

Abstract
Bacteroides thetaiotaomicron was examined to determine whether its obligate anaerobiosis is imposed by endogenous reactive oxygen species or by molecular oxygen itself. Previous analyses established that aerated B. thetaiotaomicron loses some enzyme activities due to a high rate of endogenous superoxide formation. However, the present study establishes that another key step in central metabolism is poisoned by molecular oxygen itself. Pyruvate dissimilation was shown to depend upon two enzymes, pyruvate:formate lyase (PFL) and pyruvate:ferredoxin oxidoreductase (PFOR), that lose activity upon aeration. PFL is a glycyl-radical enzyme whose vulnerability to oxygen is already understood. The rate of PFOR damage was unaffected by the level of superoxide or peroxide, showing that molecular oxygen itself is the culprit. The cell cannot repair PFOR, which amplifies the impact of damage. The rates of PFOR and fumarase inactivation are similar, suggesting that superoxide dismutase is calibrated so the oxygen- and superoxide-sensitive enzymes are equally sensitive to aeration. The physiological purpose of PFL and PFOR is to degrade pyruvate without disrupting the redox balance, and they do so using catalytic mechanisms that are intrinsically vulnerable to oxygen. In this way the anaerobic excellence and oxygen sensitivity of B. thetaiotaomicron are two sides of the same coin.
Keywords: Oxidative stress, obligate anaerobiosis, pyruvate:ferredoxin oxidoreductase, pyruvate:formate lyase, iron-sulfur clusters
Graphical abstract
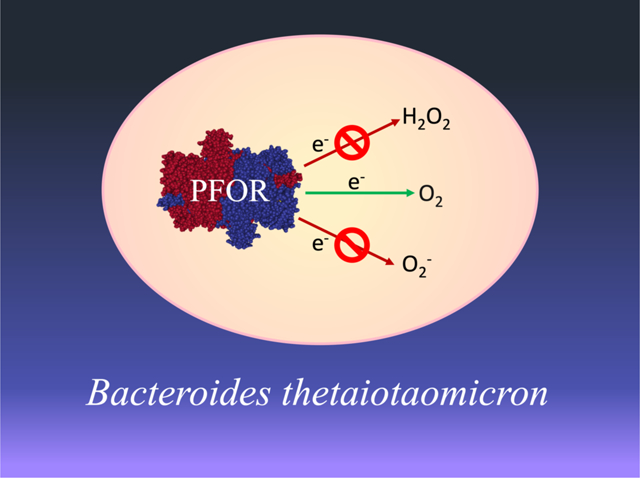
Abbreviated summary
Pyruvate:ferredoxin oxidoreductase and pyruvate:formate lyase, the key enzymes involved in pyruvate dissimilation in Bacteroides thetaiotaomicron, are irreversibly inactivated by molecular oxygen itself rather than by reactive oxygen species. Therefore, neither elevated scavenging enzymes nor repair systems could sustain the metabolism of this bacterium if substantial oxygen were present.
(Enzyme structure from Chabriere et al. 1999, PDB: 1B0P)
Introduction
Life originated and invented its core biochemical mechanisms in a world devoid of molecular oxygen. It was not until the appearance of oxygenic photosynthesis 2 byr later that oxygen began to accumulate, and only in the last 500 million years did concentrations approach their current level (Knoll & Nowak, 2017). Many contemporary organisms exploit oxygen in their respiratory schemes—but oxygen is also a reactive chemical with potentially toxic effects. Indeed, virtually all organisms suffer growth problems when they are exposed to more oxygen than is found in their native environments. This phenomenon is most obvious in the case of microbes that normally dwell in anoxic habitats. The defining characteristic of these obligate anaerobes is that their growth stops when they are exposed to full aeration, either in the lab or in natural environments.
The basis of this oxygen sensitivity has long been an open question. In 1970, shortly after their discovery of superoxide dismutase (SOD), Joe McCord and Irwin Fridovich noted that many obligate anaerobes lack SOD and catalase (McCord et al., 1971). These enzymes degrade superoxide (O2−) and hydrogen peroxide (H2O2), respectively, which are far more potent oxidants than is oxygen itself. The correlation prompted many workers to infer that it is the absence of these enzymes that constrains anaerobes to oxygen-free environments. Their data, and this interpretation, are presented in many textbooks. In 1987 Carlioz and Touati demonstrated that if SOD is genetically removed from Escherichia coli, this normally oxygen-tolerant bacterium becomes incompetent at some basic biosynthetic and catabolic functions (Carlioz & Touati, 1986). This result supported the hypothesis that reactive oxygen species (ROS) might underlie the toxicity of oxygen.
The details of ROS toxicity were subsequently worked out, primarily through studies of E. coli. In vitro experiments demonstrated that ROS can be formed when oxygen adventitiously oxidizes the flavins and metal centers of redox enzymes (Seaver & Imlay, 2004, Korshunov & Imlay, 2010, Ballou et al., 1969, Messner & Imlay, 2002b). Measurements showed that O2− and H2O2 are formed at rates of 5 and 10 μM/sec inside E. coli, although in wild-type cells the scavenging enzymes are thought to keep the steady-state levels at ~ 0.1 nM and 0.2 μM, respectively (Seaver & Imlay, 2001, Imlay, 2013). These concentrations lie slightly below the doses that create disabling injuries. At higher doses superoxide and H2O2 both oxidize the exposed iron atoms of [4Fe-4S] dehydratases and of mononuclear Fe(II) enzymes, triggering the dissociation of the catalytic metal and loss of activity (Kuo et al., 1987, Flint et al., 1993b, Jang & Imlay, 2007, Sobota & Imlay, 2011, Anjem & Imlay, 2012). Such enzymes play roles in the pentose-phosphate pathway, the TCA cycle, and several amino acid biosynthetic pathways; therefore, these processes fail in cells that contain elevated levels of either oxidant.
Those results emerged over several decades. In the meantime, biochemical and genomic inspections had revealed that obligate anaerobes do not lack scavenging enzymes after all. The bacteria and archaea that had been surveyed in the McCord study turned out to possess superoxide reductases and peroxidases rather than SODs and catalases (Imlay, 2002b, Poole, 2005). Other anaerobes contain authentic SODs and catalases. Their possession of scavenging enzymes presumably protects these organisms from episodic exposure to oxygen while transiting between hosts, during infection into erstwhile oxic tissues, and when venturing into microoxic edges of their habitats. These discoveries overturned the simple notion that anaerobes are poisoned by oxygen because they lack defenses against ROS. At the same time, as workers dissected the biochemistries of anaerobic metabolism, they encountered key enzymes that rapidly lost activity in oxic buffers. This discovery raised the prospect that anaerobes may rely on enzymes that molecular oxygen poisons directly. Some ambiguity remained, however, because ROS are easily formed in oxic reaction mixes (Gardner & Fridovich, 1991b, Gardner & Fridovich, 1991a, Liochev & Fridovich, 1993), particularly those that contain reducing substrates; and in any case it was unclear whether oxygen itself or ROS were the primary toxic species in vivo. Further complicating the picture was the realization that some enzymes that are inactivated by oxygen in vitro—such as radical SAM enzymes—retain function inside aerobic cells (Imlay et al., 2019).
To inspect this issue in living cells, we have analyzed the metabolic failure of aerated Bacteroides thetaiotaomicron, an anaerobe that is one of the most abundant residents of the human gut (Pan & Imlay, 2001). Like E. coli, B. thetaiotaomicron dwells in the colon, where it primarily ferments carbohydrates. However, whereas excreted E. coli can thrive in oxygen-containing surface waters, aerated B. thetaiotaomicron cannot grow. Similarly, when it is exposed to oxygen in the lab, its growth stops within minutes. This phenomenon has been closely examined. Substrate/product analysis revealed that upon aeration its glucose catabolism quickly stalled (Pan & Imlay, 2001). Specific bottlenecks were identified in both branches of its core metabolism: in the conversion of malate to fumarate, and in the breakdown of pyruvate [Figure 1]. These reactions were known to be catalyzed by fumarase and pyruvate:ferredoxin oxidoreductase (PFOR), respectively, and measurements confirmed that both enzymes lost activity when cells were aerated. Most other enzymes remained fully functional.
Figure 1. Primary pathways of glucose fermentation in B. thetaiotaomicron.
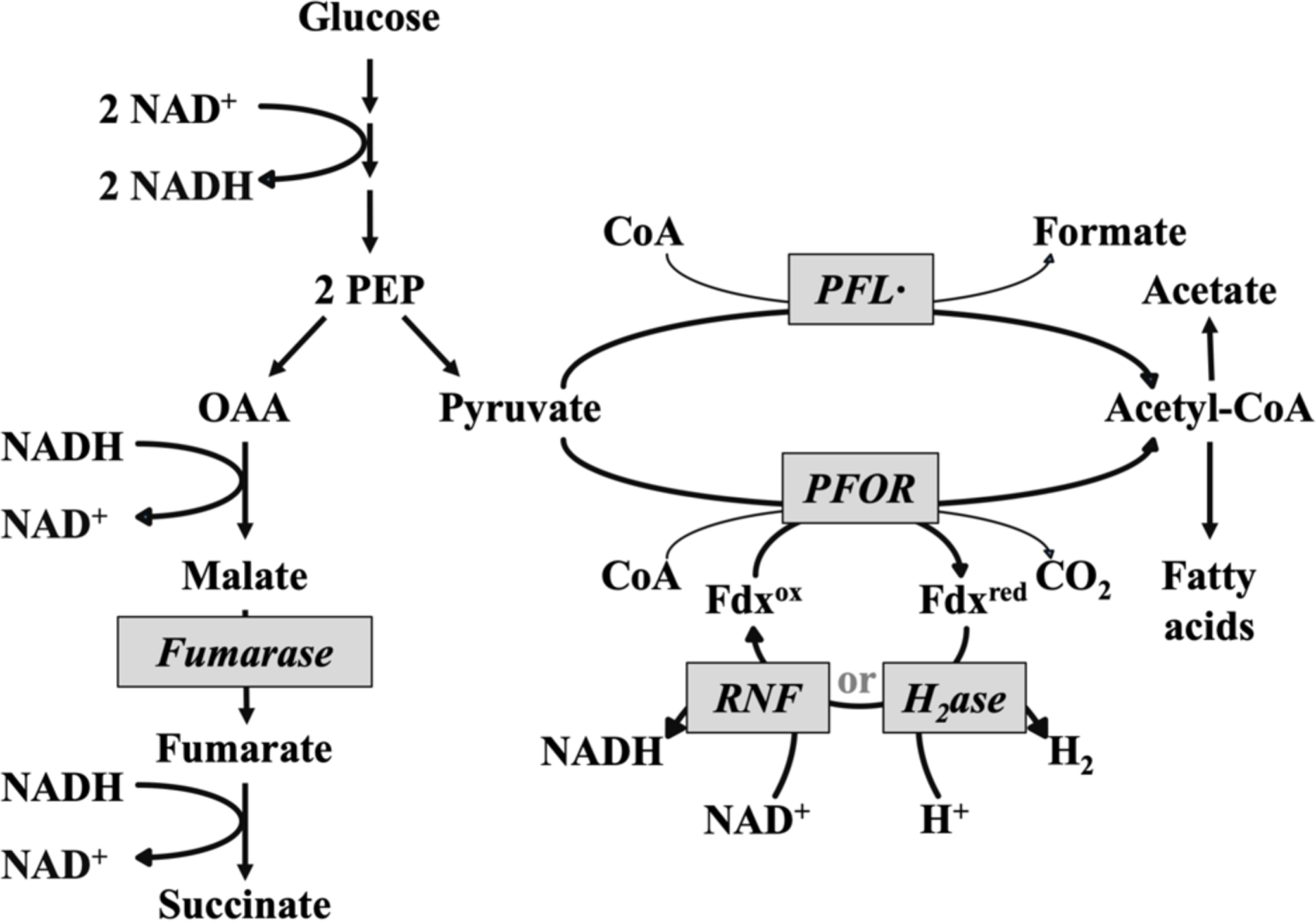
PEP: phosphoenolpyruvate, OAA, oxaloacetate; Fdx: ferredoxin; PFL, pyruvate-formate lyase; PFOR, pyruvate:ferredoxin oxidoreductase; Rnf, ferrodoxin:NAD oxidoreductase, H2ase: hydrogenase.
The inactivation of fumarase struck a chord, because it belongs to the same family of dehydratases that are poisoned by O2− and H2O2 in E. coli scavenger mutants (Liochev & Fridovich, 1992, Flint et al., 1993b, Jang & Imlay, 2007). Indeed, our recent work determined that O2− is the culprit in the anaerobe (Lu et al., 2018). The fact that fumarase loses activity in aerated B. thetaiotaomicron, but remains fully active in aerated E. coli, was ascribed to the fact that ROS production is far faster during aeration of the anaerobe (Lu et al., 2018, Mishra & Imlay, 2013). The reason is unclear. While it was originally suspected that endogenous ROS in bacteria arise from electron leaks in the respiratory chain, data from E. coli show that the chain is usually only a minor contributor (Seaver & Imlay, 2004). At present only a few enzymes, including fumarate reductase, have been shown to produce significant ROS in E. coli; the major sources of intracellular ROS remain to be determined (Imlay, 1995, Messner & Imlay, 2002b, Korshunov & Imlay, 2010). It seemed plausible that the excess ROS of B. thetaiotaomicron might be emitted by its own fumarate reductase (Meehan & Malamy, 2012, Korshunov & Imlay, 2010), which is a key enzyme in its anaerobic metabolism, but subsequent analysis indicated that the B. thetaiotaomicron enzyme is electronically configured in a way that avoids ROS formation (Lu & Imlay, 2017). The source of the ROS remains unknown.
So too is the mechanism of PFOR damage. In the present study, we demonstrate that PFOR is one of two pyruvate-dissimilating enzymes in B. thetaiotaomicron—and that both of them are directly damaged by molecular oxygen itself. Genetic analysis demonstrated that a block in either branch of central metabolism is sufficient to prohibit carbohydrate fermentation. Surprisingly, the damage to PFOR, unlike that to fumarase, cannot be repaired, making PFOR particularly vulnerable to poisoning. The SOD titer inside the cell seems to be set so that O2−- sensitive enzymes are inactivated by approximately the same oxygen concentrations that directly incapacitate PFOR. Thus in this bacterium the phenomenon of obligate anaerobiosis is driven by a combination of ROS and O2.
Results
B. thetaiotaomicron stops growing upon aeration.
When log-phase anoxic cultures were aerated, growth stopped [Figure 2]. When anoxia was restored, growth quickly resumed. This behavior mirrors what was reported before, and it reflects the fact that oxygen does not kill the cell (Lu et al., 2018). The rapid recovery from aeration may be important in natural environments.
Figure 2. B. thetaiotaomicron cells stop growing upon exposure to oxygen, but resume growth when anoxia is restored.
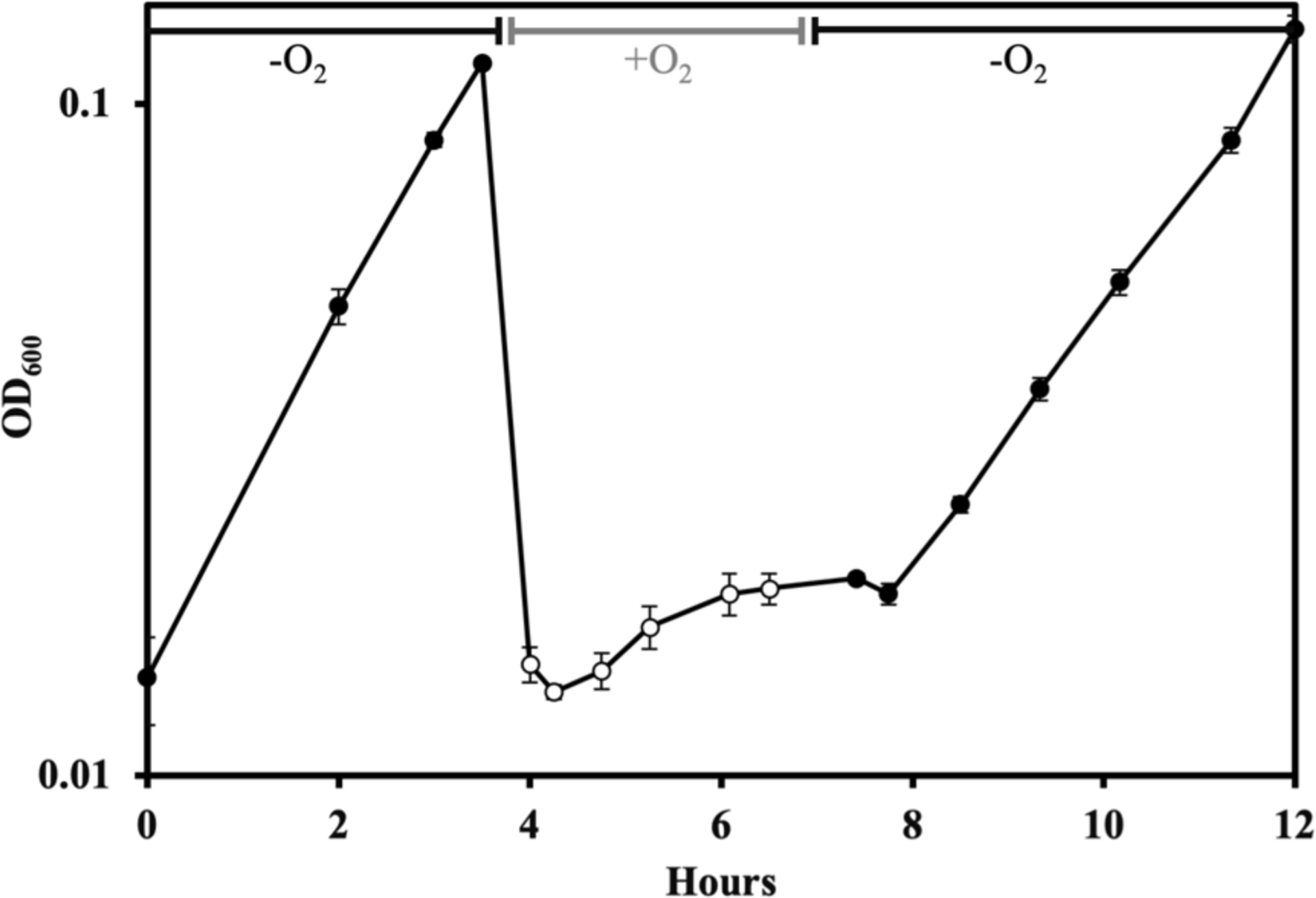
Wildtype B. thetaiotaomicron (BT5482) cells were grown in anoxic BHIS (no cysteine) media. They were then diluted and aerated for 2.5 hours (white markers). Cells were then returned to the anaerobic chamber, washed and resuspended in anoxic BHIS (no cysteine) (black markers). Error bars represent the standard error of the mean of three biological replicates. In this and some subsequent figures, error bars might be obscured by the markers.
Both branches of central metabolism are essential for carbohydrate fermentation.
The ecological niches of bacteria are typically established by features of their energy-harvesting schemes. B. thetaiotaomicron is primarily a fermenter of carbohydrates (Xu, 2003), and so we chose to examine the impact of oxygen upon the catabolism of glucose. Our previous work demonstrated that this process becomes impaired and unbalanced when B. thetaiotaomicron is exposed to oxygen (Pan & Imlay, 2001). To proceed further, we sought to update our understanding of which enzymes are necessary for B. thetaiotaomicron metabolism under our growth conditions. Figure 1 presents a map that is useful in following this argument.
Glucose is processed through glycolysis. To achieve redox-balancing, the NADH that is generated during glycolysis is recycled to NAD+ by the left-hand branch that culminates in the formation of succinate. This branch also generates proton-motive force (pmf) when electrons are transferred from NADH to fumarate, a membrane-bound process that involves the proton-translocating NADH dehydrogenase I complex and fumarate reductase. The right-hand branch involves the dissimilation of pyruvate to acetyl-CoA. Acetyl-CoA is an essential precursor for the synthesis of lipids and the 2-oxoglutarate family of amino acids, but most of it is converted to acetate, with ATP being formed by substrate-level phosphorylation. Thus succinate and acetate are the primary fermentation products during growth on glucose.
Earlier analysis had focused upon PFOR as a pyruvate dissimilating enzyme; it was assumed that reduced ferredoxin was then recycled by electron flow through hydrogenase (Frey, 2002). Genes encoding two hydrogenases are apparent in the B. thetaiotaomicron genome. More-recent work in Bacteroides fragilis, a cousin of B. thetaiotaomicron, has shown that ferredoxin can alternatively transfer electrons to NAD+ via ferredoxin:NAD+ oxidoreductase (Rnf) (Biegel et al., 2011, Hess et al., 2016). An rnf homolog is readily found in the B. thetaiotaomicron genome. Further, we observed two genes that resemble E. coli pfl, encoding the alternative pyruvate-dissimilating enzyme pyruvate:formate lyase (PFL). PFL is a glycyl-radical enzyme that splits pyruvate into acetyl-CoA and formate, thereby obviating any need for downstream electron transfer (Wagner et al., 1992, Shibata & Toraya, 2015). It could provide a route of pyruvate dissimilation independent of PFOR. We have renamed the genes for easier identification in this paper [Table 1].
Table 1: The genes and their respective proteins used in this study.
These proteins are annotated to be parts of the central metabolism of B. thetaiotaomicron. We confirmed the identity of PFOR and PFL4738 genes in this study. Fumarase activity was confirmed in a previous study (Lu et al., 2018).
| Encoded protein | Name in this study | |
|---|---|---|
| BT_1747 | Pyruvate:ferredoxin oxidoreductase | PFOR |
| BT_4738 | Pyruvate:formate lyase | PFL4738 |
| BT_2955 | Pyruvate:formate lyase homolog | PFL2955 |
| BT_2414 | Putative ferredoxin | Fdx |
| BT_0616 to BT_0622 | Putative NADH:ferredoxin oxidoreductase operon | Rnf |
| BT_1834 | Putative Fe-Fe hydrogenase subunit | Hyd1834 |
| BT_3472 | Hydrogenase subunit homolog | Hyd3472 |
| BT_2256 (fum) | Fumarase | Fumarase |
To understand the role of the above enzymes in central metabolism of B. thetaiotaomicron, clean deletion mutants were successfully generated, and the growth of each mutant in anoxic minimal glucose medium was monitored [Figure 3A]. To our surprise, the only single mutant that showed a substantial growth arrest was Δfum. We had isolated this mutant using medium containing fumarate; because B. thetaiotaomicron encodes a fumarate importer, the fumarate supplements were used to by-pass the need for endogenous fumarate production (Lu & Imlay, 2017). However, when the mutant was washed and resuspended in medium lacking fumarate, growth ceased [Figure 3B]. Thus function of the succinate pathway is essential for glucose fermentation. This result confirms what was previously suggested: that the inactivation of fumarase by oxidants would be sufficient to block growth on carbohydrates.
Figure 3. Fumarase, plus either PFL or PFOR, are essential for growth on glucose.
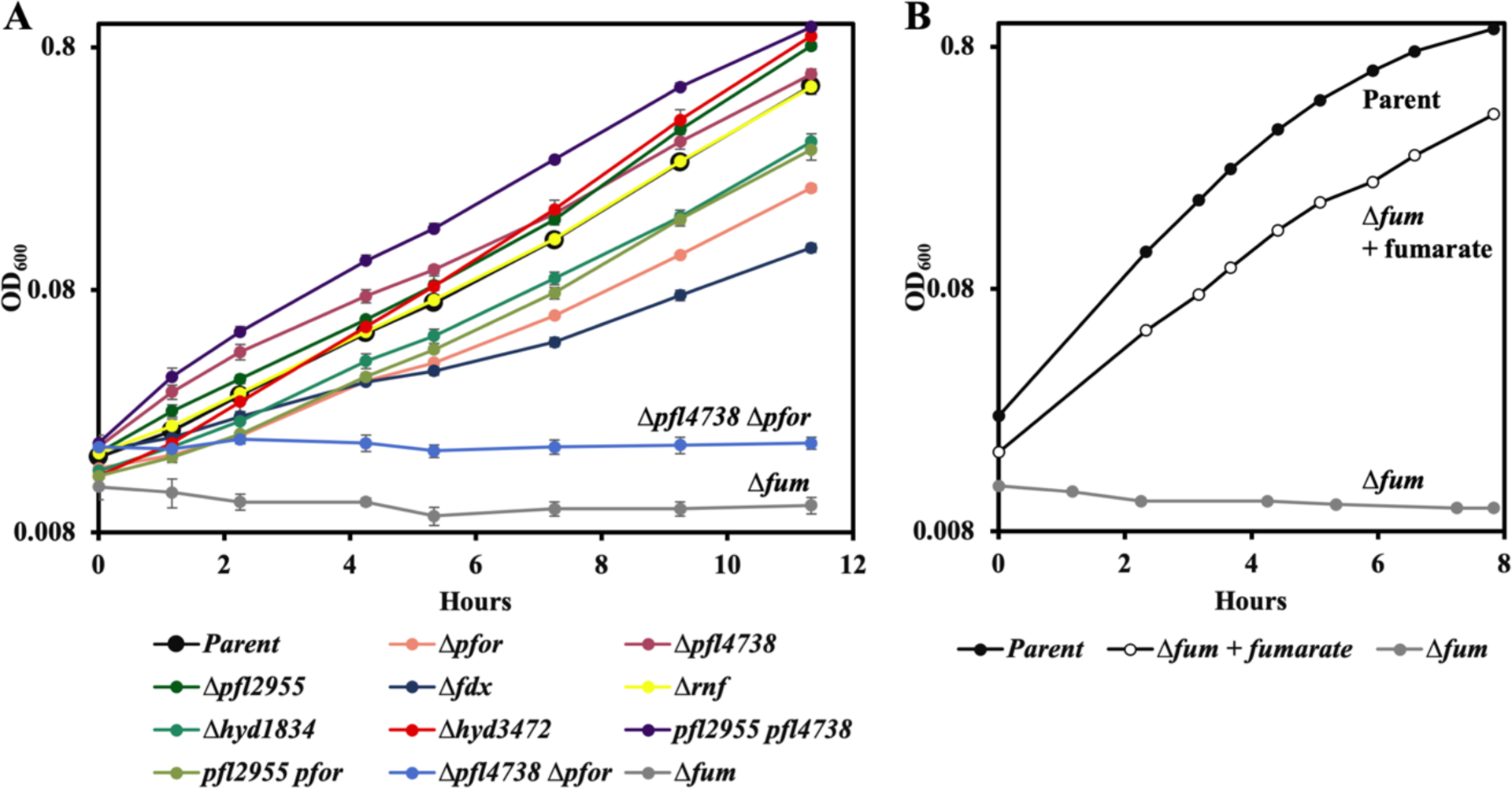
(A) Exponentially growing cells were subcultured at time zero into anoxic glucose medium. Where indicated, the Δfum mutant was supplemented with fumarate. (B) Exponentially growing cells in BHIS medium were washed and suspended at time zero into anoxic glucose medium. Strains: parent (BT5482 Δtdk), Δpfor (MK508), Δpfl4738 (MK550), Δpfl2955 (MK494), Δfdx (MK532), Δrnf (MK520), Δhyd1834 (MK540), Δhyd3472 (MK500), Δpfl4738 Δpfl2955 (MK644), Δpfl4738 Δpfor (MK635), Δpfor Δpfl2955 (MK630), and Δfum (LZ62). Error bars represent the standard error of the mean of three biological replicates.
We then evaluated the roles of PFOR and PFL. Mutants lacking either or both of the pfl alleles (pfl4738 and pfl2955) were able to grow, as were single mutants lacking pfor. A Δpfor Δpfl4738 double mutant was generated in complex medium that would allow compensatory assimilation of substrates downstream of pyruvate, thereby ensuring a route of acetyl-CoA formation. This double mutant could not grow in minimal glucose media [Figure 3A]. These data suggested that during carbohydrate fermentation, PFOR and PFL4738 catalyze alternative routes of an essential pyruvate conversion to acetyl-CoA.
Measurements confirmed that formate is secreted into the medium of wild-type cells. This formate was virtually absent from cultures of the Δpfl4738 mutant but was unchanged in that of the Δpfl2955 strain, identifying PFL4738 as the key pyruvate:formate lyase in the cell. Notably, the production of formate was more than doubled in the Δpfor mutant [Figure 4A].
Figure 4. Pyruvate dissimilation is split between PFL4738 and PFOR.
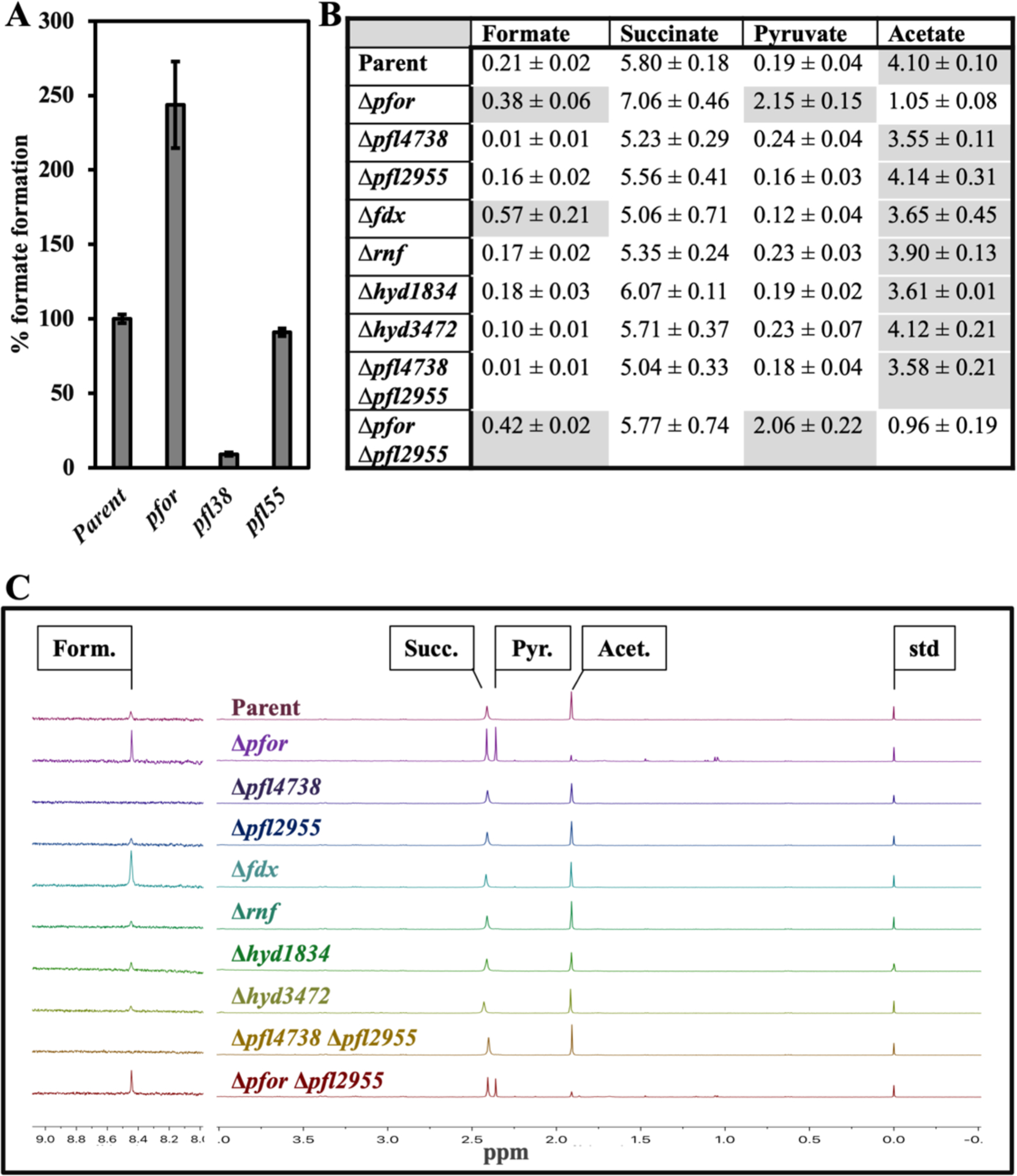
Cells were cultured overnight in defined glucose media, and the concentrations of accumulated fermentation products were determined. (A) Formate was determined by enzyme-linked assay. The data identify PFL4738 as the functional pyruvate:formate lyase. (B) The mean values of integrated peak areas from (B) for formate, succinate, pyruvate and acetate, derived from three experiments. High values of each column are shaded. (C) End products were determined by NMR analysis. The Δpfl4738 Δpfor and Δfum mutants are not included because they were unable to grow in this medium. The formate signal is amplified 13-fold here for better representation. Note that Δpfor and Δfdx strains produce more formate. Form., formate; succ., succinate; pyr., pyruvate; acet., acetate; std, DSS standard. Strains: parent (BT5482 Δtdk), Δpfor (MK508), Δpfl4738 (MK550), Δpfl2955 (MK494), Δfdx (MK532), Δrnf (MK520), Δhyd1834 (MK540), Δhyd3472 (MK500), Δpfl4738 Δpfl2955 (MK644), and Δpfor Δpfl2955 (MK630). Error bars represent the standard error of the mean of three biological replicates.
NMR analysis supported these observations: Formate was detected in the culture supernatants of all strains except those lacking pfl4738 [Figure 4B and 4C]. Again, the formate levels were elevated in Δpfor mutants. Further, the latter mutants generated less acetate and excreted excess pyruvate, suggesting that PFL4738 could not fully compensate for the absence of PFOR. Collectively, these data indicate that when wild-type cells ferment glucose there is substantial flux through both of these two enzymes.
It is interesting that neither of the hydrogenase mutants phenocopied the PFOR mutant, as they excreted normal amounts of formate, pyruvate, and acetate [Figure 4B and 4C]. Hydrogenases, including the Fe-Fe-type enzyme (Mulder et al., 2011) encoded by hyd1834, are sensitive to oxidation, but these genetic data show that hydrogenase damage by oxidants would not be enough to block central metabolism. At the same time, the rnf mutant also exhibit a wild-type product formation. We surmise that when PFOR reduces ferredoxin/flavodoxin, these electron carriers can be re-oxidized by either hydrogenase or Rnf. We did not attempt to produce the multiple mutants that would be needed to confirm this idea. We also noted that although the fdx mutant exhibited higher production of formate, consistent with a role in PFOR turnover, it did not generate elevated levels of pyruvate nor diminished levels of acetate. The result indicates that PFOR can use other moieties, possibly flavodoxins, as electron acceptors in its catalytic cycle.
For the purposes of understanding oxygen toxicity, these data establish that the inactivation of fumarase in the succinate pathway, or of both PFOR and PFL in the acetate pathway, would be sufficient to block cell growth.
Molecular oxygen itself inactivates PFOR.
As detailed in the Introduction, the aeration of B. thetaiotaomicron creates bottlenecks at two points in catabolism (Pan & Imlay, 2001). Succinate production is blocked due to the inactivation of fumarase. The iron-sulfur cluster of this enzyme is vulnerable to univalent oxidants. The second-order rate constants for inactivation are much greater for O2− (106 M−1 s−1) than for H2O2 (103 M−1 s−1) or molecular oxygen (100 M−1 s−1) (Lu et al., 2018). It was determined that in aerated B. thetaiotaomicron, fumarase is inactivated by the excessively high concentrations of superoxide (Lu et al., 2018). This superoxide also poisons other enzymes of the dehydratase class, as well as mononuclear enzymes with solvent-exposed Fe(II) cofactors.
Aeration also poisons the acetate branch of central metabolism. Pyruvate is excreted, indicating that pyruvate dissimilation is impaired (Pan & Imlay, 2001). According to the present analysis, this result must indicate that both PFL and PFOR become inactive. The oxygen sensitivity of PFL is well-understood: Molecular oxygen, which is a di-radical, rapidly adducts the glycyl radical of PFL, forming a hydroperoxyl radical species that ultimately cleaves the polypeptide (Zhang et al., 2001). Enzymes of this class cannot operate inside fully aerobic cells.
The PFOR enzymes of most (Ragsdale, 2003), but not all (Vita et al., 2008, Yoon et al., 1997, Kerscher & Oesterhelt, 1981, Baughn et al., 2009), microbes are sensitive to oxygenation. The B. thetaiotaomicron enzyme completely loses activity within 90 min of aeration [Figure 5] . We wished to determine whether the key oxidant was molecular oxygen, superoxide, or hydrogen peroxide. To do so, we compared the rates of inactivation inside B. thetaiotaomicron mutants that lacked enzymes to scavenge superoxide (SOD−) or hydrogen peroxide (Hpx−: ΔkatE ΔahpCF Δrbr1 Δrbr2) [Figure 5]. The oxidant-sensitive dehydratases and mononuclear enzymes are inactivated very rapidly in such mutants (Lu et al., 2018); however, the elevated levels of superoxide and H2O2 did not alter the rate of PFOR inactivation. As further confirmation, the fifteen-fold overexpression of SOD did not exert any protective effect. These data indicate that molecular oxygen, rather than either partially reduced species derived from it, is the oxidant that inactivates PFOR.
Figure 5. The rate of PFOR inactivation in aerated cells is independent of the intracellular concentrations of H2O2 and superoxide.
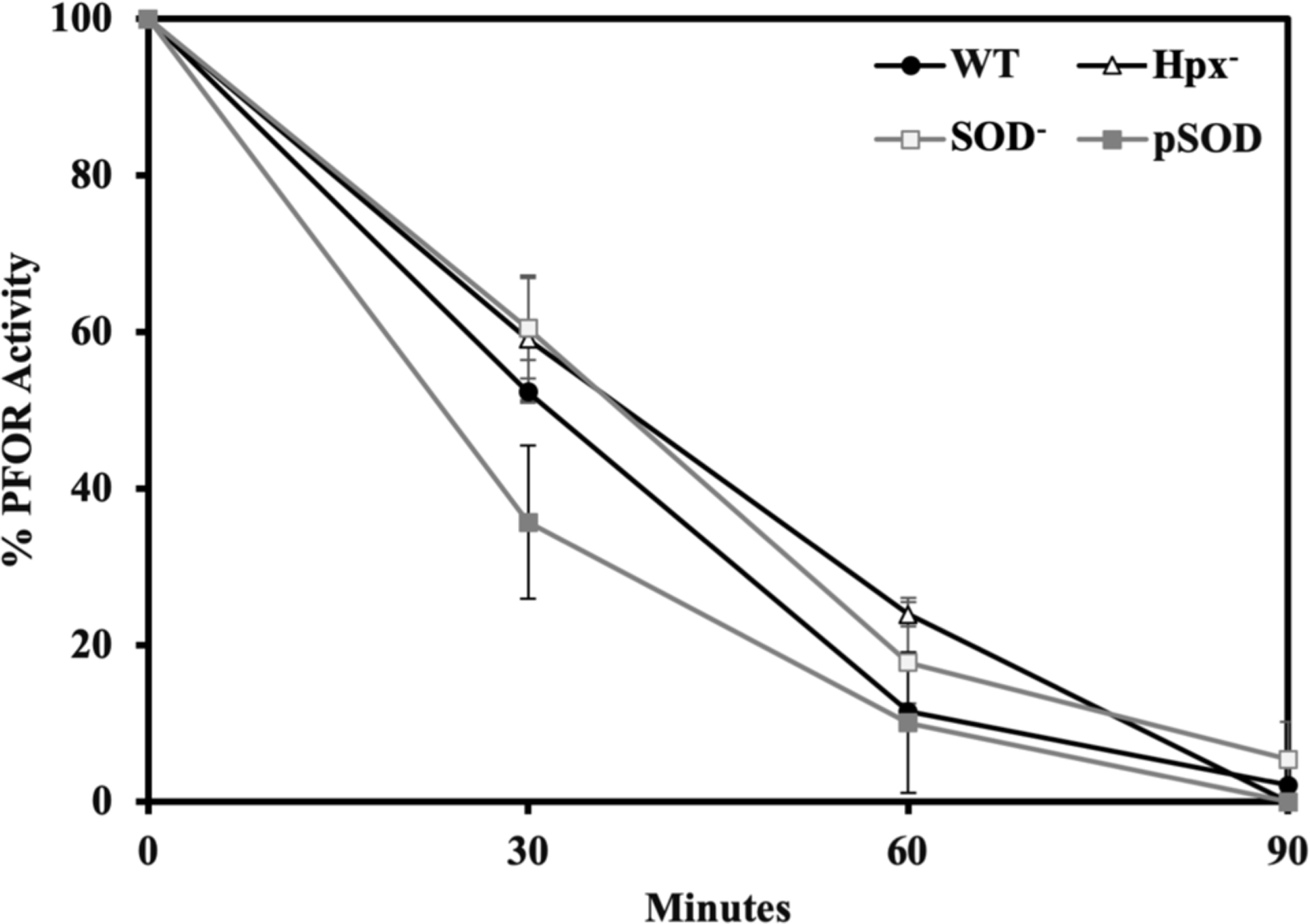
Cells growing exponentially in anoxic BHIS media were washed, resuspended in glucose buffer containing chloramphenicol, and aerated. At intervals aliquots were returned to the anaerobic chamber and PFOR activity was assayed. Strains: WT (BT5482), Hpx− (SM135), SOD− (LZ01), and pSOD (LZ200). Error bars and values after ± represent the standard error of the mean of three biological replicates.
PFOR employs a thiamine cofactor to catalyze pyruvate cleavage; the electrons from the fractured bond are then conducted through a series of three low-potential iron-sulfur clusters to the enzyme surface, where they are transferred to ferredoxin (Ragsdale, 2003). Others have noted that the inactivation of the enzyme in aerobic buffers occurs concomitant with cluster bleaching, suggesting that over-oxidation of one or more clusters may be responsible for the loss of activity (Vita et al., 2008, Menon & Ragsdale, 1997, Pieulle et al., 1995). We observed the same. Reactive oxygen species can arise in such experimental systems, making the identity of the oxidant ambiguous. However, we found that neither the addition of SOD nor of catalase was able to protect PFOR in vitro: with or without them, aeration reduced the activity by half in 20 min (data not shown).
Both H2O2 and superoxide must bind directly to an iron-sulfur cluster in order to oxidize it; the clusters of dehydratases have an open coordination site in order to bind substrate, and therefore they are vulnerable to attack by these oxidants. In contrast, molecular oxygen is able to accept electrons from a distance by outer-sphere transfer (Goldstein et al., 1993). Crystal structures of PFOR from Desulfovibrio africanus suggest that its redox clusters are fully coordinated by cysteine residues and are occluded by polypeptide (Vita et al., 2008, Chabriere et al., 1999). To examine this point under more forcing conditions, we exposed PFOR to very high doses of H2O2 both in the Hpx− mutant and in vitro [Figure 6]. Even 1 mM H2O2 produced only a modest amount of inactivation, corresponding to a rate constant of 0.8 M−1 s−1. In contrast, enzymes like fumarase with exposed iron-sulfur clusters are inactivated with rate constants near 3000 M−1 s −1 (Lu et al., 2018). Intracellular doses of H2O2 in real-world habitats probably never reach 10 micromolar (Imlay, 2013)—in fact, 0.2 micromolar is high enough to trigger stress responses in bacteria (Aslund et al., 1999, Seaver & Imlay, 2001)—and so it is clear that PFOR is effectively resistant to H2O2. Because H2O2 is an especially rapid oxidant of low-potential iron atoms, this result seems to confirm that the clusters in PFOR are not accessible to it. This feature would also explain the resistance in vivo to superoxide. (Superoxide sensitivity could not be independently tested in vitro, as O2− cannot be generated in the absence of molecular oxygen.) The slight sensitivity of PFOR to millimolar H2O2 is consistent with its direct oxidation of amino acid residues (Imlay, 2002a); this matter was not examined further.
Figure 6. Physiological (<10 μM) concentrations of H2O2 cannot inactivate purified PFOR.
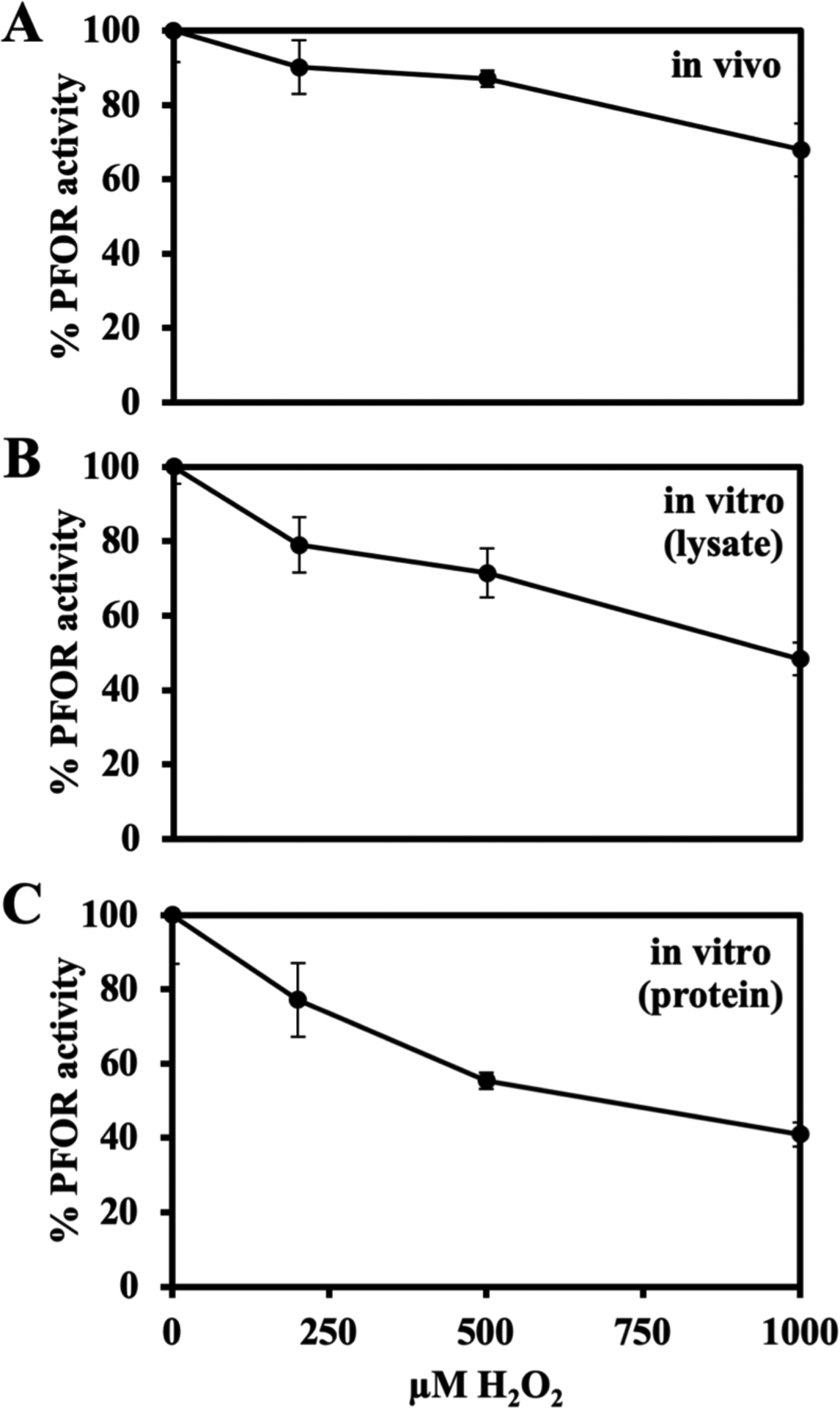
All cell growth, protein purification and enzyme assays were performed in the anaerobic chamber; see Materials & Methods for details. (A) Exponentially growing Hpx− (SM135) cells were washed with and resuspended in Tris buffer and incubated with H2O2 for 15 minutes at 4 oC.. PFOR activity was then assayed. (B) In vitro: Cell lysates were prepared from Hpx− cells. Lysates were incubated with H2O2 for 15 minutes at 4 °C, and PFOR activity was assayed. (C) The purified B. thetaiotaomicron PFOR was incubated with H2O2 for 15 minutes at 4 °C and then assayed. Error bars represent the standard error of the mean of three biological replicates.
These data show that in aerated B. thetaiotaomicron, PFOR is directly inactivated by molecular oxygen. This event, combined with the damage to PFL, eliminates the capacity to degrade pyruvate. The consequence would be a general bottleneck in the processing of carbohydrate, a diminution of ATP yield, and an inability to produce the essential metabolite acetyl-CoA. It is striking that one branch of metabolism is poisoned by superoxide, whereas the other is poisoned by oxygen itself.
Unlike fumarase, PFOR cannot be reactivated.
Previous studies in E. coli have shown that bacteria have the capacity to repair damaged iron-sulfur clusters, so that the activity of these enzymes in vivo represents the steady-state balance between the rates of damage and repair (Djaman et al., 2004, Gardner & Fridovich, 1992, Imlay, 2013). When B. thetaiotaomicron cells were aerated for different lengths of time, both fumarase and PFOR were inactivated [Figure 7]. After anoxia was restored, fumarase activity reappeared, despite the presence of chloramphenicol that blocked new protein synthesis. The activity could also be restored in vitro if the lysates of aerated cells were incubated with Fe(II) and DTT, confirming that aeration left the cluster in a [3Fe-4S]+ form (Jang & Imlay, 2007, Jang & Imlay, 2010, Gardner & Fridovich, 1992).
Figure 7. Unlike fumarase, oxidized PFOR cannot be reactivated in vitro or in vivo.
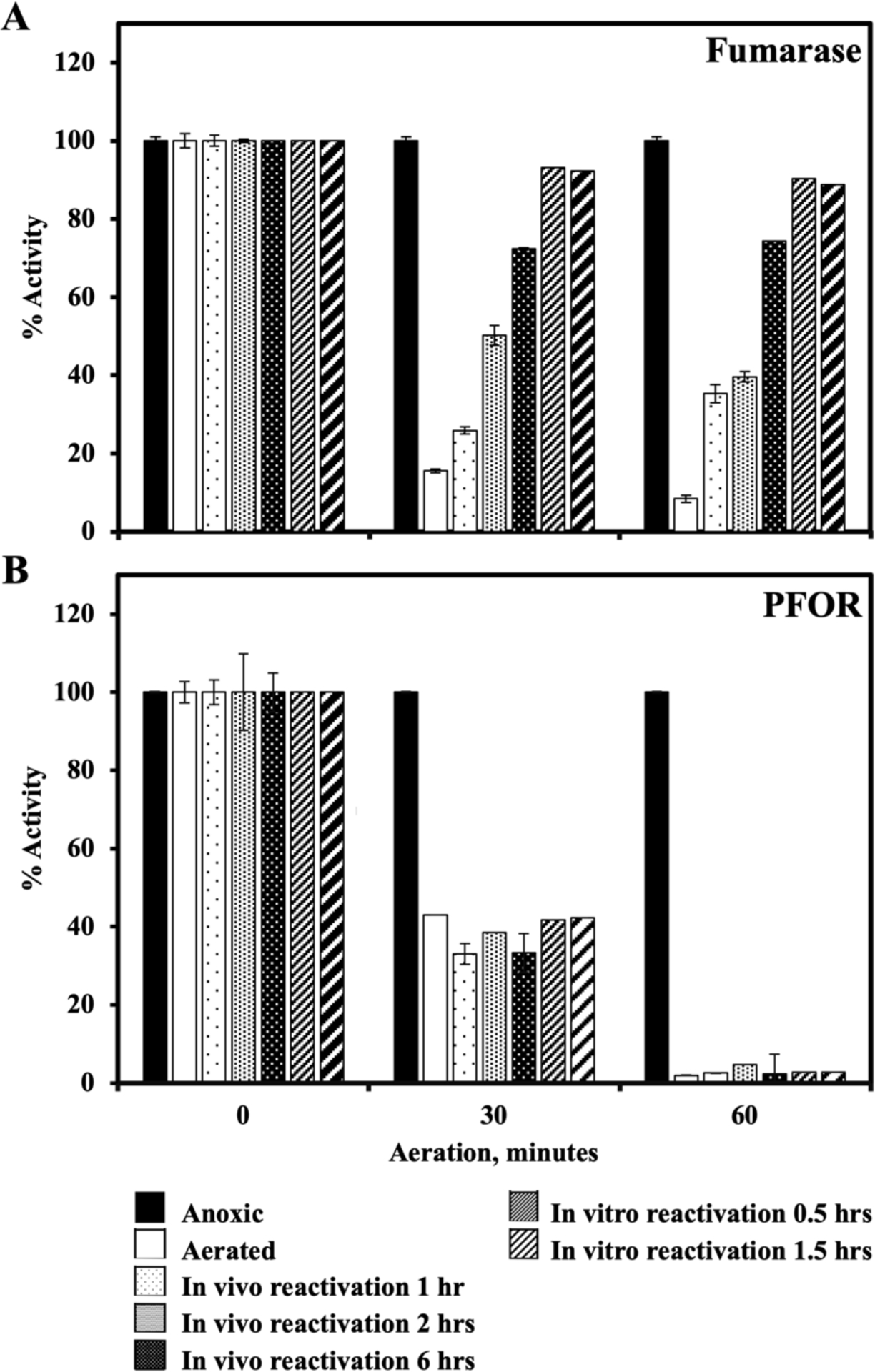
WT (BT5482) cells were grown in anoxic BHIS (no cysteine). Cells were centrifuged, suspended in Tris buffer containing glucose and chloramphenicol, and divided among 3 flasks. These were aerated for 0, 30 min or 60 min. Samples were removed to the anaerobic chamber, and the activities of fumarase (A) and PFOR (B) were measured in the same lysate. To test in vivo reactivation, the aerated cells were washed and resuspended in anoxic BHIS containing chloramphenicol, and at different time points cells were lysed and the activities of PFOR and fumarase were measured. For in vitro reactivation, the anoxic lysates of aerated cells were incubated with 0.5 mM ferrous ammonium sulfate and 5 mM DTT for 30 or 90 minutes, and the enzyme activities were measured. This experiment is representative of multiple experiments with different aeration and reactivation times, all of which yielded the same results. Error bars represent standard error of two technical replicates.
Unlike fumarase, inactivated PFOR could not be reactivated, either in vivo or in vitro [Figure 7]. A reasonable explanation is that the damaged cluster is not solvent exposed, as suggested earlier, and therefore that it is not accessible to either Fe and DTT or to any cellular repair process. Alternatively, enzyme oxidation may be accompanied by polypeptide damage. The nature of the injury remains to be determined. However, because damage is irreversible, the vulnerability of this pathway is amplified.
Oxygen concentration determines the level of PFOR activity.
If oxygen directly damages the enzyme, it is likely that the rate of damage is proportionate to oxygen concentration. This prediction was confirmed [Figure 8]. The response to damage was again approximately the same as that of fumarase. The latter enzyme is damaged by superoxide rather than oxygen itself, but the rate of superoxide production is itself proportionate to oxygen level (Lu & Imlay, 2017). One implication is that at low but non-zero levels of oxygen, both enzymes—and the pathways to which they belong—may retain a substantial degree of function.
Figure 8. PFOR inactivation depends upon the concentration of oxygen.
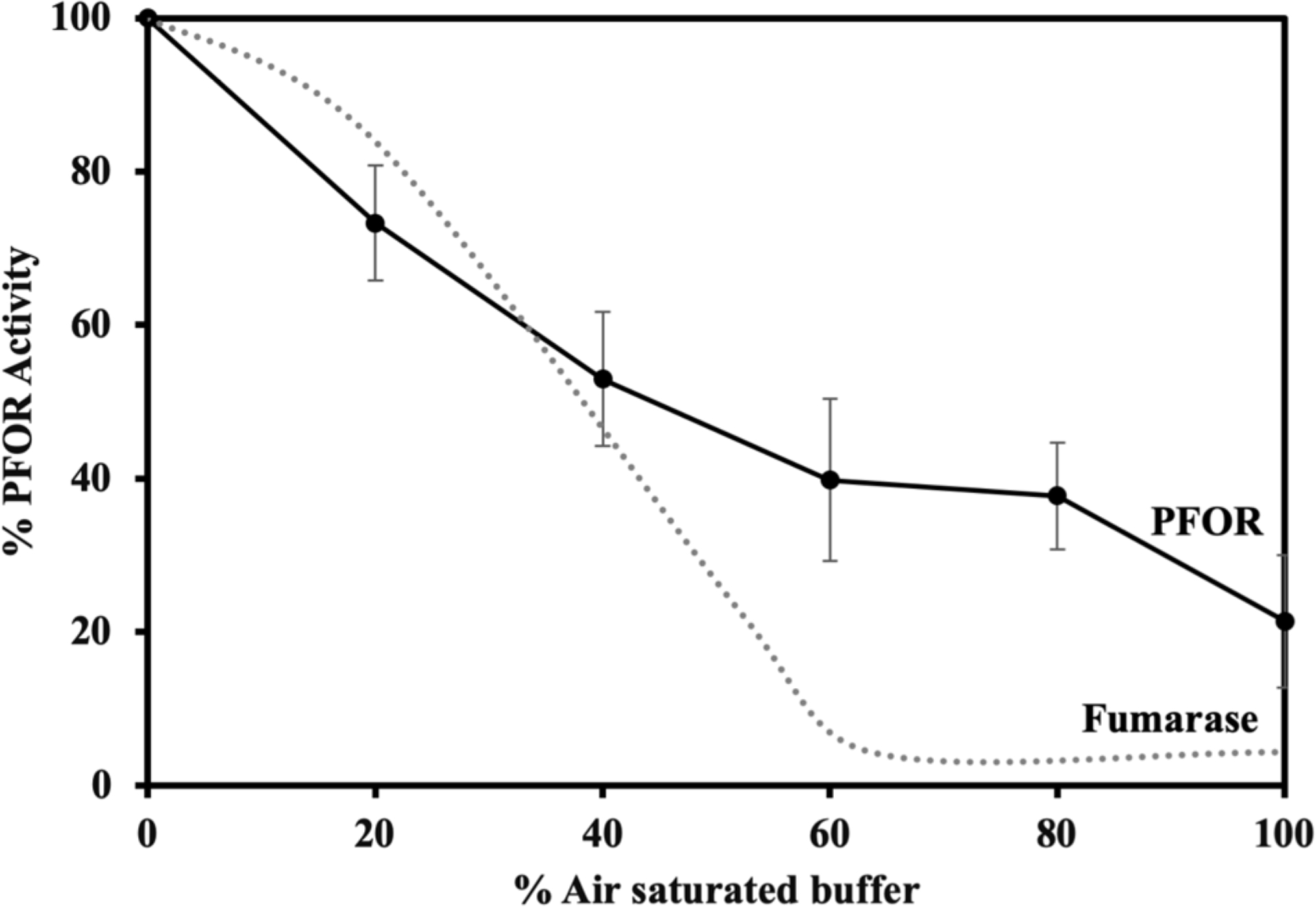
Exponentially growing WT cells (BT5482) in anoxic BHIS were washed, suspended in Tris buffer containing glucose and chloramphenicol, and incubated with the indicated percent of air saturation for 1 hr at 37 °C in tightly closed bottles. Cells were then harvested and their PFOR activities determined. The dotted line represents prior data (Lu et al., 2018) showing that the fumarase inactivation by superoxide parallels PFOR inactivation by oxygen. Error bars represent the standard error of the mean of six biological replicates.
We failed to identify the primary source of ROS inside aerated cells.
To arrive at a complete view of how oxygen poisons B. thetaiotaomicron, we will also need to understand how aeration generates disabling doses of superoxide. Endogenous superoxide formation cannot be directly quantified, but its dismutation product, H2O2, can be measured as it effluxes from Hpx− strains that cannot scavenge it (Mishra & Imlay, 2013). It this way it was determined that aerated B. thetaiotaomicron produces 10 times more H2O2 than does aerated E. coli (Lu et al., 2018, Mishra & Imlay, 2013). This likely reflects a similar increase in O2− formation, and the O2− stress is further worsened because the SOD titers in B. thetaiotaomicron are only 1/3 those of E. coli. In aggregate, a thirty-fold higher concentration of O2− may result (Lu et al., 2018).
We hypothesized that the ROS might arise primarily from the autoxidation of enzymes at the pyruvate node of central metabolism. Our logic was that this element of B. thetaiotaomicron metabolism is substantially different from that of E. coli, that the electron flux rate is high enough to accommodate the high rate of ROS formation, and that this section of metabolism involves electron movement through low-potential cofactors that might be easily oxidized to release ROS. However, we found that the yield of H2O2 was not substantially changed by mutations that eliminated PFOR, PFL, ferredoxin, hydrogenase, or Rnf [Figure 9]. This step of oxygen toxicity remains unsolved.
Figure 9. None of the tested enzymes are the primary source of H2O2 production in B. thetaiotaomicron.
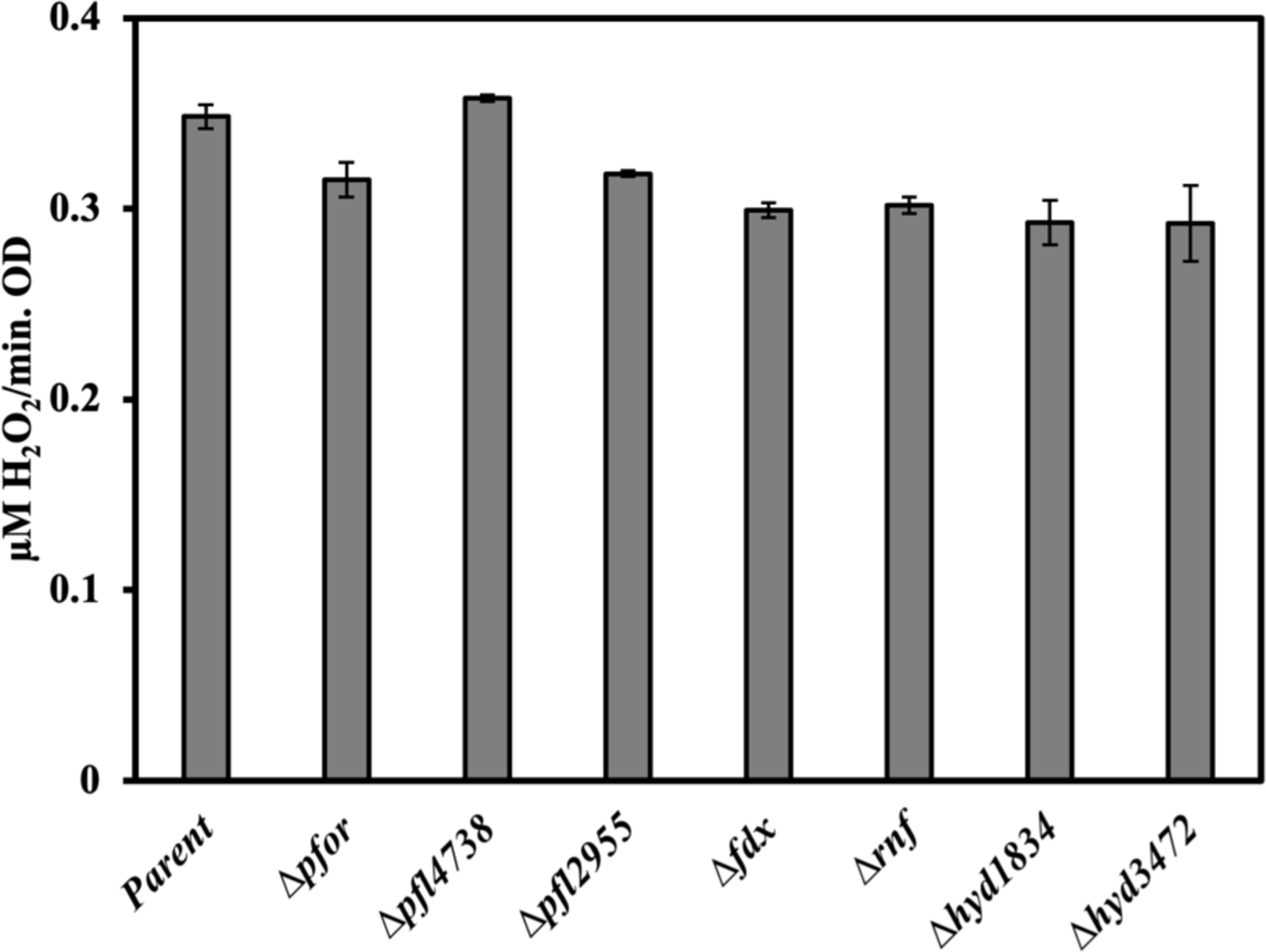
The Hpx− strain (SM135) and its mutant derivatives were grown in BHIS media, washed, resuspended in Tris buffer containing glucose and chloramphenicol, and then aerated at 37 °C. The rate of H2O2 production was measured as described. Strains: parent (SM135 aka Hpx−), Hpx− Δpfor (MK592), Hpx− Δpfl4738 (MK556), Hpx− Δpfl2955 (MK562), Hpx− Δfdx (MK654), Hpx− Δrnf (MK584), Hpx− Δhyd1834 (MK570), Hpx− Δhyd3472 (MK580). Error bars represent the standard error of the mean of three biological replicates.
Discussion
Oxidative stress and the central metabolism of B. thetaiotaomicron
While oxygenation of the atmosphere led to the emergence of aerobes, anaerobic bacteria continue to use their low potential biochemical pathways in the remaining anoxic habitats. They dominate these niches. In the gut, for example, Bacteroides can be the majority organisms, whereas facultative anaerobes are relatively niche species (Wexler & Goodman, 2017).
Nevertheless, transient oxygen exposure is likely a not-infrequent occurrence. Soil-dwelling bacteria, for example, may experience a rush of oxygen when rivulets of rainwater penetrate into the anoxic zones of soil (Šťovíček et al., 2017, Morris & Schmidt, 2013). An oxygen gradient from the epithelium into the intestinal lumen likewise confronts resident anaerobes with some degree of exposure (Albenberg, 2014, Espey, 2013). Signs of these encounters are manifest in the anaerobe genomes, which universally encode superoxide dismutases and/or reductases alongside catalases and/or peroxidases. Moreover, many presumptive anaerobes also possess oxygen-consuming cytochrome bd oxidases (Korshunov & Imlay, 2010, Morris & Schmidt, 2013) and rubredoxin oxidases (Cypionka, 2000). Indeed, Baughn and Malamy demonstrated that the respiratory action of cytochrome bd oxidase improves B. fragilis cell economy during periods of microoxia (Baughn & Malamy, 2004). The evolving view is that the familiar dichotomy of aerobe and anaerobe is more reflective of experimental considerations than of natural circumstances. In lab cultures it is far easier to establish air-saturated or oxygen-free growth conditions than it is to sustain intermediate oxygen levels, particularly as oxygen-consuming cells grow exponentially.
The Bacteroides species that we focused upon in this study must be constrained to microoxic and anoxic habitats, as signified by its reliance upon a glycyl-radical-based ribonucleotide reductase (NrdD) that cannot function in the presence of significant oxygen (Fontecave et al., 2002). We regard the absence of oxygen-tolerant NrdAB or NrdEF (Nordlund & Reichard, 2006) as a concession to the anaerobic lifestyle that is imposed upon it by its central metabolic strategy. As shown in this study, its mechanisms of pyruvate dissimilation depend upon two enzymes, PFL and PFOR, that are quickly poisoned by elevated oxygen. The virtue of these enzymes is that they can fracture pyruvate without generating NADH, which would otherwise disrupt the cellular redox balance and ultimately diminish the ATP yield of the fermentation. The features that enable this chemistry are precisely those which are incompatible with oxygen: for PFL, the glycyl radical that enables radical-based cleavage; for PFOR, the low-potential clusters that ultimately enable electron flow to hydrogenase. Thus the attributes that optimize B. thetaiotaomicron performance in anoxic environments are exactly those that forbid its growth in oxic ones.
The key finding of this study is that oxygen poisoning of B. thetaiotaomicron is simultaneously imposed by both superoxide and molecular oxygen. The superoxide stress derives from rapid endogenous O2− formation in a cell containing a modest amount of SOD. The present study tracked the inactivation of fumarase, but a previous study demonstrated simultaneous damage to aconitase, isopropylmalate isomerase, ribulose-phosphate epimerase, and peptide deformylase (Lu et al., 2018). Those injuries would impair the synthesis of the 2-oxoglutarate and branched-chain families of amino acids, NADPH production via the pentose-phosphate pathway, and the essential maturation of formylated proteins. Additional targets doubtless exist; therefore, the metabolic footprint of superoxide stress is large. These enzymes are no more intrinsically sensitive to superoxide than are their homologs in oxygen-tolerant bacteria; instead, the root issue is that superoxide is generated in such high amounts when this anaerobe is aerated. We presume that this superoxide arises when oxygen steals electrons from redox enzymes, as in E. coli. The fact that ROS formation occurs roughly in proportion to oxygen concentration means that the electron-leaking enzymes lack saturable oxygen-binding sites and that the reactions are adventitious. It also means that superoxide stress will fall within tolerable bounds at lower oxygen concentrations, suggesting that this anaerobe may continue to grow even close to the intestinal margins.
It might seem that the bacterium would be well-served if it compensated for the high superoxide flux by synthesizing at least as much SOD as does E. coli. However, it appears that the SOD titer is calibrated so that superoxide toxicity occurs at roughly the same oxygen concentration that poisons pyruvate catabolism. Therefore, no benefit would accrue from a higher scavenging rate. This logic suggests that the ultimate determinant of oxygen sensitivity is not likely to be superoxide stress but rather the impairment of PFOR and PFL. Further, because both the damage done by oxygen, and that produced by superoxide, occur at rates proportionate to oxygen concentration, the balance between ROS- and O2-mediated damage will likely pertain to all oxygen levels.
How is pyruvate dissimilation restored after a period of aeration?
In the growth conditions of this study, pyruvate was processed predominantly by PFOR but also by PFL. The value of having both enzymes was not obvious; in fact, either alone sufficed to enable good growth. We note that PFOR can direct electrons, via Rnf and transhydrogenase (Zhang et al., 2017), to supply NADPH for biosynthesis. It can also dissipate reducing equivalents via hydrogenase under acidic conditions in which formate production is undesirable. Conversely, in natural habitats hydrogenase activity can be inhibited by the accumulation of H2, either for kinetic or thermodynamic reasons (Fourmond et al., 2013)—which would furnish a raison d’etre for PFL. To understand the roles of the two enzymes, it will be valuable to quantify their respective fluxes during growth with different substrates and under a range of environmental conditions that might impinge upon this process.
Both enzymes lose activity in aerated cells. PFL is extremely sensitive to oxygen in vitro, with inactivation occurring in seconds if its buffer is fully aerated (Knappe et al., 1994). As would be expected, inactivation occurs more slowly at lower oxygen levels (Yamada et al., 1985), raising the prospect that glycyl-radical enzymes may remain functional in microoxic environments. Interestingly, groups that have studied the PFL of E. coli have suggested two routes by which the enzyme may be protected when this facultative bacterium encounters oxygen. The first is that before oxygen can cleave the enzyme, PFL may be deactivated via reduction of its glycyl radical to a standard, albeit catalytically inactive, glycyl residue. This model suggests that once oxygen levels decline, the PFL-activating system will regenerate the radical. Surprisingly, biochemical experiments implicated alcohol dehydrogenase as a deactivating system (Kessler et al., 1992, Asanuma et al., 2004); however, another group failed to reproduce that effect (Nnyepi et al., 2007). This issue remains unresolved. The second mechanism of protection is that a protein named GrcA may bind to the catalytic fragment of oxygen-cleaved PFL and serve as an ancillary radical-generating system (Wagner et al., 2001). B. thetaiotaomicron lacks any GrcA homolog, and so the fate of PFL during aeration remains uncertain.
We suspect that oxygen poisons PFOR by over-oxidizing one of its clusters to an unstable state. This appraisal is based on the bleaching of color. Both thiazolium and substrate radicals are formed during the catalytic cycle (Ragsdale, 2003) and in principle might also react with oxygen—but that cannot be the full answer, because the enzyme is sensitive to oxygen in vitro even in the absence of substrate. The [4Fe-4S] clusters normally shift between +1 and +2 valences as electrons flow through them; oxidation to a +3 valence may destabilize the clusters, as it does the clusters of the fumarase-family enzymes. Whether the notional high-valence cluster would disintegrate or abstract an electron from surrounding polypeptide is unknown. Similar uncertainties exist regarding the mechanism by which oxygen inactivates hydrogenase, despite extensive efforts (Kubas et al., 2016). The data of this study show that the physiological oxidant of PFOR is molecular oxygen itself. This fact is not predicated on thermodynamics, as molecular oxygen (Eo’ = −0.16 V) is a much poorer univalent oxidant than either H2O2 (Eo’ = +0.38 V) or O2− (Eo’ = +0.94 V) (Imlay, 2003). However, H2O2 must directly bind an iron atom in order to oxidize it, because the stability of the ferryl product pulls the reaction forward (Goldstein et al., 1993); for this reason, H2O2 is unable to oxidize clusters that are buried in polypeptide (Jang & Imlay, 2007). Superoxide, like molecular oxygen, is a radical that in principle might receive electrons by outer-sphere transfer; however, dissociated superoxide is an anion at physiological pH and in that form cannot directly accept another electron. The oxidation of dehydratase clusters by O2− probably proceeds when electrostatics drive the formation of a superoxide-iron complex, with oxidation occurring only when the complex is momentarily protonated by bulk solvent (Flint et al., 1993b). From our data we infer that the PFOR clusters are sufficiently occluded that neither H2O2 nor O2− can directly ligand them, so only molecular oxygen, which is a capable outer-sphere oxidant, can oxidize them. Interestingly, oxygen is too slow an oxidant of fumarase to be consequential in vivo (Lu et al., 2018), which reflects the higher potential of its cluster (Flint et al., 1993b). In contrast, because the working +2/+1 potentials of the PFOR clusters are set at quite low values— −390, −515, and −540 mV for the D. sulfovibrio enzyme (Pieulle et al., 1995)—the +3/+2 potentials may also be relatively low, enhancing their vulnerability to over-oxidation by oxygen. This is conjecture; detailed work will need to be done both to establish the clusters as the initial sites of oxidation, and then to relate their oxidizability to their potentials.
Very-low-potential metal centers also underpin other difficult anaerobic biochemistry— the iron-molybdenum cofactor of nitrogenase (Imlay et al., 2019), the Co(I) center of methyl-CoM reductase (Scheller et al., 2010), the [4Fe-4S] clusters of Clostridial archerases (Buckel & Golding, 2006)—and these enzymes are poisoned in oxic cells. Once again, the key question is not whether oxygen can inactivate these enzymes—it can—but whether oxygen is the primary physiological oxidant, or whether ROS overtake it. With their low potentials and occluded active sites these enzymes perhaps resemble PFOR more than fumarase. If so more damage may be generated in vivo by molecular oxygen rather than by ROS, again explaining why microbes that use such enzymes maintain relatively low titers of scavenging enzymes.
The microaerophilic behavior of Helicobacter pylori and Campylobacter jejuni may reflect the degree of oxygen tolerance of their PFOR enzymes (Taylor & Kelly, 2019, Kelly, 1998). Interestingly, not all PFOR isozymes are oxygen-sensitive. Oxygen-tolerant enzymes have been found operate in aerobic thermophilic and halophilic bacteria (Yoon et al., 1997, Kerscher & Oesterhelt, 1981), and aerobic Mycobacterium tuberculosis employs the related 2-oxoglutarate:ferredoxin oxidoreductase (Baughn et al., 2009). It is not yet clear why these versions of the enzyme are resistant. The PFOR of Desulfovibrio africanus is substantially (~80%) inhibited by oxygen, which triggers the formation of a disulfide bond that locks an extra loop of polypeptide over the cluster proximal to the thiamine moiety (Vita et al., 2008). Upon re-reduction of the disulfide, full activity is regained. The authors proposed that this maneuver protects the enzyme by obstructing the approach of oxygen to the cluster, and they showed that mutants that lack the loop are fully oxygen sensitive. The contrast with the B. thetaiotaomicron PFOR, which cannot be reactivated after oxidation, highlights the value of the reversible deactivation of the D. africanus enzyme.
Other examples exist of enzymes that have evolved some degree of oxidant resistance. These include the aerobic-type ribonucleotide reductases, certain Ni-Fe hydrogenases, and dehydratases that use [2Fe-2S] rather than labile [4Fe-4S] clusters (Imlay et al., 2019). It is likely that trade-offs exist. Data indicate that the spinach [2Fe-2S] dihydroxyacid dehydratase, for example, turns over at < 10% the rate of the conventional [4Fe-4S] enzyme (Flint & Emptage, 1988, Flint et al., 1993a). If a similar price is exacted for stabilizing PFOR, then it would make sense that bacteria that primarily dwell in anoxic habitats would retain the oxygen-sensitive enzyme and simply ride out periods of oxygen exposure.
If oxygen-exposed cells lack any capacity to reactivate PFL or PFOR, it is unclear how they so quickly resume growth when anoxia returns. While de novo enzyme synthesis would restore activity, such synthesis seems implausible in a cell whose core metabolism is frozen. We regard this question as a very important one if we are to understand the behavior of anaerobes in natural environments.
Materials and Methods
Reagents:
All antibiotics (ampicillin, erythromycin, gentamicin and chloramphenicol), 5′ fluorodeoxyuridine, D-glucose, methyl viologen, malate, hemin, pyruvate, coenzyme A, horseradish peroxidase, Candida boidinii formate dehydrogenase, and 30% hydrogen peroxide were purchased from Sigma. Amplex UltraRed reagent was obtained from Life Technologies. 3-(Trimethylsilyl)-1-propane sulfanic acid sodium salt (DSS) and deuterium oxide were purchased from Aldrich Chemistry. BBL brain heart infusion medium, bacto tryptone, bacto agar and bacto yeast extract were purchased from Difco.
Bacterial strains, plasmids and growth conditions
Bacterial strains, plasmids and primers used in this study are listed in Supplementary Tables 1–3. BT5482 Δtdk was used as the parent strain in building the deletion mutants; the partial deletion of the tdk gene (thymidine kinase) is employed in the requisite selections (Koropatkin et al., 2008). All anoxic growth and assays were done using anoxic buffers, media, and reaction components in a Coy anaerobic chamber (Coy Laboratory Products, Inc.) under 85% N2, 10% H2, and 5% CO2.
Brain heart infusion (BHIS) medium contained 37 g/L of brain-heart infusion, 7.7 μM hemin, and 1 g/L of cysteine hydrochloride monohydrate (Bacic & Smith, 2008). Where indicated, BHIS medium without cysteine was used to prevent ROS formation via cysteine autoxidation during aeration (Korshunov et al., 2019, Bagiyan et al., 2003). The pH of BHIS was adjusted to 7. After autoclaving (30 minutes) the hot medium was transferred to the anaerobic chamber and kept there for at least 48 hours before use, ensuring full deoxygenation.
Minimal defined medium was based upon the recipe from Bacic and Smith, with minor changes (Bacic & Smith, 2008). The Mineral 3B solution contained 6.6 mM potassium dihydrogen phosphate, 15 mM sodium chloride, 100 μM magnesium (II) chloride hexahydrate, 180 μM calcium (II) chloride dihydrate, 4.2 μM cobalt (II) chloride hexahydrate, 50 μM manganese (II) chloride tetrahydrate, 9 mM ammonium chloride and 1.8 mM sodium sulfate. Separate solutions of 10 mM iron (II) sulfate heptahydrate, 0.2% L-methionine (14 mM), 0.5 g/L hemin (0.8 mM), 20% D-glucose (1.1 M), and 10% sodium bicarbonate were prepared and filter sterilized. The hemin solution was kept at 4 °C. The glucose and bicarbonate solutions were maintained in the anaerobic chamber. The bicarbonate solution was used within two weeks of preparation. To prepare 1 liter of defined minimal medium, 50 ml Mineral 3B, 10 ml of hemin, 10 ml of methionine, 1.5 ml of iron sulfate and 1 g of cysteine hydrochloride monohydrate were mixed to the final volume of 970 ml. The pH was adjusted to 7.1, and the solution was autoclaved for 30 minutes. The medium was immediately transferred to the anaerobic chamber. After it cooled, 10 ml glucose and 20 ml of bicarbonate were added. Antibiotics were used as 20 μg/ml erythromycin, 200 μg/ml gentamicin, 200 μg/ml chloramphenicol, and 200 μg/ml 5′-fluorodeoxyuridine (FuDR).
B. thetaiotaomicron cultures were grown at 37 °C in the anaerobic chamber. Exponential cultures were obtained by diluting overnight cultures to 0.01 OD600 and then incubating them until the culture reached approximately 0.1 OD600. Anoxic Tris buffer (50 mM, pH 7.5) was used in all wash steps unless stated otherwise. Anaerobic cell lysates were made by centrifuging cells (typically 10 ml per time point) and then resuspending them in about 7% of the original volume in Tris buffer. They were then sonicated in the anaerobic chamber for 90 seconds (3 seconds on, 3 seconds off). The lysates were centrifuged in the chamber for 10 minutes at 12,000 rpm at 4 °C, and the supernatant was immediately used in assays. Aerated cultures were shaken at 220 rpm and 37 °C in flasks with the media volume comprising not more than 10% of the flask volume. When indicated, 0.2% glucose was included to enable metabolism, and 200 μg/ml chloramphenicol was used to block new protein synthesis. E. coli cultures were grown in Luria–Bertani medium with 10 g/L of bacto-tryptone, 5 g/L of bacto-yeast extract, and 10 g/L of NaCl. Ampicillin (100 μg/ml) was used when necessary.
Construction of deletion mutants
The B. thetaiotaomicron mutants were derived from BT5482 Δtdk using a previously described method (Koropatkin et al., 2008). Tdk is involved in the pyrimidine salvage pathway, and it is used as a selection tool in the construction of mutants. Regions that flank the target gene (approximately 700 bp each) were amplified using PCR [Supplementary Table 3] and joined with SOEing PCR (Horton, 1995). The fragment was then ligated to the pExchange-tdk suicide plasmid (Koropatkin et al., 2008) and transformed into E. coli donor strains, BW19851 or WM6026. WM6026 needs 100 μg/ml DAP (diaminopimelic acid) to grow (Blodgett, 2007). Note that the media used for growing WM6026 should not contain cysteine or cystine, as they block DAP entry into the cell (Imlay et al., 2015). The conjugation process was started by mixing approximately equal densities of the donor E. coli containing the pExchange-tdk-flanking fragment plasmid and the recipient BT5482 Δtdk. The mixture was spread on a BHI plate (+DAP if needed) and incubated aerobically for 24 hours. The bacterial mass from the conjugation plate surface was collected, washed with BHI, and plated on BHI containing gentamycin and erythromycin. Gentamycin kills the donor E. coli. Erythromycin kills the B. thetaiotaomicron cells lacking the pExchange plasmid. Twenty-four single colonies form this plate were picked and re-streaked; these cells have gone through the first recombination event. Each colony was then grown separately overnight in BHIS. Equal amounts of the 24 cultures were pooled into one tube, and 10-fold serial dilutions were plated on BHIS containing 200 μg/ml FuDR and grown for 2–3 days. FUdR counter-selection selects for clones that have gone through the second recombination event and eliminated pExchange-tdk from the genome. Single colonies from these plates were re-streaked, and the loss of the target gene was confirmed by test primers and sequencing.
B. thetaiotaomicron PFOR overexpression
The 3,552 bp pfor gene (BT_1747) was inserted into pBAD/HisA (Invitrogen, catalog number V430–01) at the XhoI/KpnI cut site and transformed into TOP10 E. coli cells (Invitrogen). Cells were grown anaerobically with ampicillin to ~ 0.5 OD600 and induced with 2% arabinose for 5 hours. After centrifugation, the pellet in a tightly capped tube was frozen on dry ice in the anaerobic chamber and then transferred to a −80 °C freezer. The next day the frozen pellet was returned to the chamber, thawed, and resuspended in 10 ml anoxic Tris buffer. Celled were lysed by sonication and loaded on a His Gravitrap column (GE Healthcare) that was pre-equilibrated by ice-cold anoxic binding buffer (20 mM Tris HCl, 500 mM NaCl, 5 mM imidazole, pH 7.9). The column was washed by two rounds of ice-cold anoxic wash buffer (20 mM Tris HCl, 500 mM NaCl, 60 mM imidazole, pH 7.9). PFOR protein was then eluted with ice-cold anoxic elution buffer (20 mM Tris HCl, 500 mM NaCl, 500 mM imidazole, pH 7.9). The protein solution was about 90% pure based upon SDS-PAGE analysis. The purified PFOR lost activity if kept on ice or at −80 °C overnight; therefore, all assays were performed on the same day as purification.
Enzyme assays
The PFOR assay was performed at room temperature in 50 mM Tris, pH 7.5, containing 5 mM pyruvate, 0.5 mM coenzyme A, 2 mM MgCl2, 0.1 mM TPP, and 2 mM methyl viologen, as described (Yoon et al., 1999). The anoxic components were mixed in the anaerobic chamber in capped cuvettes and then transferred outside for the spectroscopic measurement of reduced methyl viologen at 587 nm (absorption coefficient of 9.8 mM−1 cm−1). The activity of fumarate was measured by following the conversion of 50 mM malate to fumarate at 250 nM (absorption coefficient of 1.62 mM−1 cm−1) in anoxic 50 mM Tris, pH 7.5 (Hill & Bradshaw, 1969).
The concentration of formate was measured in overnight cultures after the cells were removed by centrifugation. The formate level was determined by measuring the rate of NADH production at 340 nm (absorption coefficient of 6.22 mM−1 cm−1) by formate dehydrogenase and correlating it to formate concentration, using Lineweaver-Burk plots derived from standard samples (Fourmond et al., 2013). The assay was performed in oxic conditions at room temperature; the reaction mixture contained 50 mM Tris, pH 7.5, 1.2 mM NAD+, and 0.05–0.15 unit formate dehydrogenase (Sigma, F8649). For accurate measurements the supernatants were diluted to contain 0–1 mM formate.
H2O2 incubation assay
The ability of H2O2 to inactivate PFOR was tested in live cells, in extracts, and with purified protein. For the in vivo experiments, Hpx− cells were grown anaerobically in BHIS (no cysteine) from OD600 0.01 to 0.2. Cells were then washed, resuspended in Tris buffer, and incubated with different concentrations of H2O2 for 15 minutes at 4 °C. The cells were then washed with Tris buffer and sonicated, and the activity of PFOR was measured. For the testing of extracts, lysates of Hpx− cells were prepared after growth described above, without incubating the cells with H2O2. Aliquots of the lysate were then incubated with different concentrations of H2O2 for 15 minutes at 4 °C. The activity of PFOR was measured immediately. The use of Hpx− cells ensured that no catalase was present to degrade the H2O2. Purified PFOR from B. thetaiotaomicron was expressed and purified in E. coli TOP10 cells as described above. The protein was then incubated with different concentrations of H2O2 for 15 minutes at 4 °C, and activity was then assayed. Notably, in the experiments with cell extracts and purified PFOR, the H2O2 was not removed with catalase prior to the assay, to avoid the evolution of oxygen; therefore, the persistence of activity indicates that H2O2 does not even inhibit the enzyme.
H2O2 production assay
The parent strain (Hpx−) and different mutant derivatives were grown anaerobically in BHIS from OD600~0.01 to 0.1. Cells were then washed two times with 50 mM RT Tris, pH 7.5, and finally resuspended in anoxic 50 mM Tris pH 7.5 containing 0.2% glucose; the culture volume was no more than 10% of the flask volume. The flasks were removed from the anaerobic chamber, and cells were aerated for 30 minutes by shaking in a 37 °C water bath. Every 10 minutes a 1-ml aliquot was centrifuged, and the supernatant was frozen on dry ice. After the sample collection was complete, the samples were thawed, and the H2O2 concentration was immediately determined in a Shimadzu RF Mini-150 fluorometer using the Amplex Red/ horseradish peroxidase method (Messner & Imlay, 2002a).
NMR spectroscopy
All 1H-NMR experiments were performed at room temperature on a Varian VXR 500 spectrometer at 600 mHz using a 5 mm Nalorac QUAD probe in the School of Chemical Sciences NMR lab at University of Illinois at Urbana-Champaign. 3-(Trimethylsilyl)-1-propane sulfanic acid sodium salt (DSS) was used as the chemical shift reference and integration standard. The parent strain (BT5482 Δtdk) and different mutants were grown on defined minimal plates, precultured in defined minimal media to OD ~0.1 OD, and then inoculated into the same medium to 0.01 OD and cultured overnight to OD ~ 1. The next day, samples were centrifuged, and 900 μl of the supernatant was mixed with 100 μl of 50 mM DSS in D2O, to a final concentration of 5 mM DSS and 10% D2O. The data were normalized to the final culture OD and are representative of at least three experiments. The solvent signal was suppressed by the pre-saturation technique. Standard additions of known metabolites were used to confirm the identities of the end products. The quantification of the NMR data was done using the peak area of DSS at zero ppm as the standard for normalization.
Supplementary Material
Acknowledgments.
We thank Bill Metcalf for providing strains, and Dr. Dean Olson of the School of Chemical Sciences NMR Laboratory for experimental support. This work was supported by grant GM049640 from the National Institutes of Health.
References.
- Albenberg L, Esipova TV, Judge CP, Bittinger K, Chen J, Laughlin A, Grunberg S, Baldassano RN, Lewis JD, Li H, Thom SR, Bushman FD, Vinogradov SA, and Wu GD (2014). Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology. 147: 1055–63.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anjem A, and Imlay JA (2012) Mononuclear iron enzymes are primary targets of hydrogen peroxide stress. J. Biol. Chem 287: 15544–15556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanuma N, Yoshii T, and Hino T (2004) Molecular characteristics and transcription of the gene encoding a multifunctional alcohol dehydrogenase in relation to the deactivation of pyruvate formate-lyase in the ruminal bacterium Streptococcus bovis. Arch. Microbiol 181: 122–128. [DOI] [PubMed] [Google Scholar]
- Aslund F, Zheng M, Beckwith J, and Storz G (1999) Regulation of the OxyR transcription factor by hydrogen peroxide and the cellular thiol-disulfide status. Proc. Natl. Acad. Sci. USA 96: 6161–6165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacic MK, and Smith CJ (2008) Laboratory maintenance and cultivation of Bacteroides species. Current Protocols Microbiol. 13C.11.11–13C.11.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagiyan GA, Koroleva IK, Soroka NV, and Ufimtsev AV (2003) Oxidation of thiol compounds by molecular oxygen in aqueous solutions. Russ. Chem. Bull 52: 1135–1141. [Google Scholar]
- Ballou D, Palmer G, and Massey V (1969) Direct demonstration of superoxide anion production during the oxidation of reduced flavin and of its catalytic decomposition by erythrocuprein. BBRC. 36: 898. [DOI] [PubMed] [Google Scholar]
- Baughn AD, Garforth SJ, Vilcheze C, and W R Jacobs J (2009) An anaerobic-type alpha-keotglutarate ferredoxin oxidoreductase completes the oxidative tricarboxylic acid cycle of Mycobacterium tuberculosis. PLoS Pathog. 5: e1000662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughn AD, and Malamy MH (2004) The strict anaerobe Bacteroides fragilis grows in and benefits from nanomolar concentrations of oxygen. Nature. 427: 441–444. [DOI] [PubMed] [Google Scholar]
- Biegel E, Schmidt S, González J, and Müller V (2011) Biochemistry, evolution and physiological function of the Rnf complex, a novel ion-motive electron transport complex in prokaryotes. Cell. Mol. Life Sci 68: 613–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blodgett JA, Thomas PM, Li G, Velasquez JE, van der Donk WA, Kelleher NL, and Metcalf WW (2007) Unusual transformations in the biosynthesis of the antibiotic phosphinothricin tripeptide. Nat. Chem. Biol 3: 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckel W, and Golding BT (2006) Radical enzymes in anaerobes. Annu. Rev. Microbiol 60: 27–49. [DOI] [PubMed] [Google Scholar]
- Carlioz A, and Touati D (1986) Isolation of superoxide dismutase mutants in Escherichia coli: is superoxide dismutase necessary for aerobic life? EMBO J. 5: 623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabriere E, Charon MH, Volbeda A, Pieulle L, Hatchikian EC, and Fontecilla-Camps JC (1999) Crystal structures of the key anaerobic enzyme pyruvate:ferredoxin oxidoreductase, free and in complex with pyruvate. Nat. Struct. Biol 6: 182–190. [DOI] [PubMed] [Google Scholar]
- Cypionka H (2000) Oxygen respiration by Desulfovibrio species. Annu. Rev. Microbiol 54: 827–848. [DOI] [PubMed] [Google Scholar]
- Djaman O, Outten FW, and Imlay JA (2004) Repair of oxidized iron-sulfur clusters in Escherichia coli. J. Biol. Chem 279: 44590–44599. [DOI] [PubMed] [Google Scholar]
- Espey MG (2013) Role of oxygen gradients in shaping redox relationships between the human intestine and its microbiota. Free Rad. Biol. Med 55: 130–140. [DOI] [PubMed] [Google Scholar]
- Flint DH, and Emptage MH (1988) Dihydroxy acid dehydratase from spinach contains a [2Fe-2S] cluster. J. Biol. Chem 263: 3558–3564. [PubMed] [Google Scholar]
- Flint DH, Emptage MH, Finnegan MG, Fu W, and Johnson MK (1993a) The role and properties of the iron-sulfur cluster in Escherichia coli dihydroxy-acid dehydratase. J. Biol. Chem 268: 14732–14742. [PubMed] [Google Scholar]
- Flint DH, Tuminello JF, and Emptage MH (1993b) The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J. Biol. Chem 268: 22369–22376. [PubMed] [Google Scholar]
- Fontecave M, Mulliez E, and Logan DT (2002) Deoxyribonucleotide synthesis in anaerobic microorganisms: the class III ribonucleotide reductase. Prog. Nucl. Acid Res 72: 95–127. [DOI] [PubMed] [Google Scholar]
- Fourmond V, Baffert C, Sybirna K, Dementin S, Abou-Hamdan A, Meynial-Sales I, Soucaille P, Bottin H, and Leger C (2013) The mechanism of inhibition by H2 of H2-evolution by hydrogenases. Chem. Commun 49: 6840–6842. [DOI] [PubMed] [Google Scholar]
- Frey M (2002) Hydrogenases: hydrogen-activating enzymes. Chem. Bio. Chem 3: 153–160. [DOI] [PubMed] [Google Scholar]
- Gardner PR, and Fridovich I (1991a) Superoxide sensitivity of the Escherichia coli 6-phosphogluconate dehydratase. J. Biol. Chem 266: 1478–1483. [PubMed] [Google Scholar]
- Gardner PR, and Fridovich I (1991b) Superoxide sensitivity of the Escherichia coli aconitase. J. Biol. Chem 266: 19328–19333. [PubMed] [Google Scholar]
- Gardner PR, and Fridovich I (1992) Inactivation-reactivation of aconitase in Escherichia coli. A sensitive measure of superoxide radical. J. Biol. Chem 267: 8757–8763. [PubMed] [Google Scholar]
- Goldstein S, Meyerstein D, and Czapski G (1993) The Fenton reagents. Free Rad. Biol. Med 15: 435–445. [DOI] [PubMed] [Google Scholar]
- Hess V, Gallegos R, Jones JA, Barquera B, Malamy MH, and Muller V (2016) Occurrence of ferredoxin:NAD+ oxidoreductase activity and its ion specificity in serveral Gram-positive and Gram-negative bacteria. PeerJ. 4: e1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill RL, and Bradshaw RA (1969) Fumarase. Methods Enzymol. 13: 91–99. [Google Scholar]
- Horton RM., (1995) PCR-mediated recombination and mutagenesis. SOEing together tailor-made genes. Mol. Biotechnol 3: 93–99. [DOI] [PubMed] [Google Scholar]
- Imlay JA (1995) A metabolic enzyme that rapidly produces superoxide, fumarate reductase of Escherichia coli. J. Biol. Chem 270: 19767–19777. [PubMed] [Google Scholar]
- Imlay JA (2002a) How oxygen damages microbes: oxygen tolerance and obligate anaerobiosis. Adv. Microb. Physiol 46: 111–153. [DOI] [PubMed] [Google Scholar]
- Imlay JA (2002b) What biological purpose is sereved by superoxide reductase? J. Biol. Inorg. Chem 7: 659–663. [DOI] [PubMed] [Google Scholar]
- Imlay JA (2003) Pathways of oxidative damage. Annu. Rev. Microbiol 57: 395–418. [DOI] [PubMed] [Google Scholar]
- Imlay JA (2013) The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat. Rev. Microbiol 11: 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay JA, Sethu R, and Rohaun SK (2019) Evolutionary adaptations that enable enzymes to tolerate oxidative stress. Free Rad. Biol. Med 140: 4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay KRC, Korshunov S, and Imlay JA (2015) The physiological roles and adverse effects of the two cystine importers of Escherichia coli. J. Bacteriol 197: 3629–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S, and Imlay JA (2007) Micromolar intracellular hydrogen peroxide disrupts metabolism by damaging iron-sulfur enzymes. J. Biol. Chem 282: 929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S, and Imlay JA (2010) Hydrogen peroxide inactivates the Escherichia coli Isc iron-sulphur assembly system, and OxyR induces the Suf system to compensate. Mol. Microbiol 78: 1448–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly DJ (1998) The physiology and metabolism of the human gastric pathogen Helicobacter pylori. Adv. Microb. Physiol 40: 137–189. [DOI] [PubMed] [Google Scholar]
- Kerscher L, and Oesterhelt D (1981) Purification and properties of 2-oxoacid:ferredoxin oxidoreductases from Halobacterium halobium. Eur. J. Biochem 116: 587–594. [DOI] [PubMed] [Google Scholar]
- Kessler D, Herth W, and Knappe J (1992) Ultrastructure and pyruvate formate-lyase radical quenching property of the multienzymic AdhE protein of Escherichia coli. J. Biol. Chem 267: 18073–18079. [PubMed] [Google Scholar]
- Knappe J, Neugebauer FA, Blaschkowski HP, and Ganzler M (1994) Post-translational activation introduces a free radical into pyruvate formate-lyase. Proc. Natl. Acad. Sci. USA 81: 1332–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll AH, and Nowak MA (2017) The timetable of evolution. Sci. Adv 3 e1603076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koropatkin NM, Martins EC, Gordon JI, and Smith TJ (2008) Starch catabolism by a prominent human gut symbiont is directed by the recognition of amylose helices. Structure. 16: 1105–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korshunov S, and Imlay JA (2010) Two sources of endogenous hydrogen peroxide in Escherichia coli. Mol. Microbiol 75: 1389–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korshunov S, Imlay KRC, and Imlay JA (2019) Cystine import is a valuable but risky process whose hazards Escherichia coli minimizes by inducing a cysteine exporter. Mol. Microbiol 103: 22–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubas A, Orain C, Sancho DD, Sensi LS, Gauquelin C, Meynial-Salles I, Soucaille P, Bottin H, Baffert C, Fourmond V, Best RB, Blumberger J, and Leger C (2016) Mechanism of O2 diffusion and reduction in FeFe hydrogenases. Nat. Chem 9: 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CF, Mashino T, and Fridovich I (1987) α,β-dihydroxyisovalerate dehydratase: a superoxide-sensitive enzyme. J. Biol. Chem 262: 4724–4727. [PubMed] [Google Scholar]
- Liochev SI, and Fridovich I (1992) Fumarase C, the stable fumarase of Escherichia coli, is controlled by the soxRS regulon. Proc. Natl. Acad. Sci. USA 89: 5892–5896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liochev SI, and Fridovich I (1993) Modulation of the fumarases of Escherichia coli in response to oxidative stress. Arch. Biochem. Biophys 301: 379–384. [DOI] [PubMed] [Google Scholar]
- Lu Z, and Imlay JA (2017) The fumarate reductase of Bacteroides thetaiotaomicron, unlike that of Escherichia coli, is configured so that it does not generate reactive oxygen species. mBio. 8: e01873–01816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Sethu R, and Imlay JA (2018) Endogenous superoxide is a key effector of the oxygen sensitivity of a model obligate anaerobe. Proc. Natl. Acad. Sci. USA 115: e3266–e3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord JM, Keele BB Jr., and Fridovich I (1971) An enzyme-based theory of obligate anaerobiosis: the physiological function of superoxide dismutase. Proc. Natl. Acad. Sci. USA 68: 1024–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meehan BM, and Malamy MH (2012) Fumarate reductase is a major contributor to the generation of reactive oxygen species in the anaerobe Bacteroides fragilis. Microbiology. 158: 539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon S, and Ragsdale SW (1997) Mechanism of the Clostridium thermoaceticum pyruvate:ferredoxin oxidoreductase: evidence for the common catalytic intermediacy of the hydroxyethylthiamine pyrophosphate radical. Biochemistry. 36: 8484–8494. [DOI] [PubMed] [Google Scholar]
- Messner KR, and Imlay JA (2002a) In vitro quantitation of biological superoxide and hydrogen peroxide generation. Meth. Enzymol 349: 354–361. [DOI] [PubMed] [Google Scholar]
- Messner KR, and Imlay JA (2002b) Mechanism of superoxide and hydrogen peroxide formation by fumarate reductase, succinate dehydrogenase, and aspartate oxidase. J. Biol. Chem 277: 42563–42571. [DOI] [PubMed] [Google Scholar]
- Metcalf WW, Jiang W, and Wanner BL (1994) Use of the rep technique for allele replacement to construct new Escherichia coli hosts for maintenance of R6Kλ origin plasmids at different copy numbers. Gene. 138: 1–7. [DOI] [PubMed] [Google Scholar]
- Mishra S, and Imlay JA (2013) An anaerobic bacterium, Bacteroides thetaiotaomicron, uses a consortium of enzymes to scavenge hydrogen peroxide. Mol. Microbiol 90: 1356–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RL, and Schmidt TM (2013) Shallow breathing: bacterial life at low O2. Nat. Rev. Microbiol 11: 205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder DW, Shepard EM, Meuser JE, Joshi N, King PW, Posewitz MC, Broderick JB, and Peters JW (2011) Insights into [FeFe]-hydrogenase structure, mechanism, and maturation. Structure. 19: 1038–1052. [DOI] [PubMed] [Google Scholar]
- Nnyepi MR, Peng Y, and Broderick JB (2007) Inactivation of E. coli pyruvate formate-lyase: role of AdhE and small molecules. Arch. Biochem. Biophys 459: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordlund P, and Reichard P (2006) Ribonucleotide reductases. Annu. Rev. Biochem 75: 681–706. [DOI] [PubMed] [Google Scholar]
- Pan N, and Imlay JA (2001) How does oxygen inhibit central metabolism in the obligate anaerobe Bacteroides thetaiotaomicron? Mol. Microbiol 39: 1562–1571. [DOI] [PubMed] [Google Scholar]
- Pieulle L, Guigliarelli B, Asso M, Dole F, Bernadac A, and Hatchikian EC (1995) Isolation and characterization of the pyruvate-ferredoxin oxidoreductase from the sulfate-reducing bacterium Desulfovibrio africanus. Biochim. Biophys. Acta 1250: 49–59. [DOI] [PubMed] [Google Scholar]
- Poole LB (2005) Bacterial defenses against oxidants: mechanistic features of cysteine-based peroxidases and their flavoprotein reductases. Arch. Biochem. Biophys 433: 240–254. [DOI] [PubMed] [Google Scholar]
- Ragsdale SW (2003) Pyruvate ferredoxin oxidoreductase and its radical intermediate. Chem. Rev 103: 2333–2346. [DOI] [PubMed] [Google Scholar]
- Scheller S, Goenrich M, Boecher R, Thauer RK, and Jaun B (2010) The key nickel enzyme of methanogenesis catalyses the anaerobic oxidation of methane. Nature. 465: 606–609. [DOI] [PubMed] [Google Scholar]
- Seaver LC, and Imlay JA (2001) Hydrogen peroxide fluxes and compartmentalization inside growing Escherichia coli. J. Bacteriol 183: 7182–7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaver LC, and Imlay JA (2004) Are respiratory enzymes the primary sources of intracellular hydrogen peroxide? J. Biol. Chem 279: 48742–48750. [DOI] [PubMed] [Google Scholar]
- Shibata N, and Toraya T (2015) Molecular architectures and functions of radical enzymes and their (re)activating proteins. J. Biochem 158: 271–292. [DOI] [PubMed] [Google Scholar]
- Sobota JM, and Imlay JA (2011) Iron enzyme ribulose-5-phosphate 3-epimerase in Escherichia coli is rapidly damaged by hydrogen peroxide but can be protected by manganese. Proc. Natl. Acad. Sci. USA 108: 5402–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šťovíček A, Kim M, Or D, and Gillor O (2017) Microbial community response to hydration-desiccation cycles in desert soil. Sci. Rep 7: 45735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AJ, and Kelly DJ (2019) The function, biogenesis and regulation of the electron transport chains in Campylobacter jejuni: new insights into the bioenergetics of a major food-borne pathogen. Adv. Microb. Physiol 74: 239–329. [DOI] [PubMed] [Google Scholar]
- Vita N, Hatchikian EC, Nouailler M, Dolla A, and Pieulle L (2008) Disulfide bond-dependent mechanism of protection against oxidative stress in pyruvate-ferredoxin oxioreductase of anaerobic Desulfovibrio bacteria. Biochemistry. 47: 957–964. [DOI] [PubMed] [Google Scholar]
- Wagner AF, Frey M, Neugebauer FA, Schafer W, and Knappe J (1992) The free radical in pyruvate formate-lyase is located on glycine-734. Proc. Natl. Acad. Sci. USA 89: 996–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AF, Schultz S, Bomke J, Pils T, Lehmann WD, and Knappe J (2001) YfiD of Escherichia coli and Y061 of bacteriophage T4 as autonomous glycyl radical cofactors reconstituting the catalytic center of oxygen-fragmented pyruvatet formate-lyase. Biochem. Biophys. Res. Commun 285: 456–462. [DOI] [PubMed] [Google Scholar]
- Wexler AG, and Goodman AL (2017) An insider’s perspective: Bacteroides as a window into the microbiome. Nat. Microbiol 2: 17026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper LV, Gordon JI (2003) A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science. 299: 2074–2076. [DOI] [PubMed] [Google Scholar]
- Yamada T, Takahashi-Abbe S, and Abbe K (1985) Effects of oxygen on pyruvate formate-lyase in situ and sugar metabolism of Streptococcus mutans and Streptococcus sanguis. Infect. Immun 47: 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KS, Hille R, Hemann C, and Tabita FR (1999) Rubredoxin from the green sulfur bacterium Chlorobium tepidum functions as an electron acceptor for pyruvate ferredoxin oxidoreductase. J. Biol. Chem 274: 29772–29778. [DOI] [PubMed] [Google Scholar]
- Yoon KS, Ishii M, Kodama T, and Igarashi Y (1997) Purification and characterization of pyruvate:ferredoxin oxidoreductase from Hydrogenobacter thermophilus TK-6. Arch. Microbiol 167: 275–279. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Padayati PS, and Leung JH (2017) Proton-translocating nicotinamide nucleotide transhydrogenase: a structural perspective. Front. Physiol 8: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wong KK, Magliozzo RS, and Kozarich JW (2001) Inactivation of pyruvate formate-lyase by dioxygen: defining the mechanistic interplay of glycine 734 and cysteine 419 by rapid freeze-quench EPR. Biochemistry. 40: 4123–4130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.