

Abstract
We propose a new plausible mechanism by mean of which SARS-CoV-2 produces extrapulmonary damages in severe COVID-19 patients. The mechanism consist on the existence of vulnerable proteins (VPs), which are (i) mainly expressed outside the lungs; (ii) their perturbations is known to produce human diseases; and (iii) can be perturbed directly or indirectly by SARS-CoV-2 proteins. These VPs are perturbed by other proteins, which are: (i) mainly expressed in the lungs, (ii) are targeted directly by SARS-CoV-2 proteins, (iii) can navigate outside the lungs as cargo of extracellular vesicles (EVs); and (iv) can activate VPs via subdiffusive processes inside the target organ. Using bioinformatic tools and mathematical modeling we identifies 26 VPs and their 38 perturbators, which predict extracellular damages in the immunologic endocrine, cardiovascular, circulatory, lymphatic, musculoskeletal, neurologic, dermatologic, hepatic, gastrointestinal, and metabolic systems, as well as in the eyes. The identification of these VPs and their perturbators allow us to identify 27 existing drugs which are candidates to be repurposed for treating extrapulmonary damage in severe COVID-19 patients. After removal of drugs having undesirable drug-drug interactions we select 7 drugs and one natural product: apabetalone, romidepsin, silmitasertib, ozanezumab, procaine, azacitidine, amlexanox, volociximab, and ellagic acid, whose combinations can palliate the organs and systems found to be damaged by COVID-19. We found that at least 4 drugs are needed to treat all the multiorgan damages, for instance: the combination of romidepsin, silmitasertib, apabetalone and azacitidine.
Keywords: SARS-CoV-2, COVID-19, Multiorgan damage, Protein-protein interaction, Biological networks, Extracellular vesicles, Exosomes, Subdiffusion
Highlights
-
•
A new mechanism for multiorgan damage in COVID-19 based on extracellular vesicle transport of proteins from the lungs and intracellular subdiffusive movement of the proteins at target organs is proposed and validated.
-
•
The new mechanism explains the main damages observed in most of organs reported in patients with severe COVID-19.
-
•
Several repurposable drugs for targeting extracellular damages in COVID-19 severe patients are identified.
1. Introduction
Since December 2019 the world is facing the pandemic COVID-19 (coronavirus disease 2019) produced by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [[1], [2], [3], [4]] COVID-19 has revealed as a complex disease which causes substantial respiratory pathology accompanied by several extrapulmonary manifestations [5,6]. These damages have been found to involve several organs/systems, such as neurologic, renal, hepatic, gastrointestinal, hematologic, cardiac, endocrine and dermatological [[7], [8], [9]]. In this sense, infection by SARS-CoV-2 resembles those produced by other coronaviruses like SARS-CoV and MERS-CoV, which were reported to affect heart, lungs, liver and central nervous system as well as kidney (SARS-CoV) [8]. The potential causes for these multiorgan damages have been reviewed by several authors [[5], [6], [7], [8], [9]]. The two most frequently mentioned causes of multiorgan damages produced by SARS-CoV-2 are: (i) the direct viral toxicity and (ii) cytokine-release syndrome or cytokine storm. The direct viral toxicity is expected by the entrance of SARS-CoV-2 in the affected organs mediated by the protein ACE2 [[10], [11], [12], [13]]), which is abundant in many human organs [14], and has been identified as the receptor mediating the endocytic cell entrance of SARS-CoV-2 [15]. This abundance of SARS-CoV-2 receptors in human cells and the isolation of viral RNA from fecal samples in high titers, and in less amount from blood and urine, has trigger the hypothesis that multiorgan injury may occur partially due to direct damage of tissues by SARS-CoV-2 [7]. It has been stressed by Gupta et al. [7] that the mechanism behind such extrapulmonary spread of the virus remains elusive. The second attributed cause for extrapulmonary injury in COVID-19 patients is believed to be produced by the dysregulation of the immune system response [7]. It is a consequence of a hyperactive immune response of the innate immune system to the viral infection, releasing levels of interferons, interleukines, tumor-necrosis factors, chemokynes and other mediators that result injurious to host cells [[16], [17], [18]]. This hypothesis is supported by the report of higher-than-normal levels of cytokines in patients blood, particularly of IL-6 [[19], [20], [21]]. Additionally, other key mechanisms of multiorgan damage mentioned in the literature include endothelial cell damage and thromboinflammation and the dysregulation of the renin-angiotensin-aldosterone system [7].
According to the review of the literature by Gupta et al. [7], hematological, gastrointestinal, and endocrine manifestations are mainly produced by the direct toxicity of the virus mediated by ACE2. Dermatologic manifestations are however due to immune response to the virus, mainly produced by the cytokine-release syndrome. The rest of manifestations, cardiovascular, renal, hepatobiliary and neurologic, are considered from multifactorial causes with main components coming from direct viral toxicity and cytokine storm. Apart from few cases reporting the isolation of the virus in myocardial tissue, as well as from gastrointestinal epithelial cells, the hypothesis of the direct virus toxicity is sustained by the abundance of ACE2 in the affected organs, i.e., these are the cases in lymphocytes, kidney, liver, pancreas, and brain. It should be added here that ocular damages have also been reported in several COVID-19 patients. These ocular manifestations are believed to be caused by the viral interaction with ACE2 receptor which is abundant in the conjunctiva and cornea inferior parts.
Here we do not rule out any of the two previously discussed mechanisms of multiorgan damage in COVID-19, which are supported in some cases either directly or indirectly. For instance, the successful use of tocilizumab–an IL-6 receptor antagonist used to treat the cytokine release syndrome (CRS)–in some cases of severe COVID-19 [22] could be indicative of cytokine involvement in these patients, although it has also failed in others where patients has progressed toward secondary hemophagocytic lymphohistiocytosis (sHLH) despite the treatment with this drug [23], possibly indicating alternative mechanisms. The comments by Hedrick et al. [24] and by Campochiaro and Dagna [25] reflect the high degree of uncertainty about the reliability of the IL-6 hypothesis. Our goal instead is to propose a plausible alternative mechanism through which damages can be extended from the lungs to other organs in COVID-19. Although we only consider the case of this novel disease here, our model is generalizable to other viral infectious diseases. It is based on modern understanding of diseases as perturbations of protein-protein interaction (PPI) networks and uses state-of-the-art knowledge about interorgan crosstalk mediated by extracellular vesicles (EVs).
2. Results
2.1. Why another mechanism for multiorgan damage?
Soon after the 2002/2003 SARS epidemic, Hamming et al. [26] studied experimentally the localization of ACE2 protein in human organs, such as oral and nasal mucose, nasopharynx, lung, stomach, small intestine, colon, skin, lymph nodes, thymus, bone marrow, spleen, liver, kidney and brain. They found that ACE2 is present in arterial and venous endothelial cells and arterial smooth muscle cells of all organs studied. A recent bioinformatic study has confirmed, at the level of RNA, that ACE2 is abundant at small intestine, testis, kidneys, heart, thyroid and adipose tissue, with medium expression levels at the lungs, colon, liver, bladder and adrenal gland, and low expression levels in the blood, spleen, bone marrow, brain, blood vessel and muscle [14]. Also in 2004, To et al. [27] analyzed the cellular tropism of six fatal cases of the SARS epidemic of 2002/2003. They found that the lungs and the intestine were effectively infected by SARS-CoV but not the heart, liver, spleen, kidney, bone marrow and lymph node. More recently, Pan et al. [28] have found no evidence of SARS-CoV-2 in semen of males recovering from COVID-19, in spite of the fact that testis is the organ with the highest RNA expression level of ACE2. This lack of matching between ACE2 expression and virus presence in human organs, prompted Hamming et al. [26] to formulate the following fundamental question. If ACE2 is abundant on endothelia and smooth muscle cell of virtually all organs, once the virus is present in the circulation, why it is not found in all these organs? This question remains still open and important today.
In addition, Manne et al. [29] have found that although circulating platelet-neutrophil, −monocyte, and -T-cell aggregates are all significantly elevated in COVID-19 patients compared to healthy donors, ACE2 was not detected by mRNA or protein in platelets. They have found important differences for platelets from COVID-19 patients, such as faster aggregation, and increased spreading on both fibrinogen and collagen. They have concluded that SARS-CoV-2 infection is associated with platelet hyperreactivity which may also contribute to COVID-19 pathophysiology.
The second general mechanism commonly assumed to explain the extrapulmonary effects of COVID-19 is based on the cytokine release syndrome (CRS). CRS was previously found as a potential major cause of morbidity in patients infected with SARS-CoV and MERS-CoV [7]. The syndrome starts with the virus infecting monocytes, macrophages and dendritic cells, which results in the secretion of proinflammatory cytokines like IL-6 and other immunomodulators [16]. The process triggers a systemic “cytokine storm” which involves secretion of more modulators, which finally produces tissues injuries in the host due to hypo- or hyperinflammation. One of the hyperinflammatory syndromes produced by CRS is the secondary hemophagocytic lymphohistiocytosis (sHLH), which produces multiorgan failures. The supporting base for the hypothesis of the CRS is mainly the reports of “elevated” plasma levels of cytokines in COVID-19 [[19], [20], [21]]. In particular, the plasma levels of IL-6 has become a marker of the severity of COVID-19.
The plasma levels of Interleukin-6 (IL-6) reported in several studies for severe cases of COVID-19 are resumed in Table 1. In a meta-analysis performed by Aziz et al. [21] for a total of nine studies including 1426 patients (mean age: 53.0 years, females: 46.6%) the mean serum IL-6 was 56.9 pg/mL (41.4–72.3 pg/mL) for severe COVID-19 and 17.3 pg/mL (13.5–21.1 pg/mL) for non-severe group. The previous values of IL-6 in plasma contrast significantly with those reported for acute respiratory distress syndrome (ARDS), which are given in Table 1. Such dramatic differences have already called the attention of specialists. Leisman et al. [30] remarked that physiologic, clinical and immunologic phenotypes of the respiratory failure produced by COVID-19 “are not consistent with either ARDS or cytokine-release syndromes”. Sinha et al. [31] have also considered this impairment in the IL-6 levels typically produced by “cytokine storm” and the ones found in COVID-19 patients. They asked whether “cytokine storm” is relevant to COVID-19 and their response is a very clear no. Their claim is supported by the fact that the median levels of IL-6 in patients with hyperinflammatory phenotype of ARDS are 10- to 200-fold higher [32] than those observed in patients with severe COVID-19 (see also the comments in [24,25]).
*Critically ill cases.
In closing, neither the direct viral toxicity nor the “cytokine storm” hypothesis can explain along or in combination the multiorgan injuries suffered by patients of severe COVID-19. Therefore, we look at an alternative hypothesis based on modern paradigm of systems biology for explaining the multiorgan propagation of damages from the lungs in COVID-19.
2.2. Model of interorgan propagation of protein perturbations
Targeting specific PPI of the host is an important strategy that viruses use to subvert host cellular processes [41]. PPIs form networks of interacting proteins which have proved to be robust to multiple random failures yet fragile to targeted perturbations. Therefore, the idea that “diseases are a consequence of perturbations” has become a paradigm of modern system biology [42,43]. In a PPI network there are dependencies between proteins connected to each other, which enable perturbations on one protein to propagate along the network and affect other proteins. The network of proteins targeted by SARS-CoV-2 has been constructed by Gysi et al. [44] on the basis of proteomic analysis carried out by Gordon et al. [45]. It consists of all human proteins which interact with those of SARS-CoV-2, such that two proteins are connected in this PPI network if they have been found to interact in the human proteome. In total there are 332 viral targets, from which 239 proteins form a connected subnetwork of viral targets (here called PPI network).
The term “perturbation” embraces several different kinds of protein modifications, ranging from missense mutations to post-translational modifications (PTMs) [[46], [47], [48]]. Here, we mainly focus on PTMs which translate into conformational changes of a protein which may modify its interactions with other proteins. This is the case, for instance, observed for influenza A in which its nonstructural protein NS1 downregulates and inhibits the function of the human Ras homolog gene family member A (RHOA) by means of the physical interaction of RHOA and NS1 [49]. The problem of how a perturbation on one protein is propagated to another in the PPI network is far from trivial. If the two proteins are located in the same cell we can figure out that they navigate the crowded intracellular space in a subdiffusive way until they find each other [50]. However, if the two proteins are in different cells, located in two distant organs, the situation is more complicated. Here we use recent finding on the role played by extracellular vesicles (EVs) in organ crosstalk to sort out this problem [[51], [52], [53], [54], [55]]. EVs are small, membrane-enclosed vesicles ranging from ∼40 nm to several microns in size, and which transport a cargo of proteins, lipids, metabolites and microRNA across organs. They include exosomes (usually 50–150 nm), microvesicles (around 0.1–1 μm), apoptotic vesicles (>1 μm) and large oncosomes (>1 μm). In particular, EVs have been recently recognized as mediators of intercellular crosstalk in lung inflammation and injury [53], which is relevant to the goal of this work. The role of EV in SARS-CoV-2 infection has been reviewed recently by Hassanpour et al. [56] on the basis of their contribution to spread the virus, based on their role in transporting receptors as CD9 and ACE2, which make recipient cells susceptible to virus docking. It is also worth mentioning that EVs play important roles in the viral spread and replication [57,58], particularly of HIV, HCV and SARS [59], Newcastle disease virus [60], HPV [61], flavivirus infection [62], as well as in the pathogenicity of certain viruses in the nervous system [63].
Let us start with one example. The protein NSD2 is in the PPI network of proteins targeted by SARS-CoV-2. It has been found to get involved in hematologic diseases, blood, hemorrhagic and hemostatic disorders, cardiovascular, vascular and immunosystem diseases as well as in lymphatic disorders. This protein is significantly expressed in several tissues outside the lungs. We will call any protein with these conditions a “vulnerable” protein. That is, a vulnerable protein is a human protein which:
(i) is mainly expressed outside the lungs;
(ii) is involved in at least one human disease;
(iii) can be perturbed directly or indirectly by SARS-CoV-2 proteins.
These proteins are vulnerable in the sense that a perturbation of their structures can trigger an extrapulmonary disease. A direct perturbation means that a SARS-CoV-2 protein directly interacts with the vulnerable protein, maybe because the virus can enter in the organ in which this protein is mainly expressed. We should have in mind that viral toxicity does not appear to be a main mechanism of protein damage in general. Therefore, we should suppose that because this protein is expressed mainly outside the lungs, i.e., in the organ O, it is not directly affected by one of the SARS-CoV-2 proteins discharged by the virus in the lung cells L during infection. We should assume then that it is “indirectly” perturbed in the following way. Suppose that there is a protein p that is mainly expressed in the lungs. Then, the human protein p can be directly perturbed by a SARS-CoV-2 protein in L. Now, let us consider that p can navigate from L to O by means of an extracellular vesicle. Once in a cell of the organ O, the protein p can diffuse in the crowded cellular environment until it reaches NSD2, interacting with it, and transmitting the “perturbation” initiated by the SARS-CoV-2 proteins. For the particular case we are considering there are at least three candidates to be perturbators of NSD2. They are p = {BRD4, HDAC2, PLEKHF2}. These three proteins are: (1) highly expressed in the lungs, (2) targeted by SARS-CoV-2 proteins, (3) found in extracellular vesicles, and (4) display high probability of perturbating NSD2 by subdiffusive processes starting at them. Thence, the proteins like BRD4, HDAC2, PLEKHF2 are “perturbators” in the sense that they:
(i) are mainly expressed in the lungs and are targeted by SARS-CoV-2;
(ii) can navigate outside the lungs by using extracellular vehicles to reach a target organ;
(iii) can perturb a vulnerable protein.
We should remark that we have limited our search of vulnerable proteins to the PPI of proteins targeted by SARS-CoV-2. However, other human proteins not in this PPI can also be indirectly targeted by SARS-CoV-2 in the way explained before. A schematic framework of the model proposed in this work is illustrated in Fig. 1 on the basis of the example provided before for the indirect perturbation of NSD2. Here we explain how the model works and in Methods we give the mathematical details of it. Once SARS-CoV-2 enters a lung cell by ACE2-mediated endocytosis it uncoates to liberate the viral RNA and proteins into the human cell cytosol (Fig. 1 Step 1). In this specific example viral protein E interacts with human protein BRD4 producing the perturbation of the last one. The perturbed protein BRD4 is then encapsulated in Step 2 into extracellular vesicles (EV), such as exosomes, which also contain microRNA, lipids and other molecules [[51], [52], [53], [54], [55],64]. It is plausible that in some cases the PPI complex of the human protein with a viral one can be encapsulated in these EVs. In Step 3, many of these EVs go out the lung cells via exocytosis, which is facilitated by an increased permeability in the lungs due to the damage produced by the virus [65]. Some of these exosomes are recognized by their cells of a target organ, which then allow their entrance in these tissues. Once inside the new organ, the exosomes, or other EVs, can liberate the perturbed human protein (or their complexes with a viral protein). The perturbed BRD4 protein finds itself in a crowded cellular environment in which it navigates in a subdiffusive way [[66], [67], [68]]. This subdiffusive displacement of the perturbed BRD4 proteins is illustrated in Step 4 and it allows to encounter NSD2, which is a vulnerable protein. This interaction between BRD4 and NSD2 produces the perturbation of the last one, which may trigger a damage of the target organ in Step 5. (See Table 1 .)
Fig. 1.
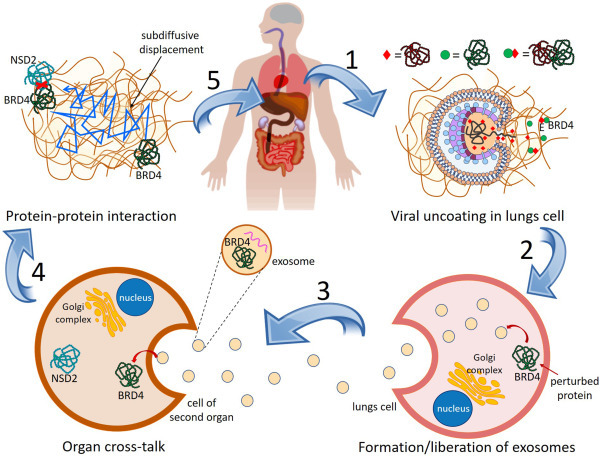
Mechanism of interorgan protein-protein perturbation transmission. Once the SARS CoV-2 enters the cells in the lungs it uncoates and discharges its RNA and proteins into the human cell cytosol (Step 1). Viral proteins interact with human proteins as illustrated for the case of the viral protein E interacting with human protein BRD4. Some of the perturbed human proteins (or its complex with a viral protein) are then encapsulated into exosomes, which are liberated from lungs cells via exocytosis (Step 2). The exosomes containing a perturbed human protein (or its complex with a viral protein) navigates to another organ where they enter via endocytosis (Step 3). Once in the interior of the cell of another organ the perturbed human protein is discharged into the cytosol and it navigates subdiffusively (Step 4) until it encounters an unperturbed protein expressed mainly in this organ. This interaction perturbs the protein NSD2 which is vulnerable and may trigger a damage at this organ (Step 5).
Table 1.
Plasma level of Interleukin-6 (IL-6) reported in severe cases of COVID-19 as well as in “hypo” and hyperinflammatory processes in acute respiratory distress syndrome (ARDS).
| severe COVID-19 | population | IL-6 level pg/mL | ||
|---|---|---|---|---|
| [33] | 84 | 7 (6–11) | ||
| [34] | 54 | 11 (8–14) | ||
| [35] | 286 | 25 (10–55) | ||
| [36] | 237 | 26 (11–69) | ||
| [37] | 85 | 64 (31–165) | ||
| [19] | 17* | 64 (25.6–111.9) | ||
| ARDS | “hypoinflamatory” | hyperinflamatory | ||
| pop. | IL-6 level pg/mL | pop. | IL-6 level pg/mL | |
| [38] | 638 | 86 (34–216) | 246 | 578 (181–2621) |
| [39] | 386 | 154 (67–344) | 135 | 1525 (584–3802) |
| [40] | 451 | 282 (111–600) | 269 | 1618 (517–3205) |
2.3. Vulnerable proteins and their perturbators
Using bioinformatic databases we searched for proteins in the PPI of proteins targeted by SARS-CoV-2 which (i) are involved in human diseases and (ii) which are significantly expressed at organs different from the lungs. In total we selected 26 proteins which are involved in 13 classes of organs/systems damages. These classes (Table S1) correspond to: (1) neurologic diseases; (2) hematologic diseases; (3) liver damage; (4) gastrointestinal diseases; (5) cardiovascular diseases; (6) endocrine system diseases; (7) dermatological diseases; (8) damage to organs in the lymphatic system; (9) immunologic system diseases; (10) musculoskeletal diseases; (11) metabolic diseases; (12) respiratory system diseases (not including specifically lung diseases), and (13) eye system. The classes selected here are very general and the reader should be aware that some of them include diseases which target only specific organs in the systems considered. For instance, in the class “endocrine system” there are diseases involving thyroid or parathyroid glands, testis, seminal vesicle, pancreas, and so forth. More details on each specific diseases and organs are given in Table S1. In assigning these proteins to organs/systems potentially damaged by their perturbations we also consider the RNA expression of the corresponding protein. For instance, protein ALG8 is reported in several metabolic diseases, thus it classifies in the class 11: metabolic diseases. Additionally, it is highly expressed in liver and testis, which also groups it in the classes (3) and (6), respectively. The 26 VPs found in this work are illustrated in Fig. 2 (central column) and the organs/systems affected are in right column of the same Figure (see also Table S1).
Fig. 2.
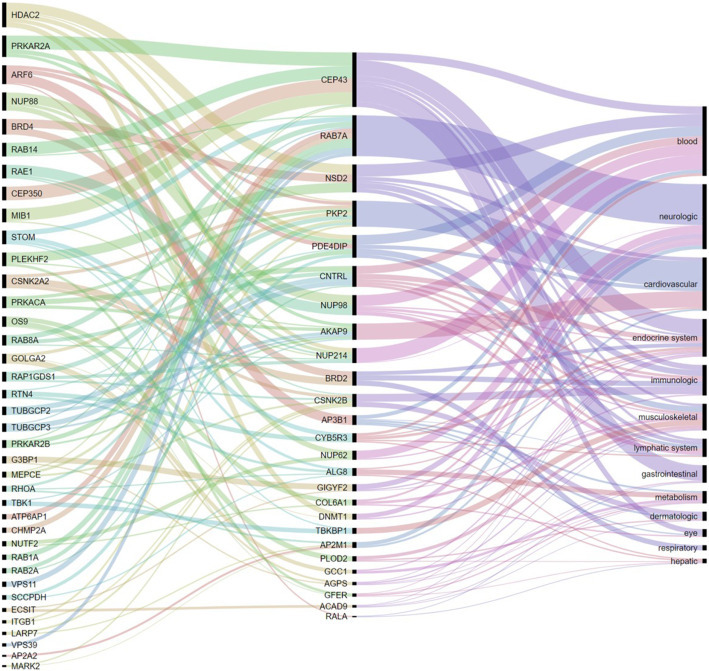
Interorgan perturbations through protein-protein interactions. Left column are proteins mainly expressed in the lungs with high probability of perturbing those with strong evidence of being involved in human diseases (central column), which are mainly expressed in the tissues given in the right column. The thickness of the connections is proportional to the number of diseases of a given class in which a protein is directly (2nd and 3rd columns) or indirectly (1st and 2nd columns) involved.
We now proceed to the identification of the potential perturbators of the 26 candidates to vulnerable proteins identified as detailed in Methods. First, we consider all proteins in the PPI network of proteins targeted by SARS-CoV-2 and find the subset of proteins significantly expressed in the lungs. Using this subset of proteins we interrogate the database “ExoCarta” to determine which of these proteins significantly expressed in the lungs have been reported in extracellular vesicles. This reduced subset of proteins consists of all human proteins that are targeted by SARS-CoV-2 in the lungs and that can navigate outside this organ as cargo of EVs. Finally, every individual protein in this subset was evaluated for its capacity to perturbate any of the vulnerable proteins previously identified. This is carried out as follows. In Fig. 3 (a) we illustrate the time evolution of a subdiffusive process on the PPI network of proteins targeted by SARS-CoV-2 initiated at the protein HDAC2, which is a protein in the subset of candidates to perturbators of vulnerable proteins. Shortly after the process starts, six proteins increase significantly their probabilities of being perturbed by the subdiffusive process initiated at HDCA2. Four of these proteins, DNMT1, NSD2, PDE4DIP and NUP214, are amount the vulnerable proteins previously identified in this work. These proteins are not necessarily adjacent to HDCA2 in the PPI network as can be seen in Fig. 3 (b). We conclude that HDCA2: (i) is significantly expressed in the lungs, (ii) forms exosomes that can transport it outside the lungs, and (iii) produces a significant increment of the perturbation probability for some vulnerable protein. Therefore, we consider HDCA2 as a perturbator of DNMT1, NSD2, PDE4DIP and NUP214.
Fig. 3.
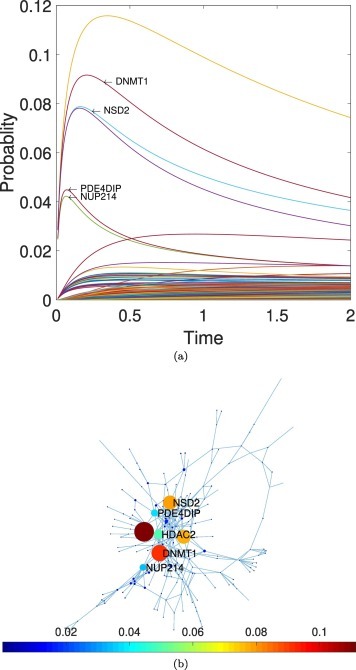
Identification of potential perturbators of vulnerable proteins. (a) Temporal evolution of a subdiffusive process initiated at the protein HDCA2, which is mainly expressed in the lungs. At short times there is a fast increase in the probability of perturbing several proteins. Four of these proteins, DNMT1, NSD2, PDE4DIP and NUP214, have been previously identified as “vulnerable proteins”. (b) Illustration of the position of the activator candidate, HDCA2, and the four vulnerable proteins potentially activated by it, in the PPI network of proteins targeted by SARS-CoV-2. The colorbar indicates the probability that a protein is perturbed at the given time from a perturbation started at HDCA2.
In total we identified 38 potential perturbators of vulnerable proteins as can be seen in Fig. 2 (left column) and Table S1. The average number of vulnerable proteins perturbed by each of these proteins is 1.79, due to the fact that 48.2% of perturbators only perturb one vulnerable protein. A bipartite network representation of the VPs and their perturbators is illustrated in Fig. 4 . The most prolific perturbators are ARF6 and HDAC2, which perturb 4 (not necessarily different) vulnerable proteins each. The protein PRKAR2A which is a hub in the PPI network only perturbs 3 vulnerable proteins. However, it appears at the second top position in the first column of Fig. 2. This is due to the fact that this protein perturbates only three VPs, but they are involved in several diseases. For instance, it perturbates AKAP9 which is involved in several cardiovascular disorders, CEP43 which is involved in several gastrointestinal and immunologic system diseases, and PDE4DIP which is involved in several hematological disorders.
Fig. 4.
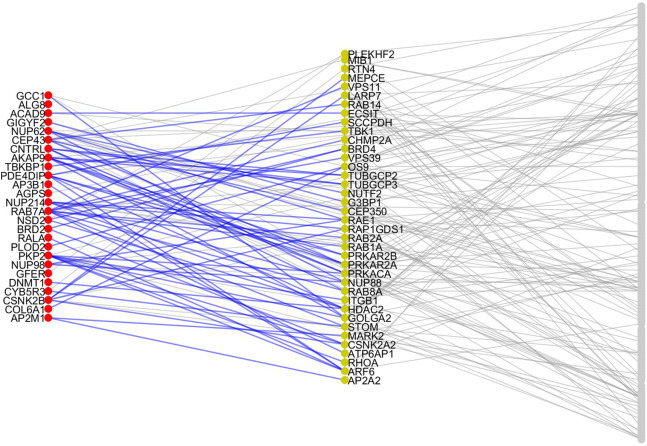
Vulnerable proteins and their activators. Representation of the PPI network of proteins targeted by SARS-CoV-2 which are detected here as VPs for producing multiorgan injuries (left column) and their perturbators (central column). The rest are proteins in the PPI network which are neither in the previous two classes.
2.3.1. VPs and their perturbators in postmortem tissues
Every protein in the PPI network of human proteins targeted by SARS-CoV-2 was interrogated against the database of postmortem tissue proteomic analysis developed by Qiu et al. [69]. These authors collected postmortem tissue samples during the autopsy of 3 patients who deceased from COVID-19 in Wuhan. The samples were from the lungs, heart, liver. Spleen, kidney, intestine, brain and muscle. Total proteins from these samples were treated with trypsin and the resulting 49,815 nonredundant peptides were analyzed with tandem mass spectrometry after being subjected to tandem mass tag. In total 5346 human proteins were quantified in at least one sample. The authors reported the differentially expressed proteins (DEPs) determined with Model-based Analysis of Proteomic data (MAP) [70]. In this case muscle and spleen samples were not considered due to the lack of the corresponding normal tissue data in the Human Proteome Map database (see Methods). Then, we interrogate all the proteins in the PPI network against the 2604, 611, 212, 173, 51, and 42 DEPs in the lungs, kidney, intestine, brain, and heart tissues. We recorded whether the corresponding protein in the PPI network was up- or downregulated in each of the tissues previously mentioned and the statistical analysis of its significance was performed as described in Statistical Analysis. Remarkably, we found that 71.8% of perturbators are altered (up or downregulated) in the postmortem proteomic analysis. In contrast, only 28.8% of all the proteins targeted by SARS CoV-2 were found up- or downregulated in the postmortem proteomic analysis. This difference is statistically significant (p=1.3426 ⋅ 10−6, χ 2=23.4) (see Table S2).
2.4. Drug repurposing for multiorgan damage
One of the main goals of the current protein-driven analysis is to find potential drugs to treat multiorgan damage in COVID-19. We searched for existing drugs that interact with those proteins which have been found to be directly involved in the potential organ damage, i.e., VPs, as well as for drugs that interact with the perturbators of VPs. In total we found 27 drugs which are inhibitors of 12 of the VPs found in this work, either directly or indirectly. The complete list is: amlexanox, apabetalone, azacitidine, belinostat, BI-2536, chlormezanone, decitabine, DI17E6, ellagic acid, entinostat, epigallocatechin gallate, etaracizumab, fedratinib, firategrast, intetumumab, Ly294002, mocetinostat, natalizumab, ozanezumab, panobinostat, procaine, romidepsin, silmitasertib, SNAP-7941, valategrast, volociximab, and vorinostat. The direct (direct path) or indirect routes of action of these drugs is illustrated in Fig. 5 and Table S3. For instance, apabetalone is an inhibitor of the protein BRD2, which is a VP found in hematological, bone marrow, immunologic, pancreas, testis and respiratory system diseases. Additionally, it also inhibits the activity of BRD4 which is a perturbator of BRD2, thus acting indirectly over the same set of diseases. Moreover, BRD4 is also a perturbator of NSD2, which is involved in blood, bone marrow, heart, thymus, immunologic, and lymphatic disorders.
Fig. 5.
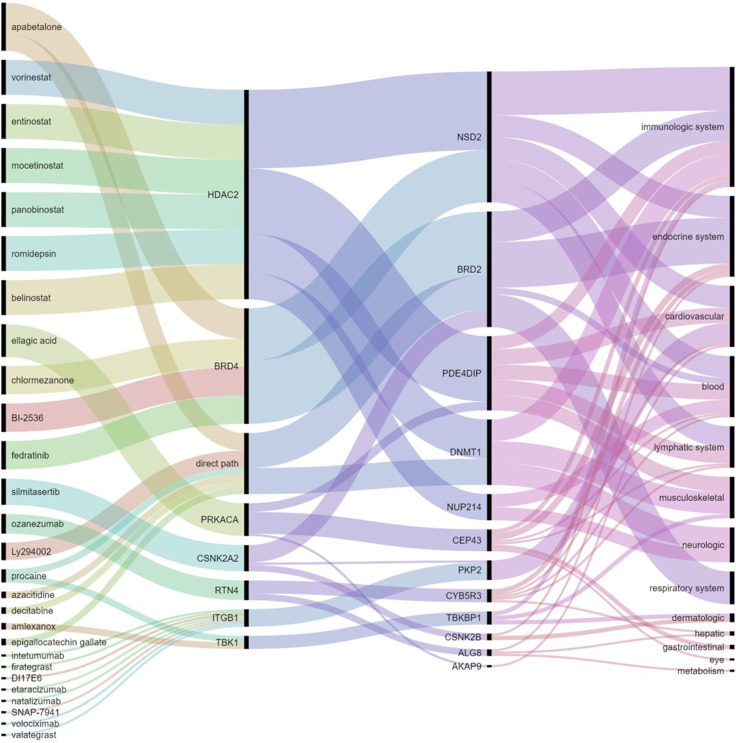
Repurposed drugs for interorgan damage. Group of known drugs (left column) which inhibit receptors mainly expressed in the lungs (second column from left) which have been found here to perturb proteins involved in human diseases (third column from the left) which are mainly expressed in several tissues or systems outside the lungs (right column).
We have interrogated the database Drugs.com for detecting any drug-drug interactions that had been reported among any pair of these 27 drugs. In total we have found 7 “major”, i.e., highly clinically significant, drug-drug interactions indicating that such combinations should be avoided because the risk of the interaction outweighs the benefit. Additionally we identified one “moderately” clinically significant drug-drug interaction, such that this combination should usually be avoided or use it only under special circumstances. The pairs of drugs displaying interactions are illustrated in the form of a graph in Fig. 6 , where major interactions are marked in dark red and the moderate one in dark blue. Three other drugs: chlomezanone, panobinostat, and fedratinib are known to have interactions with foods according to the information in the same database.
Fig. 6.
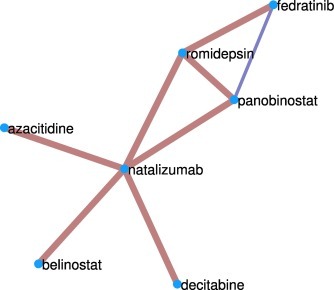
Drug-drug interaction graph. Group of the drugs found here for targeting extrapulmonary damage of COVD-19 which have been reported to have major or moderate interactions that may increase their toxicity.
Some of these 27 drugs: apabetalone (atherosclerosis and associated cardiovascular disease), romidepsin (anticancer agent), silmitasertib (inhibitor of protein kinase CK2), ozanezumab (a monoclonal antibody designed for the treatment of ALS and multiple sclerosis), procaine (local anesthetic), azacitidine (chemotherapy drug), amlexanox (anti-inflammatory antiallergic immunomodulator) and volociximab (antitumoral chimeric monoclonal antibody), form a cluster of drugs free of undesirable drug-drug interactions. The list may also include ellagic acid, which is a natural product found in several fruits and vegetables. The reason is that none of the previously mentioned drugs is known to have interactions with food. The importance of this list is that some of these drugs inhibit proteins that are involved in several of the organs/systems affected by COVID-19, while others are more specific for a smaller number of them. For instance, ellagic acid, apabalone and romidepsin interact with proteins involved in 7 of the 12 organs/systems damaged by COVID-19, while volociximab interacts with proteins involved in one of these organs/systems. In Fig. 7 (Table S3) we illustrate the connection between these organs/diseases through a few representative drugs found in this work. In this multigraph, every node correspond to one of the organs/systems affected by COVID-19 and they are connected by a colored edge if one of the nine drugs found here inhibits proteins involved in both diseases. From the point of view of the organs/systems damaged by COVID-19, the cardiovascular and immunological systems are the ones having more drugs targeting proteins which are affected by SARS-CoV-2, followed by the lymphatic and musculoskeletal ones. On the other side of the coin, the gastrointestinal, hepatic and metabolic systems have only one repurposable drug candidate each. The graph in Fig. 7 allows the selection of drug combinations for treating different sets of organs/systems damaged by COVID-19. For treating all of them it is necessary to find combination of drugs (edges of the graph) that cover all the diseases (nodes), which is achieved by mean of spanning trees. One example is given by the combination of romidepsin, silmitasertib, apabetalone and azacitidine, which is illustrated in the same Figure.
Fig. 7.
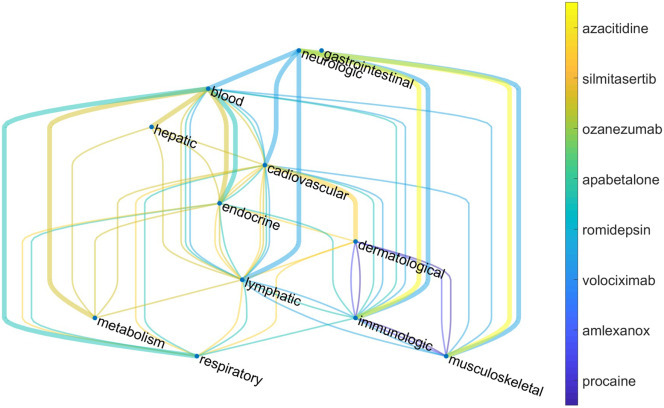
Drugs-relational graph of organs/systems affected by COVID-19. Nodes represent organs/systems targeted by COVID-19. Two nodes are connected if there is an existing drug that inhibits VP(s) involved in diseases affecting both organs/systems simultaneously. Thick lines represents an example of spanning tree which corresponds to a combination of drugs that cover all the organs/systems damaged by COVID-19.
3. Discussion
The first finding of the current work has been the identification of 26 VPs which are involved in human organs/systems diseases and disorders. These VPs have been identified in 7 out of the 8 major groups of extrapulmonary manifestations reported by Gupta et al. [7] for COVID-19. This includes diseases involving organs/systems at the neurologic, gastrointestinal, hematologic, cardiovascular, endocrine and dermatological groups. In the case of renal and hepatic manifestations, none of the VPs reported here are involved directly in diseases targeting these organs/systems. However, several proteins are significantly expressed at the liver, which could be a source of activation at this organ. Also, several of the VPs identified here are involved in metabolic disorders which may have implications in these two organs/systems. In addition, several studies have demonstrated that the liver is an active site of EV uptake [55], which may direct some of the exosomes containing perturbator proteins toward this organ. The renal damages produced by COVID-19 could also be partially explained by the presence of EV [[71], [72], [73]], which have been isolated from urine and which are composed of proteins and nucleic acids that can reflect the pathophysiological state of cells lining the nephron. This is particularly true for the cases of proteinuria occurring during COVID-19. The current work also identified several VPs involved in lymphatic, immunologic, musculoskeletal, and eye disorders [74], which may ring a bell about potential damages of these organs/systems as a consequence of COVID-19.
The main contribution of this work is the proposal of a plausible mechanism by mean of which the VPs significantly expressed in organs other than the lungs are perturbed. The protein perturbators, which are the ones responsible for perturbing VPs, are those which can be perturbed directly by SARS-CoV-2 proteins in the lungs, can navigate outside the lungs in the form of EVs, and finally can perturb VPs in a target organ by a subdiffusive process in the cell of the new organ. A novel paradigm for explaining intercellular crosstalk during lung inflammation and injury is that it occurs through EV enriched in microRNA, proteins, and lipids [53]. With lung inflammation there is a dramatic increase in vascular permeability [65]. Then, some of these EVs can be released in the extracellular space and diffuse in the bloodstream up to other organs where they finally liberate their cargoes. These EVs can reach distant organs such as the brain [75,76], as they have been shown to cross the blood-brain-barrier (BBB) and produce brain inflammation [77]. It is known that different cells in the liver can interact with EVs through specific receptors. It has been documented for instance that exosomes can regulate Hepatitis C virus infection and transfer its infection to non-infected hepatome cells [55]. Similarly, EVs have been extensively described to play important roles in cardiovascular diseases [54,78,79], including the vascular damage produced by the parvovirus B19, which is associated with elevated circulating EVs. The roles of EVs in metabolic diseases are also well-documented [52,80]. Changes in plasma concentrations of EVs and alterations in their cargo have been associated with metabolic dysfunction involving adipose tissue, pancreas, muscle and liver. It has been reported that immunological relevant molecules like cytokines are only transported inside EVs [81], and other molecules like chemokines are markedly enriched inside EVs [53]. Then, it has been observed that circulating EVs may play a role in the development of renal diseases by immunomodulation, thrombogenesis and matrix modulation [73]. Pathologies of main endocrine organs, such as pancreas, thyroid and reproductive system are mediated by EV as intercellular communication tools [82]. EVs are also found to play an increasing role in lymphatic [83] and musculoskeletal disorders [84]. For instance, there are evidence that EVs could be at the interplay between lymphatic function and atherosclerosis [83]. Also, EVs play important roles in normal tissue metabolism as well as tissue injury repair and regeneration in bone, skeletal muscle, and joint health [84]. Finally, it is important to remark that aging has a profound influence on the levels and cargo nature of EVs. It has been shown that EV levels are significantly elevated in old plasma in relation to young one [85]. Additionally, EVs derived from senescent cells have differences in the proteins contained in their cargoes in relation to healthy cells. It is known that senescent cells affect bystander cells by means of the senescence-associated secretory phenotype (SASP). There is increasing evidence that EVs secreted from senescent cells are a novel SASP factor, which have unique characteristics [85]. All these differences in the EV levels and cargo of senescent cells may be related to the observed differences in the damages produced by COVID-19 in young and elder population.
We have seen that only 28.8% of all the proteins targeted by SARS CoV-2 were reported as differentially expressed proteins (DEPs) in a postmortem proteomic analysis of various tissues reported by Qiu et al. [69]. Remarkably, 71.8% of protein perturbators were reported as DEPs (up- or downregulated) in this postmortem proteomic analysis. From those proteins that are DEPs in the lungs, 71.4% were found to be downregulated and only 28.6% were upregulated. The same study reported that ACE2 is a DEP downregulated in the lungs, while it appeared upregulated in the kidneys and it is not a DEP in any other of the tissues studied. Qiu et al. [69] used Gene Ontology analysis to find the major biological functions of the DEPs reported in their study. They reported 8 major functional groups, all of which are represented by proteins identified here as perturbators. The functional groups and the percentages of perturbators found in each of them are as follows (Table S4): Rab protein signal transduction (50%); intracellular protein transport (66.7%); blood coagulation (57.1%); vesicle-mediated transport (66.7%); protein localization to plasma membrane (16.7%); viral process (50%) neutrophil degranulation (20%); post-translational protein modification (42.8). All this together points out to the relevance of these proteins identified here as potential activators of VPs via extracellular vesicle transport from the lungs plus subdiffusion at the target organ.
The third important contribution of the current work is the identification of a group of known drugs which can be repurposed for treating the multiorgan damages produced by COVID-19. Some of these drugs (apabetalone, Ly294002, azacitidine, procaine, decitabine, and epigallocatechin gallate) inhibit directly some of the VPs found in this work, namely BRD2 and DNMT1. Therefore, they act directly over diseases involving the immunologic, endocrine and respiratory systems as well as on musculoskeletal and neurologic disorders. The other drugs may act indirectly over VPs by inhibiting their perturbators. These are the cases of romidepsin and ozanezumab, which may act over 6 different organs/systems through the inhibition of HDAC2 (romidepsin) or RTN4 (ozanezumab). HDAC2 is a perturbator of DNMT1, NSD2, NUP214, NUP214 and PDE4DIP and RTN4 perturbs ALG8 and CYB5R3. From all drugs found here as candidate to be repurposed for treating multiorgan damages produced by COVID-19, romidepsin and apabetalone (we exclude here ellagic acid, which is a natural product) are the ones acting over a larger number of organs/systems damaged by COVID-19. Apabetalone has been previously identified as a potential drug for targeting inflammation and cytokine storm in COVID-19 [86,87]. Such identification has been based on the fact that it inhibits expression of genes related to vascular inflammation via the suppression of BET-mediated transcription. The last are triggered by multiple pro-inflammatory stimuli, such as the ones occurring in COVID-19. The same works pointed out that the inhibition of BRD4 could be considered as a potential route to ameliorate hyperinflammation and cytokine storm produced by this disease. Romidepsin and vorinestat [88,89] which target HDAC2, has also been suggested as potential candidates for treating COVID-19 effects. In fact, romidepsin [88] is one of the drugs recommended by experts for immunomodulators treatment and immune based dermatological disorders produced by COVID-19.
Silmitasertib is a drug acting over 4 of the 12 organs/systems damaged by COVID-12. It does so by inhibiting CSNK2A2 that is involved in regulation of stress granules, and which is a perturbator of BRD2 and CSNK2B. It was recently found that silmitasertib has antiviral activity (IC50 = 2.34 mM) against SARS-CoV-2 [90], which may be an important addition for its use in COVID-19 patients. According to Bouhaddou et al. [90] this drug is currently being considered for human testing as a potential treatment for COVID-19 due to its combination of actions which includes its potential role in remodeling extracellular matrix upon infection (see also [91]). Procaine is a drug which acts directly and indirectly over VPs involving 4 organs/systems targeted by COVID-19. Local anesthetics, like lidocaine and procaine, have been recommended in case of acute lung injury produced by COVID-19 [92]. Another drug found here as a potential candidate for treating multiorgan damage produced by COVID-19 is amlexanox. It has been identified as a candidate for downregulating the androgen biosynthesis pathway, which is upregulated due to COVID-19, as well as for downregulating the macrophages, monocytes and dendritic cells pathway potentially linked to severe symptoms in COVID-19 patients [93]. Das et al. [93] proposed the drugs vorinostat, panobinostat, azacitidine, and clofarabine, all identified here as candidates to treat multiorgan damage produced by COVID-19, for downregulating the mitotic cell cycle-related pathways and proteasome activity, which play an important role in coronaviral replication. Azacitidine and vorinostat are proposed as drugs to upregulate the CTL or Interleukin 2 pathways, which are important for antiviral immune responses [93]. Azacitidine was indeed identified by Gordon et al. [45] as possible treatment options for COVID-19.
An interesting case from the perspective of drug repurposing is ozanezumab, which is a humanized monoclonal antibody against Nogo-A (RTN4) that was well tolerated in humans. It was though as a drug for targeting amyotrophic lateral sclerosis (ALS). The results of a randomized, double-blind, placebo-controlled, phase 2 trial concludes that ozanezumab does not “show efficacy compared with placebo in patients with ALS” [94]. The protein RTN4 was found here as an important perturbator of the VPs ALG8 and CYB5R3, which are involved in hepatic, metabolic, endocrine, blood, cardiovascular and lymphatic organs/systems damages produced by COVID-19. Therefore, conducting clinical trials with ozanezumab to target multiorgan damages in COVID-19 appears as an excellent opportunity for this drug.
Finally, we consider the analysis of ellagic acid, a natural polyphenol, which is found in fruits and vegetables. Ellagic acid is known for its antioxidant and other pharmacological properties [95]. It has been reported to inhibit the protein PRKACA, in particular to be a strong inhibitor of the catalytic subunit of the cyclic AMP-dependent protein kinase [96]. PRKACA is one of the hubs of the PPI network of human proteins targeted by SARS-CoV-2. It is a perturbator of the following VPs: AKAP9, CEP43, and PDE4DIP, which are involved in cardiovascular, blood, immunologic, gastrointestinal, endocrine, eye, lymphatic and musculoskeletal disorders. It is difficult to infer whether this natural product could be effective in the treatment of multiorgan damages produced by COVID-19 due to the lack of information about its use as a drug in humans. However, it could be interesting to design trials to assess its potential use for palliating the effects of COVID-19.
4. 3. Conclusions
We propose here a new plausible mechanism by mean of which the SARS-CoV-2 can propagate its damages from the lungs of infected individuals to other organs. First, we present evidence against the “universal” character of currently accepted mechanisms for such multiorgan damages and propose an alternative mechanism. This new mechanism considers the perturbation of host proteins expressed in the lungs by viral proteins. These perturbations are transmitted outside the lungs via the protein-protein interaction (PPI) network of the host, by a combination of extracellular transport mediated by extracellular vesicles and intracellular subdiffusion in the target organ. We identify 26 vulnerable proteins (VPs) and their 38 perturbators which are involved in potential damage of most of the organs/systems affected in COVID-19 patients. We identify several drugs that target VPs or their perturbators and represent good candidates for repurposable drugs against the multiorgan damage produced by SARS-CoV-2 in COVID-19 patients.
The main limitation of this work is that we have only considered the human proteins directly targeted by SARS-CoV-2 for the analysis. It is known that these proteins are embedded in the human PPI network which contains thousands more proteins and interactions that the ones considered here. Because the set of perturbators is formed only by proteins directly targeted by the virus, it is not altered if we consider the whole human PPI network. However, the transmission of protein perturbations from the lungs to other organs can be mediated also from other proteins and organs/systems damaged not considered here, but which are relatively close to the VPs detected here. Also it is plausible that other proteins, not targeted directly by the virus, can be perturbed by proteins directly targeted by SARS-CoV-2. This implies that the set of VPs could be wider than the one considered here and include other proteins not directly targeted by SARS-CoV-2. In this view, the current work can be seen as the first order approach to the interorgan transmission of protein perturbations involving only those directly targeted directly by the virus.
5. 4. Materials and methods
5.1. 4.1. Protein-protein interaction network
Gordon et al. [45] identified 332 human proteins which are targeted by 26 of the 29 SARS-CoV-2 proteins. These proteins were identified using affinity-purification followed by mass spectrometry. Gysi et al. [44] built a human interactome consisting of 18,508 proteins and 332,749 interactions. This PPI network of human proteins was assembled from 21 public databases compiling experimentally-derived PPI data. Then every protein targeted by SARS-CoV-2 was mapped into this interactome creating the PPI network of proteins targeted by the virus. This network is a graph G = (V, E) where the nodes are the 332 human proteins targeted by SARS-CoV-2 and the edges are formed by pairs of human proteins that have been reported to interact in the human interactome previously considered. This graph is formed by a main connected component having n=239 proteins and m=360 edges representing PPIs. The other 93 human proteins targeted by the virus form a few components disconnected from the main one. Here we consider the main connected component as the PPI network of proteins targeted by the virus.
5.2. 4.2. Identification of vulnerable proteins (VPs)
Every protein in the PPI network of proteins targeted by SARS-CoV-2 was interrogated with version 7.0 of DisGeNET ([97]) which contains 1,134,942 gene-disease associations (GDAs), between 21,671 genes and 30,170 diseases, disorders, traits, and clinical or abnormal human phenotypes, and 369,554 variant-disease associations (VDAs), between 194,515 variants and 14,155 diseases, traits, and phenotypes. We only selected proteins with “definitive” or “strong” evidence of being involved in a human disease or syndrome. Then, we interrogated each of these candidates by using the disease network database created by Menche et al. [98], which contains 299 diseases and 3173 associated genes. This database integrates disease-gene annotations from Online Mendelian Inheritance in Man (OMIM) and UniProtKB/Swiss-Prot with GWAS data from the Phenotype-Genotype Integrator database (PheGenI), using a genome-wide significance cutoff of p value ≤5 × 10−8. The resulting subset of proteins consists of 26 proteins which are involved in 105 diseases. We then grouped these diseases according to the main organs/systems targeted by them. This resulted in 13 categories: (1) neurologic, (2) blood, (3) hepatic, (4) gastrointestinal, (5) cardiovascular, (6) endocrine, (7) dermatological, (8) lymphatic, (9) immunologic, (10) musculoskeletal, (11) metabolism, (12) respiratory, and (13) eye disorders. We then used the consensus database of the Human Protein Atlas [99], which integrates several databases for the median gene expression of RNA-seq for 61 different tissues. Then, we checked every VPs previously selected for the tissues in which it has the highest expression according to the consensus database. In case that such tissues are not previously considered among the ones affected by diseases involving that protein, we include it.
5.3. 4.3. Identification of protein perturbators
We start by dividing the set of proteins in the PPI network into two subsets V V and V R, such that all previously identified VPs are in the set V V and the rest of proteins are in V R. We then identified those proteins in V R which are significantly expressed in the lungs according to The Human Protein Atlas and form the subset V L ⊂ V R. Then, we interrogate the database ExoCarta for each protein in V L. Those proteins in V L found to be reported as part of any extracellular vesicle in ExoCarta form the subset of proteins V P ⊂ V L ⊂ V R.
Then, we pick a protein i ∈ V P and set p 0, i=1 and p 0, j=0 for all j ≠ i. We then perform a simulation of the subdiffusive propagation of a perturbation from the protein i ∈ V P to the rest using the fractional diffusion equation (FDE) (see next subsubsection) and its analytic solution. If for a given value of α and a given value of t < t c, the probability that a protein j ∈ V V is perturbed increases significantly (see Fig. 3(a)) we consider that the protein i is a perturbator of j. We explored different values of 0<α<1 and observed that for small values of this parameter, e.g., for α ≤ 0.75, the increase in the probability of perturbation for some proteins occur instantaneously, which lack any physical sense [100]. Then, we selected here α=0.75 as the threshold and t c=1 for all simulations. This process is repeated for each of the proteins in V R. This creates a new subset of perturbator candidates V PC ⊆ V P which is formed by all those proteins targeted by SARS-CoV-2, which are significantly expressed in the lungs, that can navigate outside this organ via EVs, and that can activate VPs via subdiffusive dynamics in the PPI network. The mathematical details of the simulations of the subdiffusive process in the PPI is given in the next subsection.
5.3.1. 4.3.1. Protein subdiffusion in intracellular media
Protein-protein interaction occurs after the encounter of the two proteins in the intracellular environment. For such encounter to take place the proteins need to navigate in a very crowded media which is know to produce subdiffusive dynamics [[66], [67], [68]]. In this scenario, the mean square displacement 〈x 2(t)〉 of a protein scales as a power-law with the time t of diffusion, such that
| (4.1) |
where 0<κ<1 is the anomalous diffusion exponent. Experiments conducted by Gupta et al. [66] using state-of-the-art neutron spectroscopy at a resolution of up to 10−9 s has shown that the motion of (globular) proteins in a crowded media is well-described by the fractional Fokker-Planck equation (FFPE) with a periodic potential. Here we consider the case in which there is no external force in the FFPE, which reduces it to the fractional diffusion equation (FDE). Then, let G = (V, E) be the PPI network of proteins targeted by SARS-CoV-2, and let p i(t) be the probability that the protein i is perturbed at time t by a perturbation being propagated through the network. Then, the evolution of p i(t) as the time progresses is determined by the FDE on the network [67,68]:
| (4.2) |
with a given initial condition (see further), and where K α is the diffusion coefficient of the network (hereafter set to unity), and where D t α f is the time-fractional Caputo derivative of f of order α [101]. This time-fractional derivative generalizes the classical integer-order derivative of a function and it is defined for any 0<α ≤ 1 and any function f:[0, ∞ ) → ℝ, as [101]
where Γ(⋅) is the Euler gamma function, γ = ⌈α⌉ and f (γ) represents the γth derivative of the function f.
Here we consider a vector p(t) ∈ ℝ n×1 whose ith entry corresponds to p i(t). Then, the initial condition is given by the vector p(0) = p 0. If a subdiffusive process starts from a perturbation of the protein i we then set p 0, i=1 and p 0, j=0 for all j ≠ i. We can now write the FDE in matrix-vector form as
| (4.3) |
where is the graph Laplacian, i.e., , with K the diagonal matrix of node degrees and A the adjacency matrix of the network. The solution of the FDE with given initial condition p 0 is
| (4.4) |
where E α, 1(γL) is a Mittag-Leffler function of the Laplacian matrix of a graph define as [101]:
| (4.5) |
4.4. Identification of repurposable drug candidates
This experiment was carried out by interrogating each of the VPs and their perturbators against the drug–gene interaction database (DGIdb) [102]. This database normalizes content from 30 disparate sources of 56,309 interaction claims. Its main different with other popular databases like DrugBank and Therapeutic Target Database is that DGIdb aggregates many such databases into a common framework and it adds considerable functionality for efficiently searching a list of input genes against these sources. DGIdb incorporates sources of drug-gene interaction data that were previously only available in inaccessible formats (e.g. Tables in a PDF document). Then, we first interrogate each of our target proteins in this database using the “Search Interactions” facility. We double-checked each of the interactions found by using DrugBank and ChemDBL databases. We only considered interactions between the targeted proteins and approved or investigational drugs, but not for chemical compounds identified as inhibitors of these proteins but not in clinical trials.
5.4. 4.5. Statistical analysis
Statistical analysis was used to compare two proportions. In particular we compared the proportion of perturbators that were found altered (up- or downregulated) in the postmortem dataset of Qiu et al. [69] versus the proportion of VPs reported altered in the same dataset. We also used the test to compare the proportion of altered perturbators to the proportion of altered proteins in the whole PPI network, as well as the last versus the VPs. Here we used a Matlab implementation of a simple Chi-square test to compare two proportions for a 2-sided test with α=0.05.
5.5. 4.6. Resume of databases used
| RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| List of human proteins targeted by SARS-CoV-2 | [45] | |
| PPI network of proteins targeted by SARS-CoV-2 | [44] | |
| Proteins found in extracellular vesicles | [103] | http://www.exocarta.org/ |
| The Human Protein Atlas | [99] | https://www.proteinatlas.org/ |
| DisGeNET database | [97] | https://www.disgenet.org/home/ |
| ExoCarta | [103] | http://www.exocarta.org/ |
| Gene-disease relations | [98] | |
| Postmortem tissue proteomic analysis | [69] | |
| The Drug Gene Interaction Database, DGIdb | [102] | http://www.dgidb.org/ |
| DrugBank | https://www.drugbank.ca/ | |
| ChemDBL | https://www.ebi.ac.uk/chembl/ | |
| Drugs Interaction Checker | https://www.drugs.com/drug _interactions.html | |
| Software and Algorithms | ||
| MATLAB | MathWorks | http://www.mathworks.com |
| RAWGraph | DensityDesign Research Lab (Politecnico di Milano) | https://app.rawgraphs.io/ |
Conflict of interest
The author declares no conflict of interest of any kind.
Credit author statement.
EE designed the research, perform all calculations and simulations, analyzed the results and wrote the paper.
References
- 1.Estrada E. Covid-19 and sars-cov-2. Modeling the present, looking at the future. Phys Rep. 2020;869:1–51. doi: 10.1016/j.physrep.2020.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorbalenya A., Baker S., Baric R., de Groot R., Drosten C., Gulyaeva A. Coronaviridae study group of the international committee on taxonomy of viruses. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-ncov and naming it sars-cov-2. Nat Microbiol. 2020;2020:03–04. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., Song Z.-G. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang F., Deng L., Zhang L., Cai Y., Cheung C.W., Xia Z. Review of the clinical characteristics of coronavirus disease 2019 (covid-19) J Gen Intern Med. 2020;35(7798):545–1549. doi: 10.1007/s11606-020-05762-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.M. Zhou, X. Zhang, J. Qu, Coronavirus disease 2019 (covid-19): a clinical update, Frontiers of Medicine (2020) in press. doi: 10.1007/s11684–020–0767–8. [DOI] [PMC free article] [PubMed]
- 7.Gupta A., Madhavan M.V., Sehgal K., Nair N., Mahajan S., Sehrawat T.S. Extrapulmonary manifestations of covid-19. Nat Med. 2020;26(7):1017–1032. doi: 10.1038/s41591-020-0968-3. [DOI] [PubMed] [Google Scholar]
- 8.Prasanna P.L., Abilash V. 2020. Coronaviruses pathogenesis, comorbidities and multi-organ damage–a review, life sciences. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robba C., Battaglini D., Pelosi P., Rocco P.R. 2020. Multiple organ dysfunction in sars-cov-2: MODS-CoV-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bourgonje A.R., Abdulle A.E., Timens W., Hillebrands J.-L., Navis G.J., Gordijn S.J. Angiotensin-converting enzyme-2. ace2), sars-cov-2 and pathophysiology of coronavirus disease 2019 (covid-19J Pathol. 2020;251:228–248. doi: 10.1002/path.5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alqahtani S.A., Schattenberg J.M. Liver injury in covid-19: the current evidence. United European Gastroenterology Journal. 2020;8(5):509–519. doi: 10.1177/2050640620924157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.H. Su, M. Yang, C. Wan, L.-X. Yi, F. Tang, H.-Y. Zhu, F. Yi, H.-C. Yang, A. B. Fogo, X. Nie, et al., Renal histopathological analysis of 26 postmortem findings of patients with covid-19 in China, Kidney Int 98 (1) (2020) P219–227. [DOI] [PMC free article] [PubMed]
- 13.Hess D.C., Eldahshan W., Rutkowski E. Covid-19-related stroke. Transl Stroke Res. 2020;11:322–325. doi: 10.1007/s12975-020-00818-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li M.-Y., Li L., Zhang Y., Wang X.-S. Expression of the sars-cov-2 cell receptor gene ace2 in a wide variety of human tissues. Infect Dis Poverty. 2020;9:1–7. doi: 10.1186/s40249-020-00662-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S. Sars-cov-2 cell entry depends on ace2 and tmprss2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore J.B., June C.H. Cytokine release syndrome in severe covid-19. Science. 2020;368(6490):473–474. doi: 10.1126/science.abb8925. [DOI] [PubMed] [Google Scholar]
- 17.Nile S.H., Nile A., Qiu J., Li L., Jia X., Kai G. Covid-19: pathogenesis, cytokine storm and therapeutic potential of interferons. Cytokine Growth Factor Rev. 2020;53:66–70. doi: 10.1016/j.cytogfr.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coperchini F., Chiovato L., Croce L., Magri F., Rotondi M. The cytokine storm in covid-19: an overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020;53:25–32. doi: 10.1016/j.cytogfr.2020.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen X., Zhao B., Qu Y., Chen Y., Xiong J., Feng Y. Detectable serum SARS-CoV-2 viral load (rnaaemia) is closely correlated with drastically elevated interleukin 6 (il-6) level in critically ill covid-19 patients. Clin Infect Dis. 2020 doi: 10.1093/cid/ciaa449. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y., Zhang C., Huang F., Yang Y., Wang F., Yuan J. Elevated plasma level of selective cytokines in covid-19 patients reflect viral load and lung injury. Natl Sci Rev. 2020;7:1003–1011. doi: 10.1093/nsr/nwaa037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aziz M., Fatima R., Assaly R. Elevated interleukin-6 and severe covid-19: a meta-analysis. J Med Virol. 2020 doi: 10.1002/jmv.25948. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu X., Han M., Li T., Sun W., Wang D., Fu B. Effective treatment of severe covid-19 patients with tocilizumab. Proc Natl Acad Sci. 2020;117(20):10970–10975. doi: 10.1073/pnas.2005615117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Radbel J., Narayanan N., Bhatt P.J. Use of tocilizumab for covid-19 infection-induced cytokine release syndrome: a cautionary case report. Chest. 2020;158(1):E15–E19. doi: 10.1016/j.chest.2020.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hedrick T.L., Murray B.P., Hagan R.S., Mock J.R. Covid-19: clean up on il-6. American Journal of Respiratory Cell and Molecular Biology (ja) 2020 doi: 10.1165/rcmb.2020-0277LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campochiaro C., Dagna L. The conundrum of interleukin-6 blockade in covid-19. The Lancet Rheumatology. 2020 doi: 10.1016/S2665-9913(20)30287-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.I. Hamming, W. Timens, M. Bulthuis, A. Lely, G. v. Navis, H. van Goor, Tissue distribution of ace2 protein, the functional receptor for SARS coronavirus. a first step in understanding sars pathogenesis, The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland 203 (2) (2004) 631–637. [DOI] [PMC free article] [PubMed]
- 27.K. To, Tong J.H., Chan P.K., Au F.W., Chim S.S., Allen Chan K. Tissue and cellular tropism of the coronavirus associated with severe acute respiratory syndrome: an in-situ hybridization study of fatal cases. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland. 2004;202(2):157–163. doi: 10.1002/path.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan F., Xiao X., Guo J., Song Y., Li H., Patel D. No evidenceof severe acute respiratory syndrome-coronavirus 2 in semen of males recovering from coronavirus disease 2019. Fertil Steril. 2020;113(6):1135–1139. doi: 10.1016/j.fertnstert.2020.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manne B.K., Denorme F., Middleton E.A., Portier I., Rowley J.W., Stubben C.J. Platelet gene expression and function in covid-19 patients. Blood. 2020 doi: 10.1182/blood.2020007214. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leisman D.E., Deutschman C.S., Legrand M. Facing covid-19 in the icu: vascular dysfunction, thrombosis, and dysregulated inflammation. Intensive Care Med. 2020;46:1105–1108. doi: 10.1007/s00134-020-06059-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sinha P., Matthay M.A., Calfee C.S. Is a "cytokine storm" relevant to covid-19? JAMA Intern Med. 2020 doi: 10.1001/jamainternmed.2020.3313. in press. [DOI] [PubMed] [Google Scholar]
- 32.Sinha P., Delucchi K.L., McAuley D.F., O'Kane C.M., Matthay M.A., Calfee C.S. Development and validation of parsimonious algorithms to classify acute respiratory distress syndrome phenotypes: a secondary analysis of randomised controlled trials. Lancet Respir Med. 2020;8(3):247–257. doi: 10.1016/S2213-2600(19)30369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu C., Chen X., Cai Y., Zhou X., Xu S., Huang H. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020;180(7):934–943. doi: 10.1001/jamainternmed.2020.0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.F. Zhou, T. Yu, R. Du, G. Fan, Y. Liu, Z. Liu, J. Xiang, Y. Wang, B. Song, X. Gu, et al., Clinical course and risk factors for mortality of adult inpatients with covid-19 in Wuhan, China: a retrospective cohort study, The Lancet 395 (10229) (2020) P1054–1062. [DOI] [PMC free article] [PubMed]
- 35.Qin C., Zhou L., Hu Z., Zhang S., Yang S., Tao Y. Dysregulation of immune response in patients with covid-19 in Wuhan, China. Clin Infect Dis. 2020;71(15):762–768. doi: 10.1093/cid/ciaa248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.M. J. Cummings, M. R. Baldwin, D. Abrams, S. D. Jacobson, B. J. Meyer, E. M. Balough, J. G. Aaron, J. Claassen, L. E. Rabbani, J. Hastie, et al., Epidemiology, clinical course, and outcomes of critically ill adults with covid-19 in New York city: a prospective cohort study, The Lancet 395 (10239) (2020) P1763–1770. [DOI] [PMC free article] [PubMed]
- 37.Mo P., Xing Y., Xiao Y., Deng L., Zhao Q., Wang H. 2020. Clinical characteristics of refractory covid-19 pneumonia in Wuhan, China, clinical infectious diseases. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Famous K.R., Delucchi K., Ware L.B., Kangelaris K.N., Liu K.D., Thompson B.T. Acute respiratory distress syndrome subphenotypes respond differently to randomized fluid management strategy. Am J Respir Crit Care Med. 2017;195(3):331–338. doi: 10.1164/rccm.201603-0645OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calfee C.S., Delucchi K., Parsons P.E., Thompson B.T., Ware L.B., Matthay M.A. Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med. 2014;2(8):611–620. doi: 10.1016/S2213-2600(14)70097-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sinha P., Delucchi K.L., Thompson B.T., McAuley D.F., Matthay M.A., Calfee C.S. Latent class analysis of ards subphenotypes: a secondary analysis of the statins for acutely injured lungs from sepsis (sails) study. Intensive Care Med. 2018;44(11):1859–1869. doi: 10.1007/s00134-018-5378-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fan Y., Sanyal S., Bruzzone R. Breaking bad: how viruses subvert the cell cycle. Front Cell Infect Microbiol. 2018;8:396. doi: 10.3389/fcimb.2018.00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Del Sol A., Balling R., Hood L., Galas D. Diseases as network perturbations. Curr Opin Biotechnol. 2010;21(4):566–571. doi: 10.1016/j.copbio.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 43.Furlong L.I. Human diseases through the lens of network biology. Trends Genet. 2013;29(3):150–159. doi: 10.1016/j.tig.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 44.D. M. Gysi, Í. D. Valle, M. Zitnik, A. Ameli, X. Gan, O. Varol, H. Sanchez, R. M. Baron, D. Ghiassian, J. Loscalzo, et al., Network medicine framework for identifying drug repurposing opportunities for covid-19, arXiv preprint arXiv:2004.07229 (2020). [DOI] [PMC free article] [PubMed]
- 45.Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M. A sars-cov-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sahni N., Yi S., Taipale M., Bass J.I.F., Coulombe-Huntington J., Yang F. Widespread macromolecular interaction perturbations in human genetic disorders. Cell. 2015;161(3):647–660. doi: 10.1016/j.cell.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen Q., Cheng F., Song H., Lu W., Zhao J., An X. Proteome-scale investigation of protein allosteric regulation perturbed by somatic mutations in 7,000 cancer genomes. The American Journal of Human Genetics. 2017;100(1):5–20. doi: 10.1016/j.ajhg.2016.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rajasekaran N., Sekhar A., Naganathan A.N. A universal pattern in the percolation and dissipation of protein structural perturbations. The Journal of Physical Chemistry Letters. 2017;8(19):4779–4784. doi: 10.1021/acs.jpclett.7b02021. [DOI] [PubMed] [Google Scholar]
- 49.Jiang W., Wang Q., Chen S., Gao S., Song L., Liu P. Influenza a virus ns1 induces g0/g1 cell cycle arrest by inhibiting the expression and activity of rhoa protein. J Virol. 2013;87(6):3039–3052. doi: 10.1128/JVI.03176-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grimaldo M., Roosen-Runge F., Zhang F., Schreiber F., Seydel T. Dynamics of proteins in solution. Q Rev Biophys. 2019;52(e7):1–63. [Google Scholar]
- 51.Tkach M., Théry C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164(6):1226–1232. doi: 10.1016/j.cell.2016.01.043. [DOI] [PubMed] [Google Scholar]
- 52.Guay C., Regazzi R. Exosomes as new players in metabolic organ cross-talk. Diabetes Obes Metab. 2017;19:137–146. doi: 10.1111/dom.13027. [DOI] [PubMed] [Google Scholar]
- 53.Lee H., Abston E., Zhang D., Rai A., Jin Y. Extracellular vesicle: an emerging mediator of intercellular crosstalk in lung inflammation and injury. Front Immunol. 2018;9:924. doi: 10.3389/fimmu.2018.00924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sluijter J.P.G., Davidson S.M., Boulanger C.M., Buzas E.I., De Kleijn D.P.V., Engel F.B. Extracellular vesicles in diagnostics and therapy of the ischaemic heart: position paper from the working group on cellular biology of the heart of the european society of cardiology. Cardiovasc Res. 2018;114(1):19–34. doi: 10.1093/cvr/cvx211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nawaz M., Shah N., Zanetti B.R., Maugeri M., Silvestre R.N., Fatima F. Extracellular vesicles and matrix remodeling enzymes: the emerging roles in extracellular matrix remodeling, progression of diseases and tissue repair. Cells. 2018;7(10):167. doi: 10.3390/cells7100167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hassanpour M., Rezaie J., Nouri M., Panahi Y. Infection; Genetics and Evolution: 2020. The role of extracellular vesicles in covid-19 virus infection; p. 104422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bello-Morales R., Ripa I., López-Guerrero J.A. Extracellular vesicles in viral spread and antiviral response. Viruses. 2020;12(6):623. doi: 10.3390/v12060623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kumar A., Kodidela S., Tadrous E., Cory T.J., Walker C.M., Smith A.M. Extracellular vesicles in viral replication and pathogenesis and their potential role in therapeutic intervention. Viruses. 2020;12(8):887. doi: 10.3390/v12080887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Giannessi F., Aiello A., Franchi F., Percario Z.A., Affabris E. The role of extracellular vesicles as allies of hiv, hcv and sars viruses. Viruses. 2020;12(5):571. doi: 10.3390/v12050571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou C., Tan L., Sun Y., Qiu X., Meng C., Liao Y. Exosomes carry micrornas into neighboring cells to promote diffusive infection of Newcastle disease virus. Viruses. 2019;11(6):527. doi: 10.3390/v11060527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guenat D., Hermetet F., Prétet J.-L., Mougin C. Exosomes and other extracellular vesicles in hpv transmission and carcinogenesis. Viruses. 2017;9(8):211. doi: 10.3390/v9080211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reyes-Ruiz J.M., Osuna-Ramos J.F., Jesús-González D., Adrián L., Palacios-Rápalo S.N., Cordero-Rivera C.D. The regulation of flavivirus infection by hijacking exosome-mediated cell–cell communication: new insights on virus–host interactions. Viruses. 2020;12(7):765. doi: 10.3390/v12070765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kutchy N.A., Peeples E.S., Sil S., Liao K., Chivero E.T., Hu G. Extracellular vesicles in viral infections of the nervous system. Viruses. 2020;12(7):700. doi: 10.3390/v12070700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Szabo G., Momen-Heravi F. Extracellular vesicles in liver disease and potential as biomarkers and therapeutic targets. Nat Rev Gastroenterol Hepatol. 2017;14(8):455. doi: 10.1038/nrgastro.2017.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nagy J.A., Benjamin L., Zeng H., Dvorak A.M., Dvorak H.F. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis. 2008;11(2):109–119. doi: 10.1007/s10456-008-9099-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gupta S., Biehl R., Sill C., Allgaier J., Sharp M., Ohl M. Protein entrapment in polymeric mesh: diffusion in crowded environment with fast process on short scales. Macromolecules. 2016;49(5):1941–1949. [Google Scholar]
- 67.Sposini V., Chechkin A.V., Seno F., Pagnini G., Metzler R. Random diffusivity from stochastic equations: comparison of two models for brownian yet non-gaussian diffusion. New J Phys. 2018;20(4) [Google Scholar]
- 68.Barkai E., Metzler R., Klafter J. From continuous time random walks to the fractional fokker-planck equation. Physical Review E. 2000;61(1):132. doi: 10.1103/physreve.61.132. [DOI] [PubMed] [Google Scholar]
- 69.Qiu Y., Wu D., Ning W., Zhang J., Shu T., Huang C. 2020. Postmortem tissue proteomics reveals the pathogenesis of multiorgan injuries of covid-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li M., Tu S., Li Z., Tan F., Liu J., Wang Q. Map: model-based analysis of proteomic data to detect proteins with significant abundance changes. Cell Discovery. 2019;5(1):1–12. doi: 10.1038/s41421-019-0107-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Salih M., Zietse R., Hoorn E.J. Urinary extracellular vesicles and the kidney: biomarkers and beyond. American Journal of Physiology-Renal Physiology. 2014;306(11):F1251–F1259. doi: 10.1152/ajprenal.00128.2014. [DOI] [PubMed] [Google Scholar]
- 72.Zhang W., Zhou X., Zhang H., Yao Q., Liu Y., Dong Z. Extracellular vesicles in diagnosis and therapy of kidney diseases. American Journal of Physiology-Renal Physiology. 2016;311(5):F844–F851. doi: 10.1152/ajprenal.00429.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Karpman D., Ståhl A.-l., Arvidsson I. Extracellular vesicles in renal disease. Nat Rev Nephrol. 2017;13(9):545–562. doi: 10.1038/nrneph.2017.98. [DOI] [PubMed] [Google Scholar]
- 74.Lawrenson J.G., Buckley R.J. Covid-19 and the eye. Ophthalmic Physiol Opt. 2020 doi: 10.1111/opo.12708. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frühbeis C., Fröhlich D., Krämer-Albers E.-M. Emerging roles of exosomes in neuron–glia communication. Front Physiol. 2012;3:119. doi: 10.3389/fphys.2012.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rajendran L., Bali J., Barr M.M., Krämer-Albers E.-M., Picou F., Raposo G. Emerging roles of extracellular vesicles in the nervous system. J Neurosci. 2014;34(46):15482–15489. doi: 10.1523/JNEUROSCI.3258-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grapp M., Wrede A., Schweizer M., Hüwel S., Galla H.-J., Snaidero N. Choroid plexus transcytosis and exosome shuttling deliver folate into brain parenchyma. Nat Commun. 2013;4(1):1–13. doi: 10.1038/ncomms3123. [DOI] [PubMed] [Google Scholar]
- 78.Jansen F., Nickenig G., Werner N. Extracellular vesicles in cardiovascular disease: potential applications in diagnosis, prognosis, and epidemiology. Circ Res. 2017;120(10):1649–1657. doi: 10.1161/CIRCRESAHA.117.310752. [DOI] [PubMed] [Google Scholar]
- 79.Lawson C., Vicencio J.M., Yellon D.M., Davidson S.M. Microvesicles and exosomes: new players in metabolic and cardiovascular disease. J Endocrinol. 2016;228(2):R57–R71. doi: 10.1530/JOE-15-0201. [DOI] [PubMed] [Google Scholar]
- 80.Akbar N., Azzimato V., Choudhury R.P., Aouadi M. Extracellular vesicles in metabolic disease. Diabetologia. 2019;62:2179–2187. doi: 10.1007/s00125-019-05014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Konadu K.A., Chu J., Huang M.B., Amancha P.K., Armstrong W., Powell M.D. Association of cytokines with exosomes in the plasma of hiv-1–seropositive individuals. J Infect Dis. 2015;211(11):1712–1716. doi: 10.1093/infdis/jiu676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pomatto M.A., Gai C., Deregibus M.C., Tetta C., Camussi G. 2018. Noncoding rnas carried by extracellular vesicles in endocrine diseases, international journal of endocrinology 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Milasan A., Farhat M., Martel C. 2020. Extracellular vesicles as potential prognostic markers of lymphatic dysfunction, Frontiers in physiology 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Murphy C., Withrow J., Hunter M., Liu Y., Tang Y.L., Fulzele S. Emerging role of extracellular vesicles in musculoskeletal diseases. Mol Aspects Med. 2018;60:123–128. doi: 10.1016/j.mam.2017.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alibhai F.J., Lim F., Yeganeh A., DiStefano P.V., Binesh-Marvasti T., Belfiore A. Cellular senescence contributes to age-dependent changes in circulating extracellular vesicle cargo and function. Aging Cell. 2020;19(3) doi: 10.1111/acel.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huang Q., Wu X., Zheng X., Luo S., Xu S., Weng J. Targeting inflammation and cytokine storm in covid-19. Pharmacol Res. 2020;159:105051. doi: 10.1016/j.phrs.2020.105051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.M. Ruscica, A. Corsini, N. Ferri, M. Banach, C. R. Sirtori, Clinical approach to the inflammatory etiology of cardiovascular diseases, Pharmacological Research (2020) 104916. [DOI] [PMC free article] [PubMed]
- 88.Seirafianpour F., Sodagar S., Mohammad A.P., Panahi P., Mozafarpoor S., Almasi S. Cutaneous manifestations and considerations in covid-19 pandemic: a systematic review. Dermatol Ther. 2020 doi: 10.1111/dth.13986. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Alexander S., Armstrong J., Davenport A., Davies J., Faccenda E., Harding S. A rational roadmap for sars-cov-2/covid-19 pharmacotherapeutic research and development. IUPHAR review 29. Br J Pharmacol. 2020 doi: 10.1111/bph.15094. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bouhaddou M., Memon D., Meyer B., White K.M., Rezelj V.V., Marrero M.C. The global phosphorylation landscape of SARS-CoV-2 infection. Cell. 2020;182:685–712. doi: 10.1016/j.cell.2020.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tutuncuoglu B., Cakir M., Batra J., Bouhaddou M., Eckhardt M., Gordon D.E. The landscape of human cancer proteins targeted by SARS-CoV-2. Cancer Discov. 2020 doi: 10.1158/2159-8290.CD-20-0559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Papathanasiou G. Local anesthetics and covid19 associated acute respiratory distress syndrome: a new therapeutic indication. Clin Res Open Access. 2020;6(2) [Google Scholar]
- 93.Das S., Camphausen K., Shankavaram U. In silico drug repurposing to combat covid-19 based on pharmacogenomics of patient transcriptomic data. Research Square. 2020 [Google Scholar]
- 94.Meininger V., Genge A., van den Berg L.H., Robberecht W., Ludolph A., Chio A. Safety and efficacy of ozanezumab in patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled, phase 2 trial. The Lancet Neurology. 2017;16(3):208–216. doi: 10.1016/S1474-4422(16)30399-4. [DOI] [PubMed] [Google Scholar]
- 95.Ríos J.-L., Giner R.M., Marín M., Recio M.C. A pharmacological update of ellagic acid. Planta Med. 2018;84(15):1068–1093. doi: 10.1055/a-0633-9492. [DOI] [PubMed] [Google Scholar]
- 96.Wang B.H., Lu Z.X., Polya G.M. Inhibition of eukaryote serine/threonine-specific protein kinases by piceatannol. Planta Med. 1998;64(03):195–199. doi: 10.1055/s-2006-957407. [DOI] [PubMed] [Google Scholar]
- 97.Piñero J., Ramírez-Anguita J.M., Saüch-Pitarch J., Ronzano F., Centeno E., Sanz F. The disgenet knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020;48(D1):D845–D855. doi: 10.1093/nar/gkz1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Menche J., Sharma A., Kitsak M., Ghiassian S.D., Vidal M., Loscalzo J. Uncovering disease-disease relationships through the incomplete interactome. Science. 2015;347(6224) doi: 10.1126/science.1257601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A. Tissue-based map of the human proteome. Science. 2015;347(6220) doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 100.Estrada E. Fractional diffusion on the human proteome as an alternative to the multi-organ damage of SARS-CoV-2. Chaos. 2020;30 doi: 10.1063/5.0015626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mainardi F. World Scientific; 2010. Fractional calculus and waves in linear viscoelasticity: An introduction to mathematical models. [Google Scholar]
- 102.Griffith M., Griffith O.L., Coffman A.C., Weible J.V., McMichael J.F., Spies N.C. Dgidb: mining the druggable genome. Nat Methods. 2013;10(12):1209–1210. doi: 10.1038/nmeth.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Keerthikumar S., Chisanga D., Ariyaratne D., Al Saffar H., Anand S., Zhao K. Exocarta: a web-based compendium of exosomal cargo. J Mol Biol. 2016;428(4):688–692. doi: 10.1016/j.jmb.2015.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]