

Abstract
Mitochondria are cellular power plants that supply most of the ATP required in the brain to power neuronal growth, function, and regeneration. Given their extremely polarized structures and extended long axons, neurons face an exceptional challenge to maintain energy homeostasis in distal axons, synapses, and growth cones. Anchored mitochondria serve as local energy sources; therefore, the regulation of mitochondrial trafficking and anchoring ensures that these metabolically active areas are adequately supplied with ATP. Chronic mitochondrial dysfunction is a hallmark feature of major aging-related neurodegenerative diseases, and thus, anchored mitochondria in aging neurons need to be removed when they become dysfunctional. Investigations into the regulation of microtubule (MT)-based trafficking and anchoring of axonal mitochondria under physiological and pathological circumstances represent an important emerging area. In this short review article, we provide an updated overview of recent in vitro and in vivo studies showing (1) how mitochondria are transported and positioned in axons and synapses during neuronal developmental and maturation stages, and (2) how altered mitochondrial motility and axonal energy deficits in aging nervous systems link to neurodegeneration and regeneration in a disease or injury setting. We also highlight a major role of syntaphilin (SNPH) as a key MT-based regulator of axonal mitochondrial trafficking and anchoring in mature neurons.
Keywords: axonal transport, development, energy deficits, mitochondria, mitochondrial anchoring, mitochondrial trafficking, neurodegeneration, regeneration, syntaphilin
Introduction
Neurons require a huge amount of adenosine triphosphate (ATP) as cellular energy to support their various functions as well as growth and regrowth. Although the human brain accounts for only 2% of our total body mass, it consumes nearly 20% of the total ATP produced (Attwell and Laughlin, 2001). In the human cortex, 77% of ATP consumption is used by grey matter, in which approximately 96% is consumed by neurons; a single neuron is estimated to require approximately 4.7 billion ATPs per second in a resting state (Zhu et al., 2012). The major source of ATP in neurons comes from mitochondria that convert glucose and pyruvate into ATP through the electron transport chain and oxidative phosphorylation. Apart from its major role as the cellular energy powerhouse, mitochondria also help maintain Ca2+ homeostasis (Billups and Forsythe, 2002; David and Barrett, 2003; Tang and Zucker, 1997; Werth and Thayer, 1994) and are involved in a range of signaling pathways (Bock and Tait, 2019). As mitochondrial biogenesis and turnover primarily take place in the soma and ATP diffusion into distal axons is limited, it is essential for neurons to deliver and position the proper number of mitochondria in distal axons, synapses, and growth cones where energy is in high demand. However, this is quite a challenging task due to the complex geometry of neurons that have an extended long axon with extensive branches. Single nigrostriatal dopaminergic neurons in the rat brain possess widely spread and highly dense axonal arborizations that can extend to ~0.47 meter in length if all the axon fibers are summed (Matsuda et al., 2009). Likewise, in the mouse peripheral nervous system, just one cutaneous sensory neuron can branch to make contact with around 200 hair follicles, and individual neuron axon arbors in hairy skin may contain several thousand branch points, with a total length of up to one meter long (Wu et al., 2012). Besides their exceptional architecture, what makes this logistical problem more difficult is that neurons are post-mitotic, meaning that each neuron needs to tackle this task for the entire lifetime of the organism. Thus, neurons face an exceptional challenge to maintain energy homeostasis in distal terminals to meet the local energy demand. Specifically, neurons deploy specialized mechanisms to (1) recruit and re-distribute healthy mitochondria from the cell body to distal axon arbors and terminals, and (2) remove aged or damaged mitochondria from distal regions for recovery or turnover during their lifespan (Ashrafi et al., 2014; Lin et al., 2017; Zheng et al., 2019). One attractive hypothesis is that mitochondrial trafficking and distribution is tightly regulated in order to respond to changes in metabolic and growth status and synaptic activity, and to pathological stress at neuronal developmental and aging stages (Misgeld and Schwarz, 2017; Saxton and Hollenbeck, 2012; Sheng, 2017; Sheng and Cai, 2012; Devine and Kittler, 2018).
The intracellular environment changes dynamically as immature neurons undergo various developmental stages toward maturation and mature neurons gradually age towards the end-stage of the organism’s lifespan. It is no surprise that neurons’ bioenergetic status is altered accordingly, with mitochondrial trafficking and distribution in axons and synapses being differentially regulated along this trajectory. The distribution of mitochondria along axons displays a linearly interspersed pattern, which is quite distinct from that in the soma where densely populated mitochondria form a closely connected network. At certain sites along axons where energy demand is particularly high, such as presynaptic boutons, growth cones, axonal branches, and nodes of Ranvier, more mitochondria are allocated to sustain the unique functions of these specialized regions (Harris and Weinberg, 2012; Morris and Hollenbeck, 1993; Ruthel and Hollenbeck, 2003; Zhang et al., 2010; Tao et al., 2014). These distribution profiles become relatively stable after neurons mature and presynaptic structures are formed. Furthermore, when neurons encounter physiological or pathological stress, mitochondria will quickly redistribute in response to metabolic changes, thus regaining energy homeostasis. This highly dynamic redistribution of axonal mitochondria is mainly achieved by coordination of a set of MT-based trafficking and anchoring machineries, composed of motors, adaptors and anchors (MacAskill and Kittler, 2010; Guedes-Dias and Holzbaur, 2019; Misgeld and Schwarz, 2017; Saxton and Hollenbeck, 2012; Sheng, 2015; 2017). Deficiencies in these mechanisms will lead to impaired mitochondrial trafficking and positioning, consequently contributing to local energy deficits and axonal degeneration (Sheng and Cai, 2012). When an organism enters late stages of adulthood, mitochondrial biogenesis, turnover, and motility all slow down (Chen and Chan, 2009; Takihara et al., 2015; Zhou et al., 2016; Zhou et al., 2016; Morsci et al., 2016). This phenotype can also occur in younger and adult nerves that have a traumatic injury or under pathological stress in aging-linked neurodegenerative diseases.
Research into the developmental and aging-associated regulation of axonal mitochondrial trafficking, anchoring, and distribution is thus an important emerging frontier. Recent advances provide exciting lines of in vivo and in vitro evidence as to how mitochondrial trafficking and anchoring are coordinated during neuronal development, maturation, and aging. Axonal mitochondrial trafficking and anchoring rely on MTs and F-actin filaments, the latter being highly enriched at synapses (Cingolani and Goda, 2008). While MT-based kinesin and dynein motors drive long-range mitochondrial transport along axons, actin-based myosin motors mediate shorter-range local movement and anchoring (Morris and Hollenbeck, 1995; Chada and Hollenbeck, 2004; Hirokawa and Takemura, 2005; Quintero, et al., 2009). In this review, we first briefly introduce the MT-based trafficking and anchoring machineries that control axonal mitochondrial motility and distribution. Next, we provide an in-depth discussion of recent findings on aging-associated decline of axonal mitochondrial trafficking, as well as physiological and pathological relevance of altered mitochondrial motility in neurodegeneration and regeneration of mature neurons in adult nervous systems. For additional insights and alternative perspectives on dendritic mitochondrial trafficking and anchoring, actin-based regulation of mitochondrial motility, and axonal energy maintenance, we refer readers to the following reviews (Misgeld and Schwarz, 2017; Sheng 2017; Devine and Kittler, 2018; Chada and Hollenbeck, 2004; Chamberlain and Sheng, 2019).
Motor-Adaptor Complexes Driving Mitochondrial Transport
Long-distance mitochondrial transport in axons is driven by ATP-hydrolyzing motor proteins along MT tracks, which are uniformly arranged with plus ends directed towards axon terminals and minus ends directed towards the microtubule organization center in the soma (Hollenbeck and Saxton, 2005). Mitochondrial anterograde transport from the soma to distal axons is mainly driven by the kinesin-1 family (KIF5A, B, C), while their retrograde transport from distal ends to the cell body is mediated primarily by cytoplasmic dynein and the dynactin complex (Cai et al, 2005; MacAskill et al, 2009; Hirokawa et al., 2010). Motor proteins are recruited to organelles through their cargo adaptors, thus ensuring targeted trafficking and regulation. Mitochondria also deploy adaptors for linking the organelles with their transport motors. Miro1/2 and Trak1/2 (Milton in Drosophila) are two major adaptors of kinesin-1 motors (Brickley et al., 2005; Stowers et al., 2002; Glater et al., 2006). Trak1/2 link to KIF5B at one end and interact with Miro at the other end. Miro1/2 directly attach to the outer mitochondrial membrane (OMM), loading motor machineries on mitochondria. Together, KIF5, Miro1/2, and Trak1/2 comprise the mitochondrial transport machinery driving mitochondrial anterograde transport (MacAskill et al., 2009a; Wang and Schwarz, 2009). Syntabulin is an alternative KIF5 adaptor for driving mitochondrial transport. Syntabulin attaches to the OMM via its carboxyl-terminal domain and recruits KIF5 motors to mitochondria (Cai et al, 2005).
The dynein motor complex (dynein, dynactin and p150Glu) is the primary transport motor mediating retrograde transport of axonal mitochondria. Compared with Trak-Miro in the KIF5 adaptor complex, adaptors that recruit dynein motors to mitochondria require further verification. However recent evidence suggests that KIF5 and dynein motors may share a similar set of mitochondrial adaptor complexes. Loss of dmiro impairs bi-directional mitochondrial transport in Drosophila neurons (Guo et al., 2005), while overexpressing dMiro enhances both anterograde and retrograde mitochondrial transport in axons (Russo et al., 2009). In addition, Trak1/2 contain separate binding domains for KIF5 and dynein/dynactin, allowing their coupling to both motors (van Spronsen et al., 2013). These studies suggest an attractive model in which the relative activity of opposing motors could be regulated through their separate interactions with the same set of adaptors/receptors, thereby allowing mitochondria to move bi-directionally and frequently change direction.
MT-Based Anchoring of Axonal Mitochondria
In mature neurons, the majority of axonal mitochondria are in a stationary pool, with the relative percentage of anchored mitochondria depending on the maturation stage of the neurons, whether mitochondria are measured in proximal axons versus terminal axon branches, culture conditions, and in vitro and in vivo imaging tools used for measurement of motility. Thus, MT-based anchoring is essential for arresting mobile mitochondria at particular sites along axons. We previously identified syntaphilin (SNPH) as a “static anchor” that holds axonal mitochondria stationary via its docking interaction with MTs (Kang et al., 2008). SNPH targets axonal mitochondria through its carboxyl-terminal transmembrane domain that inserts into the OMM and its axon-sorting sequence, while SNPH anchors axonal mitochondria through its N-terminal MT-binding domain. Deleting the snph gene in mouse neurons results in a robust increase (70–79%) of axonal mitochondria in motile pools in both in vitro and in vivo nervous systems, while overexpressing SNPH abolishes axonal mitochondrial transport. Selectively recruiting the KIF5 motor or SNPH onto axonal mitochondria via an optogenetically controlled method changes the balance of their mobile versus stationary status (van Bergeijk et al., 2015), thus supporting the notion that moving or stopping axonal mitochondria depends on the balance of motor-adaptor and anchoring protein SNPH. MT-based mitochondrial motility is thus regulated by affecting their trafficking or anchoring mechanisms in order to sense, integrate, and respond to changes in neuronal activity, metabolic and growth status, mitochondrial function, aging, and pathological stress. By dynamic interplay of these motors, adaptors, and the anchor, axonal mitochondria undergo long-distance bi-directional transport, become paused or stationary, and are re-mobilized and re-distributed. The snph knockout mouse is an ideal model system to examine the impact of axonal mitochondrial anchoring on nerve regeneration and degeneration in mature and aging neurons, as will be discussed in the following sections.
Developmental Regulation of Axonal Mitochondrial Motility
At early developmental stages, neurons actively synthesize cellular proteins and organelles in bulk to support axon outgrowth, branching, and synaptogenesis. These developmental events require robust axonal mitochondrial trafficking and distribution that is distinct from mature stages (Chada and Hollenbeck, 2004; Morris and Hollenbeck, 1993; Ruthel and Hollenbeck, 2003). In vitro studies have revealed a significant difference in the motility and morphology of axonal mitochondria in young and mature rat cortical neurons. Chang and Reynolds reported that in young neurons at DIV5 (day in vitro), mitochondria have a higher motility compared with mature neurons at DIV14 (Chang and Reynolds, 2006). Within the same developmental stage, the number of functional synapses was also increased. Interestingly, individual mitochondrion in different aged neurons appeared to be functionally similar, as their membrane potential and Ca2+ buffering capacity were indistinquishable. The authors reasoned that the enhanced axonal mitochondrial motility in immature stages may permit faster energy dispersal to support energy-demanding synaptogenesis, and the reduction in motility at mature stages may serve as stable energy sources at synapses (Chang and Reynolds, 2006). Indeed, this is echoed by other studies that showed mitochondria are highly enriched at presynaptic sites and 30–50% of the active zones in presynaptic boutons contain one or more mitochondria (Harris and Weinberg, 2012; Kang et al., 2008; Sun et al., 2013; Smith et al., 2016; Devine and Kittler, 2018).
Active growth cones also constitute a site of high ATP consumption. In both central and peripheral neurons, distribution of mitochondria was highly skewed toward growth cones, with several fold higher density in the region adjacent to the growth cone than regions far away. This mitochondrial density gradient is attributable to a net anterograde transport entering the axon after axon specification. Blocking axonal growth led to collapse of this mitochondrial distribution gradient and reversal of the net transport (Morris and Hollenbeck, 1993; Ruthel and Hollenbeck, 2003). This biased motility and the subsequent arrest of mitochondria at growth cones is partly triggered by nerve growth factor (NGF) and regulated by the NGF-phosphoinositide 3-kinase signaling pathway (Chada and Hollenbeck, 2003; 2004). When terminal axon branching starts to develop, anterogradely moving mitochondria enter the growing branches twice as often as in the non-growing branches, where they transition into a stationary status through the LKB1 (liver kinase B1)-NUAK1 signaling pathway (Courchet et al., 2013; Ruthel and Hollenbeck, 2003). Maturation of axonal filopodia into branches is promoted by sustained generation of ATP from the stationary mitochondrial pool, as local ATP supply is necessary for axonal mRNA translation and reorganization of the cytoskeleton during axonal branching formation and maintenance. In the presence of NGF, ~84% of axonal branching sites contained one or more mitochondria in chick embryonic sensory neurons (Spillane et al., 2013). Activation of the cellular energy sensor, AMP-activated protein kinase (AMPK), is required for axonal branch formation where it serves to balance mitochondrial trafficking and anchoring (Tao et al., 2014). Activation of AMPK increases anterograde mitochondrial transport towards axonal terminals and leads to the accumulation of docked mitochondria in specific regions where new axonal branches will subsequently emerge. A SNPH-mediated anchoring mechanism has been suggested to underly AMPK-activated mitochondrial docking at axon branching sites (Courchet et al., 2013). While AMPK activation replenishes ATP supply in distal axons by recruiting and anchoring mitochondria, depleting SNPH reduces the stationary pool size of axonal mitochondria accompanied by decreased axon branching, establishing a causal correlation between SNPH-mediated anchoring and AMPK-induced axonal branching. Thus, mitochondrial trafficking and anchoring is tightly regulated during neurite outgrowth, axonal branching, and synapse formation.
After synaptogenesis, axon outgrowth and arborization, axonal mitochondria gradually enter a steady phase. Under resting conditions in vitro, approximately 20–30% of mitochondria move along axons at a velocity of ~0.2–2.0 μm/s. Among this motile pool, the anterograde portion is slightly larger than the retrograde portion, a phenotype that is consistent across different neuron types (Brickley and Stephenson, 2011; Courchet et al., 2013; Kang et al., 2008; Zhou et al., 2016). For a fully mature neuron, synaptic transmission and action potential propagation are two major functional features, both of which require high energy consumption. Accordingly, the axons of mature neurons display increased mitochondrial density at synapses and nodes of Ranvier, two metabolically active sites, instead of a uniformly distributed pattern. Through rebalancing trafficking and anchoring, mitochondria are delivered to and anchored at these axonal sites to provide sustained energy. However, even in adult and aged neurons, synaptic function and structures remain highly plastic and undergo activity-dependent remodeling, thus axonal mitochondria need to dynamically transit between stationery and mobile pools in response to changes in synaptic activity. Several works independently reported that Miro1 can bind Ca2+ through its two EF-hand motifs in order to sense Ca2+ influx during synaptic activation. Activation of this Ca2+-signaling pathway was suggested to disrupt its functional coupling with transport motor machineries, thereby arresting mitochondria at activated synapses (Macaskill et al., 2009b; Wang and Schwarz, 2009). Interestingly, deleting the snph gene in neurons abolishes activity-dependent mitochondrial arrest at synapses (Chen and Sheng 2013). SNPH can compete with Trak2 to bind KIF5 motors and inhibits motor ATPase activity; and KIF5-SNPH interaction is required in order to arrest an axonal mitochondrion in response to synaptic activity. These findings suggest an “engine-switch and brake” model: when a motile mitochondrion passes by an activated synapse, SNPH responds to elevated Ca2+ levels by switching off the engine (motor) and placing a brake on the mitochondrion, thus keeping it stationary. This model represents a molecular interplay between the KIF5-Trak-Miro trafficking complex and the anchoring protein SNPH, in which trafficking and anchoring mechanisms can be turned on or off in response to changes in Ca2+ levels and synaptic activity.
While synapses are activated by Ca2+ influx via activating voltage-dependent calcium channels or neurotransmitter receptors, the node of Ranvier is excited mainly by Na+ influx. Using adult frog sciatic nerve explants from the peripheral nervous system, Zhang et al. found that during nerve impulse, evoked Ca2+ elevation and Na+ pump activation synergistically arrest mitochondrial motility at nodes. During repetitive action potentials, the number of mobile mitochondria is reduced dramatically by ~40% in both directions, causing more mitochondria arrested at nodes. Bathing nerve explants in Ca2+ free solutions or blocking Na/K-ATPase abolished the activity-dependent arrest of axonal mitochondria (Zhang et al., 2010). Using rat cerebellar slice cultures as a model, Ohno et al. monitored mitochondria behavior in axons of Purkinje cells. Similarly, they found motile mitochondria stopped preferentially in node-paranodal areas in these myelinated CNS fibers. Following stimulation, the number of motile mitochondria and the mean velocity of mitochondrial motility were significantly decreased at nodes (Ohno et al., 2011). Although mitochondria are enriched at internodal areas in the CNS, it is agreed in both the CNS and PNS that firing of action potentials regulates mitochondrial distribution and motility (Ohno et al., 2011; Zhang et al., 2010). Thus, in mature neuron axons, mitochondria trafficking and anchoring are modulated by neuronal electrical activity coupled to increased Ca2+ levels both in presynaptic boutons and nodes of Ranvier.
While kinesin and dynein motors drive long-range mitochondrial transport in axons, actin-based myosin motors mediate short-range local movement and anchoring. Cytoskeletal filaments are uniquely organized in mature neuronal axons, where two-tier F-actin filaments are differentially organized. While stable subplasmalemmal actin rings form periodic structures that wrap around axons to support the axonal membrane, dynamic intra-axonal filaments help enrich actin at presynaptic terminals (Xu et al., 2013; Ganguly et al., 2015). It has been reported that mitochondrial receptors Miro1/2 regulate recruitment and stability of myosin-19, thus likely coordinating MT- and actin-based mitochondrial trafficking in mouse embryonic fibroblasts (MEFs) (Quintero et al., 2009; Bocanegra et al., 2019; Oeding et al., 2018; López-Doménech et al., 2018). However, the role of myosin-19 in axonal mitochondrial trafficking is not clear. At presynaptic terminals, actin filaments form highly dense mesh structures (Coles and Bradke, 2015), suggesting presynaptic F-actin filaments as a platform in capturing mitochondria. This notion was supported by studies showing that actin filaments capture mitochondria at nerve growth factor stimulation sites (Chada and Hollenbeck, 2004) and that actin depolymerization enhances axonal mitochondrial transport (Gutnick et al., 2019), although the actin-based anchoring mechanisms remain unclear. These studies highlight an opposing effect of actin filaments and MTs on axonal mitochondrial motility.
Regulation of Axonal Mitochondrial Trafficking in Developing and Adult Nervous Systems
While cultured neurons provide an experimentally tractable system for investigating the molecular mechanisms of motility events, most isolated neurons typically lack features of their whole model organisms, including (1) the complex neurite geometry and neuron-neuron or neuron-glia interactions; (2) a physiological metabolic environment with vascularization providing oxygen and nutrients; and (3) integrated circuit activity characteristic of an intact nervous system. Since mitochondria respond to their surrounding metabolic environment, status of mitochondrial trafficking and anchoring is likely influenced by both the structural and functional properties of whole model organisms. Therefore, to characterize mitochondrial trafficking in a physiological context, many have turned to in vivo systems—mostly using transgenic animal lines with fluorescently-tagged mitochondria and intravital microscopy techniques. Findings from several model organisms have provided valuable insights into the behavior of axonal mitochondria across multiple developmental stages, ranging from embryonic to adulthood.
Zebrafish are an ideal model organism for developmental studies since they are mostly transparent at both the embryonic and larval stages, allowing for mitochondria to be visualized noninvasively in intact neuronal axon arbors. Misgeld and colleagues were the first to describe axonal mitochondrial trafficking in vivo using zebrafish embryos (Plucinska et al., 2012). They measured mitochondrial flux in Rohon-Beard (RB) neurons, a type of sensory nerve cell, at both the proximal axonal segment and at distal axon branches. They revealed more mitochondria moving anterogradely than retrogradely (antero: 0.55 ± 0.04 vs retro: 0.32±0.03 mito/min) in proximal axons, corresponding to a net displacement of mitochondrial volume into the peripheral arbor. This anterograde/retrograde ratio remained constant throughout development. Interestingly, they observed a much lower percentage of moving mitochondria (~1%) in distal axon branches compared to proximal axon tracts (~16%). Similarly, using zebrafish embryos and larvae, Mandal et al. also observed a highly mobile population of mitochondria in sensory posterior lateral line axons across various developmental stages, while only ~30% remained stationary. In addition, mitochondrial velocity remained constant at ∼0.83 μm/s and ∼1.14 μm/s for anterograde and retrograde movement, respectively (Mandal et al., 2018). Overall, in vivo axonal mitochondrial motility in developing stages is quite robust, at least within the proximal shaft of axons.
Studying the relationship between axonal transport and the structure and function of neurons in adult animal models is much more challenging, especially the visualization of long-distance axonal transport longitudinally. Taking advantage of the short life-span (~2 weeks) and transparent nature of C. elegans, our group reported age-associated changes in axonal mitochondrial morphology, density, and trafficking throughout adulthood along the distal process of the anterior lateral microtubule (ALM), a specific class of mechanosensory neuron (Morsci et al., 2016). We observed significantly lower mitochondrial motility in ALM neurons in vivo compared to cultured mammalian neurons. Moreover, we found a progressive age-related decline in both motility and run length of motile mitochondria starting in young adulthood in wide-type C. elegans, whereas long-lived daf-2 mutants had no such decay, suggesting a close association between stably maintained mitochondrial trafficking rates and extended longevity. Interestingly, this age-dependent decline in mitochondrial motility is not restricted to C. elegans. The Bullock group investigated mitochondrial transport in early adulthood in Drosophila (~30 days) in vivo by tracking the axon bundles of the wing marginal nerve, which comprises both chemosensory and mechanosensory neurons. They also observed a progressive decline in mitochondrial motility from 30% at day 1 (beginning of adulthood) to ~10 fold lower at day 30, with no changes in mitochondria density (Vagnoni and Bullock, 2018). These age-dependent changes in mitochondria motility do not reflect general changes in axonal cargo transport since dense-core vesicles along the same axons did not display these changes over time. The development of transgenic mice with fluorescently-tagged mitochondria and two-photon microscopy have allowed visualization of intact nerves in adult mice with minimal invasiveness. Misgeld et al. conducted the first in vivo study of mitochondrial motility in adult mammalian neurons using intact intercostal nerves of Thy1-mitoCFP mice (Misgeld et al., 2007). They reported ~87% of axonal mitochondria are stationary with an average length of ~3 μm, and among the motile pool of mitochondria, two thirds moved in the anterograde direction and one third moved in the retrograde direction. They also noted that the number of motile mitochondria dropped in a proximal-to-distal direction as the stream of mitochondrial cargo is divided at each axonal branch point. Consistently, Smith and colleagues reported in the saphenous nerve (the largest cutaneous branch of the femoral nerve and a strictly sensory nerve without motor function) that the majority of axonal mitochondria are stationary when the nerve is in a resting state, with a mean number of 1 moving mitochondrion per imaged axon (Sajic et al., 2013). However, they observed a significant surge of anterogradely moving axonal mitochondria when impulse conductions were applied in the physiological range of stimulations, resulting in an accumulation of mitochondria at the distal terminal, but not Nodes of Ranvier. Interestingly, they observed that this increase in the number of motile mitochondria came from both the static mitochondrial population that became mobile, and portions of mitochondria that pinched off the static population by fission. The first in vivo study of mitochondrial trafficking in the mammalian central nervous system was performed by Takihara et al. using mouse retinal ganglion cells (RGCs) (Takihara et al., 2015), where they observed highly dynamic mitochondrial trafficking along the axon tracts. Notably, since the retina is a sensory organ that responds to light, the fluorescence from intravital imaging may have induced neural activity, and thereby increase mitochondrial dynamics in response to increased stimulation.
More recently, two studies independently reported a “predominantly static” mitochondrial phenotype with less than 1% motility at axonal arbors of layer 2/3 cortical pyramidal neurons in adult mice using in vivo two-photon microscopy (Lewis et al., 2016; Smit-Rigter et al., 2016). Consistent with in vitro studies, they reasoned that this decrease in mitochondrial motility is likely due to the increased number of stable presynaptic sites in mature cortical neurons as compared to developing ones, as almost half of the axonal mitochondria are localized at presynaptic terminals. Interestingly, while Smit-Rigter et al (2016) reported a predominantly stationary mitochondrial pool within the axonal arbor of layer 2/3 pyramidal neurons in the V1 neocortex region of the adult mouse brain, they also showed that merely 1.06 % of mitochondria are motile in the developing V1 during postnatal days P10–13. One explanation for these seemingly contradictory findings is that the application of two-photon microscopy on thick brain tissues in live animals prevents imaging of the main axonal tracts, but only allows access to the very terminal branches of axons or synapses. This is consistent with findings from the developing zebrafish nervous system (Plucinska et al., 2012) in which the percentage of motile mitochondria in distal axon branches is only 1%, whereas mitochondrial motility rises to 16% in proximal axonal regions. In addition, Smit-Rigter et al (2016) applied P10–13 mice as their developing model. Our previous study (Kang et al., 2008) demonstrated that SNPH is almost undetectable at embryonic stages and becomes readily detectable at P7 and peaks at P14. Thus, even in developing mouse brains at P10–13, SNPH expression is robustly increased, thus contributing to a large stationary pool of axonal mitochondria observed in vivo (Smit-Rigter et al., 2016).
Altogether, these in vivo studies have advanced our knowledge of axonal mitochondrial trafficking and anchoring in adult nervous systems under physiological conditions. It is becoming clearer that axonal mitochondrial trafficking and anchoring could vary based on neuron type, age, activity, and even neuronal compartment. Briefly, (1) neuronal activity is a potent regulator of axonal mitochondrial transport and distribution; more frequent firing rates could change mitochondrial distribution in either the main axon tract or terminal branches; (2) the density and motility of mitochondria drop when moving from the main axon tract (proximal to the cell body) towards terminal axon branches where many functional synapses exist; and (3) young neurons have more motile mitochondria than mature or aged neurons. These features indicate that we should be cautious in interpreting mitochondria trafficking and positioning data, which likely vary according to neuronal type and site of imaging. It is noteworthy that several in vitro and in vivo studies similarly revealed a slightly higher anterograde over retrograde motility under resting conditions (Misgeld et al., 2007; Takihara et al., 2015; Chang and Reynolds, 2006; Lin et al., 2017). This may suggest local degradation of stressed or aging mitochondria along the axon by active lysosomes (Ashrafi et al., 2014; Farfel-Becker et al., 2019).
Mechanisms for Mature Neuron-Associated Reduction in Axonal Mitochondrial Motility
One caveat from the above studies is that none of the live imaging was performed for longer than 12 hours, likely due to the temporal limitations surrounding the duration of live-cell imaging. Nonetheless, we can conclude that the diversity of mitochondrial trafficking data reflects the fact that mitochondrial distribution is finely-tuned in order to maintain function and metabolic homeostasis during neuronal development and maturation. One aspect revealed by both in vitro and in vivo studies across the different species described above is the progressive decline of mitochondrial motility from early stages (neurons <14 days in vitro or at developing stages in animals) to late stages (neurons ≥14 days in vitro or at adult stages in animals). However, the molecular basis of these aging-related changes remains largely elusive. It is likely that there is either a simultaneous change in the abundance of proteins regulating mitochondrial trafficking and anchoring with age, or post-translational modifications of these proteins at different developmental and maturation stages.
One prominent protein involved in this change is SNPH. SNPH-mediated anchoring is an attractive candidate mechanism contributing to developmental and aging-associated regulation of axonal mitochondrial motility, as the expression of SNPH is developmentally regulated and is progressively increased during brain maturation. Specifically, SNPH expression is hardly detectable in E18 brains and in cultured neurons before DIV3, becomes readily detectable after DIV9, and peaks at DIV22 in culture and adult mouse brains (Figure 1A) (Zhou et al., 2016). This expression pattern is unique to SNPH as we failed to detect such changes in other mitochondrial motor-adaptor proteins (KIF5, DIC, Miro1/2, and Trak2) or OMM proteins (TOM20 and VDAC). This expression pattern is consistent with our previous findings from rat brains in vivo where SNPH expression is initially undetectable at embryonic stages, yet gradually increases throughout brain development and finally peaks in adulthood (Das et al., 2003). This unique expression pattern suggests a model in which SNPH-mediated anchoring plays a critical role in controlling axonal mitochondrial motility in mature neurons. Consistent with this hypothesis, axonal mitochondria display progressively reduced motility from DIV7 (47.60 ± 4.60%) to DIV18 (22.88 ± 3.09%, p<0.001) (Figure 1B) (Zhou et al., 2016). Importantly, this reduction is not due to a general decline in axonal transport, but is specific to axonal mitochondria. This age-dependent decline in axonal mitochondrial motility and predominantly arrested mitochondria in terminal axon branches were also observed in rodent nervous systems in vitro and in vivo from several independent studies (Chen and Sheng 2013; Lewis et al., 2016; Smit-Rigter et al., 2016; Takihara et al., 2015), likely due to a robust elevation of SNPH expression in mature neurons. SNPH specifically targets mitochondria within axons but not in dendrites. Given that dendritic mitochondria are largely stationary, potential anchors for dendritic mitochondria remain to be identified.
Figure 1. Developmental regulation of SNPH-mediated anchoring of axonal mitochondria.
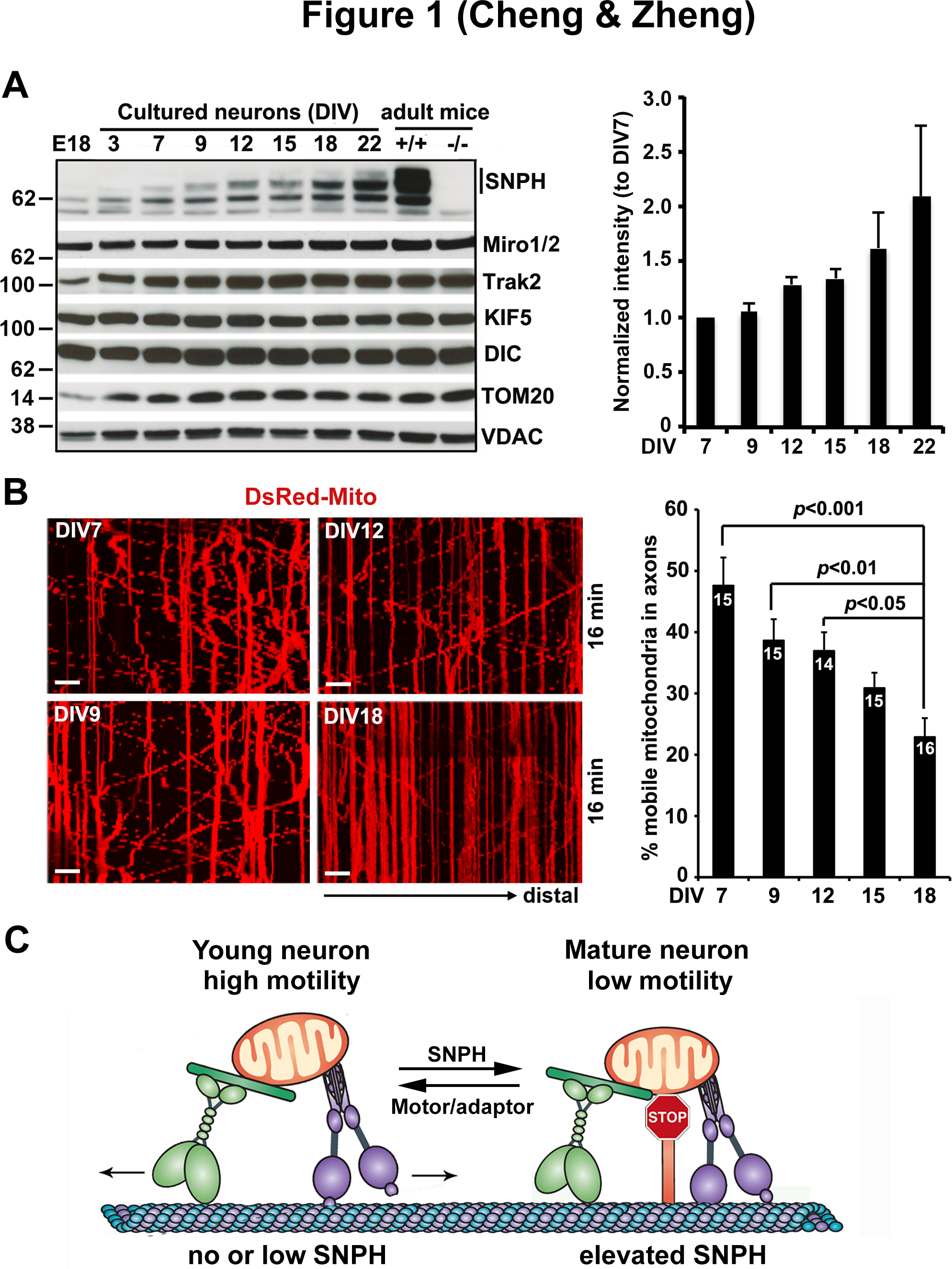
(A) Representative immunoblots and quantitative analysis showing a progressive increase in SNPH expression with neuronal development and maturation. Note that while SNPH expression is abundant in adult mouse brains, it is hardly detectable in E18 brains and in cultured neurons before DIV3, becomes readily detectable in cultured neurons after DIV9, and peaks at DIV22. This expression pattern is specific to SNPH and does not apply to mitochondrial motor-adaptor proteins (KIF5, DIC, Miro1/2, and Trak2) or outer mitochondrial membrane proteins (TOM20 and VDAC).
(B) Representative kymographs and quantification from time-lapse live imaging of mouse cortical neuron axons showing a progressive decline of mitochondrial motility with maturation from DIV7 to DIV18. In kymographs, vertical lines represent stationary mitochondria; oblique lines or curves to the right indicate anterograde transport toward distal terminals, to the left denote retrograde transport toward the cell body. Scale bars:10 μm.
(C) Model illustration of mechanistic link between mitochondria motility and SNPH expression with neuron maturation. In young neurons where SNPH expression is not detectable or extremely low, axonal mitochondria display robust motility; whereas in mature neurons with high SNPH expression, the majority of axonal mitochondria remain stationary.
Data in (A) and (B) are adapted from Zhou, B., Yu, P., Lin, M.Y., Sun, T., Chen, Y., Sheng, Z.H., 2016. Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. The Journal of Cell Biology 214, 103–119.
When the snph gene is deleted in neurons, axonal mitochondria remain highly motile. The majority (78%) of axonal mitochondria in snph KO hippocampal neurons exhibit high motility even after DIV14; similarly, 71% of mitochondria in sciatic nerves of 2-month old adult snph KO mice remain motile in axonal bundles (Zhou et al., 2016). Considering its specific role in axonal mitochondrial anchoring, we propose that SNPH is a major regulator contributing to the aging-associated decline in axonal mitochondrial motility. In developing neurons where SNPH expression is absent or minimal, axonal mitochondria display a robust motility and thus can be actively delivered to distal tips to supply energy for axonal growth and synaptogenesis. This is in stark contrast to mature neurons with elevated SNPH expression, in which the majority of axonal mitochondria are captured at sites of high energy consumption, such as synapses and axon branch sites (Figure 1C). This notion was further supported by work on demyelinated axons, a major cause of neurological disability in diseases of primary oligodendrocyte dysfunction. SNPH-mediated anchoring is required for increased mitochondria volume in demyelinated axons, which is thought to protect against axonal degeneration (Ohno et al., 2014).
In another study using Drosophila as a model system, Vagnoni and Bullock (2018) observed a protein reduction in both the catalytic subunit of protein kinase A and its direct substrate kinesin-1 (KIF5) in the wing of aged flies compared to young flies. Overexpressing kinesin-1 heavy chain (Khc) is sufficient to boost mitochondrial transport in wing neurons of aged flies in both directions (Vagnoni and Bullock, 2018). Drosophila display balanced stationary and motile mitochondria pools in axons similar to those in mammalian neurons but lack a known SNPH homolog, suggesting alternative docking mechanisms that also mediate mitochondrial anchoring in Drosophila (Vignoni and Bullock, 2018). Thus, coordinated regulation of mitochondrial trafficking and anchoring machineries is crucial to ensure that metabolically active areas are adequately supplied with ATP during neuronal development, maturation, and aging.
Altered Mitochondrial Trafficking and Anchoring in Neurodegeneration
Due to their extended axons and long lifespan, neurons face the unique challenge of maintaining mitochondrial positioning and thus local energy homeostasis in distal axons and synapses, where energy demand is high. Deficiency in mitochondrial trafficking and anchoring could cause inadequate distribution of ATP and abnormal Ca2+ buffering within these highly metabolic regions. On the other hand, damaged mitochondria are accumulated in distal regions of aging neurons due to inefficient removal, which can have severe consequences: damaged mitochondria not only supply less ATP, causing local energy deficits, but also release toxic ROS and apoptotic factors (Alvarez et al., 2008). Therefore, it is not surprising that defects in axonal mitochondrial trafficking and positioning have been associated with several major neurodegenerative diseases (Schon and Przedborski, 2011; Misgeld and Schwarz, 2017; Sheng and Cai 2012; De Vos et al., 2007; Xie et al., 2015). For example, using intravital imaging techniques, Takihara et al. revealed a dramatic reduction in the number of moving mitochondria in retinal ganglion cell axons of aging mice as compared to young mice when insulted with experimental glaucoma, suggesting increased vulnerability of mitochondria transport in aged neurons exposed to glaucoma (Takihara et al., 2015).
Chronic mitochondrial stress is a central problem associated with aging-linked neurodegenerative diseases. Early removal of stressed mitochondria from distal axons and synapses constitutes a critical step of mitochondrial quality control. While the PINK1/Parkin pathway has been well characterized as a mechanism of mitochondrial quality control, mature neurons often show delayed Parkin-mediated mitophagy in response to mitochondrial depolarization (Cai et al., 2012; Van Laar et al. 2011). This finding raises the question of whether mitophagy is the first line of quality control in mature neurons where SNPH is highly abundant and the majority of axonal mitochondria remain anchored in distal axons and synapses. Although acute mitochondrial depolarization with a high-dose of Antimycin A1 (AA, 40 μM), a respiratory complex III inhibitor, quickly induces Parkin-mediated mitophagy in axons (Ashrafi, et al., 2014), it was not known whether axonal mitophagy could be induced by mild and chronic mitochondrial stress in distal axons, a condition more relevant to chronic mitochondrial dysfunction associated with major neurodegenerative diseases. In our recent study, we sought to address whether mature neurons have an intrinsic mechanism to turn off SNPH-mediated anchoring in order to remobilize stressed mitochondria in distal axons under chronic stress conditions (Lin et al., 2017). We recapitulated chronic mitochondrial dysfunction by applying a very mild stress using 5 nM AA, a 1000-times lower dosage than reported in the literature. This mild treatment induces a slow and reversible mitochondrial stress. Surprisingly, axonal mitochondria respond to this mild stress by biased directional transport, favoring the removal of stressed mitochondria out of distal axons. We further demonstrated that this transport regulation is triggered by the bulk release of SNPH in the form of cargo vesicles from stressed mitochondria. Such a dynamic pinch out of SNPH vesicles from mildly stressed mitochondria was captured at ultrastructural levels (immuno-TEM) (Figure 2A) and by time-lapse STED super-resolution imaging (Figure 2B). Upon release, SNPH cargo vesicles undergo retrograde transport en route to the endosome-lysosome pathway for degradation. Inhibition of mitochondrial motility by overexpression of SNPH would largely preclude stressed mitochondria from recovery. Thus, SNPH may serve as a sensitive sensor of mitochondrial stress. Interestingly, the generation of SNPH cargo vesicles is robustly increased in the axons of spinal ventral root motor neurons during the early asymptomatic stages of familial amyotrophic lateral sclerosis (fALS)-linked SOD1G93A mice and in the early pathological stages of Alzheimer’s disease (AD)-related hAPP mutant neurons. Progressive mitochondrial damage depletes SNPH after disease onset and in the late stages in both fALS- and AD-linked mouse brains and postmortem human brains from AD patients. Thus, the generation of SNPH cargo vesicles ensures that axonal mitochondria can respond to chronic pathological stress, such that dysfunctional mitochondria anchored in distal axons by SNPH can be remobilized and transported to the soma for repair (recovery) or degradation, where mature lysosomes are highly enriched (Figure 2C). Since this SNPH-mediated protective pathway happens far earlier than the onset of these diseases and is independent of Pink1/Parkin-mediated mitophagy, we consider it as the first line of mitochondrial surveillance during early stages of fALS and AD. Thus, early intervention of SNPH-mediated mitochondrial anchoring at asymptomatic stages will potentially delay the clinical onset or progression of the pathology.
Figure 2. Stress-induced release of SNPH remobilizes anchored mitochondria in the early stages of neurodegenerative diseases.
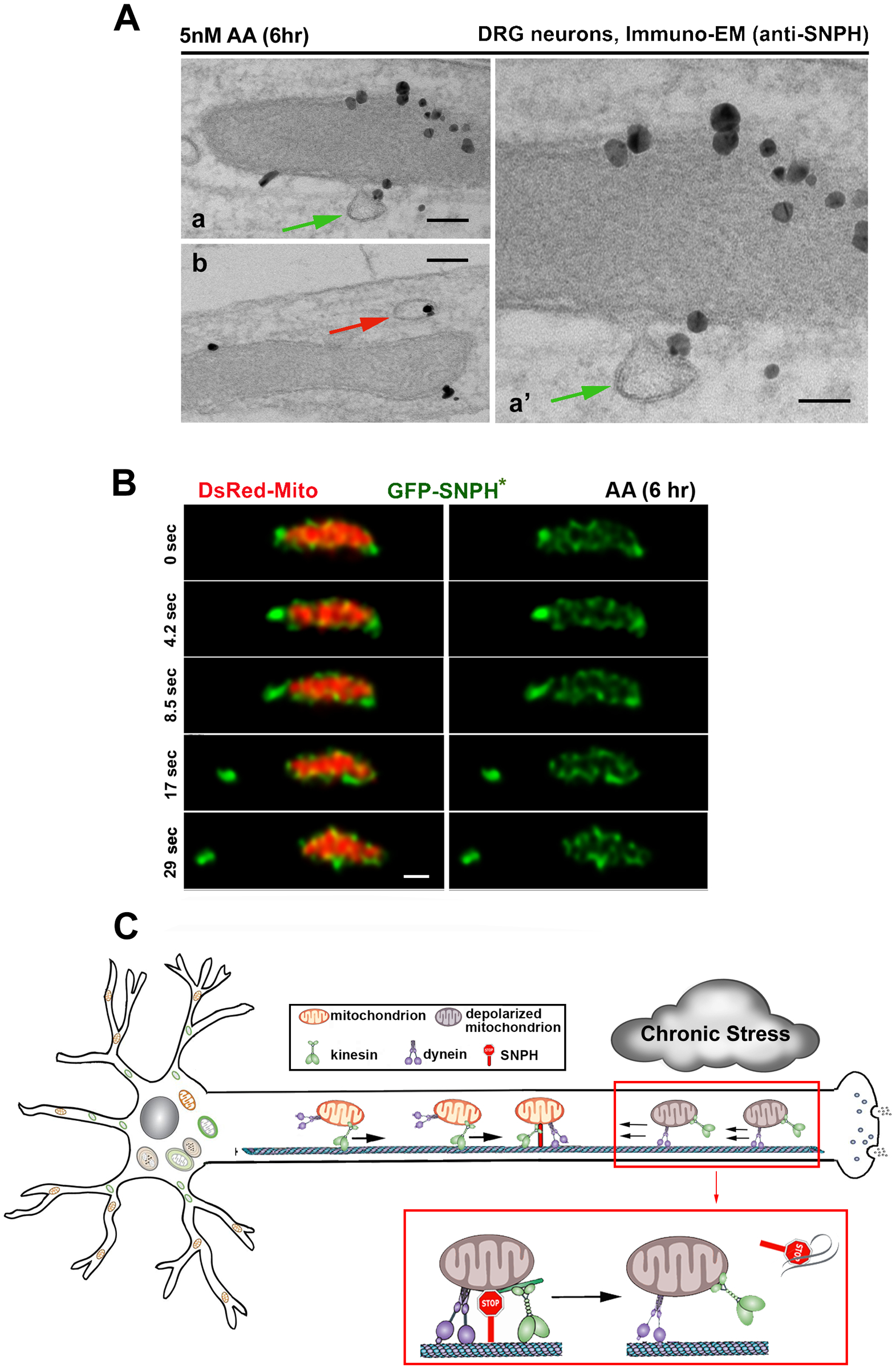
(A) Immuno-EM graphs showing the budding process of SNPH cargo vesicles from stressed mitochondria in DRG neuron axons following mild and chronic stress (5 nM Antimycin A1 for 6 hrs). Endogenous SNPH is labeled with an anti-SNPH antibody and nanogold conjugates. Green arrow denotes a budding vesicle with its limiting membrane continuous with the OMM. Red arrows point to SNPH cargo in close proximity to mitochondria. Scale bars: 100 nm (Aa, Ab), 50 nm (Aa’).
(B) Time-lapse STED super-resolution images showing a SNPH cargo vesicle (green, GFP-SNPH*) pinching out from an axonal mitochondrion (red, DsRed-Mito) in DRG neurons after mild and chronic stress (5 nM Antimycin A1 for 6 hrs). Scale bar: 500 nm.
(C) Model illustration showing an intrinsic mechanism remobilizing dysfunctional mitochondria that are anchored in the distal axon of a mature neuron by selectively removing the anchoring protein SNPH in response to mild and chronic mitochondrial stress. The generation of SNPH cargo vesicles results in the selective regulation of biased mitochondrial transport, favoring the removal of stressed mitochondria out of distal axons in a retrograde direction and delivery of healthy ones to distal terminals anterogradely. Thus, the early removal of those anchored stressed mitochondria constitutes an important pathway to maintain mitochondrial integrity in distal axons. This mechanism is likely the first line of mitochondrial surveillance during early stages of diseases.
Data in (A) and (B) are adapted from Lin, M.Y., Cheng, X.T., Tammineni, P, Xie, Y, Zhou, B, Sheng, Z.H., 2017. Releasing syntaphilin removes stressed mitochondria from axons independent of mitophagy under pathophysiological conditions. Neuron 94 (3), 595–610. Model Illustration (C) is adapted from Lin, M.Y., Cheng, X.T., Xie, Y, Zhou, B, Sheng, Z.H., 2017. Removing dysfunctional mitochondria from axons independent of mitophagy under pathophysiological conditions. Autophagy 13 (10), 1792–1794.
Our study is further supported by two independent reports. Zheng et al. investigated axonal mitochondrial quality control in an ischemic injury model and found that depleting SNPH expression in mature neurons is critical to eliminate damaged mitochondria in distal axons through enhanced retrograde transport towards the soma. Conversely, overexpressing SNPH anchored the damaged mitochondria in distal axons and blocked neuronal mitophagy in the soma, thus aggravating axonal degeneration following ischemic neuronal injury (Zheng et al., 2019). This study confirmed the critical role of SNPH-mediated anchoring in mature neurons and established that turning off SNPH anchoring benefits elimination of damaged mitochondria in distal axons. In a second study, Joshi et al. reported that deleting SNPH produces striking benefits in the Shiverer mouse, a demyelinating model of early-phase multiple sclerosis (MS), by prolonging survival, reducing cerebellar damage, suppressing oxidative stress, and improving mitochondrial health in demyelinated axons (Joshi et al., 2015).
It is thought that increased mitochondrial transport might aid the delivery of healthy mitochondria to distal axons and/or the removal of damaged mitochondria from distal synapses. However, when mitochondria are globally damaged during rapid-onset pathology such as in motor neurons of fALS-linked hSOD1G93A mutant mice (Cozzolino et al., 2013), is simply enhancing their transport sufficient to benefit mitochondrial quality control in distal axons and synapses in this mouse model? Altered transport of axonal mitochondria and the aberrant accumulation of damaged mitochondria at distal axons has been consistently reported in motor neurons of hSOD1G93A mice (Bilsland et al., 2010; De Vos et al., 2007; Magrané and Manfredi, 2009). To address this issue, we previously examined whether increasing axonal mitochondrial motility has any impact on the pathogenesis of the fALS-linked mouse model by crossing hSOD1G93A and snph−/− mice (Zhu and Sheng, 2011). Surprisingly, despite a more than 2-fold increase in total axonal mitochondrial motility in the crossed mice, we did not observe any clinical or histological difference in the disease course. Both hSOD1G93A and crossed hSOD1G93A/snph−/− mice exhibited a similar body weight loss pattern accompanied by similar deterioration of motor function beginning at approximately 17 weeks. Both mouse strains exhibited a similar lifespan of 152–154 days. Furthermore, both SOD1G93A and SOD1G93A/snph−/− mice showed similar ALS-like disease histopathology that included 55% motor neuron loss and significant gliosis at the disease end stage. This study suggests that the impairment of mitochondrial transport seen in hSOD1G93A mice is a secondary effect and may play a minor role in the rapid-onset pathology in this fALS-linked mouse model. Our findings are supported by an ex vivo study showing that deficits in mitochondrial transport are not sufficient to cause axon degeneration in mutant hSOD1 models (Marinkovic et al., 2012).
These observations raise a fundamental question: Do autophagy-lysosomal deficits impair the elimination of damaged mitochondria in distal axons, thus augmenting mitochondria pathology in spinal motor neurons of fALS-linked mice? We recently addressed this issue by providing in vitro and in vivo evidence that progressive lysosomal deficits and impaired autophagic clearance are the early fALS-linked pathological events that impair the degradation of damaged mitochondria from spinal motor neurons (Xie et al., 2015). Elucidation of this early pathological mechanism is broadly relevant because defective transport, lysosomal deficits, autophagy stress, and mitochondrial pathology are all associated with major neurodegenerative diseases. When mitochondria are globally dysfunctional in hSOD1G93A mutant mice, simply enhancing their transport by deleting SNPH (Zhu and Sheng, 2011) or turning off SNPH-mediated anchoring (Lin et al., 2017) does not benefit mitochondrial quality control in distal axons and synapses at late stages of the disease. Therefore, increasing mitochondrial motility combined with enhanced clearance capacity of damaged mitochondria may be a potential therapeutic strategy for ALS and perhaps other rapid-onset neurodegenerative diseases.
Regulation of Mitochondrial Trafficking and Anchoring in Axon Regeneration
Selective regulation of mitochondrial trafficking and anchoring is not only key to axonal degeneration, but also to nerve regeneration after injury and disease. While young neurons possess robust axon growth, mature neurons typically fail to regrow after injury; mature CNS axons lose their regrowth capacity due to an intrinsic decline in permissive conditions for regeneration. To survive an injury, neuronal membranes must be quickly re-sealed, cytoskeletal structures rearranged, and regrowth programs initiated in order to reform active growth cones (Bradke et al., 2012). All these regrowth events require high levels of energy consumption. Thus, ATP supplied by local mitochondria in the injured axons is essential for supporting nerve regeneration. In mature CNS axons and synapses, SNPH expression is highly abundant and thus the majority of mitochondria remain stationary (Sun et al., 2013; Lewis et al., 2016; Smit-Rigter et al., 2016). However, axonal injury is a strong stressor that induces local mitochondrial damage (O’Donnell et al., 2013; Cavallucci et al., 2014). These studies raise the attractive hypothesis that increased expression of SNPH and progressive decline of mitochondrial motility in mature CNS axons contributes to their low regeneration capacity. If this hypothesis is correct, then enhancing mitochondrial transport in mature neurons would remove locally damaged mitochondria and deliver healthy ones into injured axons to meet increased energy requirements during regeneration. To test this hypothesis, we recently addressed two fundamental questions: (1) Do mature neurons maintain an effective capacity to recruit healthy mitochondria to injured axons? (2) If this function declines with neuron maturation, does enhancing mitochondrial transport enable mature neurons to regain regenerative capacity? Snph KO mice provide an ideal model to investigate how enhanced mitochondrial transport influences regenerative capacity (Zhou et al., 2016). We conducted live imaging of mitochondrial transport and integrity, as well as dynamic ATP levels, in injured axons cultured in a microfluidic chamber system. We showed that axon injury is an acute stress signal that rapidly depolarizes mitochondria in the vicinity of the injured site. Enhancing mitochondrial transport in snph KO axons helps remove dysfunctional mitochondria and replenishes healthy ones to injured axons, thus reversing energy deficits. We further demonstrated that enhancing axonal mitochondrial trafficking facilitates axonal regeneration capacity in mature cortical neurons and dorsal root ganglion neurons after axotomy in culture, and in vivo regeneration of sciatic nerves after crush injury in adult mice. Conversely, inhibition of mitochondrial motility by overexpression of SNPH largely abolishes axon regeneration. Our study provides new mechanistic insights into how elevated SNPH expression in mature neurons contributes to regeneration failure.
Two in vivo studies in mouse retinal ganglion neurons (Cartoni et al., 2016) and C. elegans GABA motor neurons (Han et al., 2016) further support the notion that boosting mitochondrial transport facilitates regeneration after injury. Cartoni et al. found that Armcx1, a mitochondria-localized protein, is critical for maintaining the high regenerative capacity of retinal ganglion neurons through enhancing axonal mitochondrial transport in SOCS3 and PTEN double KO mice, an established mouse model with robust axon regeneration (Cartoni et al., 2016). Han et al. used C. elegans as a model organism and revealed that increased mitochondria density in injured axons of GABA motor neurons is correlated with higher regenerative capacity. This increased translocation of mitochondria after neuronal injury is partly due to the activation of MAP kinase dual-leucine zipper kinase 1 (DLK-1) (Han et al., 2016).
Several earlier in vivo studies consistently support the notion that mitochondrial transport to and distribution at injured axons play critical roles in nerve regeneration after injury. Misgeld et al examined the proximal segments of transected intercostal nerves and found that the injured nerve has a vigorous growth response: forming a complex tangle of axonal sprouts populated by mitochondria. This re-distribution of mitochondria at the growth cone is contributed by a sustained increase of anterograde transport; this increased mitochondrial transport following transection occurs well before axonal regrowth is initiated and can potentially serve as a readout of “injury response” (Misgeld et al., 2007). In C. elegans mutant ric-7 with impaired mitochondrial transport to distal axons, injured axons degenerate rapidly; such degeneration can be suppressed by forcing mitochondria into the axons (Rawson et al., 2014). A peripheral injury in CNS axons induces a global increase in the axonal transport of organelles including mitochondria, lysosomes, and other axonal building blocks in order to support axon regeneration (Mar et al., 2014). Therefore, activating an intrinsic “regrowth program” in mature neurons requires the coordinated recovery of energy supply via enhanced mitochondrial transport and recruitment. Development of therapeutic molecules to increase mitochondrial transport and/or local energetic metabolism will be an attractive strategy for facilitating nerve regeneration after injury and possibly in disease (Sheng 2017; Kaasik 2016).
Acknowledgments
The authors apologize to those colleagues whose work could not be cited owing to space limitations. We thank members of the Sheng lab for constructive discussion; K. A. Chamberlain and J. C. Roney for critical reading and editing. This work was supported by the Intramural Research Program of NINDS, NIH ZIA NS003029, and ZIA NS002946 (Z-H. Sheng).
Abbreviations:
- AA
antimycin A1
- AD
Alzheimer’s disease
- ALS
amyotrophic lateral sclerosis
- ATP
adenosine triphosphate
- DIV
days in vitro
- Δψm
mitochondrial membrane potential
- KO
knockout; MT: microtubule
- NGF
nerve growth factor
- OMM
outer mitochondrial membrane
- SNPH
syntaphilin
- WT
wild type
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare no competing financial interests.
References
- Alvarez S, Moldovan M, Krarup C, 2008. Acute energy restriction triggers Wallerian degeneration in mouse. Exp Neurol 212, 166–178. [DOI] [PubMed] [Google Scholar]
- Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL, 2014. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. The Journal of Cell Biology 206, 655–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Laughlin SB, 2001. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21, 1133–1145. [DOI] [PubMed] [Google Scholar]
- Billups B, Forsythe ID, 2002. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. The Journal of Neuroscience 22, 5840–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G, 2010. Deficits in axonal transport precede ALS symptoms in vivo. Proceedings of the National Academy of Sciences of the United States of America 107, 20523–20528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock FJ, Tait SWG, 2019. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. [DOI] [PubMed] [Google Scholar]
- Bradke F, Fawcett JW, Spira ME, 2012. Assembly of a new growth cone after axotomy: the precursor to axon regeneration. Nat Rev Neurosci 13, 183–193. [DOI] [PubMed] [Google Scholar]
- Brickley K, Smith MJ, Beck M, Stephenson FA, 2005. GRIF-1 and OIP106, members of a novel gene family of coiled-coil domain proteins: association in vivo and in vitro with kinesin. The Journal of Biological Chemistry 280, 14723–14732. [DOI] [PubMed] [Google Scholar]
- Brickley K, Stephenson FA, 2011. Trafficking kinesin protein (TRAK)-mediated transport of mitochondria in axons of hippocampal neurons. The Journal of Biological Chemistry 286, 18079–18092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Gerwin C, Sheng ZH, 2005. Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. The Journal of Cell Biology 170, 959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Zakaria HM, Simone A, Sheng ZH, 2012. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Current Biology 22, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartoni R, Norsworthy MW, Bei F, Wang C, Li S, Zhang Y, Gabel CV, Schwarz TL, He Z, 2016. The Mammalian-Specific Protein Armcx1 Regulates Mitochondrial Transport during Axon Regeneration. Neuron 92, 1294–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallucci V, Bisicchia E, Cencioni MT, Ferri A, Latini L, Nobili A, Biamonte F, Nazio F, Fanelli F, Moreno S, Molinari M, Viscomi MT, D’Amelio M, 2014. Acute focal brain damage alters mitochondrial dynamics and autophagy in axotomized neurons. Cell Death & Disease 5, e1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chada SR, Hollenbeck PJ, 2003. Mitochondrial movement and positioning in axons: the role of growth factor signaling. J Exp Biol 206, 1985–1992. [DOI] [PubMed] [Google Scholar]
- Chada SR, Hollenbeck PJ, 2004. Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Current Biology 14, 1272–1276. [DOI] [PubMed] [Google Scholar]
- Chamberlain KA, Sheng ZH, 2019. Mechanisms for the maintenance and regulation of axonal energy supply. Journal of Neuroscience Research 97, 897–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DT, Reynolds IJ, 2006. Mitochondrial trafficking and morphology in healthy and injured neurons. Prog Neurobiol 80, 241–268. [DOI] [PubMed] [Google Scholar]
- Chen H, Chan DC, 2009. Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Human Molecular Genetics 18, R169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Sheng ZH, 2013. Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. The Journal of Cell Biology 202, 351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani LA, Goda Y, 2008. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci 9, 344–356. [DOI] [PubMed] [Google Scholar]
- Courchet J, Lewis TL Jr., Lee S, Courchet V, Liou DY, Aizawa S, Polleux F, 2013. Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell 153, 1510–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzolino M, Ferri A, Valle C, Carri MT, 2013. Mitochondria and ALS: implications from novel genes and pathways. Molecular and cellular neurosciences 55, 44–49. [DOI] [PubMed] [Google Scholar]
- Das S, Boczan J, Gerwin C, Zald PB, Sheng ZH, 2003. Regional and developmental regulation of syntaphilin expression in the brain: a candidate molecular element of synaptic functional differentiation. Brain Res Mol Brain Res 116, 38–49. [DOI] [PubMed] [Google Scholar]
- David G, Barrett EF, 2003. Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J Physiol 548, 425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine MJ, Kittler JT, 2018. Mitochondria at the neuronal presynapse in health and disease. Nat Rev Neurosci 19, 63–80. [DOI] [PubMed] [Google Scholar]
- De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, Brownlees J, Ackerley S, Shaw PJ, McLoughlin DM, Shaw CE, Leigh PN, Miller CCJ, Grierson AJ, 2007. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Human Molecular Genetics 16, 2720–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farfel-Becker T, Roney JC, Cheng XT, Li S, Cuddy SR, Sheng ZH, 2019. Neuronal Soma-Derived Degradative Lysosomes Are Continuously Delivered to Distal Axons to Maintain Local Degradation Capacity. Cell Reports 28, 51–64 e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly A, Tang Y, Wang L, Ladt K, Loi J, Dargent B, Leterrier C, Roy S, 2015. A dynamic formin-dependent deep F-actin network in axons. The Journal of cell biology 210, 401–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glater EE, Megeath LJ, Stowers RS, Schwarz TL, 2006. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. The Journal of Cell Biology 173, 545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedes-Dias P, Holzbaur ELF, 2019. Axonal transport: Driving synaptic function. Science 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, Zinsmaier KE, 2005. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 47, 379–393. [DOI] [PubMed] [Google Scholar]
- Gutnick A, Banghart MR, West ER, Schwarz TL, 2019. The light-sensitive dimerizer zapalog reveals distinct modes of immobilization for axonal mitochondria. Nature cell biology 21, 768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SM, Baig HS, Hammarlund M, 2016. Mitochondria Localize to Injured Axons to Support Regeneration. Neuron 92, 1308–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Weinberg RJ, 2012. Ultrastructure of synapses in the mammalian brain. Cold Spring Harbor Perspectives in Biology 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, Tanaka Y, 2010. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68, 610–638. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Takemura R, 2005. Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci 6, 201–214. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM, 2005. The axonal transport of mitochondria. Journal of Cell Science 118, 5411–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi DC, Zhang CL, Lin TM, Gusain A, Harris MG, Tree E, Yin Y, Wu C, Sheng ZH, Dempsey RJ, Fabry Z, Chiu SY, 2015. Deletion of mitochondrial anchoring protects dysmyelinating shiverer: implications for progressive MS. The Journal of Neuroscience 35, 5293–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaasik A, 2016. Mitochondrial Mobility and Neuronal Recovery. The New England Journal of Medicine 375, 1295–1296. [DOI] [PubMed] [Google Scholar]
- Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, Sheng ZH, 2008. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis TL Jr., Turi GF, Kwon SK, Losonczy A, Polleux F, 2016. Progressive Decrease of Mitochondrial Motility during Maturation of Cortical Axons In Vitro and In Vivo. Current Biology 26, 2602–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MY, Cheng XT, Tammineni P, Xie YX, Zhou B, Cai Q, Sheng ZH, 2017. Releasing Syntaphilin Removes Stressed Mitochondria from Axons Independent of Mitophagy under Pathophysiological Conditions. Neuron 94, 595–610 e596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Domenech G, Covill-Cooke C, Ivankovic D, Halff EF, Sheehan DF, Norkett R, Birsa N, Kittler JT, 2018. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. The EMBO journal 37, 321–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill AF, Brickley K, Stephenson FA, Kittler JT, 2009a. GTPase dependent recruitment of Grif-1 by Miro1 regulates mitochondrial trafficking in hippocampal neurons. Molecular and Cellular Neurosciences 40, 301–312. [DOI] [PubMed] [Google Scholar]
- MacAskill AF, Kittler JT, 2010. Control of mitochondrial transport and localization in neurons. Trends in Cell Biology 20, 102–112. [DOI] [PubMed] [Google Scholar]
- Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT, 2009b. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61, 541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane J, Manfredi G, 2009. Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis. Antioxidants & redox signaling 11, 1615–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal A, Pinter K, Drerup CM, 2018. Analyzing Neuronal Mitochondria in vivo Using Fluorescent Reporters in Zebrafish. Front Cell Dev Biol 6, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mar FM, Simoes AR, Leite S, Morgado MM, Santos TE, Rodrigo IS, Teixeira CA, Misgeld T, Sousa MM, 2014. CNS axons globally increase axonal transport after peripheral conditioning. The Journal of Neuroscience 34, 5965–5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinkovic P, Reuter MS, Brill MS, Godinho L, Kerschensteiner M, Misgeld T, 2012. Axonal transport deficits and degeneration can evolve independently in mouse models of amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the United States of America 109, 4296–4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, Kaneko T, 2009. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. The Journal of Neuroscience 29, 444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, Lichtman JW, 2007. Imaging axonal transport of mitochondria in vivo. Nature Methods 4, 559–561. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Schwarz TL, 2017. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 96, 651–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ, 1993. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. Journal of Cell Science 104 (Pt 3), 917–927. [DOI] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ, 1995. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. The Journal of cell biology 131, 1315–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsci NS, Hall DH, Driscoll M, Sheng ZH, 2016. Age-Related Phasic Patterns of Mitochondrial Maintenance in Adult Caenorhabditis elegans Neurons. The Journal of Neuroscience 36, 1373–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell KC, Vargas ME, Sagasti A, 2013. WldS and PGC-1alpha regulate mitochondrial transport and oxidation state after axonal injury. The Journal of Neuroscience 33, 14778–14790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno N, Kidd GJ, Mahad D, Kiryu-Seo S, Avishai A, Komuro H, Trapp BD, 2011. Myelination and axonal electrical activity modulate the distribution and motility of mitochondria at CNS nodes of Ranvier. The Journal of Neuroscience 31, 7249–7258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno N, Chiang H, Mahad DJ, Kidd GJ, Liu L, et al. 2014. Mitochondrial immobilization mediated by syntaphilin facilitates survival of demyelinated axons. Proc Natl Acad Sci U S A 111, 9953–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plucinska G, Paquet D, Hruscha A, Godinho L, Haass C, Schmid B, Misgeld T, 2012. In vivo imaging of disease-related mitochondrial dynamics in a vertebrate model system. The Journal of Neuroscience 32, 16203–16212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintero OA, DiVito MM, Adikes RC, Kortan MB, Case LB, Lier AJ, Panaretos NS, Slater SQ, Rengarajan M, Feliu M, Cheney RE, 2009. Human Myo19 is a novel myosin that associates with mitochondria. Current biology : CB 19, 2008–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson RL, Yam L, Weimer RM, Bend EG, Hartwieg E, Horvitz HR, Clark SG, Jorgensen EM, 2014. Axons degenerate in the absence of mitochondria in C. elegans. Current Biology 24, 760–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo GJ, Louie K, Wellington A, Macleod GT, Hu F, Panchumarthi S, Zinsmaier KE, 2009. Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. The Journal of Neuroscience 29, 5443–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthel G, Hollenbeck PJ, 2003. Response of mitochondrial traffic to axon determination and differential branch growth. The Journal of Neuroscience 23, 8618–8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajic M, Mastrolia V, Lee CY, Trigo D, Sadeghian M, Mosley AJ, Gregson NA, Duchen MR, Smith KJ, 2013. Impulse conduction increases mitochondrial transport in adult mammalian peripheral nerves in vivo. PLoS Biology 11, e1001754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnoczky G, 2008. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proceedings of the National Academy of Sciences of the United States of America 105, 20728–20733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton WM, Hollenbeck PJ, 2012. The axonal transport of mitochondria. Journal of Cell Science 125, 2095–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon EA, Przedborski S, 2011. Mitochondria: the next (neurode)generation. Neuron 70, 1033–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH, 2017. The Interplay of Axonal Energy Homeostasis and Mitochondrial Trafficking and Anchoring. Trends in Cell Biology 27, 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH, Cai Q, 2012. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci 13, 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HL, Bourne JN, Cao G, Chirillo MA, Ostroff LE, Watson DJ, Harris KM, 2016. Mitochondrial support of persistent presynaptic vesicle mobilization with age-dependent synaptic growth after LTP. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit-Rigter L, Rajendran R, Silva CA, Spierenburg L, Groeneweg F, Ruimschotel EM, van Versendaal D, van der Togt C, Eysel UT, Heimel JA, Lohmann C, Levelt CN, 2016. Mitochondrial Dynamics in Visual Cortex Are Limited In Vivo and Not Affected by Axonal Structural Plasticity. Current Biology 26, 2609–2616. [DOI] [PubMed] [Google Scholar]
- Spillane M, Ketschek A, Merianda TT, Twiss JL, Gallo G, 2013. Mitochondria coordinate sites of axon branching through localized intra-axonal protein synthesis. Cell Reports 5, 1564–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowers RS, Megeath LJ, Gorska-Andrzejak J, Meinertzhagen IA, Schwarz TL, 2002. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron 36, 1063–1077. [DOI] [PubMed] [Google Scholar]
- Sun T, Qiao H, Pan PY, Chen Y, Sheng ZH, 2013. Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Reports 4, 413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takihara Y, Inatani M, Eto K, Inoue T, Kreymerman A, Miyake S, Ueno S, Nagaya M, Nakanishi A, Iwao K, Takamura Y, Sakamoto H, Satoh K, Kondo M, Sakamoto T, Goldberg JL, Nabekura J, Tanihara H, 2015. In vivo imaging of axonal transport of mitochondria in the diseased and aged mammalian CNS. Proceedings of the National Academy of Sciences of the United States of America 112, 10515–10520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Zucker RS, 1997. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron 18, 483–491. [DOI] [PubMed] [Google Scholar]
- Tao K, Matsuki N, Koyama R, 2014. AMP-activated protein kinase mediates activity-dependent axon branching by recruiting mitochondria to axon. Dev Neurobiol 74, 557–573. [DOI] [PubMed] [Google Scholar]
- Vagnoni A, Bullock SL, 2018. A cAMP/PKA/Kinesin-1 Axis Promotes the Axonal Transport of Mitochondria in Aging Drosophila Neurons. Current Biology 28, 1265–1272 e1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bergeijk P, Adrian M, Hoogenraad CC, Kapitein LC, 2015. Optogenetic control of organelle transport and positioning. Nature 518, 111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laar VS, Arnold B, Cassady SJ, Chu CT, Burton EA, Berman SB, 2011. Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Human Molecular Genetics 20, 927–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Spronsen M, Mikhaylova M, Lipka J, Schlager MA, van den Heuvel DJ, Kuijpers M, Wulf PS, Keijzer N, Demmers J, Kapitein LC, Jaarsma D, Gerritsen HC, Akhmanova A, Hoogenraad CC, 2013. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron 77, 485–502. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ, 2005. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47, 365–378. [DOI] [PubMed] [Google Scholar]
- Wang X, Schwarz TL, 2009. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell 136, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werth JL, Thayer SA, 1994. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. The Journal of Neuroscience 14, 348–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Williams J, Nathans J, 2012. Morphologic diversity of cutaneous sensory afferents revealed by genetically directed sparse labeling. Elife 1, e00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Zhou B, Lin MY, Sheng ZH, 2015. Progressive endolysosomal deficits impair autophagic clearance beginning at early asymptomatic stages in fALS mice. Autophagy 11, 1934–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Zhong G, Zhuang X, 2013. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 339, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CL, Ho PL, Kintner DB, Sun D, Chiu SY, 2010. Activity-dependent regulation of mitochondrial motility by calcium and Na/K-ATPase at nodes of Ranvier of myelinated nerves. The Journal of Neuroscience 30, 3555–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Zhang X, Wu X, Jiang L, Ahsan A, Ma S, Xiao Z, Han F, Qin ZH, Hu W, Chen Z, 2019. Somatic autophagy of axonal mitochondria in ischemic neurons. The Journal of Cell Biology 218, 1891–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Yu P, Lin MY, Sun T, Chen Y, Sheng ZH, 2016. Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. The Journal of Cell Biology 214, 103–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XH, Qiao H, Du F, Xiong Q, Liu X, Zhang X, Ugurbil K, Chen W, 2012. Quantitative imaging of energy expenditure in human brain. Neuroimage 60, 2107–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YB, Sheng ZH, 2011. Increased Axonal Mitochondrial Mobility Does Not Slow Amyotrophic Lateral Sclerosis (ALS)-like Disease in Mutant SOD1 Mice. The Journal of Biological Chemistry 286(26): 23432–40. [DOI] [PMC free article] [PubMed] [Google Scholar]