

Abstract
Background:
Myeloid cell leukemia-1 (Mcl-1) plays an important role in the clearance of Mycobacterium tuberculosis (MTB) infection. It has the effect of anti-apoptosis, protecting macrophages that have engulfed pathogens and preventing pathogen clearance. Meanwhile, the MAPK signaling pathway plays a significant role in regulating Mcl-1 expression during tuberculosis infection. In the case of latent infection and active infection, the apoptosis and polarization of macrophages have a great influence during MTB infection, so we discussed the effect of Mcl-1 on apoptosis and polarization. Then, further discussed its mechanism.
Methods:
An infected RAW264.7 macrophage model was established to investigate the regulatory role and mechanism of the Mcl-1 pathway inhibition during apoptosis and polarization of H37Rv infection. First, Mcl-1 protein and mRNA was identified by western blotting and Real-Time Polymerase Chain Reaction (RT-PCR). RAW264.7 macrophage apoptosis was detected by flow cytometry. RT-PCR was utilized to detect Bax, Caspase-3, Cyt-c and Bcl-2 mRNA expression. Next, Then the expression levels of inflammation factors CD86, CD206, iNOS, Fizz1, IL-6, IL-10, TNF-α, and TGF-β was detected by ELISA. SEM was used to observe macrophages phenotype. Finally, Bax, Bcl-2 and Bcl-xl the expression was detected by western blotting. Confocal microscopy was used to analyze mitochondrial membrane potential using the JC-10 kit.
Results:
In this study, we found that inhibiting the Mcl-1 expression signaling pathway led to infection by different virulence Mycobacterium tuberculosis, as well as changes in Mcl-1 protein and mRNA expression. Concomitantly macrophage apoptosis rate also changed, While, two phenotypic states of M1 and M2 appeared in the infected cells. We also found that the mitochondrial pathway was activated, the expression of its related genes Bax, casepase3, and Cyt-c, increased, whereas that of Bcl-2 decreased, and the mitochondrial membrane depolarization function was changed.
Conclusions:
We found that Mcl-1 affected the apoptosis and polarization of macrophages infected by Mycobacterium tuberculosis, mainly M1 in the early stage and M2 in the later stage. In addition, mitochondria played a crucial role in this process.
Keywords: Mcl-1, MAPK signaling pathway, MTB infection, macrophage, apoptosis, polarization
1. Introduction
Mycobacterium tuberculosis (MTB) is a chronic respiratory cause of human tuberculosis and is considered to be among the most harmful pathogens.[1,2] MTB is a typical intracellular parasitic pathogen, and macrophages are the first line of defense of the immune defense mechanism upon MTB infection.[3] According to the 2019 global tuberculosis report by the World Health Organization, tuberculosis epidemic still need to be closely monitored.[4] Because of its widespread distribution and high incidence in poor and remote areas, an effective treatment is of the utmost importance.
Apoptosis is initiated by 2 main pathways, namely, exogenous and internal pathways. The change in mitochondrial membrane potential induced by MTB infection is key to determine the death mode of infected macrophages.[5] Researchers[6–8] found that human neutrophil apoptosis induced by MTB depends on the MAPK signaling pathway. High Mcl-1 expression blocks by the MAPK signaling pathway, inducing apoptosis.[9–12] Our previous study[13] showed that in host macrophages infected with the H37Rv strain, small hairpin RNA molecules inhibit Mcl-1 expression and increase host apoptosis. Because the mitochondrial pathway is an important regulator of host macrophage death, we intended to elucidate the regulatory role of Mcl-1 in the MAPK signaling pathway in MTB-infected macrophages and the mechanism underlying mitochondrial-mediated apoptosis.
During MTB infection, host macrophages can show different fates and inflammatory response. The pro-inflammatory and anti-inflammatory responses to infection are extremely important to the host; thus, it is necessary to have a better understanding of the role of polarization in host macrophages.[14–18] Changes in mitochondrial function are associated with numerous human diseases, such as heart failure and metabolic diseases.[19–21] It is believed that promoting mitochondrial biosynthesis can promote M2 phenotype and reduce inflammation and apoptosis of macrophages.[13,22] However, limited information is available regarding the polarization of macrophages during MTB infection when the Mcl-1 expression signaling pathway is inhibited.
In the present study, we examined the effect of the MAPK signaling pathway on the apoptosis and polarization of macrophages infected with MTB, to explore whether the mitochondrial pathway played an important role in the process, and to further observe the fate of macrophages after MTB infection.
2. Materials and methods
2.1. MTB infection and inhibitors treatment
MTB strains were selected in this study, virulent strains including H37Rv and XJ-MTB, attenuated strains including BCG and H37Ra. Uninfected RAW264.7 was the control group. Infection RAW264.7 group was infected with different virulent MTB strains at a multiplicity of infection (MOI) of 10:1 (bacterial: cell) and incubated at 37°C in the 5% CO2 incubator. MTB infection groups were treated with specific signaling pathway inhibitors according to the instructions, AG490 (AG) to inhibit JAK/STAT, LY294002 (LY) to inhibit PI3K, PD98059 (PD) to inhibit ERK, SP600125 (SP) to inhibit JNK and SB203580 (SB) to inhibit p38. All inhibitors were applied to cells culture medium 4 hours after infection (time 0) and maintained during the course of the experiments. All inhibitors were purchased from Sigma-Aldrich.
2.2. RNA isolation and real-time PCR quantifications
Total RNA was isolated from RAW264.7 macrophages using RNeasy kit (TianGen, Beijing, China) following the manufacturer's protocol. First-strand cDNAs were synthesized using the Thermo-script RT kit (Life Technologies, Rockville, MD). Primers were synthesized by Sangon Biotech (Shanghai, China). Samples were amplified with a QuantiFast SYBR Green PCR Kit (QIAGEN, Hilden, Germany) according to the manufacturer's instructions. Relative amounts of transcripts were calculated using the 2−ΔΔCt formula. All PCR primers are reported in Table 1.
Table 1.
Primers for target genes.

2.3. Western blotting
The cells were harvested and lysed by RIPA lysis buffer (Solarbio Biotechnology, Beijing, China). RAW264.7 macrophages homogenates containing equal amounts of protein were separated by 10% to 15% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and assayed by western blotting as described previously.[3] The concentration of primary antibodies: rabbit anti-mouse Mcl-1 (Cat. No. ab32087, 1:1000), Bcl-2 (Cat. No. #2870, 1:1000), Bax (Cat. No. #14796, 1:1000), Bcl-xl (Cat. No. #2764, 1:1000) and Cx43 (Cat. No. ab79010, 1:1000) that were purchased from Cell Signaling Technology (Danvers, MA) and Abcam (CA), and mouse anti-mouse β-actin (Cat. No. TA-09, 1:1000) was purchased from ZSGB-BIO (Beijing, China). Secondary antibodies are horseradish peroxidase-conjugated goat anti-rabbit (Cat. No. ZB-2301, 1:20000) and goat anti-mouse (Cat. No. ZB-2305, 1:20000) were purchased from ZSGB-BIO (Beijing, China). Immunoblots were developed by enhanced chemiluminescent reaction (ECL, Millipore, Burlington, MA).
2.4. Annexin V-APC/ 7-AAD flow cytometry analysis
The RAW264.7 macrophages were collected and washed twice with cold PBS. Staining was performed according to the manufacturer's protocol. Cells were resuspended in cold 1x binding buffer to a concentration of 1 × 106, add 100ul of cells to each tube, then 5ul of Annexin V-APC and 10ul of 7-AAD was added. The mixture was vortexed gently and incubated for 15 minutes on ice in the dark. Cells without washing and the volume was added 380ul of cold 1x binding buffer to each tube analyzed by flow cytometer.
2.5. Enzyme-linked immunosorbent assay
Supernatant was collected at 12 hours after H37Rv infection. Mcl-1, IL-6, IL-10, TNF-α, and TGF-β were measured using ELISA kits (Elabscience, Wuhan, China) according to the manufacturer's instructions.
2.6. Mitochondrial membrane potential assay
Mitochondrial membrane potential (ΔΨm) was determined using the JC-10 kit according to the manufacturer's manual (KeyGEN BioTECH, Jiangsu, China). A total of 1 × 106 RAW264.7 macrophages were loaded with JC-10 working fluid at 37°C in the 5% CO2 incubator for 20 minutes. Polarized mitochondria with J-aggregate forms and depolarized mitochondria with monomer form were marked by red and green fluorescence staining respectively, while simultaneously being excited by 490 nm laser sources. Then observed by the confocal microscope.
2.7. Scanning electron microscope (SEM)
RAW264.7 macrophages were collected and washed by PBS for 3 times, then fixed in 2.5% glutaraldehyde solution, dehydrated through a graded tert-butanol series to 100%. Samples were coated via platinum sputtering after retaining it under vacuum condition, and imaged using a scanning electron microscope (JEOL, JSM-6390LV).
2.8. Statistical analysis
The data are expressed as mean ± SD. Statistical correlation of data was checked for significance by ANOVA and Student's t test. Differences were considered significant at the level of P < .05. These analyses were performed using SPSS 20.0 software.
3. Results
3.1. Inhibiting the MAPK signaling pathways suppressed Mcl-1 expression in MTB-infected RAW264.7 macrophages
Studies found that different virulence from MTB infection could affect Mcl-1 protein and mRNA expression in vitro. XJ-MTB and H37Rv infection could significantly induce Mcl-1 protein and mRNA expression compared to control. Additionally, Mcl-1 protein and mRNA levels were lower in macrophages exposed to H37Ra or BCG compared to the control, indicating that H37Ra or BCG infection could deduce expression of Mcl-1 in host macrophage. Next, both control and infection groups were treated with the Mcl-1 expression signaling pathway inhibitors AG490, PD98059 and LY294002. Analysis of H37Rv infection yielded the following results: Mcl-1 protein and mRNA expression levels were significantly decreased in the treated infection groups when compared to control groups, presenting more than a 67% (PD98059) (P > .05) and 18% (AG490) decrease compared to virulent strains infection groups, with the exception of in LY294002 treated groups increasing 44% (LY294002) (P > .05). Moreover, we found PD980589 to be the most powerful inhibitor for Mcl-1 expression, and the MAPK signaling pathway might play a major role in Mcl-1 expression regulation in virulent MTB strains-infected macrophages than PI3K and JAK/STAT signaling pathways. (Fig. 1, P < .05)
Figure 1.
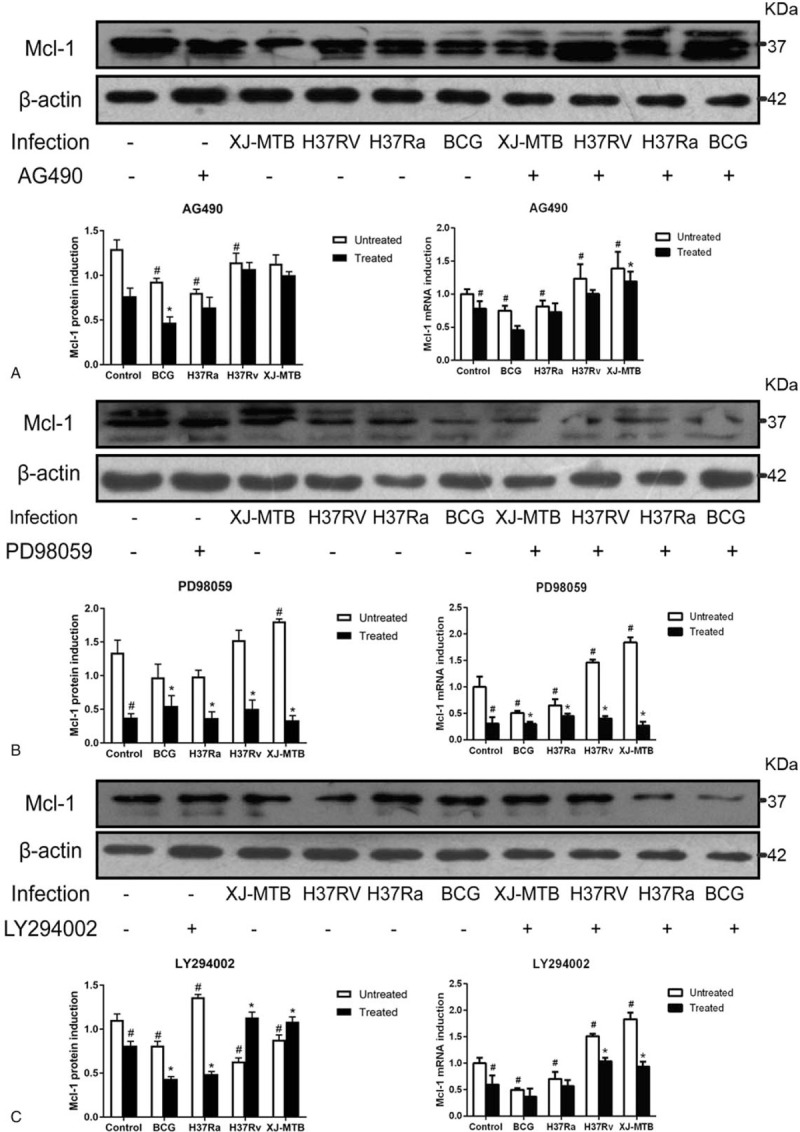
Mcl-1 protein and mRNA expression decreased in RAW264.7 cells infected with different virulent MTB when treated with Mcl-1 expression signaling pathway inhibitors. RAW264.7 cells were infected with BCG, H37Ra, H37Rv and XJ-MTB at MOI = 10 (bacterium: cell) for 24 hours, then specific signaling pathway inhibitors AG490 (Fig. 1A), PD98059 (Fig. 1B), LY294002 (Fig. 1C) were used to affect Mcl-1 expression signaling pathways. Cells lysate and total RNA was collected, Mcl-1 protein and mRNA expression were detected using western blotting and RT-PCR. The grouping of blots cropped from different gels. Values represent the mean ± SD from three independent experiments. #P < 0.05 VS uninfected control group, ##P < 0.01 VS uninfected control group; ∗P < .05 VS untreated group, ∗∗P < .01 VS untreated group.
3.2. Inhibiting the MAPK signaling pathways could induce MTB-infected RAW264.7 macrophages apoptosis
Next, we studied the role of Mcl-1 on apoptosis of RAW264.7 macrophages during MTB infection. This was achieved by binding the V-APC/7-AAD annexin to phosphatidylserine on the cell surface. Flow cytometry analysis revealed an increased apoptosis rate in the MTB-infected RAW264.7 macrophages compared with the controls. BCG and H37Ra infection could induce a higher apoptosis rate than H37Rv and XJ-MTB infection. When the infected cells were exposed to the specific signaling pathway inhibitors, analysis of H37Rv infection yielded the following results: the apoptosis rate increased more than 41% (PD98059), 7% (AG490), and 32% (LY294002) compared to virulent MTB infection groups, but apoptosis in attenuated strain infection groups was not obvious when treated with inhibitors. Additionally, the host cells seemed to be more sensitive to apoptosis when the MAPK signaling pathway was inhibited during virulent MTB infection. (Fig. 2, P < .05)
Figure 2.
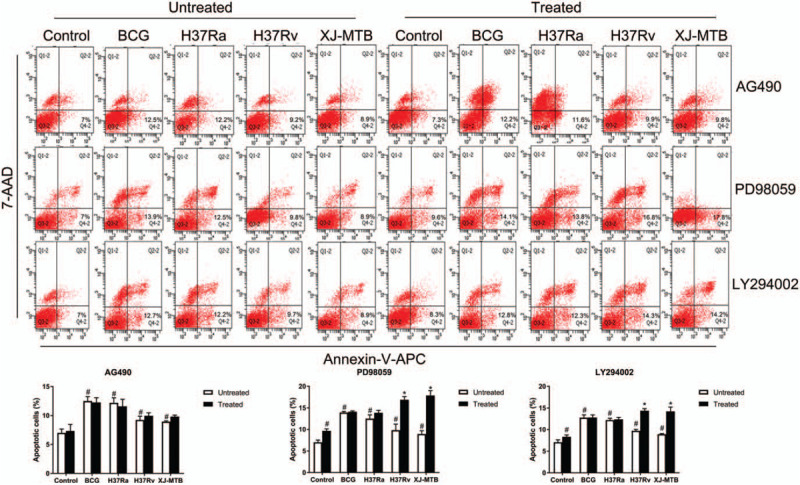
Inhibiting Mcl-1 expression signaling pathway could increase apoptosis of MTB infection RAW264.7 macrophages. Control and MTB infected RAW264.7 macrophages treated by Mcl-1 expression signaling pathway inhibitors (AG490, PD98059 and LY294002) were assessed by flow cytometry to analysis apoptosis rates by Annexin V-APC/7-AAD staining. Values represent the mean ± SD from three independent experiments (10,000 events per treatment group in each experiment). #P < .05 VS uninfected control group, ##P < .01 VS uninfected control group; ∗P < .05 VS untreated group, ∗∗P < .01 VS untreated group.
Next, RT-PCR was performed to analyze the mRNA expression of apoptosis-associated genes Bax, Bcl-2, Cyt-c, and caspase-3 (Fig. 3). The results revealed that when RAW264.7 cells were exposed to BCG and H37Ra strains, the expression of pro-apoptosis genes, such as Bax (Fig. 3A), caspase-3 (Fig. 3C) and Cyt-c (Fig. 3D), significantly increase in infection groups compared to the control. The expression of Bcl-2 increased in XJ-MTB and H37Rv infection groups and decreased in the attenuated strain infection groups when compared to the control group (Fig. 3B). When the Mcl-1 expression signaling pathways was inhibited with blockers, expression levels of the anti-apoptosis gene Bcl-2 significantly decreased whereas those of the pro-apoptosis genes Bax, Cyt-c and caspase-3 increased in XJ-MTB and H37Rv infection groups when compared to the control groups. Moreover, PD98059 treated groups showed more observable changes than AG490 and LY294002. Since Bax and Cyt-c play a major role in the mitochondrial apoptosis pathway, we speculated that the mitochondrial apoptosis pathway may be activated when Mcl-1 expression signaling pathway is inhibited through the inhibition of the MAPK signaling pathway. (Fig. 3, P < .05)
Figure 3.
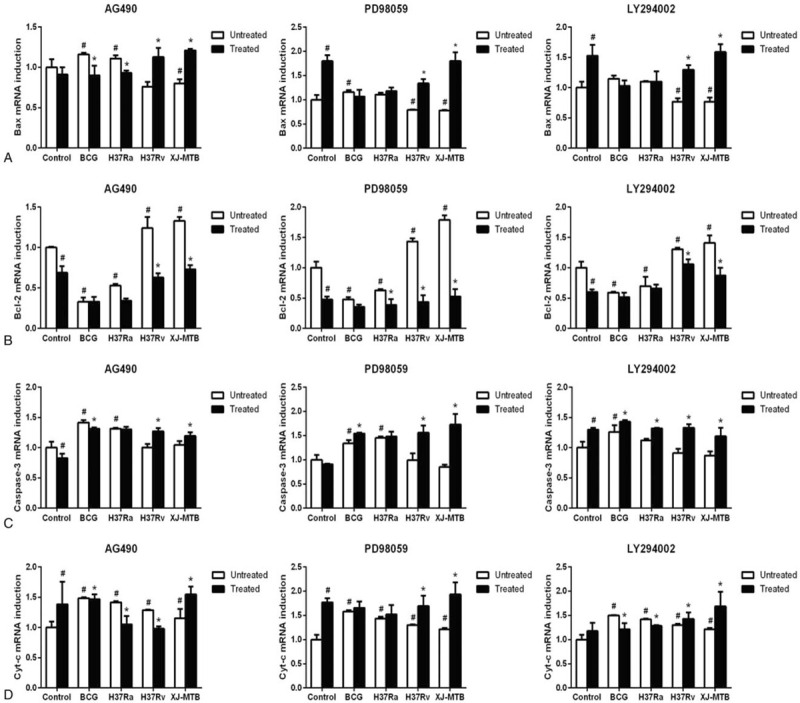
Inhibiting Mcl-1 expression signaling pathway induce host macrophage apoptosis may active mitochondrial apoptosis pathway. RT-PCR was used to detect the expression of mitochondrial apoptosis associated genes Bax (Fig. 3A), Bcl-2 (Fig. 3B), caspase-3 (Fig. 3C) and Cyt-c (Fig. 3D) in control and different virulence of MTB infection groups that treated by specific inhibitors (AG490, PD98059 and LY294002). Values represent the mean ± SD from three independent experiments. #P < .05 VS uninfected control group, ##P < .01 VS uninfected control group; ∗P < .05 VS untreated group, ∗∗P < .01 VS untreated group.
3.3. Inhibiting the Mcl-1 signaling pathway could induce the polarization of the H37Rv-infected macrophages into the M2 phenotype
To explore whether macrophage polarization phenotype changed when the Mcl-1 expression signaling pathway was inhibited during H37Rv infection, we determined the phenotypic and functional markers associated with M1 and M2 macrophages, including CD86, CD206, iNOS, and Fizz1, as well as the expression of related cytokines including IL-6, IL-10, TNF-α, and TGF-β. Figure 4 shows that following H37Rv infection, IL-6, TNF-α, and iNOS (M1 markers), and IL-10, TGF-β, and Fizz1 (M2 markers) levels significantly increased compared to control. While CD86 and CD206 had no significant increase (P > .05). Next, the infected cells were treated with MAPK signaling pathways inhibitors, and the results indicated that the expression levels of M1 markers were significantly reduced compared to the H37Rv infection group. IL-6 and TNF-α expression levels decreased differently depending on the inhibitor; ERK inhibition yielded a 27% and 9% decreased, JNK yielded a 22% and 30% decrease, and p38 yielded a 7% and 12% decrease, respectively. p38 inhibition also caused a 41% decrease in iNOS expression. Additionally, expression of M2 markers increased compared to the infection group. IL-10 and Fizz1 expression levels in ERK inhibition groups increased more (72% and 154%, respectively) than in those affected with JNK and p38 inhibition. No noticeable increase was observed in the M2-related protein TGF-β expression in p38 inhibition. Additionally, CD86 and CD206 expression was significantly increased when the JNK pathway was inhibited compared with the H37Rv infection group. These results suggested that MTB infection induces mixed M1/M2 activation. More specifically, MAPK signaling pathways inhibition induced the polarization of activated macrophages into M2, and JNK signaling pathway was found to be the main regulator of host macrophage polarization.
Figure 4.
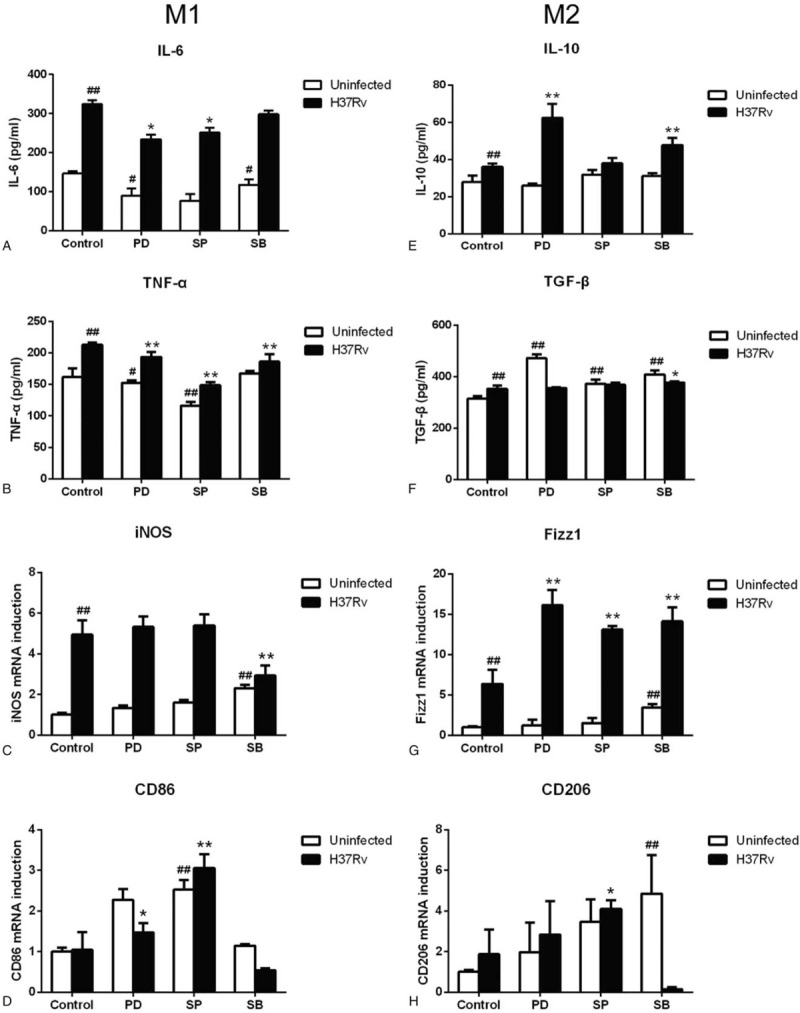
Inhibition Mcl-1 expression signaling pathway induce host macrophage polarized to M2 phenotype. H37Rv infection RAW264.7 macrophages were treated by MAPK signaling pathway inhibitors PD98059 (PD), SP600125 (SP) and SB203580 (SB) for 12 hours. The supernatant and reverse-transcribed total RNA were prepared to determine M1 and M2 macrophage markers (Fig. 4A–H) by ELISA and RT-PCR. Values represent the mean ± SD from three independent experiments. #P < .05 VS uninfected control group, ##P < .01 VS uninfected control group; ∗P < .05 VS H37Rv infection group, ∗∗P < .01 VS H37Rv infection group.
3.4. H37Rv infection induced RAW264.7 macrophage polarization
Scanning electron microscopy showed that the M0 RAW264.7 macrophage surface had rich microvilli with pseudopodia, the cells are like “spherical” in shape. After H37Rv infection, two types of macrophages with distinct shapes were observed. M1-type macrophages formed narrow, fully extended pseudopodia, with reduced microvilli, whereas the M2-type were large and flat, with an asperous cell surface, similar to the shape of a “fried egg” (Fig. 5).
Figure 5.

The polarization morphology of RAW264.7 macrophage. RAW264.7 macrophage infected by H37Rv for 12 hours, then morphology of polarization macrophage investigated by SEM.
3.5. Inhibition of the MAPK signaling pathways to induce H37Rv-infection RAW264.7 macrophages apoptosis activated the mitochondrial apoptosis pathway
To verify whether the mitochondrial pathway was activated when the MAPK signaling pathways was inhibited, western blot (Fig. 6A) and JC-10 (Fig. 7) analyses were used to assess mitochondrial dysfunction in RAW264.7 cells. Figure 6A shows that when H37Rv infection macrophages were treated with MAPK signaling pathway inhibitors, Bcl-2 and Bcl-xl expression decreased significantly, while that of Bax increased. ERK inhibition resulted in a 67% decrease in Bcl-xl expression, and Bax and Bcl-2 had no significant change compared to the H37Rv infection group. JNK inhibition resulted in a 63% and 85% decrease in Bcl-2 and Bcl-xl expression, respectively, whereas Bax expression increased 127% compared with the H37Rv infection group. Notably, in the p38 inhibition H37Rv infection group, Bcl-2 and Bcl-xl had a 77% and 86% expression decrease, and Bax expression increased 170% compared to the non-inhibited infection group. Interestingly, JNK and p38 inhibition in H37Rv infection cells caused an increase in the Bax/Bcl-2 ratio compared to the H37Rv infection group. Further, H37Rv infection induced host macrophage Mcl-1 expression; however, when treated with MAPK signaling pathways inhibitors PD98059 (inhibiting the ERK signaling pathway), SP600125 (inhibiting the JNK signaling pathway), and SB203580 (inhibiting the p38 signaling pathway), Mcl-1 expression decreased more than that in the H37Rv infection group, especially the SB203580 treated infection group (Fig. 6B).
Figure 6.
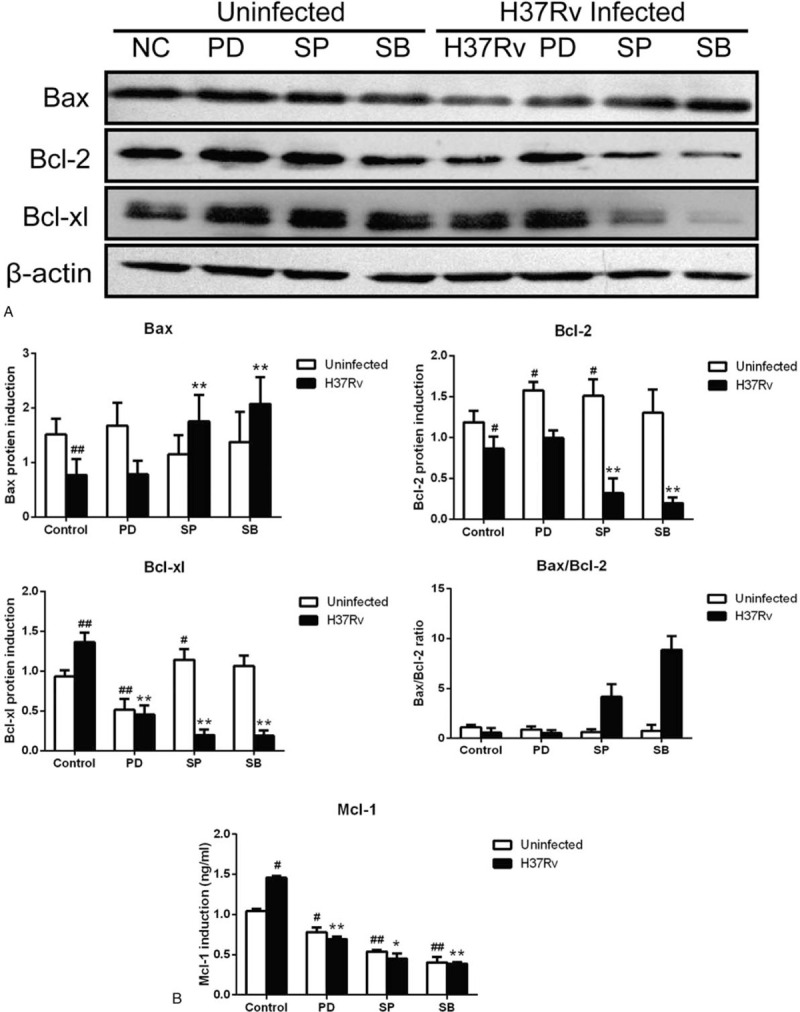
Inhibition MAPK signaling pathway to induce host macrophage apoptosis active mitochondrial apoptosis pathway. H37Rv infection RAW264.7 cells were treated by MAPK signaling pathway inhibitors PD98059 (PD), SP600125 (SP), SB203580 (SB) for 12 hours, then western blotting was used to detect Bax, Bcl-2 and Bcl-xl protein expression (Fig. 6A), and Mcl-1 expression detected by ELISA (Fig. 6B). The grouping of blots cropped from different gels. Values represent the mean ± SD from three independent experiments. #P < .05 VS uninfected control group, ##P < .01 VS uninfected control group; ∗P < .05 VS H37Rv infection group, ∗∗P < .01 VS H37Rv infection group.
Figure 7.
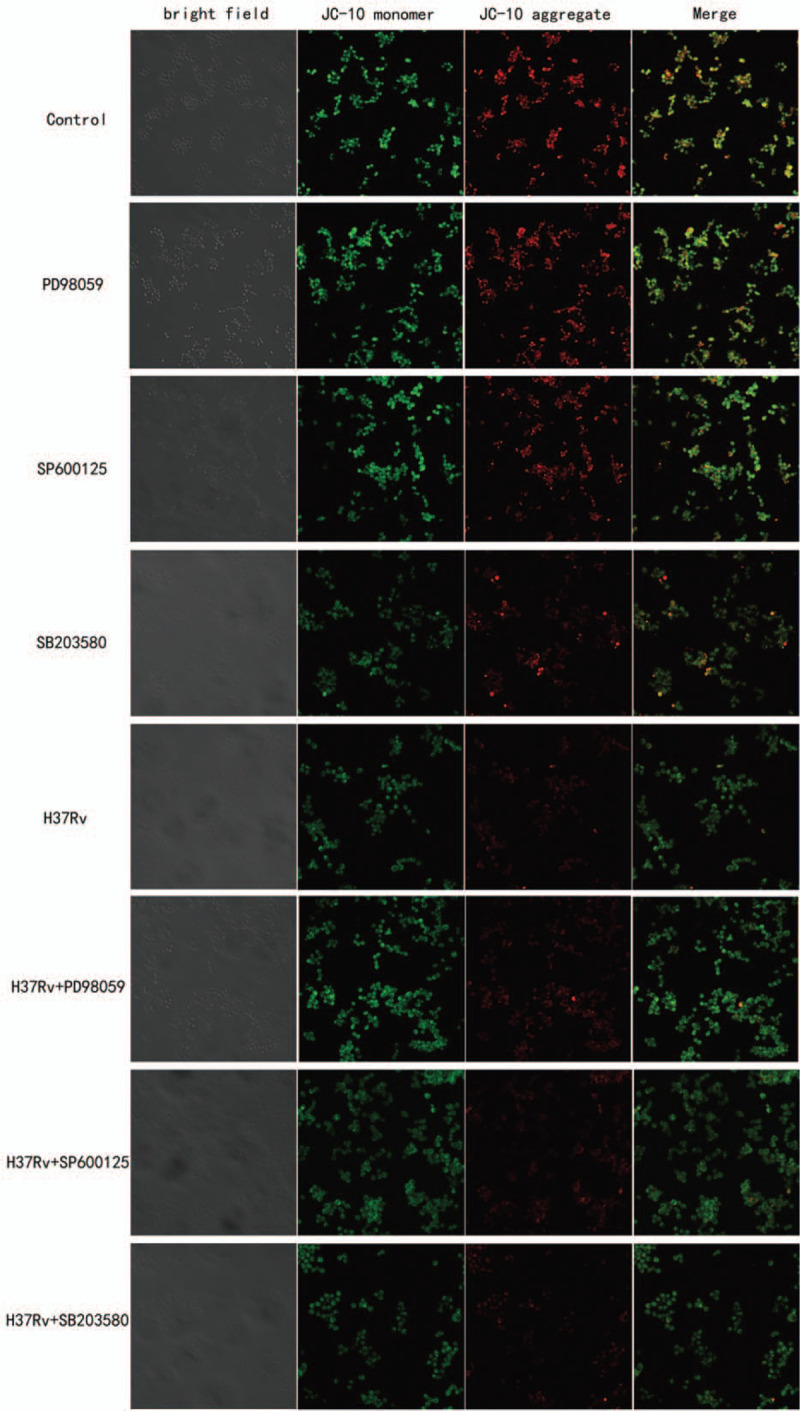
Inhibiting MAPK signaling pathway induce host macrophage mitochondrial membrane potential depolarization. H37Rv infection macrophages that treated by MAPK signaling pathway inhibitors for 12 hours, then adding JC-10 staining solution. We can see the red fluorescence significantly reduce in SB203580 treated infection group, cause obvious mitochondrial dysfunction.
Mitochondrial membrane potential was detected by JC-10 dye (Fig. 7). In healthy cells, the aggregated JC-10 exhibits red fluorescence. When the mitochondrial membrane potential was depolarized, the JC-10 monomers exhibited green fluorescence, and simultaneously, red fluorescence was reduced. In healthy RAW264.7 cells, bright yellow fluorescence was registered, in apoptotic cells, the same filter yielded green fluorescence. Red fluorescence in inhibited infection group was decreased than in infection group. In the p38 inhibition H37Rv infection group, red fluorescence decreased more than that in the infection group and other inhibition infection groups. The results demonstrated that MAPK signaling pathway inhibition can induce host macrophage mitochondrial membrane potential dysfunction during the early stages of apoptosis following H37Rv infection, and that the p38 signaling pathway plays an important role in this process.
4. Discussion
Studies[13,23,24] have confirmed that inhibiting Mcl-1 inhibition can regulate the apoptosis of macrophages infected with MTB through the MAPK signaling pathway. Additionally, the mitochondrial signaling pathway has been found to be related to the MAPK signaling pathway in the apoptotic process in various diseases,[25] but no prior studies have investigated the polarization of macrophages infected by MTB. Therefore, the present study aimed to explore the relationship between the MAPK signaling and mitochondrial pathways involved in macrophage apoptosis and polarization after MTB infection. There is an urgent need to formulate an effective treatment for latent tuberculosis infection, drug-resistant patients, and HIV / AIDS co-infection.
Previous studies have shown that mitochondria-dependent apoptosis is regulated by the Bcl-2 protein family.[26] Wu et al[27] reported that mycocide-induced apoptosis is related to mitochondria-mediated apoptosis. When mitochondrial membrane potential is lost, cytochrome c (Cyt-c) is released, Bax, Bad and caspase-3/9 are activated, and p-Bad, Bcl-xl and Bcl-2 are inhibited. In the Bcl-2 family, Mcl-1 is mainly responsible for maintaining the stability of the mitochondrial membrane and inhibiting the release of Cyt-c, thus promoting cell survival and preventing apoptosis.[28,29] Our previous experimental results[30] confirmed that down-regulation of Mcl-1 promotes Cyt-c-induced apoptosis of latent virulent MTB host macrophages, and the mitochondria-mediated “endogenous” apoptosis pathway may play a significant role in this process. In addition, the Bcl-2 family contains key regulators of cell survival, which can inhibit (Bcl-2 and Bcl-xl) or promote (Bad and Bax) apoptosis.[31] Based on these reports and hypotheses, we found that the Bax/Bcl-2 ratio of Raw264.7 cells infected with MTB H37Rv for 12 h was lower than that of untreated RAW264.7 macrophages. The expression of Bcl-2 and Bcl-xl protein decreased and the expression of Bax protein increased in H37Rv infected macrophages treated with specific MAPK signaling pathway inhibitors. The level of Bax/Bcl-2 in SB203580 inhibition group was significantly higher than that in the control group. In addition, the MAPK signaling pathway inhibition group showed the loss of mitochondrial membrane potential, and p38 inhibition led to the aggravation of mitochondrial dysfunction of host macrophages. Inhibition of Mcl-1 expression through the MAPK signaling pathway may lead to mitochondrial membrane potential depolarization by changing the expression of Bcl-2 family members, induce Cyt-c release into the cytoplasm, and activate caspase-3 to induce apoptosis of MTB infected host macrophages.
As the signal center of cells, the mitochondrial pathway is associated with the apoptosis and polarization of infected macrophages.[22] Changes in mitochondrial function are involved in numerous human diseases, such as heart failure and metabolic diseases.[19–21] The metabolism of activated macrophages changes, with M1 and M2 macrophages progressing to aerobic glycolysis and oxidative metabolism, respectively.[32,33] Formentini et al[34] observed that the inhibition of mitochondrial ATP synthase can trigger anti-inflammatory response in the small intestine of transgenic mice through mitochondrial mtROS-mediated NF- κB activation and polarize the M2 anti-inflammatory phenotype of macrophages. According to the related research results,[35,36] it has been confirmed that mitochondria can regulate immune function through the metabolic pathway, activation of inflammation, and mitosis and fusion of mitochondria. In the metabolic pathway, M1 macrophages show blocking of TCA circulation and exert a pro-inflammatory effect. In contrast, M2 macrophages undergo beta oxidation to produce an anti-inflammatory response. Therefore, it has been hypothesized that promoting mitochondrial biosynthesis can promote M2 phenotype and reduce macrophage-mediated inflammation and apoptosis. It is suggested that the mitochondrial pathway plays a major role in regulating the polarization and apoptosis of macrophages. The underlying mechanism remains unclear.
It is common knowledge that inflammatory activation and apoptosis of macrophages play a central role in the host defense mechanism in the process of MTB infection. Activated macrophages play a major role in host resistance to mycobacterial infection. MTB infection can not only induce host macrophage apoptosis but also induce host macrophage polarization.[37] Huang et al[17] focused on the polarization of macrophages at different stages of tuberculous granuloma formation. They concluded that the polarization of macrophages was mainly M1 in the early stage of tuberculous granuloma formation, while M2-related markers gradually increased in the late stage. Lim et al[38] found that when macrophages were infected by H37Rv, there were more M2 macrophages, while M1 macrophages were activated by H37Ra infection. In addition, Zhao et al[39] noted that IL-4 may prevent atherosclerosis by inhibiting the MAPK signaling pathway and inducing macrophages to polarize to the M2 phenotype for anti-inflammation, which can protect atherosclerosis by inhibiting this pathway. Although activated macrophages play a key role in host defense against mycobacterial infection, the regulatory function of the MAPK signaling pathway in M1 or M2 polarized macrophages is unclear during tuberculosis infection. Our results showed that expression levels of M1/M2-related markers were higher after H37Rv infection. During the infection of RAW264.7 macrophages following inhibition of the Mcl-1 expression signaling pathway, MTB induced an increase in the expression of pro-inflammatory cytokines, such as IL-6, TNF-α, and iNOS, as well as the expression increase of anti-inflammatory cytokines, including IL-10, TGF-β, Fizz1, and CD206, which indirectly reflected that macrophages gradually differentiated into the M2 type when the MAPK signaling pathway was inhibited. In addition, when the JNK signaling pathway was inhibited, CD86 expression increased, indicating that inhibition of the JNK signaling pathway could improve the antigen presentation ability of the host macrophages. Inflammatory injury can be alleviated by inducing apoptosis of the host macrophages by inhibiting the Mcl-1 expression signaling pathway. The three pathways of MAPK signaling are involved in several inflammatory responses, in which the release of pro-inflammatory cytokines is mainly regulated by the JNK signaling pathway, anti-inflammatory cytokines are mainly regulated by the ERK signaling pathway, and JNK signaling pathway is the main regulator of macrophage polarization.
To the best of our knowledge, the role of Mcl-1 expression signaling pathway in regulating apoptosis and polarization of RAW264.7 macrophages infected with MTB and whether mitochondria are involved in the host immune response after MTB infection, have not been reported. Therefore, in the current study, we proved for the first time that the signaling pathway that inhibits Mcl-1 expression (especially the MAPK signaling pathway) can increase the apoptosis rate of host macrophages and up regulate anti-inflammatory activity, to reduce inflammatory injury and eliminate MTB from host macrophages. In this group of host immune defense responses, the MAPK signaling pathway regulates apoptosis and polarization of host macrophages by regulating Mcl-1 expression. There may be crosstalk between the branches of MAPK signaling pathway. The P38 signaling pathway regulates host macrophage apoptosis by activating the mitochondrial apoptosis pathway, while the JNK signaling pathway mainly regulates the polarization phenotype of host macrophages. According to our findings, inhibition of the Mcl-1 expression signaling pathway may provide a novel mechanism for the treatment of tuberculosis.
Author contributions
Conceived and designed the experiments: Ling Han, Xin-min Wang, Le Zhang.
Performed the experiments: Ling Han, Yang Lu, Shu-jun Zhang.
Funding acquisition: Le Zhang.
Contributed reagents/ materials/ analysis tools: Zhi-hong Zheng, Wan-jiang, Zhang
Analyzed the data: Ling Han, Fang Wu, Ying-zi Wang.
Conceptualization: Xin-min Wang.
Data curation: Ling Han.
Investigation: Ling Han, Yang Lu, Xiao-fang Wang.
Methodology: Ling Han, Ying-zi Wang, Zhi-hong Zheng, Fang Wu.
Project administration: Ling Han.
Resources: Xin-min Wang, Fang Wu, Wan-jiang Zhang, Le Zhang.
Software: Ling Han, Yang Lu, Xiao-fang Wang.
Writing – original draft: Ling Han.
Writing – review & editing: Ling Han, Xin-min Wang, Le Zhang.
Footnotes
Abbreviations: ELISA = enzyme-linked-immunosorbent serologic assay, Mcl-1 = Myeloid cell leukemia-1, MTB = Mycobacterium tuberculosis, RT-PCR = real-time polymerase chain reaction, SEM = scanning electron microscopy.
How to cite this article: Han L, Lu Y, Wang X, Zhang S, Wang Y, Wu F, Zhang W, Wang X, Zhang L. Regulatory role and mechanism of the inhibition of the Mcl-1 pathway during apoptosis and polarization of H37Rv-infected macrophages. Medicine. 2020;99:42(e22438).
LH and XMW contributed equally to this work.
This study was an in vitro experiment. The Internal Review Board of the Shihezi University reviewed and approved the study protocol.
This research was supported by the National Natural Science Foundation of China (No.81660330).
The authors declare that they have no conflicts of interest.
All data generated or analyzed during this study are included in this published article [and its supplementary information files]
References
- [1].Wu X, Deng G, Li M, et al. Wnt/beta-catenin signaling reduces Bacillus Calmette-Guerin-induced macrophage necrosis through a ROS -mediated PARP/AIF-dependent pathway. BMC Immunol 2015;16:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Eldholm V, Rieux A, Monteserin J, et al. Impact of HIV co-infection on the evolution and transmission of multidrug-resistant tuberculosis. eLife 2016;5:e16644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wu X, Deng G, Hao X, et al. A caspase-dependent pathway is involved in wnt/β-catenin signaling promoted apoptosis in bacillus calmette-guerin infected RAW264.7 Macrophages. Int J Molr Sci 2014;15:5045–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].WHO. Global tuberculosis report 2019. http://www.who.int/tb/publications/global_report/en/. October 2019;1–27. [Google Scholar]
- [5].Divangahi M, Behar SM, Remold H. Dying to live: how the death modality of the infected macrophage affects immunity to tuberculosis. Adv Exp Med Biol 2013;783:103–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhao X, Bai X, Guan L, et al. microRNA-4331 promotes transmissible gastroenteritis virus (TGEV)-induced mitochondrial damage via targeting RB1, Upregulating Interleukin-1 receptor accessory protein (IL1RAP), and activating p38 MAPK pathway in vitro. Mol Cell Proteomics 2018;17:190–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang L, Li S, Wang R, et al. Cytokine augments the sorafenib-induced apoptosis in Huh7 liver cancer cell by inducing mitochondrial fragmentation and activating MAPK-JNK signalling pathway. Biomed Pharmacother 2019;110:213–23. [DOI] [PubMed] [Google Scholar]
- [8].Persson A, Blomgran-Julinder R, Eklund D, et al. Induction of apoptosis in human neutrophils by Mycobacterium tuberculosis is dependent on mature bacterial lipoproteins. Microb Pathog 2009;47:143–50. [DOI] [PubMed] [Google Scholar]
- [9].Pathania AS, Joshi A, Kumar S, et al. Reversal of boswellic acid analog BA145 induced caspase dependent apoptosis by PI3K inhibitor LY294002 and MEK inhibitor PD98059. Apoptosis 2013;18:1561–73. [DOI] [PubMed] [Google Scholar]
- [10].Park JS, Lee J, Lim MA, et al. JAK2-STAT3 blockade by AG490 suppresses autoimmune arthritis in mice via reciprocal regulation of regulatory T Cells and Th17 cells. J Immunol 2014;192:4417–24. [DOI] [PubMed] [Google Scholar]
- [11].Liu Z, Zhang YY, Zhang QW, et al. 3-Bromopyruvate induces apoptosis in breast cancer cells by downregulating Mcl-1 through the PI3K/Akt signaling pathway. Anti-Cancer Drugs 2014;25:447–55. [DOI] [PubMed] [Google Scholar]
- [12].Li QF, Zhu HY, Yang YF, et al. Prokineticin-1/endocrine gland-derived vascular endothelial growth factor is a survival factor for human multiple myeloma cells. Leuk Lymphoma 2010;51:1902–12. [DOI] [PubMed] [Google Scholar]
- [13].Wang FY, Zhang YQ, Wang XM, et al. A small hairpin RNA targeting myeloid cell leukemia-1 enhances apoptosis in host macrophages infected with Mycobacterium tuberculosis. J Microbiol 2016;54:330–7. [DOI] [PubMed] [Google Scholar]
- [14].Qin Y, Sun X, Shao X, et al. Macrophage-Microglia Networks Drive M1 Microglia Polarization After Mycobacterium Infection. Inflammation 2015;38:1609–16. [DOI] [PubMed] [Google Scholar]
- [15].Lopes RL, Borges TJ, Zanin RF, et al. IL-10 is required for polarization of macrophages to M2-like phenotype by mycobacterial DnaK (heat shock protein 70). Cytokine 2016;85:123–9. [DOI] [PubMed] [Google Scholar]
- [16].Liu YC, Zou XB, Chai YF, et al. Macrophage polarization in inflammatory diseases. Int J Biol Sci 2014;10:520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Huang Z, Luo Q, Guo Y, et al. Mycobacterium tuberculosis-induced polarization of human macrophage orchestrates the formation and development of tuberculous granulomas in vitro. PLoS One 2015;10: [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gengenbacher M, Kaufmann SH. Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol Rev 2012;36:514–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Van den Bossche J, Baardman J, Otto NA, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep 2016;17:684–96. [DOI] [PubMed] [Google Scholar]
- [20].Michel S, Wanet A, De Pauw A, et al. Crosstalk between mitochondrial (dys) function and mitochondrial abundance. J Cell Physiol 2012;227:2297–310. [DOI] [PubMed] [Google Scholar]
- [21].Melkov A, Baskar R, Alcalay Y, et al. A new mode of mitochondrial transport and polarized sorting regulated by Dynein. Milton Miro Dev 2016;143:4203–13. [DOI] [PubMed] [Google Scholar]
- [22].Li C, Ding XY, Xiang DM, et al. Enhanced M1 and Impaired M2 Macrophage Polarization and Reduced Mitochondrial Biogenesis via Inhibition of AMP Kinase in Chronic Kidney Disease. Cell Physiol Biochem 2015;36:358–72. [DOI] [PubMed] [Google Scholar]
- [23].Sly LM, Hingley-Wilson SM, Reiner NE, et al. Survival of Mycobacterium tuberculosis in host macrophages involves resistance to apoptosis dependent upon induction of antiapoptotic Bcl-2 family member Mcl-1. J Immunol 2003;170:430–7. [DOI] [PubMed] [Google Scholar]
- [24].Palaga T, Ratanabunyong S, Pattarakankul T, et al. Notch signaling regulates expression of Mcl-1 and apoptosis in PPD-treated macrophages. Cell Mol Immunol 2013;10:444–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang FY, Wang XM, Wang C, et al. Suppression of Mcl-1 induces apoptosis in mouse peritoneal macrophages infected with Mycobacterium tuberculosis. Microbiol Immunol 2016;60:215–27. [DOI] [PubMed] [Google Scholar]
- [26].Dzhagalov I, St John A, He YW. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood 2007;109:1620–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wu YJ, Wong BS, Yea SH, et al. Sinularin Induces Apoptosis through Mitochondria Dysfunction and Inactivation of the pI3K/Akt/mTOR Pathway in Gastric Carcinoma Cells. Marine drugs 2016;14:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Moulding DA, Akgul C, Derouet M, et al. BCL-2 family expression in human neutrophils during delayed and accelerated apoptosis. J Leukoc Biol 2001;70:783–92. [PubMed] [Google Scholar]
- [29].Floyd K, Bernardo PH, Sivaraman T, et al. Synthesis of a rhodanine-based compound library targeting Bcl-XL and Mcl-1. Pure Appl Chem 2011;83: [Google Scholar]
- [30].Wang XF, Wang XM, Zhang L, et al. The regulatory role of Mcl-1 in apoptosis of mouse peritoneal macrophage infected with M. tuberculosisstrains that differ in virulence. Int J Clin Exp Pathol 2017;10:7565–77. [PMC free article] [PubMed] [Google Scholar]
- [31].Michels J, Johnson PW, Packham G. Mcl-1. Int J Biochem Cell Biol 2005;37:267–71. [DOI] [PubMed] [Google Scholar]
- [32].Vats D, Mukundan L, Odegaard JI, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab 2006;4:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity 2013;38:633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Formentini L, Santacatterina F, Nunez de Arenas C, et al. Mitochondrial ROS Production Protects the Intestine from Inflammation through Functional M2 Macrophage Polarization. Cell Rep 2017;19:1202–13. [DOI] [PubMed] [Google Scholar]
- [35].Ramond E, Jamet A, Coureuil M, et al. Pivotal role of mitochondria in macrophage response to bacterial pathogens. Front Immunol 2019;10:2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Angajala A, Lim S, Phillips JB, et al. Diverse roles of mitochondria in immune responses: novel insights into immuno-metabolism. Front Immunol 2018;9:1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Chen D, Li G, Fu X, et al. Wnt5a deficiency regulates inflammatory cytokine secretion, polarization, and apoptosis in mycobacterium tuberculosis-infected macrophages. DNA Cell Biol 2017;36:58–66. [DOI] [PubMed] [Google Scholar]
- [38].Lim YJ, Yi MH, Choi JA, et al. Roles of endoplasmic reticulum stress-mediated apoptosis in M1-polarized macrophages during mycobacterial infections. Sci Rep 2016;6:37211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhao XN, Li YN, Wang YT. Interleukin-4 regulates macrophage polarization via the MAPK signaling pathway to protect against atherosclerosis. Genet Mol Res 2016;15:1–8. [DOI] [PubMed] [Google Scholar]