

Abstract
Liver cancer is the fourth-leading cause of cancer-related mortality worldwide and incidence is on the rise. HCC is the most common form of liver cancer, with a complex etiology and limited treatment options. The standard of care treatment for advanced HCC patients is sorafenib, a tyrosine kinase inhibitor that offers limited survival benefit. In the past years, therapeutic options for the treatment of advanced HCC have increased substantially, including additional multikinase inhibitors as well as immune-checkpoint inhibitors. Nivolumab and pembrolizumab were approved in 2017 and 2018, respectively, as second-line treatment in advanced HCC. These drugs, both targeting the programmed death-1 (PD-1) pathway, demonstrate unprecedented results, with objective response rates of approximately 20%. However, the majority of patients do not respond, necessitating the identification of biomarkers of response and resistance to immunotherapy. With the recent success of immunotherapies in oncology, mouse models that better recapitulate the human disease and anti-tumor immune response are needed. This review lists ongoing clinical trials testing immunotherapy in HCC, briefly discusses the unique immunosuppressive environment of the liver, then delves into the most applicable current murine model systems to study oncoimmunology within the context of HCC, including syngeneic, genetically-engineered, and humanized models.
Introduction
Liver cancer is the fourth-leading cause of cancer-related mortality globally, and in contrast to other solid tumors, the incidence is increasing (1). Risk factors leading to hepatocellular carcinoma (HCC), the most common type of liver cancer, include chronic viral infection, alcohol-induced cirrhosis, and non-alcoholic steatohepatitis or fatty liver disease (NASH/NAFLD)(1, 2). The standard of care for advanced HCC patients is the multi-kinase inhibitor sorafenib, which offers limited survival benefit (3). Recent clinical trials have shown a subpopulation of HCC patients exhibit unprecedented responses to PD-1/PD-L1 checkpoint blockade (4–7). Checkpoint inhibitors nivolumab and pembrolizumab are now approved as 2nd line therapies, and combinations of antiangiogenic agents with either pembrolizumab or atezolizumab have received FDA breakthrough designation following promising clinical trial results (4–8). Table 1 details notable clinical trials evaluating the efficacy of different immunotherapies in HCC (9). Modeling HCC in the laboratory presents unique challenges given significant inter-patient heterogeneity and the background of underlying chronic inflammation and fibrosis (1). When studying immunotherapy and any treatment that engages the anti-tumor immune response, applicable models are further restricted by the need to preserve a functional immune system. Here, we review liver immunity and models currently available that enable investigation of the immune responses associated with HCC.
Table 1:
Clinical trials of immunotherapy in HCC.
| Trial | Phase | Investigational Treatment | Patient Population | Status and results |
|---|---|---|---|---|
| NCT01658878 (Checkmate-040) | I/II | nivo | advanced HCC, +/− HBV or HCV, previous sora treatment allowed | ORR 20% (95% CI 15–26) in dose-expansion phase, and 15% (95% CI 6–28) in dose-escalation phase; led to FDA approval of nivo in 2nd line (4) |
| NCT02576509 (Checkmate-459) | III | nivo VS sora | 1st line, advanced HCC | Did not meet primary OS endpoint. Median OS 16.4 months for nivo, 14.7 months for sora; ORR 15% for nivo, 7% for sora; CR in 14 nivo patients, 5 sora patients (5) |
| NCT03383458 (CheckMate 9 DX) | III | nivo VS placebo | Adjuvant after resection or ablation | Currently active, recruiting |
| NCT03222076 | II | nivo + ipi | Neoadjuvant | Currently active, recruiting. 1st interim analysis: 37.5% CR (81) |
| NCT04039607 (CheckMate 9DW) | III | nivo + ipi VS sora VS lenva | 1st line, advanced HCC | Currently active, recruiting |
| NCT03299946 | I | nivo + cabo | Neoadjuvant | Currently active, not recruiting |
| NCT02702414 (Keynote-224) | II | pembro | Advanced HCC, +/− prior sora treatment | ORR 17% (95% CI 11–26), 44% with stable disease (6) |
| NCT02702401 (Keynote-240) | III | pembro VS placebo | Advanced HCC progressed on sora | Pembro improved OS (HR: 0.78, p = 0.0238) and PFS (HR: 0.78, p = 0.0209) vs placebo, but did not meet pre-determined significance threshold (7) |
| NCT03006926 (KEYNOTE-524) | Ib | pembro + lenva | Advanced HCC | Currently active, not recruiting; at interim analysis, ORR 11%; Led to breakthrough therapy designation by the FDA for leva + pembro (8) |
| NCT03713593 (LEAP-002) | III | pembro + lenva VS lenva | 1st line advanced HCC | Currently active, recruiting |
| NCT03397654 (PETAL) | I | pembro OR pembro + lenva | HCC following TACE | Currently active, recruiting |
| NCT03867084 | I | pembro OR pembro + lenva | HCC with CR after resection/ablation | Currently active, recruiting |
| NCT02715531 | Ib | atezo VS beva + atezo | Advanced and unresectable HCC | Improved PFS with combination (5.6 vs. 3.4 months, HR 0.55) ORR 36% for combination; Led to breakthrough design by FDA for atezo + beva (82) |
| NCT03434379 (IMbrave150) | III | atezo + beva VS sora | Advanced HCC, untreated | Currently active, recruiting |
| NCT03755791 (COSMIC-312) | III | atezo + cabo VS sora VS cabo | Advanced HCC, untreated | Currently active, recruiting |
| NCT03298451 (HIMALAYA) | III | trem + durva | First line | Currently active, not recruiting |
| NCT01853618 | I,II | trem + TACE or RFA | Advanced HCC or biliary tract carcinoma | Median OS 12.3 months (95% CI 9.3–15.4); median time to tumor progression of 7.4months (95% CI 4.7–19.4); significant reduction in HCV viral load (83) |
| NCT01008358 | II | trem | Advanced HCC with HCV | Time to progression 6.48 months; 17.6 % partial response rate, 76.4% disease control rate, significant drop in HCV viral load (84) |
| NCT03778957 (EMERALD-1) | III | durva + beva + TACE | Locoregional HCC | Currently active, recruiting |
| NCT03847428 (EMERALD-2) | III | durva or durva + beva | HCC with high risk of recurrence after curative treatment | Currently active, recruiting |
| NCT02694822 | III | AGEN1884 (CTLA4 inhibitor) | Advanced cancer (including HCC) with progression on anti-PD-1/PD-L1 therapy | Currently active, recruiting |
nivo = nivolumab, sora = sorafenib, ipi = ipilimumab, cabo = cabozantinib, lenva = lenvatinib, pembro = pembrolizumab, beva= bevacizumab, atezo = atezolizumab, trem = tremelimumab, durva = durvalumab, CR = complete response, ORR = objective response rate, OS = overall survival
Liver Immunity
Given the liveŕs unique arterial and portal vasculature, the latter supplying antigens derived from innocuous food and commensal gut bacteria, the liver is inherently tolerogenic (10). However, it is enriched in immune subsets that maintain a homeostatic balance between tolerizing and effector functions, and thus, the liver harbors intrinsic mechanisms for responses against pathogens and transformed or malignant cells (10).
The hepatic lymphoid compartment includes B, T, natural killer (NK), and NKT cells (11, 12). B cells represent about 5–8% of the total lymphocyte population and T cells around 23% (11, 13). The liver contains one of the largest gamma-delta T (γδT) cell reservoirs in the body, comprising approximately 15% of total T cells (11). Additionally, hepatic CD8+ T cells outnumber CD4+ T cells by about two to one, typically the opposite of that found in blood (11, 13). Immunosuppressive regulatory T (Treg) cells are also present in the liver and may play a role in HCC (14). In both humans and mice, NK and NKT cells predominate the lymphoid composition of the liver. In mice, NKT cells represent approximately 40–50% of total liver-resident lymphocytes and NK cells 10–30%, whereas in humans the proportions are reversed (11, 13). While both cell types harbor granzyme and perforin-based cytolytic activity, NKT cells also perform important immunosuppressive functions (12). Non-parenchymal liver cells are comprised of approximately 45% liver sinusoidal endothelial cells (LSECs), 33% Kupffer cells (KCs), and 22% hepatic stellate cells (HSCs) (11). While not part of the hematopoietic lineage, hepatocytes, LSECs, and HSCs each serve as important immune sentinels capable of antigen presentation (12). Additionally, KCs, positioned throughout the liver sinusoids, represent 90% of all tissue-resident macrophages in the body (12). The liver also contains an extensive dendritic cells (DC) compartment, which in mice is comprised of 29% myeloid DCs, 18% plasmacytoid DCs, 18% NK-DCs, 18% lymphoid DCs, and 17% mixed lymphoid and myeloid DCs (11). Hepatocytes, LSECs, HSCs, KCs and DCs normally promote tolerogenic responses, for example through expression of PDL1; however, they can initiate potent T cell responses depending on microenvironmental cues (15). Finally, myeloid-derived suppressor cells (MDSCs) can be found in the liver and contribute to a tolerogenic state through production of IL-10, TGF-β, arginase, and IDO (13). Figure 1 summarizes the key immune functions of the different hepatic cell populations.
Figure 1: Liver immunity overview.
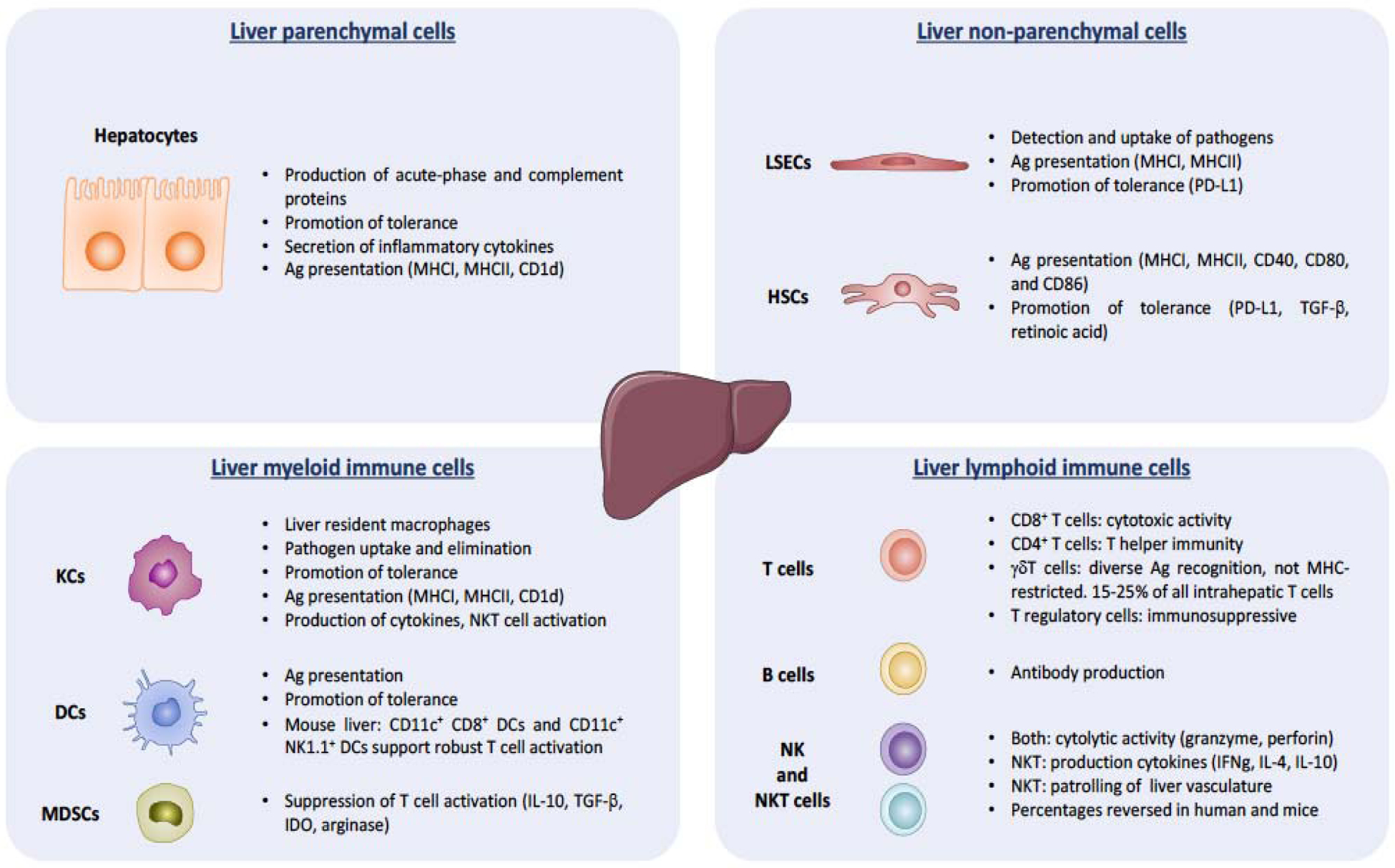
Schematic depicting key functions of the different liver cell populations, hepatic parenchymal, non-parenchymal, myeloid, and lymphoid, in maintaining a balance between tolerogenic and effector responses against pathogens and malignant cells.
In sum, hepatic immune cells generally serve a default immunosuppressive role, where resident antigen-presenting cells express inhibitory ligands and secrete anti-inflammatory cytokines, functioning to tolerize responses towards blood- and gut-derived antigens by both circulating and liver-resident T cells. However, upon receiving appropriate cues within the hepatic microenvironment, such as cytokine/chemokine signaling, these immune cells can become activated and perform pro-inflammatory effector functions. While there are many parallels between murine and human hepatic immunity, there are key differences, such as the NK compartment, that are critical to consider when modeling immune surveillance and immune escape during HCC development and immunotherapy treatment. More comprehensive reviews on liver immunology (11–13, 15) as well as how this immunology changes within the context of HCC (10, 16) have been conducted.
Genetically-engineered mouse models
Genetically-engineered mouse models (GEMMs) of cancer involve manipulation of the murine genome to induce tumor formation, typically through activating oncogenes or silencing tumor suppressor genes. In the past decades, GEMMs have proven to be powerful models for cancer research as they provide insight into the role of specific genes in tumor biology (17, 18). Typically generated in an immunocompetent setting, they represent an attractive alternative to transplantable models for the study of anti-tumor immune responses, in which autochthonous tumors gradually arise from normal cells in the intended organ (19, 20).
In general, GEMMs can be classified into two categories: germline transgenic models, in which genetic manipulation of the germline leads to spontaneous tumor development, and conditional transgenic models, in which gene expression or inactivation can be temporally and spatially regulated (19, 21). Controlling expression of the gene of interest in a spatiotemporal fashion represents a major advantage of conditional GEMMs and can be achieved through Cre-Lox recombination and tissue-specific promoters, such as the hepatocyte-specific albumin promoter (22). For example, mice with albumin-cre-mediated deletion of the tumor suppressor gene Pten (PtenΔH) develop liver tumors at approximately 8–9 months of age (23). Using this model, Lee J et al. demonstrated a role for immunostimulatory synthetic double-stranded RNAs in preventing initiation of liver tumorigenesis through modulation of innate anti-tumor functions, including induction of immunoregulatory cytokines, activation of DCs and NK cells, and reprogramming of macrophage polarization (24). Liver-specific promoter-directed GEMMs can also be used to model overexpression of oncogenes such as c-Myc and KRAS, either alone or in combination with other genes such as transcription factors or growth factors (25–27).
Generation of conditional GEMMs with inducible and reversible expression systems (e.g. tetracycline-controlled transcriptional regulation system) enables one to control both initiation and duration of the oncogenic insult, and as a consequence, of tumorigenesis. In this setting, transcription of the target gene can be reversibly turned on or off in the presence of an antibiotic (28). For example, it is possible to induce MYC overexpression in the hepatocytes of LAP-tTA/tet-O-hMYC mice by removing doxycycline treatment from the drinking water, which leads to HCC development with a mean latency of 35 weeks in adult mice (29). Lai et al. employed this model to test an mRNA-based immunotherapy with IL-12 mRNA encapsulated within lipid nanoparticles (IL-12-LNP) to facilitate delivery of this cytokine to the tumor site. Without causing animal toxicity or decreasing MYC levels, IL-12-LNP induced HCC regression through recruitment of CD3+ CD4+ CD44+ helper T cells and augmented IFNγ production (30). Another primary advantage of GEMMs over other models is that they enable study of the interplay between tumor cells and the immune system in a more physiological environment. For example, by selectively expressing model tumor antigens in hepatocytes, researchers can monitor antigen-specific T cell responses. This is demonstrated in the work of Morales-Kastresana et al. in which ovalbumin (OVA) was expressed as a model antigen in c-myc-OVA-tTALAP double transgenic mice. This model of multifocal HCC was used to prove the synergistic therapeutic effects of three immunostimulatory monoclonal antibodies (anti-CD137, anti-OX40, and anti-PD-L1) in combination with adoptive transfer of activated antigen-specific T cells (31).
Non-germline mosaic GEMMs of HCC, generated by hydrodynamic delivery of DNA plasmids into the hepatocytes, have gained increasing importance for the evaluation of immunotherapies. These models are less time consuming and less expensive than other GEMMs since injections are usually performed in wild type mice, eliminating the need for multiple crossings. With hydrodynamic injection (32), CRISPR-based plasmids or transposons can be introduced into the hepatocytes, leading to inactivation of tumor suppressor genes or overexpression of oncogenes, respectively. Many groups have used this approach to generate liver tumors harboring either single or combined genetic alterations commonly present in human HCCs such as TP53 and PTEN loss, β-catenin and NRASG12V mutation and AKT1, MET, and MYC overexpression (33–36). Recently, our group utilized this technique to develop a mouse model with customized genetic alterations and antigenicity in conjunction with luciferase reporter activity that facilitates monitoring of tumor growth. With this model, we demonstrated that β-catenin-driven HCCs presents impaired DC activity, T cell exclusion, and resistance to anti-PD-1 therapy (37). Other studies with hydrodynamic delivery-based models have investigated different aspects of anti-tumor immunity, such as T cell exhaustion or the effect of oncogenes on leukocyte composition in the liver (35, 38).
Syngeneic Implantation Models
Implantable syngeneic mouse models utilize HCC cell lines or murine tumoral tissue allografted into an immunocompetent mouse of the same genetic strain, enabling preservation of the host immune system. These models have been critical tools in the pre-clinical development of immune checkpoint blockade and in elucidating the role of anti-cancer immune response in maintaining long-term disease control (39).
Syngeneic tumor cells can be implanted orthotopically into the tissue of origin or ectopically at another site (most commonly subcutaneously). Ectopic models enhance simplicity and precision both in tumor access for interventions and tumor growth monitoring. For example, synergy between radiotherapy and immune checkpoint blockade was demonstrated using subcutaneous injections of murine HCC cell line HCa-1 on the flanks of immunocompetent mice (40). Additionally, ectopic models in combination with GEMMs have been utilized to understand interactions between tumor antigens and T cells, elucidating mechanisms of B7 superfamily member 1-driven CD8+ T cell exhaustion (41). Ectopic models, however, do not faithfully recapitulate the tumor microenvironment (TME) of the liver, as the resident immune and stromal cells differ significantly between organ systems. In contrast, orthotopic models more accurately reflect the natural TME of the organ in which tumorigenesis proceeds. Hage et al. characterized one such syngeneic orthotopic model involving hepatic implantation of Hep-55.1c cells (42). Another study harvested ectopically implanted Hepa1–6 cell tumors, then implanted sections into recipient mouse livers and tested an AFP-expressing DC-derived exosome-based vaccine therapy. The authors demonstrated that this vaccine slowed tumor growth and improved survival by modulating the immune TME (24). Orthotopic HCC models are technically challenging to establish since they involve survival surgeries and monitoring tumor growth usually requires abdominal imaging.
Syngeneic models can be generated from either commercially available murine HCC cell lines or cell lines established from murine liver tumors induced through a variety of strategies (chemotoxic agents, GEMMs, dietary models, etc.). The ability to engineer characteristics of cancer cells in vivo or in vitro prior to implantation expands the repertoire of experimental questions that can be addressed. This concept is illustrated in Figure 2, which details the experimental schematic of a recent study that used orthotopically implanted murine HCC cells transfected with PD-L1 to study alterations in T cell functionality during treatment with anti-PD-1 (43). Another group used tumorigenic hepatocytes expressing SV40 T antigen for intrasplenic inoculation to create an orthotopic model of HCC suitable for adoptive immune cell transfer experiments (44).
Figure 2: Representative murine model system for oncoimmunology study (43).
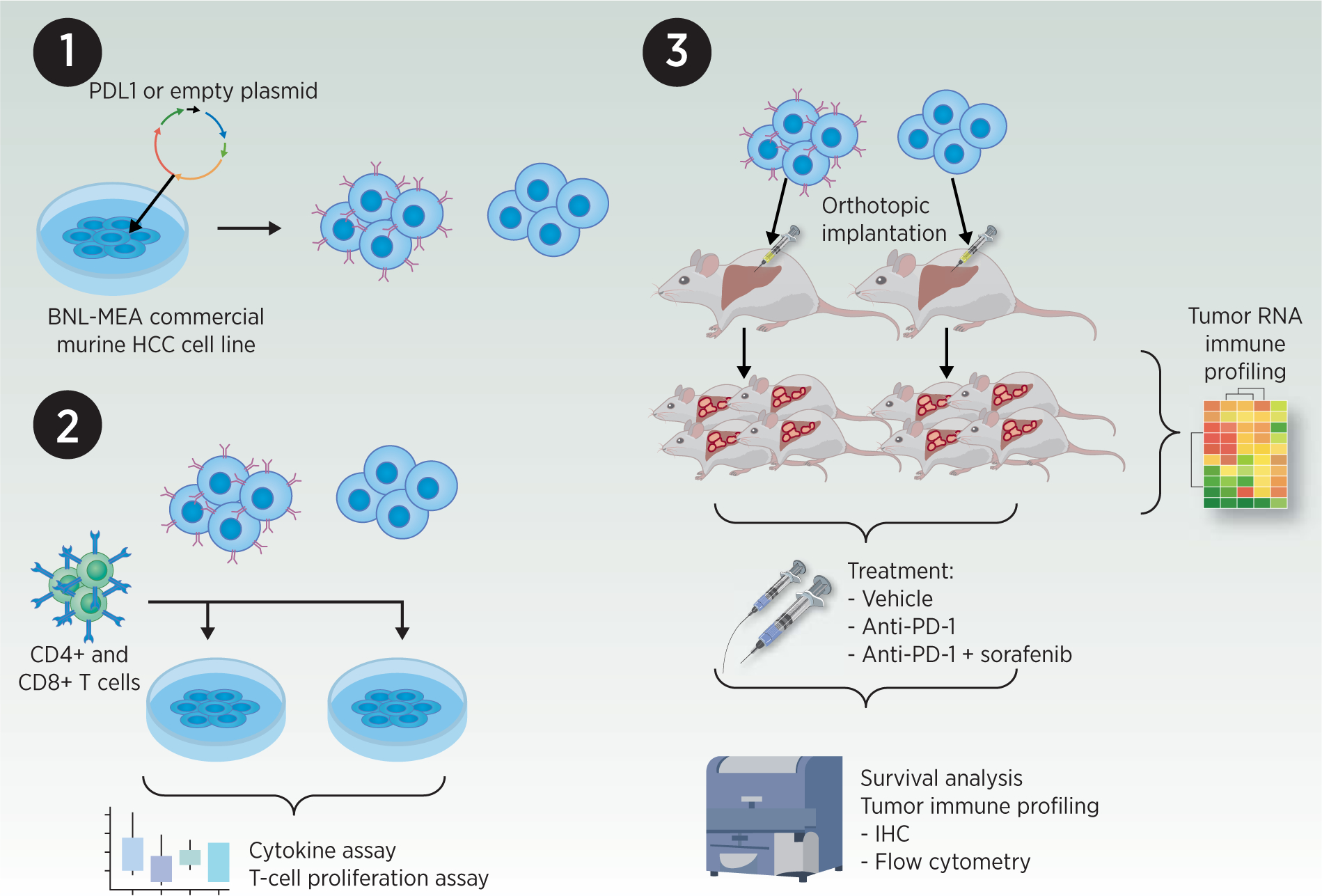
1) Transfection of a commercial murine HCC cell line with PD-L1 or control plasmid. 2) Transfected HCC cells were co-cultured with murine splenic CD4+ and CD8+ T cells; cytokine profiling and proliferation assays were performed. 3) Syngeneic orthotopic implantation of transfected HCC cells into immunocompetent mice. Resultant untreated tumors were collected and processed for RNA immune profiling. In parallel, mice harboring orthotopic tumors were treated with vehicle, anti-PD-1 or anti-PD-1+sorafenib; tumor immune profiling and survival analysis were performed.
Implantable syngeneic models are best for experiments requiring rapid tumor development, reproducibility, and large experimental groups. However, implantation of relatively uniform and already poorly differentiated cells restricts intra-tumoral heterogeneity and does not accurately recreate all naturally occurring stages of tumorigenesis (39). These models generate tumors rapidly without the chronic inflammation that normally underlies human HCC. Additionally, the inflammatory reaction to injection itself or to dead cells and their associated cellular debris can create an artificial immune response (17, 20).
Humanized Mouse Models
The aforementioned models, while irrefutably valuable, lack the complexity involved in natural tumorigenesis, including intra-tumoral genetic and histologic heterogeneity as well as intrinsic tumor architecture (17, 20). Further, these models either lack critical components of the immune system (e.g. tumor-infiltrating immune cells, cytokine/chemokine signaling within the TME) or they contain murine immune components, which do not accurately recapitulate tumor immunosurveillance and the TME in human disease, as murine and human immune systems have marked differences with regards to immune cell development and activation (18, 39, 45).
One approach to more accurately model tumor heterogeneity observed in human HCC is through implantation of patient-derived primary tumors into immunocompromised mice (46). These patient-derived xenografts (PDXs) harbor the same genetic heterogeneity, architecture, and local TME, including the tumor-associated stroma and tumor-infiltrated human immune cells, at least for a limited duration prior to replacement with mouse cells (39, 46). It is important to note that PDX-engraftment studies can restrict overall cohort size; however, PDXs can be expanded through in vivo passaging in mice to achieve a larger cohort (47). Further, numerous HCC PDX models can be commercially obtained through vendors such as Crown Biosciences, for example. To enable engraftment and prevent rejection, PDX transplantation requires NOD-scid Il2rg−/− (NSG) mice, which are also able to support the engraftment of human leukocytes; thus, this NSG-based PDX model can be further modified to integrate a more comprehensive, humanized tumor-immunity state (17, 39). Transplantable human leukocytes are commonly derived from peripheral blood mononuclear cells (PBMCs) and CD34+ hematopoietic stem cells isolated from bone marrow, fetal liver, umbilical cord blood, and GM-CSF mobilized PBMCs (17, 20, 39). “Immuno-avatar” mice are NSG-PDX mice co-transplanted with human PBMCs, preferably from the same patient, through intra-venous or intra-peritoneal injection (39, 48). This model has been used to assess anti-tumor properties of immunotherapy in different tumor types (49–51). PBMCs contain mature leukocytes, both an advantage and disadvantage as there is no lag time for these cells to undergo development; however, these leukocytes induce a strong xenogeneic graft-versus-host response and have limited viability upon transfer, restricting this model to short term studies (17, 20, 39, 49, 52, 53). MHC class I/II-deficient NOG mice have been shown to reduce this graft-versus-host response (54). Alternatively, engraftment of immature CD34+ human hematopoietic stem cells permits the leukocytes to undergo negative selection within the murine host during their maturation, thus preventing the graft-versus-host effect and enabling longer-term studies (20, 48). Effective engraftment of the CD34+ hematopoietic stem cells requires sublethal irradiation and takes approximately 10–12 weeks (20, 39). Zhao and colleagues developed a humanized HCC PDX model in NSG mice with HLA I-matched fetal liver-derived CD34+ hematopoietic stem cells (55). This model enabled evaluation of response to pembrolizumab and ipilimumab, immune changes within the TME, as well as clinically observed toxicities associated with ipilimumab (55). A limitation of the human CD34+ hematopoietic stem cell engraftment model is the mismatch between murine thymus and human T cell development requirements, leading to some deficiencies in the mature T cell compartment, such as FoxP3+ regulatory T cells (56). The humanized BLT mouse model, developed by Lan and colleagues, involves co-transplantation of human fetal liver (L), thymus (T), and CD34+ hematopoietic stem cells (bone marrow, B) into irradiated NOD/SCID mice (48, 57). It has been shown that these BLT-humanized mice demonstrate similar immune-mediated adverse effects in response to nivolumab (58). Zhai and colleagues utilized this BLT model combined with glioblastoma PDX to demonstrate that T cells regulate IDO1 expression directly in glioblastoma (59). Finally, one study reported a double-humanization with mature hepatocytes and hematopoietic stem cells in Fah−/−Rag2−/−Il2rg−/− immunodefiecient mice on NOD-strain background resulting in over 80% human liver repopulation and 40–80% human hematopoietic chimerism, notably with human Kupffer Cells observed (60). In conjunction with the three main categories of humanized PDX models, multiple groups have generated genetically humanized hosts to support survival and expansion of transplanted human leukocytes (17). Rongvaux and colleagues developed MIT(S)RG mice (Rag2−/−IL2 rγ−/− with human GM-CSF, M-CSF, IL3, TPO, +/− human SIRPα), which support monocyte, macrophage, and NK cell development upon CD34+ human stem cell engraftment and enable similar tumor infiltration seen in patients with PDX-transplanted mice (61). Similarly, Jangalwe and colleagues developed NSG-SGM3 mice (with human SCFr, GM-CSF, and IL3 knocked in) which, in the context of the BLT system, improved B cell development compared with NSG mice (62). Additionally, incorporation of HLA-A2 or HLA-DR1 into NSG mice may facilitate functional in vivo studies of antigen-specific CD8+ T cells and CD4+ T cell functions, respectively (39).
In addition to humanized PDX models, targeted humanization of individual murine immune components is commonly employed (39). This can involve transfer of human PBMCs sorted for specific populations into NSG mice. For example, Asai and colleagues transferred CD14+ cells from HCC patients into NSG mice to assess anti-tumor capacity (63). Alternatively, genetically-based humanization either of entire murine loci (e.g. replacement of murine MHC, a/b TCR, and Fc receptor loci) or individual immune targets (e.g. CTLA-4) with human counterparts can be achieved (39, 64–66). The latter becomes especially important during pre-clinical evaluation of drug efficacy and toxicities. Finally, the human hepatocyte-engrafted, HCV-infected MUP-uPA/SCID/Bg mouse has been utilized to evaluate HCV-associated HCC (67).
It is important to note that current therapeutics in HCC, such as lenvatinib and sorafenib, have also been shown to affect the immune system (68). For example, one study identified proliferating CD8+ T cells producing IFNγ as a key biomarker of response to sorafenib in HCC (69). This underscores the importance of using murine models of HCC with fully humanized immune systems, possibly within the context of a humanized PDX system.
Modeling liver inflammation and combination models
Common causes of HCC (e.g. HBV, HCV, alcohol-induced cirrhosis, NASH/NAFLD) all create a chronic inflammatory state, altering tumor-immunity and the TME (70, 71). Therefore, evaluation of tumor development and response to therapies within this context is critical for bench-to-bedside translation of findings. A number of mouse models involve carcinogen- or dietary-mediated liver inflammation, damage, and fibrosis (detailed in Table 2). Some develop HCC without further manipulation but typically with low penetrance, significant heterogeneity, and long latency. The background of inflammation and long latency enable evolution of immune, stromal, and vascular responses, better recapitulating human HCC development (39, 72, 73).
Table 2:
Models of inflammation and/or fibrosis induction: chemotoxic, dietary, and others.
| Model (References) | Details | Obesity, IR | Inflammation, Steatosis, Fibrosis, Cirrhosis | HCC development |
|---|---|---|---|---|
| Carcinogen-based HCC induction | ||||
| DEN (18, 85, 86) |
Genotoxic agent (DNA alkylation, oxidative stress). Single dose given at ~2 wks of age IP, does not require additional insult to develop HCC. Older mice need additional insult for HCC development, or long-term administration (dose and schedule can vary). |
No | No | Single Dose: Tumors 8–10 mo, 80–100% penetrance in males, 10–30% in females. Long term: Tumors at 7 months, ~60% penetrance (higher in males); metastasizes |
| Ccl4 (18, 41, 85, 87) |
Metabolite Ccl3 causes hepatocyte necrosis leading to fibrosis. Repetitive IP injection or oral gavage; varying degrees of fibrosis and cirrhosis depending on strain and dosing schedule. Often combined with DEN to accelerate tumorigenesis. |
No | Inflammation and steatosis; fibrosis in ~12 wks (self-resolving if Ccl4 discontinued) | 1–2 years for most strains. 50–94% penetrance (dose dependent), metastasizes |
| TAA (41, 85) |
Causes oxidative stress in hepatocytes. IP injection or administration in drinking water (orally less systemic toxicity). | No | Fibrosis progressing to cirrhosis in ~6 wks | Develop HCC in 6–12 mo |
| Aflatoxin B1 (86, 87) |
Metabolite causes DNA damage. IP injection in infant mice. |
No | Mild fibrosis | Early HCC in ~12 mo, high grade HCC in ~22–30 mo. Penetrance varies with strain (25–90%). Metastasizes |
| Diet-based HCC induction | ||||
| Choline deficient (CD) (85, 87) |
Oxidative damage and ER stress from defective phospholipid synthesis. Hepatic steatosis model with progression to HCC. | No | Steatohepatitis | HCC in 1–2 years, ~50% prevalence |
| CD + ethionine (CDE) (85) |
CD diet + supplementation with ethionine- a cytotoxic methionine analogue which causes inflammation and HSC activation. Accelerates tumor formation in CD model. | No | Inflammation and fibrosis | HCC by ~7 mo, 75% penetrance by 14 mo |
| Methionine choline deficient (MCDD) (18, 85, 88) |
High sucrose diet (40% sucrose, 10% fat) deficient in methionine and choline. Leads to buildup of cholesterol and fatty acid in liver. Model of NASH. High morbidity with significant weight loss. Inter-strain differences seen in immune infiltration. | No | Steatohepatitis in 1–2 wks, fibrosis in 8–10 wks | Not well studied in mice, due to long latency and toxic effects of diet |
| CD, L-amino acid defined (18, 88, 89) | CD diet with proteins replaced by semi-synthetic L-amino acids. Longer latency to pathology than MCDD. | No | Steatohepatitis and fibrosis | ~20% prevalence of HCC at 84 wks |
| High fat diet (HFD) (18, 85, 88) |
Ad libitum diet with 45% to75% fat; classic ratio 71% fat, 11% carbohydrates, 18% protein. Reproducible model of hepatic steatosis. | Yes | Fibrosis ~9 wks, Steatohepatitis ~19 wks | Usually does not progress to HCC without additional accelerating factor |
| Western / Fast food (18, 85) |
High cholesterol, high saturated fat, and high fructose. Similar phenotype to HFD + fibrosis. Histologically and transcriptomically very similar to human NASH and HCC. | Yes | Fibrosis in ~8 wks; NASH with progressive fibrosis after 6 months of diet | ~90% penetrance at 1 year |
| ALIOS (85) |
Diet high in trans-fats and high-fructose corn syrup, with removal of cage racks to promote sedentary behavior. Induction of HCC with only diet and lack of exercise. | Yes | At 16 wks, severe hepatic steatosis and inflammation. Fibrosis after 12-months | ~1 year, 60% penetrance |
| Ethanol (18) |
Lieber-DeCarli diet: ad libitum liquid diet with ethanol as 36% of calories. NIAAA model: 10 day Lieber-DeCarli diet + single binge feed of alcohol. Tsukamoto-Franch model: Infusion pump placed in stomach for continuous ethanol feeds. Do not induce HCC, but rather create a background of alcohol-induced liver disease. |
No | LD: Mild steatosis, very mild inflammation NIAAA: binge synergistically causes liver injury, inflammation, fatty liver TF: Severe steatosis, mild inflammation and fibrosis |
No; requires additional insult |
| Other models of inflammation or fibrosis | ||||
| MUP-uPA +HFD (18, 85) |
Major urinary protein (MUP)-urokinase type plasminogen activator (uPA) transgenic mice. uPA overexpression in hepatocytes causes ER stress and TNF-dependent inflammation. Model of inflammation-induced HCC. |
Yes | Steatosis from HFD, plus immune cell infiltration leading to HCC, fibrosis at ~24 wks after HFD initiation | HCC at ~ 32–40 wks, penetrance ~80% |
| Mdr2−/− (18, 20, 86) |
MDR2 knockout mice lack a phospholipid floppase responsible for biliary excretion of phospholipids; leads to buildup of bile salts, inflammation due to bile acid mediated cholestatic damage to hepatocytes. Chronic liver inflammation and fibrosis model. | No | Cholestatic hepatitis by ~3wks, fibrosis by ~8–10 wks (females>males) | Dysplastic lesions at 4–6 mo, tumors at ~1 year, ~100% penetrance. Pulmonary metastasis at ~1.5 years |
| Fah−/− (18) |
Lack fumarylacetoacetate hydrase; hepatic fumarylacetoacetate buildup causes inflammation, fibrosis, and liver dysfunction that can be controlled with NTBC. Used to model cyclic liver inflammation and injury via repeated withdrawal of NTBC. |
No | Hepatic inflammation and fibrosis | HCC in ~30% after 2 cycles of NTBC withdrawal and re-initiation; ~100% penetrance after 6 cycles |
| Liver-specific PTEN −/− (18, 85, 86) |
Albumin-Cre transgenic mice with floxed PTEN allele; model of obesity-related HCC. | No | Hepatomegaly and steatosis at ~10 wks, Inflammation and fibrosis at ~40 wks | ~1.5 years; ~80% penetrance in males, 50% in females |
| Liver specific Tak1 −/−(86) | Albumin-Cre or ALFP-Cre transgenic mice crossed with TGF-β Activated Kinase 1 conditional allele leads to hepatocyte death and resultant inflammation and fibrosis. Model of chronic hepatocyte injury. |
No | hepatic inflammation and fibrosis by 1 month | Tumors develop ~4 mo; penetrance ~80 % by 9–10 mo |
| Ob/ob and db/db (18) |
Ob/ob: no functional leptin; Db/db: mutated leptin receptor. Effectively remove leptin signaling leading to hyperphagia, obesity, hyperglycemia and insulin resistance, hyperlipidemia, fatty liver Model of metabolic syndrome. |
Yes | No spontaneous fibrosis or steatohepatitis without addition of diet (ie, HFD or MCDD). Leptin is required for fibrosis, so only db/db can develop | No; requires additional insult |
IR = Insulin resistance ; DEN = Diethylnitrosamine; IP= intraperitoneal; Ccl4 = carbon tetrachloride; wks = weeks; mo= months; TAA = Thioacetamide; ALIOS: American Lifestyle-Induced Obesity Syndrome; TNF = tumor necrosis factor; MDR2 = Multidrug resistance 2; Fah = Fumarylacetoacetate hydrase; NTBC = (2-[2-nitro-4-(trifluoromethyl) benzoyl]cyclohexane-1,3-dione or nitisinone); PTEN = Phosphatase and tensin homolog; Tak1 = TGF-β Activated Kinase
Combining the aforementioned models of HCC with those that induce a state of inflammation, fibrosis, and altered metabolic activity may be advantageous in providing a more comprehensive model to mimic the natural progression of human HCC. For example, two orthotopic models of HCC with underlying carbon tetrachloride (CCl4)- or diet-induced fibrosis were used to demonstrate the contribution of monocytic MDSCs to tumor growth through an HSC-dependent mechanism. In these models, combination therapy with anti-PD-1 and BET bromodomain inhibition was efficacious in suppressing monocytic MDSCs, increasing tumor-infiltrating lymphocytes, and slowing tumor growth (52). Another model combined CCl4 injections with either 1) subsequent HCa1 murine cancer cell orthotopic implantation, or 2) inducible liver-specific knockout of Stk3 via hydrodynamic delivery of adeno-CRE into Stk4−/−Stk3F/− mice (74). These techniques delineated how CD4+ T cell-induced normalization of vasculature mediates synergy between anti-angiogenic therapy and checkpoint inhibition (75).
Conclusions and future perspectives
While immunotherapies have revolutionized clinical management of HCC, response rates are still low, highlighting a need to elucidate mechanisms of resistance, design novel combination therapies to restore sensitivity, and identify predictive markers of response (9). Over the last years, advances in cancer research have provided a deeper understanding of liver immune surveillance and genomic characterization of HCC (76, 77). Though this has enabled development of a wide range of preclinical mouse models to study immunotherapies in HCC, some challenges remain to be addressed.
First, considering human HCC develops in a background of chronic inflammation, combining techniques of tumor induction with models that simulate underlying liver disease will allow for more reliable studies of immunotherapies. Additionally, a critical feature current approaches should consider is the incorporation of human gut microbiota. Given the gut-associated nature of the liver and the emerging role of gut microbiota in immunity and, more specifically, efficacy of immunotherapies (52), mouse models for the study of oncoimmunology would tremendously benefit from humanization of the intestinal microbiome. Additionally, controlling for factors such as animal strains, vendors, and housing conditions, as well as being conscientious of antibiotic-regulated expression use is key, since they impact gut microbiota. Furthermore, many of the humanized mouse models mentioned have been broadly used in other types of cancers but not as much in HCC; the expansion of this technology into the field of liver cancer would allow for investigation of immunotherapies in a model system that more closely resembles humans and would be more easily translatable to clinical successes.
Finally, while the focus of this review has been on mouse models of oncoimmunology due to their usefulness in translational research, in vitro models can complement the study of cancer immunotherapy. Utilization of 3D-culture systems incorporating tumor organoids with stromal cells and immune cells could elucidate mechanisms at the immune-tumor interface and changes in response to immunotherapy. HCC patient-derived organoids have been developed from both resected tumor tissue (78) and needle biopsies (79) but protocols for co-culture of HCC organoids and immune cells have not been established to the same degree as they have for other cancer types (ie, in colorectal cancer and lung cancer) (80). Additional development in this area could combine some of the benefits of 2D-cell culture (reproducible high-throughput screening at lower cost) with those of in vivo studies (preservation of the microenvironment and immune cell interactions) to generate further advances in oncoimmunology (Table 3).
Table 3:
Murine models of HCC: advantages and disadvantages
| Model | Advantages | Disadvantages |
|---|---|---|
| Germline transgenic models | Some commercially available Defined genetic alterations Autochthonous |
Time-consuming breeding and genotyping Temporal studies not feasible; mutation is present from birth Not representative of mutational heterogeneity of human HCC |
| Inducible transgenic model (Cre-lox system, TET system) | Spatiotemporal control of genetic alteration Defined genetic alterations Autochthonous |
Time consuming breeding and genotyping Not mosaic Not representative of mutational heterogeneity of human HCC |
| HDTVI (non-germline mosaic model) | Defined genetic alterations Mosaic Autochthonous Enables temporal control (inducible plasmids) Fast tumor growth Reproducible in large cohorts Good for treatment experiments |
Not representative of mutational heterogeneity of human HCC Technically challenging (HDTVI) Short-term liver damage from HDTVI |
| Syngeneic implantation: subcutaneous | Easy to monitor tumor growth Fast tumor growth Reproducible in large cohorts Accessibility of tumors for injections or radiation |
Not autochthonous Lacks hepatic immune microenvironment Bypasses initial steps of tumorigenesis Secondary inflammation from implantation Poor translatability due to high rate of false positives in treatment experiments |
| Syngeneic implantation: orthotopic | Autochthonous Fast tumor growth Reproducible Good for treatment experiments |
Reaction to injection/implantation Technically challenging (survival surgery) Bypasses initial steps of tumorigenesis Secondary inflammation from implantation |
| PDX in immunocompromised mice | Representative of mutational heterogeneity of human HCC Enables personalized therapy |
Not amenable for oncoimmunology studies Limited patient samples Technically challenging (survival surgery) |
| PDX in humanized mice | Representative of mutational heterogeneity of human HCC Enables personalized therapy Humanized immune system; amenable for oncoimmunology studies Multiple methods of humanization; suitable for many applications Model adverse effects of immunotherapy |
Limited patient samples Complicated by GVHD (when using mature lymphocytes) Time consuming engraftment process (when using CD34+ cells) |
| Carcinogen-induced models | Underlying liver disease can be modeled (fibrosis, steatosis, and cirrhosis) Cost effective |
Unpredictable genetic alterations Generally long latency unless multiple insults are combined |
| Dietary models | More accurately recreate the inflammation and steatosis seen in human liver disease Better for modeling NASH/NAFLD |
Generally long latency unless multiple insults are combined Unpredictable penetrance; not ideal for treatment experiments |
| 2D cell culture | Easily manipulated Facilitates mechanism of action studies Cost efficient High-throughput screening |
Poor translatability due to high rate of false positives in treatment experiments |
| Organoid/ 3D culture models | Facilitates mechanism of action studies Cost efficient High-throughput screening |
Poor translatability due to high rate of false positives in treatment experiments Technically challenging (establishment) |
Acknowledgments:
This work was supported by the National Cancer Institute (R37CA230636; A. Lujambio, K.E. Lindblad), the Damon Runyon Cancer Research Foundation (DR52-18; A. Lujambio, E. Bresnahan, M. Ruiz de Galarreta), and Fundación Alfonso Martín Escudero (M. Ruiz de Galarreta).
Footnotes
Disclosure of conflicts of interest
The authors have nothing to disclose.
References
- 1.Villanueva A Hepatocellular Carcinoma. N Engl J Med. 2019;380(15):1450–62. [DOI] [PubMed] [Google Scholar]
- 2.Younossi Z, Stepanova M, Ong JP, Jacobson IM, Bugianesi E, Duseja A, et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin Gastroenterol Hepatol. 2019;17(4):748–55 e3. [DOI] [PubMed] [Google Scholar]
- 3.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–90. [DOI] [PubMed] [Google Scholar]
- 4.El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yau T, Park JW, Finn RS, Cheng A-L, Mathurin P, Edeline J, et al. LBA38_PRCheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Annals of Oncology. 2019;30(Supplement_5). [Google Scholar]
- 6.Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 2018;19(7):940–52. [DOI] [PubMed] [Google Scholar]
- 7.Finn RS, Ryoo BY, Merle P, Kudo M, Bouattour M, Lim HY, et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J Clin Oncol. 2019:JCO1901307. [DOI] [PubMed] [Google Scholar]
- 8.Ikeda M, Sung MW, Kudo M, Kobayashi M, Baron AD, Finn RS, et al. A phase 1b trial of lenvatinib (LEN) plus pembrolizumab (PEM) in patients (pts) with unresectable hepatocellular carcinoma (uHCC). Journal of Clinical Oncology. 2018;36(15_suppl):4076-. [Google Scholar]
- 9.Yarchoan M, Agarwal P, Villanueva A, Rao S, Dawson LA, Llovet JM, et al. Recent Developments and Therapeutic Strategies against Hepatocellular Carcinoma. Cancer Res. 2019;79(17):4326–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnston MP, Khakoo SI. Immunotherapy for hepatocellular carcinoma: Current and future. World J Gastroenterol. 2019;25(24):2977–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bogdanos DP, Gao B, Gershwin ME. Liver immunology. Compr Physiol. 2013;3(2):567–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kubes P, Jenne C. Immune Responses in the Liver. Annu Rev Immunol. 2018;36:247–77. [DOI] [PubMed] [Google Scholar]
- 13.Robinson MW, Harmon C, O’Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol. 2016;13(3):267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langhans B, Nischalke HD, Kramer B, Dold L, Lutz P, Mohr R, et al. Role of regulatory T cells and checkpoint inhibition in hepatocellular carcinoma. Cancer Immunol Immunother. 2019;68(12):2055–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol. 2013;14(10):996–1006. [DOI] [PubMed] [Google Scholar]
- 16.Hou J, Zhang H, Sun B, Karin M. The immunobiology of hepatocellular carcinoma in humans and mice: Basic concepts and therapeutic implications. J Hepatol. 2019. [DOI] [PubMed] [Google Scholar]
- 17.Olson B, Li Y, Lin Y, Liu ET, Patnaik A. Mouse Models for Cancer Immunotherapy Research. Cancer Discov. 2018;8(11):1358–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown ZJ, Heinrich B, Greten TF. Mouse models of hepatocellular carcinoma: an overview and highlights for immunotherapy research. Nat Rev Gastroenterol Hepatol. 2018;15(9):536–54. [DOI] [PubMed] [Google Scholar]
- 19.DuPage M, Jacks T. Genetically engineered mouse models of cancer reveal new insights about the antitumor immune response. Curr Opin Immunol. 2013;25(2):192–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li E, Lin L, Chen CW, Ou DL. Mouse Models for Immunotherapy in Hepatocellular Carcinoma. Cancers (Basel). 2019;11(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanmamed MF, Chester C, Melero I, Kohrt H. Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies. Ann Oncol. 2016;27(7):1190–8. [DOI] [PubMed] [Google Scholar]
- 22.Kellendonk C, Opherk C, Anlag K, Schutz G, Tronche F. Hepatocyte-specific expression of Cre recombinase. Genesis. 2000;26(2):151–3. [DOI] [PubMed] [Google Scholar]
- 23.Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim YJ, et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected]. Proc Natl Acad Sci U S A. 2004;101(7):2082–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee J, Liao R, Wang G, Yang BH, Luo X, Varki NM, et al. Preventive Inhibition of Liver Tumorigenesis by Systemic Activation of Innate Immune Functions. Cell Rep. 2017;21(7):1870–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conner EA, Lemmer ER, Omori M, Wirth PJ, Factor VM, Thorgeirsson SS. Dual functions of E2F-1 in a transgenic mouse model of liver carcinogenesis. Oncogene. 2000;19(44):5054–62. [DOI] [PubMed] [Google Scholar]
- 26.Murakami H, Sanderson ND, Nagy P, Marino PA, Merlino G, Thorgeirsson SS. Transgenic mouse model for synergistic effects of nuclear oncogenes and growth factors in tumorigenesis: interaction of c-myc and transforming growth factor alpha in hepatic oncogenesis. Cancer Res. 1993;53(8):1719–23. [PubMed] [Google Scholar]
- 27.Xu Y, Poggio M, Jin HY, Shi Z, Forester CM, Wang Y, et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat Med. 2019;25(2):301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A. 1992;89(12):5547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beer S, Zetterberg A, Ihrie RA, McTaggart RA, Yang Q, Bradon N, et al. Developmental context determines latency of MYC-induced tumorigenesis. PLoS Biol. 2004;2(11):e332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai I, Swaminathan S, Baylot V, Mosley A, Dhanasekaran R, Gabay M, et al. Lipid nanoparticles that deliver IL-12 messenger RNA suppress tumorigenesis in MYC oncogene-driven hepatocellular carcinoma. J Immunother Cancer. 2018;6(1):125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morales-Kastresana A, Sanmamed MF, Rodriguez I, Palazon A, Martinez-Forero I, Labiano S, et al. Combined immunostimulatory monoclonal antibodies extend survival in an aggressive transgenic hepatocellular carcinoma mouse model. Clin Cancer Res. 2013;19(22):6151–62. [DOI] [PubMed] [Google Scholar]
- 32.Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol. 2014;184(4):912–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature. 2014;514(7522):380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matter MS, Marquardt JU, Andersen JB, Quintavalle C, Korokhov N, Stauffer JK, et al. Oncogenic driver genes and the inflammatory microenvironment dictate liver tumor phenotype. Hepatology. 2016;63(6):1888–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Juric V, Ruffell B, Evason KJ, Hu J, Che L, Wang L, et al. Monocytes promote liver carcinogenesis in an oncogene-specific manner. J Hepatol. 2016;64(4):881–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subleski JJ, Scarzello AJ, Alvord WG, Jiang Q, Stauffer JK, Kronfli A, et al. Serum-based tracking of de novo initiated liver cancer progression reveals early immunoregulation and response to therapy. J Hepatol. 2015;63(5):1181–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruiz de Galarreta M, Bresnahan E, Molina-Sanchez P, Lindblad KE, Maier B, Sia D, et al. beta-Catenin Activation Promotes Immune Escape and Resistance to Anti-PD-1 Therapy in Hepatocellular Carcinoma. Cancer Discov. 2019;9(8):1124–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu YT, Tseng TC, Soong RS, Peng CY, Cheng YH, Huang SF, et al. A novel spontaneous hepatocellular carcinoma mouse model for studying T-cell exhaustion in the tumor microenvironment. J Immunother Cancer. 2018;6(1):144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zitvogel L, Pitt JM, Daillere R, Smyth MJ, Kroemer G. Mouse models in oncoimmunology. Nat Rev Cancer. 2016;16(12):759–73. [DOI] [PubMed] [Google Scholar]
- 40.Kim KJ, Kim JH, Lee SJ, Lee EJ, Shin EC, Seong J. Radiation improves antitumor effect of immune checkpoint inhibitor in murine hepatocellular carcinoma model. Oncotarget. 2017;8(25):41242–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J, Lee Y, Li Y, Jiang Y, Lu H, Zang W, et al. Co-inhibitory Molecule B7 Superfamily Member 1 Expressed by Tumor-Infiltrating Myeloid Cells Induces Dysfunction of Anti-tumor CD8(+) T Cells. Immunity. 2018;48(4):773–86 e5. [DOI] [PubMed] [Google Scholar]
- 42.Hage C, Hoves S, Ashoff M, Schandl V, Hort S, Rieder N, et al. Characterizing responsive and refractory orthotopic mouse models of hepatocellular carcinoma in cancer immunotherapy. PLoS One. 2019;14(7):e0219517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ou DL, Lin YY, Hsu CL, Lin YY, Chen CW, Yu JS, et al. Development of a PD-L1-Expressing Orthotopic Liver Cancer Model: Implications for Immunotherapy for Hepatocellular Carcinoma. Liver Cancer. 2019;8(3):155–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Avella DM, Li G, Schell TD, Liu D, Zhang SS, Lou X, et al. Regression of established hepatocellular carcinoma is induced by chemoimmunotherapy in an orthotopic murine model. Hepatology. 2012;55(1):141–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ngiow SF, Loi S, Thomas D, Smyth MJ. Mouse Models of Tumor Immunotherapy. Adv Immunol. 2016;130:1–24. [DOI] [PubMed] [Google Scholar]
- 46.Blumer T, Fofana I, Matter MS, Wang X, Montazeri H, Calabrese D, et al. Hepatocellular Carcinoma Xenografts Established From Needle Biopsies Preserve the Characteristics of the Originating Tumors. Hepatol Commun. 2019;3(7):971–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mattar M, McCarthy CR, Kulick AR, Qeriqi B, Guzman S, de Stanchina E. Establishing and Maintaining an Extensive Library of Patient-Derived Xenograft Models. Front Oncol. 2018;8:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De La Rochere P, Guil-Luna S, Decaudin D, Azar G, Sidhu SS, Piaggio E. Humanized Mice for the Study of Immuno-Oncology. Trends Immunol. 2018;39(9):748–63. [DOI] [PubMed] [Google Scholar]
- 49.Sanmamed MF, Rodriguez I, Schalper KA, Onate C, Azpilikueta A, Rodriguez-Ruiz ME, et al. Nivolumab and Urelumab Enhance Antitumor Activity of Human T Lymphocytes Engrafted in Rag2−/−IL2Rgammanull Immunodeficient Mice. Cancer Res. 2015;75(17):3466–78. [DOI] [PubMed] [Google Scholar]
- 50.Lin X, Zeng T, Lin J, Zhang Q, Cheng H, Fang S, et al. Establishment of humanized tumor microenvironment mouse models based on the injection of peripheral blood mononuclear cells and IFN-gamma to evaluate the efficacy of PD-L1/PD-1-targeted immunotherapy. Cancer Biol Ther. 2019:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin S, Huang G, Cheng L, Li Z, Xiao Y, Deng Q, et al. Establishment of peripheral blood mononuclear cell-derived humanized lung cancer mouse models for studying efficacy of PD-L1/PD-1 targeted immunotherapy. MAbs. 2018;10(8):1301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng Y, Wang T, Tu X, Huang Y, Zhang H, Tan D, et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J Immunother Cancer. 2019;7(1):193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pearson T, Greiner DL, Shultz LD. Creation of “humanized” mice to study human immunity. Curr Protoc Immunol. 2008;Chapter 15:Unit 15 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yaguchi T, Kobayashi A, Inozume T, Morii K, Nagumo H, Nishio H, et al. Human PBMC-transferred murine MHC class I/II-deficient NOG mice enable long-term evaluation of human immune responses. Cell Mol Immunol. 2018;15(11):953–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao Y, Shuen TWH, Toh TB, Chan XY, Liu M, Tan SY, et al. Development of a new patient-derived xenograft humanised mouse model to study human-specific tumour microenvironment and immunotherapy. Gut. 2018;67(10):1845–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Halkias J, Yen B, Taylor KT, Reinhartz O, Winoto A, Robey EA, et al. Conserved and divergent aspects of human T-cell development and migration in humanized mice. Immunol Cell Biol. 2015;93(8):716–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lan P, Tonomura N, Shimizu A, Wang S, Yang YG. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood. 2006;108(2):487–92. [DOI] [PubMed] [Google Scholar]
- 58.Weaver JL, Zadrozny LM, Gabrielson K, Semple KM, Shea KI, Howard KE. BLT-Immune Humanized Mice as a Model for Nivolumab-Induced Immune-Mediated Adverse Events: Comparison of the NOG and NOG-EXL Strains. Toxicol Sci. 2019;169(1):194–208. [DOI] [PubMed] [Google Scholar]
- 59.Zhai L, Ladomersky E, Lauing KL, Wu M, Genet M, Gritsina G, et al. Infiltrating T Cells Increase IDO1 Expression in Glioblastoma and Contribute to Decreased Patient Survival. Clin Cancer Res. 2017;23(21):6650–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wilson EM, Bial J, Tarlow B, Bial G, Jensen B, Greiner DL, et al. Extensive double humanization of both liver and hematopoiesis in FRGN mice. Stem Cell Res. 2014;13(3 Pt A):404–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, et al. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol. 2014;32(4):364–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jangalwe S, Shultz LD, Mathew A, Brehm MA. Improved B cell development in humanized NOD-scid IL2Rgamma(null) mice transgenically expressing human stem cell factor, granulocyte-macrophage colony-stimulating factor and interleukin-3. Immun Inflamm Dis. 2016;4(4):427–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Asai A, Tsuchimoto Y, Ohama H, Fukunishi S, Tsuda Y, Kobayashi M, et al. Host antitumor resistance improved by the macrophage polarization in a chimera model of patients with HCC. Oncoimmunology. 2017;6(4):e1299301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lute KD, May KF Jr., Lu P, Zhang H, Kocak E, Mosinger B, et al. Human CTLA4 knock-in mice unravel the quantitative link between tumor immunity and autoimmunity induced by anti-CTLA-4 antibodies. Blood. 2005;106(9):3127–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Obenaus M, Leitao C, Leisegang M, Chen X, Gavvovidis I, van der Bruggen P, et al. Identification of human T-cell receptors with optimal affinity to cancer antigens using antigen-negative humanized mice. Nat Biotechnol. 2015;33(4):402–7. [DOI] [PubMed] [Google Scholar]
- 66.Bournazos S, DiLillo DJ, Ravetch JV. humanized mice to study FcgammaR function. Curr Top Microbiol Immunol. 2014;382:237–48. [DOI] [PubMed] [Google Scholar]
- 67.Wang Z, Wu N, Tesfaye A, Feinstone S, Kumar A. HCV infection-associated hepatocellular carcinoma in humanized mice. Infect Agent Cancer. 2015;10:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin YY, Tan CT, Chen CW, Ou DL, Cheng AL, Hsu C. Immunomodulatory Effects of Current Targeted Therapies on Hepatocellular Carcinoma: Implication for the Future of Immunotherapy. Semin Liver Dis. 2018;38(4):379–88. [DOI] [PubMed] [Google Scholar]
- 69.Kalathil SG, Hutson A, Barbi J, Iyer R, Thanavala Y. Augmentation of IFN-gamma+ CD8+ T cell responses correlates with survival of HCC patients on sorafenib therapy. JCI Insight. 2019;4(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. [DOI] [PubMed] [Google Scholar]
- 71.O’Rourke JM, Sagar VM, Shah T, Shetty S. Carcinogenesis on the background of liver fibrosis: Implications for the management of hepatocellular cancer. World J Gastroenterol. 2018;24(39):4436–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dow M, Pyke RM, Tsui BY, Alexandrov LB, Nakagawa H, Taniguchi K, et al. Integrative genomic analysis of mouse and human hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2018;115(42):E9879–E88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Connor F, Rayner TF, Aitken SJ, Feig C, Lukk M, Santoyo-Lopez J, et al. Mutational landscape of a chemically-induced mouse model of liver cancer. J Hepatol. 2018;69(4):840–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reiberger T, Chen Y, Ramjiawan RR, Hato T, Fan C, Samuel R, et al. An orthotopic mouse model of hepatocellular carcinoma with underlying liver cirrhosis. Nat Protoc. 2015;10(8):1264–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shigeta K, Datta M, Hato T, Kitahara S, Chen IX, Matsui A, et al. Dual Programmed Death Receptor-1 and Vascular Endothelial Growth Factor Receptor-2 Blockade Promotes Vascular Normalization and Enhances Antitumor Immune Responses in Hepatocellular Carcinoma. Hepatology. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cancer Genome Atlas Research Network. Electronic address wbe, Cancer Genome Atlas Research N. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. 2017;169(7):1327–41 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology. 2015;149(5):1226–39 e4. [DOI] [PubMed] [Google Scholar]
- 78.Broutier L, Mastrogiovanni G, Verstegen MM, Francies HE, Gavarro LM, Bradshaw CR, et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat Med. 2017;23(12):1424–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nuciforo S, Fofana I, Matter MS, Blumer T, Calabrese D, Boldanova T, et al. Organoid Models of Human Liver Cancers Derived from Tumor Needle Biopsies. Cell Rep. 2018;24(5):1363–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell. 2018;174(6):1586–98 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kaseb AO, Pestana RC, Vence LM, Blando JM, Singh S, Ikoma N, et al. Randomized, open-label, perioperative phase II study evaluating nivolumab alone versus nivolumab plus ipilimumab in patients with resectable HCC. Journal of Clinical Oncology. 2019;37(4_suppl):185-. [Google Scholar]
- 82.Lee M Randomised efficacy and safety results for atezolizumab (Atezo) bevacizumab (Bev) in patients (pts) with previously untreated, unresectable hepatocellular carcinoma (HCC). Annals of oncology.30. [Google Scholar]
- 83.Duffy AG, Ulahannan SV, Makorova-Rusher O, Rahma O, Wedemeyer H, Pratt D, et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J Hepatol. 2017;66(3):545–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sangro B, Gomez-Martin C, de la Mata M, Inarrairaegui M, Garralda E, Barrera P, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81–8. [DOI] [PubMed] [Google Scholar]
- 85.Carlessi R, Kohn-Gaone J, Olynyk JK, Tirnitz-Parker JEE. Mouse Models of Hepatocellular Carcinoma In: Tirnitz-Parker JEE, editor. Hepatocellular Carcinoma. Brisbane (AU)2019. [PubMed] [Google Scholar]
- 86.Caviglia JM, Schwabe RF. Mouse models of liver cancer. Methods Mol Biol. 2015;1267:165–83. [DOI] [PubMed] [Google Scholar]
- 87.Heindryckx F, Colle I, Van Vlierberghe H. Experimental mouse models for hepatocellular carcinoma research. Int J Exp Pathol. 2009;90(4):367–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen K, Ma J, Jia X, Ai W, Ma Z, Pan Q. Advancing the understanding of NAFLD to hepatocellular carcinoma development: From experimental models to humans. Biochim Biophys Acta Rev Cancer. 2019;1871(1):117–25. [DOI] [PubMed] [Google Scholar]
- 89.Denda A, Kitayama W, Kishida H, Murata N, Tsutsumi M, Tsujiuchi T, et al. Development of hepatocellular adenomas and carcinomas associated with fibrosis in C57BL/6J male mice given a choline-deficient, L-amino acid-defined diet. Jpn J Cancer Res. 2002;93(2):125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]