

Abstract
Molecular glues are an intriguing therapeutic modality that harness small-molecules to induce interactions between proteins that typically do not interact. However, such molecules are rare and have been discovered fortuitously, thus limiting their potential as a general strategy for therapeutic intervention. We postulated that natural products bearing one or more electrophilic sites may be an unexplored source of new molecular glues, potentially acting through multi-covalent attachment. Using chemoproteomic platforms, we show that members of the manumycin family of polyketides, which bear multiple potentially reactive sites, target C374 of the putative E3 ligase UBR7 in breast cancer cells and engage in molecular glue interactions with the neo-substrate tumor-suppressor TP53, leading to p53 transcriptional activation and cell death. Our results reveal a novel anti-cancer mechanism of this natural product family and highlight the potential for combining chemoproteomics and multi-covalent natural products for the discovery of new molecular glues.
Keywords: manumycin, asukamycin, molecular glues, E3 ligases, chemoproteomics, activity-based protein profiling, natural products, covalent ligands, undruggable, protein-protein interaction (PPI)
Introduction
The challenge of tackling the undruggable proteome has inspired the development of innovative technologies that enable functional targeting of biomolecules with new therapeutic modalities. Examples of some of these small molecule-based technologies include chemoproteomics-enabled covalent ligand screening and DNA-encoded libraries for discovering ligands against undruggable proteins, proteolysis-targeting chimeras (PROTACs) or degraders for targeted ubiquitin-proteasome system-dependent degradation of proteins, and “molecular glues” for small-molecule induced formation of protein interfaces that confer enhanced, inhibited, or new protein function 1–6. Molecular glue-based degraders, as exemplified by the IMiD-family of immunomodulatory drugs including thalidomide, are notable for their potentially lower molecular weight as compared to linker-based bifunctional molecules (e.g. PROTACS), a possible advantage for increased bioavailability and improved pharmacokinetic profiles 4. Moreover, molecular glues are also particularly interesting from a functional perspective, since the few well-characterized examples uniquely modulate protein function and downstream biology by stabilizing protein complexes between binding partners that otherwise would not interact.
Three notable molecular glues showcase diverse functional outcomes associated with novel protein complexes. The natural product rapamycin binds to FKBP12, leading to the recruitment of mTORC1, resulting in partial inhibition of mTORC1 7. The cotylenin diterpenes form interfaces between the 14-3-3 protein family and a variety of interacting partners 8. Thalidomide binds to the E3 ligase cereblon to form a new protein interface that recruits neo-substrates such as SALL4 or Ikaros transcription factors for ubiquitination and degradation 9–12. Paradoxically, degradation of the former leads to teratogenic effects while degradation of the latter is a proven chemotherapeutic strategy4,6. Moreover, analogs of rapamycin and thalidomide can still maintain binding to FKBP12 and cereblon respectively, but shift their neo-substrate binding profiles to confer new and distinct biological functions 13,14. This realization has led to the discovery of approved drugs such as lenalidomide, pomalidomide, and FK506. Unfortunately, only a handful of well-characterized molecular glues have been discovered; thus, limiting our understanding of the design principles behind their function and reducing their impact on drug discovery. Natural products have been a robust source of many known molecular glue interactions (e.g. rapamycin, FK506, auxin, brefeldin A, forskolin, and others), and their diverse and stereochemically-rich architectures possess many positive attributes for recruiting protein surfaces 6,15.
Electrophilic natural products represent a vastly underexplored subset of natural products from which molecular glues can originate, and such species could have unique and positive attributes owing to their potential for covalent bond formation16. While covalently-acting natural products have not been previously identified as molecular glues, we hypothesized that natural products which bear: 1) one or more potential sites for covalent interaction with nucleophilic amino acids on proteins; and 2) have also been shown to possess biological activity, would constitute candidates for possessing potentially unique glue-like characteristics.
The covalency of these potential natural product-based molecular glues also enables rapid mechanistic deconvolution of protein targets using chemoproteomic platforms. One such platform used herein is activity-based protein profiling (ABPP), which uses reactivity-based chemical probes to profile proteome-wide reactive, functional, and ligandable hotspots directly in complex living systems. When used in a competitive manner, covalently-acting small-molecules can be competed against the binding of reactivity-based probes to facilitate the identification of their protein targets and covalent sites of modification 1,2,17. Upon identifying the direct targets of the natural product, standard pull-down and quantitative proteomic profiling experiments can be used to identify specific molecular glue interactions enabled by the natural product.
In this study, we interrogated the Streptomyces-derived family of manumycin polyketide natural products, specifically asukamycin (1) and manumycin A (2), which have documented antibiotic and antiproliferative properties 18. Manumycin A has also been show to inhibit farnesyl transferase activity and sphingomyelinase activity, but asukamycin is not known to inhibit these targets and its anti-cancer mechanism of action is unclear 19–23. These molecules possess a suite of electrophilic sites that could potentially react covalently with nucleophilic amino acids within proteins, such as cysteines (Figure 1a) 19. We hypothesized that multiple reactive sites within these natural products may lead to specific interactions with multiple protein targets resulting in bifunctional or molecular glue-type interactions.
Figure 1. Asukamycin targets C374 of UBR7 in breast cancer cells.
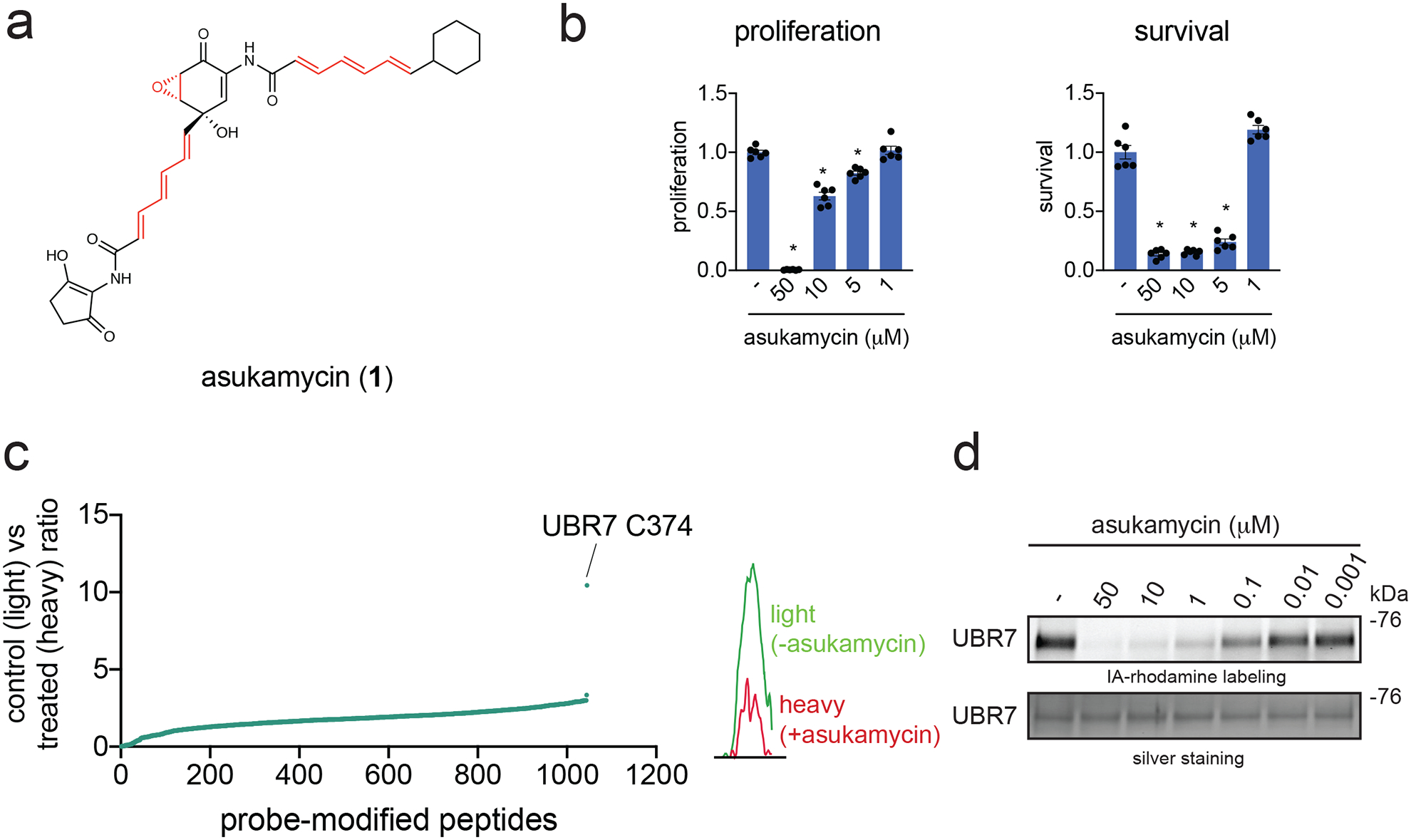
(a) Structure of asukamycin highlighting (in red) three potentially reactive sites. (b) Proliferation and serum-free cell survival in 231MFP breast cancer cells treated with DMSO vehicle or asukamycin for 48 h, assessed by Hoechst stain. (c) IsoTOP-ABPP analysis of asukamycin in situ in 231MFP cells. 231MFP cells were treated with DMSO vehicle or asukamycin (10 μM) for 3 h, and resulting cell lysates were labeled with IA-alkyne (100 μM) for 1 h after which point isotopically light (control) or heavy (asukamycin-treated)-treated TEV tag-bearing biotin-azide handles were appended by CuAAC and taken through the isoTOP-ABPP method. Shown are light to heavy ratios of probe-modified peptides. Shown are average probe-modified peptide ratios. The individual replicate values and total datasets are shown in Source Data Tables for Figure 1. On the right is a representative MS1 chromatogram of the probe-modified C374-bearing UBR7 peptide between control versus asukamycin-treated cells. This data represents data from n=3 biologically independent samples. (d) Gel-based ABPP analysis of asukamycin against pure human UBR7 protein. UBR7 protein was pre-incubated with DMSO vehicle or asukamycin for 30 min prior to IA-rhodamine (200 nM) labeling of UBR7 for 1 h at room temperature. Protein was resolved by SDS/PAGE and visualized by in-gel fluorescence and protein loading was assessed by silver staining. Data shown in (b) is shown as individual replicate values and average ± sem and are n=6 biologically independent samples/group. Gels shown in (d) are representative blots from n=3 biologically independent samples/group. Statistical significance was calculated with two-tailed unpaired Student’s t-tests and are shown as *p<0.05 compared to vehicle-treated controls within each group. Source data for cropped blots can be found in Source Data for Figure 1. Source data for bar graphs can be found in Source Data Tables for Figure 1.
Using ABPP-based chemoproteomic platforms, we demonstrate that one of the primary targets of asukamycin is C374 of the postulated E3 ubiquitin ligase UBR7. We further showed that this UBR7-asukamycin complex engages multiple proteins in breast cancer cells including the tumor suppressor TP53, and that the resulting UBR7-asukamycin-TP53 complex contributes to the anti-cancer activity of asukamycin.
Results
Anti-Cancer Activity of Asukamycin
There is significant unmet medical need for new triple-negative breast cancer (TNBC) therapies as such malignancies have worse clinical prognoses than other breast cancer subtypes. Moreover, few targeted therapies are approved for the treatment of TNBCs. New small-molecules, mechanisms, and therapeutic modalities for combatting TNBCs could greatly reduce mortalities associated with these aggressive breast cancers 24. We show that asukamycin impairs the proliferation (with full serum-containing media) and serum-free cell survival of two TNBC-derived cell lines, 231MFP and HCC38 (Figure 1b, Supplementary Figure 1), with 50 % effective concentration (EC50) values of 13.8 and 4.5 μM for 231MFP proliferation and survival, respectively. The apparent difference in sensitivity observed with asukamycin anti-proliferative and anti-survival effects in 231MFP cells were likely due to asukamycin-binding to serum in the media, since the relative levels of intracellular asukamycin were much higher in 231MFP cells in serum-free media, compared to serum-containing media (Supplementary Figure 1). The apparent low cell-based potency may also be due to low free asukamycin exposure in cells because of its high lipophilicity, as well as low cellular permeability, due to its higher molecular weight and high number of rotatable bonds. We also tested asukamycin growth inhibitory effects across 250 cancer cell lines spanning many different tumor types and show broad growth inhibitory effects across many cancer cell lines with concentrations values at which 50 % growth inhibition observed (GI50) of 0.08 to >30 μM (Extended Data Figure 1a). Among the 19 breast cancer cell lines tested from receptor positive and TNBC cell lines, 7 showed heightened sensitivity with GI50 values of 5.7–11.2 μM, 9 showed intermediate sensitivity with GI50 values of 13–16 μM, and 3 showed refractory effects with GI50 values >20 μM, in which HCC38 and the MDA-MB-231 line from which 231MFP cells were derived showed intermediate sensitivity (Extended Data Figure 1b, 1c). Given our interest in TNBCs, we focused our subsequent mechanistic studies for asukamycin in 231MFP cells, as a representative TNBC line.
ABPP to Map Asukamycin Targets
We postulated that the antiproliferative activity of asukamycin may be dependent on covalent interactions with cysteines on specific protein targets. Asukamycin could react with protein nucleophiles through either hetero-Michael addition reactions with one or both of the two polyunsaturated amide side chains or reaction with the epoxyketone moiety, a known process for similar epoxy ketone natural products. 25 We performed ABPP-based chemoproteomic profiling to identify asukamycin targets in 231MFP TNBC cells. Proteome samples treated with vehicle or asukamycin were labeled with the cysteine-reactive probe iodoacetamide-alkyne (IA-alkyne or N-hex-5-ynyl-2-iodo-acetamide), prior to further processing using a well-validated method for covalent ligand target identification—isotopic tandem orthogonal proteolysis ABPP (isoTOP-ABPP) 2,17,26. We identified C374 of UBR7 as a primary target of asukamycin with the highest control to treated (or light to heavy) ratio in 231MFP cells (Figure 1c). We confirmed that UBR7 was a direct target of asukamycin using gel-based ABPP approaches, wherein we show competition of asukamycin against rhodamine-functionalized iodoacetamide (IA-rhodamine) labeling of recombinant human UBR7 protein (Figure 1d).
Next, we investigated whether the anti-cancer effects of asukamycin were driven through asukamycin interactions with C374 of UBR7. Stable UBR7 knockdown in 231MFP cells did not compromise proliferation but conferred complete resistance to asukamycin-mediated anti-proliferative effects (Figure 2a, 2b). Expression of wild-type (WT) or C374A mutant UBR7 in shUBR7 231MFP cells led to re-sensitization of asukamycin-mediated anti-proliferative effects in WT but not C374A UBR7-expressing cells (Figure 2c). Taken together, these results demonstrate that asukamycin targets C374 of UBR7 and that this interaction is responsible for the anti-proliferative effects of asukamycin in breast cancer cells. These data also hinted that asukamycin, through targeting C374 of UBR7, may be conferring a gain-of-function effect, since UBR7 knockdown itself does not impair cell proliferation (Supplementary Figure 2). Further supporting this hypothesis, we found that overexpression of WT, but not C374A mutant FLAG-UBR7 conferred significantly heightened asukamycin-induced anti-proliferative effects with EC50 values of 10.0, 4.7, and 10.0 μM for GFP-, WT UBR7, and C374A mutant UBR7-expressing 231MFP cells, respectively (Figure 2d, 2e).
Figure 2. Asukamycin impairs breast cancer cell proliferation through targeting UBR7.

(a) Expression of UBR7 and loading control GAPDH levels in 231MFP shControl, shUBR7, or shUBR7 cells expressing FLAG-tagged wild-type (WT) or FLAG-tagged C374A mutant UBR7 protein, assessed by Western blotting. (b) Cell proliferation in 231MFP shControl or shUBR7 cells treated with DMSO vehicle or asukamycin for 24 h, assessed by Hoechst stain. (c) Cell proliferation in 231MFP shUBR7 cells expressing FLAG-WT or FLAG-C374A mutant UBR7 treated with DMSO vehicle or asukamycin (50 μM) for 24 h. (d) anti-FLAG, anti-UBR7, and loading control anti-GAPDH protein expression in 231MFP cells stably expressing FLAG-GFP, FLAG-UBR7 WT, or FLAG-UBR7 C374A mutant assessed by Western blotting. (e) Cell proliferation of 231MFP cells transiently expressing FLAG-GFP, FLAG-UBR7 WT or FLAG-UBR7 C374A mutant treated with DMSO or asukamycin (5 μM) for 48 h, assessed by Hoechst stain. Data shown in (b, c, and e) are shown as individual replicate values and average ± sem and are n=6 biologically independent samples/group. Gels shown in (a, d) are representative blots from n=3 biologically independent samples/group. Statistical significance was calculated with two-tailed unpaired Student’s t-tests and are shown as *p<0.05 compared to vehicle-treated controls within each group, #p<0.05 against respective concentrations of asukamycin-treatment in shControl groups in (b, c) or asukamycin-treatment in FLAG-GFP expressing groups in (e). Source data for cropped blots can be found in Source Data for Figure 2. Source data for bar graphs can be found in Source Data Tables for Figure 2.
While UBR7 is annotated as an E3 ubiquitin ligase and postulated to be involved in histone ubiquitination 27, its biochemical and physiological functions are poorly understood especially with regard to cancer cell proliferation. We attempted to reconstitute the activity of UBR7 in vitro, but our various efforts to demonstrate UBR7 activity failed. Instead, we next explored the potential for UBR7 to engage in asukamycin-induced interactions with other proteins.
Mapping Molecular Glue Interactions of UBR7-Asukamycin
From these results and the potential of asukamycin to achieve multi-covalency, we postulated that asukamycin may be conferring gain-of-function effects upon UBR7 through engaging in molecular glue interactions with other proteins. To substantiate this hypothesis, we performed proteomic analysis on anti-FLAG pulldown eluate from FLAG-GFP or FLAG-UBR7-expressing 231MFP cells treated with vehicle or asukamycin to identify protein-protein interactions that were dependent on both UBR7 and asukamycin. From this experiment, we identified 13 proteins that showed significant (p<0.01) and >15-fold higher pulldown in the asukamycin-treated Flag-UBR7 cells compared to vehicle-treated FLAG-UBR7 cells (Figure 3a). Among these 13 targets, 8 are UBR7-dependent, with >4-fold enrichment between asukamycin-treated FLAG-UBR7 cells versus asukamycin-treated FLAG-GFP cells (Figure 3a). These 8 targets include proteins of great importance in cancer, including DNA protein kinase (PRKDC) and the tumor-suppressor p53 (TP53) 28,29 (Figure 3b). In pulldown experiments, we confirmed the asukamycin-dependent interactions with UBR7 with Western blots showing that proteins such as TP53 and PRKDC interacted specifically in FLAG-UBR7 expressing cells treated with asukamycin (Figure 3c).
Figure 3. Mapping molecular glue interactions of asukamycin and UBR7.
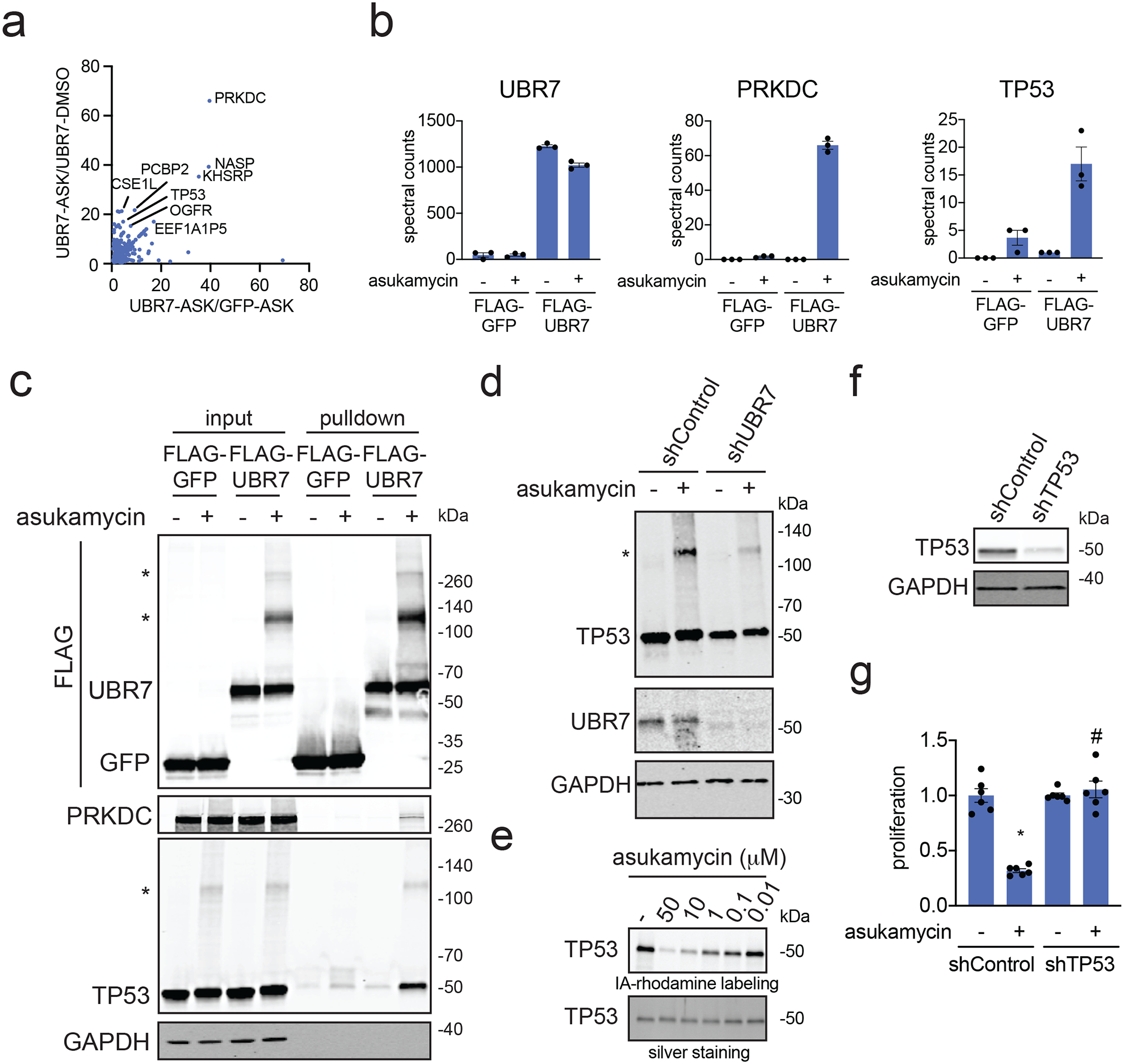
(a) Proteomics analysis of molecular glue interactions of UBR7-asukamycin. 231MFP cells stably expressing FLAG-GFP or FLAG-UBR7 were treated with DMSO or asukamycin (50 μM) for 3 h. FLAG-GFP and FLAG-UBR7 interacting proteins were subsequently enriched and then subjected to proteomic analysis. Raw and analyzed proteomic analysis is shown in Source Data Tables for Figure 3 and is quantified by spectral counting. For those proteins that showed no peptides in a particular sample group, we set those proteins to 1 to enable relative fold-change quantification to generate this figure to visually show those proteins that showed high enrichment in asukamycin (ASK)-treated FLAG-UBR7 groups compared to asukamycin-treated FLAG-GFP groups on the x-axis and ASK-treated FLAG-UBR7 groups compared to DMSO-treated FLAG-UBR7 groups. Proteins highlighted showed >15-fold enrichment comparing UBR-ASK to UBR7-DMSO groups and >4-fold enrichment comparing UBR7-ASK to GFP-ASK groups. (b) Shown are spectral counts for representative proteins in the experiment described in (a). (c) Protein levels of FLAG-tagged proteins, PRKDC, TP53, and loading control GAPDH from a repeat of the experiment described in (a) by Western blotting showing specific interactions of PRKDC and TP53 with FLAG-UBR7 only when cells are treated with asukamycin. Higher molecular weight species of FLAG-UBR7 and TP53 are noted with (*). (d) anti-TP53, anti-UBR7, and loading control anti-GAPDH Western blots in shControl or shUBR7 231MFP cells treated with vehicle DMSO or asukamycin (50 μM) for 3 h. Higher molecular species of TP53 is noted with (*). Quantification for this gel is provided in Supplementary Figure 5a. (e) Gel-based ABPP analysis of asukamycin against pure human TP53 protein. TP53 protein was pre-incubated with DMSO vehicle or asukamycin for 30 min prior to IA-rhodamine labeling of TP53 for 1 h at room temperature. Protein was resolved by SDS/PAGE and visualized by in-gel fluorescence and protein loading was assessed by silver staining. (f) TP53 protein levels in 231MFP shControl and shTP53 cells. (g) Cell proliferation of 231MFP shControl and shTP53 cells treated with DMSO or asukamycin (10 μM) for 24 h. Data shown in (b and g) are shown as individual replicate values and average ± sem and are n=3 for (b) and n=6 for (g) biologically independent samples/group. Gels shown in (c, d, e, and f) are representative blots from n=3 biologically independent samples/group. Statistical significance was calculated with two-tailed unpaired Student’s t-tests and are shown as *p<0.05 compared to vehicle-treated controls within each group, #p<0.05 compared to asukamycin-treated shControl group in (g). Source data for cropped blots can be found in Source Data for Figure 3. Source data for bar graphs can be found in Source Data Tables for Figure 3.
Interestingly, in our anti-FLAG blot from FLAG-UBR7 pulldown studies, we also observed several higher molecular weight species in a reducing SDS/PAGE gel that corresponded to the estimated combined masses of FLAG-UBR7 and TP53 as well as even higher molecular weight species that corresponded to the estimated molecular weight of PRKDC (Figure 3c). In the anti-TP53 blot from the same pulldown studies, in addition to the expected TP53 band, we also observed a distinct higher molecular weight species that corresponded to a molecular weight in line with the addition of FLAG-UBR7 and TP53 (Figure 3c). Reinforcing that this higher molecular weight FLAG-UBR7 band included the TP53 higher molecular weight species, a dual color Western blot showed overlap between the FLAG-UBR7 and TP53 higher molecular weight bands (Supplementary Figure 3). Proteomic analysis of this higher molecular weight region further confirmed significantly increased levels of TP53 and UBR7 in this 100–140 kDa range (Extended Data Figure 2). UBR7 knockdown in 231MFP cells treated with asukamycin also resulted in >80 % reduction in the higher molecular weight TP53 species compared to shControl counterparts (Figure 3d, Extended Data Figure 3a). TP53 knockdown in 231MFP cells treated with asukamycin resulted in a significant, albeit less pronounced reduction in the higher molecular weight FLAG-UBR7 species compared to shControl counterparts (Extended Data Figure 3b, 3c). While there may be additional proteins of similar molecular weight to TP53 that are included in this higher molecular weight FLAG-UBR7 species, we believe that TP53 is one of those proteins. Further confirming that this higher molecular weight TP53 species is a ternary complex between UBR7, asukamycin, and TP53, interactions with the higher molecular weight TP53 complex was completely lost and interactions with the parent molecular weight TP53 was significantly reduced from pulldown of C374A mutant FLAG-UBR7 compared to wild-type FLAG-UBR7 in 231MFP cells (Extended Data Figure 4). The higher molecular weight TP53 species in the input for both wild-type and C374A mutant FLAG-UBR7 lines is likely the ternary complex arising from endogenous wild-type UBR7 in the FLAG-UBR7 C374A mutant expressing cells.
We further confirmed that these higher molecular weight species were not polyubiquitinated TP53 since treatment of proteomes with the deubiquitinase USP2 was not able to eliminate these higher molecular weight TP53 bands (Supplementary Figure 4). These results show enrichment of both the parent molecular weight protein interaction partner and also a higher molecular weight species, suggesting that the complexes between UBR7-asukamycin and binding partners such as TP53 or PRKDC initially form through reversible interactions which eventually in-part lead to multi-covalent interactions. We also showed that abundant proteins such as GAPDH did not interact with the asukamycin-UBR7 complex, arguing for specific molecular interactions over non-specific binding events (Figure 3c). While TP53 was not identified as a target in our isoTOP-ABPP studies likely due to its low abundance, we further demonstrated that asukamycin directly interacts with TP53 by gel-based ABPP (Figure 3e).
Given the importance of PRKDC and TP53 in cancer pathogenicity, we next tested the relative contributions of these two proteins in asukamycin-mediated anti-proliferative effects. TP53 knockdown, but not PRKDC knockdown, conferred complete resistance to asukamycin-mediated anti-proliferative effects in 231MFP breast cancer cells, indicating that TP53 is the more critical target (Figure 3f, 3g, Supplementary Figure 5). Taken together, these results suggest that asukamycin has the ability to promote the formation of a gain-of-function complex between UBR7 and TP53 that results in anti-proliferative effects in TNBC cell lines.
UBR7-Asukamycin Molecular Glue Interactions with TP53
We next investigated the biochemical and functional consequences of UBR7, asukamycin, and TP53 interactions. We wondered whether the UBR7-asukamycin-TP53 complex might affect TP53 function in the TP53-mutant setting of TNBC cell lines. Consistent with asukamycin directly binding to TP53 and potentially stabilizing TP53 folding, asukamycin caused a significant increase in TP53 thermal stability in vitro in 231MFP cell lysate (Figure 4a, 4b). We also observed a higher molecular weight TP53 species, which showed even greater thermal stability compared to the asukamycin-treated TP53 parental protein (Figure 4a, 4b). This increased thermal stability of TP53 conferred by asukamycin was attenuated upon UBR7 knockdown or FLAG-UBR7 depletion (Extended Data Figure 5, Extended Data Figure 6).
Figure 4. Asukamycin acts as a molecular glue between UBR7 and TP53 and activates TP53 transcriptional activity in a UBR7-dependent manner.
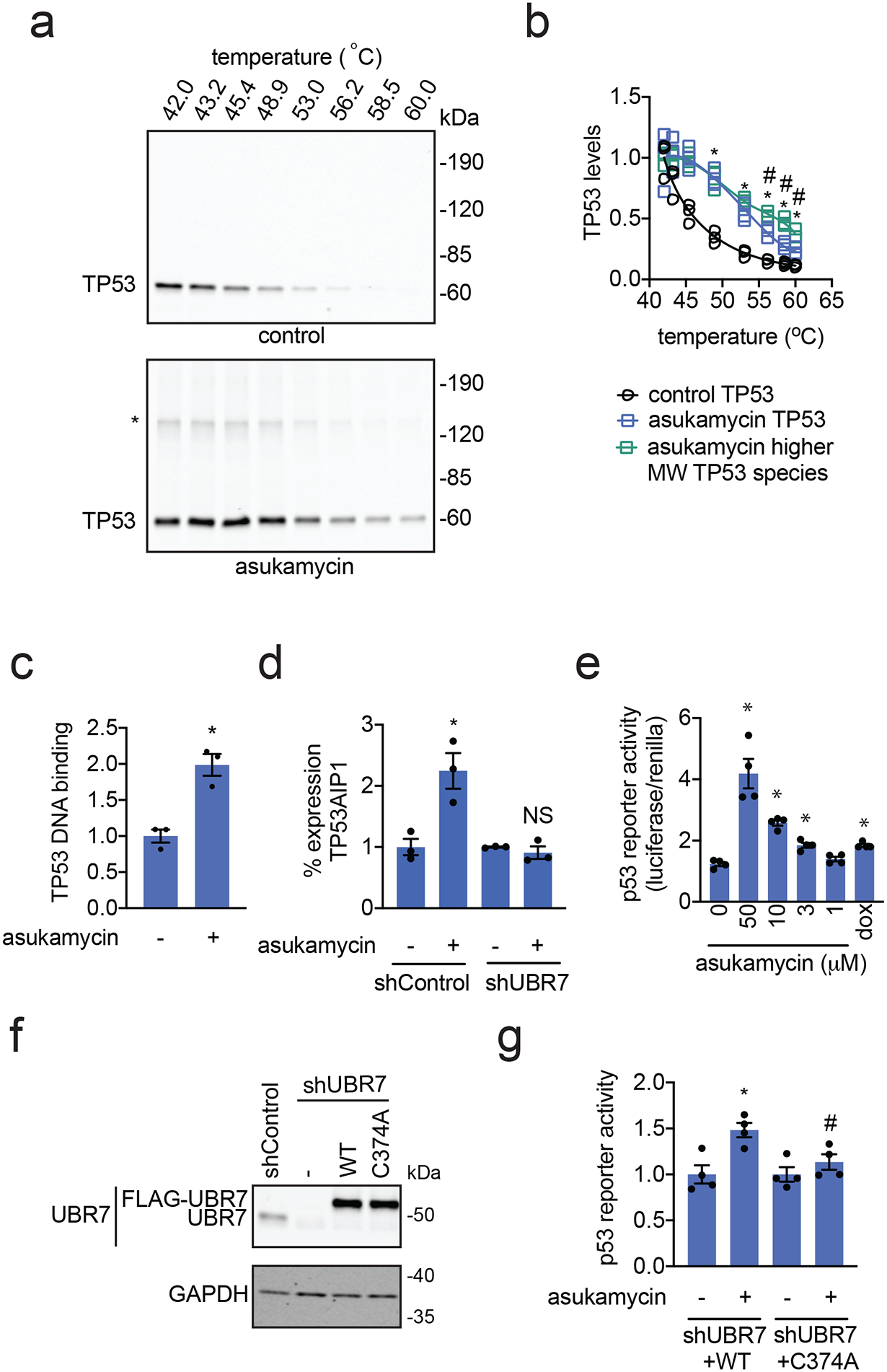
(a) Thermal shift assay in vitro in 231MFP breast cancer cell lysate treated with DMSO vehicle or asukamycin (50 μM). Lysate was heated to the designated temperature for 3 minutes, followed by 3 minutes at room temperature, centrifugation, and SDS/PAGE and Western blotting for TP53. (b) Quantification of TP53 parent and higher molecular weight levels from thermal shift assay described in (a). Data is normalized to respective 42 °C TP53 protein levels. (c) TP53 DNA binding to p53 DNA consensus sequence in vitro with TP53 spiked into 231MFP breast cancer cell lysate treated with DMSO vehicle or asukamycin (50 μM). (d) mRNA expression of TP53AIP1 in 231MFP shControl and shUBR7 cells treated with DMSO vehicle or asukamycin (50 μM) for 6 h, assessed by qPCR. (e) p53 reporter activity reported as the ratio between luciferase reporter activity versus cell number control renilla levels in shUBR7 HEK293T cells expressing WT or C374A mutant UBR7 and the p53 reporter construct treated with DMSO vehicle control, asukamycin, or doxorubicin (1 μM) for 6 h. (f) Protein expression of UBR7 and loading control GAPDH in HEK293T shControl and shUBR7 cells expressing empty vector, wild-type UBR7, or C374A mutant UBR7 assessed by Western blotting. (g) p53 reporter activity reported compared to DMSO vehicle-treated controls in each group in HEK293T shUBR7 cells expressing the p53 reporter construct treated with DMSO vehicle control or asukamycin (10 μM) for 6 h. Data shown in (b, c, d, e, and g) are shown as individual replicate values and average ± sem and are n=3 for (b, c, and d) and n=4 for (e and g) biologically independent samples/group. Gels shown in (a and f) are representative blots from n=3 biologically independent samples/group. Statistical significance was calculated with two-tailed unpaired Student’s t-tests and are shown as *p<0.05 compared to each temperature control group for (b), vehicle-treated controls for (c, d, e, and g) within each group, #p<0.05 compared to corresponding asukamycin treatment groups for each temperature for parent TP53 species in (b) and asukamycin-treated shUBR7 WT group in (g). NS denotes not significant. Source data for cropped blots can be found in Source Data for Figure 4. Source data for bar graphs and plots can be found in Source Data Tables for Figure 4.
Asukamycin also significantly increased TP53 binding to its DNA consensus sequence in vitro when TP53 was spiked into 231MFP cell lysate and this increased binding was attenuated by UBR7 knockdown, indicating that asukamycin potentially activated TP53 transcriptional activity in a UBR7-dependent manner (Figure 4c, Extended Data Figure 7). As other thiol-reactive compounds have been reported to both thermally stabilize TP53 and to rescue the activity of mutant TP53 30, we next asked if asukamycin could increase the transcriptional activity of TP53. Consistent with functional activation of p53 activity in cells, asukamycin induced the expression of the p53 target gene TP53AIP1 in 231MFP cells. The asukamycin-mediated increase in expression was attenuated in UBR7 knockdown cells (Figure 4d). Asukamycin also significantly induced p53 luciferase reporter transcriptional activity in HEK293T cells, even more so than the positive control topoisomerase inhibitor doxorubicin (dox) that is known to induce apoptosis through activating the p53 pathway (Figure 4e). This asukamycin-induction of p53 transcriptional reporter activity was significantly attenuated upon UBR7 knockdown or in cells expressing C374A mutant UBR7 compared to wild-type UBR7 in shUBR7 HEK293T cells (Figure 4f, 4g, Extended Data Figure 8). Consistent with asukamycin-mediated activation of p53 activity, quantitative proteomic analysis of protein expression changes conferred by asukamycin treatment in 231MFP cells showed many known p53 transcriptional targets upregulated in expression, including GADD45A, GADD45B, ANKRD1, HMOX1, ATF3, PMAIP1, DDIT4, DNAJB9, and PLK2 (Extended Data Figure 9) 31–37. Functional enrichment analysis of significantly upregulated proteins showed a significant enrichment in the TP53 signaling pathway, as well as pathways in protein processing, senescence, MAPK signaling (Extended Data Figure 9). One of the well-known TP53 transcriptional tumor-suppressor targets CDKN1A (p21) was not detected in our proteomic analysis, and thus we specifically measured CDKN1A levels and found that asukamycin significantly increased CDKN1A levels in 231MFP cells and that this increase was significantly attenuated upon UBR7 knockdown (Extended Data Figure 10). We also showed that asukamycin conferred anti-proliferative effects in an isogenic HCT116 colorectal cancer cell line background expressing either wild-type or R248W mutant TP53 but showed resistance in TP53 knockout cells (Supplementary Figure 6). Finally, to rule out that asukamycin was inducing TP53 transcriptional activation through DNA damage, we also demonstrated that asukamycin did not induce DNA damage compared to known DNA-damaging agents such as etoposide and hydrogen peroxide based on assessment of DNA damage markers such as H2A.X and RPA32 phosphorylation (Supplementary Figure 7).
Collectively, these results indicated that asukamycin directly targeted TP53, stabilized TP53 folding, and activated p53 transcriptional activity in a UBR7 C374-dependent manner.
Activity of related natural product manumycin A
Asukamycin belongs to a larger family of polyketide natural products known as the manumycins. While manumycin A, a close structural analog of asukamycin that also bears multiple electrophilic sites, has been shown to inhibit farnesyl transferase or sphingomyelinase 22,23 (Figure 5a), the effect of manumycin A on UBR7 or its molecular glue activity with TP53 have not been previously reported. Interestingly, manumycin A also bound to UBR7 at comparable potency to asukamycin by gel-based ABPP (Figure 5b). We also found that manumycin A, like asukamycin, activated p53 transcriptional activity more so than doxorubicin (Figure 5c). Manumycin A treatment also led to molecular glue interactions between UBR7 and TP53 and resulted in distinct higher molecular weight species which corresponded to the added molecular weight of TP53 and UBR7 (Figure 5d). Finally, manumycin A, like asukamycin, interacted directly with TP53 by gel-based ABPP (Figure 5e).
Figure 5. Manumycin A also interacts with UBR7 and engages in molecular glue activities with TP53.
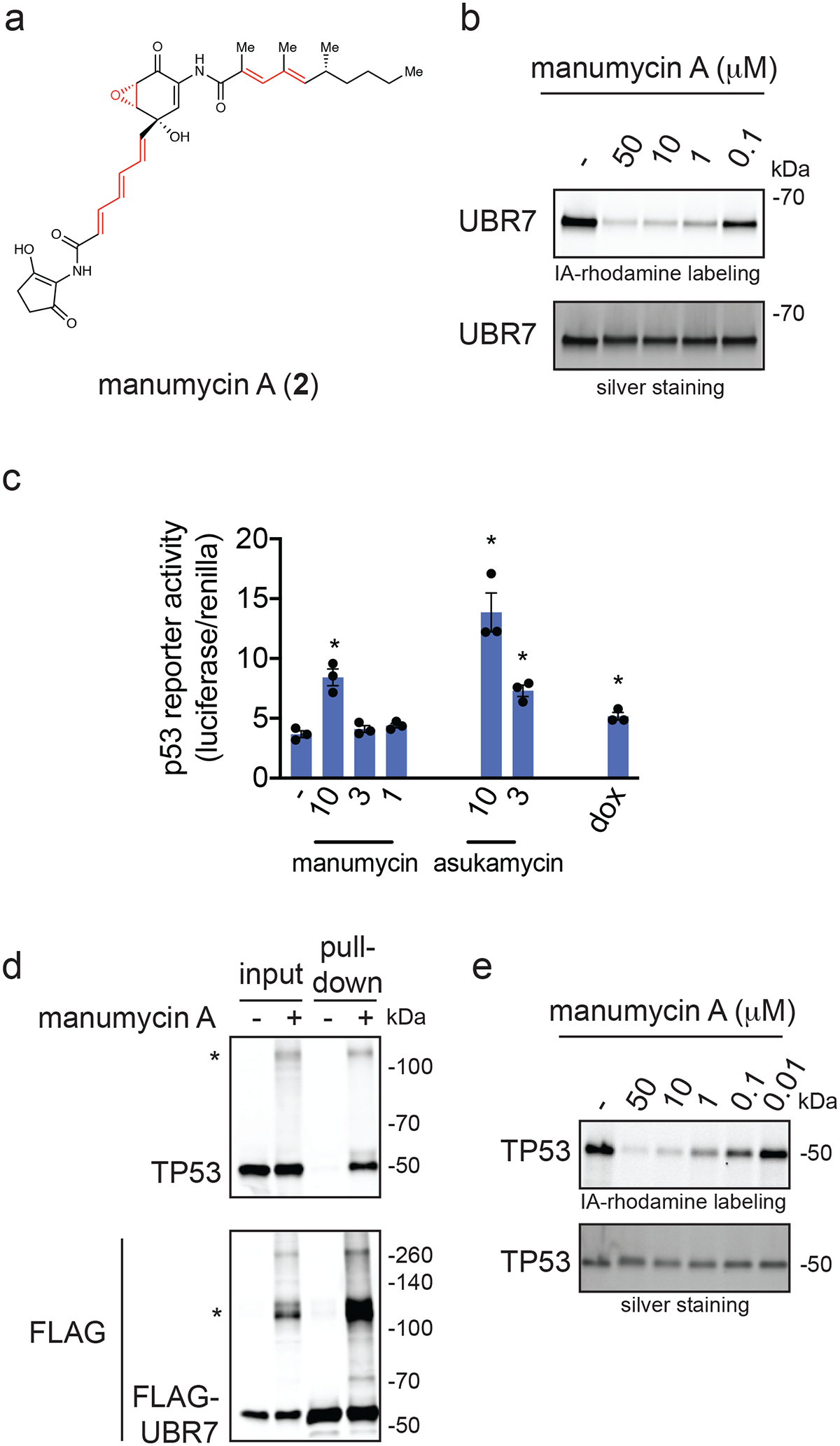
(a) Structure of manumycin A highlighting potentially reactive sites in red. (b) Gel-based ABPP analysis of manumycin A against pure human UBR7 protein. UBR7 protein was pre-incubated with DMSO vehicle or manumycin A for 30 min prior to IA-rhodamine labeling of UBR7 for 1 h at room temperature. Protein was resolved by SDS/PAGE and visualized by in-gel fluorescence and protein loading was assessed by silver staining. (c) p53 reporter activity reported as the ratio between luciferase reporter activity versus cell number control renilla levels in HEK293T cells expressing the p53 reporter construct treated with DMSO vehicle control, manumycin A, asukamycin, or doxorubicin (1 μM) for 6 h. (d) Protein levels of TP53 and FLAG-tagged proteins by Western blotting in 231MFP cells stably expressing FLAG-UBR7 treated with DMSO vehicle or manumycin A (50 μM) for 3 h, after which FLAG-UBR7 interacting proteins were subsequently enriched. Higher molecular weight TP53 and FLAG-UBR7 species are noted with a (*). (e) Gel-based ABPP analysis of manumycin A against pure human TP53 protein. TP53 protein was pre-incubated with DMSO vehicle or manumycin A for 30 min prior to IA-rhodamine labeling of TP53 for 1 h at room temperature. Data shown in (c) are shown as individual replicate values and average ± sem and are n=3 biologically independent samples/group. Gels shown in (b, d, and e) are representative blots from n=3 biologically independent samples/group. Statistical significance as calculated with two-tailed unpaired Student’s t-tests and are shown as *p<0.05 compared to vehicle-treated controls. Source data for cropped blots can be found in Source Data for Figure 5. Source data for bar graphs can be found in Source Data Tables for Figure 5.
To investigate the relative contribution of the epoxide warhead on UBR7 binding versus TP53 binding, we prepared manumycin D (3) (Figure 6a) 38, an epoxide-reduced variant, in one step from manumycin A (see Synthetic Methods) 39. Manumycin D still labeled UBR7 by gel-based ABPP, albeit weaker than manumycin A (Figure 6b). However, manumycin D bound less potently to TP53 compared to manumycin A or asukamycin by gel-based ABPP (Figure 6c). Consistent with these data, manumycin D, unlike manumycin A, was incapable of inducing interactions between UBR7 and TP53 (Figure 6d). Moreover, the anti-proliferative effects and p53 transcriptional activation observed with manumycin A were attenuated with manumycin D in 231MFP breast cancer cells (Figure 6e, 6f). These data collectively suggested that the epoxide contributed to interactions with TP53 and was necessary to engage in multi-covalent interactions between UBR7 and TP53 to exert anti-proliferative effects. These results also implied that the unsaturated side chains of manumycin A, and by analogy asukamycin, were responsible for the covalent interaction with C374 of UBR7.
Figure 6. Comparing manumycin D to manumycin A.
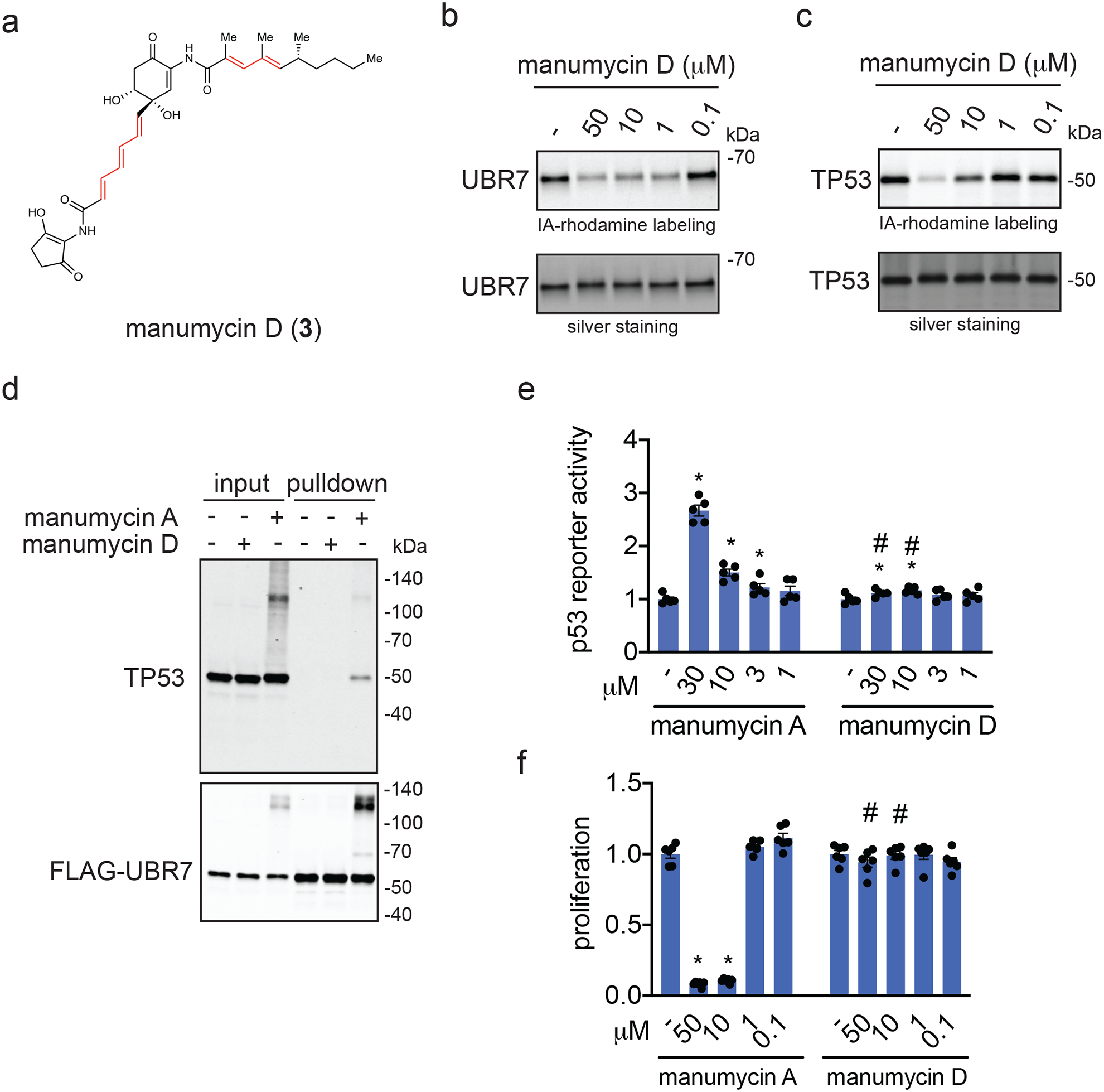
(a) Structure of manumycin D with reactive sites highlighted in red. (b) Gel-based ABPP analysis of manumycin D against pure human UBR7 protein. UBR7 protein was pre-incubated with DMSO vehicle or manumycin D for 30 min prior to IA-rhodamine labeling of UBR7 for 1 h at room temperature. (c) Gel-based ABPP analysis of manumycin D against pure human TP53 protein. TP53 protein was pre-incubated with DMSO vehicle or manumycin D for 30 min prior to IA-rhodamine labeling of TP53 for 1 h at room temperature. (d) Protein levels of TP53 and FLAG-tagged proteins by Western blotting in 231MFP cells stably expressing FLAG-UBR7 treated with DMSO vehicle or manumycin D (50 μM) for 3 h, after which FLAG-UBR7 interacting proteins were subsequently enriched. (e) Proliferation in 231MFP breast cancer cells treated with DMSO vehicle or manumycin A or manumycin D for 48 h and assessed by Hoechst staining. (f) p53 reporter activity in HEK293T cells treated with DMSO vehicle or manumycin A, or manumycin D for 3 h. Gels shown in (b, c, and d) are representative blots from n=3 biologically independent samples/group. Data shown in (e, f) as individual replicate values and average ± sem and are n=6 for (e) or n=5 for (f) biologically independent samples/group. Statistical significance was calculated with two-tailed unpaired Student’s t-tests and are shown as *p<0.05 compared to vehicle-treated controls within each group. Statistical significance as calculated with two-tailed unpaired Student’s t-tests and are shown as *p<0.05 compared to vehicle-treated controls, and #p<0.05 compared to the corresponding manumycin A treatment group. Source data for cropped blots can be found in Source Data for Figure 6. Source data for bar graphs can be found in Source Data Tables for Figure 6.
Discussion
While molecular glues are exciting therapeutic modalities that have the potential for stabilizing interactions between proteins that usually would not interact enabling exploitation of unique protein functions, discovery of new molecular glue scaffolds has been mostly serendipitous. We postulated that natural products bearing multiple electrophilic sites may engage in multi-covalent interactions that bring specific proteins into novel, functionally relevant complexes. In this study, we show that asukamycin, a manumycin polyketide bearing multiple electrophilic sites, reacts with C374 of the E3 ligase UBR7 to impair breast cancer cell proliferation through engaging in molecular glue interactions with multiple neo-substrates, including TP53. We demonstrate that asukamycin activates TP53 transcriptional activity in a UBR7 C374-dependent manner and that the anti-proliferative effects observed in breast cancer cells depends on UBR7 and TP53. This molecular glue activity is also recapitulated with a related polyketide manumycin A. Furthermore, we show that manumycin D, which lacks an epoxide, still retains binding to UBR7 but shows less potent binding to TP53 and is not able to covalently modify TP53 to the same extent, consistent with this class of molecules targeting C374 of UBR7 through a hetero-Michael addition, and then engaging TP53 likely initially through reversible binding and then through a covalent bond involving the reactive epoxide. This latter observation is based on UBR7 pulldown studies showing not only the higher molecular weight TP53 species, but also the parent TP53 molecular weight. While we seem to observe one primary higher molecular weight UBR7-TP53 band, there are also several additional higher molecular weight species observed, which may correspond to multiple multi-covalent interactions with the three potential reactive sites on asukamycin and manumycin A. These complexes could also contain additional molecular glue partners, such as those identified in the enrichment proteomic studies. Furthermore, while we show by isoTOP-ABPP that C374 of UBR7 is one of the primary targets of asukamycin and our genetic validation data shows that targeting this cysteine appears to drive the asukamycin-mediated antiproliferative effects, asukamycin and manumycin A likely possess additional targets. Previously reported targets including farnesyltransferase and sphingomyelinase may also play a role in mediating anti-cancer effects or other biological activities 22,23. Manumycin A has also been shown to interfere with IKK activity by directly binding to IKKβ and forming stable high molecular mass complexes, indicating that these alternate biological activities may also be mediated through additional molecular glue-type interactions beyond those reported here 40.
Several unanswered questions still remain. We do not understand how these manumycin polyketide/UBR7 complexes interact with p53 to activate p53 function. Our data suggest that the UBR7-asukamycin complex acts like a chaperone to improve TP53 thermal stability and folding, which likely improved its ability to bind to DNA and activate TP53 transcriptional activity. We also do not know the site or nature of the amino acid(s) on p53 wherein the suspected reaction with the epoxide takes place. We tried to map the cysteine on p53 targeted by asukmycin using LC-MS/MS, but we were not able to detect a modified peptide, likely due to instability of the asukamycin cysteine adduct under mass spectrometry conditions. Determining which cysteine on p53 is targeted by asukamycin and is responsible for the asukamycin-mediated anti-cancer effects will be important to further understand the biological actions of asukamycin. Solving the crystal structure of the UBR7-asukamycin-TP53 ternary complex will be particularly interesting to understand the biochemical underpinnings of this molecular glue activity. We tried to reconstitute the molecular glue activity of UBR7 with asukamycin and TP53 in vitro with purified proteins, but we observed asukamycin-mediated self-oligomerization of UBR7 and TP53 independently, which was not observed in cells or in complex proteomes, further complicating in vitro biochemical and structural analysis. Furthermore, we also do not understand the endogenous role of UBR7 and whether asukamycin affects this function. UBR7 belongs to the UBR family of N-recognin E3 ligases, but unlike UBR1–6, contains a plant homeodomain (PHD) rather than an F-box domain 41. It has been reported that UBR7 can ubiquitinylate histones and has a variety of interacting partners in other contexts 42. Our data currently suggest that the asukamycin-mediated phenotypes and effects upon TP53 activity are independent of ubiquitination functions of UBR7. However, we cannot exclude the normal functional role of UBR7 in conferring anti-cancer activity of asukamycin 27,42. Previous studies have suggested that UBR7 functions as a tumor suppressor in triple-negative breast cancer cells 27, through reducing monoubiquitination of histone H2B and reducing CDH4 levels, but our proteomic profiling studies show that asukamycin does not affect CDH4 expression (Source Data Tables for Extended Data Figure 9) and asukamycin broadly impairs TNBC cell growth, further indicating that asukamycin confers a gain-of-function to UBR7, independent of its potential endogenous role. Further work will be critical in better understanding the role of UBR7 and cancer.
Despite these remaining questions, our study showcases the possibility that multi-covalent small-molecules can potentially act as new molecular glues and that these interactions can be quickly deciphered using chemoproteomic approaches. Of future interest will be whether other natural products or synthetic small-molecules that bear multiple reactive centers can act as specific molecular glues to confer new protein functions. In addition, the ability of manumycin polyketides to template the interaction of UBR7 with multiple other proteins warrants the further examination of this natural product family for applications beyond cancer. Quite intriguingly, unsaturated side chains reminiscent of the manumycins appear in many other natural products, many of which also harbor diverse electrophilic warheads 43–45. Moreover, the multitude of polycyclic natural products containing one or more potentially protein-reactive functional groups is vast, implying this concept might extend across a greater section of natural product space 46,47.
Online Methods
Materials
Asukamycin and Manumycin A were obtained from Cayman Chemicals and were >95 % pure. Heavy and light TEV-biotin tags were synthesized per previously described methods 17. Manumycin D synthesis and characterization is in Synthetic Procedures. Recombinant UBR7 pure proteins were purchased from Origene or NOVUS Biologicals. Recombinant TP53 pure protein was purchased from R&D Systems.
Cell Culture
The 231MFP cells were obtained from Prof. Benjamin Cravatt and were generated from explanted tumor xenografts of MDA-MB-231 cells as previously described 48. HCC38 and HEK293T cells were obtained from American Type Culture Collection (ATCC). Isogenic HCT116 colorectal cancer cell lines were generated by Novartis. 231MFP cells were cultured in L15 medium (HyClone) containing 10% FBS and 2 mM glutamine and maintained at 37℃ with 0% CO2. HEK293T and isogenic HCT116 cell lines cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% FBS and 2 mM glutamine and maintained at 37℃ with 5% CO2. HCC38 cells were cultured in RPMI medium containing 10% FBS and maintained at 37℃ with 5% CO2.
Survival and Proliferation Assays
Cell survival and proliferation studies were performed using Hoechst 33342 dye (Invitrogen) as described previously 26. Briefly, 231MFP cells were seeded at 20,000 (proliferation) or 40,000 (survival) cells/well, respectively, in serum-containing (proliferation) or serum-free (survival) media in 96-well plates and allowed to adhere overnight. Under serum-free survival conditions, we have previously observed that the cells do not proliferate, and thus we are measuring loss of viability in these cells under serum-free conditions with pharmacological treatment 49. Under serum-containing conditions, the cells proliferate normally, and thus pharmacological effects in this assay indicate a combination of loss of proliferation and viability. The cells were treated with DMSO vehicle- or Asukamycin-containing media for 24 or 48 h before fixation and staining with 10% formalin and Hoechst 33342 dye. Studies with HCC38 cells were also performed as above but were seeded with 10,000 cells/well for proliferation and 20,000 cells/well for survival.
Growth Inhibition Assay Across Cancer Cell Line Panel
Cells in growth medium were plated into a 1536 well plate (5 μL/well; 250 cells/well) using a GNF Bottle Valve liquid handler. A Labcyte Echo acoustic transfer instrument was used to transfer 15 nL of compounds in DMSO to each well (final concentration 30 μM, 9.5 μM, 3 μM, 1 μM, 0.3 μM, 0.1 μM, 0.03 μM, and 0.01 μM). The cells were then incubated (37 °C, 95% Humidity, 5% CO2) for 3 days and 6 hours prior to addition of 4 μL of 50% Cell-Titer Glo (Promega) in water using a GNF Bottle Valve liquid handler. Plates were incubated with Cell Titer Glo for 15 minutes at room temperature prior to reading luminescence (5 s exposure) on a Perkin Elmer ViewLux. For determining GI50 values, data was normalized to a day 0 cell count measured using a cell plate copy that was not treated with compound and growth inhibition dose-response curves were calculated using Helios.
IsoTOP-ABPP of Asukamycin Targets
IsoTOP-ABPP studies were done as previously reported 26. 231MFP cells were treated with DMSO vehicle or 10 μM of Asukamycin for 3 h. Cells were then harvested and lysed by probe sonication in PBS and protein concentrations were determined by bicinchoninic acid (BCA) assay (Pierce). Proteomes were subsequently labeled with 100μM of iodoacetamide (IA)-alkyne (CHESS Gmbh.) at room temperature (RT) for 1 h. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) was performed by sequential addition of tris(2-carboxyethyl) phosphine (1 mM, Sigma), tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (34 mM, Sigma), copper (II) sulfate (1 mM, Sigma), and biotin-linker-azide, the linker functionalized with a TEV protease recognition sequence along with an isotopically light or heavy valine for treatment of vehicle- or Asukamycin-treated proteome, respectively. After CuAAC, proteomes were precipitated by centrifugation at 6500×g, washed in ice-cold methanol, combined in a 1:1 vehicle/Asukamycin ratio, washed again, then denatured and resolubilized by heating in 1.2 % SDS/PBS at 80℃ for 5 min. Insoluble components were precipitated by centrifugation at 6500×g and soluble proteome was diluted with PBS to be 0.2% of SDS. Labeled proteins were bound to streptavidin-agarose beads (Pierce) while rotating overnight at 4℃. After washing three times each in PBS and water, the bead-linked proteins were resuspended in 6 M urea/PBS and reduced in 1 mM of TCEP, alkylated with 18 mM of IA (Sigma), then washed and resuspended in 2 M urea and trypsinized overnight at 37℃ with 2 ug/sample sequencing grade trypsin (Promega). After washing three times each in PBS and water, the beads were resuspended in TEV buffer solution (1×TEV buffer containing 100 μM of dithiothreitol) and incubated overnight at 29 °C with Ac-TEV protease (Invitrogen). Peptides were diluted in water and acidified with 1.2 M of formic acid (Spectrum) for isoTOP-ABPP analysis. Subsequent steps of the isoTOP-ABPP and mass spectrometry analysis were performed using the same methods as we have described previously26.
Data was extracted in the form of MS1 and MS2 files using Raw Extractor 1.9.9.2 (Scripps Research Institute) and searched against the Uniprot human database using ProLuCID search methodology in IP2 v.3 (Integrated Proteomics Applications, Inc) 50. Cysteine residues were searched with a static modification for carboxyaminomethylation (+57.02146) and up to two differential modifications for methionine oxidation and either the light or heavy TEV tags (+464.28596 or +470.29977, respectively). Peptides were required to be fully tryptic peptides and to contain the TEV modification. ProLUCID data was filtered through DTASelect to achieve a peptide false-positive rate below 5%. Only those probe-modified peptides that were evident across two out of three biological replicates were interpreted for their isotopic light to heavy ratios. For those probe-modified peptides that showed ratios >2, we only interpreted those targets that were present across all three biological replicates, were statistically significant, and showed good quality MS1 peak shapes across all biological replicates. Light versus heavy isotopic probe-modified peptide ratios are calculated by taking the mean of the ratios of each replicate paired light vs. heavy precursor abundance for all peptide spectral matches (PSM) associated with a peptide. The paired abundances were also used to calculate a paired sample t-test p-value in an effort to estimate constancy within paired abundances and significance in change between treatment and control. P-values were corrected using the Benjamini/Hochberg method. Data processing and statistical analysis algorithms from our lab can be found on our lab’s Github site: https://github.com/NomuraRG, and we can make any further code from this study available at reasonable request.
Gel-Based ABPP
Recombinant pure protein (0.1μg/sample) was pre-treated with either DMSO vehicle or natural products at 37℃ for 30 min in 25 μL of PBS, and subsequently treated with 200 nM of IA-Rhodamine (Setareh Biotech) at RT for 1 h. The reaction was stopped by addition of 4×reducing Laemmli SDS sample loading buffer (Alfa Aesar). After boiling at 95℃ for 5 min, the samples were separated on precast 4–20% Criterion TGX gels (Bio-Rad). Probe-labeled proteins were analyzed by in-gel fluorescence using a ChemiDoc MP (Bio-Rad).
Constructing Knockdown Lines and Reinforced Expression or Overexpression Lines
Short-hairpin oligonucleotides were used to knock down the expression of UBR7 or TP53 in 231MFP cells using previously described methods 26. For lentivirus production, lentiviral plasmids and packaging plasmids (pMD2.5G, Addgene nos. 12259 and psPAX2, Addgene no. 12260) were transfected into HEK293T cells using Lipofectamine 2000 (Invitrogen). Lentivirus was collected from filtered cultured medium and used to infect the target cell line with 1:1000 dilution of polybrene. Target cells were selected over 3 days with 1 μg/ml of puromycin. The short-hairpin sequences which were used for generation of the knockdown lines were:
UBR7: CCGGGATGATGTCCGGGAGGTTAAACTCGAGTTTAACCTCCCGGACATCATCTTTTTG (Sigma UBR7 MISSION shRNA Bacterial Glycerol Stock, TRCN0000294293).
TP53: CCGGCGGCGCACAGAGGAAGAGAATCTCGAGATTCTCTTCCTCTGTGCGCCGTTTTT (Sigma TP53 MISSION shRNA Bacterial Glycerol Stock, TRCN0000003753).
PRKDC: CCGGCCTGAAGTCTTTACAACATATCTCGAGATATGTTGTAAAGACTTCAGGTTTTTTG (Sigma PRKDC MISSION shRNA Bacterial Glycerol Stock, TRCN0000194719).
MISSION TRC1.5 pLKO.1- or TRC2 pLKO.5-puro Non-Mammalian shRNA Control (Sigma) was used as a control shRNA.
For expression of WT or C374A mutant UBR7 upon UBR7 knockdown, cells were transiently transfected with UBR7 expression plasmid using Lipofectamine 2000. Wild-type human UBR7 expression plasmid with C-terminal FLAG tag was purchased from Origene (RC218298). The UBR7 C374A mutant was generated with Q5 Site-Directed Mutagenesis Kit (New England Biolabs) according to manufacturer’s protocols.
For making stable overexpression lines, lentivirus was used as with knockdown lines described above. To make lentiviral constructs for GFP and UBR7, Gibson assembly was performed using a Gibson Assembly Cloning Kit (New England Biolabs) according to manufacturer’s protocols. The primer sequences which were used to amplify the ORFs with desired overlaps, or to linearize the pLenti-Entry backbone (PS100069, Origene) were:
GFP Forward: GCCGCCGCGATCGCCatggtgagcaagggcgagg
GFP Reverse: CGGCCGCGTACGCGTcttgtacagctcgtccatgc
UBR7 Forward: GCCGCCGCGATCGCCatggccggagccgagggcg
UBR7 Reverse: CGGCCGCGTACGCGTgctgcagtaatactgcatcg
pLenti-Entry Forward: ACGCGTACGCGGCCGCTCGAG
pLenti-Entry Reverse: GGCGATCGCGGCGGCAGATC
For transient knockdown of UBR7 with siRNA (Dharmacon), cells were seeded in 6-well plates overnight (200,000 cells/well for 231MFP cells and 300,000 cells/well for HEK293T cells) and then transfected with either non-targeting siRNA oligonucleotide (siControl, D-001810–01, Dharmacon) or siUBR7 oligonucleotides (J-016489–11, Dharmacon) using Dharmafect 1 (Dharmacon). Cells were harvested 48 h after transfection for seeding for luciferase reporter assays or were treated with DMSO vehicle or asukamycin 72 h after transfection to see protein expression by Western blotting.
Western Blotting
FLAG antibody (M2) was obtained from Sigma. Antibodies to UBR7 (PA5–31559) and p53 (DO-1) were obtained from Thermo Fisher Scientific. Antibodies to GAPDH (D16H11), Ubiquitin (P4D1), FLAG (D6W5B), p21 (12D1), Histone H2A II (2578S), p-Histone H2A.X (2577S), RPA32 (4E4), and p-RPA32/RPA2 (83745S) were obtained from Cell Signaling Technology.
Proteins were resolved by SDS/PAGE and transferred to nitrocellulose membranes using the iBlot system (Invitrogen). Membranes were blocked with 5 % nonfat milk or BSA in Tris-buffered saline containing Tween 20 (TBS-T) solution for 30 min at RT, washed in TBS-T, and probed with primary antibody diluted in recommended diluent per manufacturer overnight at 4℃. After 3 times washes with TBS-T, the membranes were incubated in the dark with IR800-conjugated secondary antibodies at 1:10,000 dilution in 5 % nonfat milk in TBS-T at RT for 1 h. Blots were visualized using an Odyssey Li-Cor fluorescent scanner. The membranes were stripped using ReBlot Plus Strong Antibody Stripping Solution (EMD Millipore) when additional primary antibody incubations were performed. For dual color Western blotting, anti-mouse IR680 and anti-rabbit IR800 were used to detect TP53 (DO-1) and FLAG (D6W5B), respectively.
Quantitative PCR
Total RNA was extracted from cultured cells with TRIzol reagent (Thermo Fisher Scientific), and cDNA was synthesized with the Maxima Reverse Transcriptase (Thermo Fisher Scientific). Gene expression was measured from the cDNA using DyNAmo HS SYBR Green qPCR Kit (Thermo Fisher Scientific) on a BioRad CFX Connect PCR system. Samples were measured in technical triplicate and PRKDC expression was normalized to GAPDH levels and calculated using the comparative CT method. The following primer sequences were used for qPCR.
PRKDC Forward: CTGTGCAACTTCACTAAGTCCA
PRKDC Reverse: CAATCTGAGGACGAATTGCCT
GAPDH Forward: GGAGCGAGATCCCTCCAAAAT
GAPDH Reverse: GGCTGTTGTCATACTTCTCATGG
Anti-FLAG Pulldown
231MFP cells with stable expression of FLAG-GFP or FLAG-UBR7 were treated with DMSO vehicle or 50 μM of Asukamycin for 3 h. Cells were collected, washed twice with PBS, and lysed by probe sonication in TBS. After centrifugation at 12,000×g for 10 min, the supernatant was incubated with Anti-DYKDDDDK G1 affinity resin (GenScript) at 4℃ for 3 h. Beads were washed two times with 1 mL of cold TBS (10 min per incubation), and the samples were eluted two times by 250 μg/mL of 3×FLAG-peptide solution. Eluent was subsequently prepared for proteomic analysis as described below.
Proteomic Analysis of Pulldown Samples
The samples were precipitated by addition of trichloroacetic acid (TCA) at a final concentration of 20% and incubated at −80℃ for 1 h. The samples were then centrifuged at 14,800 rpm for 10 min and supernatant was carefully removed. After washing two times with ice cold 0.01 M HCl/90 % acetone solution, the precipitated protein was resuspended in 4 M urea containing 0.1 % Protease Max (Promega) and diluted in 40 mM ammonium bicarbonate buffer. The samples were reduced with 10 mM TCEP at 60℃ for 30 min. The samples were then diluted with PBS and incubated overnight at 37℃ with sequencing grade trypsin. After centrifugation at 13,200 rpm for 30 min, the supernatant was acidified with 5 % formic acid and was subsequently analyzed by LC-MS/MS. Data was analyzed by spectral counting. Only those proteins that showed >2 peptides in at least one sample was subsequently interpreted. While this filtered and original list was interpreted for average, sem, and p-values, for visual representation of this proteomic data in Figure 3a, we set those proteins that showed averages <1 within a particular group as 1 to generate fold-changes that could be plotted in the figure.
In-gel Digestion of Higher Molecular Weight Region for Pulldown Products
Products from anti-Flag pulldown were resolved by 4–20% SDS/PAGE using Tris-Glycine precast Midi-PROTEAN TGX gel (BioRad). The molecular weight region between 100 and 140 kDa was excised from the gel. Samples were then reduced, alkylated, and tryptically digested in-gel as previously described 51.
Tryptic peptides were analyzed using a Q-Exactive Plus mass spectrometer with the same method for isoTOP-ABPP.
FLAG-UBR7 Immunodepletion
231MFP cells with stable expression of FLAG-UBR7 were washed twice with PBS, and lysed by probe sonication in TBS containing protease inhibitor cocktail (Pierce), and protein concentrations were determined by BCA assay. Cell lysates (1 mg/mL) were incubated with FLAG antibody (Sigma, M2, 20 μg) on ice for 1 h. Subsequently samples were incubated with 100 μL of Pierce Protein G Agarose (Pierce) by rotating overnight at 4 °C. Samples were centrifuged, and the unbound supernatant was collected for thermal shift assay.
In Vitro Deubiquitination Assay
231MFP cells were treated with either DMSO vehicle or 50 μM of Asukamycin for 3 h. Cells were then harvested and lysed by probe sonication in TBS, and protein concentrations were determined by BCA assay. Cell lysates were incubated with deubiquitinating enzyme USP2 (R&D Systems) at 37℃ for 1 h. The reaction was stopped by addition of 4× reducing Laemmli SDS sample loading buffer. The reaction products were analyzed by Western blotting.
Thermal Shift Assay
231MFP cells were lysed by probe sonication in TBS containing complete protease inhibitors cocktail (Roche), and protein concentrations were determined by BCA assay. Cell lysates (1 mg/mL) were treated with either DMSO vehicle or 50 μM of Asukamycin for 1 h at 37℃. Cell lysates were then separated into 8 fractions for thermal profiling. Fractions were heated at the indicated temperatures (42–60℃) for 3 min using a thermal cycler (T100 Thermal Cycler, Bio-Rad), followed by 3 minutes at room temperature. Samples were then centrifuged at 16,000 × g for 15 min to separate protein aggregates from soluble proteins. Supernatants were collected and analyzed by Western blotting.
In Vitro Transcription Factor Assay for TP53
Recombinant TP53 protein (0.05 μg/sample) was spiked into 40 μL of 231MFP breast cancer cell lysate (0.5 mg/mL in TBS) and then treated with either DMSO vehicle or 50 μM of Asukamycin for 1 h at 37℃. TP53 DNA binding to p53 DNA consensus sequence was then assessed by using the p53 transcription factor assay kit (Cayman) according to manufacturer’s protocols.
Luciferase Reporter Assay
HEK293T cells were seeded at 30,000 cell/well in 96-well plates. After 24 h, the cells were transiently co-transfected with 0.1 μg /well of pGL4.38 [luc2P/p53 RE/Hygro] (Promega) and 0.02 μg/well of pRL-TK (renilla luciferase control vector) using Lipofectamine 2000. After treating the cells with compounds for 6 h, the expression levels of the firefly and renilla luciferase reporter genes were examined by a Dual-Glo luciferase assay system (Promega) according to the manufacturer’s protocols. The luminescent signals were measured using a SpectraMax i3 plate reader (Molecular Devices).
LC-MS/MS Analysis of Intracellular Asukamycin Level
231MFP cells were seeded at 1,000,000 cells/dish in serum-containing L-15 media in 6-cm dishes and allowed to adhere overnight. Media was than replaced with serum-containing or serum-free L-15 containing DMSO or Asukamycin. 1.5 h later cells were washed twice with PBS, and Asukamycin was extracted from the cells using chloroform:methanol:PBS solution mixture (2:1:1, v/v/v; 4 ml total) with 3-O-Dodecyl-sn-glycerol (10 nmol, Santa Cruz Biotechnology) as an internal standard. The organic layer was collected, evaporated under stream of N2, resolubilized in chloroform and analyzed by multiple-reaction monitoring-based targeted liquid chromatography-tandem mass spectrometry on an Agilent 6430 QQQ using a Luna reverse-phase C5 column (50 × 4.6 mm with 5 mm diameter particles, Phenomenex) as described previously 52. Parent/daughter ion multiple-reaction monitoring transitions used to determine asukamycin levels were 547.2/ 189.2 with collision energy of 15 V. Peak area of asukamycin to that of internal standard was determined.
Quantitative TMT Proteomics Analysis
Quantitative TMT-based proteomic analysis was performed as previously described 26. Acquired MS data was processed using Proteome Discoverer v. 2.2.0.388 software (Thermo) utilizing Mascot v 2.5.1 search engine (Matrix Science, London, UK) together with Percolator validation node for peptide-spectral match filtering 53. Data was searched against Uniprot protein database (canonical human and mouse sequences, EBI, Cambridge, UK) supplemented with sequences of common contaminants. Peptide search tolearances were set to 10 ppm for precursors, and 0.8 Da for fragments. Trypsin cleavage specificity (cleavage at K, R except if followed by P) allowed for up to 2 missed cleavages. Carbamidomethylation of cysteine was set as a fixed modification, methionine oxidation, and TMT-modification of N-termini and lysine residues were set as variable modifications. Data validation of peptide and protein identifications was done at the level of the complete dataset consisting of combined Mascot search results for all individual samples per experiment via the Percolator validation node in Proteome Discoverer. Reporter ion ratio calculations were performed using summed abundances with most confident centroid selected from 20 ppm window. Only peptide-to-spectrum matches that are unique assignments to a given identified protein within the total dataset are considered for protein quantitation. High confidence protein identifications were reported using a Percolator estimated <1% false discovery rate (FDR) cut-off. Differential abundance significance was estimated using a background-based ANOVA with Benjamini-Hochberg correction to determine adjusted p-values.
Gene enrichment analysis was performed using gProfiler. Statistical analysis was performed using the g:SCS algorithm for computing multiple testing correction for p-values gained from GO and pathway enrichment analysis. It corresponds to an experiment-wide threshold of a=0.05. Given a fixed input query size, g:SCS analytically approximates a threshold t corresponding to the 5% upper quantile of randomly generated queries of that size. All actual p-values resulting from the query are transformed to corrected p-values by multiplying these to the ratio of the approximate threshold t and the initial experiment-wide threshold a=0.05 54–56.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code Availability Statement
Data processing and statistical analysis algorithms from our lab can be found on our lab’s Github site: https://github.com/NomuraRG, and we can make any further code from this study available at reasonable request.
Extended Data
Extended Data Fig. 1. Testing asukamycin growth inhibitory effects across 250 cancer cell lines.
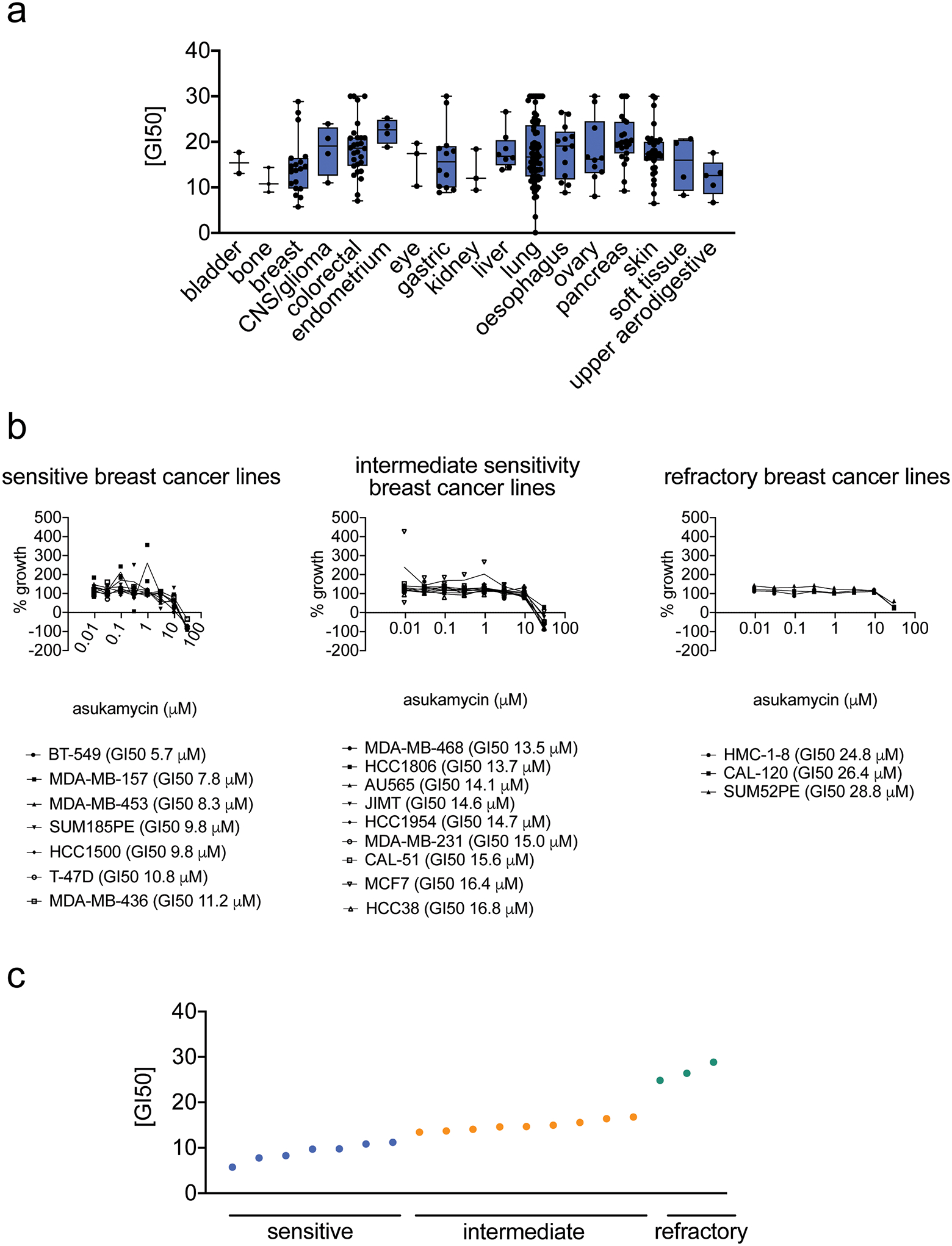
(a) GI50 values for asukamycin across 250 cancer cell lines by cancer tissue origin. The individual cell line data can be found in Source Data Tables for Extended Data Figure 1. The box plot shows median, standard deviation, and minimum and maximum ranges. For bladder cancer, there were 2 cell lines; for bone cancer, 3 cell lines; breast cancer, 19 cell lines; CNS/glioma cancers, 4 cell lines; colorectal cancer, 28 cell lines; endometrial cancer, 4 cell lines; eye cancers, 3 cell lines; gastric cancers, 12 cell lines; kidney cancer, 3 cell lines; liver cancer, 8 cell lines; liver cancer, 8 cell lines; lung cancer, 73 cell lines; esophageal cancer, 13 cell lines, ovarian cancer, 10 cell lines; pancreatic cancer, 23 cell lines; skin cancer, 35 cell lines; soft tissue cancers, 4 cell lines; upper aeodigestive cancers, 5 cell lines. (b, c) Dose-response curves for % growth of 19 breast cancer cell lines tested (b) and GI50 values (c) for asukamycin treatment, separated by relative sensitivity to asukamycin. Data from (a-c) were calculated in each cell line from n=2 biologically independent samples/group.
Extended Data Fig. 2. TP53 levels in 100–140kDa range in SDS/PAGE gel from FLAG-UBR7 pulldown.
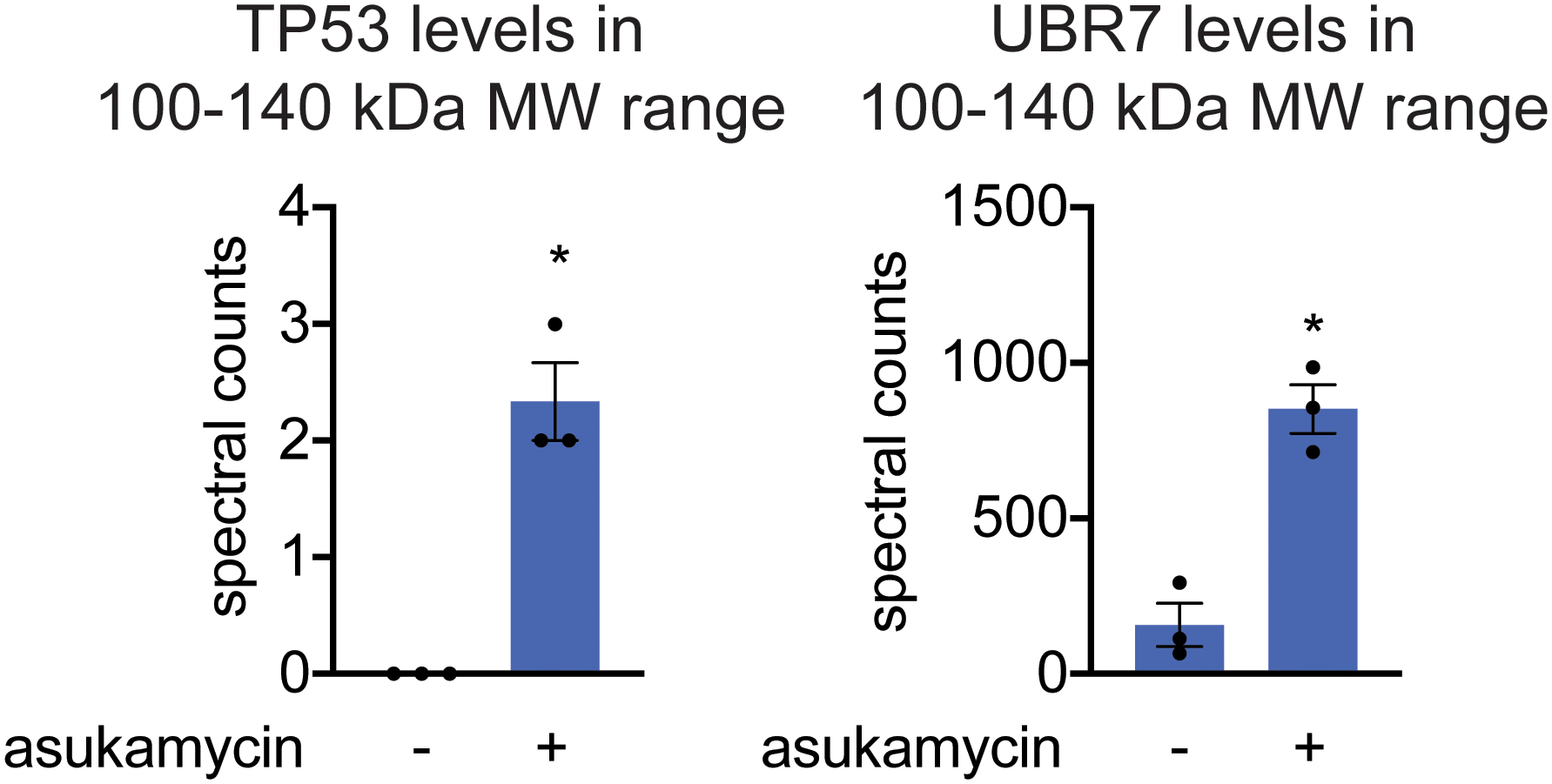
LC-MS/MS proteomic analysis of 100–140 kDa range tryptic digests of SDS/PAGE gels from FLAG-UBR7 pulldown from 231MFP cells treated with DMSO vehicle or asukamycin (50 μM) for 3 h. Data shown as individual replicate spectral count values and average ± sem and are n=3 biologically independent samples/group. Significance was calculated by a Student’s two-tailed t-test and is shown as *p<0.05 in asukamycin-treated pulldown samples compared to vehicle-treated controls. Source data can be found in Source Data Tables for Extended Figure 2.
Extended Data Fig. 3. Understanding the composition of the asukamycin-mediated higher molecular weight band.
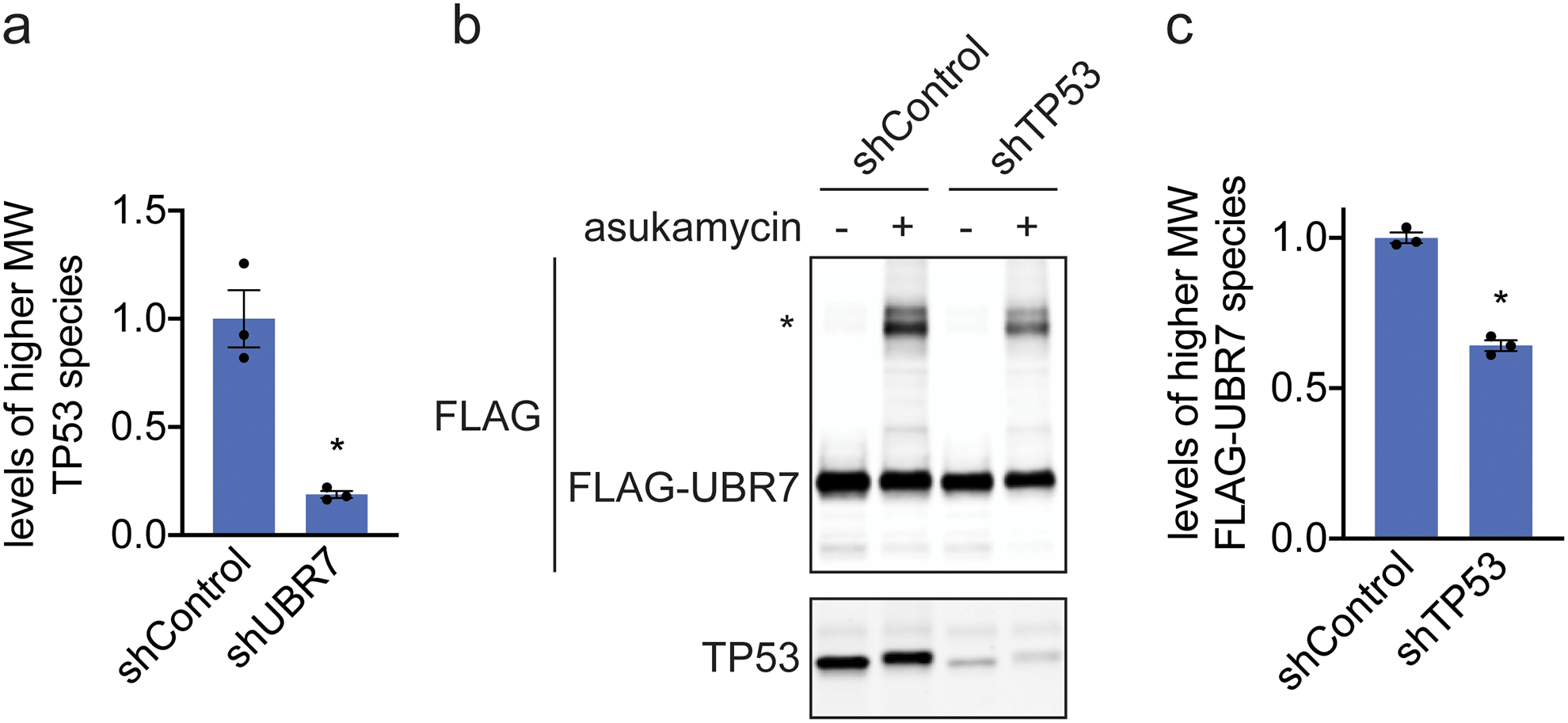
(a) Quantification of TP53 levels normalized to GAPDH levels in Western blot shown in Figure 2d. (b) Anti-FLAG and anti-TP53 blot in shControl and shTP53 231MFP breast cancer cells expressing FLAG-UBR7 treated with vehicle DMSO or asukamycin (50 μM) for 3 h. * notes the higher molecular FLAG-UBR7 band. (c) Quantification of higher molecular weight FLAG-UBR7 band noted with * in (b). Data shown in (a, c) as individual replicate values and average ± sem and are n=3 biologically independent samples/group. Gel shown in (b) is a representative gel of n=3 biologically independent samples/group. Statistical significance was calculated with two-tailed unpaired Student’s t-tests and are shown as *p<0.05 compared to shControl cells treated with asukamycin in (a, c). Uncropped blots can be found in Source Data for Extended Figure 3. Source data for bar graphs can be found in Source Data Tables for Extended Figure 3.
Extended Data Fig. 4. TP53 pulldown in wild-type or C374A mutant UBR7-expressing cells.
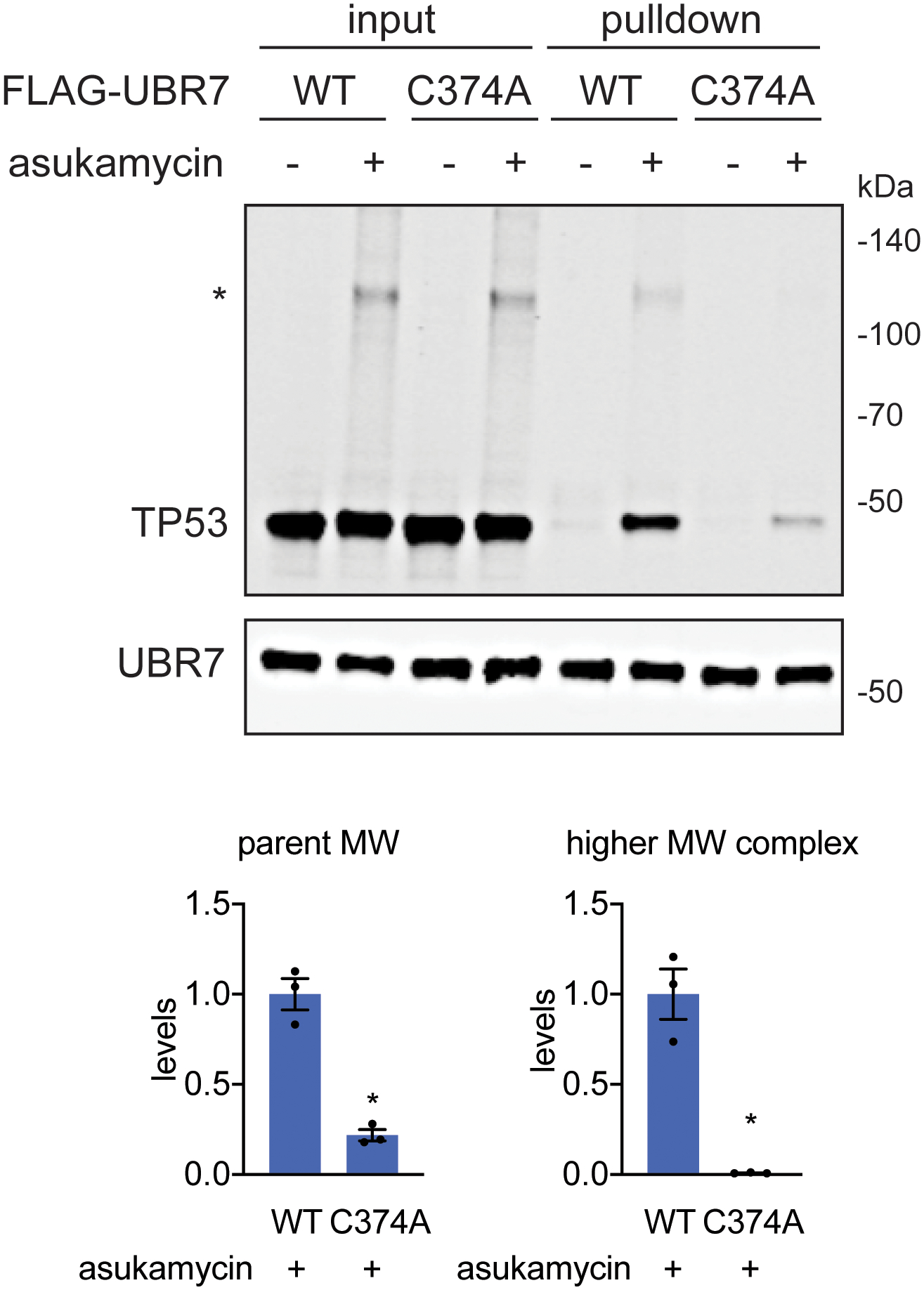
TP53 and UBR7 levels in input and pulled down eluate from FLAG-UBR7 pulldown in 231MFP cells stably expressing wild-type or C374A mutant FLAG-UBR7 in cells treated with DMSO vehicle or asukamycin (50 μM) treatment for 3 h, assessed by Western blotting. The “*” indicates the higher molecular weight (MW) TP53-asukamycin-UBR7 ternary complex. The gel is representative of n=3 biologically independent samples/group. Bar graphs below show quantification of parent and higher molecular weight TP53 levels in asukamycin-treated FLAG-pulldown samples from wild-type and C374A-FLAG-UBR7 expressing cells. Statistical significance was calculated with two-tailed unpaired Student’s t-tests and are shown as *p<0.05 compared to WT asukamycin-treated groups. Uncropped blots can be found in Source Data for Extended Figure 4. Source data for bar graphs can be found in Source Data Tables for Extended Figure 4.
Extended Data Fig. 5. Thermal stability of TP53 in 231MFP cell lysate.
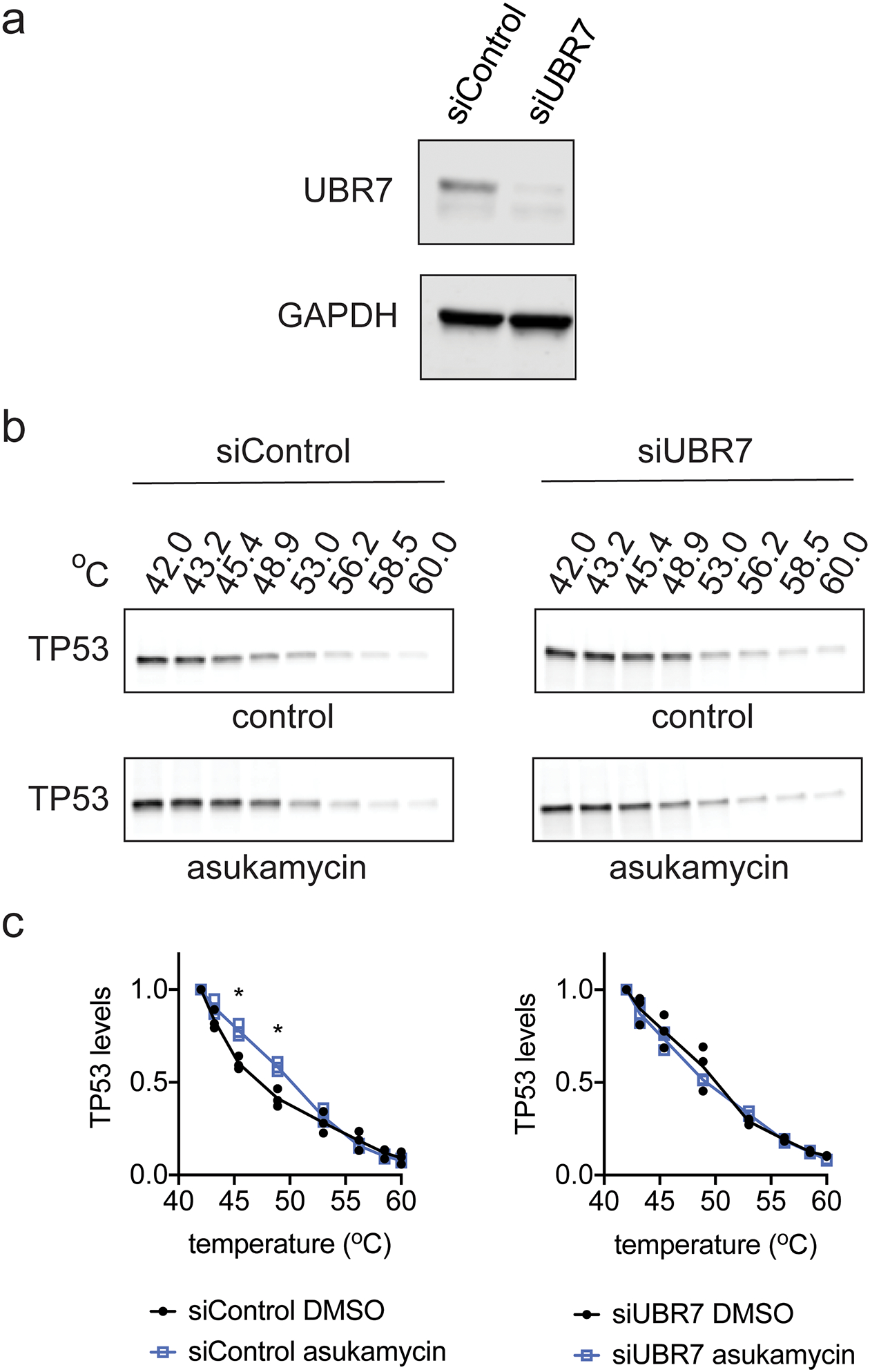
(a) UBR7 and loading control GAPDH expression in siControl and siUBR7 231MFP cells. (b) Thermal stability of TP53 in DMSO vehicle or asukamycin (50 μM, 1 h) treated cell lysate from 231MFP siControl or siUBR7 cells. Gels are representative of n=3 biologically independent samples/group. (c) Quantification of thermal shift assay from (b). Data in (c) shown as individual replicate values and average ± sem and are n=3 biologically independent samples/group. Statistical significance in (c) was calculated with two-tailed unpaired Student’s t-tests and are shown as *p<0.05 compared to vehicle-treated siControl groups. Data was not significant (p>0.05) for all asukamycin versus DMSO comparisons in siUBR7 groups. Uncropped blots can be found in Source Data for Extended Figure 5. Source data for plots can be found in Source Data Tables for Extended Figure 5.
Extended Data Fig. 6. TP53 thermal stability.

(a) FLAG-UBR7, UBR7, and loading control GAPDH levels from 231MFP cells expressing FLAG-UBR7 after mock depletion or FLAG-UBR7 depletion as assessed by Western blotting. (b) TP53 thermal stability in cell lysate from 231MFP FLAG-UBR7-expressing cells treated with DMSO vehicle or asukamycin (50 μM, 1 h) after mock depletion or FLAG-UBR7 depletion. (c) Quantification of experiments described in (b). Gels in (a) are representative of n=3 biologically independent samples/group. Gels in (b) are representative of n=3 biologically independent samples/group which are quantified and shown as individual replicate values in (c). Statistical significance in (c) was calculated with two-tailed unpaired Student’s t-tests and are shown as *p<0.05 comparing UBR7-depleted asukamycin-treated groups to mock-depleted asukamycin-treated groups. Uncropped blots can be found in Source Data for Extended Figure 6. Source data for plots can be found in Source Data Tables for Extended Figure 6.
Extended Data Fig. 7. TP53 binding to DNA consensus sequence.
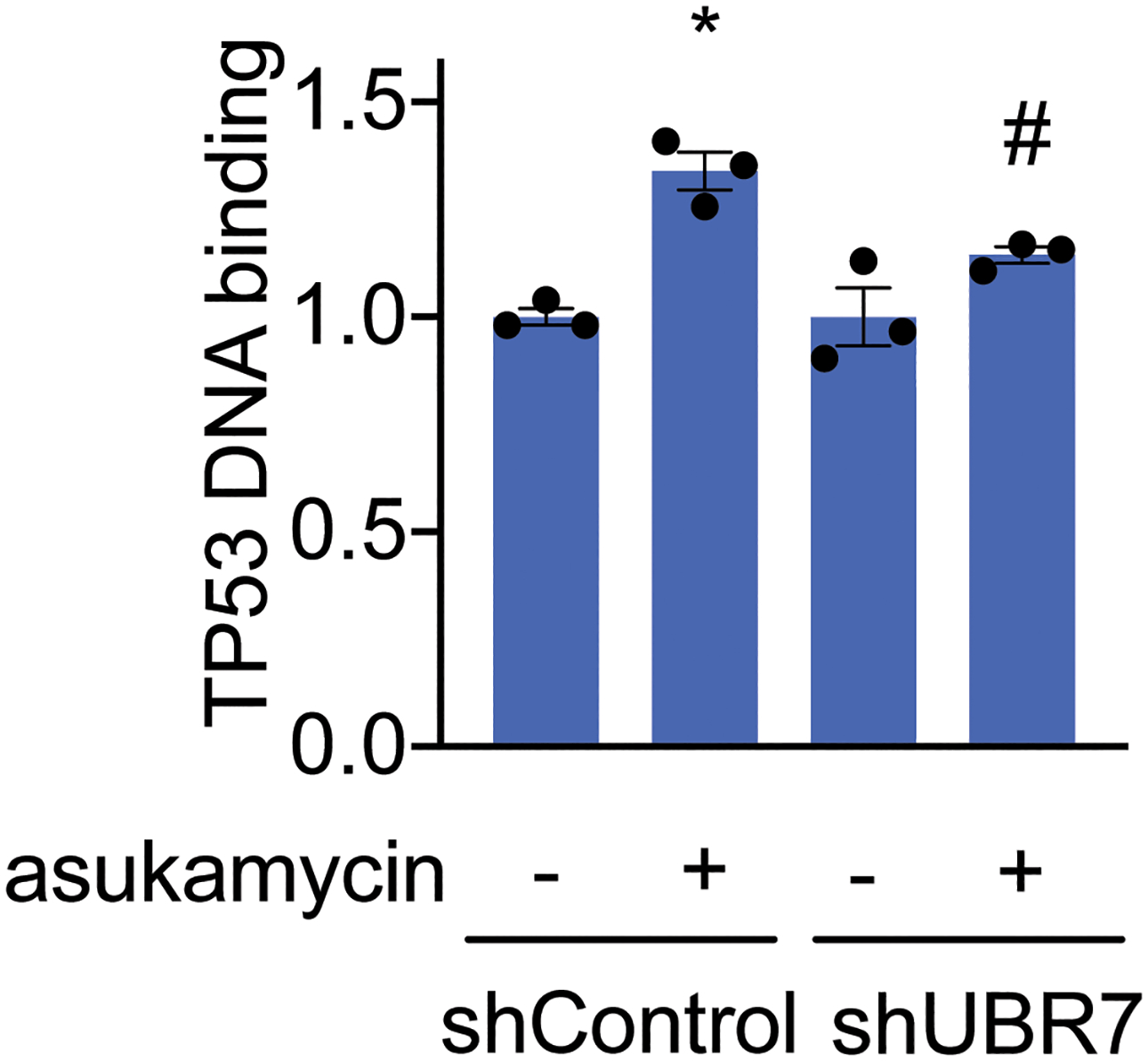
TP53 DNA binding to p53 DNA consensus sequence in vitro with TP53 spiked into 231MFP shControl or shUBR7 breast cancer cell lysate treated with DMSO vehicle or asukamycin (50 μM). Data shown are individual replicate values and average ± sem from n=3 biologically independent samples/group. Statistical significance is calculated with two-tailed unpaired Student’s t-tests and are shown as *p<0.05 compared to shControl vehicle-treated groups and #p<0.05 compared to shControl asukamycin-treated groups. Source data for bar graph can be found in Source Data Tables for Extended Figure 7.
Extended Data Fig. 8. TP53 reporter activity.
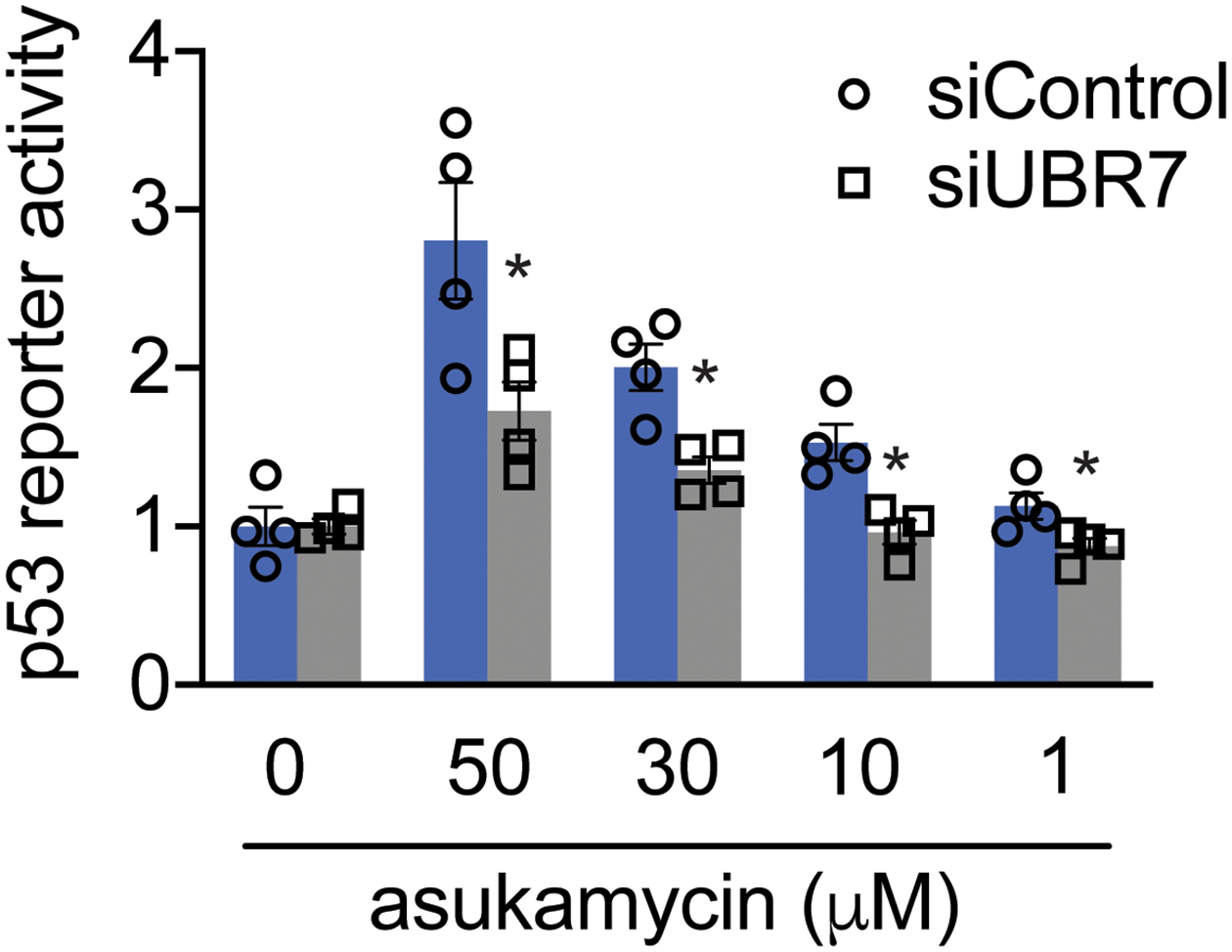
TP53 reporter activity in HEK293T siControl and siUBR7 cells treated with DMSO vehicle or asukamcyin for 3 h. Data shown are individual replicate values and average ± sem from n=4 biologically independent samples/group. Statistical significance is calculated with two-tailed unpaired Student’s t-tests and are shown as *p<0.05 compared to corresponding siControl treatment groups. Source data for bar graph can be found in Source Data Tables for Extended Figure 8.
Extended Data Fig. 9. Quantitative proteomic analysis of asukamycin treatment in 231MFP cells.
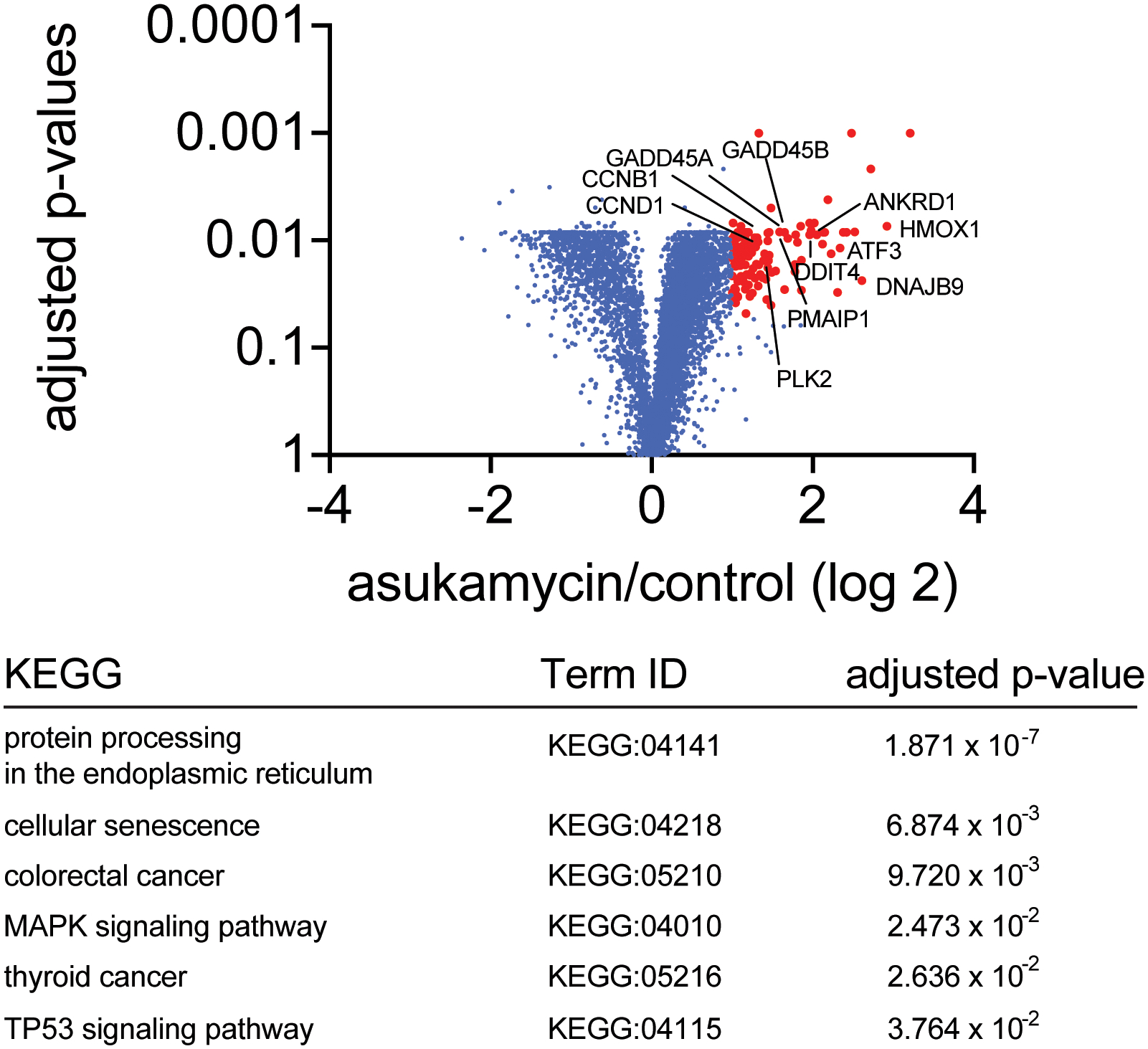
231MFP cells were treated with DMSO vehicle or asukamycin (50 mM, 12 h), and protein expression changes were assessed by quantitative TMT-based proteomics. Data shown are from n=3 biologically independent samples/group. Highlighted in red are proteins in which their expression was heightened by >2-fold with adjusted p-value<0.05. The full dataset can be found in Source Data Tables for Extended Data Figure 9. Highlighted are representative known p53 transcriptional targets. The lower table shows pathways that were significantly enriched in a functional enrichment analysis of the proteins that were upregulated by >2-fold with adjusted p-value<0.05, which includes TP53 signaling. Differential abundance significance from TMT-based proteomic data estimated using a background-based ANOVA with Benjamini-Hochberg correction to determine adjusted p-values. Statistical analysis for Pathway Enrichment Analysis was performed using the g:SCS algorithm for computing multiple testing correction for p-values gained from GO and pathway enrichment analysis. It corresponds to an experiment-wide threshold of a=0.05. Given a fixed input query size, g:SCS analytically approximates a threshold t corresponding to the 5% upper quantile of randomly generated queries of that size. All actual p-values resulting from the query are transformed to corrected p-values by multiplying these to the ratio of the approximate threshold t and the initial experiment-wide threshold a=0.05.
Extended Data Fig. 10. CDKN1A level changes with asukamycin treatment.
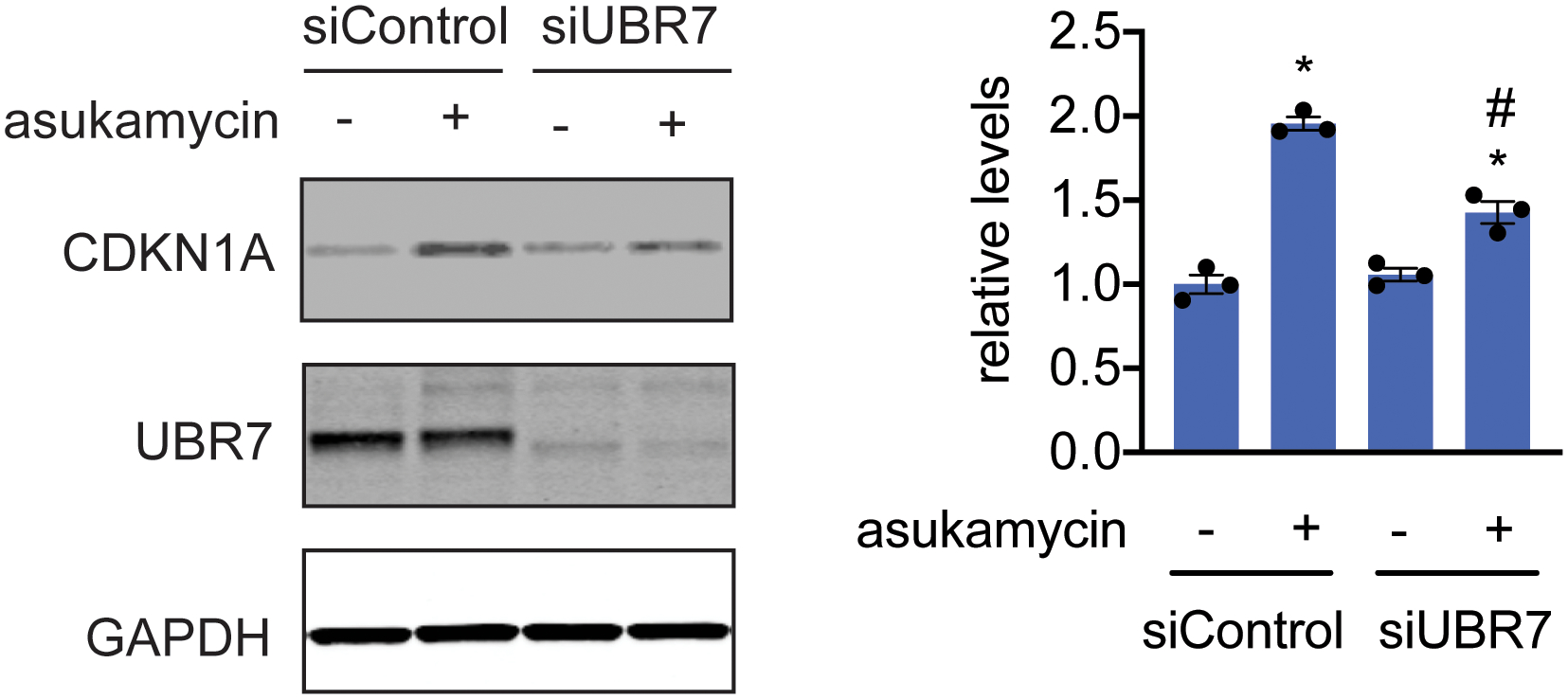
Tumor suppressor CDKN1A (p21), UBR7, and loading control GAPDH levels in 231MFP siControl and siUBR7 cells treated with DMSO vehicle or asukamycin (50 μM, 12 h), assessed by Western blotting, and quantified by densitometry and normalized to GAPDH levels. Gel on the left is representative of n=3 biologically independent samples/group. Bar graph on the right shows individual replicate values and average ± sem from n=3 biologically independent samples/group. Statistical significance is calculated with two-tailed unpaired Student’s t-tests and are shown as *p<0.05 compared to shControl vehicle-treated groups and #p<0.05 compared to shControl asukamycin-treated groups. Uncropped blots can be found in Source Data for Extended Figure 10. Source data for plots can be found in Source Data Tables for Extended Figure 10.
Supplementary Material
Acknowledgement
We thank the members of the Nomura Research Group, the Maimone Research Group, and Novartis Institutes for BioMedical Research for critical reading of the manuscript. This work was supported by Novartis Institutes for BioMedical Research and the Novartis-Berkeley Center for Proteomics and Chemistry Technologies (NB-CPACT) for all listed authors. We would like to thank Felipa Mapa, Peter Aspesi, Jessi Ambrose, Justin Oborski, Zhao Bin Kang, Matthew Shum, and Benjamin Cornett for their work profiling asukamycin in a panel of cancer cell lines. This work was also supported by the Nomura Research Group and the Mark Foundation for Cancer Research and Chordoma Foundation ASPIRE Award for DKN, YI. This work was also supported by grants from the National Institutes of Health (R01CA240981 for DKN, TJM, YI). YI and MO were also supported by the Japanese Society for the Promotion of Science (JSPS) postdoctoral fellowships.
Competing Financial Interests Statement
JAT, JMK, LM, MS, SMB, MDJ, XL, WF are employees of Novartis Institutes for BioMedical Research. This study was funded by the Novartis Institutes for BioMedical Research and the Novartis-Berkeley Center for Proteomics and Chemistry Technologies. DKN is a co-founder, shareholder, and adviser for Artris Therapeutics and Frontier Medicines.
References
- 1.Roberts AM, Ward CC & Nomura DK Activity-based protein profiling for mapping and pharmacologically interrogating proteome-wide ligandable hotspots. Curr. Opin. Biotechnol 43, 25–33 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Backus KM et al. Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burslem GM & Crews CM Small-Molecule Modulation of Protein Homeostasis. Chem. Rev 117, 11269–11301 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Hughes SJ & Ciulli A Molecular recognition of ternary complexes: a new dimension in the structure-guided design of chemical degraders. Essays Biochem. 61, 505–516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andrei SA et al. Stabilization of protein-protein interactions in drug discovery. Expert Opin. Drug Discov 12, 925–940 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Milroy L-G, Grossmann TN, Hennig S, Brunsveld L & Ottmann C Modulators of protein-protein interactions. Chem. Rev 114, 4695–4748 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Schreiber SL Chemistry and biology of the immunophilins and their immunosuppressive ligands. Science 251, 283–287 (1991). [DOI] [PubMed] [Google Scholar]
- 8.Ottmann C et al. A structural rationale for selective stabilization of anti-tumor interactions of 14-3-3 proteins by cotylenin A. J. Mol. Biol 386, 913–919 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Lu G et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chamberlain PP et al. Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat. Struct. Mol. Biol 21, 803–809 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Matyskiela ME et al. SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat. Chem. Biol (2018) doi: 10.1038/s41589-018-0129-x. [DOI] [PubMed] [Google Scholar]
- 12.Donovan KA et al. Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray Syndrome. eLife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo Z et al. Rapamycin-inspired macrocycles with new target specificity. Nat. Chem 11, 254 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sievers QL et al. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 362, eaat0572 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andrei SA et al. Stabilization of protein-protein interactions in drug discovery. Expert Opin. Drug Discov 12, 925–940 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Lagoutte R & Winssinger N Following the Lead from Nature with Covalent Inhibitors. Chimia 71, 703–711 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Weerapana E et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sattler I, Thiericke R & Zeeck A The manumycin-group metabolites. Nat. Prod. Rep 15, 221–240 (1998). [DOI] [PubMed] [Google Scholar]
- 19.Omura S, Kitao C, Tanaka H, Oiwa R & Takahashi Y A new antibiotic,, asukamycin, produced by Streptomyces. J. Antibiot. (Tokyo) 29, 876–881 (1976). [DOI] [PubMed] [Google Scholar]
- 20.Hu Y & Floss HG New type II manumycins produced by Streptomyces nodosus ssp. asukaensis and their biosynthesis. J. Antibiot. (Tokyo) 54, 340–348 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Shipley PR, Donnelly CCA, Le CH, Bernauer AD & Klegeris A Antitumor activity of asukamycin, a secondary metabolite from the actinomycete bacterium Streptomyces nodosus subspecies asukaensis. Int. J. Mol. Med 24, 711–715 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Arenz C et al. Manumycin A and its analogues are irreversible inhibitors of neutral sphingomyelinase. Chembiochem Eur. J. Chem. Biol 2, 141–143 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Hara M et al. Identification of Ras farnesyltransferase inhibitors by microbial screening. Proc. Natl. Acad. Sci. U. S. A 90, 2281–2285 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bianchini G, Balko JM, Mayer IA, Sanders ME & Gianni L Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol 13, 674–690 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kozawa I, Kato K, Teruya T, Suenaga K & Umezawa K Unusual intramolecular N-->O acyl group migration occurring during conjugation of (−)-DHMEQ with cysteine. Bioorg. Med. Chem. Lett 19, 5380–5382 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Spradlin JN et al. Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat. Chem. Biol 15, 747–755 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adhikary S et al. Atypical plant homeodomain of UBR7 functions as an H2BK120Ub ligase and breast tumor suppressor. Nat. Commun 10, 1398 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goodwin JF & Knudsen KE Beyond DNA repair: DNA-PK function in cancer. Cancer Discov. 4, 1126–1139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levine AJ & Oren M The first 30 years of p53: growing ever more complex. Nat. Rev. Cancer 9, 749–758 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Q, Bergman J, Wiman KG & Bykov VJN Role of Thiol Reactivity for Targeting Mutant p53. Cell Chem. Biol 25, 1219–1230.e3 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Zhan Q Gadd45a, a p53- and BRCA1-regulated stress protein, in cellular response to DNA damage. Mutat. Res 569, 133–143 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Kim Y-A et al. Gadd45β is transcriptionally activated by p53 via p38α-mediated phosphorylation during myocardial ischemic injury. J. Mol. Med. Berl. Ger 91, 1303–1313 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Fan F et al. ATF3 induction following DNA damage is regulated by distinct signaling pathways and over-expression of ATF3 protein suppresses cells growth. Oncogene 21, 7488–7496 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Oda E et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288, 1053–1058 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Du F et al. DDIT4 promotes gastric cancer proliferation and tumorigenesis through the p53 and MAPK pathways. Cancer Commun. 38, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee HJ et al. Genotoxic stress/p53-induced DNAJB9 inhibits the pro-apoptotic function of p53. Cell Death Differ. 22, 86–95 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matthew EM et al. The p53 target Plk2 interacts with TSC proteins impacting mTOR signaling, tumor growth and chemosensitivity under hypoxic conditions. Cell Cycle Georget. Tex 8, 4168–4175 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohno J et al. TMC-1 A, B, C and D, new antibiotics of the manumycin group produced by Streptomyces sp. Taxonomy, production, isolation, physico-chemical properties, structure elucidation and biological properties. J. Antibiot. (Tokyo) 49, 1212–1220 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Grové JJC, Wei X & Taylor RJK The first total synthesis of a type II manumycin antibiotic, (+)-TMC-1 A: the total syntheses of (–)-LL-C10037β and (+)-manumycin B. Chem. Commun 421–422 (1999) doi: 10.1039/A900261H. [DOI] [Google Scholar]
- 40.Bernier M et al. Binding of manumycin A inhibits IkappaB kinase beta activity. J. Biol. Chem 281, 2551–2561 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Tasaki T et al. The substrate recognition domains of the N-end rule pathway. J. Biol. Chem 284, 1884–1895 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kleiner RE, Hang LE, Molloy KR, Chait BT & Kapoor TM A Chemical Proteomics Approach to Reveal Direct Protein-Protein Interactions in Living Cells. Cell Chem. Biol 25, 110–120.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haritakun R, Srikitikulchai P, Khoyaiklang P & Isaka M Isariotins A-D, alkaloids from the insect pathogenic fungus Isaria tenuipes BCC 7831. J. Nat. Prod 70, 1478–1480 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Hammerschmidt L et al. Cytotoxic acyl amides from the soil fungus Gymnascella dankaliensis. Bioorg. Med. Chem 23, 712–719 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Amagata T, Minoura K & Numata A Gymnastatins F-H, cytostatic metabolites from the sponge-derived fungus Gymnascella dankaliensis. J. Nat. Prod 69, 1384–1388 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Gersch M, Kreuzer J & Sieber SA Electrophilic natural products and their biological targets. Nat. Prod. Rep 29, 659–682 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Zeng T et al. Exploring Chemical and Biological Space of Terpenoids. J. Chem. Inf. Model 59, 3667–3678 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Jessani N et al. Carcinoma and stromal enzyme activity profiles associated with breast tumor growth in vivo. Proc. Natl. Acad. Sci. U. S. A 101, 13756–13761 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nomura DK et al. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140, 49–61 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu T et al. ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J. Proteomics 129, 16–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shevchenko A, Tomas H, Havlis J, Olsen JV & Mann M In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc 1, 2856–2860 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Chung CY-S et al. Covalent targeting of the vacuolar H+-ATPase activates autophagy via mTORC1 inhibition. Nat. Chem. Biol 15, 776–785 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Käll L, Canterbury JD, Weston J, Noble WS & MacCoss MJ Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Ashburner M et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Raudvere U et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.