

Introduction
Migraine is the second-most disabling disease worldwide [27], characterized by unilateral throbbing pain, cutaneous allodynia, often accompanied by nausea and sensitivity to light and sound. Attacks can be caused by triggers that include changes in hormones or sleep patterns, skipping meals, consumption of alcohol and certain foods [32]. Susceptibility to attacks following exposure to these common events suggests maladaptive changes have occurred within migraine-related nociceptive pathways. The most commonly-reported trigger for migraine is stress [32], which can increase the duration of headache [57] and may play a role in the development of chronic headache disorders [50]. Attacks often do not occur while stress is ongoing, but rather once the stressful event has passed [53; 54] with greatest susceptibility 6 to 18 hours following resolution of stress [38]. Additionally, multiple days of intense stress are more likely to trigger attacks than a single day [38]. Despite the close correlation between stress and migraine, mechanistic links between the two remain poorly understood.
Preclinical migraine studies have used methods of stress including restraint, bright light, unpredictable sounds, wet bedding, exposure to predators, or a combination of stressors. Acute or chronic stress decreases the threshold for cortical-spreading depression (CSD) [6; 58], increases nitric-oxide synthase expression [59], alters immune cell properties in the dura [44; 45], and regulates hypothalamic modulation of the trigeminal nucleus caudalis [48]. Behaviorally, restraint stress increases rat tail-flick responses to high-doses (10 mg/kg) of the nitric oxide (NO) donor nitroglycerin (NTG) [17] and increases eye blinking and shuddering behavior in a mouse model of familial-hemiplegic migraine [15]. Additionally, a 14-day social-defeat stress exposure and 40-day chronic-variable stress protocol caused anxiety-like responses, hindpaw hypersensitivity, and increased responses to high-dose NTG (10 mg/kg) [31]. These prior studies were limited to the effects of stress either during or in the acute time frame following exposure, not whether stress causes lasting changes that may influence responses to future events i.e. hyperalgesic priming. Hyperalgesic priming (or latent sensitization) was shown previously following repeated exposure to sumatriptan which caused priming to subsequent bright-light stress or injection of PGE2 [4; 20; 21]; priming was also caused by application of interleukin-6 (IL-6) to the dura, which primed rodents to dural stimulation with a pH 7.0 solution [11; 13]. However, these studies did not evaluate whether stress itself causes priming and thus whether exposure to stress might influence the threshold for future attacks. Given the major contributions of stress to migraine, more work is necessary to better understand how stress contributes to the disorder.
In the studies described here, we explored whether repeated stress in mice produces hyperalgesic priming to doses of NO donors much lower than those used in previous stress models (0.1 mg/kg here compared to 10 mg/kg in prior work) and whether it also caused priming to normally subthreshold stimulation of the dura. Additionally, we evaluated whether two common migraine therapeutics, a triptan and a monoclonal antibody to calcitonin gene-related peptide (CGRP), could decrease the response to the NO donor in stress-primed mice.
Methods
Animals
Female and male ICR mice ages 6–8 weeks (Envigo; Livermore, CA) were used for most of the experiments described here. ICR mice are used as they are outbred which provides increased genetic diversity between animals over standard inbred mouse strains. A mixed strain (C57BL6 and 129J background; bred in-house at UT Dallas) were also used to determine whether findings are specific to the ICR strain (see Supplementary Figure 1). Animal weights varied between 19 g up to a maximum of 30 g. Animals were housed on a 12-hour light-dark cycle and had access to food and water ad libitum. Animals were allowed a minimum of 72 hours in the animal facility to acclimate to their new environment after arrival from Envigo. The stage of the female estrous cycle was determined at the end of some experiments, but no influence of cycle was observed for any findings. In all experiments, investigators were blinded to treatment groups. All procedures were conducted with prior approval of the Institutional Animal Care and Use Committee at the University of Texas at Dallas.
Measurement of facial mechanical hypersensitivity
Mice were handled for a single 5-min session at 24-hours prior to habituation to the behavior chambers. During each session of habituation animals were placed in 4 oz paper cups (Choice) for 2 hours a day for 3 consecutive days as previously described [13]. Habituation was done in the rooms where all further behavioral testing occurred to acclimate animals to the room and light conditions. von Frey testing of the periorbital skin [13] was used to assess baseline values following habituation prior to stress as well as mechanical hypersensitivity that resulted from restraint stress and drug treatments. von Frey thresholds were not measured on the days that animals were subject to restraint stress. Testing began 24 hours after the third day of restraint unless otherwise noted. Prior to stress, mice were subjected to baseline tests of cutaneous facial sensitivity for approximately 3–4 days. Baselined animals were defined as animals that exhibited a withdrawal threshold approximately 0.5 g - 0.6 g. Filaments greater than 0.6 g were not used. Mice with a baseline threshold lower than 0.5 g at the end of 4 days were excluded from experiments. Mechanical thresholds were determined by applying von Frey filaments to the periorbital region of the face (the midline of the forehead at the level of the eyes) in an ascending/descending manner starting from the 0.07 g filament. Briefly, if an animal did not respond, increasing filament forces were applied until the 0.6 g filament was reached or until a response was observed. If the animal responded to a specific filament, decreasing filament forces were applied until the 0.008 g filament was reached or until there were no responses. A response was defined as a mouse actually removing/swiping the filament away from its face during application. All animals were numbered and randomly allocated to experimental groups by drawing from pre-labeled paper slips.
Repetitive stress paradigm
Animals were subjected to restraint stress 24 hours following baseline. Sensory threshold values were determined with von Frey filaments. Animals were placed in cylindrical tail access rodent restrainers designed for animals 15–30 g (Stoelting 51338). Animals were placed in these restraint devices so that their tail was threaded through the moveable disk and their faces project out of the hole in the acrylic front face of the tube. Animals were introduced to the tube by placing the restrainer in front of the animals on the first day of stress and guiding them into the restrainer with the animal facing the acrylic front. Once the animal was in position the tail was threaded through the moveable disk, the disk was moved toward the animal and tightened to ensure that the animal was incapable of movement. Care was taken to avoid any trauma to the mice due to injuries from moving the disk or from threading the tail. Mice were also restrained at a level that still allowed normal respiration. Animals were placed in the restraint tube so that the moveable disk faced upward and an opening on the tube was on the bottom. The animals were restrained for 2 hours a day for 3 consecutive days unless otherwise noted. Restraint stress started no earlier than 9:30 am and stress ended prior to 12:00pm in all cases to account for natural rising in corticosterone levels that have been shown occur in rodents in the afternoon starting at 1:00 pm [18]. Sham animals were left in their home cages without access to water or food to ensure that water/food deprivation alone did not contribute to the stress responses. Sham animals were kept in a separate room from stressed animals for the duration of stress. Once animals were in restraint devices, checks were made every 15–20 minutes to ensure that animals had not altered their position; if an animal altered their position, they were readjusted by the experimenter by loosening the movable disk without completely removing the animal from the restrainer.
Animal weight was taken into consideration to ensure that all animals were restrained equivalently. Animals above 34 g were not used for stress due to the maximum weight of the restrainer and animals that weighed under 22 g had a custom 3D printed 1 mm thick plastic insert fit into the restrainer. Animals weighing 18 g or less were not used for restraint stress as in pilot studies, animals at this weight were capable of exiting through the hole in the acrylic front of the restrainer. Animals subjected to stress were not co-housed with sham animals to avoid the transfer of a stressed phenotype between mice.
Measurement of grimacing pain behavior
Grimace was performed according to previously published methods [5; 13; 35] prior to von Frey testing for all time points. Assessment of 5 characterized pain behaviors on portions of the face (orbital tightening, nose bulging, cheek bulging, flattening of ears, and flattening of whiskers). These behaviors were scored on a scale of 0–2 (0 = not present, 1 = somewhat present, 2 = clearly present).
Dural Injections of reagents/drugs
For experiments where dural stimulation was applied, dural injections were performed according to previously published methods [13]. Briefly, animals were anesthetized for < 2 minutes under isoflurane using a nose cone. While under anesthesia, stimuli were administered onto the dura through the junction of the lambdoid and sagittal sutures via a modified internal cannula (Invivo1, part #8IC313ISPCXC, Internal Cannula, Standard, 28 gauge, fit to 0.5 mm) in a volume of 5 μl. Following injection, animals were placed back in their respective cups in the testing chamber for 1 hour prior to von Frey testing.
Drugs
Drugs applied via dural injection were made in synthetic interstitial fluid (SIF) consisting of 135 mM NaCl, 5 mM KCl, 10 mM HEPES, 2 mM CaCl2, 10 mM glucose, 1 mM MgCl2 (pH 7.4, 310 mOsm). Solutions at pH 7.0 were made using SIF and adjusting pH with HCl. Sodium Nitroprusside (Sigma Aldrich) and sumatriptan (suma; Sigma Aldrich) were dissolved in sterile 1 X PBS. Drug concentrations and administration routes are reported in Table 1.
Table 1.
| Drugs | Source | Doses | Administration |
|---|---|---|---|
| SNP | Sigma-Aldrich | 0.1 mg/kg | Intraperitoneal |
| Sumatriptan | Bachem | 0.6 mg/kg | Intraperitoneal |
| ALD405 | Alder Biopharmaceuticals | 10 mg/kg | Intraperitoneal |
| Isotype Control | Alder Biopharmaceuticals | 10 mg/kg | Intraperitoneal |
Statistical analysis
All data here are shown as mean ± SEM. These data were analyzed among groups for each time point via two-way repeated measures (RM) ANOVA and then followed by Bonferroni post hoc analysis. All graphs were analyzed in two phases: 1. A two-way RM ANOVA was done on the intial acute phase starting from baseline to the return to baseline after stress. 2. Another two-way RM ANOVA was done only on the time points during the priming phase. Data was analyzed using Prism 8.0 (Graph-Pad Software). Significance was set at p < 0.05 for all analyses. The resulting F and P values are reported in Table 2.
Table 2.
Statistical Analyses Results
| Figure | Analysis | Statistics | |
|---|---|---|---|
| Fig. 1A | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(10, 115)=12.04, p<0.0001 | ||
| BL-BL | Time Factor | F(3.008, 69.19)=50.31, p<0.0001 | |
| Treatment Factor | F(2, 23)=28.16, p<0.0001 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 1A | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(4, 46)=3.862, p=0.0087 | ||
| SNP 1–24hr | Time Factor | F(1.854, 42.63)=6.911, p=0.0031 | |
| Treatment Factor | F(2, 23)=1.685, p=0.2075 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 1B | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(10, 89)=36.19, p<0.0001 | ||
| BL-BL | Time Factor | F(2.822, 50.23)=180.0, p<0.0001 | |
| Treatment Factor | F(2, 18)=149.7, p<0.0001 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 1B | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(6, 54)=3.952, p=0.0024 | ||
| SNP 1–24hr | Time Factor | F(1.799, 32.37)=2.446, p=0.1074 | |
| Treatment Factor | F(2, 18)=14.38, p=0.0002 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 1C | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(5, 130)=35.53, p<0.0001 | ||
| BL-BL | Time Factor | F(2.574, 66.93)=42.04, p<0.0001 | |
| Treatment Factor | F(1, 26)=124.6, p<0.0001 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 1C | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(3, 78)=1.571, p=0.2032 | ||
| SNP 1–24hr | Time Factor | F(2.6, 67.60)=7.452, p=0.0004 | |
| Treatment Factor | F(1, 26)=4.448, p=0.0447 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 2A | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(12, 138)=1.608, p=0.0961 | ||
| Time Factor | F(3.257, 74.91)=4.811, p=0.0032 | ||
| Treatment Factor | F(2, 23)=0.1607, p=0.8525 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 2B | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(12, 108)=0.7945, p=0.6652 | ||
| Time Factor | F(3.763, 67.74)=0.9845, p=0.4186 | ||
| Treatment Factor | F(2, 18)=2.249, p=0.1343 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 3A | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(16, 176)=33.03, p<0.0001 | ||
| BL-35D | Time Factor | F(4.232, 93.11)=144.2, p<0.0001 | |
| Treatment Factor | F(2, 22)=170.6, p<0.0001 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 3A | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(6, 66)=1.526, p=0.1834 | ||
| SNP1–72hr | Time Factor | F(2.334, 51.35)=0.7346, p=0.5043 | |
| Treatment Factor | F(2, 22)=0.2865, p=0.7537 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 3B | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(18, 198)=16.84, p<0.0001 | ||
| BL-35D | Time Factor | F(4.475, 98.44)=76.59, p<0.0001 | |
| Treatment Factor | F(2, 22)=78.94, p<0.0001 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 3B | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(8, 88)=0.8713, p=0.5439 | ||
| SNP 1–72hr | Time Factor | F(2.849, 62.69)=2.019, p=0.1233 | |
| Treatment Factor | F(2, 22)=0.5833, p=0.5664 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 4A | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(20, 130)=59.77, p<0.0001 | ||
| BL-14D | Time Factor | F(3.744, 97.34)=222.8, p<0.0001 | |
| Treatment Factor | F(2, 26)=392.2, p<0.0001 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 4A | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(10, 130)=3.467, p=0.0005 | ||
| SNP 16D-72hr | Time Factor | F(3.448, 89.64)=15.26, p<0.0001 | |
| Treatment Factor | F(2, 26)=10.56, p=0.0004 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 4B | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(10, 105)=76.44, p<0.0001 | ||
| BL-14D | Time Factor | F(3.171, 66.58)=408.3, p<0.0001 | |
| Treatment Factor | F(2, 21)=278.4, p<0.0001 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 4B | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(10, 105)=5.245, p<0.0001 | ||
| SNP 16D-72hr | Time Factor | F(3.279, 68.86)=6.289, p=0.0005 | |
| Treatment Factor | F(2, 21)=17.62, p<0.0001 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 5 | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(18, 156)=28.96, p<0.0001 | ||
| BL-14D | Time Factor | F(3.536, 91.93)=264.0, p<0.0001 | |
| Treatment Factor | F(3, 26)=341.9, p<0.0001 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 5 | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(12, 108)=4.287, p<0.0001 | ||
| SNP+Suma 1–72hr | Time Factor | F(3.430, 92.62)=8.103, p<0.0001 | |
| Treatment Factor | F(27, 108)=, p=0.0105 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 6A | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(12, 172)=26.66, p<0.0001 | ||
| BL-14D | Time Factor | F(3.108, 133.6)=94.04, p<0.0001 | |
| Treatment Factor | F(3, 43)=64.93, p<0.0001 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 6A | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(6, 86)=3.244, p=0.0064 | ||
| pH 1–24hr | Time Factor | F(1.915, 82.33)=1.063, p=0.3478 | |
| Treatment Factor | F(3, 43)=27.50, p<0.0001 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 6B | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(12, 112)=14.27, p<0.0001 | ||
| BL-14D | Time Factor | F(2.616, 73.25)=55.79, p<0.0001 | |
| Treatment Factor | F(3, 28)=60.03, p<0.0001 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Fig. 6B | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(6, 56)=1.494, p=0.1970 | ||
| pH 1–24hr | Time Factor | F(1.929, 54.01)=5.457, p=0.0075 | |
| Treatment Factor | F(3, 28)=2.769, p=0.0478 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Suppl. Fig. A. | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(7, 63)=12.15, p<0.0001 | ||
| BL-14D | Time Factor | F(2.811, 25.30)=11.19, p<0.0001 | |
| Treatment Factor | F(1,9)=80.78, p<0.0001 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Suppl. Fig. A. | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(4, 36)=1.241, p=0.3110 | ||
| SNP 1–72hr | Time Factor | F(2.242, 20.18)=1.263, p=0.3075 | |
| Treatment Factor | F(1, 9)=2.299, p=0.0011 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Suppl. Fig. B. | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(7, 70)=7.490, p<0.0001 | ||
| BL-14D | Time Factor | F(3.211, 32.11)=9.698, p<0.0001 | |
| Treatment Factor | F(1, 10)=55.15, p<0.0001 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) | |||
| Suppl. Fig. B. | Two-way repeated measure ANOVA | ||
| Interaction Factor | F(4, 40)=7.378, p=0.0002 | ||
| SNP 1–72hr | Time Factor | F(2.829, 28.29)=2.544, p=0.0791 | |
| Treatment Factor | F(1, 10)=31.81, p=0.0002 | ||
| Bonferroni’s multiple comparisons between treatments (see figure legend) |
Results
Repeated stress using restraint causes facial hypersensitivity and priming to an NO donor in mice
Prior studies using restraint as a model of stress show wide variations in protocols for mice with some using 30 minutes for 1 day [41] while others use 1 hour a day for 2 weeks [14], 2 hours a day for 3 days [33] or 5 hours a day for 14 days [52]. Due to this variation between stress protocols, we first aimed to determine the amount of stress required to produce facial hypersensitivity in mice. Because the durations of a single session of stress are varied, we decided to use 2-hour sessions as an intermediate duration of each stress session. Because consecutive days of stress are more likely to trigger attacks in migraine patients than is a single day, we decided to test this stress across a 3 day paradigm [33; 38]. Males and females were restrained for 2 hours for 3 consecutive days, and von Frey testing was then conducted 24 hours after the end of the final stress session. The 24-hour time point was chosen as the earliest von Frey test after stress in order to avoid the potential of additional stress from behavioral testing as well as stress-induced acute analgesia, both of which have been documented immediately following restraint in rats [26]. Three consecutive days of stress produced significant facial hypersensitivity in both males (Fig. 1A) and females (Fig. 1B) that lasted from 24 hours following stress out to between 7 and 10 days in both males and females. Mice returned to baseline withdrawal thresholds typically by 14 days following the last session of stress. In a few instances, animals did not return to baseline until longer than 14 days, with a few animals even remaining hypersensitive out to 21 days after stress (data not shown as these animals were not used for the figures presented in this manuscript). In order to measure spontaneous non-evoked pain in these animals, we assessed grimace scores in female mice following stress. Females showed significant grimacing that lasted from 24 hours post stress to 7 days’ post stress (Fig. 1C).
Figure 1. The repeated stress paradigm primes male and female mice to subthreshold doses of a NO donor.
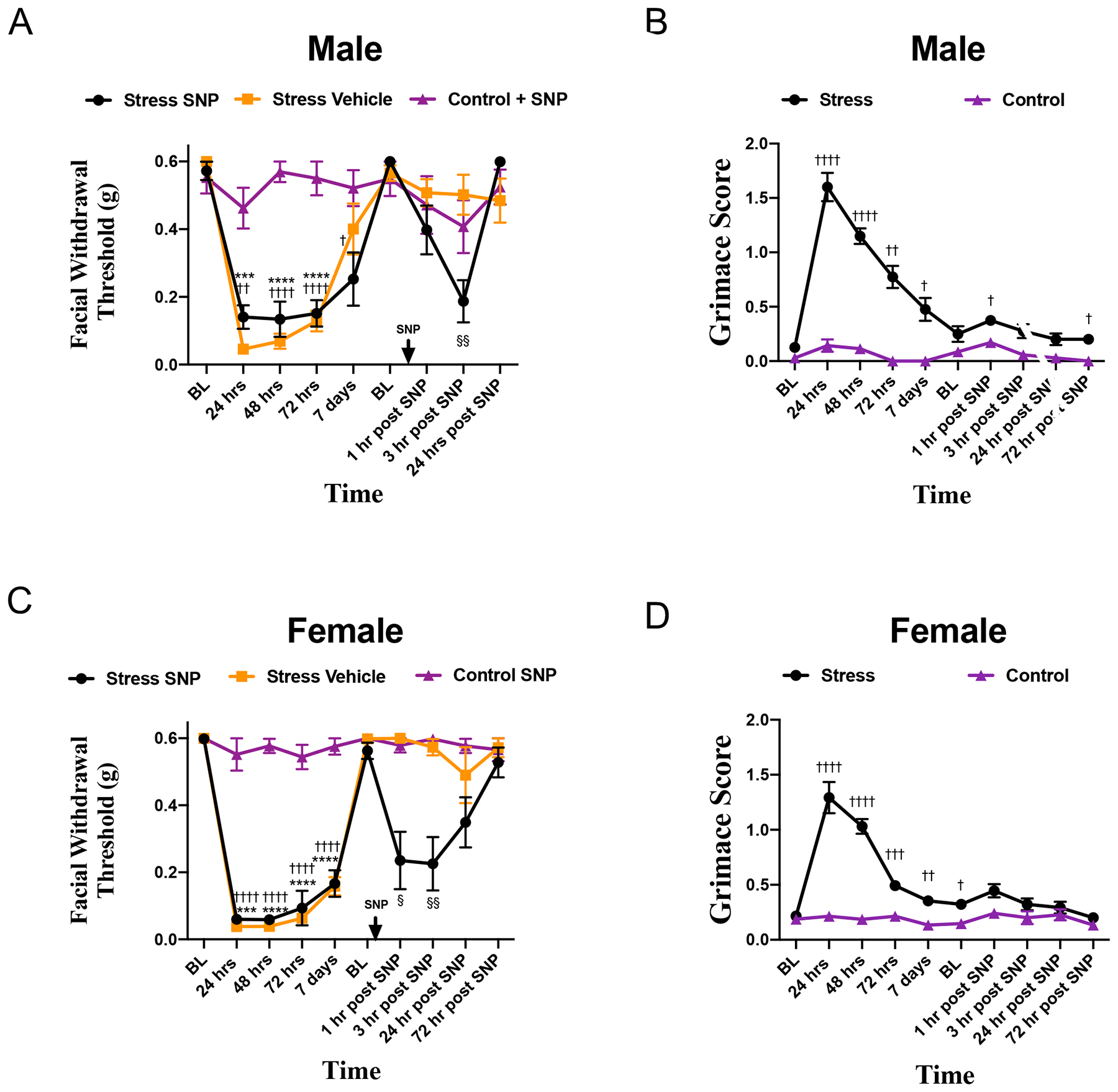
Facial withdrawal thresholds were measured in male (A) and female (C) mice following repeated restraint stress. Upon returning to baseline thresholds at 14 days following the final day of stress, stressed mice were administered either 0.1 mg/kg SNP (n = 8 males, n = 8 females) or vehicle (n = 10 males, n = 6 females). All control mice were given SNP (n = 8 males, n = 7 females). (†) denotes statistical significance between stressed mice that received SNP and control mice; (*) denotes statistical significance between stressed mice that received vehicle and control mice. (§) denotes significance between stressed mice that received SNP and stressed mice that received vehicle. In a separate cohort of mice, grimace responses (B, D) were measured for stressed (n = 15) and control (n = 13) mice following acute stress and administration of 0.1 mg/kg SNP. Two-way RM ANOVA followed by Bonferroni multiple comparison analysis indicated significant differences between stressed and control mice following SNP in both males and females. Data are represented as means ± SEM. †,§p<0.05, ††,§§p<0.01, †††,***p<0.001, ††††,****p< 0.0001. See Table 2 for additional results of analysis.
Prior studies have shown that in contrast to acute restraint, repeated restraint stress in rats is capable of inducing hypersensitivity to nitroglycerin in the tail-flick test [17]. In order to determine whether these repeated stress exposures caused priming to the NO-donor, animals were injected with the normally subthreshold dose of 0.1 mg/kg SNP following a return to baseline. Both male and female mice that received SNP had significantly reduced facial withdrawal thresholds at 1 and 3 hours following SNP injection. Female mice were also observed for facial grimace responses following SNP injection. While females that received the NO donor showed significantly reduced withdrawal thresholds, these animals showed no significant grimacing following SNP (Fig. 1C).
To ensure that stress responses to this paradigm were maintained across strains, mice with a mixed background of both C57BL6 and 129J were exposed to stress (Supplementary fig. 1). Both male and female mice off this background responded to initial stress from 24 hours following stress out to 14 days following stress. Additionally, male and female mice were primed to respond to 0.1 mg/kg SNP. These data demonstrate that this stress paradigm is not specific to a single mouse strain.
As previously mentioned, stress across consecutive days is more likely to cause migraine in migraine patients when compared with 1 day of stress [38]. In order to explore the potential differences in repeated sessions of stress in this model male and female mice underwent 1 session of 2-hour restraint stress. One 2-hour session of restraint stress failed to produce significantly reduced facial withdrawal thresholds in both male (Fig. 2A) or female mice (Fig 2B). Mice were tested out to 5 days following stress to ensure that delayed hypersensitivity did not occur. While the stress produced no acute facial hypersensitivity, our prior work demonstrated that a single stimulus (dural stimulation with IL-6 or CGRP) can cause priming to subthreshold doses of a common migraine trigger i.e. an NO donor [5; 12] so it was also important to test for potential priming following a single stress exposure. Prior work using the tail-flick test in rats found that 1 session of acute stress caused stress-induced analgesia and resulted in no response the NO-donor nitroglycerin [17]. We thus tested whether acute stress would cause priming to a subthreshold dose of the NO-donor sodium nitroprusside (SNP). Following the fifth day of testing, neither males nor females that received 0.1 mg/kg SNP showed facial hypersensitivity. These data demonstrate that a single session of stress is not sufficient to cause acute hypersensitivity nor is it sufficient to induce priming to SNP. The 3-day stress paradigm was used for all subsequent experiments
Figure 2. Single-restraint stress does not produce facial allodynia or priming to SNP.
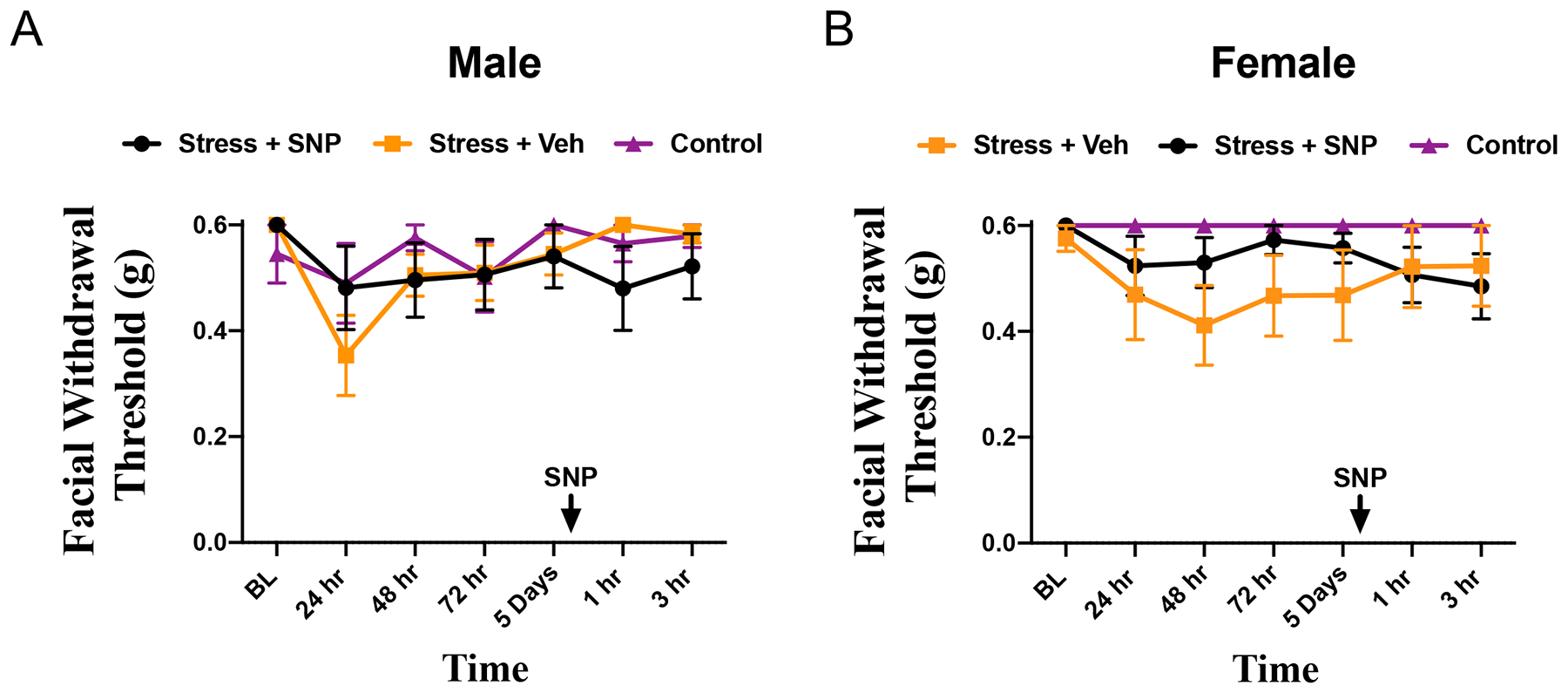
(A) Facial withdrawal thresholds of male mice following a single session of stress and administration of either SNP(n=10) or vehicle (n=7) after 5 days of testing or naïve mice (n=9). (B) Facial withdrawal thresholds of female mice following a single session of stress and administration of either SNP (n=7) or vehicle (n=6) after 5 days of testing or naïve mice (n=6). Two-way RM with Bonferroni multiple comparison analysis indicates there was no statistical difference among control mice and all cohorts that were stressed in both the acute phase and the priming phase (mean ±SEM). See table 2 for additional statistical analysis.
The previous experiments show that 3 days of restraint stress cause priming to SNP when given relatively shortly after mice return to baseline. In order to determine whether stress-induced priming is extinguished over time, animals underwent the 3-day stress protocol and were tested with von Frey filaments until they returned to baseline at approximately 14 days after stress. Following a return to baseline, animals were then tested once weekly until they reached 35 days post stress (i.e. 35 days following the last session of restraint). Animals were then injected with 0.1 mg/kg SNP at this late time point. Neither males (Fig. 3A) nor females (Fig. 3B) that received SNP at 35-days post stress showed reduced facial withdrawal thresholds when compared with controls. These data indicate that while repeated stress causes priming to SNP, the primed state is not permanent.
Figure 3. Repeated stress induces transient priming to SNP in male and female mice.
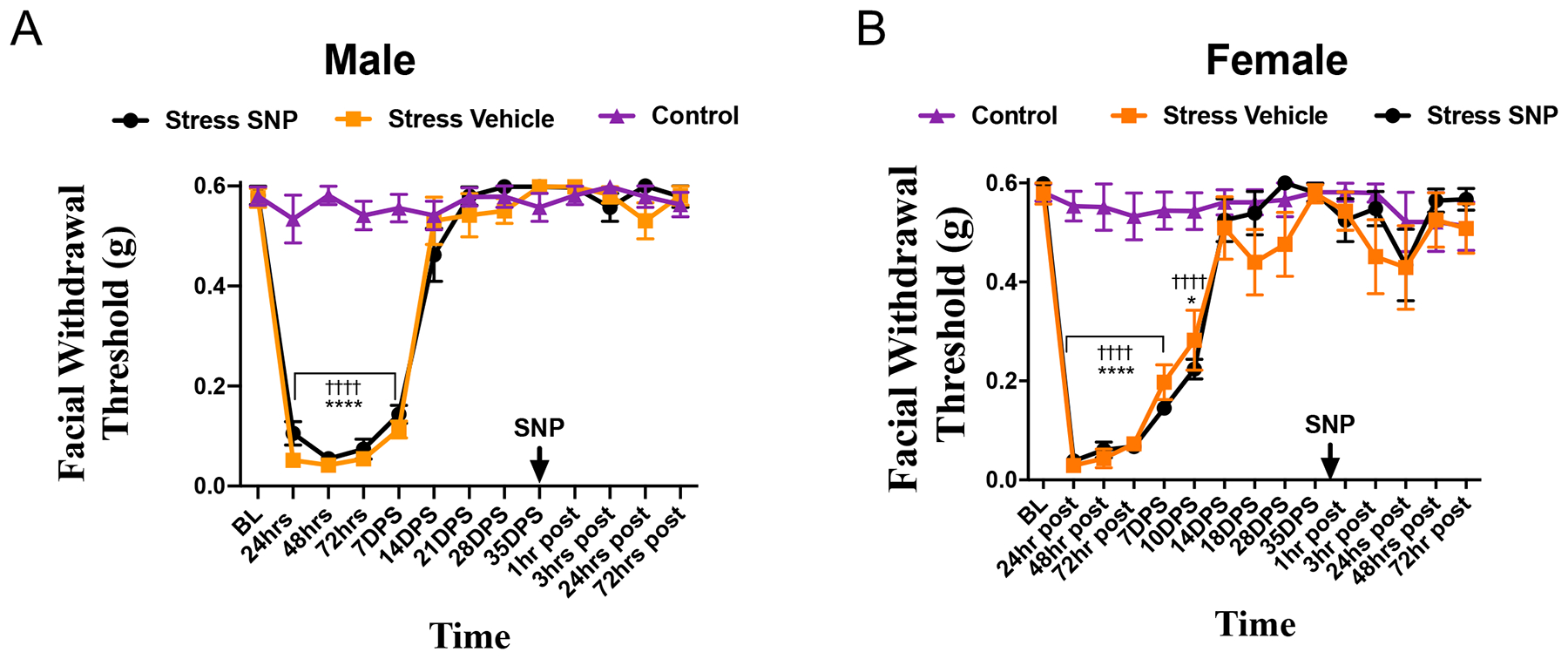
Facial withdrawal thresholds were measured in (A) male and (B) female mice following acute stress (n = 8 male, 8 female). Following the final day of stress, animals were tested out to 35 days and received either 0.1 mg/kg SNP (n = 8 males, n = 9 females) or vehicle (n = 9 males, n = 8 females). All controls received SNP. (†) denotes statistical significance between stressed mice will receive SNP and control mice. (*) denotes statistical significance between stressed mice that will receive vehicle and control mice. (§) denotes statistical significance between stressed mice that received vehicle and stressed miced that received SNP. Two-way RM ANOVA with Bonferroni multiple comparison analysis indicated a significant difference between stressed and control mice in the acute phase, but revealed no significant differences following administration of SNP. Data are represented as means ± SEM. *p<0.05, ††††,****p<0.0001. See Table 2 for additional results of analysis.
Efficacy of human migraine therapeutics against priming to SNP following stress
CGRP has been strongly implicated in the pathology of migraine, and the use of monoclonal antibodies against CGRP and the CGRP receptor have now been demonstrated to be efficacious in the preventive-treatment of migraine [22; 23; 47; 49]. To determine the possible role for CGRP in stress-induced hypersensitivity, both male and female animals underwent the 3-day restraint stress protocol and were tested until they returned to baseline withdrawal thresholds. At 24 hours following return to baseline, animals received 10 mg/kg i.p. of ALD405, a monoclonal antibody against CGRP, or an isotype control. At 24 hours after dosing of the antibody, 0.1 mg/kg SNP was given systemically as described above. ALD405 significantly blocked the effects of SNP in females (Fig. 4A) from 1–72 hours following SNP injection. Whereas males (Fig. 4B) that received ALD405 only experienced decreased mechanical sensitivity at 48 hours following SNP administration. Females that received the active form of the antibody showed no significant differences from controls in response to SNP. These findings show that a monoclonal antibody against CGRP is capable of blocking the primed response to SNP in a sex-specific manner. This suggests that not only does CGRP have a prominent role in a stress induced priming to NO donors, but that this role may be sexually dimorphic.
Figure 4. Effects of CGRP monoclonal antibodies in repeated stress induced priming to SNP in mice.
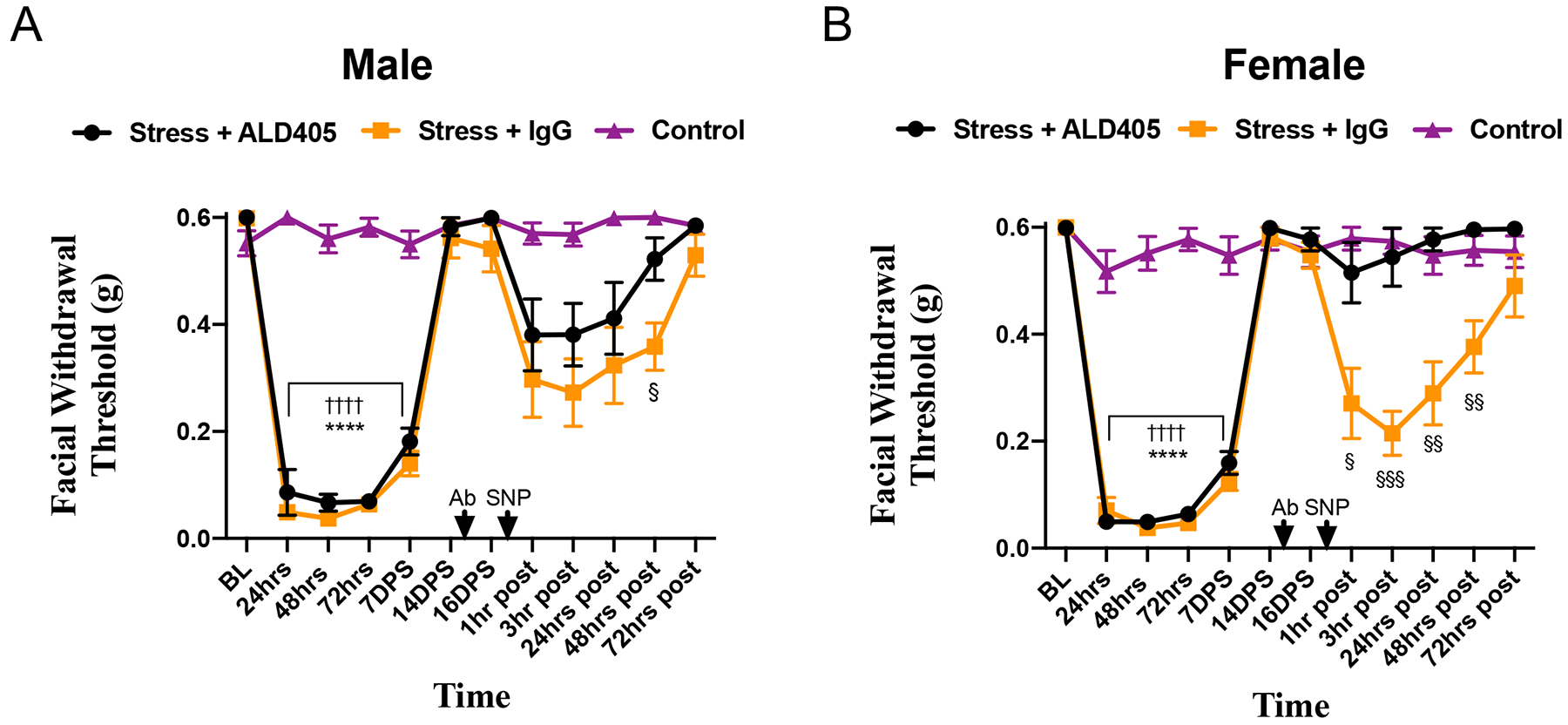
Facial withdrawal thresholds were measured in male (A) and female (B) mice following acute stress (n = 10 males, n = 7 females). 15 days following the final day of stress, animals received either the anti-CGRP antibody ALD405 (n = 10 males, n = 8 females) or an isotype control (n = 9 males, n = 9 females). 24 hours following administration of antibody, all animals received 0.1 mg/kg SNP. In the acute phase, (†) denotes statistical significance between control mice and stressed mice that will receive ALD405 and SNP. (*) denotes statistical significance between control mice and stressed mice that will receive isotype control IgG. In the priming phase, (§) denotes statistical significance between stressed mice that received ALD405 and those that received the isotype control prior to SNP. Two-way ANOVA followed by Bonferroni multiple comparison analysis revealed significant differences in the priming phase between female stressed mice that received ALD405 and those that received the isotype. Statistical significance between these groups was observed in males at 48 hours following SNP. Data are represented as means ± SEM. §p<0.05,§§p < 0.01, §§§p<0.001, ††††,****p<0.0001. See Table 2 for additional results of analysis.
Sumatriptan, as well as other triptans, are among the most commonly used acute migraine therapeutics. It was thus important to determine whether this stress priming model is sensitive to the effects of sumatriptan. To test this question, we examined the efficacy of sumatriptan against the hypersensitivity caused by SNP in stress-primed mice. Female mice underwent the restraint stress protocol and were allowed to return to baseline as described for the prior experiments. At 24-hours following return to baseline, mice received either vehicle (1X PBS), 0.1 mg/kg SNP, 0.6 mg/kg sumatriptan (the standard dose used throughout preclinical migraine studies [10; 20; 42; 46]), or a co-injection of SNP and sumatriptan (Fig. 5). Animals that received only sumatriptan were not significantly different from baseline at any time point. As in prior experiments, animals that received SNP showed significantly reduced withdrawal thresholds when compared with controls out to 24 hours. Animals that received a co-injection of SNP and sumatriptan exhibited significantly reduced withdrawal thresholds when compared to either control animals or those that received sumatriptan alone out to 3 hours following injection. There were no significant differences between the SNP + sumatriptan group and animals that received only SNP at any time point. These data demonstrate that repeated stress leads to a state of priming where sumatriptan lacks efficacy against subsequent exposure to an NO donor.
Figure 5. Sumatriptan does not block SNP responses in stress-primed mice.
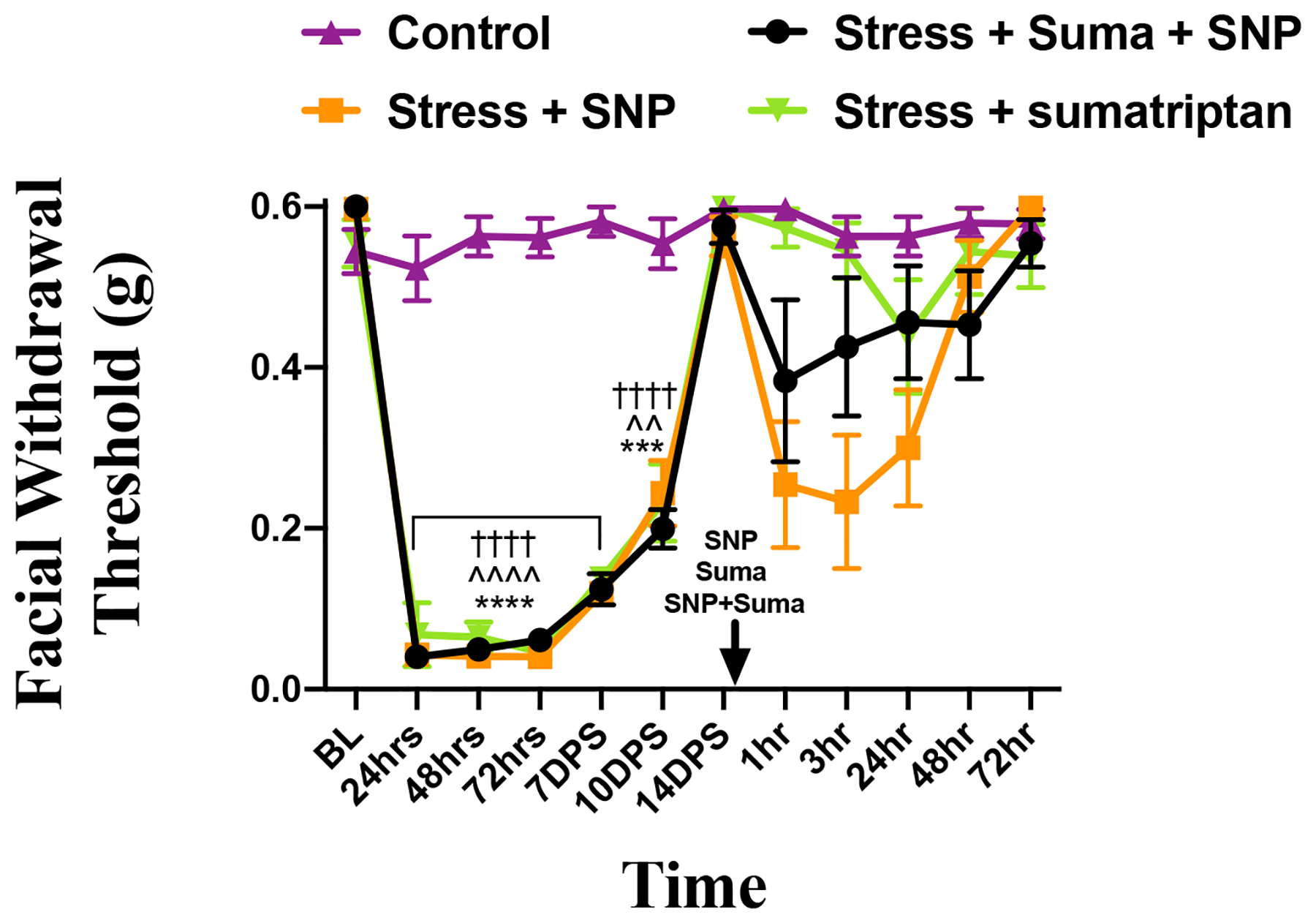
Facial withdrawal thresholds of female mice either naïve (n=8) or following acute stress and administration of either SNP (0.1 mg/kg; n=8), suma (0.6 mg/kg; n=7), or a coadministration of suma and SNP (n=7). In the acute phase, (*) denotes significance between control mice and stressed mice that will receive SNP; (^) denotes significance between control mice and stressed mice that will receive suma; (†) denotes significance between control mice and stressed mice that will receive suma and SNP. For 24hr-7DPS in the acute phase, the statistical significance is the same for each time point. Two-way RM with Bonferroni multiple comparison analysis indicates a statistical difference among control mice and all cohorts that were stressed. In the priming phase there was no statistical difference detected between stressed mice that received SNP and those that received suma and SNP (mean ±SEM, ^^p<0.01, ***p<0.001, ****,^^^^, ††††p<0.0001). See table 2 for additional statistical analysis.
Repeated stress primes mice to dural stimulation with a subthreshold pH
These studies demonstrate that repeated stress primes mice to subthreshold doses of NO-donors. However, it is not clear from these studies whether stress and NO donors have sensitizing actions within the meninges as both stimuli are non-specific to any location within the body. We thus investigated whether repeated restraint stress could prime animals to a direct dural stimulus. Female mice were subject to the repeated stress protocol and allowed to return to baseline withdrawal thresholds as in the prior experiments. At 24 hours after return to baseline, mice received dural injections of either physiological pH of 7.4 or a slightly decreased, but normally non-noxious pH 7.0, the latter a stimulus that we have previously shown animals become primed to by prior dural application of IL-6 or CGRP[5; 12]. Stressed females that received dural pH 7.0 experienced significant allodynia at 1 hour, 3hours, and 24 hours post injection when compared with stressed animals that received pH 7.4 and control non-stressed animals that received either dural pH 7.0 and pH 7.4 (Fig. 6A). In addition, these animals showed significant grimacing in response to stress out to 7 days (Fig. 6B), however none of the animals showed any significant grimace in response to any secondary stimulus. These data demonstrate that repeated stress primes mice to both systemic migraine triggers as well as direct stimuli applied to the dura.
Figure 6. Repeated stress primes female mice to decreased dural pH.
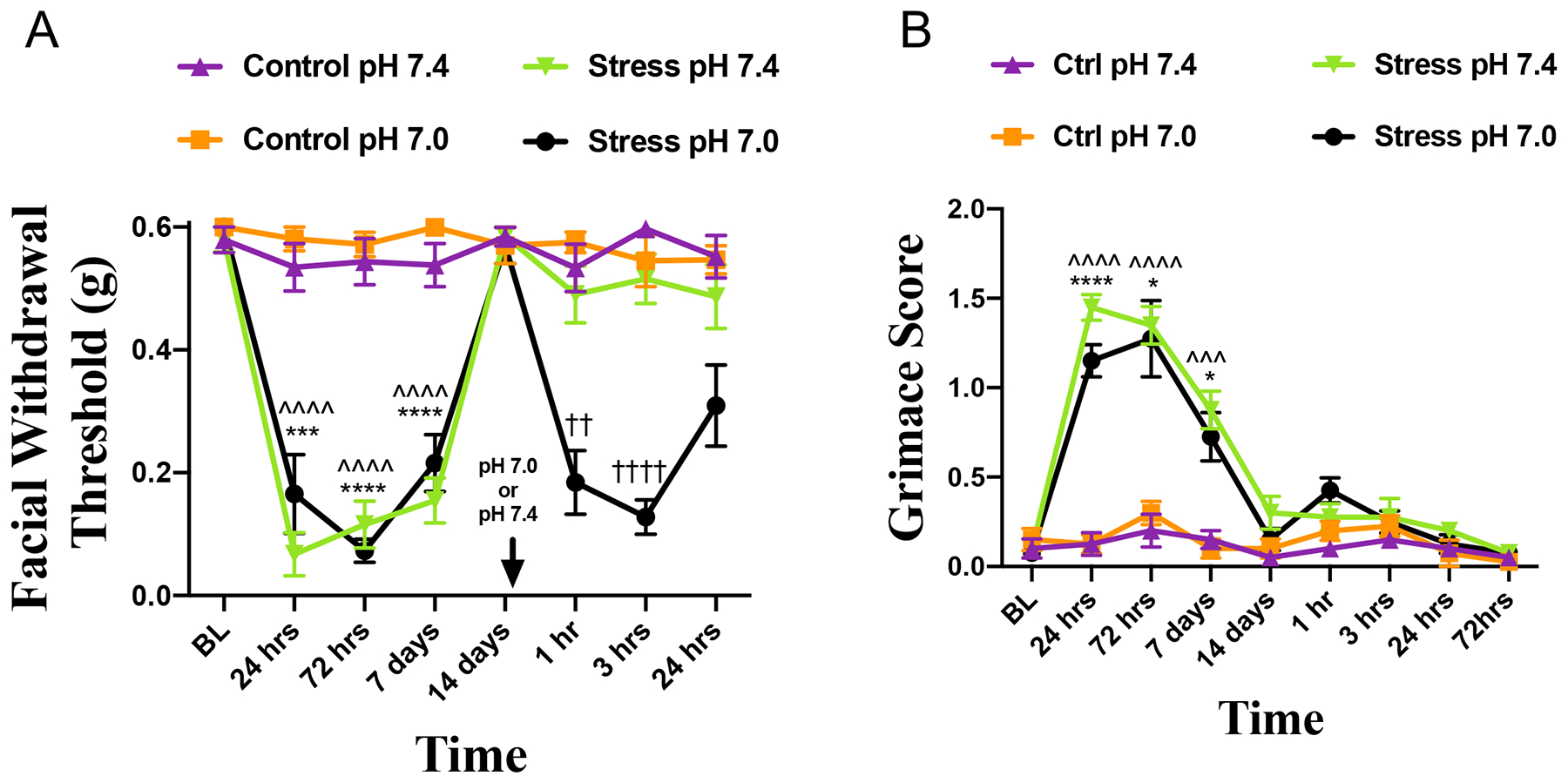
(A) Facial withdrawal thresholds of female mice following acute stress and priming to dural pH 7.0 (n=12) or 7.4 (n=12) and control mice given pH 7.0 (n=12) and pH 7.4 (n=11). (B) Grimace scores of mice that were first stressed and then administered either pH 7.0 (n=8) or pH 7.4 (n=8) or mice that were naïve and then administered either pH 7.0 (n=8) or pH 7.4 (n=8). For the acute phase, (*) denotes significance between stressed and control mice that will receive dural pH 7.0 ; (^) denotes significance between stressed and control mice that will receive dural pH 7.4. For the priming phase, (†) denotes significance between stressed mice that received pH 7.0 and stressed mice that received pH 7.4. Dural pH stimuli are given at the time point indicated by the arrows in A; prior to dural pH injections, animals were only exposed to either stress or control. Two-way ANOVA RM with Bonferroni multiple comparison analysis indicate a significant difference between stressed and control mice and stressed mice that received pH 7.0 compared to stressed mice that received pH 7.4 (mean ±SEM, *p<0.05, ††p<0.01, ***, ^^^p<0.001, ****,^^^^,††††p<0.0001). See table 2 for additional statistical analysis.
Discussion
The mechanisms by which stress contributes to migraine are poorly understood. Better understanding of these mechanisms may lead to insights for new therapeutic targets as well as other potential approaches. Here we show that repeated restraint stress results in significant facial hypersensitivity and grimace in mice, starting approximately 24 hours after the end of stress, the earliest time point tested. Repeated stress also primes mice to subthreshold doses of the nitric oxide donor SNP as well as subthreshold dural stimulation with pH 7.0. Mice remained primed to SNP for 2 weeks following stress but priming was no longer present by 5 weeks. Responses to subthreshold doses of SNP in primed animals were blocked by a CGRP monoclonal antibody, implicating CGRP in the mechanisms that contribute to low-dose NO donors responses in the primed state. This was not the case when sumatriptan was used, demonstrating that priming due to stress may lead to a “triptan-unresponsive state.” Together, these data support the use of a stress priming model to help further study the mechanisms by which stress contributes to migraine. Importantly, this model can be induced without significant tissue injury.
Calcitonin gene-related peptide (CGRP) has long been implicated in the pathology of migraine [24; 30; 36]. The observation that the response to SNP following repeated stress is blocked by a CGRP monoclonal antibody (although to a greater extent in females) suggests that exposure to a subthreshold dose of SNP in the primed state is similar to migraine attacks. However, sumatriptan had no effect on SNP-donor induced responses in primed mice. This is in contrast to humans, where sumatriptan reduces nitroglycerin-induced headaches [1; 29]. In rodent studies, both sumatriptan and the monoclonal antibody ALD405 were shown to reduce pain from repeated glyceryl trinitrate administration [16]. In other rodent studies, sumatriptan was not efficacious in blocking NO donor induced facial hypersensitivity when given prior to repeated NO-donor administration across 5 days [19]. Neither of these studies used stress however. The inability of sumatriptan to prevent NO-donor mediated hypersensitivity following repeated stress may be able to provide insight into mechanisms that are present in the approximately 40% of migraine patients that take oral triptans and do not experience relief [25] and also how stress may contribute to triptan response/non-response. Further, this model may be helpful in identifying mechanisms by which NO donors contribute to migraine since only primed animals respond to low-dose SNP (thus mimicking the human observation that NO donors do not trigger attacks in controls). Our data show that the mechanism of NO in this model is dependent on CGRP since ALD405 blocked the response to SNP in primed mice. This suggests that stress increases NO donor-evoked CGRP release. Whether this is due to increased sensitivity to NO or increased CGRP expression is not clear but prior studies found increased CGRP expression or binding following stress in several locations throughout the body [28; 51; 56]. Since the increased sensitivity to SNP is not permanent i.e. it was not present 5 weeks following the end of the stress paradigm, this model may also be valuable for identifying mechanisms by which sensitivity to the effects of stress are extinguished.
Migraine affects women disproportionately to men; mechanisms underlying this dimorphism are not known. There were no sex differences in this study in the acute response to stress, nor were there sex differences in the response to SNP given after the resolution of the stress-induced behaviors. While we did not test stress-induced priming to dural pH 7.0 in both sexes, our prior work using dural IL-6 as a priming agent did not find any differences between males and females in the primed response to dural pH 7.0 [13]. We also did not test whether there are sex differences in the efficacy of sumatriptan against the SNP response in stress-primed mice, but there is little prior evidence to suggest that sex differences in the efficacy of sumatriptan exist in animals or humans. There are sex differences in the ability of sumatriptan to cause acute hyperalgesia or to cause priming [3; 4], but not in its ability to block behavior caused by other stimuli such as SNP. There was a sexually-dimorphic response observed here for efficacy of ALD405. We have recently shown that female mice are more sensitive than males to the pain-promoting effects of CGRP in the meninges [5]. Consistent with the prior finding of female-specific CGRP effects, ALD405 was more effective in blocking responses to SNP following stress in females than in males. While anti-CGRP therapeutics are effective in many migraine patients, both female and male, factors that predict efficacy are not currently known. Additionally, mechanisms by which stress and NO donors contribute to migraine are poorly understood. While far more investigation is needed, our work is a substantial advance and suggests that stress combined with NO donors may act through CGRP-dependent mechanisms in females but not in males, that stress may increase NO donor-evoked CGRP release selectively in females, or that stress may exacerbate the hypersensitivity of female blood vessels to vasodilators [34]. In contrast, studies using traumatic-brain injury (TBI) in rodents have found greater efficacy of the CGRP monoclonal antibody in males compared to females [8; 9]. These latter studies highlight the potentially complex actions of CGRP in headache disorders supporting the need moving forward to examine all CGRP-based effects in both sexes. The model system that we have developed, and similar model systems, may start to draw mechanistic connections between psycho-social migraine findings, precipitating events like stress or TBI, and specific molecular mechanisms that can predict who will respond to certain types of therapeutics, like CGRP-targeting drugs or triptans.
How and where CGRP and NO contribute to migraine pathology is still unclear. We propose that the repeated stress-induced priming model is a valuable tool in addressing this question. In the case CGRP, the antibodies are highly unlikely to cross the blood-brain barrier and thus the location where this drug blocks the effect of SNP is within the peripheral compartment. Since NO freely diffuses throughout the body, there could be many potential locations where it causes behavioral responses at low doses in stressed mice (one of which is trigeminal ganglion neurons [7]). Nonetheless, we show that stress causes priming to stimulation of the dura, an important location for the development of headache. These data suggest that CGRP and/or NO could be acting in this tissue, but more work is needed to better address this question. Whether stress would cause sensitized responses to direct dural application of either CGRP or NO is not yet clear, but is the subject of future studies.
The findings described here demonstrate that repeated stress causes a grimace response in mice, lasting out to 7-days following the end of the stress protocol. Grimace is typically used in preclinical studies as a measure of the affective component of pain [2; 35; 37; 55]. The presence of grimace following stress suggests that a similar affective state is present to those that occur following surgical incisions and nerve injuries. The combination of grimace with cutaneous hypersensitivity is suggestive of pain, but it is naturally difficult to determine the exact state of the mice under these conditions. Grimace may nonetheless capture the state of unpleasantness that is caused by stress and thus offers a valuable additional endpoint to use in these studies. In contrast to the acute post-stress phase, there was no grimace response to SNP in the primed phase in these studies. While surprising, this may indicate that SNP causes cutaneous hypersensitivity (a characteristic present in migraine patients, particularly those with the greatest risk of progression to chronic migraine [39; 40]), but not “pain” or “unpleasantness”. Alternatively, the lack of grimace may rather be due to compensatory suppression of grimace over time during the early post-stress phase. Suppression of grimace over time as a protective mechanism to hide the presence of pain from predators has been suggested to occur in rodent pain models [43] and may occur here during the initial responses to stress. If this is indeed the case, mice may no longer exhibit a grimace response when presented with a subsequent noxious challenge as they have previously adapted to the presence of pain by suppressing grimace.
The data presented here support the use of repeated stress-induced priming as a new preclinical model of migraine in which to further investigate the mechanisms of the disorder and their regulation by stress. It may also be a valuable tool to investigate efficacy of novel therapeutics, especially those that are likely to act within mechanisms or pathways regulated by stress. Finally, it offers the opportunity to study conditions related to migraine in the absence of tissue injury. While several preclinical behavioral models of migraine currently exist, it is unlikely that any single model will accurately capture the complexity that is human migraine. This model may be an important addition to the current list of preclinical tools, and may be able to capture aspects of the disorder not currently represented in the currently-available models.
Supplementary Material
Supplementary Figure 1 Facial withdrawal thresholds of mixed background C57BL6/129J (A) male (n=5) and (B) female (n=6) mice that were stressed or male (n=6) and female (n=6) mice that were naïve. had withdrawal thresholds determined prior to and following stress. Two-way RM ANOVA with Bonferroni multiple comparison analysis indicates a statistical difference in control male and female mice compared to stressed male and female mice over time (mean ±SEM, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). See table 2 for additional statistical analysis.
Acknowledgements
The authors would like to thank Nandita Ramkumar and Gianna Maggiore for technical support. This work was supported by National Institutes of Health grants NS072204 (GD) and NS104200 (GD and AA). This work was supported in part by a grant from Alder Biopharmaceuticals. Drs. Dussor and Price are co-founders of CerSci Therapeutics and Ted’s Brain Science Products. These relationships are outside the scope of the current work.
References
- [1].Akerman S, Karsan N, Bose P, Hoffmann JR, Holland PR, Romero-Reyes M, Goadsby PJ. Nitroglycerine triggers triptan-responsive cranial allodynia and trigeminal neuronal hypersensitivity. Brain 2019;142(1):103–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Akintola T, Raver C, Studlack P, Uddin O, Masri R, Keller A. The grimace scale reliably assesses chronic pain in a rodent model of trigeminal neuropathic pain. Neurobiol Pain 2017;2:13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Araldi D, Ferrari LF, Green P, Levine JD. Marked sexual dimorphism in 5-HT1 receptors mediating pronociceptive effects of sumatriptan. Neuroscience 2017;344:394–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Araldi D, Ferrari LF, Levine JD. Gi-protein-coupled 5-HT1B/D receptor agonist sumatriptan induces type I hyperalgesic priming. Pain 2016;157(8):1773–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Avona A, Burgos-Vega C, Burton MD, Akopian AN, Price TJ, Dussor G. Dural Calcitonin Gene-Related Peptide Produces Female-Specific Responses in Rodent Migraine Models. J Neurosci 2019;39(22):4323–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Balkaya M, Seidel JL, Sadeghian H, Qin T, Chung DY, Eikermann-Haerter K, van den Maagdenberg A, Ferrari MD, Ayata C. Relief Following Chronic Stress Augments Spreading Depolarization Susceptibility in Familial Hemiplegic Migraine Mice. Neuroscience 2019;415:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bellamy J, Bowen EJ, Russo AF, Durham PL. Nitric oxide regulation of calcitonin gene-related peptide gene expression in rat trigeminal ganglia neurons. The European journal of neuroscience 2006;23(8):2057–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bree D, Levy D. Development of CGRP-dependent pain and headache related behaviours in a rat model of concussion: Implications for mechanisms of post-traumatic headache. Cephalalgia 2018;38(2):246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bree D, Mackenzie K, Stratton J, Levy D. Enhanced post-traumatic headache-like behaviors and diminished contribution of peripheral CGRP in female rats following a mild closed head injury. Cephalalgia 2020:333102420907597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Burgos-Vega CC, Ahn DD, Bischoff C, Wang W, Horne D, Wang J, Gavva N, Dussor G. Meningeal transient receptor potential channel M8 activation causes cutaneous facial and hindpaw allodynia in a preclinical rodent model of headache. Cephalalgia 2016;36(2):185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Burgos-Vega CC, Quigley LD, Avona A, Price T, Dussor G. Dural stimulation in rats causes BDNF-dependent priming to subthreshold stimuli including a migraine trigger. Pain 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Burgos-Vega CC, Quigley LD, Avona A, Price T, Dussor G. Dural stimulation in rats causes brain-derived neurotrophic factor-dependent priming to subthreshold stimuli including a migraine trigger. Pain 2016;157(12):2722–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Burgos-Vega CC, Quigley LD, Trevisan Dos Santos G, Yan F, Asiedu M, Jacobs B, Motina M, Safdar N, Yousuf H, Avona A, Price TJ, Dussor G. Non-invasive dural stimulation in mice: A novel preclinical model of migraine. Cephalalgia 2018:333102418779557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Carneiro de Oliveira PE, Zaniboni CR, Carmona IM, Fonseca AR, Canto-de-Souza A. Preliminary behavioral assessment of cagemates living with conspecifics submitted to chronic restraint stress in mice. Neurosci Lett 2017;657:204–210. [DOI] [PubMed] [Google Scholar]
- [15].Chanda ML, Tuttle AH, Baran I, Atlin C, Guindi D, Hathaway G, Israelian N, Levenstadt J, Low D, Macrae L, O’Shea L, Silver A, Zendegui E, Mariette Lenselink A, Spijker S, Ferrari MD, van den Maagdenberg AM, Mogil JS. Behavioral evidence for photophobia and stress-related ipsilateral head pain in transgenic Cacna1a mutant mice. Pain 2013;154(8):1254–1262. [DOI] [PubMed] [Google Scholar]
- [16].Christensen SL, Petersen S, Kristensen DM, Olesen J, Munro G. Targeting CGRP via receptor antagonism and antibody neutralisation in two distinct rodent models of migraine-like pain. Cephalalgia 2019:333102419861726. [DOI] [PubMed] [Google Scholar]
- [17].Costa A, Smeraldi A, Tassorelli C, Greco R, Nappi G. Effects of acute and chronic restraint stress on nitroglycerin-induced hyperalgesia in rats. Neurosci Lett 2005;383(1–2):7–11. [DOI] [PubMed] [Google Scholar]
- [18].D’Agostino J, Vaeth GF, Henning SJ. Diurnal rhythm of total and free concentrations of serum corticosterone in the rat. Acta Endocrinol (Copenh) 1982;100(1):85–90. [DOI] [PubMed] [Google Scholar]
- [19].Dallel R, Descheemaeker A, Luccarini P. Recurrent administration of the nitric oxide donor, isosorbide dinitrate, induces a persistent cephalic cutaneous hypersensitivity: A model for migraine progression. Cephalalgia 2018;38(4):776–785. [DOI] [PubMed] [Google Scholar]
- [20].De Felice M, Ossipov MH, Wang R, Dussor G, Lai J, Meng ID, Chichorro J, Andrews JS, Rakhit S, Maddaford S, Dodick D, Porreca F. Triptan-induced enhancement of neuronal nitric oxide synthase in trigeminal ganglion dural afferents underlies increased responsiveness to potential migraine triggers. Brain 2010;133(Pt 8):2475–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].De Felice M, Ossipov MH, Wang R, Lai J, Chichorro J, Meng I, Dodick DW, Vanderah TW, Dussor G, Porreca F. Triptan-induced latent sensitization: a possible basis for medication overuse headache. Ann Neurol 2010;67(3):325–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dodick DW, Goadsby PJ, Silberstein SD, Lipton RB, Olesen J, Ashina M, Wilks K, Kudrow D, Kroll R, Kohrman B, Bargar R, Hirman J, Smith J, investigators ALDs. Safety and efficacy of ALD403, an antibody to calcitonin gene-related peptide, for the prevention of frequent episodic migraine: a randomised, double-blind, placebo-controlled, exploratory phase 2 trial. Lancet Neurol 2014;13(11):1100–1107. [DOI] [PubMed] [Google Scholar]
- [23].Dodick DW, Silberstein SD, Bigal ME, Yeung PP, Goadsby PJ, Blankenbiller T, Grozinski-Wolff M, Yang R, Ma Y, Aycardi E. Effect of Fremanezumab Compared With Placebo for Prevention of Episodic Migraine: A Randomized Clinical Trial. JAMA 2018;319(19):1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Edvinsson L, Goadsby PJ. Neuropeptides in migraine and cluster headache. Cephalalgia 1994;14(5):320–327. [DOI] [PubMed] [Google Scholar]
- [25].Ferrari MD, Roon KI, Lipton RB, Goadsby PJ. Oral triptans (serotonin 5-HT(1B/1D) agonists) in acute migraine treatment: a meta-analysis of 53 trials. Lancet 2001;358(9294):1668–1675. [DOI] [PubMed] [Google Scholar]
- [26].Gamaro GD, Xavier MH, Denardin JD, Pilger JA, Ely DR, Ferreira MB, Dalmaz C. The effects of acute and repeated restraint stress on the nociceptive response in rats. Physiol Behav 1998;63(4):693–697. [DOI] [PubMed] [Google Scholar]
- [27].Group GBDNDC. Global, regional, and national burden of neurological disorders during 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol 2017;16(11):877–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Guidobono F, Netti C, Pecile A, Gritti I, Mancia M. Stress-related changes in calcitonin gene-related peptide binding sites in the cat central nervous system. Neuropeptides 1991;19(1):57–63. [DOI] [PubMed] [Google Scholar]
- [29].Iversen HK, Olesen J. Headache induced by a nitric oxide donor (nitroglycerin) responds to sumatriptan. A human model for development of migraine drugs. Cephalalgia 1996;16(6):412–418. [DOI] [PubMed] [Google Scholar]
- [30].Jang MU, Park JW, Kho HS, Chung SC, Chung JW. Plasma and saliva levels of nerve growth factor and neuropeptides in chronic migraine patients. Oral Dis 2011;17(2):187–193. [DOI] [PubMed] [Google Scholar]
- [31].Kaufmann D, Brennan KC. The Effects of Chronic Stress on Migraine Relevant Phenotypes in Male Mice. Front Cell Neurosci 2018;12:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kelman L The triggers or precipitants of the acute migraine attack. Cephalalgia 2007;27(5):394–402. [DOI] [PubMed] [Google Scholar]
- [33].Koh SJ, Kim JW, Kim BG, Lee KL, Kim JS. Restraint stress induces and exacerbates intestinal inflammation in interleukin-10 deficient mice. World J Gastroenterol 2015;21(28):8580–8587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Krause DN, Duckles SP, Pelligrino DA. Influence of sex steroid hormones on cerebrovascular function. J Appl Physiol (1985) 2006;101(4):1252–1261. [DOI] [PubMed] [Google Scholar]
- [35].Langford DJ, Bailey AL, Chanda ML, Clarke SE, Drummond TE, Echols S, Glick S, Ingrao J, Klassen-Ross T, Lacroix-Fralish ML, Matsumiya L, Sorge RE, Sotocinal SG, Tabaka JM, Wong D, van den Maagdenberg AM, Ferrari MD, Craig KD, Mogil JS. Coding of facial expressions of pain in the laboratory mouse. Nat Methods 2010;7(6):447–449. [DOI] [PubMed] [Google Scholar]
- [36].Lassen LH, Haderslev PA, Jacobsen VB, Iversen HK, Sperling B, Olesen J. CGRP may play a causative role in migraine. Cephalalgia 2002;22(1):54–61. [DOI] [PubMed] [Google Scholar]
- [37].Leach MC, Klaus K, Miller AL, Scotto di Perrotolo M, Sotocinal SG, Flecknell PA. The assessment of post-vasectomy pain in mice using behaviour and the Mouse Grimace Scale. PLoS One 2012;7(4):e35656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lipton RB, Buse DC, Hall CB, Tennen H, Defreitas TA, Borkowski TM, Grosberg BM, Haut SR. Reduction in perceived stress as a migraine trigger: testing the “let-down headache” hypothesis. Neurology 2014;82(16):1395–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lipton RB, Fanning KM, Buse DC, Martin VT, Hohaia LB, Adams AM, Reed ML, Goadsby PJ. Migraine progression in subgroups of migraine based on comorbidities: Results of the CaMEO Study. Neurology 2019;93(24):e2224–e2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lipton RB, Fanning KM, Buse DC, Martin VT, Reed ML, Manack Adams A, Goadsby PJ. Identifying Natural Subgroups of Migraine Based on Comorbidity and Concomitant Condition Profiles: Results of the Chronic Migraine Epidemiology and Outcomes (CaMEO) Study. Headache 2018. [DOI] [PubMed] [Google Scholar]
- [41].Long CC, Sadler KE, Kolber BJ. Hormonal and molecular effects of restraint stress on formalin-induced pain-like behavior in male and female mice. Physiol Behav 2016;165:278–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mason BN, Kaiser EA, Kuburas A, Loomis MM, Latham JA, Garcia-Martinez LF, Russo AF. Induction of Migraine-Like Photophobic Behavior in Mice by Both Peripheral and Central CGRP Mechanisms. J Neurosci 2017;37(1):204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Matsumiya LC, Sorge RE, Sotocinal SG, Tabaka JM, Wieskopf JS, Zaloum A, King OD, Mogil JS. Using the Mouse Grimace Scale to reevaluate the efficacy of postoperative analgesics in laboratory mice. J Am Assoc Lab Anim Sci 2012;51(1):42–49. [PMC free article] [PubMed] [Google Scholar]
- [44].McIlvried LA, Borghesi LA, Gold MS. Sex-, Stress-, and Sympathetic Post-Ganglionic Neuron-Dependent Changes in the Expression of Pro- and Anti-Inflammatory Mediators in Rat Dural Immune Cells. Headache 2015;55(7):943–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].McIlvried LA, Cruz JA, Borghesi LA, Gold MS. Sex-, stress-, and sympathetic post-ganglionic-dependent changes in identity and proportions of immune cells in the dura. Cephalalgia 2017;37(1):36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nation KM, Dodick DW, Navratilova E, Porreca F. Sustained exposure to acute migraine medications combined with repeated noxious stimulation dysregulates descending pain modulatory circuits: Relevance to medication overuse headache. Cephalalgia 2019;39(5):617–625. [DOI] [PubMed] [Google Scholar]
- [47].Reuter U, Goadsby PJ, Lanteri-Minet M, Wen S, Hours-Zesiger P, Ferrari MD, Klatt J. Efficacy and tolerability of erenumab in patients with episodic migraine in whom two-to-four previous preventive treatments were unsuccessful: a randomised, double-blind, placebo-controlled, phase 3b study. Lancet 2018. [DOI] [PubMed] [Google Scholar]
- [48].Robert C, Bourgeais L, Arreto CD, Condes-Lara M, Noseda R, Jay T, Villanueva L. Paraventricular hypothalamic regulation of trigeminovascular mechanisms involved in headaches. The Journal of neuroscience : the official journal of the Society for Neuroscience 2013;33(20):8827–8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rosen N, Pearlman E, Ruff D, Day K, Jim Nagy A. 100% Response Rate to Galcanezumab in Patients With Episodic Migraine: A Post Hoc Analysis of the Results From Phase 3, Randomized, Double-Blind, Placebo-Controlled EVOLVE-1 and EVOLVE-2 Studies. Headache 2018;58(9):1347–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Scher AI, Stewart WF, Buse D, Krantz DS, Lipton RB. Major life changes before and after the onset of chronic daily headache: a population-based study. Cephalalgia 2008;28(8):868–876. [DOI] [PubMed] [Google Scholar]
- [51].Shimozawa N, Okajima K, Harada N, Arai M, Ishida Y, Shimada S, Kurihara H, Nakagata N. Contribution of sensory neurons to sex difference in the development of stress-induced gastric mucosal injury in mice. Gastroenterology 2006;131(6):1826–1834. [DOI] [PubMed] [Google Scholar]
- [52].Son SW, Lee JS, Kim HG, Kim DW, Ahn YC, Son CG. Testosterone depletion increases the susceptibility of brain tissue to oxidative damage in a restraint stress mouse model. J Neurochem 2016;136(1):106–117. [DOI] [PubMed] [Google Scholar]
- [53].Sorbi MJ, Maassen GH, Spierings EL. A time series analysis of daily hassles and mood changes in the 3 days before the migraine attack. Behav Med 1996;22(3):103–113. [DOI] [PubMed] [Google Scholar]
- [54].Spierings EL, Sorbi M, Maassen GH, Honkoop PC. Psychophysical precedents of migraine in relation to the time of onset of the headache: the migraine time line. Headache 1997;37(4):217–220. [DOI] [PubMed] [Google Scholar]
- [55].Tuttle AH, Molinaro MJ, Jethwa JF, Sotocinal SG, Prieto JC, Styner MA, Mogil JS, Zylka MJ. A deep neural network to assess spontaneous pain from mouse facial expressions. Mol Pain 2018;14:1744806918763658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Vegas O, Poligone B, Blackcloud P, Gilmore ES, VanBuskirk J, Ritchlin CT, Pentland AP, Walter SA, Nousari Y, Tausk F. Chronic social stress Ameliorates psoriasiform dermatitis through upregulation of the Hypothalamic-Pituitary-Adrenal axis. Brain Behav Immun 2018;68:238–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wacogne C, Lacoste JP, Guillibert E, Hugues FC, Le Jeunne C. Stress, anxiety, depression and migraine. Cephalalgia 2003;23(6):451–455. [DOI] [PubMed] [Google Scholar]
- [58].Yapici-Eser H, Donmez-Demir B, Kilic K, Eren-Kocak E, Dalkara T. Stress modulates cortical excitability via alpha-2 adrenergic and glucocorticoid receptors: As assessed by spreading depression. Exp Neurol 2018;307:45–51. [DOI] [PubMed] [Google Scholar]
- [59].Zinck T, Illum R, Jansen-Olesen I. Increased expression of endothelial and neuronal nitric oxide synthase in dura and pia mater after air stress. Cephalalgia 2006;26(1):14–25. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Facial withdrawal thresholds of mixed background C57BL6/129J (A) male (n=5) and (B) female (n=6) mice that were stressed or male (n=6) and female (n=6) mice that were naïve. had withdrawal thresholds determined prior to and following stress. Two-way RM ANOVA with Bonferroni multiple comparison analysis indicates a statistical difference in control male and female mice compared to stressed male and female mice over time (mean ±SEM, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). See table 2 for additional statistical analysis.