian neoplasm who underwent surgery from January 2012 to September 2017 at the Keio University Hospital. The study protocol was approved by the ethics committee of Keio University (2007081, 20180214). All study participants provided informed consent. This study was performed following all relevant guidelines and regulations.
2.2. Next‐generation sequencing‐based multiplex gene assay (PleSSision‐160)
Frozen tissue samples collected from the patients who underwent surgeries were fixed using the PAXgene Tissue System (QIAGEN, Germantown, MD, USA) and embedded in paraffin. A pathologist evaluated tumor cell content by staining slides with hematoxylin and eosin, and macro‐dissected, if necessary. Genomic DNA was extracted and purified, and the DNA quality was checked based on the DNA integrity number (DIN) score calculated using the Agilent 2000 TapeStation (Agilent Technologies, Waldbronn, Germany). Thereafter, DNA libraries were prepared for genome sequencing of DNA with a DIN score over 3.1. Subsequently, we performed targeted amplicon exome sequencing for 160 cancer‐related genes (Table S1) using the Illumina MiSeq sequencing platform (Illumina, San Diego, CA, USA). Genome annotation and curation for analyzing the sequencing data were performed using an original bioinformatics pipeline called GenomeJack (Mitsubishi Space Software, Tokyo, Japan). We identified cancer‐specific somatic gene alterations, such as single nucleotide variations (SNVs), insertions/deletions (indels), and copy number variations (CNVs), as previously described. 7 Furthermore, the tumor mutation burden (TMB) and copy number alterations (CNAs) were calculated using these data. The detailed method used for counting CNA is as follows; for calculating the baseline data used for count correction per amplicon, the number of leads sequenced in each of the 160‐panel amplicon probe design domains was counted to calculate the reads per million (RPM) value (ie, the number of leads per one million sequence leads). Then, the RPM coefficient of variation (CV), mean, and median value per amplicon in at least 100 formalin‐fixed paraffin‐embedded (FFPE) samples were counted. Thereafter, the RPM median of the amplicons with CV < 0.32 and mean > 10 was set as the baseline. The number of leads sequenced in each of the 160‐panel amplicon probe design domains was counted in the sample to calculate the copy number (CN) of each sample (calculating CN value to calculate the RPM value). Then, the baseline ratio {log2 ratio [= log2 (sample RPM/baseline RPM. median)]} in the amplicons that satisfied the conditions of CV < 0.32 and mean > 10 were counted, and the overall SD and median value of log2 ratio for each gene were calculated. The genes with a log2 ratio median value exceeding SD were categorized as amplification (amp)‐like, and those exceeding 2SD were categorized as amp. In addition, the genes with a log2 ratio median value below ‐SD were categorized as loss‐like, and those below −2SD were categorized as loss. As a control, 50 genomic DNA samples were used to normalize the read depth per amplicon. Only amplicons with CV of the depth ≤ 1.5 were used. CNA was calculated for the genes that had more than six amplicons, and it was measured as the median value of each amplicon per gene. The analysis reports were discussed and reviewed in a conference of genome experts consisting of medical oncologists, molecular oncologists, pathologists, medical geneticists, clinical laboratory technicians, and bioinformaticians. Hypermutation was defined as ≥ 10 SNVs/Mbp, and ultramutation was defined as ≥ 100 SNVs/Mbp.
2.3. Statistical analyses
Statistical analyses were performed with GraphPad Prism 7 (GraphPad Software, Inc, San Diego, CA, USA).
3. RESULTS
3.1. Patient characteristics
Sequencing was performed for 88 ovarian malignant neoplasms (HGSC: 27, EC: 15, CCC: 30, MC: 6, undifferentiated carcinoma (UC): 4, and others: 6 (immature teratoma: 1, carcinosarcoma: 3, squamous cell carcinoma: 1, and mixed (serous + clear cell): 1)). Overall, the median age was 55 (range: 28‐86), clinical stages were I: 41, II: 6, III: 32, IV: 9, and the tumors with recurrence were present in 30 cases (34.1%). The median PFS and median overall survival (OS) were 1172 days (range: 35‐2695) and 1396.5 days (range: 111‐2695) respectively. Advanced stage (III or IV) was 96.3% of HGSC, 13.3% of EC, 20.0% of CCC, 33.3% of MC, 75.0% of UC, and 33.3% of others (Table 1).
Table 1.
Patient characteristics
| HGSC (n = 27) | EC (n = 15) | CCC (n = 30) | MC (n = 6) | UC (n = 4) |
Others (immature teratoma 1, caricinosarcoma 3, SCC 1, mixed 1) (n = 6) |
All (n = 88) |
|
|---|---|---|---|---|---|---|---|
| Age | 55 (36‐85) | 51 (34‐86) | 50 (41‐77) | 63.5 (48‐67) | 58 (28‐65) | 55.5 (37‐69) | 55 (28‐86) |
| Stage |
I: 0 II: 1 III: 20 IV: 6 |
I: 12 II: 1 III: 1 IV: 1 |
I: 23 II: 1 III: 6 IV: 0 |
I: 4 II: 0 III: 1 IV: 1 |
I: 1 II: 0 III: 2 IV: 1 |
I: 1 II: 3 III: 2 IV: 0 |
I: 41 II: 6 III: 32 IV: 9 |
| Recurrence |
Yes: 18 No: 9 |
Yes: 1 No: 14 |
Yes: 6 No: 24 |
Yes: 2 No: 4 |
Yes: 1 No: 3 |
Yes: 2 No: 4 |
Yes: 30 No: 58 |
| PFS (Days) |
712 (35‐2423) |
1813 (494‐2695) |
1545.5 (266‐2602) |
960.5 (56‐1871) |
2001 (256‐2690) |
2086 (377‐2623) |
1172 (35‐2695) |
| OS (Days) |
895 (111‐2423) |
1813 (594‐2695) |
1562.5 (432‐2602) |
960.5 (114‐1871) |
2001 (760‐2690) |
2086 (760‐2623) |
1396.5 (111‐2695) |
3.2. Actionable gene alterations and druggable gene alterations in malignant ovarian tumors
We successfully sequenced genomic DNA from 88 patients with a mean sequencing depth of 978.5x (717x–1352x). Actionable gene alterations were identified in 80 of 88 samples (90.9%): HGSC, 92.6%; EC, 100.0%; CCC, 90.0%; MC, 83.3%; UC, 75.0%; and others, 83.3% (Table 2). Druggable gene alterations were identified in 36 of 88 samples (40.9%): HGSC, 14.8%; EC, 66.7%; CCC, 43.3%; MC, 66.7%; UC, 25.0%; and others, 66.7% (Table 2). Among the patients, 3.4% were identified for Level A drug recommendation, 17.0% were identified for Level B drug, 19.3% were identified for Level C drug, 1.1% were identified for Level D drug, and 59.1% had no drug recommendation (Figure 1). 8
Table 2.
Actionable and druggable alterations identified
| . | HGSC (n = 27) | EC (n = 15) | CCC (n = 30) | MC (n = 6) | UC (n = 4) | Others (n = 6) |
Total (n = 88) |
|---|---|---|---|---|---|---|---|
| Actionable alterations | |||||||
| Yes (n (%)) | 25 (92.6%) | 15 (100.0%) | 27 (90.0%) | 5 (83.3%) | 3 (75.0%) | 5 (83.3%) | 80 (90.9%) |
| No (n (%)) | 2 (7.4%) | 0 (0.0%) | 3 (10.0%) | 1 (16.7%) | 1 (25.0%) | 1 (16.7%) | 8 (9.1%) |
| Druggable alterations | |||||||
| Yes (n (%)) | 4 (14.8%) | 10 (66.7%) | 13 (43.3%) | 4 (66.7%) | 1 (25.0%) | 4 (66.7%) | 36 (40.9%) |
| No (n (%)) | 23 (85.2%) | 5 (33.3%) | 17 (56.7%) | 2 (33.3%) | 3 (75.0%) | 2 (33.3%) | 52 (59.1%) |
Figure 1.
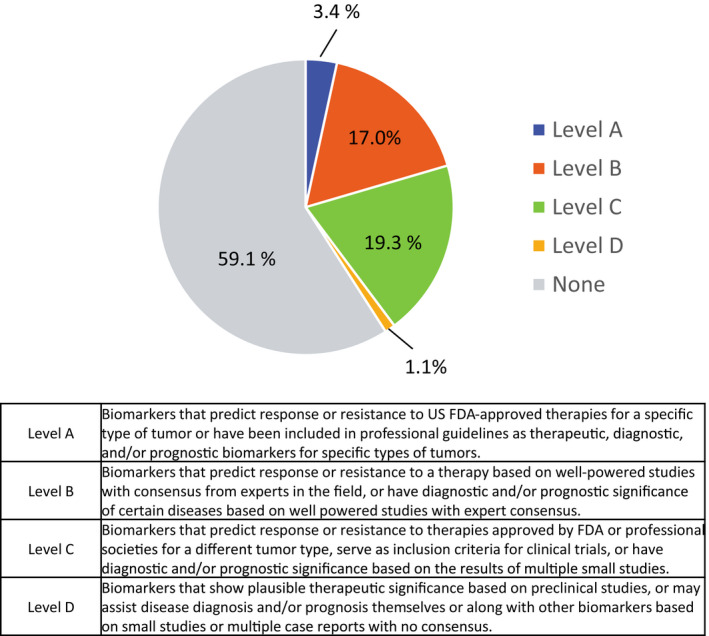
Categories of drug recommendation levels for all patients
Regarding the actionable gene variants in mismatch repair (MMR) genes, the MSH6 actionable gene variant was identified in 3.3% of CCC and MSH2 was identified in 6.7% of EC (Figure 2A, B). Actionable MLH1 or PMS2 gene variant was not observed in all the subtypes analyzed. For the genes responsible for HRD, ATM actionable gene variant was identified in 33.3% of EC, BRCA1 was identified in 14.8% of HGSC, and BRCA2 was identified in 20.0% of EC and 3.3% of CCC. BRIP1 and PALB2 actionable gene variants were not identified; however, a variant of unknown significance (VUS) was found in some cases (Figure 2A, B).
Figure 2.
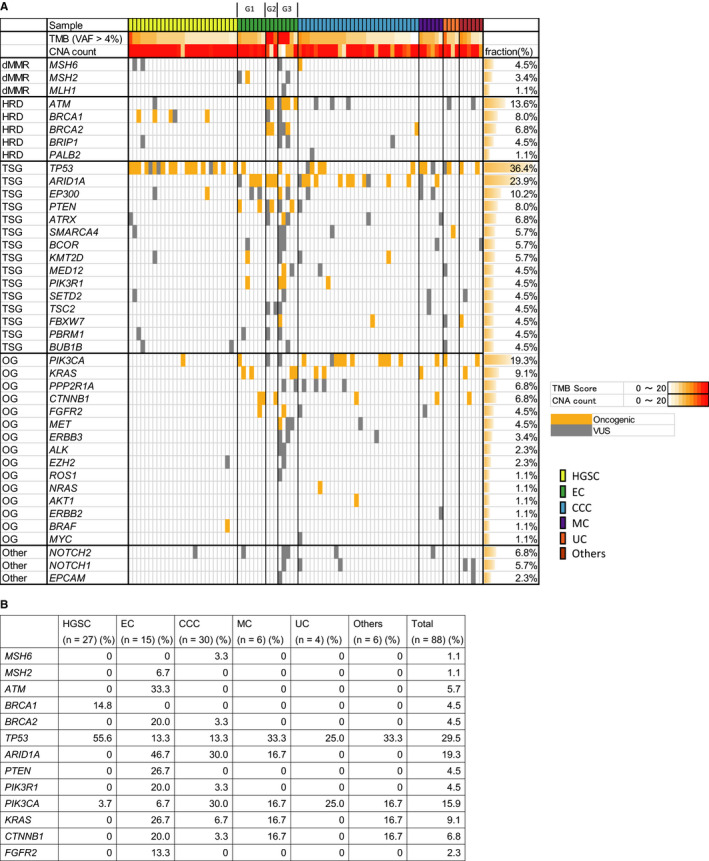
Actionable gene variants identified in malignant ovarian tumors. Oncogenic variant and variant of unknown significance (VUS) are depicted (A). List of extracted actionable gene variants in malignant ovarian tumors (B). TMB: tumor mutation burden; VAF: variant allele frequency; CNA: copy number alteration; dMMR: deficient of mismatch repair; HRD: homologous recombination deficiency; TSG: tumor suppressor gene; OG: oncogene; HGSC: high‐grade serous carcinoma; EC: endometrioid carcinoma; CCC: clear cell carcinoma; MC: mucinous carcinoma; UC: undifferentiated carcinoma
TP53 actionable gene variant was identified in 55.6% of HGSC, 13.3% of EC, 13.3% of CCC, 33.3% of MC, 25.0% of UC, and 33.3% of others. If VUS was included, TP53 alteration was identified in 66.7% of HGSC. ARID1A actionable gene variant was identified in 0% of HGSC, 46.7% of EC, 30.0% of CCC, 16.7% of MC, 0% of UC, and 0% of others. PTEN actionable gene variant was identified only in EC (26.7%). PIK3CA actionable gene variant was identified in 3.7% of HGSC, 6.7% of EC, 30.0% of CCC, 16.7% of MC, 25.0% of UC, and 16.7% of others. KRAS actionable gene variant was identified in 26.7% of EC, 6.7% of CCC, 16.7% MC, and 16.7% of others. FGFR2 actionable gene variant was identified only in EC (13.3%) (Figure 2A, B).
CNV showed frequent loss of MSH6 (43.2%), CTNNB1 (53.4%), APC (73.9%), and BRCA2 (46.6%) (Figure 3). ERBB3 amplification was identified in 11.4% of cases, of which 1 MC case showed CN = 12.5. Druggable ERBB3 amplification was identified in 16.7% of MC (Figure 3, Table 3). ERBB2 amplification was identified in 15.9% of cases, of which 1 MC case showed CN = 8.6 and 1 EC case showed CN = 38.0. Druggable ERBB2 amplification was identified in 33.3% of MC (Figure 3, Table 3). SMARCA4 (BRG1) amplification was identified in 28.4% of the overall cases (Figure 3). CNV of 160 genes showed frequent gain of genes in 3q, 6p, 7q, and 12p and frequent loss in 6q, 9q, 13q, and 17q in HGSC, which was concordant with The Cancer Genome Atlas (TCGA) data 9 (Figure S1). Focal loss of NF1 was identified in 48.1% of HGSC (Figure 3), which was also reported in TCGA as focal deletion. KRAS was amplified in 44.4% of HGSC and 20% of CCC (Figure 3).
Figure 3.
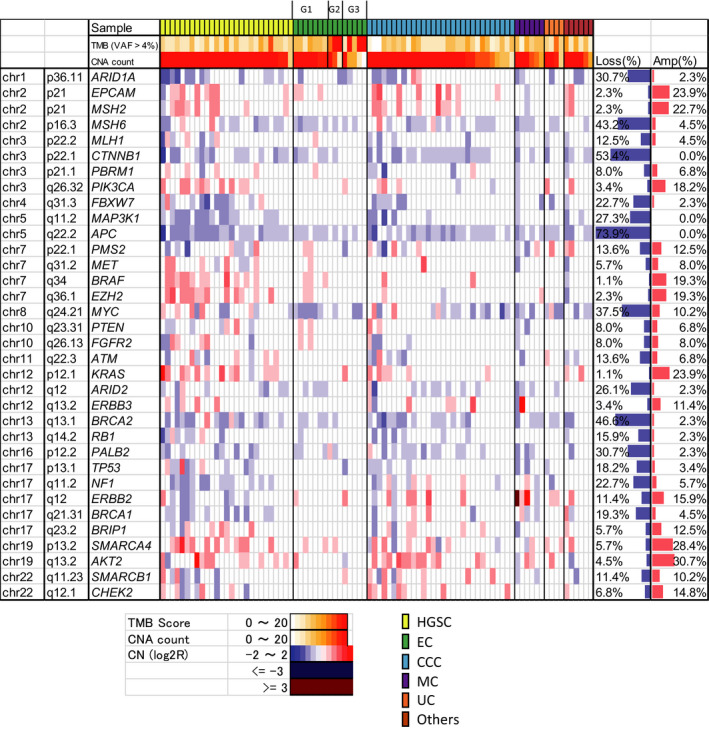
Copy number alterations (CNA) identified in malignant ovarian tumors. TMB: tumor mutation burden; VAF: variant allele frequency; CNA: copy number alteration; chr: chromosome; amp: amplification; CN: copy number; HGSC: high‐grade serous carcinoma; EC: endometrioid carcinoma; CCC: clear cell carcinoma; MC: mucinous carcinoma; UC: undifferentiated carcinoma
Table 3.
Druggable alterations in malignant ovarian tumors
| HGSC (n = 27) (n (%)) | EC (n = 15) (n (%)) | CCC (n = 30) (n (%)) | MC (n = 6) (n (%)) | UC (n = 4) (n (%)) | Others (n = 6) (n (%)) |
Total (n = 88) (n (%)) |
Drugs (Level of recommendation) | |
|---|---|---|---|---|---|---|---|---|
| Hypermutation | 2 (7.4%) | 6 (40%) | 3 (10.0%) | 0 (0%) | 1 (25.0%) | 2 (33.3%) | 14 (15.9%) | ICI (B) |
| Ultramutation | 0 (0%) | 1 (6.7%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (1.1%) | ICI (B) |
| BRCA1 | 1 (3.7%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (1.1%) | PARP inhibitor (A), Platinum (B) |
| BRCA2 | 0 (0%) | 0 (0%) | 1 (3.3%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (1.1%) | PARP inhibitor (A), Platinum (B) |
| PIK3CA | 0 (0%) | 1 (6.7%) | 8 (26.7%) | 1 (16.7%) | 1 (25.0%) | 1 (16.7%) | 12 (13.6%) | mTOR inhibitor (C), PI3K inhibitor (C), AKT inhibitor (C) |
| CTNNB1 | 0 (0%) | 3 (20.0%) | 1 (3.3%) | 1 (16.7%) | 0 (0%) | 1 (16.7%) | 6 (6.8%) | Imatinib (C), CWP232291 (C) |
| MSH2 | 0 (0%) | 1 (6.7%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (1.1%) | ICI (A) |
| FGFR2 | 0 (0%) | 1 (6.7%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (1.1%) | FGFR inhibitor (D) |
| PTEN | 0 (0%) | 1 (6.7%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (1.1%) | mTOR inhibitor (C), AKT inhibitor (3A) |
| AKT1 | 0 (0%) | 0 (0%) | 1 (3.3%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (1.1%) | mTOR inhibitor (C), AKT inhibitor (C) |
| AKT2 amplification | 1 (3.7%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (1.1%) | mTOR inhibitor (C), AKT inhibitor (C) |
| MET amplification | 0 (0%) | 0 (0%) | 1 (3.3%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (1.1%) | MET inhibitor (C) |
| ERBB2 amplification | 0 (0%) | 0 (0%) | 0 (0%) | 2 (33.3%) | 0 (0%) | 0 (0%) | 2 (2.3%) | HER2 inhibitor (C) |
| ERBB3 amplification | 0 (0%) | 0 (0%) | 0 (0%) | 1 (16.7%) | 0 (0%) | 0 (0%) | 1 (1.1%) | U3‐1402 (D) |
Categories of drug recommendation levels are depicted in Figure 1. ICI, immune checkpoint inhibitors.
Druggable gene alterations were identified in 40.9% of the cases. Among them, hypermutation was found in 7.4% of HGSC, 40.0% of EC, 10.0% of CCC, 0% of MC, 25.0% of UC, and 33.3% of others; while ultramutation was found only in EC (6.7%). Druggable BRCA1 alteration was found in only HGSC (3.7%), and druggable BRCA2 variant was found in only CCC (3.3%). For the tumor suppressor genes, variants were not considered druggable if loss of heterogeneity or uniparental disomy did not occur. Druggable PIK3CA variant was identified in 0% of HGSC, 6.7% of EC, 26.7% of CCC, 16.7% of MC, 25.0% of UC, and 16.7% of others. Druggable MSH2 variant was found in only 1 case of EC (6.7%). Druggable FGFR2 gene variant was identified in 6.7% of EC AKT2 amplification was identified in 3.7% of HGSC, and MET amplification was identified in 3.3% of CCC (Table 3).
3.3. TMB and CNA of malignant ovarian tumors
The median TMB was as follows: HGSC, 6.7 (range 2.7‐54); EC, 9.4 (5.4‐159.8); CCC, 6.7 (1.3‐12.1); MC, 8.1 (2.7‐10.7); UC, 6.7 (4‐14.8); and others, 6.75 (2.7‐12.1). EC had significantly higher TMB than the other histologic subtypes (P = .0012, one‐way ANOVA) (Figure 4A). The median CNA count was as follows: HGSC, 53 (range: 6‐93); EC, 18 (1‐55); CCC, 24 (5‐74); MC, 24.5 (11‐108); UC, 24.5 (7‐42); and others, 36 (10‐58). HGSC had significantly higher CNA count than the other tumor types (P = .0028, one‐way ANOVA) (Figure 4B). CNAs ≥ 40 were identified in 63.0% (17/27) of HGSC, 6.7% (1/15) of EC, 30% (9/30) of CCC, 33.3% (2/6) of MC, 25.0% (1/4) of UC, and 50% (3/6) of others. In CCC, advanced stage (II, III, or IV) was significantly correlated with high CNA count (P = .0187, t‐test) (Figure 5A), and high CNA count (CNAs ≥ 30) CCC tumors showed worse survival (PFS: Hazard Ratio (HR) 0.10, P = .0087; OS: HR 0.06, P = .0078, log‐rank test) (Figure 5B). In addition, the advanced stage (II, III, or IV) was correlated with high CNA count (P = .0004, t‐test) when all malignant ovarian tumors were included in the analysis (Figure 5C). High CNA count was correlated with worse survival in all malignant tumors (PFS: HR 0.28, P = .0007; OS: HR 0.30, P = .0146) (Figure 5D). In HGSC, high CNA count was insignificantly correlated with worse survival (P = .16 for PFS, P = .14 for OS) (Figure S2). In EC and MC, high CNA count was not related to survival, possibly due to the low rate of relapse and an insignificant number of cases analyzed (Figure S2).
Figure 4.
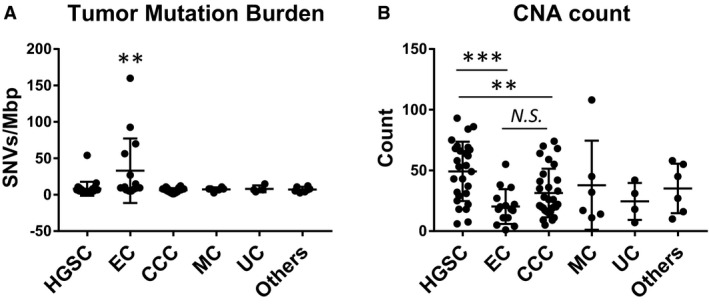
Tumor mutation burden (TMB) of malignant ovarian tumors. SNVs/Mbp. **, P < .01. (A). Copy number alteration (CNA) count of malignant ovarian tumors. **, P < .01; ***, P < .005; N.S.: not significant. (B). HGSC: high‐grade serous carcinoma; EC: endometrioid carcinoma; CCC: clear cell carcinoma; MC: mucinous carcinoma; UC: undifferentiated carcinoma
Figure 5.
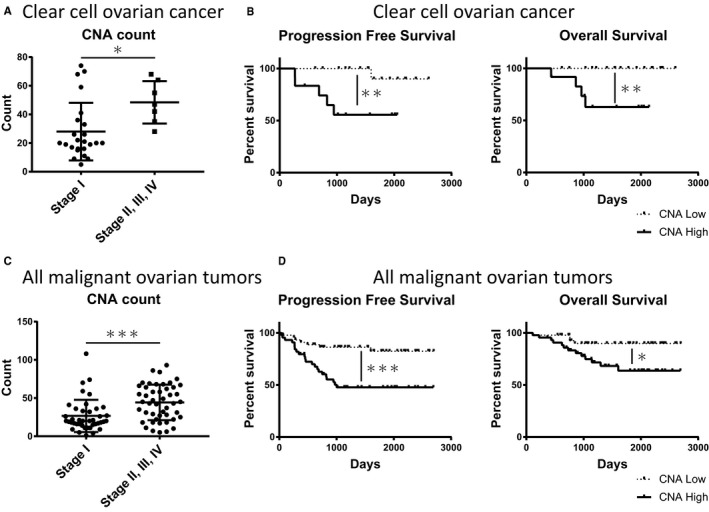
Copy number alteration (CNA) count of clear cell ovarian cancers in stage I and advanced stage (II, III, IV). *, P < .05. (A). Progression‐free survival (PFS) and overall survival (OS) analysis in CNA high (CNA count ≥ 30) and CNA low (CNA count < 30) clear cell ovarian cancers. **, P < .01. (B). CNA count of all malignant ovarian tumors in stage I and advanced stage (II, III, IV). ***, P < .005. (C). PFS and OS analysis of CNA high (CNA count ≥ 30) and CNA low (CNA count < 30) for all malignant ovarian tumors. *, P < .05; ***, P < .005. (D)
4. DISCUSSION
Paclitaxel and carboplatin (platinum) combination therapy is primarily used for ovarian cancers regardless of the histologic subtype. However, advanced CCC and MC are resistant to standard platinum combination regimens compared with HGSC or EC. 10 , 11 Even in the same pathological diagnosis of ovarian cancer, genomic alterations are shown to be different. 9 , 12 Thus, treating ovarian cancer patients based on genomic biomarkers is necessary to improve outcomes. In our study using targeted capture sequencing of 160 cancer‐related genes, we found that 40.9% of malignant ovarian tumors had druggable alterations; this is similar to the result of MSK‐IMPACT in which clinically actionable gene alteration was detected in 37% of solid tumors. 5
Actionable variants in MMR genes (MLH1, MSH2, MSH6, and PMS2) were not common in ovarian cancer. Only 1 CCC (3.3%) had MSH6 variant, and only 1 EC (6.7%) had MSH2 variant. Overall, in our study, 2.3% of malignant ovarian tumors harbored MMR gene variant (excluding VUS), which is almost concordant with a previous report that ovarian cancer has 1.3%–5% of MMR gene variant. 13 , 14 Actionable ATM gene variant was found in 33.3% of EC and was not detected in other histological types. ATM germline variant was found in only 0.9% of ovarian cancers 15 ; our study indicates that ATM somatic variant may be common in EC. Actionable BRCA1 variant was found in only HGSC (14.8%), while actionable BRCA2 variant was found in 20.0% of EC and 3.3% of CCC. It was reported that 8.9% and 3.0% of ovarian cancer have BRCA1 and BRCA2 somatic variants, respectively. 16 Sugino et al reported BRCA1 or BRCA2 (BRCA1/2) somatic variants in 5% of both CCC and EC. 13 Our study also revealed that BRCA2 somatic variant might contribute to the development of nonserous ovarian cancer except MC.
TCGA showed high CNAs, which is a hallmark of HGSC. 9 In the present study, CNA count ≥ 40 was identified in 63.0% of HGSC and 30.0% of CCC, indicating that some populations in CCC have high CNAs. CCC with high CNA correlated with advanced‐stage tumor and worse survival, this was consistent with previous findings that CNA‐high breast and prostate tumors indicate worse survival. 17 , 18 One comparative genomic hybridization analysis of CCC found that amplification in 3 of the 4 chromosomal regions (8p11.21, 8p11.22, 12p13.31, and 20q13.2) had shorter OS. 19 Our findings revealed that CNAs across the genome might be biomarkers of malignant ovarian tumors, including CCC. In addition, we found that the clinical target sequencing of 160 genes effectively allows the calculation of the CNA count, which can be a prognostic biomarker. PARP inhibitor was shown to be effective, especially in ovarian cancer with HRD or BRCA1/2 mutation. 20 , 21 These clinical trials mainly targeted HGSC and EC. It has not been clarified if PARP inhibitors are also effective in treating CCC or MC. As CNAs can be considered a reflection of HRD, these CNA high CCCs may be treatable with PARP inhibitors. Germline or somatic HRD gene mutation was identified in 44% of HGSC, 28% of CCC, 23% of EC, and 16% of MC. 13
SWItch/sucrose non‐fermentable (SWI/SNF) gene variants are common in endometriosis‐associated ovarian cancer, including CCC and EC. We found actionable ARID1A variant in 46.7% of EC and 30.0% of CCC, which is consistent with previous findings. 22 PIK3CA actionable variant was identified in 3.7% of HGSC, 6.7% of EC, 30.0% of CCC, 16.7% of MC, 25.0% of UC, and 16.7% of others. It was reported that 2.3% of HGSC, 20.0% of EC, 20.0% of CCC, 0% of MC, and 7.1% of UC had PIK3CA variant. 23 For EC, 61% have mutations in the PI3K pathway genes, including PIK3CA, PTEN, PIK3R1, PIK3R2, and AKT1. 24 Our results confirmed that PIK3CA is druggable in certain populations and mostly targetable in nonserous ovarian cancer.
KRAS actionable mutation was found in 0% of HGSC, 26.7% of EC, 6.7% of CCC, 16.7% of MC, 0% of UC, and 16.7% of others. It was reported that 79.0% of MC and 0.6% of HGSC have KRAS mutation. 25 Recently, KRAS mutation was found in 42% of EC. 24 Although KRAS is the most frequently mutated oncogene in human cancers, no therapeutic agent directly targeting RAS has yet been approved. Our results show that KRAS was amplified in 44.4% of HGSC, and it was reported to be significantly locally amplified in TCGA. 9 The development of KRAS inhibitor is under progress 26 , 27 ; thus, targeting KRAS will be a future option for ovarian cancer.
Druggable ERBB2 amplification was found in 33.3% of MC, and druggable ERBB3 amplification was found in 16.7% of MC. In MC, 19% were reported to have ERBB2 amplification 28 ; thus, HER2 inhibitors, such as HER2 ADC (trastuzumab‐deruxtecan), may be effective for such tumors. Our results indicate that targeting HER2 (ERBB2) or HER3 (ERBB3) can be considered in certain populations of MC. Druggable MET amplification was found only in 3.3% of CCC. MET inhibitor showed high response rate (50%) in a genomic matched trial of gastric cancer, which indicates that MET inhibitor can be considered for tumors with MET amplification. 29
Hypermutation or ultramutation was found in 7.4% of HGSC, 46.7% of EC, 10.0% of CCC, 0% of MC, 25.0% of UC, and 33.3% of others. Although microsatellite instability‐high (MSI‐H) tumors are rarely seen in ovarian cancer, TMB indicates that about 50% of EC and 10% of HGSC or CCC can be treated with immune checkpoint blockade. A clinical trial on urothelial carcinoma revealed that TMB is associated with response rate to a greater extent than with PD‐L1 expression. 30
Overall, targeted capture‐based genomic sequencing identified druggable alterations in about 40% of malignant ovarian tumors. It was reported that patients who received genomically matched therapy have longer PFS than the previous PFS 3 ; thus, genomic testing can be applied in clinical settings to provide treatment strategies for malignant ovarian tumors. Our findings also demonstrate that CNA count could be a useful marker in clinical settings for risk assessment of patients with malignant ovarian tumors.
CONFLICT OF INTEREST
ST, SN, and CO are employees of Mitsubishi Space Software Co., Ltd. All other authors declare no conflict of interest regarding this manuscript.
AUTHOR CONTRIBUTION
KS carried out experiments, performed data analysis, and wrote the manuscript. TC contributed to the study design, experiments, results interpretation, and manuscript writing. EA, KN, HH, and HN contributed to the interpretation of results and data analysis. ST, SN, and CO performed data analysis. YK, HN, NS, TI, WY, FK, and DA contributed the study design and sample collection. All authors reviewed and collected the manuscript.
Supporting information
Fig S1
Fig S2
Table S1
ACKNOWLEDGMENTS
This work was supported in part by the Japan Society for the Promotion of Science (JSPS), KAKENHI Grant Number 16K11156, 17K19613, and 19K18651. The authors thank Emmy Yanagita and Ryohei Matsuoka for technical assistance with preparing the NGS library.
Saotome K, Chiyoda T, Aimono E, et al. Clinical implications of next‐generation sequencing‐based panel tests for malignant ovarian tumors. Cancer Med. 2020;9:7407–7417. 10.1002/cam4.3383
DATA AVAILABILITY STATEMENT
The datasets generated/analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Vaughan S, Coward JI, Bast RC, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011;11(10):719‐725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sunami K, Ichikawa H, Kubo T, et al. Feasibility and utility of a panel testing for 114 cancer‐associated genes in a clinical setting: a hospital‐based study. Cancer Sci. 2019;110(4):1480‐1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dalton WB, Forde PM, Kang H, et al. Personalized medicine in the oncology clinic: implementation and outcomes of the johns hopkins molecular tumor board. JCO Precis Oncol. 2017;2017(1):1‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hayashi H, Tanishima S, Fujii K, et al. Genomic testing for pancreatic cancer in clinical practice as real‐world evidence. Pancreatology. 2018;18(6):647‐654. [DOI] [PubMed] [Google Scholar]
- 5. Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hainsworth JD, Meric‐Bernstam F, Swanton C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from mypathway, an open‐label, phase IIa multiple basket study. J Clin Oncol. 2018;36(6):536‐542. [DOI] [PubMed] [Google Scholar]
- 7. Tsumura K, Arai E, Tian Y, et al. Establishment of permutation for cancer risk estimation in the urothelium based on genome‐wide DNA methylation analysis. Carcinogenesis. 2019;40(11):1308‐1319. [DOI] [PubMed] [Google Scholar]
- 8. Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the association for molecular pathology, American society of clinical oncology, and college of American pathologists. J Mol Diagn. 2017;19(1):4‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Integrated genomic analyses of ovarian carcinoma . Nature. 2011;474:609‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sugiyama T, Kamura T, Kigawa J, et al. Clinical characteristics of clear cell carcinoma of the ovary: a distinct histologic type with poor prognosis and resistance to platinum‐based chemotherapy. Cancer. 2000;88(11):2584‐2589. [PubMed] [Google Scholar]
- 11. Perren TJ. Mucinous epithelial ovarian carcinoma. Ann Oncol. 2016;27(Suppl 1):i53‐i57. [DOI] [PubMed] [Google Scholar]
- 12. Itamochi H, Oishi T, Oumi N, et al. Whole‐genome sequencing revealed novel prognostic biomarkers and promising targets for therapy of ovarian clear cell carcinoma. Br J Cancer. 2017;117(5):717‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sugino K, Tamura R, Nakaoka H, et al. Germline and somatic mutations of homologous recombination‐associated genes in Japanese ovarian cancer patients. Sci Rep. 2019;9(1):17808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Song H, Cicek MS, Dicks E, et al. The contribution of deleterious germline mutations in BRCA1, BRCA2 and the mismatch repair genes to ovarian cancer in the population. Hum Mol Genet. 2014;23(17):4703‐4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hirasawa A, Imoto I, Naruto T, et al. Prevalence of pathogenic germline variants detected by multigene sequencing in unselected Japanese patients with ovarian cancer. Oncotarget. 2017;8(68):112258‐112267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hennessy BT, Timms KM, Carey MS, et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol. 2010;28(22):3570‐3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang L, Feizi N, Chi C, Hu P. Association analysis of somatic copy number alteration burden with breast cancer survival. Front Genet. 2018;9:421‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hieronymus H, Murali R, Tin A, et al. Tumor copy number alteration burden is a pan‐cancer prognostic factor associated with recurrence and death. eLife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Morikawa A, Hayashi T, Kobayashi M, et al. Somatic copy number alterations have prognostic impact in patients with ovarian clear cell carcinoma. Oncol Rep. 2018;40(1):309‐318. [DOI] [PubMed] [Google Scholar]
- 20. Moore K, Colombo N, Scambia G, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2018;379(26):2495‐2505. [DOI] [PubMed] [Google Scholar]
- 21. González‐Martín A, Pothuri B, Vergote I, et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2019;381(25):2391‐2402. [DOI] [PubMed] [Google Scholar]
- 22. Wiegand KC, Shah SP, Al‐Agha OM, et al. ARID1A mutations in endometriosis‐associated ovarian carcinomas. N Engl J Med. 2010;363(16):1532‐1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Campbell IG, Russell SE, Choong DY, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004;64(21):7678‐7681. [DOI] [PubMed] [Google Scholar]
- 24. Cybulska P, Paula ADC, Tseng J, et al. Molecular profiling and molecular classification of endometrioid ovarian carcinomas. Gynecol Oncol. 2019;154(3):516‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mueller JJ, Schlappe BA, Kumar R, et al. Massively parallel sequencing analysis of mucinous ovarian carcinomas: genomic profiling and differential diagnoses. Gynecol Oncol. 2018;150(1):127‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kessler D, Gmachl M, Mantoulidis A, et al. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci USA. 2019;116(32):15823‐15829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Janes MR, Zhang J, Li L‐S, et al. Targeting KRAS mutant cancers with a covalent G12C‐specific inhibitor. Cell. 2018;172(3):578‐589.e17. [DOI] [PubMed] [Google Scholar]
- 28. Mishra R, Hanker AB, Garrett JT. Genomic alterations of ERBB receptors in cancer: clinical implications. Oncotarget. 2017;8(69):114371‐114392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee J, Kim ST, Kim K, et al. Tumor genomic profiling guides patients with metastatic gastric cancer to targeted treatment: The VIKTORY umbrella trial. Cancer Discov. 2019;9(10):1388‐1405. [DOI] [PubMed] [Google Scholar]
- 30. Rosenberg JE, Hoffman‐Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum‐based chemotherapy: a single‐arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909‐1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Table S1
Data Availability Statement
The datasets generated/analyzed during the current study are available from the corresponding author on reasonable request.