

Abstract
The outcomes of patients with primary plasma cell leukemia (pPCL) after undergoing hematopoietic cell transplantation (HCT) in the novel agent era are unknown. We report outcomes of 348 patients with pPCL receiving autologous (auto-) HCT (n= 277) and allogeneic (allo-) HCT (n=71) between 2008 and 2015. Median age was 60 years and 56 years for auto- and allo-HCT respectively. For auto-HCT, the 4-year outcomes were: non-relapse mortality (NRM) 7% (4-11%), relapse (REL) 76% (69-82%), progression-free survival (PFS) 17% (13-23%), and overall survival (OS) 28% (22-35%). Karnofsky performance status (KPS) >90 and ≥very good partial response (VGPR) predicted superior OS in multi-variate analysis for auto-HCT. For allo-HCT, the 4-year outcomes were: NRM 12% (5-21%), REL 69% (56-81%), PFS 19% (10-31%), and OS 31% (19-44%). Compared to prior CIBMTR pPCL patients (1995-2006), inferior survival was noted in the current cohort (3 -year OS, 39% vs. 38% in allo-HCT, and 62% vs. 35% in auto-HCT) respectively. However, we noted an increased HCT utilization, from 12% (7-21%) in 1995 to 46% (34-64%) in 2009 using SEER data (available till 2009). Despite modern induction translating to higher proportion receiving HCT, the outcomes remain poor in pPCL patients, mainly derived by high relapse rates post-HCT.
Keywords: Primary plasma cell leukemia, Hematopoietic cell transplantation, Novel agents
Introduction:
Plasma cell leukemia is an aggressive malignancy that comprises 1-2% of all plasma cell neoplasms. It may present as de novo (primary plasma cell leukemia, pPCL) disease, in the absence of antecedent multiple myeloma (MM) or may occur as a leukemic transformation of underling MM (secondary plasma cell leukemia, sPCL) (1, 2). The diagnosis of PCL requires >2 x 103 /μL of circulating plasma cells (CPCs) in the blood and CPCs accounting for >20% of white cell count (3, 4).
The prognosis of PCL is poor; historically the reported median survival has ranged between 2 to 11 months, with even shorter survival in cases of sPCL(5). In contrast to MM, the prognosis in PCL remains poor despite the availability of novel therapies (6–9). Older studies suggest that high dose chemotherapy followed by autologous hematopoietic cell transplantation (auto-HCT) or allogeneic hematopoietic cell transplantation (allo-HCT) may improve outcomes(7, 10). Given the rarity of the disease, PCL data are limited to single center retrospective and small prospective studies(11–14). Only one prospective trial has investigated the role of proteasome inhibitors (PIs) and immunomodulatory drugs (IMIDs) with consolidative HCT in the upfront treatment of pPCL(14).
Given the poor prognosis of pPCL, auto-HCT is routinely performed and allo-HCT is sometimes considered as consolidation therapy although the outcomes are not well established in the novel agent era. A previous study from the Center for International Bone and Marrow Transplant Research (CIBMTR®) examined these outcomes but the majority of did not receive PIs or IMIDs (7). We therefore conducted this analysis using the CIBMTR database to examine the role of HCT for pPCL from 2008 to 2015, reflecting an era with widespread use of PIs and/or IMIDs in the first line setting.
Methods:
Data Sources:
The CIBMTR is a research collaboration between The National Marrow Donor Program/Be The Match and the Medical College of Wisconsin. It comprises of a voluntary working group of more than 450 transplantation centers worldwide. Participating centers are required to report all transplants consecutively; compliance is monitored by on-site audits and patients are followed longitudinally. Computerized checks for discrepancies, physicians’ review of submitted data, and on-site audits of participating centers ensure data quality. Studies conducted by the CIBMTR are performed in compliance with all applicable federal regulations pertaining to the protection of human research participants. Protected health information used in the performance of such research is collected and maintained in CIBMTR’s capacity as a Public Health Authority under the HIPAA Privacy Rule.
Patient Selection:
Patients who met criteria for pPCL that underwent HCT for pPCL from 2008-2015 were included in the study. A supplemental data form for disease-specific questions was requested to the centers that contributed more than 10 patients/center for a total of 152 patients who were either in TED (Transplant Essential Data) track or in CRF (Comprehensive Report Form) track but missing the information asked in the supplemental data. We received response from 126 patients (28 Allogeneic; 98 Autologous). These data were merged with the database from CIBMTR. The supplemental data form included the following variables: cytogenetics, extramedullary disease at diagnosis, pre-transplant therapy regimens, number of lines of therapy, and the best response to pre-transplant therapy. The details of patients’ selection are shown in consort diagram in the supplement (Figure S1).
Definitions and Responses:
As response criteria for PCL have not been defined, we used the definitions developed by the International Myeloma Working Group (IMWG) for MM(15). High risk cytogenetics were defined as t (4;14), t (14:16), t (14:20), deletion 17p, chromosome 1q amplification, 1p deletion, and the presence of hypodiploidy. Non-relapse mortality (NRM) was defined as death from any cause within the first 90 days or thereafter in the absence of relapse or progression. Progression free survival (PFS) was defined as time from transplantation to relapse, progression or death from any cause. Overall survival (OS) was defined as the time from transplantation to death from any cause. For patients undergoing allo-HCT, acute-graft-versus-host-disease (aGVHD) was defined and graded using established criteria(16). Chronic-graft-versus-host-disease (cGVHD) was defined as limited and extensive(17). Neutrophil engraftment was defined as time to neutrophils > 0.5 x 109/L sustained for 3 consecutive days. Platelet engraftment was defined as time to platelet count >20 x 109/l independent of platelet transfusion for 7 consecutive days
Statistical analysis:
Patient-, disease- and transplant-related factors were summarized using descriptive statistics. Probabilities of PFS and OS were calculated using Kaplan-Meier product limit estimate using the log-rank test. The cumulative incidence of NRM and disease relapse/progression were estimated accounting for competing risks. The Cox proportional hazard regression model was to understand the association between patient-, disease- and transplant-related factors with PFS and OS. Multivariate analysis was performed only for auto-HCT given the small sample size in the allo-HCT cohort and the variables included were: age at transplant (by decades), Karnofsky performance status at transplant (KPS) (≥90 vs. <90), gender, disease status at transplant (stringent and/or complete remission (sCR/CR) vs. very good partial response (VGPR) vs. partial response (PR) vs. stable disease (SD) vs. progressive disease or relapse (PD), time from diagnosis to transplant (<6 vs. 6-12 vs. 18 months), melphalan dose (140 mg/m2 vs. 200 mg/m2), type of transplant (single versus tandem) and post-transplant maintenance therapy (yes vs. no).
Variable selection at a 0.05 significance level was used to identify covariates associated with the main outcome. In the model, the assumption of proportional hazards was tested for each variable using a time-dependent covariate and graphical methods. The overall outcomes (1- and 3-year estimates of NRM, relapse/progression, PFS and OS) of this cohort were compared with the previously published CIBMTR cohort of pPCL undergoing HCT from 1995-2006(7). The HCT utilization rate was calculated by comparing the numbers of HCT performed per year and the incidence of pPCL from the Surveillance, Epidemiology and End Results (SEER) (http://www.seer.cancer.gov/) database till 2009 as the SEER stopped coding for PCL separately from 2010 onwards. All variables considered in the multivariate analysis satisfied the proportionality assumption. All tests were two-sided and p-values <0.05 was considered significant. All computations were made using the statistical package SAS version 9.4.
Results:
Autologous HCT cohort:
Two hundred and seventy-seven patients received an auto-HCT during the time period (Table 1). The median age was 60 years (range, 25-77) and 52% were males. Single auto-HCT was performed in 249 (90%) patients, while the remaining 29 (10%) underwent tandem auto-HCT. The median time from diagnosis to transplant was 5.4 (range, 2.1 – 81.2) months; 61% underwent transplant within 6 months of diagnosis. Only 19 (7%) underwent transplant greater than 12 months after diagnosis. Data on induction chemotherapy was available in 124 patients; bortezomib-based therapy was used in 83% (the most common being bortezomib, cyclophosphamide and dexamethasone CyBorD followed by bortezomib, lenalidomide and dexamethasone VRD) of patients and 76% received one line of therapy. Extramedullary disease was present in 4% of the patients.
Table 1:
Baseline characteristics of pPCL patients undergoing upfront autologous and allogeneic transplantation
| Characteristics | Autologous (N, %) | Allogeneic (N (%) |
|---|---|---|
| Number of patients | 277 (80) | 71 (20) |
| Number of centers | 86 | 36 |
| Median age, years (range) | 60 (25-77) | 53 (22-68) |
| Male Sex | 144 (52) | 34 (48) |
| Karnofsky Score ≥90 | 145 (52) | 41 (58) |
| Race, Caucasian | 215 (78) | 53 (75) |
| HCT-CI | ||
| 0 | 80 (29) | 18 (25) |
| 1 | 26 (9) | 6 (8) |
| ≥2 | 171 (61) | 44 (63) |
| missing | 1 (<1) | 3 (4) |
| Induction Chemotherapya: | ||
| VDPACE/similar | 28 (10) | 15 (21) |
| Quadruplets | 10 (4) | 4 (6) |
| Triplets | 50 (18) | 11 (15) |
| Doublets | 17 (6) | 4 (6) |
| Others | 13 (5) | 3 (4) |
| Not captures | 159 (57) | 34 (48) |
| Disease status at transplantb | ||
| CR/sCR | 53 (19) | 15 (21) |
| VGPR | 78 (28) | 19 (27) |
| PR | 112 (40) | 24 (34) |
| SD | 16 (6) | 5 (7) |
| PD/Relapse | 13 (5) | 8 (11) |
| Missing | 5 (2) | - |
| Cytogeneticsc* | ||
| No abnormality | 21 (8) | 7 (10) |
| High risk | 79 (29) | 23 (32) |
| Standard risk | 50 (18) | 12 (17) |
| Unknown | 128 (46) | 29 (41) |
| Conditioning regimen | ||
| Melphalan (140/200) | 277 (47/230) | 12 (17) |
| Melphalan+ TBI | - | 10 (14) |
| TBI-No Melphalan | - | 26 (37) |
| Bu/Cy | - | 4 (6) |
| Others | - | 18 (25) |
| Missing | - | 1 (1) |
| Time from diagnosis to transplant | ||
| < 6 months | 169 (61) | 27 (38) |
| 6-12 months | 89 (32) | 36 (51) |
| ≥ 12 months | 19 (7) | 8 (12) |
| Donor type | - | |
| HLA identical | - | 29 (41) |
| Other related | - | 9 (13) |
| Unrelated | - | 23 (32) |
| Cord Blood | - | 4 (6) |
| Other | - | 6 (9) |
| Year of transplant | ||
| 2008-2010 | 78 (28) | 31 (44) |
| 2011-2013 | 103 (37) | 18 (25) |
| 2014-2015 | 96 (35) | 22 (31) |
| Median follow up of survivors, months (range) | 48 (3-84) | 60 (6-92) |
| Planned post HCT therapy | 75 (27) | 8 (12) |
| Revlimid | 34 (12) | 3 (4) |
| Velcade | 16 (6) | 2 (3) |
| Velcade+Revlimid | 14 (5) | - |
| Velcade + Cyclcophosphamide | 1 (<1) | - |
| Carfilzomib | 2 (<1) | - |
| Other | 8 (3) | 3 (4) |
| Transplant Type | ||
| Single Auto or Allo | 249 (90) | 43 (61) |
| Tandem Auto or Tandem Auto-Allo | 29 (10) | 28 (39) |
| Extramedullary disease at diagnosis | ||
| No extramedullary disease | 88 (32) | 24 (34) |
| CNS ±Other | 2 (1) | 4 (6) |
| Other | 9 (3) | - |
| Not reported | 178 (64) | 43 (60) |
For auto [ VTD (n=3); VRD (n=23); VCD (n=24); VD (n=12); RD (n=2); TD (n=3) VAD/similar (n=6); lpam (n=4); MVP (n=1); Other (n=2), carfilzomib 2] and For allo [VTD (n=2); VRD (n=6); VCD (n=3); VD (n=2); RD (n=1); TD (n=1); VAD/similar (n=2); Other (n=1), carfilzomib 3].
CR/sCR: complete response/stringent complete response, VGPR: very good partial response, PR: partial response, PD: progressive disease;
High risk cytogenetics (HR): 79 in auto [ t (4;14) 7; t(14:16) 9; t (14:20) 1; del 17p 7, chromosome 1q 11, 1p deletion 1, and ≥2HR 43; 23 in allo [ t (4;14) 1; t(14:16) 3; t (14:20) 1; del 17p 2, chromosome 1q 13, hypodiploidy 1 and ≥2HR 5.
High-risk cytogenetics was present in 29% of the patients but was unknown in 46% of patients, with 8% reporting no abnormality and a non-high risk marker in 12%. Disease status at transplant in auto-HCT group were as follows: 19% CR, 28% VGPR, 40% PR, 6% SD, and PD in 5%. Approximately 44% of patients had an HCT comorbidity index (HCT-CI) ≥ 3. The majority of the patients (83%) received melphalan at a dose of 200 mg/m2. The cumulative incidence of neutrophil and platelet engraftment at day 28 after HCT was 99% (95% confidence interval CI 98-100) and 87% (95% CI 82-91), respectively (Table S1).
Only 27% of patients received planned post-auto HCT maintenance therapy. The median follow up of survivors was 48 months (range, 3-84 months). Twenty-one (11%) received salvage transplants including 14 (5%) salvage auto-HCT and 17 (5%) salvage allo-HCT. At the time of last follow up, 169 (61%) patients died; of those 143 (85%) died from progressive disease (Table 2). At 4 years post HCT, PFS and OS was 17% (95% CI 13%-23%) and 28% (95% CI 22%-35%) (Table S1, Figure 1A and 1B) respectively; NRM and incidence of relapse/progression was 7% (95% CI 4%-11%) and 76% (95% CI 69%-82%) respectively (Table S1, Figure 2A and 2B). In the multivariate analysis, achievement of ≥VGPR prior to HCT predicted superior PFS, OS and reduced risk of relapse (Table 3). Other significant predictors included: melphalan 200 mg/m2 for superior PFS and reduced risk of relapse/progression, while KPS ≥ 90 was associated with superior OS.
Table 2:
Causes of Death
| Causes | Autologous (277) | Allogeneic (71) |
|---|---|---|
| Total number of deaths Causes | 169 | 45 |
| Primary disease | 143 (85) | 34 (76) |
| GVHD | NA | 2 (4) |
| Infection | 11 (7) | 4 (9) |
| IPS/ARDS | 0 | 1 (2) |
| Organ Failure | 3 (2) | 2 (4) |
| Secondary malignancy | 1 (<1) | 0 |
| Vascular | 3 (2) | 1 (2) |
| Unknown | 8 (5) | 1 (2) |
IPS/ARDS: Idiopathic pneumonia syndrome/Adult respiratory distress syndrome
Figure 1A.
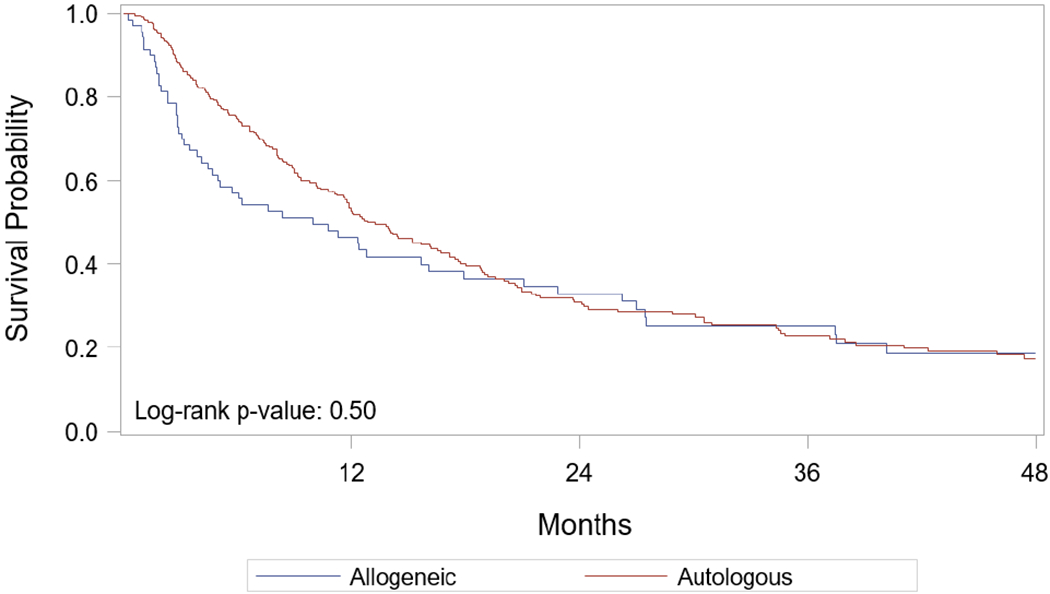
Probability of progression-free survival after HCT – by transplant type
Figure 1B.

Probability of overall survival after HCT – by transplant type
Figure 2A.

Cumulative incidence of non-relapse mortality after HCT – by transplant type
Figure 2B.

Cumulative incidence of relapse after HCT – by transplant type
Table 3:
Multivariate analysis of factors predicting outcomes after auto-HCT
| Characteristic | Progression free survival | Relapse/Progression | Overall survival | |||
|---|---|---|---|---|---|---|
| HR (95% CI) | p-value | HR (95% CI) | p-value | HR (95% CI) | p-value | |
| Disease status at transplant | <0.001 | <0.001 | <0.001 | |||
| ≥ VGPR | Reference | Reference | Reference | |||
| PR | 1.53(1.11-2.11) | 0.008 | 1.4 (1.01-1.96) | 0.04 | 1.55 (1.09-2.2) | 0.01 |
| SD/PD/relapse | 3.6 (2.3-5.8) | <0.001 | 3.3 (2.04-5.45) | <0.001 | 4.3 (2.6-7.2) | <0.001 |
| Missing | 1.8 (0.63- 5.22) | 0.26 | 1.4 (0.44-5.8) | 0.53 | 2.7 (0.99-7.8) | 0.05 |
| Melphalan dose | 0.04 | 0.08 | - | - | ||
| 140 mg/m2 | Reference | Reference | ||||
| 200 mg/m2 | 0.65 (0.44-0.98) | 0.68 (0.45-1.04) | ||||
| Karnofksy performance status | - | - | 0.0018 | |||
| ≥ 90 | Reference | |||||
| < 90 | 1.80 (1.3- 2.49) | 0.004 | ||||
| Missing | 1.79 (0.43- 7.3) | 0.41 | ||||
Allogeneic cohort:
Seventy-one patients received an allo-HCT between 2008-2015 (Table 1). The median age was 53 years (range, 22-68) and 48% of patients were males. A single allo-HCT (without prior auto HCT) was performed in 43 (61%), while the remaining patients underwent tandem auto-allo HCT. The median time from diagnosis to transplant was 6.7 months (range, 2.9 -30.7); about half underwent transplant between 6-12 months and 38% underwent HCT within 6 months of diagnosis. Only 8 (11%) patients underwent transplant greater than 12 months after diagnosis. Data on induction therapy were available in 37 patients of which 86% of patients received bortezomib-based therapy (VRD most commonly, followed by bortezomib, dexamethasone, cisplatin, doxorubicin, cyclophosphamide and etoposide, VDPACE), with 70% receiving only one line of therapy. Extramedullary disease was present in 6% of the patients.
High-risk cytogenetics were present in 32% of the patients and were unknown in 41%. Disease status at transplant was as follows: CR 21%, VGPR 27%, PR 34%, SD 7% and PD in 11%. Thirty-one (44%) patients received myeloablative conditioning and 39 (55%) patients received non-myeloablative/reduced intensity (NMA/RIC) conditioning; 51% received total body irradiation. Majority of the patients received peripheral blood graft, and 11% and 6% of the patients received bone marrow and cord blood, respectively. Tacrolimus/Methotrexate (TAC/MTX) (31%) was the most commonly prescribed GVHD prophylaxis regimen. The cumulative incidence of neutrophil and platelet engraftment at day 28 was 91% (95% CI 83-97) and 77% (95% CI 65-87), respectively.
Planned post-HCT therapy was reported to be given in 12% of patients. The median follow-up of the survivors was 60 months (range, 6-92). Acute GVHD (grade II-IV) occurred in 21 (30%) of the patients, and the cumulative incidence of chronic GVHD at 2 years was 45% (95% CI 33% -58%) (Supplemental Table 1 and 2). At the time of last follow up, 45 (63%) patients were dead; of those 34 (76%) died from progressive/relapsed disease (Table 2). PFS at 4 years was 19% (95% CI 10%-31%) and OS at 4 years was 31% (95% CI 19%-44%) (Table S1, Figure 1A and 1B). NRM at 4 years was 12% (95% CI 5%-21%) and relapse/progression in 69% (95% CI 50%-81%) (Table S1, Figure 2A and 2B).
Comparison with CIBMTR MM 09-01 study and HCT utilization
A comparison of outcomes of pPCL patients with the previous CIBMTR study (MM09-01)(7) is presented in Table 4. This showed improved NRM after allo-HCT. Relapse/progression and PFS were worse for 2008-2015 period compared to 1995-2006 after auto and allo HCT. Overall survival from transplant was worse after auto-HCT for the current cohort compared to the older study. No difference was observed in OS from diagnosis between auto- and allo-HCT (4-year OS 28% (95% CI 17%-40%) auto-HCT vs. 31% (95 CI 24%-37%) allo-HCT; p=0.68), post-relapse survival (4-year survival rates, 8% for both auto- and allo-HCT), and by prior bortezomib use (Table S3, S4 and S5).
Table 4:
Comparison of outcomes with previous CIBMTR study
| Outcomes | Allogeneic HCT | Autologous HCT | ||
|---|---|---|---|---|
| MM09-01 | MM 17-01 | MM09-01 | MM17-01 | |
| Non-relapse mortality | ||||
| 1 year | 26 (15 - 39) % | 9 (3-16) % | 2 (0 - 5) % | 3 (1-6) % |
| 3 years | 40 (27 - 55) % | 12 (5-21) % | 5 (1 - 10) % | 7 (4-10) % |
| Relapse/Progression | ||||
| 1 year | 23 (12 - 35) % | 45 (33-57) % | 30 (21 – 40) % | 44 (38-50) % |
| 3 years | 40 (26 - 54) % | 63 (50-74) % | 61 (49 - 72) % | 70 (64-76) % |
| Progression free survival | ||||
| 1 year | 51 (37 - 65) % | 47 (35-58) % | 68 (58 - 76) % | 53 (47-59) % |
| 3 years | 20 (10 - 33) % | 25 (15-37) % | 34 (24 - 46) % | 23 (18-29) % |
| Overall Survival | ||||
| 1 year | 58 (44 - 71) % | 61 (50-72) % | 86 (79 - 92) % | 70 (64-75) % |
| 3 years | 39 (25 - 53) % | 38 (26-50) % | 62 (51 - 73) % | 35 (28-41) % |
MM09-01: CIBMTR study (1995-2006)
MM17-01: CIBMTR study (2008-2015)
The number of centers performing HCT for pPCL from 2008-2015 is shown in Figure S2. The frequency of patients who received HCT for pPCL by year from 1995-2015 is also shown in Figure S3. The HCT utilization rate showed a steady increase from 1995 to 2009 (Table S6).
Discussion:
Using CIBMTR data, we report the largest study assessing the outcomes of HCT in pPCL in the novel agent era and make several important observations: 1) higher proportion of patients receiving HCT owing to improved induction regimens 2) significant improvement in NRM after allo-HCT, likely due to better patient selection and improved transplant practice, however this did not translate to improved overall outcomes compared to auto-HCT, 3) overall poor post-HCT outcomes despite the upfront use of PIs/IMIDs and increased HCT utilization owing to high post-HCT relapse. This study provides a benchmark for future HCT studies for pPCL and highlights the need for novel combinatorial approaches in conjunction with HCT to improve survival.
In a study using the SEER database from 1975-2009, there was a trend in improvement in OS in pPCL patients with reduction of early mortality from 25% to 15% before and after 2006 respectively(6), likely reflecting the use of novel agents, most of which were available for use in the upfront setting after 2006. The SEER dataset stopped reporting pPCL separately following 2009. Use of bortezomib and/or lenalidomide in PCL has resulted in overall response rates (ORR) of >70%(11, 14, 18–21), a significant improvement compared to the conventional agents with response rates of <30%(22). The availability of these induction regimens has likely contributed to an increased HCT utilization rate observed after 2003, as historically these patients carried a 30% risk of mortality within the first month of diagnosis (2, 4, 5). As observed in our study, there is a wide variability in the type of induction regimens in clinical practice, but the majority include bortezomib. Bortezomib-based induction therapy was adopted early despite paucity of data in pPCL, extrapolated from the myeloma literature, given the poor outcomes with other therapies(14, 19, 23, 24).
Given the aggressive course of the disease, and the need for early and sustained response, HCT has been incorporated in the treatment armamentarium for pPCL especially with the survival benefit observed in MM. The low incidence of pPCL makes prospective, randomized trials difficult to execute, thus a registry-based retrospective study such as ours is invaluable to elucidate the role of HCT. Prior to this study, three large retrospective studies mainly using non-novel based induction, evaluated the role of HCT in pPCL (7, 9, 10). The European Group for Blood and Marrow Transplantation (EBMT) compared outcomes of MM to pPCL (N=272) from 1980 to 2006. In those who underwent auto-HCT, patients with pPCL had an inferior median PFS (14 versus 27 months) and OS (26 versus 62 months) compared to MM(10). The previous CIBMTR study compared the role of both auto- (N=97) and allo-HCT (N=50) within 18 months of diagnosis from 1995-2006 (7). The estimated rates of 3-year PFS, relapse, NRM, and OS were 34, 61, 5, and 64 percent, respectively after auto-HCT. Corresponding rates following allo- HCT were 20, 38, 41, and 39 percent, respectively, indicating inferior outcome with allo-HCT compared to auto-HCT. In the current era, combination of both PIs and/or IMiDs in induction and maintenance, in conjunction with auto-HCT consolidation has shown to result in superior survival, but the evidence is limited to single center retrospective studies and small prospective studies(13, 19, 20, 25, 26). The EBMT presented data about 460 patients who underwent HCT from 1998 to 2012 in abstract form, finding that an upfront allo-HCT was associated with worse OS; the final manuscript has not yet been published(27). In this context, our data represent the most updated and highest quality evidence available regarding the role of HCT in pPCL in novel agent era, even with the caveat of patient selection bias. As patients in deep remission at transplant derived maximal benefit from auto-HCT, strategies for achieving deeper responses upfront with novel combinations should be further studied.
When compared to the previous CIBMTR study in PCL(7), some results were as expected, while others were surprising and raises several critical questions for consideration. Most strikingly, there has not been an improvement in OS with either transplant modality. For auto-HCT, post-HCT outcomes appear worse in the current versus pre-novel era (3 years (OS 35% vs. 65%). We conducted additional analyses to better understand this. We found that when compared to SEER incidence data, more patients are undergoing HCT in recent years (note, SEER stopped reporting PCL separately after 2009). The rising HCT utilization rate suggests the following. First, novel agents may be increasing response rates and hence transplant eligibility. Thus, juxtaposed to MM09-01, the MM17-01 population includes patients who would not have achieved remission without novel agents. Second, patients who may have been deemed ineligible for HCT prior to 2008 underwent HCT in this study. This may include patients who were more frail or those who were already progressing at the time of HCT. Despite deeper remission at that time of auto-HCT, as well as presumed higher rates of maintenance therapy (maintenance therapy was not documented in MM09-01), the 1-year relapse rates and PFS were worse in this current study, suggesting selection of more aggressive biologic disease. The differences in the rates of relapse and PFS are not as significant by 3 years. This is further strengthened by the lack of difference in survival from diagnosis between the 2 time periods. Lastly, we considered that patients who relapsed after prior use of bortezomib represented a more aggressive subgroup (similar to lymphoma outcomes after failure of rituximab-based regimens) (8, 28, 29). Though we did not observe any difference in survival based on prior bortezomib use either, we did not have sufficient power to study this adequately given the small sample size. The improvement in NRM, particularly in the allo-HCT arm, does highlight the potential improvement in supportive care, patient selection, and transplant experience, though this did not translate to improved PFS or OS.
For both modalities, relapse was the dominant cause of death in ~80 % after transplant. Several studies have shown superior outcomes with post-HCT maintenance strategies, with one study showing impressive OS of 36 months with bortezomib and lenalidomide maintenance(13). The use of maintenance was 27% and 12% post auto- and allo-HCT respectively in our study, perhaps reflecting the practice pattern at the time or disease progression while recovering post HCT. It is possible that the high relapse rate observed in these patients is due to the low rate of maintenance used; however, given the inherently high- risk nature of disease resulting in early relapse despite maintenance is also possible. Currently, the European Myeloma network is conducting a study in pPCL (EMN12/HOVON129 PCL) in which patients will be treated with carfilzomib, lenalidomide, and dexamethasone in induction, consolidation, and maintenance will hopefully shed light on the role of maintenance. The Bone and Marrow Transplant Clinical Trials Network BMT CTN 1302 study (NCT02440464) would have provided further insight into the role of allo-HCT and the use of ixazomib maintenance for patients with pPCL, but unfortunately closed prematurely due to poor accrual.
There are several limitations with our study. Firstly, cytogenetics data were not known in 46% of patients. Deletions or mutations of p53 that are known to confer poor prognosis are present in >50% of patients with pPCL and >80% in sPCL(2, 30). High risk cytogenetics comprised 29% in our cohort but deletion 17p was present in <10% of patients. The most prevalent translocation in pPCL is t (11;14) at a frequency of 25-50% (2, 28, 31, 32), but lower with what was observed in our study (17% in auto- and 23% in allo-HCT of available data). We could not calculate HCT utilization for pPCL after 2009 as the SEER cancer registry stopped coding PCL separately from 2010 onwards. Third, despite our best attempts, we were able to obtain additional supplemental data in only 43% and 51% of auto- and allo-HCT respectively, limiting our ability to study other disease- and treatment-related factors that may be important, for example, induction therapy was only available in 57% of auto-HCT and 48% of allo-HCT, respectively. Lastly, because the CIBMTR is a transplant registry, we have no data on pPCL patients who did not undergo transplant. The strength of our study is that this is the largest study in a contemporaneous period and captures the majority (~80%) of practice in the US, thus providing a benchmark of outcomes in the current treatment period.
We conclude that despite the availability of newer and rapidly effective plasma cell-directed therapies translating into a higher proportion of pPCL patients receiving HCT in recent years, post-HCT outcomes need further improvements. Our data highlight the increasing need for prospective multi-center randomized clinical trials that includes combinatorial immune and non-immune approaches in conjunction with HCT for eligible patients. For example, given the excellent single agent activity of bcl-2 inhibition in t (11;14) myeloma(33) and few case reports in pPCL(34, 35), future trials using bcl-2 inhibitors either as a single agent or in combination could be considered in this subgroup of pPCL. Ongoing efforts should include correlative studies to better understand the disease biology such that cutting-edge therapeutic options and individualized therapy can be instituted to improve survival in this rare but fatal disease.
Supplementary Material
Acknowledgements
The CIBMTR is supported primarily by Public Health Service grant/cooperative agreement U24CA076518 with the National Cancer Institute (NCI), the National Heart, Lung and Blood Institute (NHLBI) and the National Institute of Allergy and Infectious Diseases (NIAID); grant/cooperative agreement U24HL138660 with NHLBI and NCI; grant U24CA233032 from the NCI; grants OT3HL147741, R21HL140314 and U01HL128568 from the NHLBI; contract HHSH250201700006C with Health Resources and Services Administration (HRSA); grants N00014-18-1-2888 and N00014-17-1-2850 from the Office of Naval Research; subaward from prime contract award SC1MC31881-01-00 with HRSA; subawards from prime grant awards R01HL131731 and R01HL126589 from NHLBI; subawards from prime grant awards 5P01CA111412, 5R01HL129472, R01CA152108, 1R01HL131731, 1U01AI126612 and 1R01CA231141 from the NIH; and commercial funds from Actinium Pharmaceuticals, Inc.; Adaptive Biotechnologies; Allovir, Inc.; Amgen, Inc.; Anonymous donation to the Medical College of Wisconsin; Anthem, Inc.; Astellas Pharma US; Atara Biotherapeutics, Inc.; BARDA; Be the Match Foundation; bluebird bio, Inc.; Boston Children’s Hospital; Bristol Myers Squibb Co.; Celgene Corp.; Children’s Hospital of Los Angeles; Chimerix, Inc.; City of Hope Medical Center; CSL Behring; CytoSen Therapeutics, Inc.; Daiichi Sankyo Co., Ltd.; Dana Farber Cancer Institute; Enterprise Science and Computing, Inc.; Fred Hutchinson Cancer Research Center; Gamida-Cell, Ltd.; Genzyme; Gilead Sciences, Inc.; GlaxoSmithKline (GSK); HistoGenetics, Inc.; Immucor; Incyte Corporation; Janssen Biotech, Inc.; Janssen Pharmaceuticals, Inc.; Janssen Research & Development, LLC; Janssen Scientific Affairs, LLC; Japan Hematopoietic Cell Transplantation Data Center; Jazz Pharmaceuticals, Inc.; Karius, Inc.; Karyopharm Therapeutics, Inc.; Kite, a Gilead Company; Kyowa Kirin; Magenta Therapeutics; Mayo Clinic and Foundation Rochester; Medac GmbH; Mediware; Memorial Sloan Kettering Cancer Center; Merck & Company, Inc.; Mesoblast; MesoScale Diagnostics, Inc.; Millennium, the Takeda Oncology Co.; Miltenyi Biotec, Inc.; Mundipharma EDO; National Marrow Donor Program; Novartis Oncology; Novartis Pharmaceuticals Corporation; Omeros Corporation; Oncoimmune, Inc.; OptumHealth; Orca Biosystems, Inc.; PCORI; Pfizer, Inc.; Phamacyclics, LLC; PIRCHE AG; Regeneron Pharmaceuticals, Inc.; REGiMMUNE Corp.; Sanofi Genzyme; Seattle Genetics; Shire; Sobi, Inc.; Spectrum Pharmaceuticals, Inc.; St. Baldrick’s Foundation; Swedish Orphan Biovitrum, Inc.; Takeda Oncology; The Medical College of Wisconsin; University of Minnesota; University of Pittsburgh; University of Texas-MD Anderson; University of Wisconsin - Madison; Viracor Eurofins and Xenikos BV. The views expressed in this article do not reflect the official policy or position of the National Institute of Health, the Department of the Navy, the Department of Defense, Health Resources and Services Administration (HRSA) or any other agency of the U.S. Government.
CONFLICTS: The following authors have conflict of interest: Dr. Dhakal reports honorarium from Celgene; Advisory board for Takeda and Amgen. Dr. Lazarus reports Bristol Myers Squibb and Celgene. Dr. Gale reports Celgene Corporation. Dr. Stockerl-Goldstein reports Abbot and AbbVie Dr. Assal reports research funding from Incyte Corporation; Advisory board for Boston Biomedical and Incyte Corporation. Consulting for Alpha Insights. Dr. Holmberg reports Up-To-Date. Dr. Shah reports funding from Celgene, Bluebird Bio, Sutro Biopharma; Advisory role for Genentech, Seattle Genetics, Oncopeptides, Karoypharm, Surface Oncology, Precision Biosciences, GSK, Nektar, Amgen, Indapta Therapeutics, Sanofi; Stock Ownership for Indapta Therapeutics. Dr. D’Souza reports funding from Merck, Prothena, Sanofi, Mundipharma EDO, TeneoBio, Takeda and has received consulting fees from Prothena, Pfizer, Imbrium, and Akcea.
REFERENCES:
- 1.Fernandez de Larrea C, Kyle RA, Durie BG, Ludwig H, Usmani S, Vesole DH, et al. Plasma cell leukemia: consensus statement on diagnostic requirements, response criteria and treatment recommendations by the International Myeloma Working Group. Leukemia. 2013;27(4):780–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tiedemann RE, Gonzalez-Paz N, Kyle RA, Santana-Davila R, Price-Troska T, Van Wier SA, et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia. 2008;22(5):1044–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kyle RA, Maldonado JE, Bayrd ED. Plasma cell leukemia. Report on 17 cases. Arch Intern Med. 1974;133(5):813–8. [DOI] [PubMed] [Google Scholar]
- 4.Noel P, Kyle RA. Plasma cell leukemia: an evaluation of response to therapy. Am J Med. 1987;83(6):1062–8. [DOI] [PubMed] [Google Scholar]
- 5.Ramsingh G, Mehan P, Luo J, Vij R, Morgensztern D. Primary plasma cell leukemia: a Surveillance, Epidemiology, and End Results database analysis between 1973 and 2004. Cancer. 2009;115(24):5734–9. [DOI] [PubMed] [Google Scholar]
- 6.Gonsalves WI, Rajkumar SV, Go RS, Dispenzieri A, Gupta V, Singh PP, et al. Trends in survival of patients with primary plasma cell leukemia: a population-based analysis. Blood. 2014;124(6):907–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahindra A, Kalaycio ME, Vela-Ojeda J, Vesole DH, Zhang MJ, Li P, et al. Hematopoietic cell transplantation for primary plasma cell leukemia: results from the Center for International Blood and Marrow Transplant Research. Leukemia. 2012;26(5):1091–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Usmani SZ, Nair B, Qu P, Hansen E, Zhang Q, Petty N, et al. Primary plasma cell leukemia: clinical and laboratory presentation, gene-expression profiling and clinical outcome with Total Therapy protocols. Leukemia. 2012;26(11):2398–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jurczyszyn A, Radocha J, Davila J, Fiala MA, Gozzetti A, Grzasko N, et al. Prognostic indicators in primary plasma cell leukaemia: a multicentre retrospective study of 117 patients. Br J Haematol. 2018;180(6):831–9. [DOI] [PubMed] [Google Scholar]
- 10.Drake MB, Iacobelli S, van Biezen A, Morris C, Apperley JF, Niederwieser D, et al. Primary plasma cell leukemia and autologous stem cell transplantation. Haematologica. 2010;95(5):804–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Arena G, Valentini CG, Pietrantuono G, Guariglia R, Martorelli MC, Mansueto G, et al. Frontline chemotherapy with bortezomib-containing combinations improves response rate and survival in primary plasma cell leukemia: a retrospective study from GIMEMA Multiple Myeloma Working Party. Ann Oncol. 2012;23(6):1499–502. [DOI] [PubMed] [Google Scholar]
- 12.Gowda L, Shah M, Badar I, Bashir Q, Shah N, Patel K, et al. Primary plasma cell leukemia: autologous stem cell transplant in an era of novel induction drugs. Bone Marrow Transplant. 2019;54(7):1089–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mina R, Joseph NS, Kaufman JL, Gupta VA, Heffner LT, Hofmeister CC, et al. Survival outcomes of patients with primary plasma cell leukemia (pPCL) treated with novel agents. Cancer. 2019;125(3):416–23. [DOI] [PubMed] [Google Scholar]
- 14.Royer B, Minvielle S, Diouf M, Roussel M, Karlin L, Hulin C, et al. Bortezomib, Doxorubicin, Cyclophosphamide, Dexamethasone Induction Followed by Stem Cell Transplantation for Primary Plasma Cell Leukemia: A Prospective Phase II Study of the Intergroupe Francophone du Myelome. J Clin Oncol. 2016;34(18):2125–32. [DOI] [PubMed] [Google Scholar]
- 15.Kumar S, Paiva B, Anderson KC, Durie B, Landgren O, Moreau P, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17(8):e328–e46. [DOI] [PubMed] [Google Scholar]
- 16.Przepiorka D, Weisdorf D, Martin P, Klingemann HG, Beatty P, Hows J, et al. 1994 Consensus Conference on Acute GVHD Grading. Bone Marrow Transplant. 1995;15(6):825–8. [PubMed] [Google Scholar]
- 17.Shulman HM, Sullivan KM, Weiden PL, McDonald GB, Striker GE, Sale GE, et al. Chronic graft-versus-host syndrome in man. A long-term clinicopathologic study of 20 Seattle patients. Am J Med. 1980;69(2):204–17. [DOI] [PubMed] [Google Scholar]
- 18.Lebovic D, Zhang L, Alsina M, Nishihori T, Shain KH, Sullivan D, et al. Clinical outcomes of patients with plasma cell leukemia in the era of novel therapies and hematopoietic stem cell transplantation strategies: a single-institution experience. Clin Lymphoma Myeloma Leuk. 2011;11(6):507–11. [DOI] [PubMed] [Google Scholar]
- 19.Katodritou E, Terpos E, Kelaidi C, Kotsopoulou M, Delimpasi S, Kyrtsonis MC, et al. Treatment with bortezomib-based regimens improves overall response and predicts for survival in patients with primary or secondary plasma cell leukemia: Analysis of the Greek myeloma study group. Am J Hematol. 2014;89(2):145–50. [DOI] [PubMed] [Google Scholar]
- 20.Benson DM Jr., Smith MK. Effectiveness of lenalidomide (Revlimid) for the treatment of plasma cell leukemia. Leuk Lymphoma. 2007;48(7):1423–5. [DOI] [PubMed] [Google Scholar]
- 21.Guglielmelli T, Merlini R, Giugliano E, Saglio G. Lenalidomide, melphalan, and prednisone association is an effective salvage therapy in relapsed plasma cell leukaemia. J Oncol. 2009;2009:867380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vela-Ojeda J, Garcia-Ruiz Esparza MA, Rosas-Cabral A, Padilla-Gonzalez Y, Garcia-Chavez J, Tripp-Villanueva F, et al. Intermediate doses of melphalan and dexamethasone are better than vincristine, adriamycin, and dexamethasone (VAD) and polychemotherapy for the treatment of primary plasma cell leukemia. Ann Hematol. 2002;81(7):362–7. [DOI] [PubMed] [Google Scholar]
- 23.Grassinger J, Sudhoff T, Andreesen R, Hennemann B. Complete remission and successful stem cell mobilization after treatment of refractory plasma cell leukemia with bortezomib. Ann Hematol. 2006;85(2):132–3. [DOI] [PubMed] [Google Scholar]
- 24.Esparis-Ogando A, Alegre A, Aguado B, Mateo G, Gutierrez N, Blade J, et al. Bortezomib is an efficient agent in plasma cell leukemias. Int J Cancer. 2005;114(4):665–7. [DOI] [PubMed] [Google Scholar]
- 25.Gowda L, Shah M, Badar I, Bashir Q, Shah N, Patel K, et al. Primary plasma cell leukemia: autologous stem cell transplant in an era of novel induction drugs. Bone Marrow Transplant. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Musto P, Simeon V, Martorelli MC, Petrucci MT, Cascavilla N, Di Raimondo F, et al. Lenalidomide and low-dose dexamethasone for newly diagnosed primary plasma cell leukemia. Leukemia. 2014;28(1):222–5. [DOI] [PubMed] [Google Scholar]
- 27.Sea Lawless. Comparison of Haematopoietic Stem Cell Transplantation Approaches in Primary Plasma Cell Leukaemia. Blood. 2016;128(22):2293. [Google Scholar]
- 28.Avet-Loiseau H, Roussel M, Campion L, Leleu X, Marit G, Jardel H, et al. Cytogenetic and therapeutic characterization of primary plasma cell leukemia: the IFM experience. Leukemia. 2012;26(1):158–9. [DOI] [PubMed] [Google Scholar]
- 29.Gisselbrecht C, Glass B, Mounier N, Singh Gill D, Linch DC, Trneny M, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010;28(27):4184–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gertz MA. Managing plasma cell leukemia. Leuk Lymphoma. 2007;48(1):5–6. [DOI] [PubMed] [Google Scholar]
- 31.Chang H, Qi X, Yeung J, Reece D, Xu W, Patterson B. Genetic aberrations including chromosome 1 abnormalities and clinical features of plasma cell leukemia. Leuk Res. 2009;33(2):259–62. [DOI] [PubMed] [Google Scholar]
- 32.Chiecchio L, Dagrada GP, White HE, Towsend MR, Protheroe RK, Cheung KL, et al. Frequent upregulation of MYC in plasma cell leukemia. Genes Chromosomes Cancer. 2009;48(7):624–36. [DOI] [PubMed] [Google Scholar]
- 33.Kumar S, Kaufman JL, Gasparetto C, Mikhael J, Vij R, Pegourie B, et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood. 2017;130(22):2401–9. [DOI] [PubMed] [Google Scholar]
- 34.Gonsalves WI, Buadi FK, Kumar SK. Combination therapy incorporating Bcl-2 inhibition with Venetoclax for the treatment of refractory primary plasma cell leukemia with t (11;14). Eur J Haematol. 2018;100(2):215–7. [DOI] [PubMed] [Google Scholar]
- 35.Jelinek T, Mihalyova J, Kascak M, Duras J, Popkova T, Benkova K, et al. Single-agent venetoclax induces MRD-negative response in relapsed primary plasma cell leukemia with t(11;14). Am J Hematol. 2019;94(1):E35–E7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.