

Abstract
Background:
We recently reported a role for the circadian rhythm protein Period 2 (PER2) in midazolam induced cognitive dysfunction. Based on previous studies showing a critical role for the adenosine A2B receptor (ADORA2B) in PER2 regulation, we hypothesized that hippocampal ADORA2B is crucial for cognitive function.
Methods:
Midazolam treated C57BL/6J mice were analyzed for Adora2b hippocampal mRNA expression levels, and spontaneous T-maze alternation was determined in Adora2b−/− mice. Using the specific ADORA2B agonist BAY-60–6583 in midazolam treated C57BL/6J mice, we analyzed hippocampal Per2 mRNA expression levels and spontaneous T-maze alternation. Finally, Adora2b−/− mice were assessed for mRNA expression of markers for inflammation or cognitive function in the hippocampus.
Results:
Midazolam treatment significantly downregulated Adora2b or Per2 mRNA in the hippocampus of C57BL/6J mice, and hippocampal PER2 protein expression or T-maze alternation was significantly reduced in Adora2b−/− mice. ADORA2B agonist BAY-60–6583 restored midazolam mediated reduction in spontaneous alternation in C57BL/6J mice. Analysis of hippocampal Tnf-α or Il-6 mRNA levels in Adora2b−/− mice did not reveal an inflammatory phenotype. However, C-fos, a critical component of hippocampus-dependent learning and memory, was significantly downregulated in the hippocampus of Adora2b−/− mice.
Conclusion:
These results suggest a role of ADORA2B in midazolam induced cognitive dysfunction. Further, our data demonstrate that BAY-60–6583 treatment restores midazolam induced cognitive dysfunction, possibly via increases of Per2. Additional mechanistic studies hint towards C-FOS as another potential underlying mechanism of memory impairment in Adora2b−/− mice. These findings suggest the ADORA2B agonist as a potential therapy in patients with midazolam induced cognitive dysfunction.
Keywords: Adora2b, cognitive function, Per2, T maze, C-fos, Bdnf, delirium, BAY-60–6583
Introduction
Critical ill patients who develop cognitive dysfunction in the setting of delirium are at an increased risk of dying [1, 2]. As such delirium associated cognitive dysfunction is a significant public health concern, and specific therapies are urgently needed [1, 3–10]. Moreover, delirium in the intensive care unit is an independent predictor of long-term cognitive impairment after critical illness [11]. However specific treatments for cognitive dysfunction in the setting of delirium have not been established yet.
We recently found evidence that the circadian rhythm protein Period 2 (PER2) plays a critical role in midazolam-induced cognitive dysfunction that resembled key characteristics of delirium in humans [2]. We demonstrated that midazolam-induced changes in cognitive function are associated with a significant downregulation of PER2 in the hippocampus. Furthermore, genetic deletion of Per2 resulted in a phenotype that shared several features with acute delirium [2].
Based on studies demonstrating a critical role for the adenosine A2B receptor (ADORA2B) in PER2 regulation [12], we hypothesized that hippocampal ADORA2B is also critical for cognitive function during midazolam-induced behavioral changes. In fact, adenosine is a neuromodulator acting through four types of membrane-bound G-protein-coupled adenosine receptors, named ADORA1, ADORA2A, ADORA2B, and ADORA3 receptors [13]. Central effects of adenosine mostly involve ADORA1 and ADORA2A receptors [13], where ADORA1 receptors play a prominent role in the control of emotional stability and ADORA2A receptors control locomotion and cognition [14, 15]. While studies have shown that ADORA2Bs are present in hippocampal glutamatergic terminals [16], the role of ADORA2B in brain function has been less investigated.
Therefore, we tested our hypothesis that hippocampal ADORA2B is critical for cognitive function in a murine model of midazolam-induced behavioral impairment. We found that midazolam-induced changes in behavior are associated with a significant downregulation of Adora2b mRNA in the hippocampus – like studies on PER2. Furthermore, genetic deletion of Adora2b resulted in cognitive dysfunction as seen in Per2−/− mice. Finally, we successfully restored midazolam induced cognitive deficits using the specific ADORA2B agonist BAY-60–6583 [12, 17–22].
Methods
Mouse experiments:
Experimental protocols were approved by the Institutional Review Board (Institutional Animal Care and Use Committee [IACUC]) at the University of Colorado Denver, USA. They were in accordance with the AAALAC regulations, the US Department of Agriculture Animal Welfare Act, and the Guide for the Care and Use of Laboratory Animals of the NIH. Mice were housed in a 14/10-h light-dark cycle, and all mouse experiments were conducted at the same time point (ZT3, Zeitgeber Time 3 corresponding to 9 AM based on lights ‘on’ at 6 AM). To eliminate gender- and age-related variations, we routinely used 12- to 16-week-old, 24g male mice. Mice were bred in the vivarium at Denver for optimal acclimatization and housed in cages of 5 at 21 °C with food (Harlan diets, formulation 2920x, soy-free) and water ad libitum. One week before the experiments, the mice were singly housed and brought to the testing room for one hour daily to acclimate being handled and moved. The testing room had the same conditions as the housing area (e.g. lighting, temperature, etc. [12, 23]).
Animals:
C57BL/6J (wildtype mice) were obtained from the Jackson Laboratories and bred in the animal vivarium in Denver. Adorab2loxp/loxp [24] mice were generated by Ozgene (Perth, Australia) and Germline Cre [B6.C-Tg(CMV-cre)1Cgn/J] were purchased from Jackson laboratories. To obtain Adora2b−/− mice, we crossbred Adorab2loxp/loxp with the Germline Cre mouse [25].
Compounds:
Midazolam (5mg/ml, preservative free, Pfizer, USA, dose: 10 mg/kg i.p.), BAY-60–6583 (Tocris, USA, dose: 1 μg/kg i.p. [2, 12, 17]).
BAY 60–6583 Specificity.
Chinese hamster ovary (CHO) cells stably expressing recombinant human A1AR, A2AAR or A2BAR and a CRE-luciferase construct as a read-out system for intracellular cAMP modulation were grown in 384-well plates for 2 d at 37°C. Cells were incubated with increasing concentrations of BAY 60–6583. Cells were lysed with Triton X-100 and luciferin was added. Relative light units (RLU) were measured in a charge-coupled device camera system over 30 sec. To demonstrate specificity against the A3AR, an adenosine A3-Gα16 assay was performed. Briefly, a stably transfected CHO luc9AQ Adenosine A3-Gα16 pcDNA3 cell line grown in DMEM-F12/10% FCS at 37° C and 5% CO2 was used. On the day of testing, the medium was discarded and replaced by CAFTY/2 mM Ca2+/11 μM coelenterazine; after 3 h of incubation, cells were used for bioluminescent measurement of intracellular Ca2+ release due to the specific A3AR agonist IB-MECA. Compounds were tested for agonistic or antagonistic effects after 10 min preincubation by the addition of an EC50 concentration of IB-MECA (appr. 300 nM) followed by a second addition of 15 μM ATP. Antagonists will suppress the first and increase the second signal, agonists will decrease both signals. No agonistic effect of BAY 60–6583 was detected using a concentration of up to 10 μM (data not shown)[18].
Experimental groups:
Mice were randomly selected for i.p. (intraperitoneal) injections of 1. NaCl 0.9% (0.1 ml i.p., control group), 2. Midazolam (0.05 ml of midazolam 5mg/ml i.p. [10 mg/kg]), 3. Midazolam+BAY i.p. (0.05 ml of BAY-60–6583 0.48 μg/ml (1 μg/kg). 2 hours after saline, midazolam or midazolam+BAY treatment, mRNA levels were determined. T-maze alternation was determined 24 hours after saline, midazolam or midazolam+BAY treatment.
Transcriptional analysis:
Total RNA was isolated from liquid nitrogen shock frozen murine brain tissue using the RNeasy Mini Kit (QIAGEN, Germantown, MD). Primers were Quantitect from Qiagen (mouse Adora2b, QT01543444; mouse Per2, QT00198366; mouse Tnf-α, QT00104006; mouse Il-6, QT00098875; mouse Bdnf, QT00097118; mouse c-Fos, QT00147308). Primers for mouse β-actin were from in Invitrogen (custom DNAoligos, 5’-CTA GGC ACC AGG GTG TGA T −3’, 5’-TGC CAG ATC TTC TCC ATG TC-3’). cDNA from mRNA was generated using iScript (Bio-Rad), and transcript levels were determined by real-time RT-PCR (iCycler IQ; Bio-Rad Laboratories Inc.) as described [2, 12, 24–31]. qPCR reactions contained 1x final primer concentration (QIAGEN primers, Germantown, MD) or 1 μM sense and 1 μM antisense oligonucleotides (Invitrogen custom DNAoligos, Carlsbad, CA) with SYBR Green (Bio-Rad, Hercules, CA). Each target sequence was amplified using increasing numbers of cycles of 94°C for 1 min, 58°C for 0.5 min, 72°C for 1 min. Data were calculated relative to the ‘housekeeping’ gene β-actin and were expressed as fold change compared to controls [2, 23–32].
T-maze alternation:
T-maze alternation is a very sensitive test used to determine hippocampal functioning [33] and to detect cognitive impairment [34]. The T-maze was made of ABS plastic acrylic. Animals started from the base (long arm) of the T and allowed to choose one of the goal arms abutting the other end of the stem. Using a plastic panel, the mouse was blocked in the chosen arm for 30 seconds. The panel was removed, and the mouse was again placed in the long arm of the T. In ten trials in quick succession, the rodent’s choice was recorded (‘continuous trial procedure of spontaneous alternation’). If the mouse did not choose an arm within 15 minutes, the experiment was considered failed and over. With repeated trials, the animals should show less of a tendency to enter a previously visited arm. The percentage of alternation (number of turns in each goal arm) and total trial duration are recorded. This test is used to quantify cognitive deficits in transgenic strains of mice and evaluate chemical entities for their effects on cognition. Typical alternation results in alternation rates of 60–70%. The reduction of alternation below 50% indicates cognitive impairment [2, 33]. Because of its simplicity of construction and use, combined with its sensitivity, T-maze alternation has almost universal applicability in detecting cognitive dysfunction. The spontaneous alternation is very sensitive to the dysfunction of the hippocampus, but other brain structures are also involved [33].
Immunoblotting experiments:
Protein was isolated from liquid nitrogen shock frozen murine brain-hippocampus tissue using N-Per following the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA) and included a protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA). Protein was quantified using a 660nm protein assay reagent (Thermo Fisher Scientific, Waltham, MA). 20 μg of protein was denatured at 95°C in Laemmli sample buffer for 5 min. Samples were resolved on a 4–10% polyacrylamide gel and transferred to nitrocellulose membranes, which were blocked for 1h at room temperature in either 3% BSA / TBST. The membranes were incubated in primary antibody overnight at 4°C. The primary antibodies used were rabbit polyclonal PER2 (AB2202, Millipore Sigma, Burlington, MA; antibody verified for PER2 specificity [35]), mouse monoclonal β-actin (8H10D10, Cell signaling, Danvers, MA), The secondary antibodies used were goat anti-rabbit IgG (Thermo Fisher Scientific, Waltham, MA) and goat anti-mouse IgG (Thermo Fisher Scientific, Waltham, MA).
Data analysis:
Data were compared by one-way ANOVA with Tukey’s posthoc test, or by Student’s t-test where appropriate. Values are expressed as mean (±SD). P<0.05 was considered statistically significant. For all statistical analysis, GraphPad Prism 6.0 software was used. The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
Midazolam downregulates Adora2b mRNA in the hippocampus.
Based on studies showing a role for the circadian rhythm protein Period 2 (PER2) in midazolam induced cognitive dysfunction [2] and based on studies implicating the adenosine A2B receptor (ADORA2B) in PER2 regulation [12], we first evaluated brain Adora2b transcript levels after midazolam treatment. As we had seen a significant downregulation of hippocampal Per2 mRNA in mice 2 hours after intraperitoneal midazolam administration [2], we analyzed hippocampal Adora2b transcript levels using the same administration strategy (Figure 1A). We demonstrated that two hours after midazolam administration, hippocampal Adora2b mRNA was significantly downregulated in wildtype mice (Figure 1B). Taken together, midazolam downregulates hippocampal Adora2b transcript levels like studies on Per2, suggesting that ADORA2B might have a similar function as PER2 in midazolam induced cognitive dysfunction.
Figure 1. T-maze spontaneous alternation in Adora2b−/− mice.
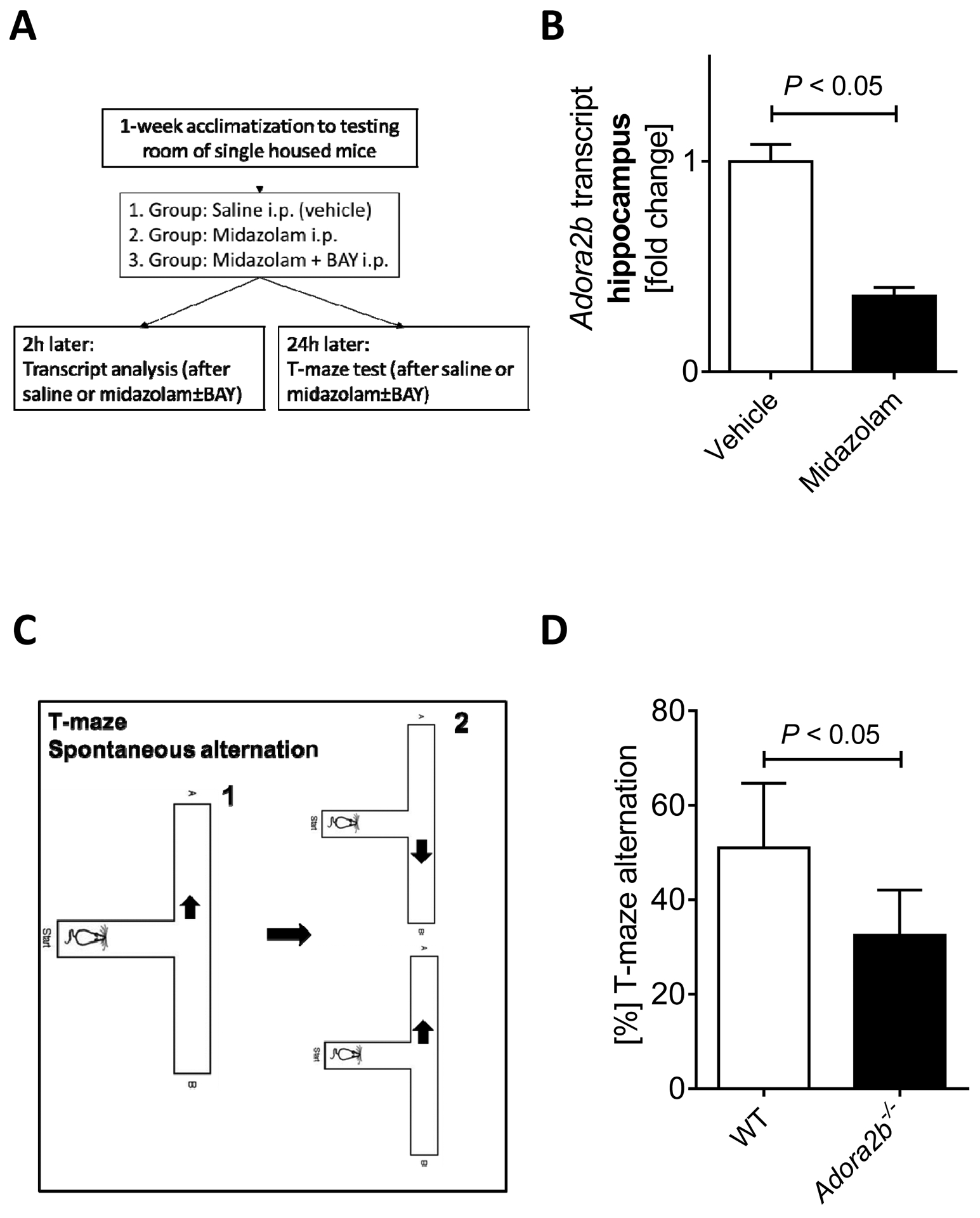
Wildtype mice (C57BL6/J) were injected with midazolam (10mg/kg i.p.) and compared to saline-treated controls. (A) Experimental setup overview. (B) Adora2b transcript levels in the hippocampus. Brain hippocampus tissue was harvested, mRNA was isolated using RNeasy Mini Kit (Qiagen), cDNA was generated using miScript RT II kits (Qiagen), and Adora2b transcript levels were determined by quantitative real-time RT-PCR (iCycler or iCycler IQ; Bio-Rad Laboratories Inc.), n=5, mean±SD; p<0.05. (C) T maze spontaneous alternation test. (D) T-maze alternation in % from Adora2b−/− mice, n=4–10, mean±SD; p<0.05.
Adora2b deficiency results in reduced spontaneous alternation using a T-maze.
After we had demonstrated that midazolam administration downregulated Adora2b transcript levels in the hippocampus, we next studied spontaneous alternation using a T-maze (Figure 1C) in mice lacking the ADORA2B receptor (Adora2b−/−). T-maze alternation is a very sensitive test used to determine hippocampal functioning [33] and to detect cognitive impairment [34]. Indeed, we found significantly reduced spontaneous alternation in Adora2b−/− mice when compared to wildtype controls (Figure 1D). Taken together, Adora2b−/− mice show cognitive impairment, as seen in previous studies on Per2−/− mice.
Adora2b−/− mice have less hippocampal expressed PER2 protein.
After we had confirmed a cognitive deficit in Adora2b−/− mice, we next studied hippocampal PER2 protein levels in Adora2b−/− mice. As shown in Figure 2A–B, western blot analysis from Adora2b−/− mice revealed significantly less PER2 protein in the hippocampus. Taken together, Adora2b−/− mice lack PER2 protein in the hippocampus, suggesting Per2 deficiency as the underlying mechanism of cognitive impairment in Adora2b−/− mice.
Figure 2. Hippocampal PER2 levels in Adora2b−/− mice.
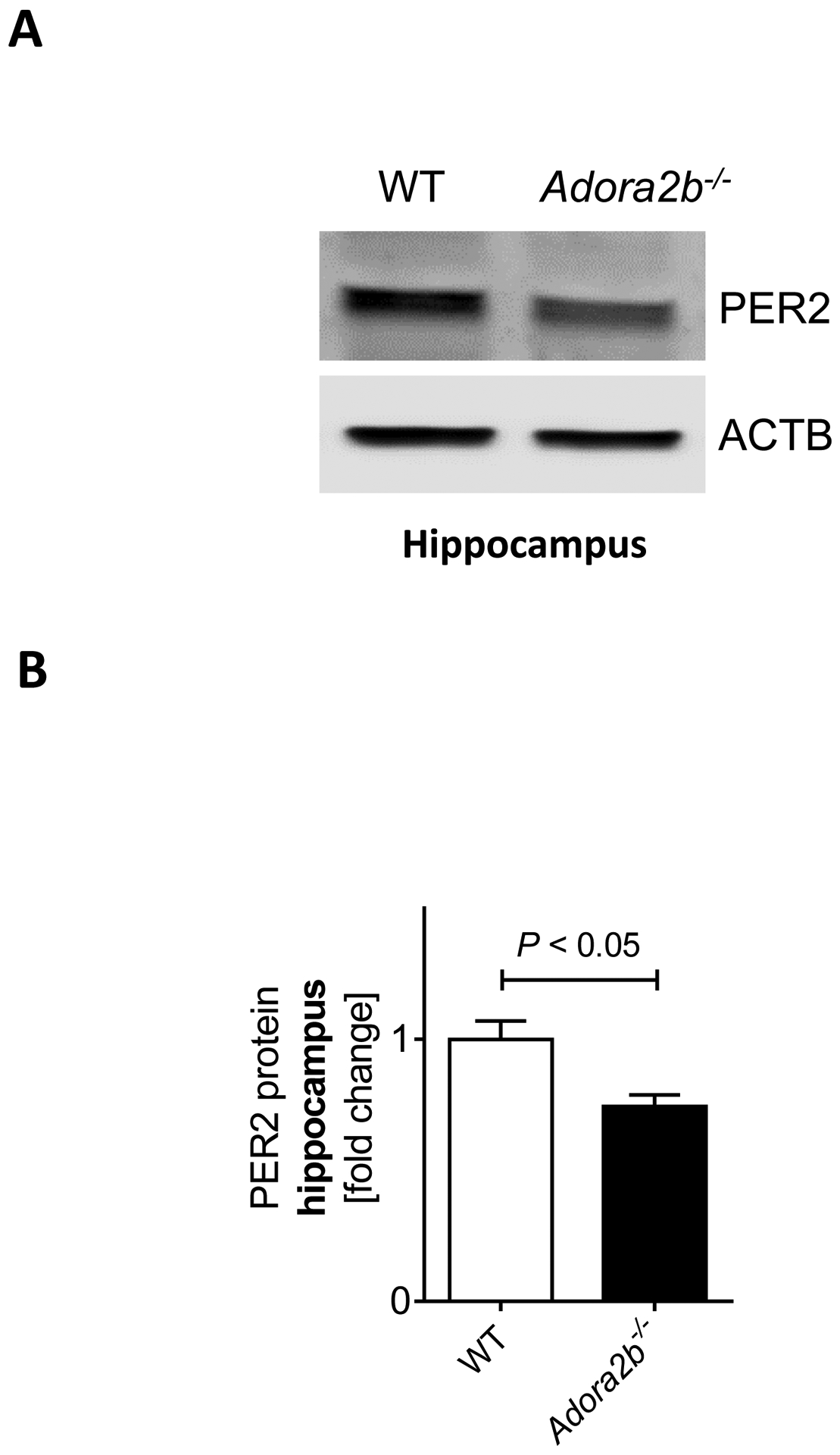
Wildtype (C57BL6/J) or Adora2b−/− mice were analyzed for hippocampal PER2 protein levels at baseline. (A) Immunoblot and (B) quantification for hippocampal PER2 protein in wildtype and Adora2b−/− mice, n=4–5, mean±SD; p<0.05.
The ADORA2B agonist BAY-60–6583 prevents midazolam induced hippocampal Per2 downregulation and restores midazolam mediated reduction in spontaneous alternation.
After we had seen impaired cognitive function and reduced hippocampal PER2 protein expression in Adora2b−/− mice, we next investigated the specific ADORA2B agonist BAY-60–6583 (Figure 3A) in midazolam induced cognitive dysfunction. We first analyzed the hippocampal Per2 transcript after midazolam administration with and without BAY-60–6583 co-administration in wildtype mice. As shown in Figure 3B, BAY-60–6583 prevented midazolam mediated downregulation of Per2 mRNA in the hippocampus. Moreover, midazolam induced cognitive dysfunction, as indicated by spontaneous alternation in a T maze test, was fully restored in mice that received BAY-60–6583 (Figure 3C). Taken together, the ADORA2B agonist BAY-60–6583 can be used to prevent midazolam induced cognitive dysfunction in mice.
Figure 3. BAY-60–6583 reverses midazolam induced cognitive deficits.
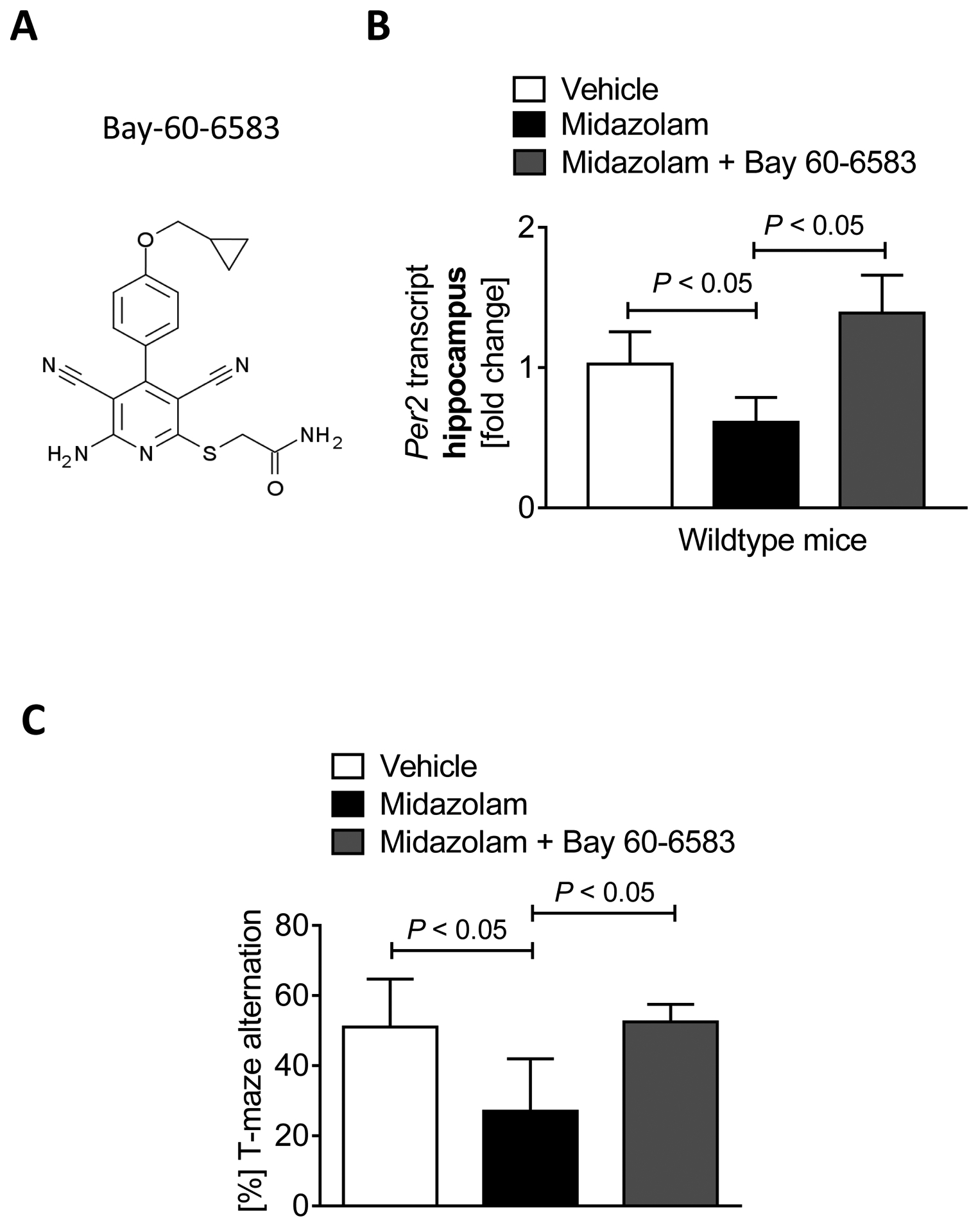
Wildtype (C57BL6/J) mice were injected with either saline, midazolam (10mg/kg i.p.), or midazolam + BAY 60–6583 (1 μg/kg i.p.) and compared to saline-treated controls. 2 hours later mice underwent brain PER2 mRNA analysis. 24 hours later, mice underwent behavioral studies using T-maze alternation tests. (A) The molecular formula of BAY 60–6583, a specific ADORA2B agonist. (B) Hippocampal PER2 transcript levels 24hours after midazolam or midazolam + BAY 60–6583 administration (n=5; p<0.05). (C) T-maze alternation in % 24 hours after a single dose of midazolam or midazolam + BAY 60–6583 (n=4–10; p<0.05). All data are presented as mean ± SD.
Adora2b−/− mice have less hippocampal expressed C-fos.
We next investigated possible additional mechanisms of ADORA2B in cognitive function. Based on studies showing a significant proinflammatory phenotype in Adora2b−/− mice [36], we first analyzed inflammatory markers in the hippocampus. As seen in Figure 4A–B, Adora2b−/− mice had similar hippocampal expressed Tnf-α and Il6 mRNA levels as wildtype mice. We, therefore, analyzed the markers for cognitive function, BDNF (brain-derived neurotrophic factor) and C-FOS, in the hippocampus. We found that Adora2b−/− had lower Bdnf or C-fos transcript levels in the hippocampus (Figure 4C–D). However, only C-fos mRNA levels were significantly lower in Adora2b−/− mice when compared to controls. Taken together, C-fos might represent another possible mechanism for the observed cognitive dysfunction in Adora2b−/− mice.
Figure 4. Adora2b−/− mice have less hippocampal C-fos levels.
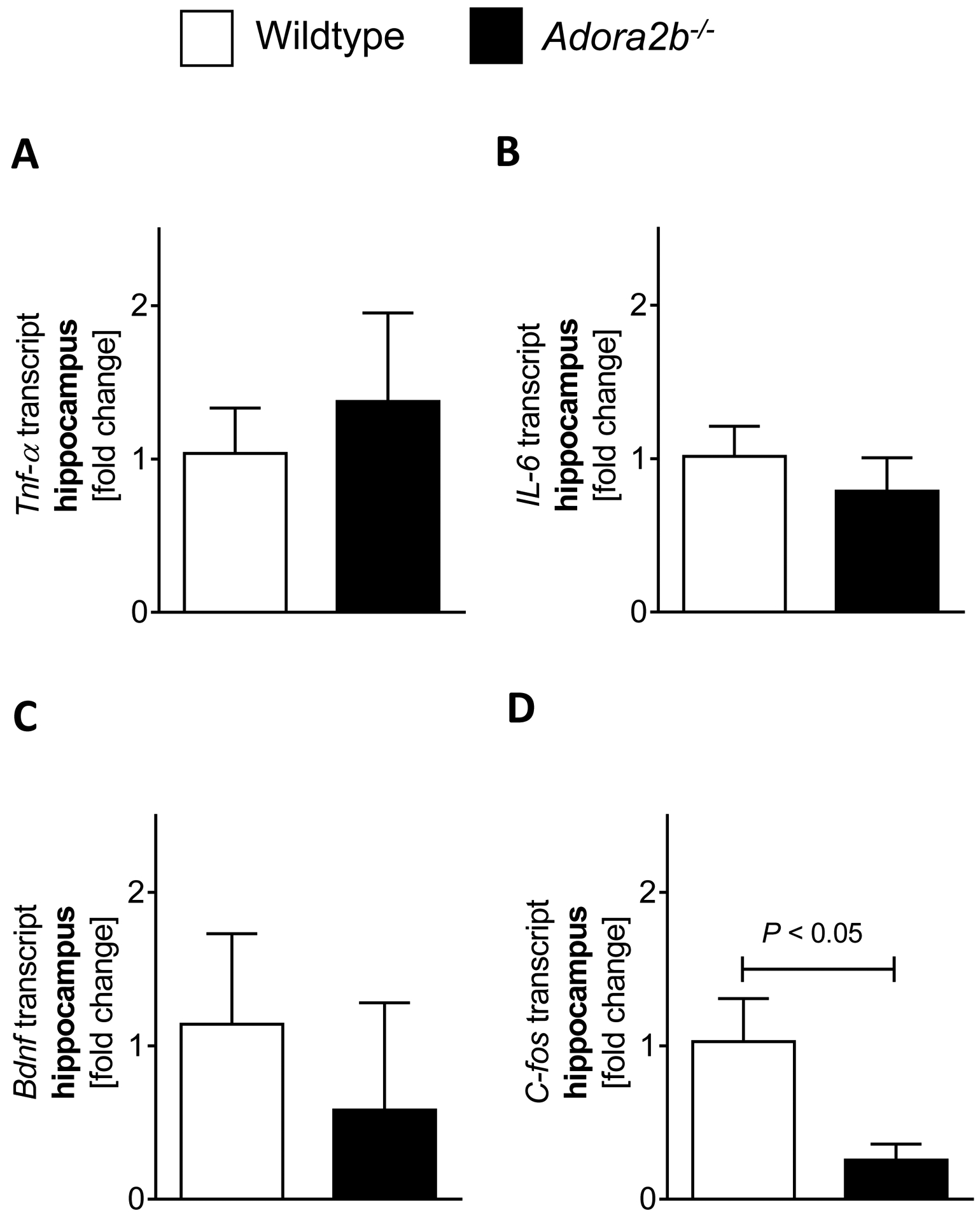
(A-D) Brain hippocampus tissue was harvested from wildtype (C57BL6/J) or Adora2b−/− mice, mRNA was isolated using RNeasy Mini Kit (Qiagen), cDNA was generated using miScript RT II kits (Qiagen), and Tnf-α. (A), Il-6 (B), Bdnf (C) and C-fos (D) transcript levels were determined by quantitative real-time RT-PCR (iCycler or iCycler IQ; Bio-Rad Laboratories Inc.), n=4–5, p<0.05, all data are presented as mean ± SD.
Discussion
In the present study, we investigated the role of the adenosine A2B receptor (ADORA2B) in the pathogenesis of midazolam induced cognitive dysfunction. We identified a role for hippocampal expressed ADORA2B and could restore hippocampal mediated cognitive deficits in midazolam induced delirium-like mouse model using the specific ADORA2B agonist BAY-60–6583. Taken together, these studies identify a possible role of ADORA2B in delirium-like cognitive alterations in mice.
Adenosine is a neuromodulator acting via four types of G-protein-coupled adenosine receptors, named ADORA1, ADORA2A, ADORA2B, and ADORA3. In the brain, studies have shown that the ADORA1 is involved in emotional stability [14], and the ADORA2A influences locomotion and cognition [15]. While expressed throughout the brain [37], the role of the ADORA2B has dominantly been investigated in the setting of organ protection [12, 17–20, 22, 24, 29, 30, 38–41]. Some studies on ADORA2B in the brain have focused on pathological conditions, which is explained by the observation that ADORA2Bs display a lower affinity for adenosine than the other adenosine receptors, suggesting that ADORA2B may be preferentially activated under noxious conditions associated with higher extracellular levels of adenosine. However, some studies suggest that ADORA2B might also control neurotransmitter release in the hippocampus [16].
The ADORA2B signals via cAMP, which is a core component of circadian rhythm regulation [42]. In fact, studies on ADORA2B signaling in the heart identified the circadian rhythm protein Period 2 (PER2) 2 as a critical downstream target, and light elicited PER2-cardioprotection was abolished in Adora2b−/− mice [12, 25]. PER2 is expressed in the SCN, the primary pacemaker in the mammalian brain [43]. The SCN controls the circadian rhythm of locomotion and behavior [5]. Per2−/− mice show disrupted circadian oscillations and loss of behavioral rhythms. Recent evidence from mice suggests that PER2 might also play a role in learning and memory due to a robust PER2 expression in the hippocampus [44]. Interestingly, studies in humans suggest a critical role of the hippocampus in the pathogenesis of delirium [45].
Moreover, a specific PER2 point mutation in humans leads to a significant disruptive sleep disorder, which can be recapitulated in mice [46]. Interrupted sleep can cause circadian disruption resulting in cognitive deficits, and has been linked to delirium in humans [47]. Mouse models with disrupted circadian rhythms show similar delirium-like phenotypes [48]. We recently studied the role of PER2 in the context of midazolam induced cognitive dysfunction, which revealed that Per2−/− mice have a phenotype that shares similarities with acute delirium [2]. In these studies, we found that midazolam-induced changes in behavior are associated with a significant downregulation of PER2 in the hippocampus. Furthermore, genetic deletion of Per2 in mice resulted in a phenotype that shared several features with acute delirium, but which was not worsened by midazolam treatment. Moreover, we successfully attenuated midazolam induced cognitive deficits using a novel PER2 enhancer.
Midazolam, a benzodiazepine, enhances the effect of the neurotransmitter GABA on the GABAA receptors. GABA is an inhibitory neurotransmitter in the CNS, and the release of GABA is associated with circadian rhythms [49, 50]. As such, studies have shown that GABA regulates PER2 expression through GABAA receptor activation in the SCN [51, 52]. In fact, treatment with muscimol, a GABAA receptor agonist in the SCN, decreased PER2 expression [51, 53], while treatment with a GABA antagonist increased PER2 expression [54]. This specific connection between GABA and PER2 signaling further supports our recent findings on a critical role of PER2 in midazolam induced cognitive dysfunction. While there are no reports to date of midazolam affecting PER2 expression in humans, benzodiazepines do alter sleep patterns and increase the risk of circadian disruption and delirium in humans [55, 56].
In the current study, we hypothesized that ADORA2B could be a possible upstream pathway of PER2 in controlling cognitive function. Indeed, we found that midazolam pretreatment downregulated Adora2b in the hippocampus as seen in our previous studies with hippocampal PER2 [2]. Moreover, using a T maze alternation test, a sensitive tool to identify hippocampal dysfunction, we found cognitive deficits in Adora2b−/− mice, which was like our previous findings in Per2−/− mice [2]. In fact, following analysis demonstrated significantly lower hippocampal PER2 protein levels in Adora2b−/− mice, suggesting an underlying Per2 deficiency as a possible mechanism. While these data are in support of our hypothesis, there are also obvious limitations such as the use of whole-body knockout animals, transcript analysis for some studies only, and limited behavioral testing in Adora2b−/− mice. E.g., we have not investigated the effects of midazolam in mice lacking the ADORA2B receptor, and therefore we have no genetic evidence that the effects of midazolam are ADORA2B specific. Furthermore, we did not test a PER2 enhancer such as nobiletin [26] in our Adora2b−/− mice, which possibly could restore a wildtype phenotype through bypassing the upstream pathways. However, using the highly specific ADORA2B agonist BAY-60–6583 [12, 17, 18], we were able to prevent midazolam mediated hippocampal PER2 downregulation or midazolam induced cognitive dysfunction in mice. These data strongly suggest a potential role for ADORA2B in cognitive function.
While there are only a few reports on ADORA2B function in the brain, there is a large body of literature on the role of ADORA2B during hypoxia or inflammation [38]. Adora2b−/− mice have been described as having dominantly a proinflammatory phenotype, and ADORA2B agonism has been well documented to be anti-inflammatory in different disease mouse models [17, 20–22, 36, 40]. Interestingly, delirium studies suggest inflammation in the brain as a possible mechanism for altered cognitive function [57–59]. However, our analysis of inflammatory markers in the hippocampus from Adora2b−/− mice did not reveal increased inflammation compared to controls. These negative findings might be explained by the fact that we analyzed the mice under baseline conditions but did not expose the mice to inflammatory agents such as lipopolysaccharide as done in our previous studies on PER2 in delirium [2]. As such, the observed phenotype in our Adora2b−/− mice might become more pronounced under inflammatory conditions.
As we did not see a proinflammatory phenotype in Adora2b−/− mice, we analyzed markers for cognitive function to further investigate the cause of cognitive dysfunction in Adora2b−/−. Essential regulators of short term or long term memory in the hippocampus are BDNF [60, 61] and C-FOS [62]. BDNF has been implicated in long term potentiation and memory. A very intriguing study has shown that BDNF is fundamental for synaptic plasticity [63]. C-FOS is part of a family of transcription factors and has been found to be important for hippocampus-dependent learning. Adult mice lacking C-FOS in the CNS show normal general and emotional behavior but are impaired explicitly in hippocampus-dependent spatial and associative learning tasks [62].
Moreover, several studies have shown the strong upregulation of C-FOS during learning tasks such as the T-maze [64, 65]. Also, the literature indicates that C-FOS might be involved in the development of postoperative delirium following general anesthesia [45]. If C-FOS, however, also is a mechanism of cognitive dysfunction in our Adora2b−/− mice will require further mechanics studies.
Interestingly, CREB, a constitutive transcription factor that regulates the transcription of genes with a CRE site in their promoter region, also regulates C-FOS [66]. Studies using truncated PER2 promoter constructs identified a CREB binding site responsible for ADORA2B-inducibility of PER2 [12]. As ADORA2B signals via the cAMP-CREB pathway, these studies are firmly supporting our findings of reduced C-FOS or PER2 levels in the brain from Adora2b−/− mice. Moreover, PER2 and C-FOS are both light-inducible genes, most likely via CRE elements in their promoter regions [28, 67, 68]. If PER2 and C-FOS are both downstream targets of ADORA2B signaling or if they are interconnected seems unclear. In fact, our recent studies on light elicited PER2 indicated that light-mediated increases of cAMP were abolished in Per2−/− mice [25]. This would suggest that also Per2−/− mice could have lower C-FOS levels and that PER2 is similarly involved in the regulation of C-FOS, like ADORA2B.
Regardless, our findings on a role for ADORA2B for cognitive function and the general lack of studies on ADORA2B function in the brain strongly indicate that additional mechanistic studies are warranted.
Conclusion
In conclusion, using gene-targeted mice for the Adora2b, our studies provide evidence that murine ADORA2B plays a role in midazolam induced cognitive dysfunction. Using the highly specific ADORA2B agonist BAY-60–6583, we reversed the cognitive dysfunction observed in mice following midazolam exposure. Future work should further explore the role of ADORA2B in brain function at baseline or during pathological conditions. If future studies confirm our findings, ADORA2B agonists could become a promising strategy to treat or prevent midazolam induced cognitive dysfunction in a clinical setting.
SOURCE OF FINANCIAL SUPPORT FOR THE WORK
National Heart, Lung, and Blood Institute (NIH-NHLBI) 5R01HL122472 Grant to T.E.; American Heart Association (AHA) Postdoctoral Fellowship 19POST34380105 Grant to Y.O.
Footnotes
Conflict of Interest
The authors declare there are no conflicts of interest.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Experimental protocols were approved by the Institutional Review Board (Institutional Animal Care and Use Committee [IACUC]) at the University of Colorado Denver, USA. They were following the NIH guidelines for the use of live animals and were compliant with the principles of the Declaration of Helsinki of the World Medical Association.
HUMAN AND ANIMAL RIGHTS
Experimental protocols were approved by the Institutional Review Board (Institutional Animal Care and Use Committee [IACUC]) at the University of Colorado Denver, USA. They were following the NIH guidelines for the use of live animals and were compliant with the principles of the Declaration of Helsinki of the World Medical Association.
AVAILABILITY OF DATA AND MATERIALS
Data are available at figshare (https://figshare.com/s/ffebf2aa985f9246ab74)
References
- [1].Salluh JI, Wang H, Schneider EB, Nagaraja N, Yenokyan G, Damluji A, Serafim RB, Stevens RD. Outcome of delirium in critically ill patients: systematic review and meta-analysis. BMJ, 2015; 350: h2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gile J, Scott B, Eckle T. The Period 2 Enhancer Nobiletin as Novel Therapy in Murine Models of Circadian Disruption Resembling Delirium. Crit Care Med, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Scott B, Eckle T. The impact of sedation protocols on outcomes in critical illness. Ann Transl Med, 2016; 4: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bellapart J, Boots R. Potential use of melatonin in sleep and delirium in the critically ill. Br J Anaesth, 2012; 108: 572–80. [DOI] [PubMed] [Google Scholar]
- [5].Fitzgerald JM, Adamis D, Trzepacz PT, O’Regan N, Timmons S, Dunne C, Meagher DJ. Delirium: a disturbance of circadian integrity? Med Hypotheses, 2013; 81: 568–76. [DOI] [PubMed] [Google Scholar]
- [6].Hatta K, Kishi Y, Wada K, Takeuchi T, Odawara T, Usui C, Nakamura H, Group D-J. Preventive effects of ramelteon on delirium: a randomized placebo-controlled trial. JAMA Psychiatry, 2014; 71: 397–403. [DOI] [PubMed] [Google Scholar]
- [7].Inouye SK, Westendorp RG, Saczynski JS. Delirium in elderly people. Lancet, 2014; 383: 911–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Madrid-Navarro CJ, Sanchez-Galvez R, Martinez-Nicolas A, Marina R, Garcia JA, Madrid JA, Rol MA. Disruption of Circadian Rhythms and Delirium, Sleep Impairment and Sepsis in Critically ill Patients. Potential Therapeutic Implications for Increased Light-Dark Contrast and Melatonin Therapy in an ICU Environment. Curr Pharm Des, 2015; 21: 3453–68. [DOI] [PubMed] [Google Scholar]
- [9].Yang J, Choi W, Ko YH, Joe SH, Han C, Kim YK. Bright light therapy as an adjunctive treatment with risperidone in patients with delirium: a randomized, open, parallel group study. Gen Hosp Psychiatry, 2012; 34: 546–51. [DOI] [PubMed] [Google Scholar]
- [10].Scott BK. Disruption of Circadian Rhythms and Sleep in Critical Illness and its Impact on the Development of Delirium. Curr Pharm Des, 2015; 21: 3443–52. [DOI] [PubMed] [Google Scholar]
- [11].Girard TD, Jackson JC, Pandharipande PP, Pun BT, Thompson JL, Shintani AK, Gordon SM, Canonico AE, Dittus RS, Bernard GR, Ely EW. Delirium as a predictor of long-term cognitive impairment in survivors of critical illness. Crit Care Med, 2010; 38: 1513–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Eckle T, Hartmann K, Bonney S, Reithel S, Mittelbronn M, Walker LA, Lowes BD, Han J, Borchers CH, Buttrick PM, Kominsky DJ, Colgan SP, Eltzschig HK. Adora2b-elicited Per2 stabilization promotes a HIF-dependent metabolic switch crucial for myocardial adaptation to ischemia. Nat Med, 2012; 18: 774–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. Adenosine and brain function. Int Rev Neurobiol, 2005; 63: 191–270. [DOI] [PubMed] [Google Scholar]
- [14].Cunha RA, Agostinho PM. Chronic caffeine consumption prevents memory disturbance in different animal models of memory decline. J Alzheimers Dis, 2010; 20 Suppl 1: S95–116. [DOI] [PubMed] [Google Scholar]
- [15].Shen HY, Canas PM, Garcia-Sanz P, Lan JQ, Boison D, Moratalla R, Cunha RA, Chen JF. Adenosine A(2)A receptors in striatal glutamatergic terminals and GABAergic neurons oppositely modulate psychostimulant action and DARPP-32 phosphorylation. PLoS One, 2013; 8: e80902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Goncalves FQ, Pires J, Pliassova A, Beleza R, Lemos C, Marques JM, Rodrigues RJ, Canas PM, Kofalvi A, Cunha RA, Rial D. Adenosine A2b receptors control A1 receptor-mediated inhibition of synaptic transmission in the mouse hippocampus. Eur J Neurosci, 2015; 41: 878–88. [DOI] [PubMed] [Google Scholar]
- [17].Koeppen M, Harter PN, Bonney S, Bonney M, Reithel S, Zachskorn C, Mittelbronn M, Eckle T. Adora2b signaling on bone marrow derived cells dampens myocardial ischemia-reperfusion injury. Anesthesiology, 2012; 116: 1245–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Eckle T, Krahn T, Grenz A, Kohler D, Mittelbronn M, Ledent C, Jacobson MA, Osswald H, Thompson LF, Unertl K, Eltzschig HK. Cardioprotection by ecto-5’-nucleotidase (CD73) and A2B adenosine receptors. Circulation, 2007; 115: 1581–90. [DOI] [PubMed] [Google Scholar]
- [19].Eckle T, Faigle M, Grenz A, Laucher S, Thompson LF, Eltzschig HK. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood, 2008; 111: 2024–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Eckle T, Grenz A, Laucher S, Eltzschig HK. A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J Clin Invest, 2008; 118: 3301–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hart ML, Jacobi B, Schittenhelm J, Henn M, Eltzschig HK. Cutting Edge: A2B Adenosine receptor signaling provides potent protection during intestinal ischemia/reperfusion injury. J Immunol, 2009; 182: 3965–8. [DOI] [PubMed] [Google Scholar]
- [22].Schingnitz U, Hartmann K, Macmanus CF, Eckle T, Zug S, Colgan SP, Eltzschig HK. Signaling through the A2B adenosine receptor dampens endotoxin-induced acute lung injury. J Immunol, 2010; 184: 5271–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bonney S, Kominsky D, Brodsky K, Eltzschig H, Walker L, Eckle T. Cardiac Per2 functions as novel link between fatty acid metabolism and myocardial inflammation during ischemia and reperfusion injury of the heart. PLoS One, 2013; 8: e71493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Seo SW, Koeppen M, Bonney S, Gobel M, Thayer M, Harter PN, Ravid K, Eltzschig HK, Mittelbronn M, Walker L, Eckle T. Differential Tissue-Specific Function of Adora2b in Cardioprotection. J Immunol, 2015; 195: 1732–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Oyama Y, Bartman CM, Bonney S, Lee JS, Walker LA, Han J, Borchers CH, Buttrick PM, Aherne CM, Clendenen N, Colgan SP, Eckle T. Intense Light-Mediated Circadian Cardioprotection via Transcriptional Reprogramming of the Endothelium. Cell Rep, 2019; 28: 1471–1484 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Oyama Y, Bartman CM, Gile J, Sehrt D, Eckle T. The circadian PER2 enhancer Nobiletin reverses the deleterious effects of midazolam in myocardial ischemia and reperfusion injury. Curr Pharm Des, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Koeppen M, Lee JW, Seo SW, Brodsky KS, Kreth S, Yang IV, Buttrick PM, Eckle T, Eltzschig HK. Hypoxia-inducible factor 2-alpha-dependent induction of amphiregulin dampens myocardial ischemia-reperfusion injury. Nat Commun, 2018; 9: 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bartman CM, Oyama Y, Brodsky K, Khailova L, Walker L, Koeppen M, Eckle T. Intense light-elicited upregulation of miR-21 facilitates glycolysis and cardioprotection through Per2-dependent mechanisms. PLoS One, 2017; 12: e0176243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Eckle T, Kewley EM, Brodsky KS, Tak E, Bonney S, Gobel M, Anderson D, Glover LE, Riegel AK, Colgan SP, Eltzschig HK. Identification of hypoxia-inducible factor HIF-1A as transcriptional regulator of the A2B adenosine receptor during acute lung injury. J Immunol, 2014; 192: 1249–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Eckle T, Hughes K, Ehrentraut H, Brodsky KS, Rosenberger P, Choi DS, Ravid K, Weng T, Xia Y, Blackburn MR, Eltzschig HK. Crosstalk between the equilibrative nucleoside transporter ENT2 and alveolar Adora2b adenosine receptors dampens acute lung injury. FASEB J, 2013; 27: 3078–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Eckle T, Brodsky K, Bonney M, Packard T, Han J, Borchers CH, Mariani TJ, Kominsky DJ, Mittelbronn M, Eltzschig HK. HIF1A reduces acute lung injury by optimizing carbohydrate metabolism in the alveolar epithelium. PLoS Biol, 2013; 11: e1001665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Frank A, Bonney M, Bonney S, Weitzel L, Koeppen M, Eckle T. Myocardial ischemia reperfusion injury: from basic science to clinical bedside. Semin Cardiothorac Vasc Anesth, 2012; 16: 123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Deacon RM, Rawlins JN. T-maze alternation in the rodent. Nat Protoc, 2006; 1: 7–12. [DOI] [PubMed] [Google Scholar]
- [34].Griffin EW, Skelly DT, Murray CL, Cunningham C. Cyclooxygenase-1-dependent prostaglandins mediate susceptibility to systemic inflammation-induced acute cognitive dysfunction. J Neurosci, 2013; 33: 15248–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].LeSauter J, Lambert CM, Robotham MR, Model Z, Silver R, Weaver DR. Antibodies for assessing circadian clock proteins in the rodent suprachiasmatic nucleus. PLoS One, 2012; 7: e35938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yang D, Zhang Y, Nguyen HG, Koupenova M, Chauhan AK, Makitalo M, Jones MR, St Hilaire C, Seldin DC, Toselli P, Lamperti E, Schreiber BM, Gavras H, Wagner DD, Ravid K. The A2B adenosine receptor protects against inflammation and excessive vascular adhesion. J Clin Invest, 2006; 116: 1913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Aherne CM, Kewley EM, Eltzschig HK. The resurgence of A2B adenosine receptor signaling. Biochim Biophys Acta, 2011; 1808: 1329–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Eltzschig HK, Bonney SK, Eckle T. Attenuating myocardial ischemia by targeting A2B adenosine receptors. Trends Mol Med, 2013; 19: 345–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gile J, Eckle T. ADORA2b Signaling in Cardioprotection. J Nat Sci, 2016; 2. [PMC free article] [PubMed] [Google Scholar]
- [40].Grenz A, Osswald H, Eckle T, Yang D, Zhang H, Tran ZV, Klingel K, Ravid K, Eltzschig HK. The reno-vascular A2B adenosine receptor protects the kidney from ischemia. PLoS Med, 2008; 5: e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Koeppen M, Eckle T, Eltzschig HK. Interplay of hypoxia and A2B adenosine receptors in tissue protection. Adv Pharmacol, 2011; 61: 145–86. [DOI] [PubMed] [Google Scholar]
- [42].O’Neill JS, Maywood ES, Chesham JE, Takahashi JS, Hastings MH. cAMP-dependent signaling as a core component of the mammalian circadian pacemaker. Science, 2008; 320: 949–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ripperger JA, Albrecht U. The circadian clock component PERIOD2: from molecular to cerebral functions. Prog Brain Res, 2012; 199: 233–45. [DOI] [PubMed] [Google Scholar]
- [44].Wang LM, Dragich JM, Kudo T, Odom IH, Welsh DK, O’Dell TJ, Colwell CS. Expression of the circadian clock gene Period2 in the hippocampus: possible implications for synaptic plasticity and learned behaviour. ASN Neuro, 2009; 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Maldonado JR. Pathoetiological model of delirium: a comprehensive understanding of the neurobiology of delirium and an evidence-based approach to prevention and treatment. Crit Care Clin, 2008; 24: 789–856, ix. [DOI] [PubMed] [Google Scholar]
- [46].Xu Y, Toh KL, Jones CR, Shin JY, Fu YH, Ptacek LJ. Modeling of a human circadian mutation yields insights into clock regulation by PER2. Cell, 2007; 128: 59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kamdar BB, Needham DM, Collop NA. Sleep deprivation in critical illness: its role in physical and psychological recovery. J Intensive Care Med, 2012; 27: 97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Fujioka A, Fujioka T, Tsuruta R, Izumi T, Kasaoka S, Maekawa T. Effects of a constant light environment on hippocampal neurogenesis and memory in mice. Neurosci Lett, 2011; 488: 41–4. [DOI] [PubMed] [Google Scholar]
- [49].Ralph MR, Menaker M. GABA regulation of circadian responses to light. I. Involvement of GABAA-benzodiazepine and GABAB receptors. J Neurosci, 1989; 9: 2858–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kim M, de la Pena JB, Cheong JH, Kim HJ. Neurobiological Functions of the Period Circadian Clock 2 Gene, Per2. Biomol Ther (Seoul), 2018; 26: 358–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ehlen JC, Novak CM, Karom MC, Gamble KL, Paul KN, Albers HE. GABAA receptor activation suppresses Period 1 mRNA and Period 2 mRNA in the suprachiasmatic nucleus during the mid-subjective day. Eur J Neurosci, 2006; 23: 3328–36. [DOI] [PubMed] [Google Scholar]
- [52].Matsuo I, Iijima N, Takumi K, Higo S, Aikawa S, Anzai M, Ishii H, Sakamoto A, Ozawa H. Characterization of sevoflurane effects on Per2 expression using ex vivo bioluminescence imaging of the suprachiasmatic nucleus in transgenic rats. Neurosci Res, 2016; 107: 30–7. [DOI] [PubMed] [Google Scholar]
- [53].Novak CM, Ehlen JC, Paul KN, Fukuhara C, Albers HE. Light and GABA)(A) receptor activation alter period mRNA levels in the SCN of diurnal Nile grass rats. Eur J Neurosci, 2006; 24: 2843–52. [DOI] [PubMed] [Google Scholar]
- [54].Aton SJ, Huettner JE, Straume M, Herzog ED. GABA and Gi/o differentially control circadian rhythms and synchrony in clock neurons. Proc Natl Acad Sci U S A, 2006; 103: 19188–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Brainard J, Gobel M, Scott B, Koeppen M, Eckle T. Health implications of disrupted circadian rhythms and the potential for daylight as therapy. Anesthesiology, 2015; 122: 1170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Smith HAB, Gangopadhyay M, Goben CM, Jacobowski NL, Chestnut MH, Thompson JL, Chandrasekhar R, Williams SR, Griffith K, Ely EW, Fuchs DC, Pandharipande PP. Delirium and Benzodiazepines Associated With Prolonged ICU Stay in Critically Ill Infants and Young Children. Crit Care Med, 2017; 45: 1427–1435. [DOI] [PubMed] [Google Scholar]
- [57].Valero J, Mastrella G, Neiva I, Sanchez S, Malva JO. Long-term effects of an acute and systemic administration of LPS on adult neurogenesis and spatial memory. Front Neurosci, 2014; 8: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hoogland IC, Houbolt C, van Westerloo DJ, van Gool WA, van de Beek D. Systemic inflammation and microglial activation: systematic review of animal experiments. J Neuroinflammation, 2015; 12: 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ming Z, Sawicki G, Bekar LK. Acute systemic LPS-mediated inflammation induces lasting changes in mouse cortical neuromodulation and behavior. Neurosci Lett, 2015; 590: 96–100. [DOI] [PubMed] [Google Scholar]
- [60].Alonso M, Vianna MR, Depino AM, Mello e Souza T, Pereira P, Szapiro G, Viola H, Pitossi F, Izquierdo I, Medina JH. BDNF-triggered events in the rat hippocampus are required for both short- and long-term memory formation. Hippocampus, 2002; 12: 551–60. [DOI] [PubMed] [Google Scholar]
- [61].Leal G, Bramham CR, Duarte CB. BDNF and Hippocampal Synaptic Plasticity. Vitam Horm, 2017; 104: 153–195. [DOI] [PubMed] [Google Scholar]
- [62].Fleischmann A, Hvalby O, Jensen V, Strekalova T, Zacher C, Layer LE, Kvello A, Reschke M, Spanagel R, Sprengel R, Wagner EF, Gass P. Impaired long-term memory and NR2A-type NMDA receptor-dependent synaptic plasticity in mice lacking c-Fos in the CNS. J Neurosci, 2003; 23: 9116–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].An JJ, Gharami K, Liao GY, Woo NH, Lau AG, Vanevski F, Torre ER, Jones KR, Feng Y, Lu B, Xu B. Distinct role of long 3’ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell, 2008; 134: 175–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Vann SD, Brown MW, Erichsen JT, Aggleton JP. Fos imaging reveals differential patterns of hippocampal and parahippocampal subfield activation in rats in response to different spatial memory tests. J Neurosci, 2000; 20: 2711–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].He J, Yamada K, Nabeshima T. A role of Fos expression in the CA3 region of the hippocampus in spatial memory formation in rats. Neuropsychopharmacology, 2002; 26: 259–68. [DOI] [PubMed] [Google Scholar]
- [66].Gandolfi D, Cerri S, Mapelli J, Polimeni M, Tritto S, Fuzzati-Armentero MT, Bigiani A, Blandini F, Mapelli L, D’Angelo E. Activation of the CREB/c-Fos Pathway during Long-Term Synaptic Plasticity in the Cerebellum Granular Layer. Front Cell Neurosci, 2017; 11: 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Porterfield VM, Piontkivska H, Mintz EM. Identification of novel light-induced genes in the suprachiasmatic nucleus. BMC Neurosci, 2007; 8: 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Shearman LP, Weaver DR. Photic induction of Period gene expression is reduced in Clock mutant mice. Neuroreport, 1999; 10: 613–8. [DOI] [PubMed] [Google Scholar]