

Abstract
Birt-Hogg-Dubé (BHD) and tuberous sclerosis (TS) syndromes share many clinical features. These two diseases display distinct histologic subtypes of renal tumors: chromophobe renal cell carcinoma and renal angiomyolipoma, respectively. Early work suggested a role for mTOR dysregulation in the pathogenesis of these two diseases, however their detailed molecular link remains elusive. Interestingly, a growing number of case reports describe renal angiomyolipoma in BHD patients, suggesting a common molecular origin. The BHD-associated proteins FNIP1/2 and the TS protein Tsc1 were recently identified as regulators of the molecular chaperone Hsp90. Dysregulation of Hsp90 activity has previously been reported to support tumorigenesis, providing a potential explanation for the overlapping phenotypic manifestations in these two hereditary syndromes.
Keywords: Birt-Hogg-Dubé (BHD), FLCN, Tuberous Sclerosis Complex (TSC), Renal Angiomyolipoma, Tsc1 (Hamartin), Tsc2 (Tuberin), Heat Shock Protein-90, Chaperones
1. Introduction
Mutations in several distinct tumor suppressor genes are capable of causing hereditary kidney cancer. Numerous genes have been implicated in the dozen categorized histologic subtypes of hereditary kidney cancer, a topic that has recently been reviewed in detail [1–3]. Due to the well-established role of the nutrient-sensing kinase mammalian target of rapamycin (mTOR) signaling in normal kidney function [4], kidney cancer research has converged on dysregulation of this pathway. There are, however, unique phenotypic manifestations between diseases, which imply the existence of distinguishing molecular characteristics that may serve as novel therapeutic targets. Many of these hereditary renal cell carcinoma (RCC) syndromes lead to other clinical manifestations in addition to the predisposition to kidney tumor development. Two such syndromes, Birt-Hogg-Dubé syndrome and tuberous sclerosis (TS) syndrome, share many clinical and molecular commonalities.
2. Two Interrelated Subtypes of Kidney Cancer
Birt-Hogg-Dubé (BHD) syndrome is caused by germline mutations in the FLCN gene, located on chromosome 17p11.2 [5]. BHD manifests most commonly as cutaneous fibrofolliculomas, pulmonary cysts, spontaneous pneumothorax, and bilateral, multifocal renal cell carcinoma [6]. The most common histologic subtypes of BHD-associated RCC are chromophobe and renal oncocytoma, which occur in approximately one-third of patients [7].
FLCN is characterized as a tumor suppressor protein, though its precise cellular role remains unknown. Loss of FLCN has been previously shown to lead to mTOR activation, and opposing works show lysosomal-FLCN is a Rag-interacting protein with GAP activity for RagC/D or RagA/B [8–11]. However, the existence of conflicting data suggests a context-dependent role in mTOR regulation [12, 13]. Interestingly, facial fibrofolliculomas in patients with BHD failed to respond to topical rapamycin treatment in a recent trial [14], reinforcing an mTOR-independent role for FLCN-mediated pathogenesis. Additionally, a trial to evaluate the effect of mTOR inhibition on BHD-associated RCC began recruitment in 2015, however patient enrollment was scant, resulting in premature study termination (ClinicalTrials.gov Identifier: NCT02504892). There are currently no FDA-approved treatments specific to BHD-associated RCC.
Tuberous sclerosis (TS) syndrome is the result of germline mutations in the TSC1 (chromosome 9q34) or, more commonly, TSC2 (16p13.3) tumor suppressor genes [15]. Afflicted patients may suffer from TS-associated neurological disorders (TAND) including intellectual disability, autism, seizures, and subependymal giant cell astrocytomas [16] [16], as well as facial angiofibromas, pulmonary lymphangioleiomyomatosis [17], and renal angiomyolipoma (AML) in 50-80% of patients [18, 19].
TSC2 mutations are found in 70-90% of TS cases, identifying Tsc2 as the crucial signaling hub in this disease [20]. Tsc2 exerts its tumor suppressor function by acting as a GTPase activating protein for the mTOR activator Rheb [21]. Loss of Tsc2 promotes mTOR activation and a hyperproliferative phenotype [22]. The recent approval of the mTORC1 inhibitor everolimus (Afinitor®) for TS symptoms is based largely on the findings of the EXIST series of clinical trials and is currently approved as a first-line therapy for SEGA [23], AML [16, 17, 24, 25] and TS-associated seizures [26]. There are also six ongoing clinical trials assessing new therapeutic interventions in TS. Three unique topical mTOR inhibitors in Phase 2 and 3 trials are under evaluation for the treatment of facial angiofibromas (ClinicalTrials.gov Identifier: NCT03363763, NCT03826628, NCT02860494). Three other studies are examining different strategies for the relief of epilepsy, including vigabatrin for epilepsy prevention in infants (Phase 2; NCT02849457), a Phase 2 trial of everolimus in treatment-resistant epilepsy (NCT02451696) and oral cannabidiol for seizure relief (Phase 3; NCT02544750). Despite this, a subset of patients do not clinically benefit from mTOR inhibition [26], suggesting additional opportunities for therapeutic intervention.
3. Co-incidence of BHD and TS
As described above, BHD and TS syndromes share many similarities in their clinical manifestations and both commonly affect the skin, lung, and kidney. This overlap suggests mutations of FLCN and TSC1/2 contribute to a common pathway dysregulation capable of affecting these organs. In agreement with this, several groups have reported the occurrence of renal angiomyolipoma in patients with BHD [27–30]. The clinical observation of angiofibromas and Koenen’s tumor in BHD [31, 32] and fibrofolliculomas in TS [33] further highlight the intimate interconnectivity of these two syndromes (Figure 1). Recent work has demonstrated a role for the molecular chaperone heat shock protein-90 (Hsp90) in the regulation of the pathways implicated in these syndromes. This connection provides a potential explanation for the significant overlap between these diseases.
Figure 1.
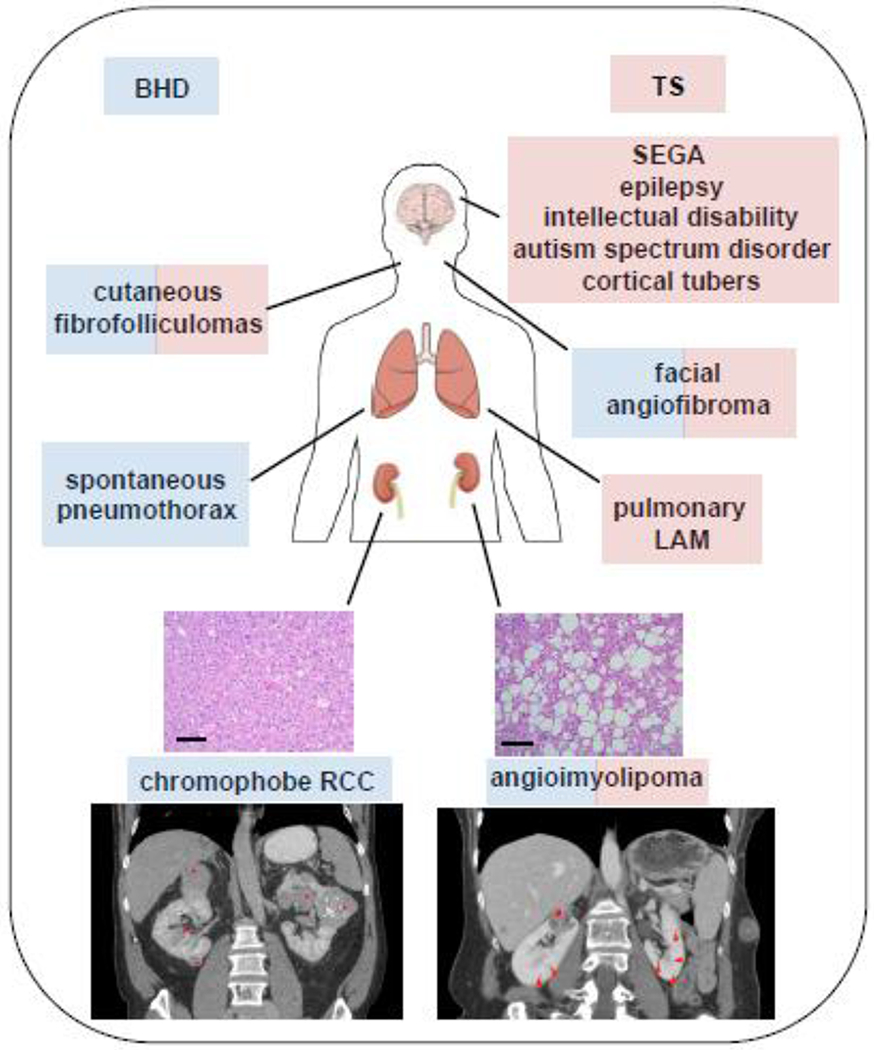
Clinical manifestations of BHD and TS. Birt-Hogg-Dubé syndrome (BHD) is commonly characterized by cutaneous fibrofolliculomas, spontaneous pneumothorax, and multifocal, bilateral renal tumors of chromophobe histology (blue), while Tuberous Sclerosis (TS) is most often associated with neurological impairment and epilepsy, facial angiofibromas, pulmonary lymphangioleiomyomatosis [17], and renal angiomyolipoma (AML) (red). The identification of angiofibromas and angiomyolipoma in BHD patients and fibrofolliculomas in a TS patient suggest a molecular overlap between these two syndromes. Histology: Kidney tissue stained with hematoxylin and eosin. Chromophobe (BHD), angiomyolipoma (TS). Scale bar = 50μM. Imaging: Abdominal CT with contrast. BHD: Asterisks represent bilateral, multifocal renal cell carcinoma. TS: A hash (#) denotes the renal AML. This lesion contains some enhancing components but also areas of macroscopic fat (~−30 HU on non-contrasted scan). Arrowheads denote numerous subcentimeter hypodense lesions through the kidneys bilaterally which likely represent renal cysts.
4. FNIP1/2 and Tsc1 Regulate Hsp90
Heat shock protein-90 (Hsp90) is an ATP-dependent molecular chaperone involved in the stabilization, activation, and maturation of more than 200 intracellular proteins termed ‘clients’ [34]. The activity and function of Hsp90 is primarily regulated by a class of interacting proteins called co-chaperones. A co-chaperone is broadly defined as a protein that interacts with Hsp90 and influences client binding or activity, but is not itself dependent on Hsp90 [35]. Recent work has identified a critical role for Hsp90 in supporting the stability of the tumor suppressors FLCN and Tsc2. Specifically, the BHD- and TS-associated proteins Folliculin-Interacting Proteins1 and 2 (FNIP1/2) and Tsc1 function as co-chaperones of Hsp90. Broadly, this co-chaperone activity provides Hsp90 functional regulation and results in the stabilization of FLCN and Tsc2. These co-chaperones appear to affect Hsp90 similarly, both by decreasing the rate of ATP hydrolysis as well as providing a scaffold for Hsp90-client interactions [36–40].
FNIP1/2 were first identified in association with FLCN and were shown to be essential for FLCN stability [41, 42]. FNIP1/2 form stable dimers, and the resulting complex binds to the C-terminus of FLCN [41, 42]. Mutations in the FLCN gene commonly result in premature truncations of FLCN protein [43, 44], precluding the necessary binding of FNIP1/2. Recent work has unraveled that FNIP1/2 mediate FLCN stability through the Hsp90 chaperone system by acting as regulatory co-chaperones of Hsp90 and providing a scaffold for the chaperoning and stability of FLCN [36].
Tsc1 has long been known to stabilize Tsc2 [45] and was subsequently found to provide protection from HERC1-mediated ubiquitination and proteasomal degradation [46]. As Hsp90 is a crucial mediator of protein stability, these findings suggested a potential role for Hsp90 in the formation of the Tsc1/Tsc2 complex. Indeed, Tsc1 binds to Hsp90 and decreases its ATPase activity, promoting the folding and activity of client proteins. Tsc1 scaffolds Tsc2 to Hsp90, enhancing Tsc2 stability and preventing its ubiquitination and degradation [37]. These data also provide an explanation for the observed tumor suppressive role for Tsc1, as mutations in TSC1 have been shown to cause TS [47]. In this model, Tsc1 loss prevents Tsc2 binding to Hsp90, leading to its degradation and loss of Tsc1/2 complex tumor suppressor activity.
5. Molecular Underpinnings of Clinical Overlap
An outstanding question in the regulation of Hsp90 concerns the dynamics of co-chaperone occupancy. Due to the sheer number of Hsp90 co-chaperones, there must be both spatial and temporal regulation of their binding to Hsp90. Indeed, many co-chaperones have been shown to either work in concert with or antagonize binding of other co-chaperones to Hsp90 [36, 37]. In light of the relative abundance of clinical reports on BHD and TS co-incidence, our group revisited the idea that Tsc1-Tsc2 and FNIP1/2-FLCN are dedicated, exclusive complexes. In fact, crosstalk between Tsc1 and FNIP1 affects Hsp90-mediated chaperoning of a patient-derived mutant FLCN, which in turn affects chaperoning of Tsc2 [30].
FLCN-L460QsX25 is a disease-associated destabilizing mutation of FLCN; a two base pair deletion leads to a frame-shift and premature stop codon resulting in C-terminal truncation [44, 48]. As such, this mutated protein is unable to bind FNIP1/2. Interestingly however, FLCN-L460QsX25 is capable of interacting with Tsc1, which partially compensates for FNIP1/2 binding and facilitates a low level of FLCN expression. Artificially increasing the expression of mutant FLCN compromises Tsc2 stability likely by occupying Tsc1 thereby, decreasing its availability for chaperoning of Tsc2 [30]. Decreased Tsc2 stability (and thus decreased tumor suppressor activity) potentially explains the finding of renal AML in this BHD patient (Figure 2). Notably, loss of both FNIP1 and TSC1 has been shown to synergistically induce mTOR activity [49], a finding that can be explained in part by their roles as Hsp90 co-chaperones [38].
Figure 2.
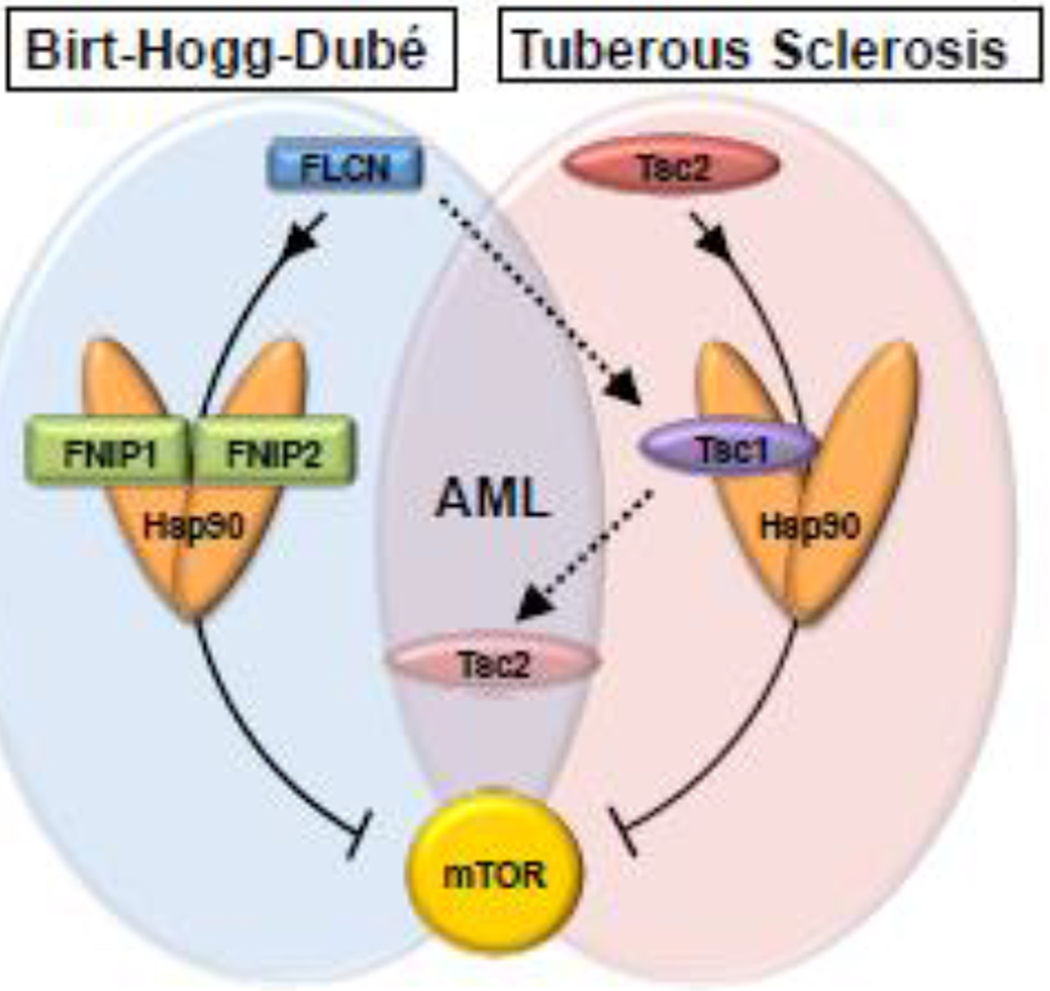
Tumor suppressor regulation in renal cancer. The tumor suppressive activity of FLCN and Tsc2 is supported by the Hsp90 chaperone. FNIP1/2 and Tsc1 co-chaperones scaffold these Hsp90 clients, and together these complexes act to inhibit mTOR. Mutations in the Hsp90 clients FLCN and Tsc2 cause Birt-Hogg-Dubé (BHD) and Tuberous Sclerosis (TS) syndromes, respectively. The patient mutation FLCN-L460Qfsx25 cannot bind to FNIP1/2 and is degraded, though its expression is partially rescued by the Tsc1-Hsp90 chaperone complex. This compromises Tsc2 stability, leading to the clinical manifestation of angiomyolipoma in BHD patients.
6. Hsp90 is a Potential Therapeutic Target in RCC
Drugs targeting Hsp90 have had a prolonged courtship with therapeutic relevance [50, 51]. Despite their pre-clinical promise, Hsp90 inhibitors have been doomed by poor patient selection, a result of a lack of biomarkers to predict clinical applicability [52]. It has previously been shown that FNIP1/2 and Tsc1 can promote Hsp90 binding to its inhibitors [36, 37, 39]. In fact, recent work has demonstrated that Tsc1 expression promotes acetylation of Hsp90 and sensitizes bladder cancer cells to Hsp90 inhibitors [40], providing further evidence that biomarker discovery of is paramount to the success of Hsp90 inhibitors. As previously described, the requirement of the FLCN-L460QsX25 mutant for Tsc1 co-chaperone activity likely leads to increased association of Tsc1 with Hsp90. It then follows that these BHD-associated tumors may be hypersensitive to Hsp90 inhibition, though this remains untested. Interestingly, FLCN mutation has also been identified in colon and breast cancers [53]. Perhaps a similar phenomenon contributes to the pathogenesis of these FLCN-deficient diseases.
7. Concluding Remarks
Despite the early characterization of BHD and TS as diseases of mTOR dysregulation, the role each protein plays in this process is unclear. Mounting evidence suggests the mTOR suppressive effects of the FNIP/FLCN and Tsc1/2 systems are potentially mediated by the action of molecular chaperones. Further, it is possible that the action of FNIP1/2 and Tsc1 as Hsp90 co-chaperones is independent of mTOR altogether. In either case, the phenotypic convergence of BHD and TS suggest a shared underlying molecular pathology instrumental in the homeostasis of multiple organ systems. Uncovering this link is likely to present more broadly applicable therapeutic options.
Acknowledgments
This work was partly supported by the National Institute of General Medical Sciences of the NIH (grant R01GM124256 to M.M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. This work was also supported by funds from SUNY Upstate Medical University, the Upstate Foundation, and in part by the Urology Care Foundation Research Scholar Award Program and American Urological Association (M.M.). The authors are grateful to their colleagues Laura S. Schmidt, W. Marston Linehan for their stimulating discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- [1].Ball MW, Pinto PA. Familial and Hereditary Syndromes in Renal Cell Cancer. Renal Cancer 2020. p. 35–48. [Google Scholar]
- [2].Linehan WM, Schmidt LS, Crooks DR, Wei D, Srinivasan R, Lang M, et al. The Metabolic Basis of Kidney Cancer. Cancer Discov. 2019;9:1006–21. [DOI] [PubMed] [Google Scholar]
- [3].Carlo MI, Hakimi AA, Stewart GD, Bratslavsky G, Brugarolas J, Chen YB, et al. Familial Kidney Cancer: Implications of New Syndromes and Molecular Insights. Eur Urol. 2019;76:754–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fantus D, Rogers NM, Grahammer F, Huber TB, Thomson AW. Roles of mTOR complexes in the kidney: implications for renal disease and transplantation. Nat Rev Nephrol. 2016;12:587–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schmidt LS, Warren MB, Nickerson ML, Weirich G, Matrosova V, Toro JR, et al. Birt-Hogg-Dubé syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. Am J Hum Genet. 2001;69:876–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674–7. [PubMed] [Google Scholar]
- [7].Schmidt LS. Birt-Hogg-Dubé syndrome, a genodermatosis that increases risk for renal carcinoma. Curr Mol Med. 2004;4:877–85. [DOI] [PubMed] [Google Scholar]
- [8].Lawrence RE, Fromm SA, Fu Y, Yokom AL, Kim DJ, Thelen AM, et al. Structural mechanism of a Rag GTPase activation checkpoint by the lysosomal folliculin complex. Science. 2019;366:971–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Petit CS, Roczniak-Ferguson A, Ferguson SM. Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of Rag GTPases. J Cell Biol. 2013;202:1107–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tsun ZY, Bar-Peled L, Chantranupong L, Zoncu R, Wang T, Kim C, et al. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell. 2013;52:495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shen K, Rogala KB, Chou HT, Huang RK, Yu Z, Sabatini DM. Cryo-EM Structure of the Human FLCN-FNIP2-Rag-Ragulator Complex. Cell. 2019;179:1319–29 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hudon V, Sabourin S, Dydensborg AB, Kottis V, Ghazi A, Paquet M, et al. Renal tumour suppressor function of the Birt-Hogg-Dubé syndrome gene product folliculin. J Med Genet. 2010;47:182–9. [DOI] [PubMed] [Google Scholar]
- [13].Khabibullin D, Medvetz DA, Pinilla M, Hariharan V, Li C, Hergrueter A, et al. Folliculin regulates cell-cell adhesion, AMPK, and mTORC1 in a cell-type-specific manner in lung-derived cells. Physiol Rep. 2014;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gijezen LM, Vernooij M, Martens H, Oduber CE, Henquet CJ, Starink TM, et al. Topical rapamycin as a treatment for fibrofolliculomas in Birt-Hogg-Dubé syndrome: a double-blind placebo-controlled randomized split-face trial. PLoS One. 2014;9:e99071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Henske EP, Jozwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016;2:16035. [DOI] [PubMed] [Google Scholar]
- [16].Kwiatkowski DJ, Palmer MR, Jozwiak S, Bissler J, Franz D, Segal S, et al. Response to everolimus is seen in TSC-associated SEGAs and angiomyolipomas independent of mutation type and site in TSC1 and TSC2. Eur J Hum Genet. 2015;23:1665–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kingswood JC, Jozwiak S, Belousova ED, Frost MD, Kuperman RA, Bebin EM, et al. The effect of everolimus on renal angiomyolipoma in patients with tuberous sclerosis complex being treated for subependymal giant cell astrocytoma: subgroup results from the randomized, placebo-controlled, Phase 3 trial EXIST-1. Nephrol Dial Transplant. 2014;29:1203–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kingswood JC, Belousova E, Benedik MP, Carter T, Cottin V, Curatolo P, et al. Renal angiomyolipoma in patients with tuberous sclerosis complex: findings from the TuberOus SClerosis registry to increase disease Awareness. Nephrol Dial Transplant. 2019;34:502–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rakowski SK, Winterkorn EB, Paul E, Steele DJ, Halpern EF, Thiele EA. Renal manifestations of tuberous sclerosis complex: Incidence, prognosis, and predictive factors. Kidney Int. 2006;70:1777–82. [DOI] [PubMed] [Google Scholar]
- [20].Rosset C, Netto CBO, Ashton-Prolla P. TSC1 and TSC2 gene mutations and their implications for treatment in Tuberous Sclerosis Complex: a review. Genet Mol Biol. 2017;40:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].El-Hashemite N, Zhang H, Henske EP, Kwiatkowski DJ. Mutation in TSC2 and activation of mammalian target of rapamycin signalling pathway in renal angiomyolipoma. Lancet. 2003;361:1348–9. [DOI] [PubMed] [Google Scholar]
- [23].Turner SG, Peters KB, Vredenburgh JJ, Desjardins A, Friedman HS, Reardon DA. Everolimus tablets for patients with subependymal giant cell astrocytoma. Expert Opin Pharmacother. 2011;12:2265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bissler JJ, Kingswood JC, Radzikowska E, Zonnenberg BA, Frost M, Belousova E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381:817–24. [DOI] [PubMed] [Google Scholar]
- [25].Bissler JJ, Kingswood JC, Radzikowska E, Zonnenberg BA, Belousova E, Frost MD, et al. Everolimus long-term use in patients with tuberous sclerosis complex: Four-year update of the EXIST-2 study. PLoS One. 2017;12:e0180939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet. 2016;388:2153–63. [DOI] [PubMed] [Google Scholar]
- [27].Byrne M, Mallipeddi R, Pichert G, Whittaker S. Birt-Hogg-Dubé syndrome with a renal angiomyolipoma: further evidence of a relationship between Birt-Hogg-Dubé syndrome and tuberous sclerosis complex. Australas J Dermatol. 2012;53:151–4. [DOI] [PubMed] [Google Scholar]
- [28].Tobino K, Seyama K. Birt-Hogg-Dubé syndrome with renal angiomyolipoma. Intern Med. 2012;51:1279–80. [DOI] [PubMed] [Google Scholar]
- [29].Dow E, Winship I. Renal angiomyolipoma in Birt-Hogg-Dubé syndrome: A case study supporting overlap with tuberous sclerosis complex. Am J Med Genet A. 2016;170:3323–6. [DOI] [PubMed] [Google Scholar]
- [30].Sager RA, Woodford MR, Shapiro O, Mollapour M, Bratslavsky G. Sporadic renal angiomyolipoma in a patient with Birt-Hogg-Dubé: chaperones in pathogenesis. Oncotarget. 2018;9:22220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Schaffer JV, Gohara MA, McNiff JM, Aasi SZ, Dvoretzky I. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dubé syndrome. J Am Acad Dermatol. 2005;53:S108–11. [DOI] [PubMed] [Google Scholar]
- [32].DiCicco B, Johnson W, Allred J, Soldano AC, Ramsdell WM. Koenen’s tumor and facial angiofibromas in a case of Birt-Hogg-Dube syndrome: A cutaneous contribution to growing evidence of a relationship with tuberous sclerosis complex. JAAD Case Rep. 2016;2:196–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Misago N, Joh K, Yatsuki H, Soejima H, Narisawa Y. A BHD germline mutation identified in an Asian family with Birt-Hogg-Dubé syndrome. Acta Derm Venereol. 2008;88:423–5. [DOI] [PubMed] [Google Scholar]
- [34].Schopf FH, Biebl MM, Buchner J. The HSP90 chaperone machinery. Nat Rev Mol Cell Biol. 2017;18:345–60. [DOI] [PubMed] [Google Scholar]
- [35].Miyata Y, Nakamoto H, Neckers L. The therapeutic target Hsp90 and cancer hallmarks. Curr Pharm Des. 2013;19:347–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Woodford MR, Dunn DM, Blanden AR, Capriotti D, Loiselle D, Prodromou C, et al. The FNIP co-chaperones decelerate the Hsp90 chaperone cycle and enhance drug binding. Nat Commun. 2016;7:12037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Woodford MR, Sager RA, Marris E, Dunn DM, Blanden AR, Murphy RL, et al. Tumor suppressor Tsc1 is a new Hsp90 co-chaperone that facilitates folding of kinase and non-kinase clients. EMBO J. 2017;36:3650–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sager RA, Woodford MR, Mollapour M. The mTOR Independent Function of Tsc1 and FNIPs. Trends Biochem Sci. 2018;43:935–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sager RA, Woodford MR, Backe SJ, Makedon AM, Baker-Williams AJ, DiGregorio BT, et al. Post-translational Regulation of FNIP1 Creates a Rheostat for the Molecular Chaperone Hsp90. Cell Rep. 2019;26:1344–56 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Woodford MR, Hughes M, Sager RA, Backe SJ, Baker-Williams AJ, Bratslavsky MS, et al. Mutation of the co-chaperone Tsc1 in bladder cancer diminishes Hsp90 acetylation and reduces drug sensitivity and selectivity. Oncotarget. 2019;10:5824–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Baba M, Hong SB, Sharma N, Warren MB, Nickerson ML, Iwamatsu A, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. 2006;103:15552–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hasumi H, Baba M, Hong SB, Hasumi Y, Huang Y, Yao M, et al. Identification and characterization of a novel folliculin-interacting protein FNIP2. Gene. 2008;415:60–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nahorski MS, Reiman A, Lim DH, Nookala RK, Seabra L, Lu X, et al. Birt Hogg-Dube syndrome-associated FLCN mutations disrupt protein stability. Hum Mutat. 2011;32:921–9. [DOI] [PubMed] [Google Scholar]
- [44].Schmidt LS, Nickerson ML, Warren MB, Glenn GM, Toro JR, Merino MJ, et al. Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dubé syndrome. Am J Hum Genet. 2005;76:1023–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Benvenuto G, Li S, Brown SJ, Braverman R, Vass WC, Cheadle JP, et al. The tuberous sclerosis-1 (TSC1) gene product hamartin suppresses cell growth and augments the expression of the TSC2 product tuberin by inhibiting its ubiquitination. Oncogene. 2000;19:6306–16. [DOI] [PubMed] [Google Scholar]
- [46].Chong-Kopera H, Inoki K, Li Y, Zhu T, Garcia-Gonzalo FR, Rosa JL, et al. TSC1 stabilizes TSC2 by inhibiting the interaction between TSC2 and the HERC1 ubiquitin ligase. J Biol Chem. 2006;281:8313–6. [DOI] [PubMed] [Google Scholar]
- [47].Hoogeveen-Westerveld M, Ekong R, Povey S, Karbassi I, Batish SD, den Dunnen JT, et al. Functional assessment of TSC1 missense variants identified in individuals with tuberous sclerosis complex. Hum Mutat. 2012;33:476–9. [DOI] [PubMed] [Google Scholar]
- [48].Toro JR, Wei MH, Glenn GM, Weinreich M, Toure O, Vocke C, et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet. 2008;45:321–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Centini R, Tsang M, Iwata T, Park H, Delrow J, Margineantu D, et al. Loss of Fnip1 alters kidney developmental transcriptional program and synergizes with TSC1 loss to promote mTORC1 activation and renal cyst formation. PLoS One. 2018;13:e0197973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18:64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sanchez J, Carter TR, Cohen MS, Blagg BS. Old and New Approaches to Target the Hsp90 Chaperone. Curr Cancer Drug Targets. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chehab M, Caza T, Skotnicki K, Landas S, Bratslavsky G, Mollapour M, et al. Targeting Hsp90 in urothelial carcinoma. Oncotarget. 2015;6:8454–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Palmirotta R, Savonarola A, Ludovici G, Donati P, Cavaliere F, ML DEM, et al. Association between Birt Hogg Dubé syndrome and cancer predisposition. Anticancer Res. 2010;30:751–7. [PubMed] [Google Scholar]