

Abstract
The complexity of integrating microbiota into clinical pharmacology, environmental toxicology, and opioid studies arises from bidirectional and multiscale interactions between humans and their many microbiota, notably those of the gut. Hosts and each microbiota are governed by distinct central dogmas, with genetics influencing transcriptomics, proteomics, and metabolomics. Each microbiota's metabolome differentially modulates its own and the host's multi‐omics. Exogenous compounds (e.g., drugs and toxins), often affect host multi‐omics differently than microbiota multi‐omics, shifting the balance between drug efficacy and toxicity. The complexity of the host‐microbiota connection has been informed by current methods of in vitro bacterial cultures and in vivo mouse models, but they fail to elucidate mechanistic details. Together, in vitro organ‐on‐chip microphysiological models, multi‐omics, and in silico computational models have the potential to supplement the established methods to help clinical pharmacologists and environmental toxicologists unravel the myriad of connections between the gut microbiota and host health and disease.
One of the grand challenges facing both clinical pharmacology and environmental toxicology in the decade of the 2020s will be to integrate into their intellectual frameworks the explosion of knowledge regarding microbial communities residing within many human tissues and fluids, individually described as a microbiota and collectively as the microbiome. 1 To quote a recent review, “the human microbiome … is widely accepted as a major factor that drives the interpersonal variation in therapeutic response.” 2 Microbial cells and human cells have about a 1:1 numeric ratio, but there is a 1:100 ratio of human‐to‐microbial genes (~ 20,000 human genes compared with 1 million microbial genes 2 ). Organs‐on‐chips (OoCs), that is, 2D and 3D human tissue constructs typically grown in perfused, microfluidic devices using primary, tissue‐resident adult stem cells, or induced pluripotent stem cells (iPSCs) to create microphysiological systems (MPS), 3 , 4 are one of the newest tool sets being introduced into clinical pharmacology and toxicology. 5 , 6 Although they are increasingly recognized as being able to recapitulate enough tissue or organ functions to recapitulate a functional unit and serve as useful in vitro human models for quantitative systems pharmacology, 4 their full potential for studying host‐microbiome interactions has yet to be realized. This review first examines microbiota‐host interactions, exogenously driven microbiota composition perturbations, and treatment outcomes that depend upon microbiota composition, all from the perspective of clinical pharmacology. The review then discusses efforts and opportunities to use the emerging methods of in silico microbiota modeling, multi‐omics, and organ‐on‐chip technology to clarify the factors that govern human‐microbiome‐drug interactions and to improve microbiota‐dependent treatments. (Given the speed with which these three fields are growing, we were unable to include full citations and in‐depth discussion of several subtopics in the body of this review. Additional discussion and references are included in the Supplementary Information , which is structured to parallel the main sections in the body of the review.)
The human microbiotas (skin, 7 oral, different regions in the gut, the male and female genitourinary systems, the vascular 8 and lymph 9 circulations, and possibly the brain 10 ) contain various microorganisms (bacteria, fungi, viruses, and archaea), some of which are benign and others pathogenic. The gut microbiota is of particular interest in pharmacology due to the manner in which gastrointestinal (GI) microbes are associated with a multitude of diseases 11 , 12 and interact with xenobiotics. 13 , 14 Drug designers may need to consider the high spatial heterogeneity and complexity of the gut microbiota: going from stomach to large intestine, there are gradients in pH, pressure, and the densities of different microbiota populations 13 that could alter drug absorption, distribution, and metabolism either directly or through microbial effects on host metabolism. 2 Gut microbiota are able to affect behavior, mood, and decision making. 15 There is growing evidence of strong connections between the microbiome and serious diseases of the central nervous system (CNS), most notably Alzheimer's and Parkinson's diseases, among many 16 , 17 (see the Supplementary Information for a more extensive list); hence, the pressing need to understand the microbiome‐gut‐liver‐immune‐brain axis (M‐GLIBA).
Historically, clinical pharmacologists have largely focused on optimizing only the human side of the equation as they work with medicinal chemists to develop drugs for the efficacious treatment and prevention of human disease with minimal toxic, off‐target effects. Environmental toxicology addresses how toxic industrial and consumer chemicals and materials adversely alter the development, growth, well‐being, and aging of humans, as well as the local and global ecosystems comprising microbes, plants, and animals. The microbiome is beginning to play a larger role in both clinical pharmacology and environmental toxicology for understanding the mechanisms behind harmful effects of exogenous compounds. Understanding of microbiota‐dependent processes is expected to increase significantly in the coming years. Figure 1 , inspired by Kaddurah‐Daouk et al., 18 , 19 , 20 provides a Vitruvian overview of how the mechanisms of action of drugs and toxins must now be expanded to include the microbiome, how the body's various microbiota are affected by both diet, lifestyle, and environment, and the ever‐increasing appreciation for the metabolome. As an example, for the CNS, it is important to recognize that the vascular system (the red and blue Vitruvian human) and the nervous system (the light green human) together support multiple connections between the human and its microbiome, as does the lymphatic system (not shown).
Figure 1.
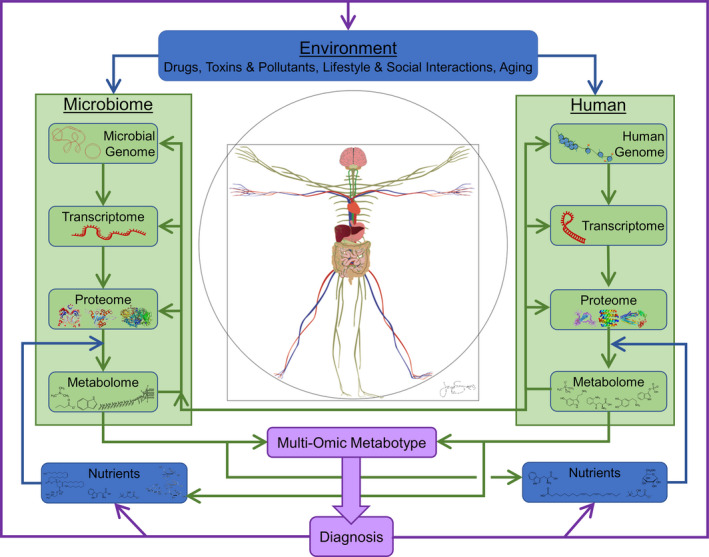
The Vitruvian human, with separate interacting central dogmas for the microbiome and human. The metabolites of one can serve as nutrients and signaling modulators for the other. The inputs to the system are external nutrients for both the human and the microbiome, and a variety of environmental factors, including drugs and toxins. The information gleaned from the combined metabolomes constitutes a metabotype that can provide diagnostic information, which then can be used to adjust the inputs to the system. Proteins shown for the microbiome are, left to right, the EcoRV restriction endonuclease and the flavin nitroreductase protein from Escherichia coli, and the enzyme beta‐gal. The microbiome nutrients are phosphatidylcholine (produces trimethylamine (TMA)), tryptophan, carnitine (produces TMA), and inulin. The microbiome metabolites are trimethylamine, butyrate, the tryptophan derivative indole, and a lipopolysaccharide. The human proteins are tau protein, the dopamine receptor DRD2, and p‐glycoprotein. The human nutrients are tryptophan, alpha‐linolenic acid, carnitine, and glucose (α‐D‐Glucopyranose). The human metabolites are trimethylamine N‐oxide, serotonin, kynurenine, dopamine, and indoxyl sulfate. (Inspired by R. Kaddurah‐Daouk's group 18 ).
In the classical concept of the central dogma of biology, 21 the human genome directs the flow of information to the human transcriptome, which, in turn, controls the human proteome, such that the flow of genetic information is almost always unidirectional, as shown by two downward arrows on the right side of Figure 1 . However, the role of the metabolome on the central dogma was not appreciated when it was first postulated in 1958. Human proteins metabolize nutrients, drugs, and toxins, and these metabolites affect not only human transcription, translation, and post‐translational modification (upward arrows on the right side), but also the corresponding processes in the microbiome (on the left side of the figure). Hence, when considering the microbiome and humans, there are multiple central dogmas, one for the human and many for the microbiome. The microbial and human biochemistries interlink at many levels, including the metabolites of one serving as nutrients for the other, with the signals between the gut and the brain and back involving both the neural and vascular systems. From the perspective of diagnosis, this picture also shows how the readily accessible metabolome of the human and many of the microbes in its microbiome can be combined to create a metabolomic profile, termed a metabotype, 18 , 19 , 20 that can drive a disease diagnosis. Even with the increasing expectation that a patient's entire coding and noncoding human genome might be known, whether the genotype of sequestered microbiota could be determined is problematic. The transcriptome is specific to the cells from which the RNA is derived and may have diagnostic value in a particular clinical assay, but it is not of general utility. Thus, the metabotype alone might provide a rapid, low‐cost means to assess the state of patients and their microbiome, which in combination with their genetics could motivate adjustments to external variables, including nutrients, drugs, toxins, pollution, and lifestyle and social interactions. There are, however, clear challenges in developing the statistical tools to identify high‐dimensional biomarkers 22 that are worthy of serious attention.
The clinical pharmacology and environmental toxicology communities would benefit from tools that would enable them to understand in detail the depth of interactions between the gut microbiota and the host that arise from metabolites that bacteria secrete. The soluble factors secreted by bacteria, termed postbiotics, include, but are not limited to, enzymes, proteins, polysaccharides, organic acids, and short‐chain fatty acids (SCFAs). Postbiotics can act locally on the gut or have systemic effects on other organs. For example, the dietary exposure of mice to the environmental toxin perfluorooctane sulfonic acid altered the balance of many bacteria in the gut microbiome and amino acids and SCFA metabolism in a manner consistent with the development of metabolic syndrome. 23 Postbiotics have been considered as a pharmacologically attractive alternative to probiotics, because postbiotics are more stable and avoid issues with horizontal gene transfer‐mediated antibiotic resistance. 14 , 24 The metabolic signaling between gut microbiota and human host has a complex connection with human physiology such that the influence of the gut microbiota extends well beyond the gut to the liver, brain, kidneys, skin, and other organs. 25 In parallel, the human metabolites of drugs, toxins, and nutrients affect the microbiome, which, in turn, can alter human physiology, resulting in multiple, intertwined feedback loops, as suggested by Figure 1 . There is an almost perfect concordance in the role of the human microbiome in clinical pharmacology and environmental toxicology—drugs and toxins affect both the human and the microbiome, and the bidirectional interactions through metabolism and signaling must become an integral component of both fields. Surely, the full integration of the microbiome will lead to a paradigm shift in how we discover, understand, utilize, or avoid drugs and toxins.
From the perspective of clinical pharmacology, the statistics describing the microbiome are daunting. As of 2017, only 4,500 of the ~ 21,000 human genes are considered druggable, 26 but fewer than 700 proteins and other human biomolecules are targeted by drugs approved by the US Food and Drug Administration (FDA). 27 , 28 In contrast, the microbiome was previously thought to have 3 million genes, 29 but the inclusion of previously overlooked small proteins 30 might add 23 million nonredundant genes to the oral microbiome and 22 million to the gut, for a total increase in microbiome genes of 45 million, 31 providing ample opportunity for new drug candidates. An expansion of the gene search space by this magnitude will require new tools and techniques, ranging from a breadth of omic technologies, to MPS and their constituent microfluidic‐enabled tissue chips or OoCs that yield data not previously available from humans, to computational models that seamlessly connect, for example, the genome, transcriptome, proteome, lipidome, and metabolome.
To appreciate fully the forthcoming paradigm shift associated with such an expansion of the possible pharmacological interactions, we need to look at the development of the relevant fields. As shown in Figure 2 , a rudimentary search for “pharmacology” in PubMed yields over six million publications since 1873. A similar search for “toxicology” yields almost 200,000 since 1830. Focusing on topics of specific interest, both clinical pharmacology and environmental toxicology have enjoyed long and productive histories, as presented in Figure 2a . The microbiome and OoCs are recent entrants to the arena, with small literatures that are rapidly growing by ~ 10,000 and ~ 1,000 articles a year, respectively. To correct for the differences in the size of the fields, the relative growth plots in Figure 2b show that over the past decade, there has been faster growth in the scientific literature on the microbiome and OoCs as compared with the more established fields, in part because the new entrants were virtually unheard of 2 decades ago, and these fields can expand into a breadth of existing scientific disciplines. The absolute rates in Figure 2c and the fit to exponential curves indicate that the microbiome literature has an exponential growth rate of 0.21/year (which corresponds to a 3.3 year literature doubling time), whereas for OoCs the rate is 0.12/year (5.7 year doubling time). In contrast, the growth rates in annual publications on clinical pharmacology and environmental toxicology are both 0.05/year and may even have peaked, but confirmation of this trend would require a more extensive analysis of search terms. In light of these trends, this review provides arguments why the microbiome should factor heavily into clinical pharmacology and environmental toxicology—a natural result as studies of the microbiome become mainstream.
Figure 2.
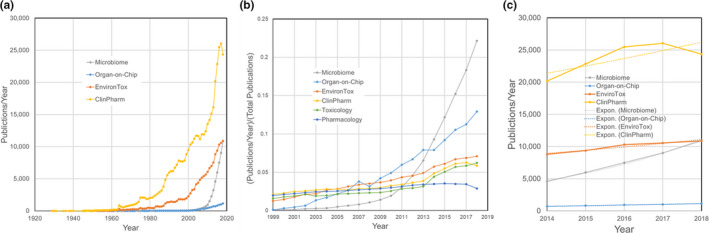
Trends in the publication rates for related fields as revealed by an informal PubMed Search through 2018. (a) Publications per year. (b) Fractional growth in publications per year. (c) Growth in publications over the past 5 years. The data are as follows: pharmacology: PubMed search for “pharmacology”: 6,150,000 publications since 1873, a recent publication rate of ~ 19,000/year, and an exponential growth rate of −0.04/year (R 2 = 0.56) for the past 5 years. Toxicology: PubMed search for “toxicology”: 188,000 publications since 1830, a recent publication rate of ~ 10,000/year, and an exponential growth rate of −0.04/year (R 2 = 0.08) for the past 5 years. Clinical Pharmacology: PubMed search for “clinical pharmacology”: 415,000 publications since 1929, a recent publication rate of ~ 25,000/year, and an exponential growth rate of 0.05/year (R 2 = 0.61) for the past 5 years. Environmental Toxicology: PubMed search for “Environmental AND (Toxicology OR Toxicity)”: 153,000 publications since 1945, a recent publication rate of ~ 11,000/year, and an exponential growth rate of 0.05/year (R 2 = 0.94) for the past 5 years. Microbiome: PubMed search for “microbiome”: 49,000 publications since 1956, a recent publication rate of ~ 10,000/year, and an exponential growth rate of 0.21/year (R 2 = 0.99) for the past 5 years. Organs on Chips: PubMed search for “((organ OR tissue) AND (chip or microfluidic)) OR (organ‐on‐chip) OR (microphysiological system)”: 8,906 publications since 1945, a recent publication rate of ~ 1,000/year, and an exponential growth rate of 0.12/year (R 2 = 0.99) for the past 5 years.
We now turn our attention to the microbiome and human disease. 11 , 12 Sharma et al. 2 provide an excellent review of clinical applications of microbiome‐based therapeutics. Numerous reports of differences between the gut microbiota of healthy and diseased patients are appearing. Furthermore, the relative abundance of various bacterial strains has been investigated in relation to their effects on drug efficacy. 13 , 14 One such instance showed that patients with relatively elevated levels of bacteria from the Faecalibacterium genus responded more positively to PD‐1 checkpoint therapy, whereas patients with higher levels from the Bacteroidales order responded poorly to PD‐1 checkpoint therapy. 32 The gut microbiota's influence has been implicated in various CNS diseases, such as Parkinson's disease, depression and anxiety, autism spectrum disorder, attention‐deficit hyperactivity disorder, schizophrenia, bipolar disorder, multiple sclerosis, Alzheimer's disease (AD), glioblastomas, substance use disorders, which include opioid use disorders (OUDs), and various non‐CNS diseases, such as obesity, type 2 diabetes, glucose intolerance, insulin resistance, acne, atopic dermatitis, psoriasis, colorectal cancer, non‐alcoholic fatty liver disease, irritable bowel syndrome, Crohn's disease, ulcerative colitis, fibromyalgia, chronic pain, stroke, lung disease, celiac disease, and metabolic syndrome. Please refer to the Supplementary Information for representative references and additional examples.
Developers of drugs to treat any of these diseases should be mindful of the role of the gut microbiota. The gut microbiota has no established mechanistic links to CNS diseases, because the primary evidence is only in the form of correlations of various bacteria populations with individuals with the CNS diseases, as compared with normal, healthy individuals. 17 Recent studies have been aimed at understanding how diet, drugs, surgery, and environmental toxins affect gut microbiota. Such knowledge has implications for establishing the missing mechanistic connection for diseases caused by dysbiosis (i.e., a change in microbial composition or function that has an adverse impact on the host‐microbiome relationship), as can be induced by antibiotics during a Clostridium difficile infection, 33 or as can occur following an ileocecal resection and antibiotics. 34
Given the complexity of the connections between the GI microbiome and the CNS, which are affected by how the microbiome interacts with nutrients, drugs, and environmental toxins, and the difficulty in obtaining and maintaining the individual human microbiota whose compositions vary along the GI tract, there are sound arguments favoring the creation of in vitro coupled human cell organ‐on‐chip models that represent different sections of the GI tract and can also be interfaced to other organ chips, such as liver, kidneys, and brain. In general, organ‐on‐chip models excel at controllability and observability, but it is not yet known the extent to which they can approximate the true gut microbiota‐host interactions. When compared to in vivo models, such as mouse or stool samples from patients, organ‐on‐chip models offer many advantages, including (i) restricted extracellular volumes that prevent autocrine, paracrine, juxtacrine, and endocrine signals from being diluted below both physiological effectiveness and detectability; (ii) long‐term support of heterogeneous populations of cocultured cells in 2D or 3D microenvironments; (iii) a high level of control of variables, such as the input of nutrient‐rich media or drugs; (iv) compatibility with high‐content imaging; (v) ready removal of secreted media, cells, and cellular waste for off‐line analysis without damaging the cells; (vi) quantification of drug absorption, distribution, metabolism, excretion, and toxicity (ADMET); and (vii) the ability to visualize host‐microbiome interactions. 3 , 6 , 35 , 36 , 37 Such systems are being utilized to create disease models and ultimately to enable personalized models for precision medicine. These in vitro models would benefit from and support multi‐omics analyses and in silico computational models. In balance, organ‐on‐chip models now require a level of technical skill well beyond conventional cell culture, and a long‐term study can cost almost as much as an animal study, but as the scientific community and industry refine their designs and techniques, these models are becoming easier to use and less expensive.
The breadth of the topics touched upon in this review indicates the importance of the microbiome in the next decade of clinical pharmacology. The integration of the microbiome into clinical pharmacology and environmental toxicology should expand our understanding of human development, disease, and aging, identify important environmental toxicants, and create opportunities for the development of new drugs. We now examine significant factors that affect the human‐microbiome interaction, explore external influences on the microbiome, and discuss decoding microbiome‐human interactions, with particular emphasis on the M‐GLIBA.
Significant Factors in the Human‐Microbiome Interaction
Biochemical signaling from gut microbiota to the host
As shown in Figure 1 , the interactions between the microbiome and the human can occur at levels that span the genome, transcriptome, proteome, metabolome, cells, tissues, organs, and organ systems. The signals that connect humans and their microbiomes are carried by physical contact or the vascular, nervous, gastrointestinal, and lymphatic systems. The signals can go in either or both directions. The metabolome of the microbiome comprises chemicals that are termed postbiotics. In the Supplementary Information , we discuss in detail the primary postbiotics that alter the host's physiological state: tryptophan metabolites, bile acids, SCFAs, and immune system signaling.
Gut microbiota and host CNS interactions
The gut microbiota postbiotics have multiple means for influencing the CNS through the enteric and vagus nerves, the hypothalamic‐pituitary‐adrenal (HPA) axis, and microglia. 17 , 38 , 39 , 40
The vagus and enteric nervous systems
The enteric nervous system (ENS) provides local control of digestive functions and an intimate connection between the stromal cells of the GI tract and the autonomic nervous system. The Supplementary Information provides citations to studies that show how (i) the ENS regulates enteric processes that include immune response, detecting nutrients, motility, microvascular circulation, intestinal barrier function, and epithelial secretion of fluids, ions, and bioactive peptides, (ii) the gut microbiota regulates maturation of the adult ENS via enteric serotonin networks, and (iii) how the vagus nerve, connected to all of the layers of the digestive wall, serves as the functional connection between the CNS and the ENS.
Inflammasomes
Inflammasomes are cytosolic signaling complexes in the innate immune response and epithelial barrier tissues that activate in response to microbial and endogenous threats. NLRP3 inflammasomes are activated in the gut epithelium through microbiota‐fermented SCFAs. In addition, activated NRLP6 in the gut epithelium modulates microbiota composition, which may induce dysbiosis. Activated NLRP3 inflammasomes, activated NLRP6 inflammasomes, and increased amounts of pro‐inflammatory cytokines, such as IL‐1β, IL‐6, and IL‐18 proteins, are implicated in the development of major depressive disorders. 38
Microglia
Microglia dysfunction is associated with AD and Parkinson's disease, autism spectrum disorder, and depression. Microglia overactivation by environmental toxins and endogenous proteins promotes the synthesis of reactive oxygen species, which can lead to neurotoxicity and the development of neurodevelopmental diseases. 17 For example, microglia influence the development of AD by inhibiting amyloid‐β clearance while promoting amyloid‐β deposition, which, in turn, leads to release of pro‐inflammatory mediators, such as reactive oxygen species and NF‐κB. 38 Important for development is the effect of gut microbiota on the morphology and gene expression of microglia. Gut microbiota‐derived SCFAs, particularly butyrate, alter the maturation and function of microglia and the permeability of the blood‐brain barrier (BBB). 41
Hypothalamic‐pituitary‐adrenal axis
The HPA axis controls the body's reactions to stressors. 39 , 42 Diet, stress, or antibiotic‐induced inflammation of the GI tract triggers the release of cytokines and neurotransmitters, which stresses the microbiome and leads to increased permeability of both the intestinal epithelium and the BBB to drugs and toxins. Pro‐inflammatory cytokines activate the HPA axis, which acts to reduce inflammation. The hypothalamus releases corticotropin‐releasing factor, which then triggers the release of adrenocorticotropic hormone in the adenohypophysis of the pituitary gland. Adrenocorticotropic hormone acts to reduce the pro‐inflammatory cytokine signal. Hyperactivity and dysregulation of the HPA axis are associated with major depression disorder and anxiety. 39 , 42
Development
One can appreciate the complexity of the human‐microbiome relationship by studying how the microbiome evolves after birth and affects maturation of the GI epithelium during the establishment of the normal symbiotic relationship. 43 Molecular signals from the gut can modify a breadth of neurodevelopmental processes in the enteric system and the CNS. 44 , 45 It has been shown that the maternal gut microbiota can influence the prenatal development of the BBB of germ‐free mice, resulting in an increase in BBB permeability that persists into adulthood, and that introduction of a pathogen‐free microbiome would tighten the BBB. 46 The initial colonization of the gut microbiome modulates brain development and adult behavior. 47 One must also appreciate the potential implications of the disruption of these developmental processes by drugs or environmental toxins that affect the microbiome, which, in turn, could complicate precision medicine unless adequate quantification tools are developed.
Gut microbiota spatial heterogeneity
As we stated in the introduction, the composition of the microbiota varies between different layers and regions of the GI tract. Specific examples are provided in the Supplementary Information .
External Influences on the Microbiome
Central to this new frontier in clinical pharmacology is the need to understand how to effect changes in the microbiome for the good of the human host, and how conventional drugs and both environmental and industrial toxins may alter the microbiome to the detriment of the human.
Xenobiotics and the exposome
Learning how nutrients and xenobiotics, such as drugs, probiotics, and bioactive compounds in the diet or the exposome, 48 affect the ENS, autonomic nervous system, and CNS is of utmost importance for understanding how the gut microbiota influence CNS disorders. Xenobiotic metabolism has potential effects on gut microbiota composition and health outcomes. Microbes can metabolize drugs, and drugs can alter the composition of the gut microbiota. 49 The gut microbiota can be affected by exposure to heavy metals, pesticides, nanoparticles, polycyclic aromatic hydrocarbons, dioxins, furans, and polychlorinated biphenyls. 50 There are two primary ways that the gut microbiome influences the metabolism of xenobiotics: direct metabolism in the gut and indirect control of liver xenobiotic metabolism. The direct mechanisms include activation (i.e., prodrugs, detoxification or deactivation of xenobiotics, and direct binding of bacteria to xenobiotics). The indirect mechanisms include enterohepatic cycling, the process by which xenobiotics are inactivated in the liver and reactivated in the gut by microbes by deconjugation, postbiotic interactions with enzyme kinetics, and alteration of the expression of various cytochrome P450s in the host liver. 13 Please refer to the Supplementary Information for additional details.
Probiotics, prebiotics, synbiotics, and the FDA
Probiotics are bacteria that are introduced to the gut for beneficial effects, typically administered orally or through stool transplantation. Prebiotics are nutrients that are indigestible to the host but promote the growth of beneficial bacteria populations. Synbiotics act to incorporate both probiotics and prebiotics to further promote the beneficial effects of probiotics. When used for therapy, these three supplements can positively regulate gut microbiota composition for improved treatment outcomes. 14 The Supplementary Information contains specific examples of probiotics, prebiotics, and synbiotics.
The FDA is beginning to address the “science and regulation of live microbiome‐based products used to prevent, treat, or cure diseases in humans.” 51 This is distinct from the possibility that the genotype of a patient's existing microbiome might affect the patient's response to a drug. 2 Already, the FDA includes pharmacogenomic information in drug labeling to inform, for example, drug exposure and clinical response variability, risk for adverse events, and drug mechanisms of action. 52 A 2008 analysis of pharmacogenomic biomarker information in FDA‐approved drug labels for 1945–2005 found that 10% of the 1,200 drug labels contained pharmacogenomic information, and 52 (4% of all labels) referred to microbial genomic biomarkers. 53 It is not clear from this study how many of these were related to infectious agents rather than the commensal microbiome, but it is reasonable to assume that the latter were in the minority. It is inevitable that as the field of pharmacometabonomics matures, biomarkers will be identified that could indicate or contraindicate the use of a drug based upon a particular metabotype (i.e., reflecting the metabolic activity of both the patient and the commensal microbiota). Whether this added complexity will represent a regulatory challenge remains to be seen.
Diet
Diet and lifestyle are two of the largest factors controlling gut microbiota composition. Diet has the potential to rapidly alter microbiota composition based on fat, protein, and fiber intake. 54 High consumption of red meat is associated with increased levels of carnitine and choline (molecules included in Figure 1 ), which are converted to trimethylamine (TMA) by the gut microbiota, which is, in turn, converted to trimethylamine N‐oxide (TMAO) by flavin‐containing monooxygenase in the liver. Other factors associated with increased TMA/TMAO production with red meat consumption include enhanced nutrient density of dietary TMA precursors and reduced renal excretion of TMAO. 55 Increased levels of TMAO are linked to nonalcoholic fatty liver disease, coronary artery disease, chronic kidney disease, and type 2 diabetes. 56 , 57 Western diets, with their high meat content, show an increase in activity of β‐glucuronidase, an enzyme involved with rescuing liver‐metabolized morphine and returning it to its original neuroactive form. 58 The Supplementary Information discusses the Western and Mediterranean diets, intermittent fasting, the ketogenic diet, and the microbiome of professional athletes.
Although diet modulates microbiota composition, it does not fully control how microbiota compositions evolve. The microbiota composition of individuals has low convergence between individuals (i.e., beta diversity), even those on the same diet, 59 which, in turn, may constrain the success of precision medicine until there are better methods to both characterize the complexity of the gut microbiota 60 and predict specific microbiota‐drug‐host interactions. 61
Drugs
Drugs are one the largest xenobiotics of interest. When exposed to 21 drug cocktails of different combinations of 271 drugs, 76 gut microbiota species were able to metabolize two‐thirds of the applied drugs. 29 The host metabolizes drugs primarily in the liver through oxidative and conjugative reactions, whereas gut bacteria utilize hydrolytic and reduction reactions. (Drug metabolism in other organs complicates the situation, but this is beyond the scope of our review.) Gut microbiota and drugs interact in a variety of ways. In addition to direct drug‐induced alteration of the gut microbiota composition or function, the gut microbiota can activate, inactivate, or reactivate drugs, or generate toxic metabolites from drugs. In addition, the gut microbiota can have indirect effects on the host's bioavailability and response to a drug, primarily through modulation of a host's immune and metabolic systems. Genetics and exogenous factors, like diet, also play a role in how the gut microbiota interacts with and metabolizes drugs. 62 The gut microbiota affects drug efficiency both directly and indirectly through multiple mechanisms, 2 , 63 as discussed in greater detail in the Supplementary Information .
Antibiotics tend to decrease microbiota diversity in the host and cause a shift in the ratio of different species. For example, fluoroquinolones and β‐lactams both significantly decreased microbial diversity (by 25%) and increased the ratio of Bacteroidetes to Firmicutes. 64 The connection between diet and antibiotics is further illustrated by mice raised on a high‐fat diet and given a subtherapeutic antibiotic treatment. The microbiome shifts in the high‐fat diet with antibiotics led to insulin resistance and hepatic steatosis. 64
Furthermore, as discussed in detail in the Supplementary Information , there are differences in how drugs and postbiotics are absorbed in the gut. Pharmacology studies have recognized that there are regional differences in how drugs are absorbed within the GI tract, including spatial variations in passive permeability and active drug transport and absorption, osmolality, the unstirred water layer, mucosal composition and function, and the presence of other fluids.
Gut microbiota drug metabolism can affect the efficacy of a drug by decreasing bioavailability. An example of this is levodopa (L‐dopa). 65 Normally, L‐dopa is converted into dopamine after crossing the BBB. In an alternative pathway, Enterococcus faecalis secretes a pyridoxal‐phosphate‐dependent tyrosine decarboxylase that converts L‐dopa to dopamine. As a second interspecies step, Eggerthella lenta, which secretes molybdenum‐dependent dehydroxylase, has 11 strains that convert dopamine to m‐tyramine with 100% conversion and 13 strains with < 11% conversion. (E. lenta also inactivates the cardiac drug digoxin. 2 ) However, this process can be blocked with a second drug: (S)‐α‐fluoromethyltyrosine attenuates mice gut microbiota enzyme production, leading to an increase in L‐dopa bioavailability 65 —another demonstration of the added complexity that the microbiome can bring to clinical pharmacology. In addition, E. lenta generates a cardiac glycoside reductase operon that metabolizes digoxin into inactive metabolites. 2 Hydrolases, lyases, transferases, and oxidoreductases are the primary microbial enzymes that act on ingested xenobiotics. So‐called “TIMER” effects (translocation, immunomodulation, metabolism, enzymatic degradation, and reduced diversity) modulate efficacy and toxicity of drug therapies. Drug‐induced leaky gut and villi shortening can cause translocation of bacteria through the gut membrane and induce sepsis and dysbiosis. Drugs can induce bacteria to modulate the immune system, which enables other species to induce an inflammatory response. Metabolism and enzyme degradation act on drugs to generate toxic metabolites, reactivate metabolized drugs, and decrease drug bioavailability. Drug‐induced dysbiosis is associated with disrupted gut microbiota composition that leads to malignant effects, such as reduction of Streptococci abundance by methotrexate, causing villi shortening and diarrhea. 66 Clarke et al. 13 provide more information regarding xenobiotic metabolism by the gut microbiota.
Gut microbiota can increase a drug's toxicity in the gut. For example, the colon cancer chemotherapeutic CPT‐11 can cause severe diarrhea due to reactivation by bacterial β‐glucuronidases. This can be addressed with β‐glucuronidase inhibitors that attenuate the toxic response. 67 Specific bacteria could evoke extreme drug reactions: mice colonized with M. morganii produced phenethylamine that accumulated systemically and crossed the BBB. The administration of a monoamine oxidase inhibitor, as might be prescribed for depression, social phobias, or panic disorders, could then lead to lethal phenethylamine poisoning. 68 This is a sobering testimony of the pharmacological power of the gut microbiome and the variability that might challenge precision medicine.
Clearly, the microbiome presents new pharmaceutical opportunities. As a harbinger of a possible explosion in microbiome pharmacology, a recent news story presents 15 examples of small molecules being developed by a dozen or more companies to target microbes or host microbe interactions. 69
Opioids
Opioids provide important and timely examples of how drugs can interact with multiple tissues, different cellular phenotypes, and gut microbiota, thereby contributing to the complex system of overlapping feedback loops shown in Figure 1 . The spinal cord and vagus nerve carry visceral pain signals that arise from nociceptor activation in the thorax, abdominal and pelvic visceras, and the nervous system and that can become sensitized under chronic pain conditions. 70 Decades of research have focused on the mechanisms of opioid analgesia and tolerance within the CNS, implicating glial cells and the release of inflammatory cytokines in an anti‐analgesic role.
An emerging literature describes a large contribution of peripheral tissues, both of the ENS in the gut and of the primary afferent relay neurons that transmit sensation from the gut to the spinal cord, whose cell bodies make up the dorsal root ganglion, in the modulation of pain and opioid responses. 71 Chronic opioid exposure activates toll‐like receptors on epithelial cells of the gut, disrupting the gut epithelial barrier and allowing bacterial translocation. 72 As bacteria pass through, immune cells and enteric glia are activated and cytokines are released, setting in motion an inflammatory response cascade that is anti‐analgesic and promotes worsening pain. This is the situation in inflammatory bowel disease, where patients treated with chronic opioids for Crohn's disease‐related pain may experience worsened pain, increased rates of infection, and increased risk of death. 73
Misunderstandings and excesses in the treatment of chronic pain with opioids have created a national crisis of opioid misuse and OUD, and have many in the field wondering where it all went wrong. Although opioids are effective analgesics for acute pain, prolonged use and/or high doses increase the risk of opioid‐related adverse events, 74 including a decline of efficacy due to tolerance, the potential establishment of opioid‐induced hyperalgesia, 75 and possible contribution to inflammatory states that promote the acute‐to‐chronic pain transition, 76 , 77 all of which can contribute to OUD. Relatively little is known about how acute vs. chronic opioid exposure or stress (e.g., after trauma or surgery) differentially affect opioid transport and metabolism along the M‐GLIBA. Even less is known about how acute or chronic opioid use changes the core functions of the M‐GLIBA.
An issue is that other than the brain, one of the organs that most expresses opioid receptors is the gut, 78 which includes the neurons of the ENS. 79 Activation of the μ opioid receptor is implicit with attenuating GI motility. The μ opioid receptor interacts biochemically with adenylate cyclase, β‐arrestin recruitment, and receptor phosphorylation by protein kinases C and A, and ERK 1/2. 71 Thus, not only does long‐term opioid use cause a reduction in gut motility, we are now beginning to appreciate that it also plays a causal role in changes to the gut microbiota 79 , 80 , 81 that can, in turn, escalate inflammation in both the gut and CNS. 80 , 82 This effect of long‐term opioid use on the gut microbiota also contributes to the physiologic changes that underlie addictive behavior, dysregulates immune response, and increases intestinal barrier permeability, bacterial translocation, the risk of enteric infection, morphine tolerance, and gut‐derived sepsis. 58 , 82 , 83 , 84 This is consistent with experiments with germ‐free mice and bacterial depletion and probiotics in mice. 80 , 83 , 84
Stress, opioids, and injury can trigger central neuroimmune activation, which involves activation of the cells that provide an interface between blood and the nervous system, as well as parenchymal astrocytes and microglia. 85 Stress can also directly affect OUD. Opioids are well known to inhibit corticotrophin‐releasing hormone and, therefore, cortisol levels. 86 Microbiota influence pro‐inflammatory and anti‐inflammatory responses in both the gut and brain. 87 In patients with chronic pain without associated depression, cortisol demonstrates low levels and blunted diurnal variation. 88 Opioid‐related inflammation of gastric epithelium may contribute to chronic pain syndromes. One might wonder whether reported changes in the gut microbiome associated with chronic pelvic pain are cause or effect. 89
Disruption of the gut microbiome by chronic opioid use has an impact on liver function, sustains a systemic inflammatory state, 90 and worsens the disease, potentially contributing to the chronification of pain. 77 Opioid usage is associated with gut dysbiosis such that there is an increased alpha diversity (within a sample) and altered beta diversity (between samples). 41 Morphine gradually changes the gut microbiota, leading to changes in the gut metabolic profile by decreasing bile acids while simultaneously increasing phosphatidylethanolamines and saturated fatty acids. The gut microbiota's primary interaction with morphine is through rescuing morphine metabolites morphine‐3‐glucuronide (M3G) and morphine 6‐glucuronide (M6G). M3G and M6G are generated through morphine glucuronidation in the liver. The liver detoxifies the morphine and the gut microbiota then retoxifies it, providing a possible source of interindividual variation in the response to opioids. Although M3G exhibits no analgesic effect, it has an important role in morphine tolerance, most likely through activation of toll‐like receptor 4 (TLR4) that mediates morphine‐induced cytokine release. 91 In the gut, M3G and M6G are hydrolyzed back to morphine by β‐glucuronidase, which is produced by Bacteroides and Bifidobacteria. 58 Dysbiosis leads to greater morphine tolerance, as the morphine‐induced gut microbiota is inefficient at hydrolyzing M3G and M6G. 82 Introducing the probiotics Bifidobacteria and Lactobacillaceae can attenuate morphine tolerance. 80 Opioid use has been linked to bile dysregulation and gut microbiota changes that then compromise the gut barrier. 77 These changes alter the metabolites coming from the liver in yet another feedback loop, 82 contributing to the system‐wide issues experienced by opioid users.
In addition to the clear role of inflammation in driving the M‐GLIBA interactions, cytochrome P450s are a critical molecular link in the M‐GLIBA. This very large family of proteins is expressed extensively in the liver, gut, and the BBB, 92 and it is critical in how humans process opioids and other drugs. 93 Opioid receptors are commonly expressed in a variety of immune cells, but the responses of macrophages to morphine are opposite to those of buprenorphine and oxycodone. 94 Morphine also suppresses NF‐κB activation in macrophages 58 and modulates the activity of glia in the spinal cord, 95 and morphine‐induced gut dysbiosis causes TLR2 and TLR4 activation, resulting in a significant increase of IL‐6 protein levels. 80
It is important to note that allosteric modulators of muscarinic acetylcholine receptors are being developed that may enable regulation of the addictive and executive dysfunction aspects of substance use disorders. 96 How these drugs interact with the M‐GLIBA and the microbiome would be a worthy area of inquiry.
Decoding Microbiome‐Human Interactions
The interactions we have described provide ample opportunity for clinical pharmacologists to develop new microbiome‐related drugs to treat CNS diseases. The readily accessible postbiotics and the chemicals in the human metabolome together provide signals for nondestructive and noninvasive assessment of the state of the Vitruvian human for both diagnosis and evaluation of pharmacological control.
Pharmacogenomics/microbiomics
In the context of precision medicine, genetics are a large factor in adverse drug reactions, which can vary due to drug, patient, and disease. 97 Human genetics account for 20–95% of variation of drug response, which is dependent on the context of treatment: the type of disease and the prescribed medication. 2 Variance of drug action, fate, efficacy, and toxicity between different individuals is a major concern for pharmacology and personalized medicine. The postgenomic revolution popularized the practice of using pharmacogenetics, where treatment variance is explained by genetic variance between patients. Akin to pharmacogenetics, the emerging field of pharmacomicrobiomics explains treatment variance by variance in a patient's gut microbiota composition. 62 , 98 Host genetics play a role in gut microbiota composition, active microbial enzymes, and effects of dietary intake. 66 Today, pharmacomicrobiomics is currently limited by inadequately validated causal relationships between gut microbiota and host and the difficulty of culturing different bacterial species in the laboratory. 66
Figure 3 illustrates the variability of patient response due to differences in host metabolic phenotype and microbiota composition. Toxicity‐efficacy trade‐off contours are a convenient means to explain the range of responses that might be encountered by patients with different microbiome compositions. 99 , 100 When a population of patients is given a drug, the observed probabilities of efficacy and toxicity can be used to create toxicity‐efficacy trade‐off contours, as shown in Figure 3a . These show the desirability of different treatment outcomes due to a specific treatment, which is defined as a dose of one or more drugs. The desirability, d, of a treatment outcome is scaled between 0 and 1 such that d increases with higher efficacy and lower toxicity. With this definition of desirability, treatments with high desirability have a high therapeutic index. If d = 0 then the treatment is highly toxic and not efficacious. Similarly, if d = 1, the treatment is efficacious and has low toxicity. The axes of a toxicity‐efficacy trade‐off contour indicate the likelihood of toxic and efficacious treatment outcomes. The desirability is constant along trade‐off contours (dashed lines in Figure 3a ). Toxicity‐efficacy trade‐off contours are typically used to quantify the dose‐dependent effects of treatments, but the diagram can be adapted to illustrate the variance in a specific treatment (a singular dosage of a drug or combination of drugs) between different patients, namely due to host metabolism and microbiota metabolism.
Figure 3.
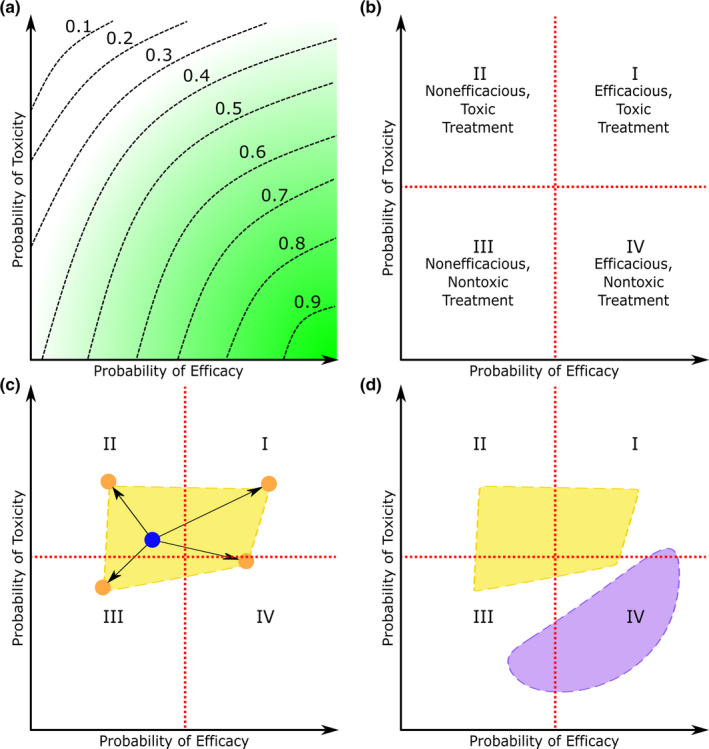
Schematic representation of the effects of the microbiome on the tradeoff between the probability of drug efficacy and probability for a patient population. (a) Toxicity‐efficacy trade‐off contours express the desirability of different treatments outcomes for a population of patients. The green gradient indicates how desirability changes with probability of efficacy and probability of toxicity. The dashed lines are trade‐off contours with constant desirability, where the respective desirability values are listed above the trade‐off contour. (b) The toxicity‐efficacy trade‐off diagram partitioned into quadrants (I–IV) representing different treatment outcomes, with quadrant boundaries shown by red dashed lines. (c) A hypothetical example of how differences in microbiota could affect treatment outcomes of oral drugs. The blue dot indicates the desirability of the drug response due to the host alone, whereas the shift in treatment outcome due to four different microbiota compositions is shown by orange dots. The microbiota‐dependent treatment outcome region, shown in yellow, marks the possible treatment outcomes due to the metabolism of all possible microbiota compositions coupled with that host's metabolism. (d) How the treatment outcome regions might differ between two patients, with the possibility of a patient's microbiome shifting into any quadrant of the toxicity‐efficacy plot.
Figure 3b simplifies toxicity‐efficacy trade‐off contours by omitting the trade‐off contours and dividing the plot into four quadrants. Treatment outcome desirability is highest in quadrant IV and decreases toward quadrant II, where it is lowest. Here, the toxicity‐efficacy trade‐off diagram has been adapted to show variability due to different drug dosages and factors, such as microbiota composition. Consider the treatment outcome due to a specific dose of an oral drug (Figure 3c ). The patient's metabolism leads to a single treatment outcome, represented by a blue dot. The patient's gut microbiota can modulate the drug's efficacy and toxicity, leading to a shift in the treatment outcome. The effect the microbiota has on treatment outcome is dependent on microbiota composition, so the treatment outcomes due to the patient's metabolism and the microbiota's metabolism will vary. To represent this variance, consider four different microbiota compositions generating four different shifts in treatment outcome, represented by orange dots. Realistically, there is a multitude of microbiota compositions with associated treatment outcomes. Let the microbiota‐dependent treatment outcome region refer to every treatment outcome possible due to the interactions of a host's metabolism and the metabolism of all possible microbiota compositions. An example of a treatment outcome region is plotted in yellow in Figure 3c . The exact shape of the region would have to be determined empirically by observing treatment outcome and microbiota composition. Treatment outcome varies between patients, however. This variance can be represented by multiple treatment outcome regions (yellow and purple in Figure 3d ). Ultimately, these plots serve to illustrate that pharmacomicrobiomics and pharmacogenomics are important considerations for treatment outcome. Two patients can have dramatically different treatment outcomes due to genetic factors, but microbiota composition can also affect treatment outcome. An important question would be the extent to which a patient's metabotype could be used to predict treatment outcome for the existing microbiome as well as another that reflected the use of therapeutic probiotics.
Drug metabolism in the host is dependent on the complex, tripartite interaction of gut microbiota, host lifestyle, and host genetics. 62 , 101 Integrating pharmacogenomics with pharmacomicrobiomics enables a higher understanding of patient‐specific adverse reactions to drugs. Genetic influences cannot be changed, but microbiomic influences can be modified by altering the gut microbiota composition to the extent allowed by host genetics. Researchers focusing on how metabolic phenotype can lead to variability in treatment outcomes have coined the term “pharmacometabonomics,” 102 defined as “the study of the metabolic response of organisms to disease, environmental change, or genetic modification.” 103 The numerous interactions in Figure 1 manifest the complexity and suggest the challenge of inferring the nature of the interactions from measurement of the metabotype.
Mathematical modeling of the microbiome‐host interaction
There is a lack of mechanistic understanding of how the host and microbiome interact. Many microbial species cannot be cultivated in the laboratory and, hence, are difficult to observe. Metabolic interactions, as reported by the metabotype, are also currently difficult to observe and hard to interpret, so quantitative modeling has been used to elucidate these interactions. Computational methods, such as ordinary differential equation modeling, statistical comparison of microbiota, constraint‐based reconstruction and analysis, Boolean networks, and agent‐based modeling have been used to understand microbiota metabolism, microbiota perturbations due to drugs, and microbiota‐host interactions. 61 Partial differential equation models could include spatial gradients in nutrients, metabolites, and metabolic activity, as has already been done for the distributed electrical activity in the intestine. 104 Modeling the M‐GLIBA is clearly a multiscale problem with multidimensional complexity, spanning molecular interactions, intracellular and extracellular signaling, cells, tissues, organs, and organ systems, with times spanning from nanoseconds for molecular dynamics to years for aging. A systems approach is necessary for understanding and treating CNS diseases, and the merging of pharmacology with systems biology has created the field of quantitative systems pharmacology. 4 , 5 , 105 The problems only become harder with the need to incorporate the microbiome. Fritz et al. 106 review the current status of in silico models. More details and references are provided in the Supplementary Information .
The advantage of the computational approach is that once mechanism‐based models of host‐microbiome interactions and microbiome‐microbiome interactions have been created, it will be straightforward to examine a large number of combinations of host and microbiota genomes to address pressing questions in pharmacomicrobiomics and pharmacometabonomics that will arise when precision medicine is applied to a single patient rather than a patient population. One hopes that the mechanisms that drive the model will be sufficiently detailed to support appropriate interactions between the species that might have been modeled in isolation—at present, there is no guarantee that the interactions will exhibit linear, commutative, associative, or other properties that would support simple overlaying of individual models to create one for the whole patient. As with many computational models of biological systems, the greater challenge is to obtain the data required first to parameterize and then validate the model as accurately representing clinical physiology. 4 Notably, there is the obvious difficulty in obtaining detailed, quantitative descriptions of microbiota sequestered deep within that patient's GI system that might be addressed using single or coupled organ chips. Hence, our discussion turns to assays and OoCs.
Assays for understanding host‐microbiome interactions
Detailed characterization of the interactions in Figure 1 between the microbiome and the GLIBA will be demanding, to say the least, and will involve genomics, transcriptomics, proteomics, lipidomics, interactomics, etc. Today, untargeted metabolomics can identify the metabolites of microbial species, and enrichment analysis allows mapping the bacterial genome to drug metabolism. 29 The human protein‐protein interactome alone is daunting, and the full capture of Figure 1 will require specification of the relationship of large numbers of both human and microbial proteins and metabolites. Although untargeted multi‐omic assays are advancing rapidly, the mere connection between the human proteome and metabolome is in its infancy. At the most reductionist level, the annotation of the interactions in Figure 1 represents a scientific grand challenge that would require a decade or more of effort because of the diversity of the interactions of totally different classes of chemical species. Obviously, work will proceed with whatever level of annotation of the interactions is available.
Frameworks are being established to develop and curate the metagenomic profiles of the microbiome 107 ; however, even limited assays are demanding. Analysis of gut microbiota involves identification of present taxa, analysis of alpha (within the same sample) and beta diversity (between samples) within the microbiota, statistical analysis of gut microbiota with sample metadata, network analysis, biomarker discovery, and functional analysis of the microbiome and metabolomics. 108 It is difficult to identify the relevant bacterial taxa in different diseases. For example, there is no consensus on which bacterial taxa are most prevalent for major depression disorder. This disparity has roots in methodologic differences, sample differences, and the complex microbiota‐host interactions. 59 However, multiple assays exist for measuring the ADMET and other properties of the gut microbiota‐drug interaction. 109 Culture collection and ex vivo incubations are two methods for collecting a gut microbiota sample. Fecalase preparation, a cell‐free fecal extract with microbial enzymes, enables analysis of microbiota‐mediated drug metabolism. Microbial RNA‐seq can be used to identify which pathways are active in microbes, and host RNA‐seq can be used for identifying which pathways are modulated by microbiota action. Genetics are difficult to study in the gut microbiota, given the inability to genetically manipulate most microbial species present in the gut. Two methods that are commonly applied are comparative genomics and functional genomics. Gene knockouts can be used for model gut microbes, such as Escherichia and Bacteroides. Microbiota profiling can be utilized for identifying drug‐responsive microbes. Cell culture models, gnotobiotic models, and antibiotic‐knockdown models are used to observe microbiota interactions. Cell cultures are often inaccurate, so in vivo gnotobiotic models, which have well‐defined microbiotas, are preferred. The creation of a gnotobiotic model involves putting specific microbes into germ‐free animals to enable investigation of the drug interactions of specific microbes in vivo. Gnotobiotic models are expensive, so antibiotic knockdown can be used as an alternative, but there are issues with reproducibility and the disparity between nonhuman host microbiota and human microbiota. 101 An important question is the extent to which some of these limitations can be overcome with in vitro, organ‐on‐chip microphysiological models.
OoCs for in vitro studies of microbiome‐organ‐organ interactions
The rationale for MPS models of the M‐GLIBA
Many studies of the gut microbiota are hard to reproduce due to the complications of sample acquisition and preparation, analytical methods, variability between individual human and animal models, dosage, duration of exposure, and cost. 50 The most important feature required by an M‐GLIBA model is two key barriers (at the minimum): the GI epithelium and the BBB. Fritz et al. 106 provide a compact but very useful review of experimental models. Accepted models for the M‐GLIBA include germ‐free mice, antibiotic‐treated mice, and clinical data. 41 Of these, germ‐free mice are the most expensive to maintain due to the required sterile conditions for the life of the animal. 41 Studying how postbiotics affect drugs taken orally can be accomplished with clinical data, but this is challenging due to the high variance of the gut microbiota and external factors, such as use of other drugs, tobacco, alcohol, and undeclared substance of abuse. 41 In vitro gut chemostats can be used to culture and analyze microbial communities. 110 , 111 Given the limitations of the existing models and the expanding needs of clinical pharmacology and environmental toxicology, there is an obvious need for compact, sterile, interconnected in vitro models of the M‐GLIBA using human cells and suitable for high‐content optical screening and untargeted multi‐omic analyses.
Since the early work by Shuler et al., some of which was directly related to toxicology, 112 there has been a growing literature on OoCs, tissue chips, and microphysiological systems (Figure 2 and Supplementary Information ). Progress in the field accelerated after the publication of a lung chip by the Ingber group, 113 the launching in 2011 of the FDA Advancing Regulatory Science Initiative 114 that led to an FDA/National Institutes of Health (NIH) award to Ingber for a heart‐lung model, and large joint programs funded by the Defense Advanced Research Projects Agency (DARPA), the NIH National Center for Advancing Translational Sciences (NCATS), and the FDA. Other major efforts were launched by the Defense Threat Reduction Agency (DTRA) and the Environmental Protection Agency (EPA). A large number of original papers and reviews are listed in the Supplementary Information , particularly with regard to the substantial progress that has been made, with multiple implementations of each of the four organs of the GLIBA: the gut, the liver, and the neurovascular unit, and/or the BBB, some with innate immune cells. There is a small but growing number of demonstrations of functionally or physically coupled organs.
Although there are notable challenges that have yet to be fully overcome for both individual and coupled chips, 3 , 35 , 115 , 116 the future for this technology is promising. In this section, we highlight MPS used for drug discovery and toxicology, briefly review the application of pharmacokinetics (PKs) to MPS studies, discuss an MPS model that recapitulates important aspects of gut‐liver‐kidney‐brain pharmacodynamics (PDs), and speculate on the future.
The cells used in MPS models are either primary ones from animals or humans (which can often be propagated as either organoids or patient‐derived xenografts), from immortal cell lines, or derived from embryonic or iPSCs. It is fully appreciated that tissue chips with iPSC‐derived cells could recapitulate familial CNS diseases. 117 , 118 Ultimately, vascularized organoids may supplement or replace tissue chips, but many multi‐omic assays require more cell mass or effluent volume than can be obtained from conventional organoid well plates or very small organ chips. 3
There are several different ways to apply OoC technologies to study the M‐GLIBA. Figure 4a is a schematic diagram of the key physiological systems involved in the M‐GLIBA, including the pancreas and the enteric and autonomic nervous systems. Were all these systems needed to recapitulate experimentally the desired physiology, pathology, or pharmacology, one would have little choice but to use animal or human models.
Figure 4.
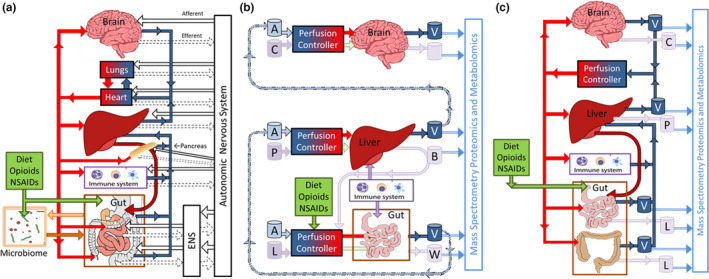
Schematic representations of the M‐GLIBA. (a) The connected organs with the greatest roles in the M‐GLIBA, including the physiological connections between both the humoral, immune, and enteric and central (autonomic) nervous systems, as well as a pancreas, heart, and lungs. (b) A schematic representation of a functionally coupled gut‐liver‐immune‐brain axis. The dashed lines represent transport of aliquots of bioreactor effluent between separate organs, possibly at different institutions. Fluid key: A = “arterial” culture media, V = “venous” cell‐conditioned media, C = cortical interstitial, L = luminal, B = parenchymal (bile), W = waste. (c) A coupled tissue‐chip gut‐immune‐liver‐brain axis model with limited complexity to study humoral signaling with high‐content mass spectrometry, as might be realized with tubing connecting separate organs or as a single body‐on‐chip. Fluid key: V = vascular, C = cortical interstitial, P = parenchymal, L = luminal. ENS, enteric nervous system; M‐GLIBA, microbiome‐gut‐liver‐immune‐brain axis; NSAIDs, nonsteroidal anti‐inflammatory drugs.
One of the driving motivations for OoCs is that the cellular microenvironment for the seeded cells, often as 3D matrices, more closely replicates the organ than a 2D plastic Petri dish or well plate. Another is to have an appropriate match between cellular and perfusate volumes so as to support realistic paracrine and juxtacrine signaling and external quantification of these signals. 35 This emerging technology is well suited to the study of individual organs (as well as the GLIBA). Figure 4b shows how a limited number of independent organ chips can be functionally interconnected by manual or auto‐pipetted transfer of conditioned media from one bioreactor to the input of another, something that has been accomplished with remarkable success. 119 The third approach, shown in Figure 4c , is to have each OoC physically interconnected to the others. The bioreactor effluent from one is automatically directed to another, and a universal culture medium is recirculated with appropriate exchange of oxygen and carbon dioxide and continually refreshed at as low a rate as is required to support organ‐chip metabolism: without a kidney to excrete toxins and metabolites, the same fraction of media would have to be regularly withdrawn and replaced, possibly confounding the concentration of key circulating factors.
Single organ chips needed for the M‐GLIBA
The addition of a microbiome to a gut‐on‐a‐chip is critical for understanding the interactions in Figure 1 . The first demonstration of this was by the Ingber group, 120 whose gut‐on‐a‐chip supported periodic stretching of the Caco‐2 GI epithelial cells that were cultured on a porous elastomeric polydimethylsiloxane (PDMS) membrane. Their devices supported the aerobic culture of Lactobacillus rhamnosus GG for longer than a week. Subsequent studies 37 , 121 provided a more robust characterization of the structure and function of the cultured Caco2 cells, their transformation to a complex villi‐like surface topography, and reconstitution into multiple cell types (absorptive, enteroendocrine, Paneth, and goblet cells) with appropriate polarization, and mucous production. The devices supported coculture with both commensal and pathogenic, noncommensal strains of Escherichia coli bacteria.
More recently, Shah et al. 122 constructed a gut‐on‐a‐chip bioreactor with a different geometry that supported a much larger area of epithelia cells and cocultured a facultative anaerobe, Lactobacillus rhamnosus GG, which was grown solely under aerobic or anaerobic conditions, or grown in combination with an obligate anaerobe, Bacteroides caccae, under anaerobic conditions. Their chip supported an oxygen gradient across the GI barrier by using different levels of oxygenation in the media on opposite sides. The chip had optodes for sensing oxygen tension in the device. They were also able to conduct transcriptomics, metabolomics, and immunological analyses that demonstrated differences between culturing the cells with L. rhamnosus GG alone vs. with B. caccae.
The most recent work from the Ingber group extends their earlier studies, 36 and the current implementation of their intestine chip has a number of useful features, including a coculture of Caco2 cells on one side of the porous PDMS membrane and human intestinal microvascular endothelial cells on the other, and the maintenance of an oxygen gradient monitored by on‐chip optical oxygen sensors. A number of different microbiotas were tested under both aerobic and anaerobic conditions, as were primary human epithelial cells and stool. In their perspective on this work, Poceviciute and Ismagilov provide a balanced and forward‐looking analysis of the human gut‐microbiome‐on‐a‐chip, pointing out the value of coupling the gut chip to other chips, particularly the liver, and the importance of the gut microbiota in transforming liver bile acids for reabsorption and regulation. 123
Hence, it is reasonable to conclude that each of the organ chips required to reconstitute the M‐GLIBA has been demonstrated individually. The next challenge is to successfully couple multiple chips while maintaining flow rates, concentrations, and sterility. 3 , 35
Multiple‐organ MPS models
Although many of the corporate ventures in OoCs are focusing on single‐organ chips for drug screening, there is much interest in using coupled‐chip MPS models for ADMET to study situations in which metabolism of a metadrug or drug by one organ creates a drug that treats or toxifies another organ. For first‐pass metabolism studies, it is also important to include the gut. 124 , 125 Other multi‐organ MPS models, including recent studies that include a gut chip, are summarized in the Supplementary Information .
One might ask whether such an artificial set‐up is preferred, or whether in silico modeling could better integrate the different parts of the envisioned microbiome and GLIBA for the CNS? As discussed above, there are two challenges with in silico modeling: parameterization and validation. Although one might be able to create a detailed computational model of an extended or coupled human organ system, it will be difficult to obtain confirmatory data from more than a limited number of sites in a small number of human subjects, and even harder to study the response of the human to a variety of drug, nutrient, and toxin challenges that could be used to test and refine the model. Possibly the greatest strength of a coupled MPS‐computational model of the GLIBA is its ability to realize the “paradigm of iterative experimental and computational modeling [that] provides testable mechanistic hypotheses serving to connect the actual pathogenesis to the ensemble of modules comprising quantitative systems pharmacology, despite the large spatiotemporal scales they encompass. 4
There is a growing recognition that coupled OoC systems can avoid what is known as the “universal media problem” 35 by providing each organ chip with separate stromal (or epithelial) and vascular compartments, separated by an endothelial barrier specific to that organ. 113 What is not yet known is the extent to which an appropriately configured GI epithelial/endothelial barrier with its associated mucosal layers will be able to maintain, over the long term, proper balance in a complex microbiome. One could expect that microbes difficult to culture in vitro might grow better in a coupled‐MPS microenvironment that provides the appropriate luminal flow of nutrients and metabolites, pH, and oxygen. The extent to which coupled MPS can support a stable microbiome is unknown, but clearly worth intense study.
We note that there has been minimal discussion in the MPS community about the challenges of adding more than a single immune cell type (macrophages in the gut, Kupffer cells in the liver, tissue‐resident lymphocytes in the kidneys, microglia in the brain, etc.), 126 and there are significant, unresolved issues with creating innate immune systems from a single line of iPSCs, as would be required to avoid HLA/MHC incompatibilities that might trigger a graft‐vs.‐host response in an immune‐enabled MPS. That said, the innate immune system, in the form of tissue‐resident macrophage‐like cells, can reproduce many important inflammatory and oxidative‐stress responses.
MPS for drug discovery
Recent reviews have well summarized the current state of lab‐on‐a‐chip and pharm‐on‐a‐chip in the determination of PK, PD, toxicokinetic, and toxicodynamic parameters of target drugs. 127 , 128 Although the initial motivation for the DARPA, NCATS, and FDA MPS programs was to address unanticipated toxicity, drug failures due to toxicity are now less common than failures in efficacy, and, as a result, it is recognized that MPS approaches will provide a greater return on investment when focused on drug mechanism of action and PK/PD. 3 Much of the effort by the MPS community to date has been to demonstrate and validate specific tissue chips. As listed in the Supplementary Information , there have been a number of papers addressing drug toxicity and pharmacology, but only a few of the many groups working on OoCs are developing the PD tools that will be needed by clinical pharmacologists and toxicologists to fully utilize tissue chips. This should improve because MPS models are beginning to move from the realm of engineers to that of pharmacologists and toxicologists.
MPS models of gut‐liver‐kidney‐brain PDs
As a final example, we discuss briefly a demonstration of how functionally coupled gut, liver, kidney, and brain tissue chips can inform PDs, shown in Figure 5 . 119 In this experiment, the liver chip was located at the University of Pittsburgh, the gut chip at the Baylor College of Medicine in Houston, the kidney chip at the University of Washington in Seattle, and the neurovascular unit (NVU) chip at Vanderbilt University in Nashville. Although a number of groups have manually or automatically pipetted conditioned media from one tissue chip to another, for this demonstration the investigators simply froze the tissue‐chip effluent and used overnight shipping to transport it to the next organ. Figure 5a shows the various steps in the workflow, including, as appropriate, addition of blank media, mixing of conditioned media, and withdrawal of samples for mass spectrometric analysis.
Figure 5.
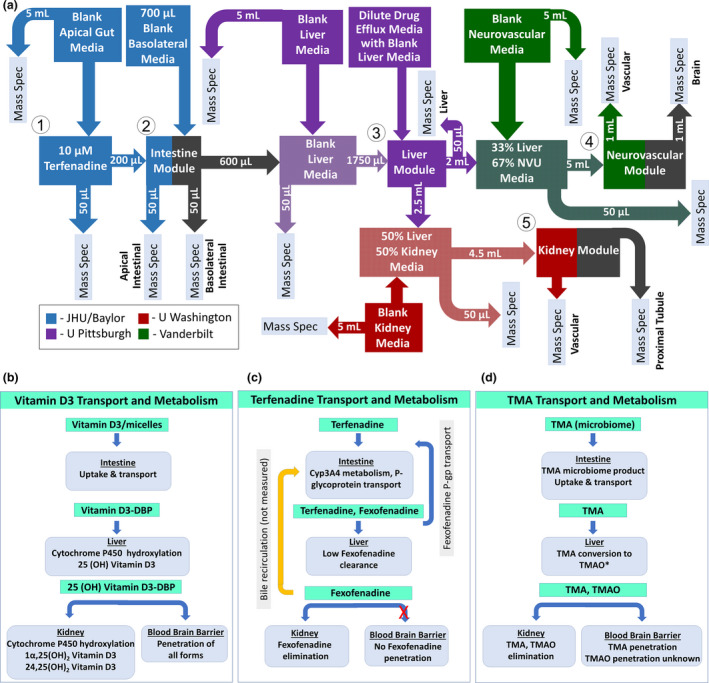
A multi‐organ MPS experiment to study in vitro the pharmacodynamics of vitamin D3, terfenadine, and trimethylamine (TMA). 119 (a) The workflow showing the functional coupling wherein the drug (1) is delivered to the intestine (2), whose effluent is directed to the liver (3). The liver effluent is directed to both the neurovascular unit (brain) (4) and kidney (5). The other boxes and arrows indicate various mixing of media and their delivery to mass spectrometry. Functional analysis of transport and metabolism of (b) vitamin (d, c) terfenadine, and (d) TMA. Adapted with permission from Vernetti et al., Scientific Reports 2017, 7, Article 42296, under a Creative Commons Attribution 4.0 International License (see http://creativecommons.org/licenses/by/4.0/). MPS, microphysiological systems; NVU, neurovascular unit; TMAO, trimethylamine N‐oxide.
Three experiments were conducted and demonstrate what types of studies are well suited for coupled tissue‐chip experiments. Figure 5b shows the sequence of events associated with vitamin D3 (VD3) transport and metabolism in human organs. Absorption and activation of VD3 requires uptake and transport in the intestine. The addition of a vitamin D binding protein to the media avoided the absorption of the hydrophobic vitamin into the PDMS, just as D binding protein in vivo limits its absorption by the lipid membranes of vascular cells. Sequential metabolism in the liver and kidney chips led to a series of active metabolites. The in vitro study confirmed that there was no metabolism of VD3 in the intestine, the liver chip produced the clinically observed VD3 metabolite as well as another, and the kidney chip produced one of the clinically observed metabolites, but the other was below the limit of detection. In the NVU, both VD3 and its appropriate metabolite crossed the BBB. 119
The second experiment, shown in Figure 5c , tested terfenadine transport and metabolism. Oral terfenadine is absorbed and metabolized to fexofenadine in the intestine. Counter‐transport carries fexofenadine back to the apical side of the intestinal wall. The remaining terfenadine is metabolized to fexofenadine in the liver. Fexofenadine cannot cross the BBB and is excreted by the kidneys. The kidney chip was in concurrence with the clinical data, except with a lower excretion of fexofenadine by the kidneys (91% rather than 11%). 119
The third experiment (Figure 5d ) examined the metabolism and transport of trimethylamine (TMA), discussed in detail earlier. In humans, TMA is produced in the intestine by the endogenous microbiome, where it is taken up and transported to the liver, where it is metabolized to TMAO. Kidney disease can lead to increased plasma concentrations of TMAO, with significant medical side effects. The intestine in this experiment was without a microbiome, so instead, TMA was added to the luminal (basal) media of the intestine chip. The liver chip metabolized the TMA into TMAO, but with a lower efficiency (< 5% in vivo vs. < 1% in the chip). The ~ 46% excretion by the kidney chip was less than the clinical excretion of > 95%. The clinical penetration of TMAO across the BBB was unknown at the time of the study, and 26% of the TMAO penetrated the NVU. 119 The significance of this study was that TMAO's penetration of the BBB was subsequently confirmed clinically by the analysis of spinal‐tap fluid from > 50 patients, 129 constituting what is believed to be the first MPS prediction later confirmed in humans.
Of course, this study, conducted > 3 years ago, would be much better were it to be repeated with today's more advanced perfusion control and bioreactor technologies and with an appropriate microbiome. However, the work flow would still resemble that in Figure 5A , whether the mixing and transfer steps were performed manually or automatically. These are complicated assays that will undoubtedly benefit from the coupled‐organ technologies currently under commercial development by TissUse, Hesperos, and CN Bio Innovations, among others.
Summary of the Challenges for CNS Clinical Pharmacology Applied to the M‐GLIBA
The growth in our understanding of the microbiome and the absence of any end in sight in the breadth and volume of its literature add urgency to understanding the complexity that the microbiome brings to clinical pharmacology and environmental toxicology. Forsythe et al. 130 provide an excellent review of and commentary on the state of research into the biology, biochemistry, and neuroscience that govern the microbiota‐gut‐brain axis. They discuss in detail the neuroscience of the bidirectional information transfer from the microbiome through the enteric nervous system, vagal and spinal nerves, and the brain, emphasizing the importance of the identification of a nicotinic intramural sensory synapse between the sensory neurons in the wall of the gut and the vagal neurons. They present a reasoned analysis of the relative merits and disadvantages of human vs. animal models. They do not address in any depth clinical pharmacology, environmental toxicology, or opioid/inflammation, or the possible role of tissue chips as in vitro models that are the subjects of this review. They do offer an important cautionary warning: “Communication between gut and brain depends on both humoral and nervous connections. Since these are bi‐directional and occur through complex communication pathways, it is perhaps not surprising that while striking observations have been reported, they have often either not yet been reproduced or their replication by others has not been successful.”
One limitation of GI chips that we have discussed is that they lack an enteric nervous system, as shown in Figure 4a . Enteric neurons have already been integrated into iPSC‐derived intestinal organoids, 131 so it is just a matter of time to the development of an innervated gut chip. Until then, it will be worthwhile to either restrict oneself to humeral M‐GLIBA questions, or consider creating hybrid in silico/electronic and biological models, in which the neural functions of the ENS are provided by a real‐time system of sensors, an in silico physiological control model, and an actuator. The recently developed microformulator, which is a system of computer‐controlled microfluidic pumps and valves that can deliver different PK and other concentration profiles to one or more organ chips and each well of a 12‐well, 24‐well, or 96‐well plate, could play a central role in the control of multiple chemical or microbial species. 132 If upon delivery of a bolus of nutrients to the basal side of the GI epithelium of a smart gut‐on‐a‐chip, a sensor were to detect a departure of a system variable from the targeted range, such as pH or osmolarity, it would prompt the model‐based controller 133 to adjust the epithelial secretions into the luminal space by ordering a simple pump or the more sophisticated microformulator to add or reduce an appropriate signaling molecule in the luminal or vascular media for the gut chip. Although such a cell‐electronic hybrid might sound far‐fetched, the appropriate technology has been used for many years. 134 , 135 , 136 , 137 , 138 , 139 As the need for pumps and valves in MPS becomes more widely appreciated, 35 it will be straightforward to have an electrochemical neurotransmitter sensor in the brain (e.g., glutamate 140 or oxygen), 141 send via a physiological‐model controller a signal to a pump that would deliver the efferent neurotransmitter in the gut chip. Ultimately, the sensing system could be a combination of electrochemical sensors and mass spectrometers. 138
Given the limitations of existing GLIBA models, 50 , 130 a coupled‐organ‐chip MPS M‐GLIBA model might provide better controlled conditions, increased reproducibility, better temporal and spatial resolution, and lower cost than animal or human models, but this has yet to be demonstrated. It has already been shown that a properly functioning epithelial barrier in an intestine‐on‐a‐chip can block microbes from entering the stromal compartment, 36 , 37 , 120 , 122 so it is simply a matter of time, money, and effort until multiple regions of the intestines, selected from the stomach, duodenum, jejunum, ileum, cecum, and the ascending, transverse, and descending and sigmoid colon, are seeded with the appropriate microbiota harvested from each region using ingested smart sampling capsules. 142 , 143 , 144 , 145 Mechanical pumps could move chyme between sections, and samples could be withdrawn at several locations. With a single‐OoC MPS, different drugs, dietary compounds, or environmental factors can be delivered to either stromal cells or the gut microbiota, with ready access to samples for multi‐omic analyses. The effluent from one OoC could be delivered to a downstream OoC to recreate the environmental and microbial differences along the GI tract. Circadian influences observed in the M‐GLIBA 146 might well be recapitulated in single or coupled tissue chips using a microformulator to drive hormonal rhythms specific to each organ. 147 Although a coupled MPS M‐GLIBA might never be able to model many brain responses, it may well be able to study in depth the interactions between microbiota and stress, 16 as would be particularly appropriate for studies of opioids and their misuse.
A coupled‐MPS system is definitely more complicated and harder to use than simple 2D cell culture on plastic. As pointed out by Taylor et al., 4 human experimental models span a broad range, with 2D cell culture at one end and coupled MPS at the other. This continuum reflects a transition from simplicity that supports high‐throughput screening to a sacrifice of throughput to obtain increased biomimetic complexity. Obviously, one should never use more complexity than required, but for those problems where the physiology and pathology involve multiple organs, coupled organ‐chips with their microbiota, as appropriate, may play an important role. We have already discussed the use of experimental MPS models coupled iteratively to in silico computational models 4 and believe that this is an area of remarkable potential for disease biology and pharmacology, particularly physiologically‐based pharmacokinetics and ADMET‐PK clinical pharmacology. 3
As the number of academic and commercial MPS device builders continues to grow and the recognized challenges to the field are addressed, 35 the ever‐increasing commercial availability of MPS models and recognition of the need for simple recapitulations of the biology of complex diseases suggest that over the next 5 years there will be an explosion of at least single‐organ MPS studies of normal human tissue‐level physiology and disease biology by academe, pharma, tox‐avoidant industries, environmental organizations, and the Department of Defense. The underlying complexity of physiology, disease, and toxicology will, in turn, trigger demand for more coupling of MPS chips. One might expect a rebirth of isolated organ physiology, which dominated physiology and medicine from the 1880s through the 1950s, but can now be implemented with human cells and remarkable analytical and computational breadth. 116 , 148 An extrapolation of the data in Figure 2 would suggest that in 5 years there might be 20,000 MPS papers each year. Undoubtedly, given the even faster growth of publications related to the microbiome, it is reasonable to assume a significant growth in microbiome‐MPS papers. Possibly one of the most important areas for growth will be systems‐based experimental/computational iterations offered by quantitative systems pharmacology. 4 Support of this growth will require attention to many of the details discussed in this review.
It is highly likely that personalized tissue‐chip assays for drug selection will be forthcoming in the next several years, first using organoids and then OoCs. Ultimately, these studies will be used prospectively to optimize pharmaceutical selection. Given the role of the microbiome in response to cancer therapies, 67 one might imagine tissue chips that include not only a patient's primary or metastasized tumor cells, but also the patient's gut with his or her microbiome. When and if this will come to pass will depend on many factors, including efficient production of iPSC‐derived intestinal 149 and other cells, economics, time delays, and regulatory approval that would have to be addressed before the prospective use of personalized tissue‐chip drug‐response assays. These are nontrivial issues. In the meantime, M‐GLIBA assays could provide new information regarding mechanism of action and PK/PD for classes of genotypes for both humans and microbiota.
This lengthy review and its detailed Supplementary Information attempt to bring to clinical pharmacology and environmental toxicology a preview of what microphysiological systems and mathematical models might deliver over the next decade. To Forsythe et al.'s cautionary statement, we add another: “The fundamental challenge to any attempt to model a biological system in vitro is the complexity of biology, as best exemplified by the hundreds of thousands of different chemicals that are active in any large biological organism. An MPS should be viewed as an approximation of reality, not as an accurate reconstruction. We should not fall into the trap of unrelenting detail and accuracy, lest we fail to recognize that ‘[t]he best material model for a cat is another, or preferably the same cat.’ 150 It is important to recognize that, following Einstein's apocryphal advice to ‘make one's theory as simple as possible but not too simple,’ we should make our organ constructs and integrated MPS [models] as simple as possible but not too simple. Were we to succeed in fully recapitulating a [µHuman] on a chip, it would be too complicated for us to understand! So the role of these systems is to provide us readily fabricated and easily studied model systems to test hypotheses.” 148 The addition of the microbiome to the gut‐liver‐brain axis will only make the experimental problems more challenging, so the experiments need to be simple and well designed, and their shortcomings appreciated, to achieve commensurate rewards.
Funding
This work was supported in part by the Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD), the National Center for Advancing Translational Sciences (NCATS), and the National Institute of Neurological Disorders and Stroke (NINDS) of the National Institutes of Health (NIH) under Award Number UG3/UH3TR002097, by NIH/NCATS under Award U01TR002383, by NIH/NCATS Contract HHSN271201700044C through CFD Research Corporation, and by the U.S. Environmental Protection Agency (EPA) under Assistance Agreement 83573601. Support for K.H. includes the NIH Training Programs in Biomedical Informatics (T15LM007450) and Environmental Toxicology (T32ES007028). The views expressed in this paper are solely those of the authors and do not necessarily reflect those of the funding agencies, nor do the funding agencies endorse any products mentioned in the paper.
Conflict of Interest
The authors declare no competing interests for this work.
Supporting information
Supplementary Material
Acknowledgments
The authors are indebted to Alan J. Simmons for his drawing of the Vitruvian human, Allison Price for her editorial contributions, and Don Berry for his bibliographic assistance. We are also indebted to the classmates of C.C. and K.H. in J.W.'s Spring 2019 biophysics seminar (Cody Heiser, Joey Simmons, Danielle Liu, Vineet Desai, Rick Li, XJ Xu, Jason Hughes, Louis Schatzki, Gabby Carlini, and Kylie Balotin), who with C.C. and K.H. performed our initial analysis of using microphysiological systems to study the M‐GLIBA. We thank Brian Bachmann, David Weaver, and Nicole Muszynski for suggesting important references. Licenses to the Vanderbilt pump and valve technologies have been issued to CN Bio Innovations Ltd., which has also licensed the MicroFormulator. Vanderbilt's MicroClinical Analyzer patents have been licensed to Agilent. J.W. is an Inaugural Member of the Scientific Advisory Board of BiOasis Technologies, Inc.
References
- 1. Lloyd‐Price, J. et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 550, 61–66 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sharma, A. , Das, P. , Buschmann, M. & Gilbert, J.A. The future of microbiome‐based therapeutics in clinical applications. Clin. Pharmacol. Therapeut. 107, 123–128 (2020). [DOI] [PubMed] [Google Scholar]
- 3. Watson, D.E. , Hunziker, R. & Wikswo, J.P. Fitting tissue chips and microphysiological systems into the grand scheme of medicine, biology, pharmacology, and toxicology. Exp. Biol. Med. 242, 1559–1572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Taylor, D.L. et al. Harnessing human microphysiology systems as key experimental models for quantitative systems pharmacology In: Concepts and Principles of Pharmacology: 100 Years of the Handbook of Experimental Pharmacology, Vol. 260 (eds. Barrett J.E., Page C.P. & Michel, M.C.) 327–367 (Springer International Publishing, Cham, 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Geerts, H. et al. Quantitative systems pharmacology for neuroscience drug discovery and development: current status, opportunities and challenges. CPT Pharmacomet. Systems Pharmacol. 9, 5–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hutson, M.S. et al. Organs‐on‐chips as bridges for predictive toxicology. Appl. In Vitro Toxicol. 2, 97–102 (2016). [Google Scholar]
- 7. Byrd, A.L. , Belkaid, Y. & Segre, J.A. The human skin microbiome. Nat. Rev. Microbiol. 16, 143–155 (2018). [DOI] [PubMed] [Google Scholar]
- 8. Schierwagen, R. et al. Circulating microbiome in blood of different circulatory compartments. Gut 68, 578–580 (2019). [DOI] [PubMed] [Google Scholar]
- 9. Zwirzitz, B. et al. Microbiota of the gut‐lymph node axis: depletion of mucosa‐associated segmented filamentous bacteria and enrichment of methanobrevibacter by colistin sulfate and linco‐spectin in pigs. Front. Microbiol. 10, 599 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roberts, R.C. , Farmer, C.B. & Walker, C.K. The human brain microbiome; there are bacteria in our brains! Program No. 594.08. In 2018 Neuroscience Meeting Planner <https://www.abstractsonline.com/pp8/#!/4649/presentation/32057>.
- 11. Durack, J. & Lynch, S.V. The gut microbiome: relationships with disease and opportunities for therapy. J. Exp. Med. 216, 20–40 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kho, Z.Y. & Lal, S.K. The human gut microbiome ‐ a potential controller of wellness and disease. Front. Microbiol. 9, 1835 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clarke, G. , Sandhu, K.V. , Griffin, B.T. , Dinan, T.G. , Cryan, J.F. & Hyland, N.P. Gut reactions: breaking down xenobiotic‐microbiome interactions. Pharmacol. Rev. 71, 198–224 (2019). [DOI] [PubMed] [Google Scholar]
- 14. Nichols, R.G. , Peters, J.M. & Patterson, A.D. Interplay between the host, the human microbiome, and drug metabolism. Human Genomics 13, 27 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sylvia, K.E. & Demas, G.E. A gut feeling: microbiome‐brain‐immune interactions modulate social and affective behaviors. Horm. Behav. 99, 41–49 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cryan, J.F. & Dinan, T.G. Mind‐altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 13, 701–712 (2012). [DOI] [PubMed] [Google Scholar]
- 17. Wang, Y.L. et al. The gut‐microglia connection: implications for central nervous system diseases. Front. Immunol. 9, 2325 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Beger, R.D. et al. Metabolomics enables precision medicine: "A White Paper, Community Perspective". Metabolomics 12, 149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Neavin, D. , Kaddurah‐Daouk, R. & Weinshilboum, R. Pharmacometabolomics informs pharmacogenomics. Metabolomics 12, 121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kaddurah‐Daouk, R. et al. Pretreatment metabotype as a predictor of response to sertraline or placebo in depressed outpatients: a proof of concept. Transl. Psychiatry 1, e26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Crick, F. Central dogma of molecular biology. Nature 227, 561–563 (1970). [DOI] [PubMed] [Google Scholar]
- 22. Pirmohamed, M. Acceptance of biomarker‐based tests for application in clinical practice: criteria and obstacles. Clin. Pharmacol. Ther. 88, 862–866 (2010). [DOI] [PubMed] [Google Scholar]
- 23. Lai, K.P. et al. Dietary exposure to the environmental chemical, pfos on the diversity of gut microbiota, associated with the development of metabolic syndrome. Front. Microbiol. 9, 2552 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aguilar‐Toala, J.E. et al. Postbiotics: an evolving term within the functional foods field. Trends Food Sci. Technol. 75, 105–114 (2018). [Google Scholar]
- 25. Schroeder, B. & Backhed, F. Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 22, 1079–1089 (2016). [DOI] [PubMed] [Google Scholar]
- 26. Finan, C. et al. The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 9, eaag1166 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Santos, R. et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16, 19–34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rodgers, G. et al. Glimmers in illuminating the druggable genome. Nat. Rev. Drug Discov. 17, 301–302 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zimmermann, M. , Zimmermann‐Kogadeeva, M. , Wegmann, R. & Goodman, A.L. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 570, 462–467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sberro, H. et al. Large‐scale analyses of human microbiomes reveal thousands of small, novel genes. Cell 178, 1245–1259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tierney, B.T. et al. The landscape of genetic content in the gut and oral human microbiome. Cell Host Microbe 26, 283–295.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gopalakrishnan, V. et al. Gut microbiome modulates response to anti‐PD‐1 immunotherapy in melanoma patients. Science 359, 97–103 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rupnik, M. , Wilcox, M.H. & Gerding, D.N. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat. Rev. Microbiol. 7, 526–536 (2009). [DOI] [PubMed] [Google Scholar]
- 34. Devine, A.A. et al. Impact of ileocecal resection and concomitant antibiotics on the microbiome of the murine jejunum and colon. PLoS One 8, e73140 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wikswo, J.P. et al. Engineering challenges for instrumenting and controlling integrated organ‐on‐chip systems. IEEE Trans. Biomed. Eng. 60, 682–690 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jalili‐Firoozinezhad, S. et al. A complex human gut microbiome cultured in an anaerobic intestine‐on‐a‐chip. Nat. Biomed. Eng. 3, 520–531 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim, H.J. , Li, H. , Collins, J.J. & Ingber, D.E. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut‐on‐a‐chip. Proc. Natl. Acad. Sci. USA 113, E7–E15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ma, Q.Q. , Xing, C.S. , Long, W.Y. , Wang, H.Y. , Liu, Q. & Wang, R.F. Impact of microbiota on central nervous system and neurological diseases: the gut‐brain axis. J. Neuroinflamm. 16, 53 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Clapp, M. , Aurora, N. , Herrera, L. , Bhatia, M. , Wilen, E. & Wakefield, S. Gut microbiota's effect on mental health: the gut‐brain axis. Clin. Pract. 7, 131–136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Klingelhoefer, L. & Reichmann, H. Pathogenesis of Parkinson disease‐the gut‐brain axis and environmental factors. Nat. Rev. Neurol. 11, 625–636 (2015). [DOI] [PubMed] [Google Scholar]
- 41. Meckel, K.R. & Kiraly, D.D. A potential role for the gut microbiome in substance use disorders. Psychopharmacology 236, 1513–1530 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Felice, V.D. & O'Mahony, S.M. The microbiome and disorders of the central nervous system. Pharmacol. Biochem. Behav. 160, 1–13 (2017). [DOI] [PubMed] [Google Scholar]
- 43. Hill, D.R. et al. Bacterial colonization stimulates a complex physiological response in the immature human intestinal epithelium. Elife 6, e29132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mu, C.L. , Yang, Y.X. & Zhu, W.Y. Gut microbiota: the brain peacekeeper. Front. Microbiol. 7, 345 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sharon, G. , Sampson, T.R. , Geschwind, D.H. & Mazmanian, S.K. The central nervous system and the gut microbiome. Cell 167, 915–932 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Braniste, V. et al. The gut microbiota influences blood‐brain barrier permeability in mice. Sci. Transl. Med. 6, 263ra158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heijtza, R.D. et al. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 108, 3047–3052 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Niedzwiecki, M.M. , Walker, D.I. , Vermeulen, R. , Chadeau‐Hyam, M. , Jones, D.P. & Miller, G.W. The exposome: molecules to populations. Annu. Rev. Pharmacol. Toxicol. 59, 107–127 (2019). [DOI] [PubMed] [Google Scholar]
- 49. Brown, D.G. et al. The microbiota protects from viral‐induced neurologic damage through microglia‐intrinsic TLR signaling. eLife 8, e47117 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tsiaoussis, J. et al. Effects of single and combined toxic exposures on the gut microbiome: current knowledge and future directions. Toxicol. Lett. 312, 72–97 (2019). [DOI] [PubMed] [Google Scholar]
- 51. Gottlieb, S. Statement from FDA Commissioner Scott Gottlieb, M.D., on advancing the science and regulation of live microbiome‐based products used to prevent, treat, or cure diseases in humans <https://www.fda.gov/news‐events/press‐announcements/statement‐fda‐commissioner‐scott‐gottlieb‐md‐advancing‐science‐and‐regulation‐live‐microbiome‐based> (2018). Accessed January 12, 2020. [Google Scholar]
- 52. US Food and Drug Administration (FDA) . Table of pharmacogenomic biomarkers in drug labeling <https://www.fda.gov/drugs/science‐and‐research‐drugs/table‐pharmacogenomic‐biomarkers‐drug‐labeling> (2018). Accessed January 12, 2020. [Google Scholar]
- 53. Frueh, F.W. et al. Pharmacogenomic biomarker information in drug labels approved by the United States Food and Drug Administration: prevalence of related drug use. Pharmacotherapy 28, 992–998 (2008). [DOI] [PubMed] [Google Scholar]
- 54. David, L.A. et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang, Z.N. et al. Impact of chronic dietary red meat, white meat, or non‐meat protein on trimethylamine N‐oxide metabolism and renal excretion in healthy men and women. Eur. Heart J. 40, 583–594 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Orman, M. et al. Structure‐guided identification of a small molecule that inhibits anaerobic choline metabolism by human gut bacteria. J. Am. Chem. Soc. 141, 33–37 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tang, W.H.W. et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 368, 1575–1584 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang, F.Y. & Roy, S. Gut homeostasis, microbial dysbiosis, and opioids. Toxicol. Pathol. 45, 150–156 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cheung, S.G. , Goldenthal, A.R. , Uhlemann, A.C. , Mann, J.J. , Miller, J.M. & Sublette, M.E. Systematic review of gut microbiota and major depression. Front. Psychiatry 10, 34 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cani, P.D. Human gut microbiome: hopes, threats and promises. Gut 67, 1716–1725 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kumar, M. , Ji, B. , Zengler, K. & Nielsen, J. Modelling approaches for studying the microbiome. Nat. Microbiol. 4, 1253–1267 (2019). [DOI] [PubMed] [Google Scholar]
- 62. Doestzada, M. et al. Pharmacomicrobiomics: a novel route towards personalized medicine? Protein Cell 9, 432–445 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Thiele, I. , Clancy, C.M. , Heinken, A. & Fleming, R.M.T. Quantitative systems pharmacology and the personalized drug–microbiota–diet axis. Curr. Opin. Syst. Biol. 4, 43–52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dudek‐Wicher, R.K. , Junka, A. & Bartoszewicz, M. The influence of antibiotics and dietary components on gut microbiota. Gastroenterol. Rev. 13, 85–92 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Maini Rekdal, V. , Bess, E.N. , Bisanz, J.E. , Turnbaugh, P.J. & Balskus, E.P. Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 364, eaau6323 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sharma, A. , Buschmann, M.M. & Gilbert, J.A. Pharmacomicrobiomics: the holy grail to variability in drug response? Clin. Pharmacol. Ther. 106, 317–328 (2019). [DOI] [PubMed] [Google Scholar]
- 67. Wallace, B.D. et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 330, 831–835 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen, H.W. et al. A forward chemical genetic screen reveals gut microbiota metabolites that modulate host physiology. Cell 177, 1217–1231.e18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cully, M. Microbiome therapeutics go small molecule. Nat. Rev. Drug Discov. 18, 569–572 (2019). [DOI] [PubMed] [Google Scholar]
- 70. O'Mahony, S.M. , Dinan, T.G. & Cryan, J.F. The gut microbiota as a key regulator of visceral pain. Pain 158, S19–S28 (2017). [DOI] [PubMed] [Google Scholar]
- 71. Akbarali, H.I. & Dewey, W.L. The gut‐brain interaction in opioid tolerance. Curr. Opin. Pharmacol. 37, 126–130 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Meng, J.J. et al. Morphine induces bacterial translocation in mice by compromising intestinal barrier function in a TLR‐dependent manner. PLoS One 8, e54040 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jones, J.L. & Loftus, E.V. Avoiding the vicious cycle of prolonged opioid use in Crohn's disease. Am. J. Gastroenterol. 100, 2230–2232 (2005). [DOI] [PubMed] [Google Scholar]
- 74. Els, C. et al. Adverse events associated with medium‐ and long‐term use of opioids for chronic non‐cancer pain: an overview of Cochrane Reviews. Cochrane Database Syst. Rev. 10, CD012509 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Roeckel, L.A. , Le Coz, G.M. , Gaveriaux‐Ruff, C. & Simonin, F. Opioid‐induced hyperalgesia: cellular and molecular mechanisms. Neuroscience 338, 160–182 (2016). [DOI] [PubMed] [Google Scholar]
- 76. Cairns, B.E. , Arendt‐Nielsen, L. & Sacerdote, P. Perspectives in pain research 2014: neuroinflammation and glial cell activation: the cause of transition from acute to chronic pain? Scand. J. Pain 6, 3–6 (2015). [DOI] [PubMed] [Google Scholar]
- 77. Banerjee, S. et al. Opioid‐induced gut microbial disruption and bile dysregulation leads to gut barrier compromise and sustained systemic inflammation. Mucosal Immunol. 9, 1418–1428 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pol, O. & Puig, M.M. Expression of opioid receptors during peripheral inflammation. Curr. Top. Med. Chem. 4, 51–61 (2004). [DOI] [PubMed] [Google Scholar]
- 79. Holzer, P. Opioids and opioid receptors in the enteric nervous system: from a problem in opioid analgesia to a possible new prokinetic therapy in humans. Neurosci. Lett. 361, 192–195 (2004). [DOI] [PubMed] [Google Scholar]
- 80. Zhang, L. et al. Morphine tolerance is attenuated in germfree mice and reversed by probiotics, implicating the role of gut microbiome. Proc. Natl. Acad. Sci. USA 116, 13523–13532 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Taylor, A. Revealing a brain‐gut microbiome connection following chronic opioid treatment. J. Pain 19, S1 (2018). [Google Scholar]
- 82. Wang, F.Y. , Meng, J.J. , Zhang, L. , Johnson, T. , Chen, C. & Roy, S. Morphine induces changes in the gut microbiome and metabolome in a morphine dependence model. Sci. Rep. 8, 3596 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lee, K. , Vuong, H.E. , Nusbaum, D.J. , Hsiao, E.Y. , Evans, C.J. & Taylor, A.M.W. The gut microbiota mediates reward and sensory responses associated with regimen‐selective morphine dependence. Neuropsychopharmacology 43, 2606–2614 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kang, M. et al. The effect of gut microbiome on tolerance to morphine mediated antinociception in mice. Sci. Rep. 7, 42658 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Deleo, J.A. , Tanga, F.Y. & Tawfik, V.L. Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. Neuroscientist 10, 40–52 (2004). [DOI] [PubMed] [Google Scholar]
- 86. Fountas, A. , Chai, S.T. , Kourkouti, C. & Karavitaki, N. Mechanisms of endocrinology: endocrinology of opioids. Eur. J. Endocrinol. 179, R183–R196 (2018). [DOI] [PubMed] [Google Scholar]
- 87. Lee, Y.K. , Menezes, J.S. , Umesaki, Y. & Mazmanian, S.K. Proinflammatory T‐cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 108, 4615–4622 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Generaal, E. et al. Reduced hypothalamic‐pituitary‐adrenal axis activity in chronic multi‐site musculoskeletal pain: partly masked by depressive and anxiety disorders. BMC Musculoskelet. Disord. 15, 227 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Shoskes, D.A. , Wang, H. , Polackwich, A.S. , Tucky, B. , Altemus, J. & Eng, C. Analysis of gut microbiome reveals significant differences between men with chronic prostatitis/chronic pelvic pain syndrome and controls. J. Urol. 196, 435–440 (2016). [DOI] [PubMed] [Google Scholar]
- 90. Hilburger, M.E. , Adler, M.W. & Truant, A.L. Morphine induces sepsis in mice. J. Infect. Dis. 176, 183–188 (1997). [DOI] [PubMed] [Google Scholar]
- 91. Lewis, N.D. et al. Arginase II restricts host defense to Helicobacter pylori by attenuating inducible nitric oxide synthase translation in macrophages. J. Immunol. 184, 2572–2582 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chang, G.W.M. & Kam, P.C.A. The physiological and pharmacological roles of cytochrome P450 isoenzymes. Anaesthesia 54, 42–50 (1999). [DOI] [PubMed] [Google Scholar]
- 93. Wilson, I.D. & Nicholson, J.K. Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Transl. Res. 179, 204–222 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Filipczak‐Bryniarska, I. , Nazimek, K. , Nowak, B. , Kozlowski, M. , Wsik, M. & Bryniarski, K. In contrast to morphine, buprenorphine enhances macrophage‐induced humoral immunity and as oxycodone, slightly suppresses the effector phase of cell‐mediated immune response in mice. Int. Immunopharmacol. 54, 344–353 (2018). [DOI] [PubMed] [Google Scholar]
- 95. Eidson, L.N. & Murphy, A.Z. Inflammatory mediators of opioid tolerance: implications for dependency and addiction. Peptides 115, 51–58 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Teal, L.B. , Gould, R.W. , Felts, A.S. & Jones, C.K. Selective allosteric modulation of muscarinic acetylcholine receptors for the treatment of schizophrenia and substance use disorders. Adv. Pharmacol. 86, 153–196 (2019). [DOI] [PubMed] [Google Scholar]
- 97. Alfirevic, A. & Pirmohamed, M. Genomics of adverse drug reactions. Trends Pharmacol. Sci. 38, 100–109 (2017). [DOI] [PubMed] [Google Scholar]
- 98. Saad, R. , Rizkallah, M.R. & Aziz, R.K. Gut pharmacomicrobiomics: the tip of an iceberg of complex interactions between drugs and gut‐associated microbes. Gut Pathog. 4, 16 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Yan, F. , Thall, P.F. , Lu, K.H. , Gilbert, M.R. & Yuan, Y. Phase I‐II clinical trial design: a state‐of‐the‐art paradigm for dose finding. Ann. Oncol. 29, 694–699 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Thall, P.F. & Cook, J.D. Dose‐finding based on efficacy‐toxicity trade‐offs. Biometrics 60, 684–693 (2004). [DOI] [PubMed] [Google Scholar]
- 101. Bisanz, J.E. , Spanogiannopoulos, P. , Pieper, L.M. , Bustion, A.E. & Turnbaugh, P.J. How to determine the role of the microbiome in drug disposition. Drug Metab. Dispos. 46, 1588–1595 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Clayton, T.A. , Baker, D. , Lindon, J.C. , Everett, J.R. & Nicholson, J.K. Pharmacometabonomic identification of a significant host‐microbiome metabolic interaction affecting human drug metabolism. Proc. Natl. Acad. Sci. USA 106, 14728–14733 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Everett, J.R. , Loo, R.L. & Pullen, F.S. Pharmacometabonomics and personalized medicine. Ann. Clin. Biochem. 50, 523–545 (2013). [DOI] [PubMed] [Google Scholar]
- 104. Aliev, R.R. , Richards, W.O. & Wikswo, J.P. Jr. A simple nonlinear model of electrical activity in the intestine. J. Theor. Biol. 204, 21–28 (2000). [DOI] [PubMed] [Google Scholar]
- 105. Sorger, P.K. et al. Quantitative and systems pharmacology in the post‐genomic era: new approaches to discovering drugs and understanding therapeutic mechanisms. An NIH white paper by the QSP Workshop Group <https://www.nigms.nih.gov/training/documents/systemspharmawpsorger2011.pdf> (2011).
- 106. Fritz, J.V. , Desai, M.S. , Shah, P. , Schneider, J.G. & Wilmes, P. From meta‐omics to causality: experimental models for human microbiome research. Microbiome 1, 14 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. The Human Microbiome Project Consortium . A framework for human microbiome research. Nature 486, 215–221 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Dovrolis, N. , Kolios, G. , Spyrou, G.M. & Maroulakou, I. Computational profiling of the gut–brain axis: microflora dysbiosis insights to neurological disorders. Brief. Bioinformatics 20, 825–841 (2019). [DOI] [PubMed] [Google Scholar]
- 109. Lynch, S.V. & Pedersen, O. The human intestinal microbiome in health and disease. N. Engl. J. Med. 375, 2369–2379 (2016). [DOI] [PubMed] [Google Scholar]
- 110. McDonald, J.A.K. et al. Evaluation of microbial community reproducibility, stability and composition in a human distal gut chemostat model. J. Microbiol. Methods 95, 167–174 (2013). [DOI] [PubMed] [Google Scholar]
- 111. McDonald, J.A.K. et al. Simulating distal gut mucosal and luminal communities using packed‐column biofilm reactors and an in vitro chemostat model. J. Microbiol. Methods 108, 36–44 (2015). [DOI] [PubMed] [Google Scholar]
- 112. Sweeney, L.M. , Shuler, M.L. , Babish, J.G. & Ghanem, A. A cell culture analogue of rodent physiology: application to naphthalene toxicology. Toxicol. In Vitro 9, 307–316 (1995). [DOI] [PubMed] [Google Scholar]
- 113. Huh, D. , Matthews, B.D. , Mammoto, A. , Montoya‐Zavala, M. , Hsin, H.Y. & Ingber, D.E. Reconstituting organ‐level lung functions on a chip. Science 328, 1662–1668 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Fitzpatrick, S. & Sprando, R. Advancing regulatory science through innovation. In vitro microphysiological systems. Cell. Mol. Gastroenterol. Hepatol. 7, 239–240 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Wikswo, J. et al. Scaling and systems biology for integrating multiple organs‐on‐a‐chip. Lab Chip 13, 3496–3511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wikswo, J.P. & Porter, A.P. Biology coming full circle: joining the whole and the parts. Exp. Biol. Med. 240, 3–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Young, J.E. & Goldstein, L.S.B. Alzheimers disease in a dish: promises and challenges of human stem cell models. Hum. Mol. Genet. 21, R82–R89 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Costa, V. et al. mTORC1 Inhibition corrects neurodevelopmental and synaptic alterations in a human stem cell model of tuberous sclerosis. Cell Rep. 15, 86–95 (2016). [DOI] [PubMed] [Google Scholar]
- 119. Vernetti, L. et al. Functional coupling of human microphysiology systems: intestine, liver, kidney proximal tubule, blood‐brain barrier and skeletal muscle. Sci. Rep. 7, 42296 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Kim, H.J. , Huh, D. , Hamilton, G. & Ingber, D.E. Human gut‐on‐a‐chip inhabited by microbial flora that experiences intestinal peristalsis‐like motions and flow. Lab Chip 12, 2165–2174 (2012). [DOI] [PubMed] [Google Scholar]
- 121. Kim, H.J. & Ingber, D.E. Gut‐on‐a‐chip microenvironment induces human intestinal cells to undergo villus differentiation. Integrative Biol. 5, 1130–1140 (2013). [DOI] [PubMed] [Google Scholar]
- 122. Shah, P. et al. A microfluidics‐based in vitro model of the gastrointestinal human‐microbe interface. Nat. Commun. 7, 11535 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Poceviciute, R. & Ismagilov, R.F. Human‐gut‐microbiome on a chip. Nat. Biomed. Eng. 3, 500–501 (2019). [DOI] [PubMed] [Google Scholar]
- 124. Esch, M.B. , Ueno, H. , Applegate, D.R. & Shuler, M.L. Modular, pumpless body‐on‐a‐chip platform for the co‐culture of GI tract epithelium and 3D primary liver tissue. Lab Chip 16, 2719–2729 (2016). [DOI] [PubMed] [Google Scholar]
- 125. Maschmeyer, I. et al. Chip‐based human liver‐intestine and liver‐skin co‐cultures ‐ a first step toward systemic repeated dose substance testing in vitro. Eur. J. Pharm. Biopharm. 95, 77–87 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Giese, C. & Marx, U. Human immunity in vitro ‐ Solving immunogenicity and more. Adv. Drug Del. Rev. 69, 103–122 (2014). [DOI] [PubMed] [Google Scholar]
- 127. Ai, Y. , Zhang, F. , Wang, C. , Xie, R. & Liang, Q. Recent progress in lab‐on‐a‐chip for pharmaceutical analysis and pharmacological/toxicological test. TrAC Trend. Anal. Chem. 117, 215–230 (2019). [Google Scholar]
- 128. Francesko, A. , Cardoso, V.F. & Lanceros‐Méndez, S. Lab‐on‐a‐chip technology and microfluidics In Microfluidics for Pharmaceutical Applications(eds. Santos H.A., Liu D. & Zhang H.), 3‐36 (William Andrew Publishing, Norwich, NY, 2019). [Google Scholar]
- 129. Del Rio, D. et al. The gut microbial metabolite trimethylamine‐N‐oxide is present in human cerebrospinal fluid. Nutrients 9, 1053 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Forsythe, P. , Kunze, W. & Bienenstock, J. Moody microbes or fecal phrenology: what do we know about the microbiota‐gut‐brain axis? BMC Med. 14, 58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Workman, M.J. et al. Engineered human pluripotent‐stem‐cell‐derived intestinal tissues with a functional enteric nervous system. Nat. Med. 23, 49–59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Wikswo, J.P. , Markov, D.A. , Samson, P.C. , Block, F.E. III , Schaffer, D.K. & Reiserer, R.S. Interconnections of multiple perfused engineered tissue constructs and microbioreactors, multi‐microformulators and applications of the same, US Patent 10,023,832 B2 (Vanderbilt University, USA, Jul. 17, 2018). [Google Scholar]
- 133. LeDuc, P.R. , Messner, W.C. & Wikswo, J.P. How do control‐based approaches enter into biology? Annu. Rev. Biomed. Eng. 13, 369–396 (2011). [DOI] [PubMed] [Google Scholar]
- 134. Szucs, A. , Varona, P. , Volkovskii, A.R. , Abarbanel, H.D.I. , Rabinovich, M.I. & Selverston, A.I. Interacting biological and electronic neurons generate realistic oscillatory rhythms. NeuroReport 11, 563–569 (2000). [DOI] [PubMed] [Google Scholar]
- 135. Kovacs, G.T.A. Electronic sensors with living cellular components. Proc. IEEE 91, 915–929 (2003). [Google Scholar]
- 136. Wiley, C. & Beeson, C. Continuous measurement of glucose utilization in heart myoblasts. Anal. Biochem. 304, 139–146 (2002). [DOI] [PubMed] [Google Scholar]
- 137. Sin, A. , Chin, K.C. , Jamil, M.F. , Kostov, Y. , Rao, G. & Shuler, M.L. The design and fabrication of three‐chamber microscale cell culture analog devices with integrated dissolved oxygen sensors. Biotechnol. Prog. 20, 338–345 (2004). [DOI] [PubMed] [Google Scholar]
- 138. Wikswo, J.P. , Prokop, A. , Baudenbacher, F. , Cliffel, D. , Csukas, B. & Velkovsky, M. Engineering challenges of BioNEMS: the integration of microfluidics, and micro‐ and nanodevices, models, and external control for systems biology. IEE Proc. Nanobiotechnol. 153, 81–101 (2006). [DOI] [PubMed] [Google Scholar]
- 139. Eklund, S.E. et al. Metabolic discrimination of select list agents by monitoring cellular responses in a multianalyte microphysiometer. Sensors 9, 2117–2133 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Miller, D.R. , McClain, E.S. & Cliffel, D.E. Electrochemical microphysiometry detects cellular glutamate uptake. J. Electrochem. Soc. 165, G3120–G3124 (2018). [Google Scholar]
- 141. Lizama‐Manibusan, B.N. , Klein, S. , McKenzie, J.R. , Cliffel, D.E. & McLaughlin, B. Analysis of a nitroreductase‐based hypoxia sensor in primary neuronal cultures. ACS Chem. Neurosci. 7, 1188–1191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Gupta, A. et al. Encapsulated fusion protein confers "sense and respond" activity to chitosan‐alginate capsules to manipulate bacterial quorum sensing. Biotechnol. Bioeng. 110, 552–562 (2013). [DOI] [PubMed] [Google Scholar]
- 143. Becker, D. et al. Novel orally swallowable intellicap (R) device to quantify regional drug absorption in human GI tract using diltiazem as model drug. AAPS PharmSciTech. 15, 1490–1497 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Soderlind, E. , Abrahamsson, B. , Erlandsson, F. , Wanke, C. , Iordanov, V. & von Corswant, C. Validation of the IntelliCap (R) system as a tool to evaluate extended release profiles in human GI tract using metoprolol as model drug. J. Control. Release 217, 300–307 (2015). [DOI] [PubMed] [Google Scholar]
- 145. Jin, G. , Wang, G. , Liu, X. , Cao, H. & Wang, B. 977 – Intestine microbiome aspiration (IMBA) capsule: a new autonomous and minimally‐invasive device for whole gut microbiome sampling and mapping. Gastroenterology 156, S‐205 (2019). [Google Scholar]
- 146. Bauer, K.C. , Huus, K.E. & Finlay, B.B. Microbes and the mind: emerging hallmarks of the gut microbiota‐brain axis. Cell. Microbiol. 18, 632–644 (2016). [DOI] [PubMed] [Google Scholar]
- 147. Cyr, K.J. , Avaldi, O.M. & Wikswo, J.P. Circadian hormone control in a human‐on‐a‐chip: in vitro biology's ignored component? Exp. Biol. Med. 242, 1714–1731 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Wikswo, J.P. The relevance and potential roles of microphysiological systems in biology and medicine. Exp. Biol. Med. 239, 1061–1072 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Negoro, R. et al. Efficient generation of small intestinal epithelial‐like cells from human iPSCs for drug absorption and metabolism studies. Stem Cell Rep. 11, 1539–1550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Rosenblueth, A. & Wiener, N. The role of models in science. Philos. Sci. 12, 316–321 (1945). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material