

Abstract
Regulation of photoreceptor phosphodiesterase (PDE6) activity is responsible for the speed, sensitivity, and recovery of the photoresponse during visual signaling in vertebrate photoreceptor cells. It is hypothesized that physiological differences in the light responsiveness of rods and cones may result in part from differences in the structure and regulation of the distinct isoforms of rod and cone PDE6. Although rod and cone PDE6 catalytic subunits share a similar domain organization consisting of tandem GAF domains (GAFa and GAFb) and a catalytic domain, cone PDE6 is a homodimer whereas rod PDE6 consists of two homologous catalytic subunits. Here we provide the x-ray crystal structure of cone GAFab regulatory domain solved at 3.3 Å resolution, in conjunction with chemical cross-linking and mass spectrometric analysis of conformational changes to GAFab induced upon binding of cGMP and the PDE6 inhibitory γ-subunit (Pγ). Ligand-induced changes in cross-linked residues implicate multiple conformational changes in the GAFa and GAFb domains in forming an allosteric communication network. Molecular dynamics simulations of cone GAFab revealed differences in conformational dynamics of the two subunits forming the homodimer and allosteric perturbations on cGMP binding. Cross-linking of Pγ to GAFab in conjunction with solution NMR spectroscopy of isotopically labeled Pγ identified the central polycationic region of Pγ interacting with the GAFb domain. These results provide a mechanistic basis for developing allosteric activators of PDE6 with therapeutic implications for halting the progression of several retinal degenerative diseases.
Keywords: x-ray crystallography, chemical cross-linking, mass spectrometry, photoreceptor, molecular dynamics simulations
Graphical Abstract
Introduction
GAF domains are a large protein domain family that have been identified in every kingdom of life [1,2] and whose functions include ligand binding, protein–protein interactions, and other functions [3]. GAF domains are so named because they were first identified in cGMP-binding phosphodiesterases, cyanobacterial Adenylyl cyclases and transcription factor FhlA [1]. In mammals, the occurrence of GAF domains is restricted to 5 of the 11 class I cyclic nucleotide phosphodiesterase (PDE) families [4,5], namely PDE2, PDE5, PDE6, PDE10, and PDE11. In all five instances, the highly conserved catalytic domain is preceded by N-terminal tandem GAF domains (individually referred to as GAFa and GAFb). The tandem GAFab domains are believed to serve several functions for PDEs, including enhancing dimerization of PDE catalytic subunits and allosterically communicating with the catalytic domain to regulate catalytic activity [3]. Whereas structural determinations have revealed the domain organization and dimerization interface of PDE2 and PDE5 GAFab dimers [6,7], nearly full-length PDE2 [8], and the rod photoreceptor PDE6 holoenzyme [9–11], much remains to be learned about the allosteric mechanisms underlying GAF domain regulation of PDE catalytic activity. This gap in knowledge arises from challenges in comparing the conformational dynamics of PDEs in various liganded states, thereby hindering the design of allosteric modulators targeting the regulatory GAF domains of PDEs for therapeutic applications [12].
All five GAF-containing PDEs are believed to bind only a single cyclic nucleotide molecule per catalytic subunit. For PDE5, PDE6, and PDE11, cGMP (or cGMP analogs) selectively bind to the first (GAFa) domain, whereas PDE2 and PDE10 selectively bind cGMP and cAMP, respectively, to the second (GAFb) domain [13–15]. For PDE2 and PDE5 GAF domains, binding of cGMP allosterically stimulates catalytic activity at the active site [16,17]. A similar allosteric activation of catalysis upon binding of ligand to the GAF domains occurs for PDE10 and PDE11 as well [15].
In the case of rod and cone photoreceptor PDE6, the only PDE family that is directly activated by a heterotrimeric G-protein (transducin) [18], cGMP occupancy of the GAFa domain does not directly alter catalytic activity of the rod photoreceptor PDE6 isozyme [19] or cause significant hydrodynamic changes in rod PDE6 conformation as is the case for PDE5 [20,21]. However, it is well documented that cGMP binding to rod PDE6 GAFa domains enhance the binding affinity of the PDE6 inhibitory γ-subunit (Pγ) [19,22,23]. This allosteric communication between the GAF and catalytic domains is reciprocal, in that binding of rod Pγ (or truncated fragments of Pγ) induces conformational changes that result in enhancement of cGMP binding affinity to the GAFa domain [19]. In addition, direct allosteric communication between the GAF and catalytic domains of rod PDE6 has been inferred based on an increase in cGMP binding affinity when the active sites of PDE6 are occupied with PDE5/6 inhibitors [24]; a similar phenomenon was reported for PDE5 [25]. Although recent advances in determining the domain organization of the rod PDE6 heterotetramer (consisting of PDE6A and PDE6B catalytic subunits and two rod Pγ subunits (PDE6G)) have generated hypotheses about the allosteric communication pathway between the regulatory and catalytic domains [10,11], experimental evidence is lacking on the structural changes that are induced by binding of cGMP or Pγ to PDE6. Much less is known about the allosteric regulation of the homologous cone PDE6 enzyme (composed of two identical catalytic subunits (PDE6C) and two cone Pγ subunits (PDE6H)).
The present study was undertaken to determine the molecular mechanism whereby binding of signaling molecules to cone PDE6 regulatory GAF domains induces allosteric conformational changes that contribute to regulating the extent and duration of PDE6 activation during visual transduction in cone photoreceptors. In this study, we first report the 3.3 Å x-ray crystal structure of the cone photoreceptor PDE6 GAFab domains in their unliganded (apo) state. To characterize the conformational changes occurring to GAFab upon cGMP and/or Pγ binding, chemical cross-linking and mass spectrometry analysis (XL-MS) were used in conjunction with integrative structural modeling to detect conformational changes observed in the four possible liganded states (apo, cGMP bound, Pγ bound, and cGMP and Pγ bound; Figure 1). Molecular dynamics simulations of the apo- and cGMP-bound structures substantiated our hypothesis that cGMP binding induces conformational changes that are allosterically communicated from the GAFa to the GAFb domain, and also supported the idea that the two subunits of the GAFab homodimer are non-identical. XL-MS analysis of Pγ bound to GAFab in conjunction with NMR spectroscopic analysis of isotopically labeled Pγ in the presence of GAFab identified the surface of interaction of Pγ with GAFab, and highlighted the central polycationic region of Pγ as the primary site of interaction with the GAFb domain. This study of the cone PDE6 GAFab regulatory domain provides the most comprehensive analysis to date of allosterically induced conformational changes in GAF-containing PDEs. This work also advances our understanding of the differences between rod and cone PDE6 regulation that likely underlie the physiological differences in light sensitivity and photoresponse kinetics observed for rod and cone photoreceptors.
Figure 1.
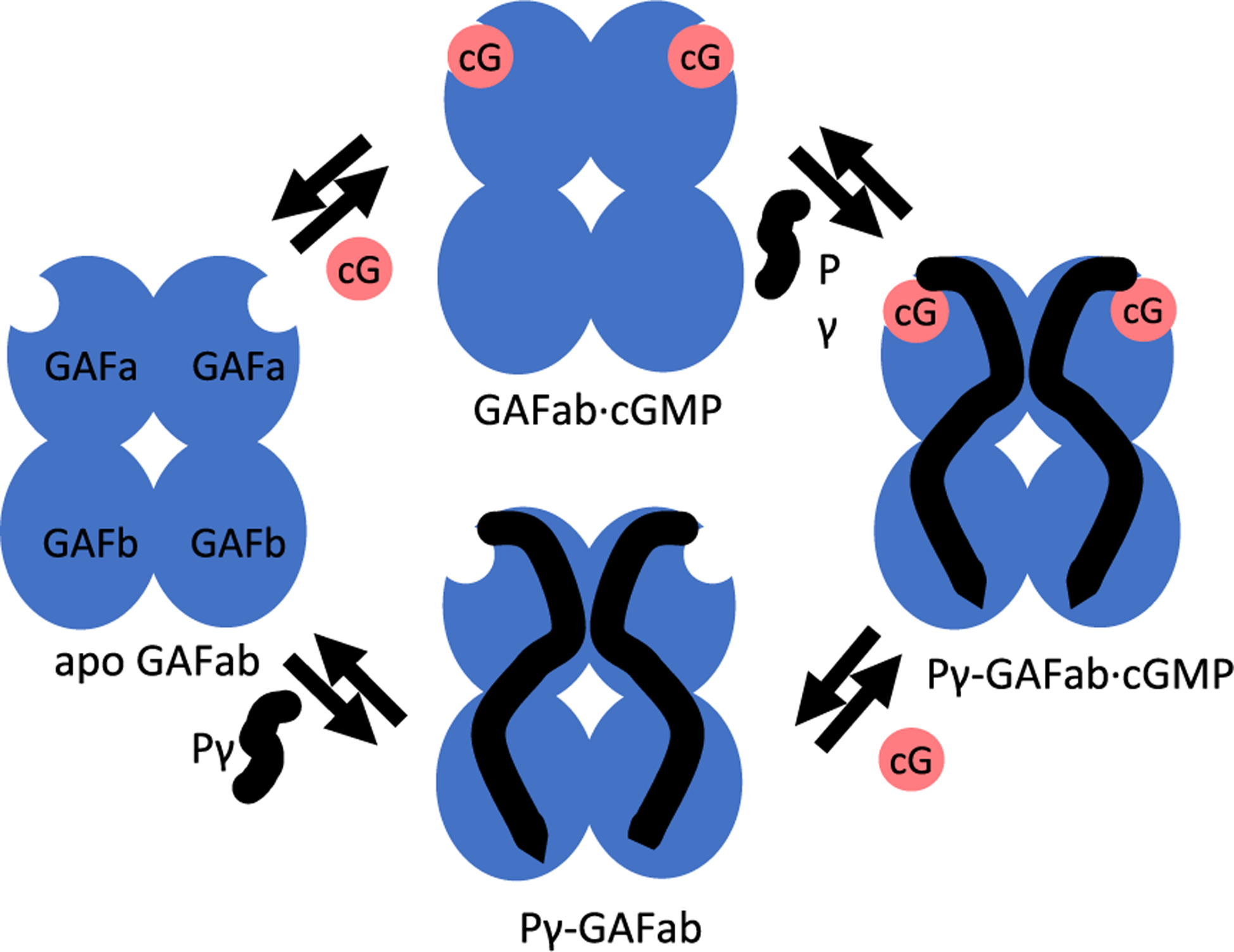
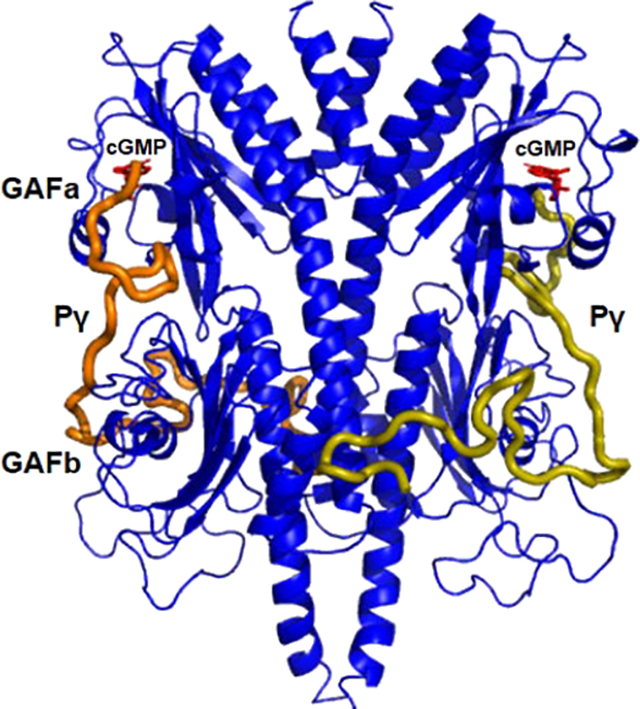
Multiple equilibria of ligand binding to PDE6 regulatory GAFab domains. The GAFab regulatory domain of cone PDE6 consists of two identical subunits each of which has tandem GAFa and GAFb domains (blue ovals). Noncatalytic cGMP binding sites are depicted on GAFa, and the inhibitory Pγ subunit is represented in a linearly extended conformation as interacting with both GAFa and GAFb.
Results
X-ray structure of the cone PDE6 regulatory GAF domains
Because of the inability to heterologously express full-length cone PDE6 catalytic subunits in sufficient quantities for structural studies [26], we relied on the previous successful expression of PDE5 GAFab [7] and the isolated GAFa domain of chicken cone PDE6 [27] to construct expression vectors for bacterial expression and purification of residues 42–458 of chicken cone PDE6 containing a 6-His tag (Figure S1). We also expressed and purified the corresponding chicken cone Pγ subunit consisting of its first 58 residues (Figure S1). Purified recombinant GAFab was judged to be properly folded, based on its apparent MW observed by gel filtration as well as its ability to bind cGMP to the GAFa domain with a KD value (22 nM; Figure S1), similar to values reported previously [28].
Purified cone PDE6 GAFab was then crystallized, and its structure determined from crystals in the space group of P65 with cell dimensions of a = b = 148.5, and c = 93.7 Å (Table 1). The structure was solved by using the PDE5 GAFab structure [7] as the initial model, and refined to R-factor of 0.207 for 16,489 reflections at 3.2–50 Å resolution (Table 1). Residues 48–451 of the primary sequence of cone PDE6 were traceable, except for residues Lys286 to Thr309 (Figure 2(a), arrows), corresponding to the GAFb β1/β2 loop. Figure S2 provides domain boundaries and secondary structure elements for the PDE6 GAFab structure.
Table 1.
Statistics on diffraction data and structure refinement of unliganded PDE6C GAFaba, b, c
| Data collection | |
| Space group | P65 |
| Unit cell (a = b, c, Å) | 148.5, 148.5, 93.7 |
| Resolution (Å) | 3.2 |
| Wavelength (Å) | 1.0 |
| Unique reflections | 16,737 |
| Redundancy | 4.8 fold |
| Completeness (%) | 86.0 (87.7)a |
| Average I/σ | 6.2 (1.9)a |
| Rmerge | 0.142 (0.712)a |
| Refinement | |
| R-factor | 0.207 |
| R-free | 0.308 (5.0%)b |
| Resolution (Å) | 50–3.2 |
| Bond (Å) | 0.011 |
| Angle | 1.65° |
| Protein | 72.5 (6251)c |
| Ramachandran plot | |
| Most favored | 94.0 |
| Allowed | 5.8 |
| Generally allowed | 0.2 |
The numbers in parentheses are for the highest-resolution shell.
The percentage of reflections omitted for calculation of R-free.
The number of atoms in the crystallographic asymmetric unit.
Figure 2.
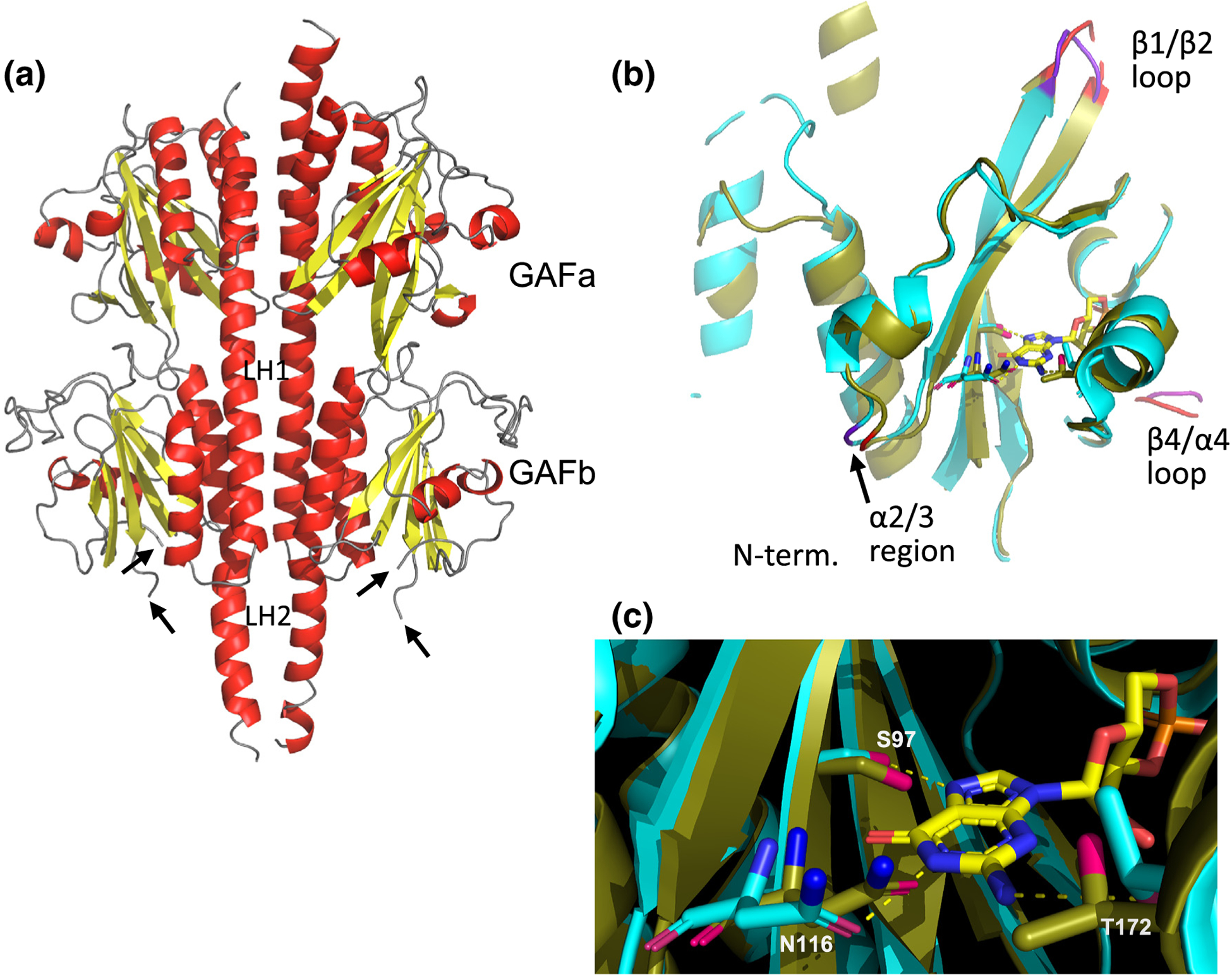
Structure of cone PDE6 regulatory domains. (a) X-ray structure of chicken cone PDE6 GAFab at 3.3 Å resolution; α-helices colored red, β-strands colored yellow, and loop regions colored gray. Arrows indicate location of the unresolved β1/β2 loop structure in the GAFb domain. (b) Structural alignment of the unliganded cone GAFa domain of the GAFab x-ray structure with the GAFa crystal structure (cyan) with bound cGMP [27]. (c) Structural alterations of residues within the cGMP binding site. Cyan sticks represent the residues in the crystal structure of cGMP-bound GAFa structure [27], while the corresponding residues of unliganded cone PDE6 are shown as olive sticks. The dotted lines represent hydrogen bonds.
The cone PDE6 GAFab molecule is a homodimeric structure (Figure 2(a)) that has the same fold as the homodimeric GAFab domains of PDE2 [8] and PDE5 [7], as well as with heterodimeric rod PDE6 [10,11]. Each GAF domain consists of an anti-parallel β-sheet, with the α2 and α5 helices positioned behind the β-sheet on the opposite face to the cGMP binding pocket, while the α4 helix and the β3 strand (and adjacent residues) are situated at the entrance to the cGMP binding pocket as previously described [27].
The dimer interface of PDE6 GAFab extends over the entire long axis of the molecule, with major stabilizing contributions from the two long α-helices (LH1 and LH2) that link GAFa to GAFb and GAFb to the catalytic domain, respectively. Interaction analysis revealed that ~20% of the residues in the GAFa α1 helix also contribute to the interaction surface of the homodimer, consistent with the proposed role of the N-terminal region of rod PDE6 catalytic subunits in stabilizing the catalytic dimer [10]. Overall, the two subunits of the cone GAFab homodimer are highly symmetric (root mean square fluctuations, RMSD = 0.74 Å for Cα atoms; Figure S1), with conformational differences located primarily in regions lacking secondary structure.
Structural superposition of the unliganded PDE6 GAFa domain with the previously determined crystal structure of the cGMP-bound PDE6 GAFa domain [27] revealed an average shift of 0.8 Å for the Cα atoms, suggesting that neither dimerization, cGMP binding nor the presence of the GAFb domain induces dramatic conformational changes within the GAFa domain (Figure 2(b)). The RMSD analysis (Figure S3) revealed major differences in the β1/β2 loop and smaller differences in the loop near α2/3 and the so-called lid region (β4/α4 loop) of GAFa (Figure S3). The largest movement among the previously identified cGMP binding residues [27] is 1.1 Å for the Cα Asn116 in the β2 strand whose side-chain forms a hydrogen bond with the base nitrogen of cGMP (Figure 2(c)).
Integrative structural modeling of the unliganded state of GAFab determined by chemical cross-linking and mass spectrometry
To investigate the allosteric regulation of the cone PDE6 GAFab regulatory domain, we first carried out chemical cross-linking coupled with liquid chromatography/mass spectrometry (XL-MS) to evaluate the structure of GAFab in aqueous solution and to create a structural model for the missing residues that were not resolved in the crystal structure. Figure 3(a) shows a typical experiment using the cross-linker bis(sulfosuccinimidyl)suberate (BS3) that shows the appearance of gel-shifted bands corresponding to the cross-linked GAFab dimer. We also conducted cross-linking experiments in the presence of cGMP and/or truncated cone Pγ (Pγ1–58, described below). The gel bands corresponding to the GAFab dimer (~100 kDa) were excised, proteolyzed, and analyzed by MS. Cross-linked peptides were used as spatial restraints for Modeller v9.11 [29] using the crystal structure as the template (see Materials and Methods).
Figure 3.
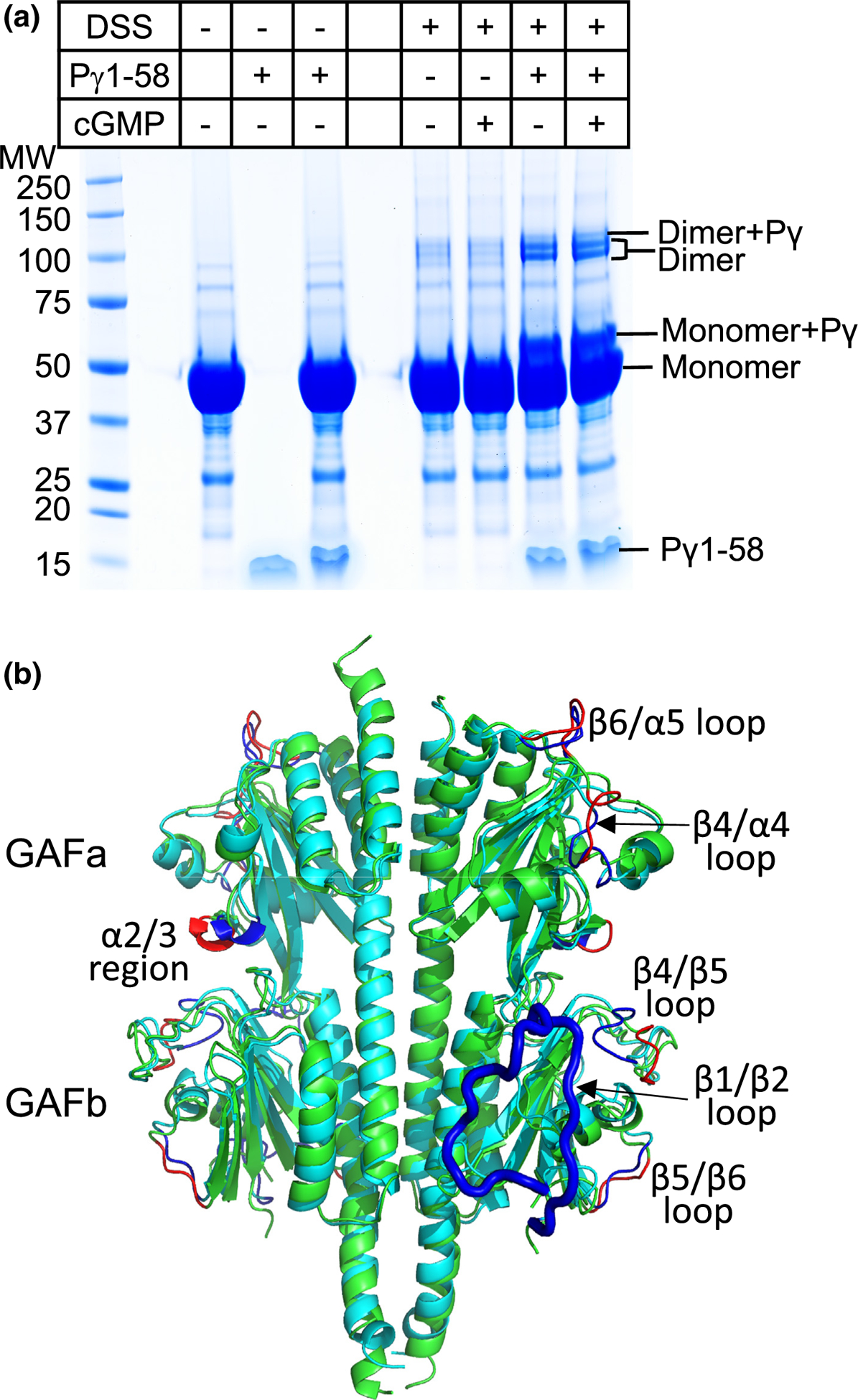
Structural model of the apo state of cone PDE6 GAFab determined by chemical cross-linking and mass spectrometry. (a) SDS-PAGE of a cross-linking experiment in which 12 μM GAFab was incubated in the presence or absence of cGMP (10-fold molar excess) and/or 120 μM Pγ1–58 prior to addition of a 50-fold molar excess of the chemical cross-linker BS3. (b) Structural alignment of cross-linked refined apo-GAFab (cyan) with the x-ray structure (green), with major differences indicated for the x-ray (red) and apo (dark blue) structures; the GAFb β1/β2 loop (residues 286 to 310) that is missing in the x-ray structure is highlighted as a thick blue loop in the apo structure.
Figure 3(b) demonstrates that the structural model of the apo state of GAFab, refined by spatial restraints imposed by 84 cross-links (Table S1), was very similar to the x-ray structure in Figure 2(a). In particular, most of the α-helical and β-strand secondary structural features of the cross-link-based structural model superimposes well with the crystal structure. Our cross-linking results also allowed us to generate a structural model for residues 288 to 310 (a segment including the β1/β2 loop) that was missing in the GAFab crystal structure.
Structural alignment of the cross-link refined, apo state of GAFab and the crystal structure (Figure 3(b)) show that the secondary structural elements align closely for both subunits (RMSD = 2.0 Å; Figure S4). However, the spatial restraints imposed by the cross-linking data result in significant differences in the GAFa α2/3 helix loop (near the cGMP binding site) as well as the β4/α4 loop (residues 155 to 163) and β6/α5 loop (residues 198 to 205). The GAFb domain of the structural model in aqueous solution also closely superimposes on the crystal structure (Figure 3(b)), except for the β4/β5 and β5/β6 loops where spatial restraints imposed by the cross-linking results show significant deviation from the x-ray structure (Figure S4). In addition, the model for the GAFb β1/β2 loop extends up toward the GAFa domain and makes close contact (<7 Å) with the GAFa β1/β2 loop. In the following sections, we use the structural model of the cross-link-refined apo state of GAFab in aqueous solution as the reference for evaluating conformational changes induced by cGMP and/or Pγ.
Conformational changes induced by cGMP binding to GAFab revealed by chemical cross-linking/MS analysis
Cross-links obtained from XL-MS analysis of GAFab incubated with cGMP (Table S2) were used to create a structural model for cGMP-bound GAFab (GAFab·cGMP; Figure 4(a)). RMSD analysis of the GAFab·cGMP versus apo GAFab structures (Figure S5A) reveals the greatest conformational changes in the GAFb β1/β2 loop, along with smaller changes in the GAFa β4/α4 “lid region” and its loop, and also the GAFb β4/β5 loop (Figure 4(a)). Closer examination of the GAFa domain structures of apo and GAFab·cGMP reveals movement of the GAFa α4 helix and the β1/β2 loop upon cGMP binding (Figure 4(b)), regions previously proposed to be part of the allosteric communication network of rod PDE6 [10]. Several cross-links that were only observed when cGMP occupied the GAFa binding pocket (Table S2) support conformational changes in the GAFb β1/β2 and β6/α5 loops upon cGMP binding (Figure 4(c)), consistent with cGMP-dependent allosteric communication between the GAFa and GAFb domains. These structural.
Figure 4.
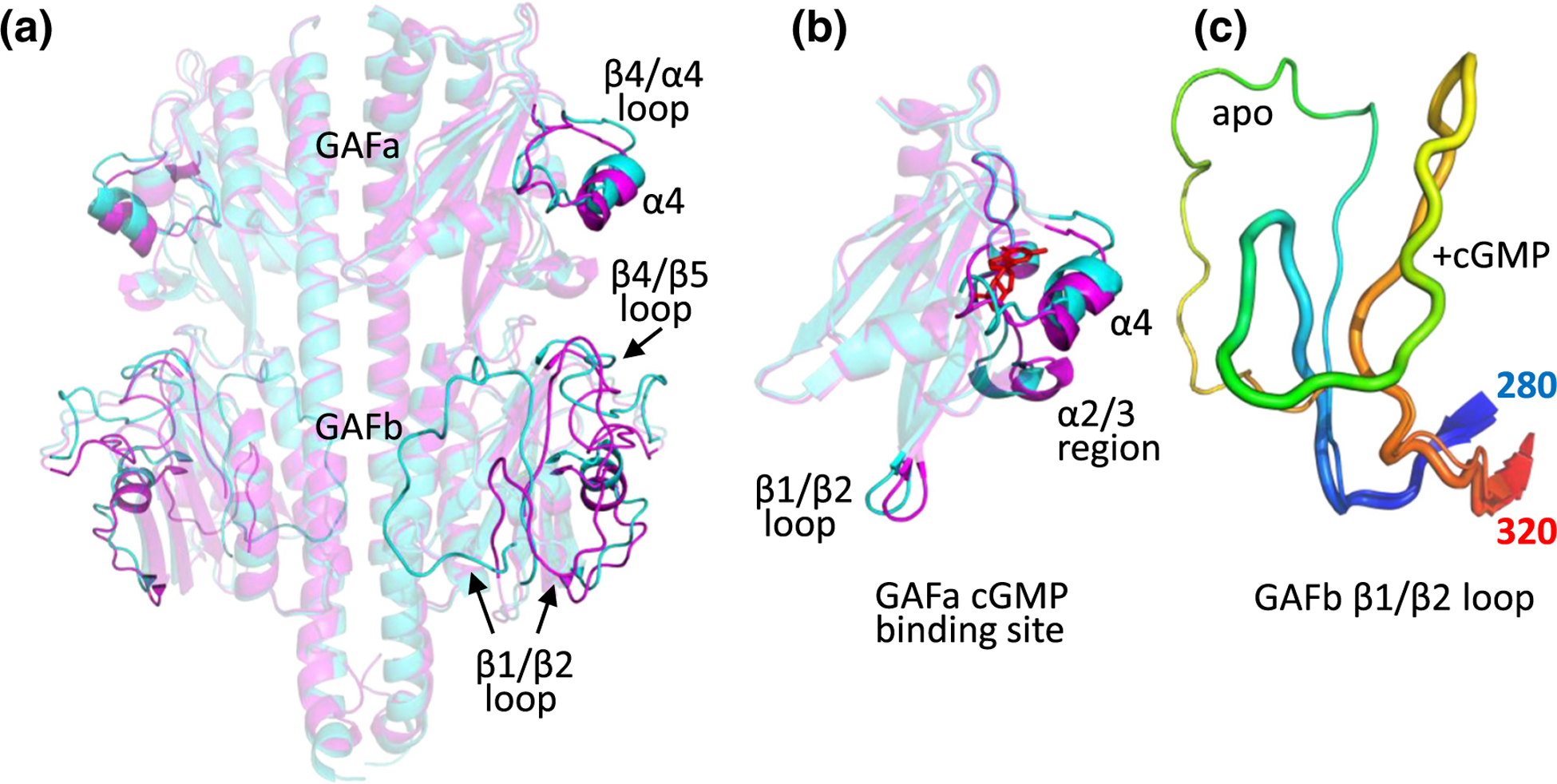
Structural changes in PDE6 GAFab upon binding of cGMP. (a) Comparison of apo GAFab (cyan) with GAFab·cGMP (magenta) with significant conformational differences highlighted. (b) Comparison of the conformational states of the GAFa domain in its apo (cyan) and cGMP-bound state (magenta, with cGMP (red) docked). (c) Comparison of the conformation of the GAFb β1/β2 loop (residues 280 (blue) to 320 (red)) in the apo (thin loop) and GAFab·cGMP (thick loop) states.
elements have been previously hypothesized to participate in allosteric communication from the GAF domains to the catalytic domains of mammalian rod PDE6 [10,11].
Molecular dynamics simulations of unliganded and GAFab·cGMP reveal subunit asymmetry and allosteric communication
To evaluate the conformational dynamics, subunit asymmetry, and allosteric communication of the cone PDE6 GAFab homodimer, we carried out three independent long time-scale molecular dynamics (MD) simulations (see Materials and Methods and Table S6) of the GAFab crystal structure as well as of the cross-link-refined structural models of GAFab in its apo and cGMP-liganded states. These MD simulations of the GAFab homodimer permitted analysis of the flexibility of individual sub-domains (root mean squared deviation (RMSD)) and individual amino acid residues (RMSF), changes in buried surface area (BSA), and correlated motions (dynamic residue–residue cross correlation (DCC)).
Assessment of the stability of the crystal structure
We first analyzed the conformational metrics of GAFab based on MD simulations of our crystal structure of the GAFab homodimer (Figure S6). The RMSF and RMSD results (Figure S6) show that the region preceding the GAFa domain as well as the GAFa and LH1 subdomains in each subunit are the least flexible, whereas the GAFb and LH2 subdomains show bimodal RMSD distributions reflecting two distinct conformational states; the higher flexibility of the LH2 subdomain (RMSD values of 0–10 Å) may be due in part to the GAFab regulatory domains not being attached to the catalytic domains of cone PDE6. Figure S6 shows that the highest BSA occurs between the LH1 (~1100–1500 Å2), LH2 (~500–1300 Å2) and the N-terminal α1 (~300–800 Å2) motifs, and likely accounts for the major interactions stabilizing dimer formation. From these results, we conclude that the overall subdomain folds and interfacial contact areas between subdomains in the GAFab crystal structure (Figure 2(a)) are stably maintained in solution states of GAFab explored by MD simulations.
Analysis of subunit asymmetry and allosteric communication of GAFab in its apo and cGMP-bound states
In comparison to the high degree of structural similarity of the two GAFab subunits in the x-ray structure (Figure 2(a), Figure S1), MD simulations of both apo and cGMP-bound GAFab (Figures S7 and S8) provide evidence that the conformational dynamics evolve in an asymmetric manner for the two subunits. As seen in the DRMSF plots in Figure 5, differences in the conformational dynamics of the two subunits of apo GAFab (Figure 5(a)) or cGMP-liganded GAFab (Figure 5(b)) are observed in the flexible loop regions, particularly the GAFb β1/β2 and β4/β5 loops, whereas the secondary structural elements are highly symmetrical in the two subunits. Although the XL-MS derived structural models represent a single conformation of the β1/β2 (and other) loops based on the distance restraints of the observed crosslinks, these atomistic simulations illustrate that these unstructured loops can exist in multiple conformations.
Figure 5.
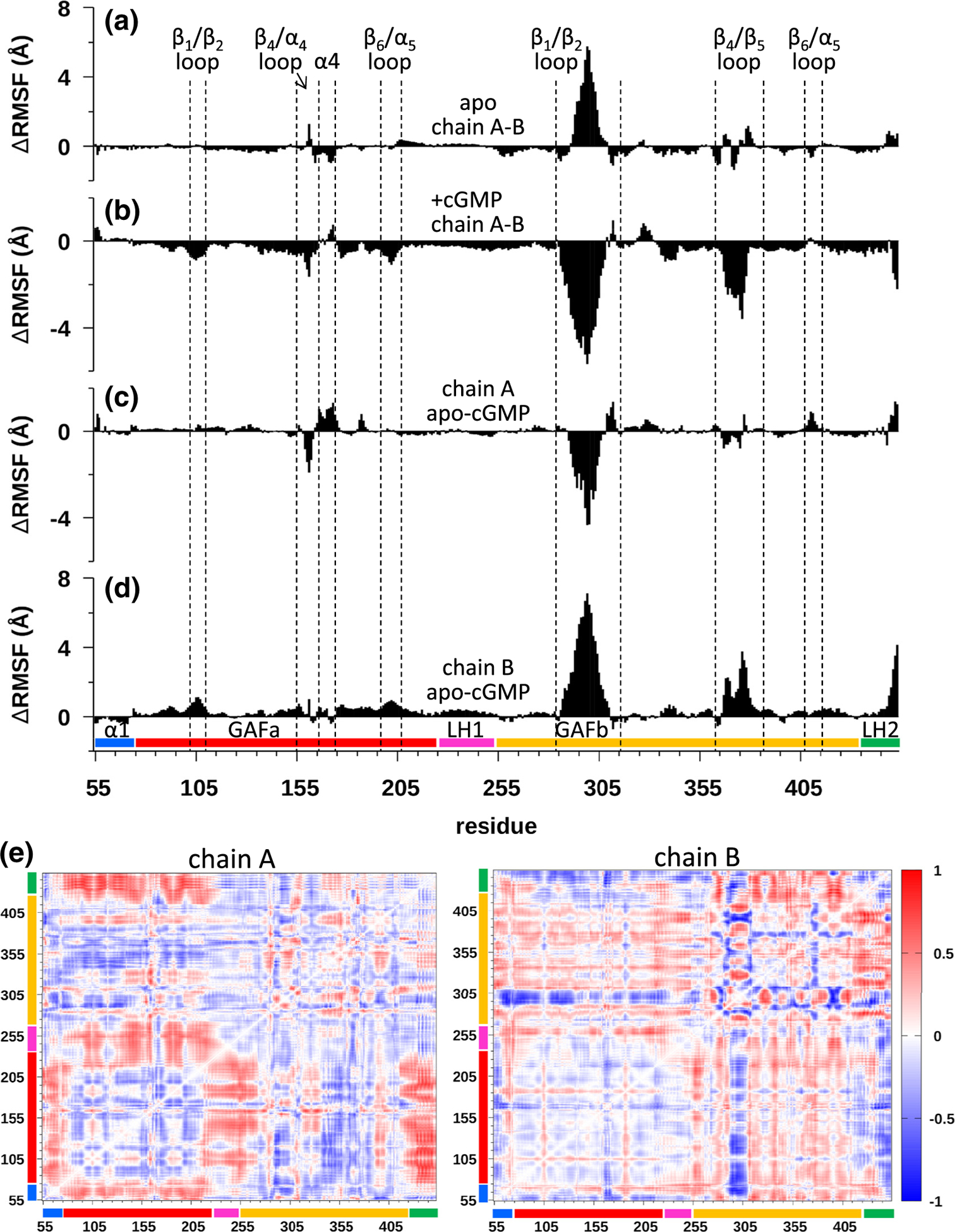
Molecular dynamics (MD) simulations of the apo and cGMP-liganded states of cone PDE6 GAFab. (a)–(d) The ΔRMSF per residue were analyzed separately for each sub-domain of GAFab (N-terminal region (α1) preceding GAFa (residues 55–74), GAFa (residues 75–224), long helix-1 (LH1, residues 225–255), GAFb (residues 256–443), and long helix-2 (LH2, residues 434–453)) to identify differences in protein dynamics of the two chains in the apo and cGMP-bound states for each subunit. (a and b) Evaluation of asymmetry in protein dynamics of the two GAFab subunits in the apo (a) and cGMP-bound (b) state. (c and d) Changes in protein dynamics upon cGMP binding to GAFab were evaluated by plotting the ΔRMSF per residue for the apo and cGMP-liganded states for subunit A (a) and subunit B (d). (e) Difference dynamic cross-correlation (DDCC) analysis of the differences in correlated motions between the apo and cGMP-liganded states of GAFab for subunits A (left) and B (right). Heat map bar indicates the range of correlations from −1 (highly decreased correlation) to 0 (no change in correlation) to +1 (highly increased correlation).
When comparing the protein dynamics of the unliganded to the cGMP-bound GAFab (Figure 5(c) and (d)), each subunit showed increased DRMSF values in several GAFa loop regions (e.g., β1/β2 loop, β4/α4 loop) as well as in the α4 helix that directly participates in cGMP binding [27]. In addition, cGMP binding to the GAFab homodimer results in differences in the behavior of the two subunits, with one subunit showing a more global increase in conformational dynamics in the GAFa domain and in the GAFb β4/β5 and β1/β2 loops (Figure 5(d)), whereas the other subunit showed more local changes in flexibility in the GAFa β4/α4 loop and α4 helix (Figure 5(c)). These DRMSF analyses of the cone PDE6 GAFab homodimer support the notion that the two subunits undergo different allosteric perturbations to several flexible loops in both the GAFa and GAFb domains upon binding of cGMP to the GAFa binding sites.
Examination of changes in the BSA profiles for the apo and cGMP-bound atomistic simulations (Figures S7D and S8D) reveal a change from a bimodal to unimodal profile upon cGMP binding to the GAFab homodimer. Analysis of each subdomain indicates that the increases in BSA upon cGMP binding occur primarily in the N-terminal α1 helix region preceding the GAFa domain as well as in the LH2 region that normally links GAFb to the catalytic domains of PDE6 (Figures S7D and S8D). Analysis of changes in BSA further supports the idea that cGMP binding to its GAFa binding pocket is predicted to induce conformational changes in several loop regions in both GAF domains, and also implicate cGMP-dependent perturbations of two helical elements that represent a portion of the “backbone” of the GAFab dimerization interface.
Finally, we compared the difference DCC (DDCC) maps of Cα–Cα motions between the apo and cGMP-liganded states of the two GAFab subunits (Figure 5(e)), which provides additional evidence for asymmetric behavior of the two subunits as well as correlated motions between several structural elements upon cGMP binding. For example, upon cGMP binding, one subunit (labeled chain A) showed a strong pattern of increased coordinated movements between the GAFa domain and the N-terminal region, the LH1 helix connecting GAFa to GAFb, and the LH2 region (attached to the catalytic domain of the intact PDE6 catalytic subunits). The other subunit (chain B) exhibited a very different DDCC map, with an alternating pattern of increased and decreased correlated motions between the GAFb β1/β2 loop and the rest of the GAFb domain as well a strong decrease in correlated movements between the GAFa domain and the GAFb β1/β2 loop (Figure 5(e)).
Taken together, these atomistic simulations provided evidence for the stability of the solution state fold of the PDE6 conformation observed in the crystal structure, differences in the conformational dynamics of each subunit, and highly coordinated allosteric perturbations upon cGMP binding.
Docking of Pγ to GAFab
Pγ-induced conformational changes of GAFab in the presence of cGMP
In order to determine the interface of interaction between cone GAFab and cone Pγ, we carried out XL-MS analysis and identified 89 intramolecular GAFab cross-links (Table S4) as well as 35 inter-molecular cross-links of a truncated cone Pγ subunit (Pγ1–58) with the GAFab dimer containing bound cGMP (Table S5). Because cone PDE6 is homodimeric (unlike rod PDE6), we were unable to uniquely assign inter-molecular cross-linked peptides of Pγ and GAFab to one of the two subunits; hence, all of the inter-molecular cross-links observed when GAFab was incubated with both Pγ1–58 and cGMP were initially applied to both subunits (see Materials and Methods). The resulting structural model using the rod Pγ as an initial template shows that both Pγ subunits interact similarly with GAFab (Figure 6(a)). The N-terminal residues of Pγ interact with the GAFa domain, specifically the α2/3 region and the α4 helix that is in close proximity to the cGMP binding site. The middle region of Pγ (amino acids 16–42) interacts with the GAFb domain, forming close associations with the β4/β5 loop, the α3 helix and the β1/β2 loop of GAFb. The remaining Pγ residues that could be resolved (amino acids 43–51) cross over the central helix to make contacts with the α2 and LH2 helices of the GAFb domain of the opposite chain (Figure 6(a)).
Figure 6.
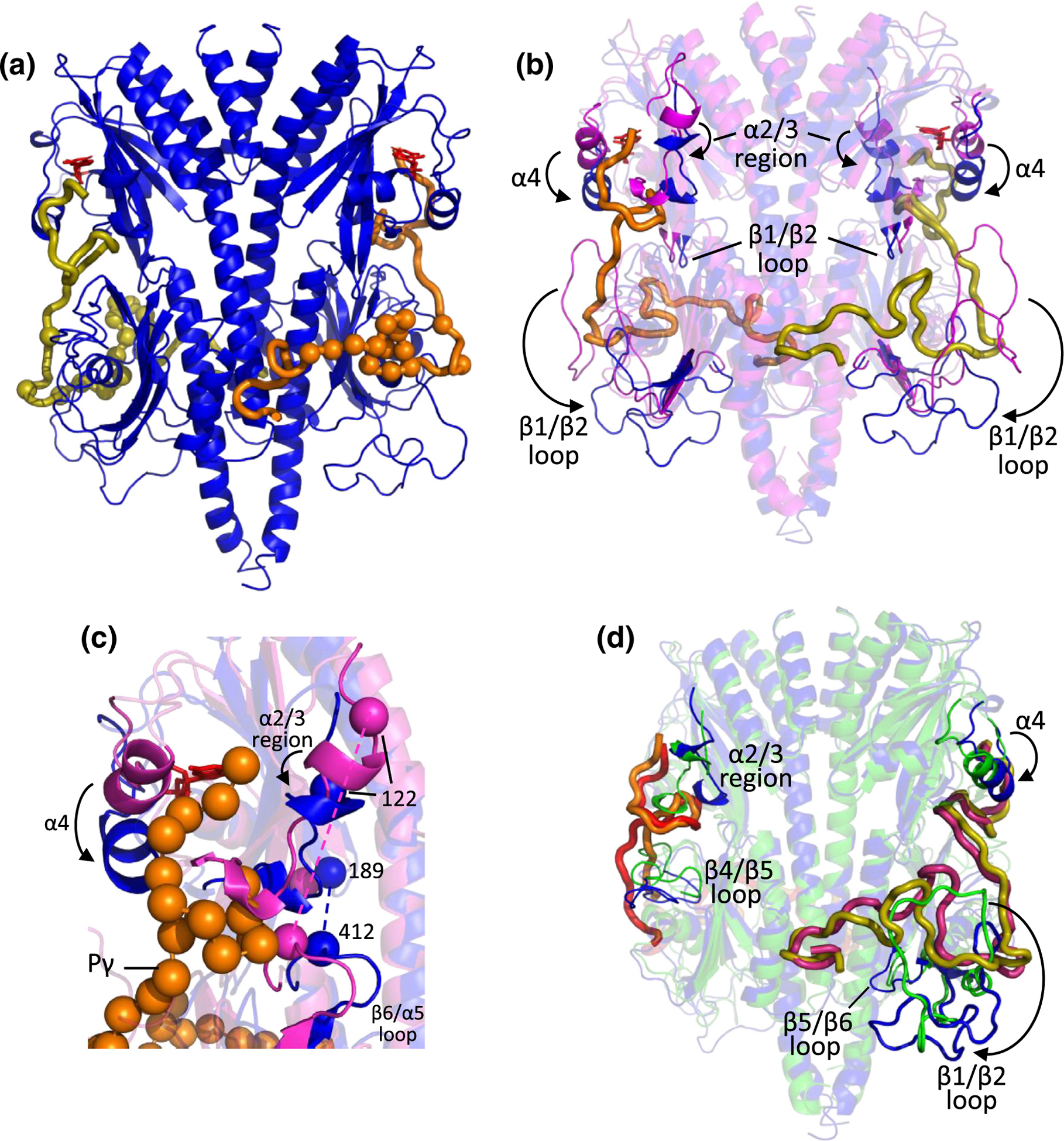
Integrative structural models of cone Pγ complexed with cone PDE6 GAFab in the presence and absence of bound cGMP. IMP was used to dock cone Pγ (residues 1 to 51; shown as thick lines) to the GAFab dimer, as described in Materials and Methods. (a) One Pγ (orange) primarily interacts with Chain A of GAFab·cGMP, while the other Pγ (olive) primarily interacts with Chain B. Spheres represent Pγ residues interacting with GAFab based on NMR results described in Figure 7. (b) Comparison of the cGMP-liganded GAFab structure in the absence (magenta) or presence of Pγ (blue; 180° rotation of (a)). Arrows indicate movement of structural elements upon Pγ binding. (c) Expanded view of the GAFa domain from panel B with orange spheres representing the Cα atoms of Pγ and blue and magenta spheres showing cross-linked residues described in the text. (d) Comparison of the Pγ-liganded GAFab structure in the presence of cGMP (blue; 180° rotation of (a)) or its absence (green, with Pγ colored red and magenta).
Structural alignment of GAFab·cGMP lacking Pγ (Figure 4(a)) with the structural model of Pγ docked to the GAFab·cGMP dimer (Figure 6(a)) showed a major displacement of the α4 helix and the α2/3 region of GAFa toward the GAFb domain resulting from Pγ binding (Figure 6(b); see also Figure S5), consistent with previous biochemical and mutagenesis studies of rod PDE6 [19,22,23,30,31]. As shown in Figure 6(c), a change in cross-linking of Lys412 in the GAFb β6/α5 loop to Lys122 (in the absence of Pγ) to Lys188 (in the presence of Pγ) highlights the potential for allosteric communication between the α2/3 region of GAFa and the β6/α5 loop of the GAFb domain upon Pγ binding. In addition, the binding of Pγ to GAFab·cGMP results in a major downward movement of GAFb β1/β2 loop (Figure 6(b)). These conformational changes in GAFab can be attributed in part to physical displacement of several structural elements of GAFab to accommodate the binding of Pγ to its sites of interaction in both the GAFa and GAFb domains.
Changes in the Pγ-GAFab complex in the presence or absence of cGMP
To identify cGMP-dependent changes in GAFab when Pγ is present, we also performed crosslinking of Pγ1–58 to GAFab lacking bound cGMP (Table S3 and S5). When compared with the fully liganded complex (Pγ-GAFab·cGMP), the Pγ-GAFab structural model demonstrated a similar trajectory of Pγ in its interactions with GAFab, with the exception of two regions (Pγ residues 21–27 and 34–38) that deviated from each other by up to ~12 Å in a cGMP-dependent manner (Figure 6(d)).
cGMP binding to Pγ-GAFab induced several significant conformational changes in both the GAFa and GAFb domains (Figure 6(d); Figure S5). Occupancy of the cGMP binding pocket in GAFa is accompanied by local changes in both the α2/3 region and the α4 helix that have been shown to form a “lid” over the cGMP binding site [27]. In the GAFb domain, cGMP binding to Pγ-GAFab resulted in a large downward movement of the β1/β2 loop away from the core of the GAFb domain. This conformational change upon cGMP binding allows several Pγ residues to interact with the β1/β2 loop which cannot occur in the Pγ-GAFab liganded state due to steric clashes. Also noted were significant cGMP-induced changes in the β4/β5 loop and in the β5/β6 loop adjacent to the LH2 helix near its C terminus (Figure 6(d)).
Characterization of cone Pγ1–58 and its binding to GAFab by solution NMR spectroscopy
Solution NMR spectroscopy was utilized to further characterize Pγ and its interactions with GAFab. Isotopically enriched Pγ1–58 (13C, 15N) was expressed in Escherichia coli, purified, and standard 2-dimensional (2D) and 3-dimensional (3D) solution NMR experiments were performed for Pγ assignments. Based on 3D HNCA, HNCACB, and HNCO spectra, 47 of the 58 Pγ 1HN and 15N backbone resonances were assigned (Figure 7(a)), with the exception of the 9 prolines and the first 2 amino acids of the sequence. A representative 3D HNCA strip plot is shown in Figure S10A.
Figure 7.
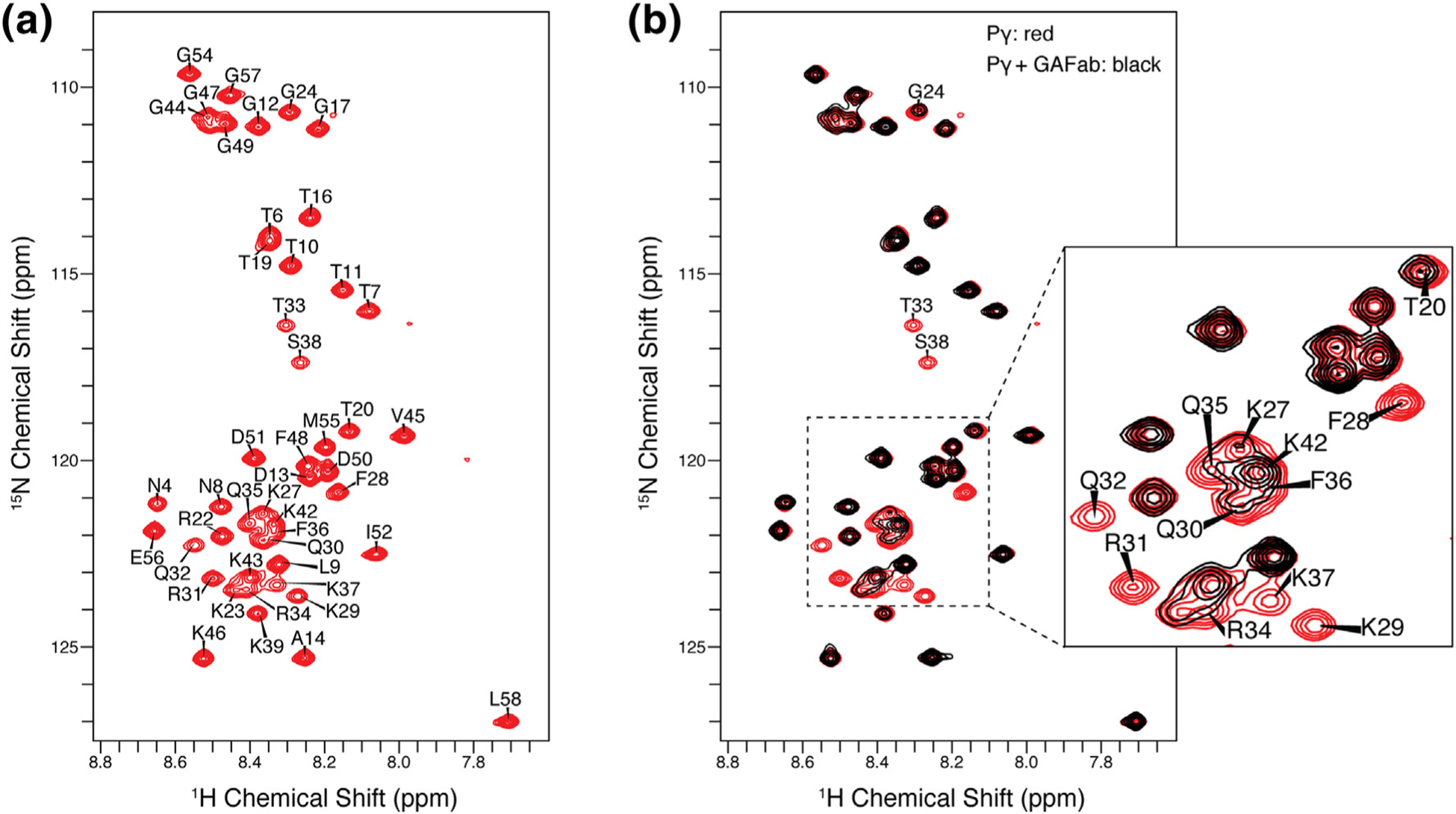
Pγ1–58 NMR resonance assignments and binding studies. (a) 2D 1H–15N HSQC NMR spectrum of Pγ in its unbound state. Narrow dispersion of 1H chemical shifts between 9.0 and 7.5 ppm is indicative of an intrinsically disordered protein. (b) The overlay of the 2D 1H–15N HSQC NMR spectra of the unbound Pγ (red spectrum) and Pγ bound to GAFab (black spectrum) illustrates that the addition of GAFab induced significant changes in the Pγ spectrum. The most significant line broadening was observed in the F28–S38 region of Pγ, indicative of the binding interface. Smaller signal attenuation was seen for neighboring residues and the C-terminal region.
The narrow dispersion of backbone 1H chemical shifts between 7.5 and 9.0 ppm in the 2D 1H–15N heteronuclear single quantum correlation (HSQC) spectrum is indicative of the intrinsically disordered nature of Pγ. In contrast, well-ordered protein spectra are typically characterized by a wider dispersion of backbone 1H resonances well beyond the 1.5-ppm range of intrinsically disordered proteins. The lack of well-defined secondary structure in Pγ1–58 is further supported by the absence of consecutive combined 13C secondary chemical shifts (Δδ13Cα–Δδ13Cβ) of ±2 ppm or more [32], as shown in Figure S10C.
To probe the binding interface of Pγ, we mixed the chicken cone Pγ fragment (isotopically enriched) with the regulatory GAFab domain (unlabeled, natural abundance). The addition of GAFab induced significant changes in the Pγ1–58 1H–15N HSQC spectrum (Figure 7(b)). The Pγ1–58 residues that make contact with GAFab are expected to exhibit the greatest chemical shift perturbations and NMR signal broadening resulting in signal attenuation. We observed that the N-terminal residues (N4–D13) were least affected in the presence of GAFab, as inferred not only by the lack of chemical shift perturbations but also by the similar NMR signal intensity of free and bound Pγ1–58 in this region of Pγ. All other Pγ residues exhibited some signal attenuation, with the most significant effects observed in the F28–S38 region of Pγ where the peaks were barely detectable and close to the baseline. Smaller attenuation was seen for neighboring residues (e.g. T20, G24, K27, and K42) and the C-terminal region, which suggests that these residues either interact with lower affinity or they are not directly interacting with GAFab yet are in close enough proximity to the binding interface to cause signal attenuation due to indirect effects. Only modest or no chemical shift perturbations of the Pγ1–58 spectra were observed upon GAFab addition, suggesting that the Pγ-GAFab interaction is in the intermediate exchange regime.
To investigate cGMP-induced conformational changes in binding of Pγ to GAFab, we also examined the NMR spectra under conditions where 0.9 mM cGMP was included with isotopically labeled Pγ1–58 and GAFab. While addition of cGMP to the Pγ-GAFab complex did not result in any major chemical shift perturbations to the spectra (Figure S10B), it did cause a global decrease in signal intensities, and some of the Pγ-interacting residues underwent a slight differential signal broadening and attenuation when compared to the Pγ-GAFab complex (Figure S10B and S10D).
Discussion
Allosteric communication pathway in the regulatory GAF domains of PDEs
This paper reports the first atomic-level crystal structure for the tandem GAF domains of cone PDE6 in conjunction with integrative structural modeling that leverages the distinct capabilities of XL-MS, NMR spectroscopy, and MD simulations to elucidate the protein dynamics and the allosteric communication pathway of PDE6 that are modulated by cGMP and Pγ binding to GAFab. For example, XL/MS is a medium-resolution approach requiring relatively small amounts of purified protein complexes and suitable for comparing different liganded states to understand the conformational changes induced by binding of cGMP and/or Pγ to GAFab. NMR spectroscopy, in contrast, was employed to obtain atomic-level determination of the structure of the small, intrinsically disordered Pγ subunit free in solution and upon its interaction with GAFab. In addition, we carried out MD simulations based on the x-ray structure and XL-MS structural models to gain insights into conformational dynamics and cGMP-induced changes in the GAFab structure.
The x-ray structure of the apo state of the GAFab dimer depicts the prototypical GAFa and GAFb domains [1] with parallel organization of the two subunits (Figure 2) consistent with other GAF-containing PDEs [3]. The PDE6 GAFab regulatory domains show the greatest structural and sequence homology with the PDE5 GAFab structure [7]. The cGMP-dependent conformational changes in the α4 helix, the β4-α4 loop, the β1/β2 loop, and the α2/3 region of PDE6 GAFa (Figure 4) are similar to those reported for cGMP binding to PDE5 GAFa and to PDE2 GAFb [7,8], but more evident when Pγ is also bound to PDE6 GAFab (Figure 6). The latter observation may explain why PDE5 undergoes direct allosteric activation of catalysis upon cGMP binding to GAFa [17], whereas for PDE6, direct allosteric communication between the GAF and catalytic domains is mediated by both cGMP binding to GAFa and by Pγ binding to both the regulatory and catalytic domains of PDE6 [24].
Figure 8 presents a working model for the allosteric communication network that signals binding of cGMP and/or changes in the multiple interactions of Pγ with the regulatory GAFab dimer of cone PDE6. Our XL-MS structural models and MD simulations predict that binding of cGMP to GAFa induces local conformational changes in the vicinity of the cGMP binding pocket (α2/3 region, β4/α4 loop, and α4 helix; Figure 4) as well as correlated conformational changes throughout the GAFa domain that include the GAFa β1/β2 loop (Figures 4(b) and 5(d)) and the LH1 helix that links GAFa to GAFb (Figure 5(e)). Although our.
Figure 8.
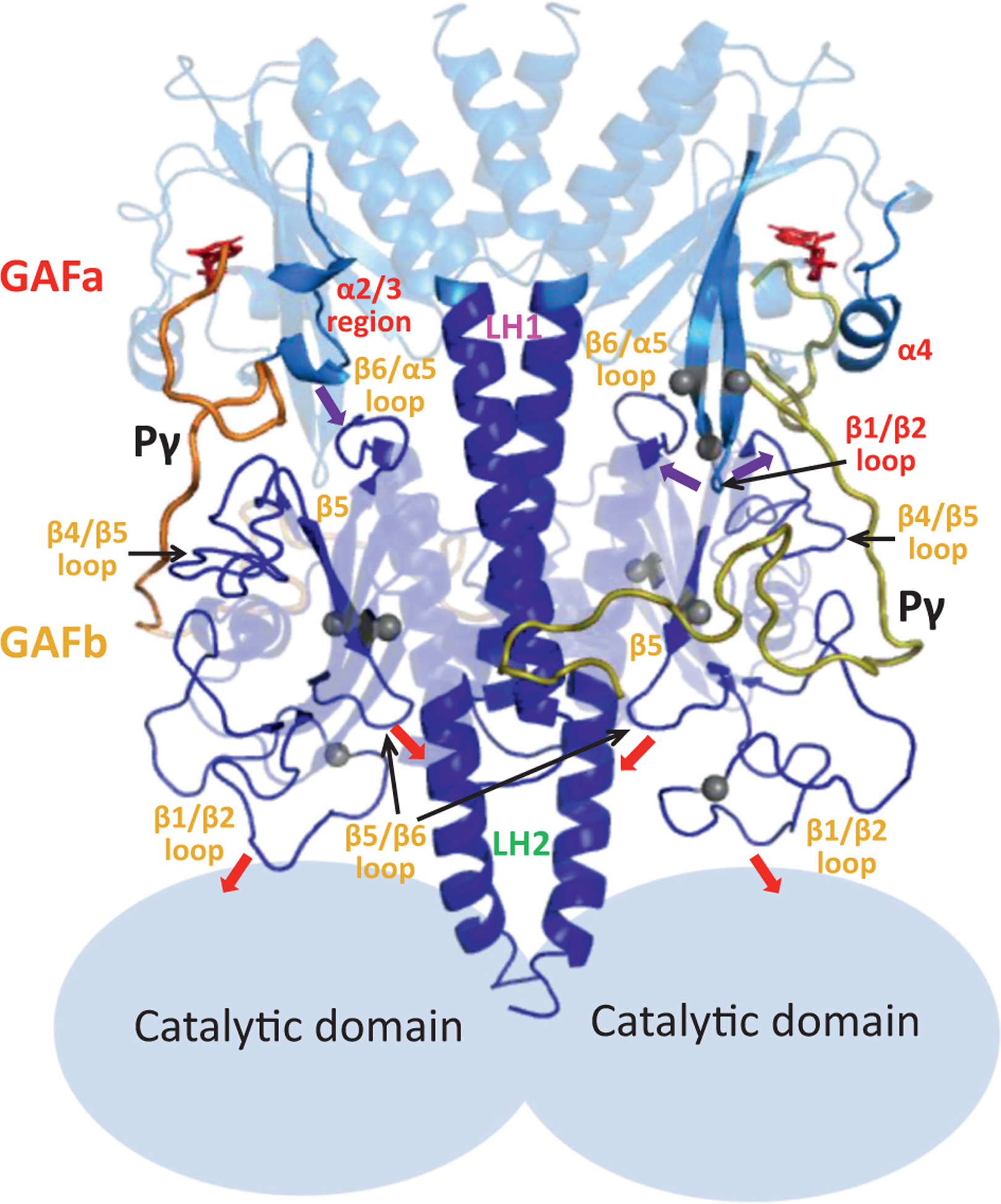
Model of allosteric communication within the regulatory tandem GAF domains. The GAFab structural model for the cGMP- and Pγ-bound state (Figure 6(a)) is shown with the major structural elements undergoing ligand-dependent conformational changes in the GAFa (red labels) and GAFb (orange labels). Pγ subunits are indicated by thick yellow and orange lines, while the catalytic domains are represented as light blue ovals based on the overall domain arrangement of rod PDE6 holoenzyme. Arrows indicate the proposed interactions that convey allosteric changes upon cGMP and/or Pγ binding, and gray spheres represent the sites of disease-causing mutations in human cone PDE6. See Discussion for details.
XL-MS and NMR results cannot distinguish between the two identical subunits of GAFab, the atomistic simulations predict that the GAFa domains of the two subunits behave differently upon cGMP binding (Figure 5(c)–(e)). It is noteworthy that conformational changes to GAFa upon cGMP binding are more pronounced when Pγ is bound, including the α2/3 region, α4 helix, and the β1/β2 loop of GAFa (Figure 6). We hypothesize that these elements reflect the structural basis for the reciprocal positive cooperativity between cGMP and Pγ binding to PDE6 [19,22,23].
Allosteric signal relay from GAFa to GAFb is proposed to occur at multiple sites at the interface of the two subdomains, notably the GAFa β1/β2 loop with the GAFb β4/β5 and β6/α5 loops, and the GAFa α2/3 region with the GAFb β6/α5 loop (Figure 8, purple arrows). These GAFb structural elements are hypothesized to propagate allosteric signals throughout the GAFb domain, including to the large β1/β2 loop and the β5/β6 loop. The extensive interactions of the central region of Pγ with GAFb (Figures 6 and 7) support the idea that the large cGMP-induced conformational changes observed in the GAFb β1/β2 loop and the β5/β6 loop (Figure 6(d)) may be mediated primarily by changes in Pγ interactions in this region.
Because the cone PDE6 catalytic domains are absent in this study, we can only speculate about how allosteric signals are conveyed from the GAFb domain to the catalytic domain. In analogy to PDE2 [8], we postulate that one allosteric pathway is via changes in conformational dynamics in the LH2 helix (Figures S7 and S8) induced by interactions with the GAFb β5/β6 loop or with the Pγ subunit (red arrows, Figure 8). We also point out that the dimerization interface between the two subunits (including the LH1 and LH2 “backbone” as well as interactions in both GAFa and GAFb between the α1/α2 loops of one subunit and the β5/β6 loops of the other subunit) may laterally transmit allosteric signals from one subunit to the other, as well as propagating conformational changes longitudinally from GAFa to GAFb (via the LH1 helix) and subsequently to the catalytic domains of the holoenzyme (via the LH2 helix). Another possible allosteric pathway has been postulated that links conformational changes in the GAFb β1/β2 loop (induced by cGMP or Pγ binding) with neighboring residues on the catalytic domain of rod PDE6 holoenzyme [10,11].
In summary, although rod and cone PDE6 may share with other GAF-containing PDEs homologous structural elements that likely serve to allosterically relay the binding of cyclic nucleotides from the GAF domains to the catalytic domains [7,8,13,33], we hypothesize that the binding of the intrinsically disordered Pγ with both the regulatory and catalytic domains of PDE6 has evolved with the vertebrate retina to meet the complex regulatory requirements of the visual transduction pathway in rod and cone photoreceptors [34,35].
Relevance of allosteric regulation of PDE6 to the visual signaling pathway in rod and cone photoreceptors
Upon photoexcitation of rhodopsin and subsequent activation of the photoreceptor G-protein (transducin), the transducin α-subunit (Gα*-GTP) rapidly binds to PDE6 and relieves the inhibitory constraint of the Pγ subunits; the resulting activation of PDE6 catalysis leads to a drop in cGMP levels, closure of cGMP-gated ion channels, and hyperpolarization of the membrane [18,36]. Rod and cone photoreceptors express homologs of these phototransduction proteins, yet the speed and sensitivity of the photoresponse are markedly different in rods and cones; differences in the regulation of rod and cone PDE6 activity are hypothesized to account for some of these differences [37,38].
Structural alignment of cone Pγ-GAFab·cGMP (Figure 6(a)) with the liganded state of rod PDE6 holoenzyme [10,11] reveals a high extent of conservation of overall secondary structure of rod and cone PDE6, with greater structural similarity in the GAFa (RMSD = 2.8 Å) than GAFb domains (RMSD = 7.8 Å; Figure S9). Within the GAFa domains, the α4 helix and the α2/3 helix (and adjacent residues) of cone GAFa are displaced away from the cGMP binding pocket compared with rod GAFa, providing a structural basis for the reported lower binding affinity of cGMP for the cone PDE6 isoform [39–41]. One notable difference between the GAFb domains of rod and cone PDE6 is the presence of an α-helical segment within the β1/β2 loop of rod, but not cone, GAFb that has been hypothesized to relay allosteric signals from the GAFb of one rod catalytic subunit to the catalytic domain of the other subunit [10,11]. We also note that the absence of the catalytic domains attached to the GAFb domains in this study of cone PDE6 does not permit us to evaluate the overall allosteric relay from GAFa through GAFb to the catalytic domains of the intact PDE6 holoenzyme that have been inferred from biochemical [24] and structural [42] studies of the rod PDE6 isoform.
Examination of the binding interface of rod and cone Pγ with their respective GAFab regulatory domains (Figure S9) reveals greater similarity within the GAFa domain compared to the GAFb domain, especially for the rod PDE6B subunit (Figure S9). Furthermore, the XL-MS (Figure 6) and NMR (Figure 7) results identifying multiple sites of interaction between the central region of Pγ and the GAFb domain in conjunction with conformational dynamics of several GAFb loops suggest that the GAFb domain serves as the central hub of allosteric communication within the PDE6 catalytic subunits. These structural differences in the binding of Pγ to the GAFb domain may contribute to the observed functional differences in the stoichiometry and mechanism of activation of rod and cone PDE6 by activated transducin α-subunit [11,38,43].
Understanding the molecular etiology of retinal diseases attributed to mutations in PDE6 catalytic subunits
Several inherited retinal diseases have been shown to be associated with mutations in the genes coding for rod PDE6A and PDE6B catalytic subunits (e.g., retinitis pigmentosa [44], congenital stationary night blindness [45]) or cone PDE6C catalytic subunit (autosomal recessive achromatopsia [46], cone dystrophy [47]). While missense mutations located within the catalytic domain of PDE6C can be directly attributed to disruption of the catalytic mechanism [48], the mechanisms underlying disease-causing mutations in the PDE6C GAFab domain remain unclear. Our identification of structural elements in cone GAFab that undergo ligand-dependent changes in conformation (Figure 8) provides a molecular basis for categorizing some of the known disease-causing mutations in PDE6C (see Figure 8, gray spheres). For example, several mutations have been identified in the vicinity of the GAFa β1/β2 loop, including Arg102Trp [49,50], Arg104Trp [48,51], and Glu109Val [50] that are likely to disrupt the function of this element of the allosteric communication network. The disease-causing mutation Leu298-His [50] is located in the GAFb β1/β2 loop (Figure 8) that undergoes major ligand-dependent conformational changes that have been proposed to relay allosteric signals to the rod PDE6 catalytic domain [10,11]. Our structural model also locates the Tyr323Asn (GAFb β2 strand [52]) and the P391L (GAFb β5 strand [51]) mutations in close proximity to the central region of Pγ (Figure 8), consistent with a report that heterologous expression of the Tyr323Asn mutant impairs Pγ affinity for the cone PDE6 catalytic dimer [48]. The structure of the PDE6 GAFab regulatory domains presented herein provides a molecular framework for understanding the molecular etiology of diseases arising from mutations in PDE6 and other GAF-containing PDEs [53,54] with the potential to design allosteric activators of PDEs targeting the regulatory GAF domains as candidates to intervene in a variety of human diseases [12].
Materials and Methods
Expression and purification of the tandem GAF domains of cone PDE6 and of truncated cone Pγ
The nucleotide sequence corresponding to amino acid residues 42–458 of the chicken cone PDE6C catalytic subunit (UniProtKB P52731) was obtained from a chicken retinal cDNA library (kindly provided by Dr. Susan Semple-Rowland [55]). The sequence-verified cone GAFab construct was found to have one amino acid difference from the published sequence at position 93 with an alanine instead of an arginine (see Figure S2); all other available bird PDE6C sequences also have alanine at this position. Cone GAFab was subcloned into pET47b (containing a C-terminal 6-His fusion tag), transformed into E. coli Rosetta cells, and grown at 37 °C in Luria Broth media to an OD600 of ~0.8. Then, 50 μM isopropyl-β-D-1-thiogalactopyranoside was added and the cells were incubated at 18 °C for 18 h. The cell pellet was resuspended in 20 mM Tris, 100 mM NaCl (pH 8.0) and disrupted by sonication. The recombinant protein was purified from the cell extract using a 1 ml HisTrap HP column with the GAFab protein being eluted from the resin with a buffer consisting of 100 mM imidazole, 100 mM NaCl and 20 mM Tris (pH 7.5). The affinity-purified protein was buffer exchanged with 50 mM Tris, 100 mM NaCl, 1 mM β-mercaptoethanol, 1 mM EDTA (pH 7.5) prior to Superdex 200 gel filtration chromatography. The apparent molecular weight and purity of GAFab were evaluated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Protein concentrations were determined by the bicinchoninic acid protein assay [56] using bovine gamma globulin as standard. For measurements of cGMP binding to GAFab, 1.5 nM GAFab was incubated with various amounts of radiolabeled cGMP (3 to 200 nM) for 1 h at room temperature followed by a filter binding assay as described previously [57].
The DNA sequence coding for the first 58 residues of chicken cone Pγ (Pγ1–58; PDE6H UniprotKB Q802E3) was inserted into the NdeI and BamHI sites of the pET11a vector, followed by transformation into the E. coli BL21(DE3) strain. A Pγ1–58 site-directed mutant in which lysines were substituted at Thr7 and at Thr11 was also constructed. Both constructs were transformed into E. coli BL21(DE3), grown on 2X-TY media at 37 °C until reaching an OD600 of ~0.6, and then protein expression induced with 0.3 mM isopropylthiogalactoside. Cells were then grown at 30 °C for 4 h after induction. Following cell harvesting, the bacterial extract was purified by HiTrap SP FF column (GE Healthcare). The Pγ construct was further purified by C18 reverse-phase high pressure liquid chromatography following standard procedures [58]. The purity of these proteins was determined to be >95% as evaluated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Protein concentrations were evaluated by the bicinchoninic protein assay.
X-ray structure determination
Purified chicken cone GAFab protein (8 mg/ml) was crystallized by the hanging drop method at 4 °C. The drop was prepared by mixing 2 μl GAFab sample with 2 μl well buffer that contains 0.1 M Na-Hepes (pH 7.5), 21–27% PEG3350, 0.2 M ammonium sulfate, and 20% glycerol. The crystals showed up in 10 days and grew to the maximum size in 4 weeks. The crystals have the space group of P65 with cell dimensions of a = b = 148.5, and c = 93.7 Å. The diffraction data of the unliganded GAFab were collected on the SERCAT beam line of APS and processed by HKL2000 [59] (Table 1). The structure determination was solved by the AutoBuild module of PHENIX [60], using the PDE5 GAF domains as the initial model [7]. The raw model of GAFab was rebuilt with program COOT [61] and refined by program REFMAC [62] (Table 1) to an R-factor of 0.207 for 16,489 reflections in the resolution of 50–3.2 Å.
Chemical cross-linking, in-gel digestion, and MS analysis
The manufacturer’s protocols were followed to perform chemical cross-linking reactions for each cross-linker. Chicken cone PDE6 GAFab was buffer exchanged into Hepes buffer (20 mM Hepes, 100 mM NaCl, 5 mM MgCl2 (pH 7.5)) and incubated with Pγ and/or cGMP. After incubating with cross-linker at RT for 1 h, proteins were separated with NuPAGE 4–12% Bis–Tris gels and visualized with Coomassie Brilliant Blue G-250.
Bands representing cross-linked proteins were in-gel digested and analyzed by LC–MS and LC–MS/MS essentially as described previously [9,11]. Briefly, protein bands were excised from the SDS-PAGE gel, washed with 25 mM NH4HCO3/50% acetonitrile, and treated with 10 mM DTT; sulfhydryl group were then alkylated with 55 mM iodoacetamide in 25 mM NH4HCO3. Dried gel pieces were treated with trypsin and asp-N (Promega) and the digested peptides sequentially extracted with 50% acetonitrile, 20% formic acid, and finally 100% acetonitrile; the samples were then concentrated to ~7 μl. One microliters of aliquots was injected into the Dionex Ultimate 3000 RSLC nano UHPLC system (Dionex Corporation Sunnyvale, CA) as previously described [11]. The eluant was introduced into the nano-electrospray ionization source of an LTQ Orbitrap XL mass spectrometer (Thermo Scientific, Waltham, MA), and LC–MS data acquired in an information-dependent acquisition mode. Full MS spectra were acquired in the Orbitrap (m/z 315–2000) with a resolution of 30,000 at m/z 400. The five most intense ions were selected for collision-induced dissociation (CID) fragmentation in the linear ion trap for MS/MS data acquisition [42].
Cross-linked peptide identification and structural modeling
Peak lists were created using PAVA for input into Protein Prospector. Data were initially searched against the full Swiss-Prot database to verify the proteins, and then the search was restricted to cone PDE6 subunits. In experiments where mutant or truncated proteins were used, all protein sequences were input as user-defined proteins. Cross-linked peptides were identified using an integrated module in Protein Prospector, based on a bioinformatic strategy described previously [42,63,64].
The GAFab x-ray crystal structure was used as the template, and cross-linking data were applied as distance restraints in Modeller [29] to obtain the cross-link refined integrative structural models of GAFab under all four condition as discussed above. Symmetry was enforced for the two subunits. The comparative model with lowest DOPE score (out of 10) was selected.
To perform docking of Pγ to GAFab incubated with cGMP (Figure 6(a)), the Integrated Modeling Platform [65] (IMP) was used, with the following segments of Pγ treated as individual rigid bodies: residues 1–6, 11–23, 29–31, 37–43, and 46–51. Pγ fragments 1–6 and 11–23 were based on a homology model of cone Pγ created using rod Pγ from the rod PDE6 cryo-EM structure as a template [10]. The remaining Pγ peptide fragments were generated in silico [66] assuming a linearly extended conformation. The absence of reactive lysine residues in the first 23 amino acid residues of the cone Pγ sequence led us to create a site-directed Pγ1–58 mutant that introduced lysine residues at Thr7 and at Thr11. With this Pγ1–58 double mutant, additional cross-linked peptides between GAFab and Pγ could be identified.
All of the inter-molecular cross-links observed when GAFab was incubated with both Pγ1–58 and cGMP were initially applied to both subunits. In three instances (GAFab residues Lys249, Lys409, and Lys412), cross-links between a single Pγ and the GAFab dimer were assigned to the other chain of GAFab either because they violated the distance restraint or because of steric hindrance.
To dock the Pγ fragments to GAFab, IMP was run as described previously [11]. The best-fitting model obtained from IMP was run in Modeller using the same cross-linking restraints in order to fill in missing Pγ residues and to add missing atoms to each subunit.
For the case where cross-linking data were collected for the GAFab dimer incubated with Pγ1–58 in the absence of cGMP (Figure 6(d)), the docking of Pγ to GAFab relied on the Pγ conformation determined for the cGMP-liganded GAFab dimer as the initial template for Modeller.
To quantify potential differences between the two subunits of GAFab and to assess conformational changes occurring upon ligand binding, analysis of the root mean square deviations (RMSD) of our structural model with other available structures was carried out using Visual Molecular Dynamics (VMD) software version 1.9.3 [67].
System setup and MD simulation details
VMD [67] was used to prepare all systems and analyze simulation trajectories. All simulations were performed using the NAMD software [68] with three different structures of cone PDE6 GAFab, namely the x-ray structure and the structural models (consisting of residues 42–458) of the unliganded (apo) and cGMP-bound GAFab states. We note that the x-ray structure for GAFab does not contain a portion of the GAFb β1/β2 flexible loop (residues 286–310), whereas the two cross-link refined structural models include this loop. In the GAFab·cGMP structural model, a cGMP molecule was docked in each GAFa binding site based on the atomic coordinates of cGMP reported for the published chicken cone GAF structure (PDB ID: 3DBA).
The three structures were solvated with TIP3P water molecules and the systems were neutralized with NaCl and MgCl2. After 500 steps of conjugate-gradient minimization, we equilibrated the volume of the simulation domain for each system by conducting a short 1 ns MD simulation in the NPT ensemble, after which we conducted three independent long time-scale MD simulations of each system (Table S6). In all simulations, we used a time-step of 2 fs and the CHARMM force-field for all molecules [69–71]. The temperature was maintained (at 310 K) using a Langevin thermostat and the pressure (at 1 atm) using a Nosé–Hoover barostat.
MD conformational metrics
RMSD/RMSF
To understand the domain-level flexibility of GAFab in various states (crystal structure, apo, and cGMP-bound states), we computed the RMSD as a metric based on the Cα atoms, where RMSD was measured relative to the initial structure in each simulation. A higher RMSD relative to the initial structure would indicate increased flexibility and vice versa. The probability distributions of RMSDs of various domains of GAFab are shown in Figures S6–S8. To quantify the conformational flexibility of each residue, we further computed RMSF per residue for each subunit of GAFab; the two identical subunits were distinguished by labeling the second subunit with an apostrophe. The RMSF calculations were based on all atoms in each residue. The RMSF data are shown in Figures S6–S8.
BSA
To characterize interfacial area between a pair of domains within PDE6, we calculated BSA between domains using the following equation: BSA = SASAa + SASAa′ − SASAaa′ where SASAa, SASAa′, and SASAaa′ are the solvent-accessible surface areas (SASA) of each domain individually or both domains taken together. We used a probe radius of 1.4 Å for SASA calculations.
DCC Analysis
We also carried out residue–residue (Cα–Cα) DCC analysis for the apo and cGMP-bound states of GAFab using MD-TASK [72]. Specifically, for a pair of atoms i and j, the correlation coefficient Cij was computed using time-averaged displacements (Δri and Δrj) from the mean positions.
NMR spectroscopy
Uniformly 13C, 15N-enriched Pγ1–58 was produced by expressing the protein in M9 minimal medium supplemented with 15NH4Cl and 13C6-glucose (for uniform labeling with nitrogen-15 and carbon-13), respectively. After purification, Pγ1–58 was resuspended in a buffer containing 20 mM Tris, 50 mM NaCl, 1 mM MgCl2 (pH 7.5). D2O, NaN3, and DSS were added for a final concentration of 5% v/v, 1 mM, and 200 μM, respectively. NMR spectra were collected on a Bruker Avance III HD 700 MHz NMR spectrometer equipped with a quadruple resonance inverse QCI-F CryoProbe at the City University of New York Advanced Science Research Center (CUNY ASRC) Biomolecular NMR Facility and on a Bruker Avance NEO 700 MHz NMR spectrometer equipped with a 5-mm triple-resonance inverse TCI CryoProbe at the University of New Hampshire Instrumentation Center NMR facility. For backbone and side-chain assignments, 2D 1H–15N HSQC, and 3D HNCA, HNCACB, and HNCO spectra were collected of 50 μM uniformly 13C,15N-enriched Pγ1–58. To investigate the binding properties of Pγ and GAFab, uniformly 13C,15N-enriched Pγ1–58 (20 μM) and natural abundance GAFab (72 μM) were mixed, and 2D 1H–15N HSQC spectra of Pγ1–58 were acquired (with and without GAFab) at 21 °C. To probe the effects of cGMP binding to GAFab on GAFab–Pγ interactions, 0.9 mM cGMP was added and spectra collected as above. All NMR data were processed using NMRPipe [73]. Analysis and assignments of the 2D and 3D data sets were carried out using NMRFAM-Sparky [74]. The assignment process was facilitated by using the PINE server for initial automated assignments [75,76] before completing the assignments manually. 13C chemical shifts are sensitive reporters of protein secondary structure [77], and secondary chemical shifts are defined as the observed chemical shift deviation from average random coil values. Secondary chemical shifts were generated using NMRFAM-Sparky [74]. Changes in peak intensity upon addition of GAFab and, subsequently, cGMP were defined as the ratio of the peak volume after (I) and before (I0) addition of unlabeled GAFab with or without cGMP to isotopically enriched Pγ1–58 [78].
Accession numbers
The crystal structure and diffraction data have been deposited in the RCSB Protein Data Bank with accession number PDBID: 6×88. Chemical shifts have been deposited in the Biological Magnetic Resonance Data Bank (BMRB) with accession number 28133. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository [79] with the dataset identifier PXD020817.
Supplementary Material
Acknowledgments
We thank Suzanne L. Matte for assistance with the preparation of proteins used in this study, Dr. Karyn B. Cahill for the initial work on optimizing expression of the GAFab protein, Michael Irwin for assistance with integrative structural modeling, and Dr. James Aramini (City University of New York Advanced Science Research Center (CUNY ASRC) Biomolecular NMR Facility) for his assistance with the NMR data collection. This work was supported by the National Eye Institute grant R01 EY05798 (R.H.C.), the National Institute of General Medical Sciences grants P20 GM113131 (R.H.C.) and GM059791 (H.K.), P20GM103449 (Vermont Genetics Network Proteomics Core Facility), National Science Foundation grant CLF 1307367 (F.C.), National Institute of Child Health and Human Development grant R01 HD093783 (F.C.), and National Science Foundation Major Research Instrumentation Award DBI-1828319 (K.V.). The computations were performed in part using the NSF-supported (ACI-1548562) Extreme Science and Engineering Discovery Environment (XSEDE) Comet resource at San Diego Supercomputer Center under grant TGMCB160183 (H.V.) and the National Science Foundation EPSCoR grant OIA-1757371 (H.V). Institutional support was provided through UNH CoRE grant (H.V., F.C, R.H.C., and K.V.) and through the UNH Research Computing Center central shared HPC Premise cluster (H.V.).
Abbreviations used:
- PDE
phosphodiesterase
- Pγ
inhibitory γ-subunit of photoreceptor PDE6
- XL-MS
chemical cross-linking-mass spectrometry
- RMSD
root mean square deviations
- RMSF
root mean square fluctuations
- MD
molecular dynamics
- BSA
buried surface area
- DCC
dynamic residue-residue cross correlation
- BS3
bis(sulfosuccinimidyl)suberate
- HSQC
heteronuclear single quantum correlation
- CID
collision-induced dissociation
- IMP
Integrated Modeling Platform
Footnotes
Declaration of Competing Interests
The authors declare no competing financial or non-financial interests.
Appendix A. Supplementary Data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2020.08.026.
References
- 1.Aravind L, Ponting CP, (1997). The GAF domain: an evolutionary link between diverse phototransducing proteins. Trends Biochem. Sci, 22, 458–459. [DOI] [PubMed] [Google Scholar]
- 2.Anantharaman V, Koonin EV, Aravind L, (2001). Regulatory potential, phyletic distribution and evolution of ancient, intracellular small-molecule-binding domains. JMB, 307, 1271–1292. [DOI] [PubMed] [Google Scholar]
- 3.Zoraghi R, Corbin JD, Francis SH, (2004). Properties and functions of GAF domains in cyclic nucleotide phosphodiesterases and other proteins. Mol. Pharmacol, 65, 267–278. [DOI] [PubMed] [Google Scholar]
- 4.Conti M, Beavo J, (2007). Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu. Rev. Biochem, 76, 481–511. [DOI] [PubMed] [Google Scholar]
- 5.Francis SH, Blount MA, Corbin JD, (2011). Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol. Rev, 91, 651–690. [DOI] [PubMed] [Google Scholar]
- 6.Martinez SE, Wu AY, Glavas NA, Tang XB, Turley S, Hol WG, et al. , (2002). The two GAF domains in phosphodiesterase 2A have distinct roles in dimerization and in cGMP binding. Proc. Natl. Acad. Sci. U. S. A, 99, 13260–13265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H, Robinson H, Ke H, (2010). Conformation changes, N-terminal involvement, and cGMP signal relay in the phosphodiesterase-5 GAF domain. J. Biol. Chem, 285, 38149–38156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pandit J, Forman MD, Fennell KF, Dillman KS, Menniti FS, (2009). Mechanism for the allosteric regulation of phosphodiesterase 2A deduced from the X-ray structure of a near full-length construct. Proc. Natl. Acad. Sci. U. S. A, 106, 18225–18230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeng-Elmore X, Gao XZ, Pellarin R, Schneidman-Duhovny D, Zhang XJ, Kozacka KA, et al. , (2014). Molecular architecture of photoreceptor phosphodiesterase elucidated by chemical cross-linking and integrative modeling. J. Mol. Biol, 426, 3713–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gulati S, Palczewski K, Engel A, Stahlberg H, Kovacik L, (2019). Cryo-EM structure of phosphodiesterase 6 reveals insights into the allosteric regulation of type I phosphodiesterases. Sci Adv, 5, eaav4322,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Irwin MJ, Gupta R, Gao XZ, Cahill KB, Chu F, Cote RH, (2019). The molecular architecture of photoreceptor phosphodiesterase 6 (PDE6) with activated G protein elucidates the mechanism of visual excitation. J. Biol. Chem, 294, 19486–19497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baillie GS, Tejeda GS, Kelly MP, (2019). Therapeutic targeting of 3′,5′-cyclic nucleotide phosphodiesterases: inhibition and beyond. Nat. Rev. Drug Discov, 18, 770–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heikaus CC, Pandit J, Klevit RE, (2009). Cyclic nucleotide binding GAF domains from phosphodiesterases: structural and mechanistic insights. Structure, 17, 1551–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gross-Langenhoff M, Hofbauer K, Weber J, Schultz A, Schultz JE, (2006). cAMP is a ligand for the tandem GAF domain of human phosphodiesterase 10 and cGMP for the tandem GAF domain of phosphodiesterase 11. J. Biol. Chem, 281, 2841–2846. [DOI] [PubMed] [Google Scholar]
- 15.Jager R, Russwurm C, Schwede F, Genieser HG, Koesling D, Russwurm M, (2012). Activation of PDE10 and PDE11 phosphodiesterases. J. Biol. Chem, 287, 1210–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martins TJ, Mumby MC, Beavo JA, (1982). Purification and characterization of a cyclic GMP-stimulated cyclic nucleotide phosphodiesterase from bovine tissues. J. Biol. Chem, 257, 1973–1979. [PubMed] [Google Scholar]
- 17.Rybalkin SD, Rybalkina IG, Shimizu-Albergine M, Tang XB, Beavo JA, (2003). PDE5 is converted to an activated state upon cGMP binding to the GAF A domain. EMBO J, 22, 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cote RH, (2006). Photoreceptor phosphodiesterase (PDE6): a G-protein-activated PDE regulating visual excitation in rod and cone photoreceptor cells in: Beavo JA, Francis SH, Houslay MD (Eds.), Cyclic Nucleotide Phosphodiesterases in Health and Disease, CRC Press, Boca Raton, FL: 2006, pp. 165–193. [Google Scholar]
- 19.Mou H, Cote RH, (2001). The catalytic and GAF domains of the rod cGMP phosphodiesterase (PDE6) heterodimer are regulated by distinct regions of its inhibitory g subunit. J. Biol. Chem, 276, 27527–27534. [DOI] [PubMed] [Google Scholar]
- 20.Matte SL, Laue TM, Cote RH, (2012). Characterization of conformational changes and protein–protein interactions of rod photoreceptor phosphodiesterase (PDE6). J. Biol. Chem, 287, 20111–20121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heikaus CC, Stout JR, Sekharan MR, Eakin CM, Rajagopal P, Brzovic PS, et al. , (2008). Solution structure of the cGMP binding GAF domain from phosphodiesterase 5: insights into nucleotide specificity, dimerization, and cGMP-dependent conformational change. J. Biol. Chem, 283, 22749–22759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamazaki A, Bartucci F, Ting A, Bitensky MW, (1982). Reciprocal effects of an inhibitory factor on catalytic activity and noncatalytic cGMP binding sites of rod phosphodiesterase. Proc. Natl. Acad. Sci. U. S. A, 79, 3702–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cote RH, Bownds MD, Arshavsky VY, (1994). cGMP binding sites on photoreceptor phosphodiesterase: role in feedback regulation of visual transduction. Proc. Natl. Acad. Sci. U. S. A, 91, 4845–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang XJ, Cahill KB, Elfenbein A, Arshavsky VY, Cote RH, (2008). Direct allosteric regulation between the GAF domain and catalytic domain of photoreceptor phosphodiesterase PDE6. J. Biol. Chem, 283, 29699–29705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turko IV, Ballard SA, Francis SH, Corbin JD, (1999). Inhibition of cyclic GMP-binding cyclic GMP-specific phosphodiesterase (type 5) by sildenafil and related compounds. Mol. Pharmacol, 56, 124–130. [DOI] [PubMed] [Google Scholar]
- 26.Gopalakrishna KN, Boyd K, Yadav RP, Artemyev NO, (2016). Aryl hydrocarbon receptor-interacting protein-like 1 is an obligate chaperone of phosphodiesterase 6 and is assisted by the gamma-subunit of its client. J. Biol. Chem, 291, 16282–16291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez SE, Heikaus CC, Klevit RE, Beavo JA, (2008). The structure of the GAF A domain from phosphodiesterase 6C reveals determinants of cGMP binding, a conserved binding surface, and a large cGMP-dependent conformational change. J. Biol. Chem, 283, 25913–25919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang D, Hinds TR, Martinez SE, Doneanu C, Beavo JA, (2004). Molecular determinants of cGMP-binding to chicken cone photoreceptor phosphodiesterase. J. Biol. Chem, 279, 48143–48151. [DOI] [PubMed] [Google Scholar]
- 29.Webb B, Sali A, (2016). Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein. Sci, 86, 2.9.1–2.9.37. [DOI] [PubMed] [Google Scholar]
- 30.Muradov KG, Granovsky AE, Schey KL, Artemyev NO, (2002). Direct interaction of the inhibitory g-subunit of rod cGMP phosphodiesterase (PDE6) with the PDE6 GAFa domains. Biochemistry, 41, 3884–3890. [DOI] [PubMed] [Google Scholar]
- 31.Muradov H, Boyd KK, Artemyev NO, (2004). Structural determinants of the PDE6 GAF A domain for binding the inhibitory gamma-subunit and noncatalytic cGMP. Vis. Res, 44, 2437–2444. [DOI] [PubMed] [Google Scholar]
- 32.Marsh JA, Singh VK, Jia Z, Forman-Kay JD, (2006). Sensitivity of secondary structure propensities to sequence differences between alpha- and gamma-synuclein: implications for fibrillation. Protein Sci, 15, 2795–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schultz JE, (2009). Structural and biochemical aspects of tandem GAF domains. Handb. Exp. Pharmacol, 93–109. [DOI] [PubMed] [Google Scholar]
- 34.Lagman D, Franzen IE, Eggert J, Larhammar D, Abalo XM, (2016). Evolution and expression of the phosphodiesterase 6 genes unveils vertebrate novelty to control photo-sensitivity. BMC Evol. Biol, 16, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamb TD, Hunt DM, (2017). Evolution of the vertebrate phototransduction cascade activation steps. Dev. Biol, 431, 77–92. [DOI] [PubMed] [Google Scholar]
- 36.Arshavsky VY, Burns ME, (2012). Photoreceptor signaling: supporting vision across a wide range of light intensities. J. Biol. Chem, 287, 1620–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Korenbrot JI, (2012). Speed, sensitivity, and stability of the light response in rod and cone photoreceptors: facts and models. Prog. Retin. Eye Res, 31, 442–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Plachetzki DC, Cote RH, (2019). The N termini of the inhibitory g-subunits of phosphodiesterase-6 (PDE6) from rod and cone photoreceptors differentially regulate transducin-mediated PDE6 activation. J. Biol. Chem, 294, 8351–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gillespie PG, Beavo JA, (1988). Characterization of a bovine cone photoreceptor phosphodiesterase purified by cyclic GMP-Sepharose chromatography. J. Biol. Chem, 263, 8133–8141. [PubMed] [Google Scholar]
- 40.Gillespie PG, Beavo JA, (1989). cGMP is tightly bound to bovine retinal rod phosphodiesterase. Proc. Natl. Acad. Sci. U. S. A, 86, 4311–4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mou H, Grazio HJ, Cook TA, Beavo JA, Cote RH, (1999). cGMP binding to noncatalytic sites on mammalian rod photoreceptor phosphodiesterase is regulated by binding of its g and d subunits. J. Biol. Chem, 274, 18813–18820. [DOI] [PubMed] [Google Scholar]
- 42.Chu F, Hogan D, Gupta R, Gao XZ, Nguyen HT, Cote RH, (2019). Allosteric regulation of rod photoreceptor phosphodiesterase 6 (PDE6) elucidated by chemical cross-linking and quantitative mass spectrometry. J. Mol. Biol, 243, 3677–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qureshi BM, Behrmann E, Schoneberg J, Loerke J, Burger J, Mielke T, et al. , (2018). It takes two transducins to activate the cGMP-phosphodiesterase 6 in retinal rods. Open Biol, 8, 180075, 10.1098/rsob. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daiger SP, Sullivan LS, Bowne SJ, (2013). Genes and mutations causing retinitis pigmentosa. Clin. Genet, 84, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsang SH, Sharma T, (2018). Congenital stationary night blindness. Adv. Exp. Med. Biol, 1085, 61–64. [DOI] [PubMed] [Google Scholar]
- 46.Remmer MH, Rastogi N, Ranka MP, Ceisler EJ, (2015). Achromatopsia: a review. Curr. Opin. Ophthalmol, 26, 333–340. [DOI] [PubMed] [Google Scholar]
- 47.Tsang SH, Sharma T, (2018). Progressive cone dystrophy and cone-rod dystrophy (XL, AD, and AR). Adv. Exp. Med. Biol, 1085, 53–60. [DOI] [PubMed] [Google Scholar]
- 48.Gopalakrishna KN, Boyd K, Artemyev NO, (2017). Mechanisms of mutant PDE6 proteins underlying retinal diseases. Cell. Signal, 37, 74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weisschuh N, Stingl K, Audo I, Biskup S, Bocquet B, Branham K, et al. , (2018). Mutations in the gene PDE6C encoding the catalytic subunit of the cone photoreceptor phosphodiesterase in patients with achromatopsia. Hum. Mutat, 39, 1366–1371. [DOI] [PubMed] [Google Scholar]
- 50.Georgiou M, Robson AG, Singh N, Pontikos N, Kane T, Hirji N, et al. , (2019). Deep phenotyping of PDE6C-associated achromatopsia. Invest. Ophthalmol. Vis. Sci, 60, 5112–5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grau T, Artemyev NO, Rosenberg T, Dollfus H, Haugen OH, Cumhur SE, et al. , (2011). Decreased catalytic activity and altered activation properties of PDE6C mutants associated with autosomal recessive achromatopsia. Hum. Mol. Genet, 20, 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thiadens AA, den Hollander AI, Roosing S, Nabuurs SB, Zekveld-Vroon RC, Collin RW, et al. , (2009). Homozygosity mapping reveals PDE6C mutations in patients with early-onset cone photoreceptor disorders. Am. J. Hum. Genet, 85, 240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mencacci NE, Kamsteeg EJ, Nakashima K, R’Bibo L, Lynch DS, Balint B, et al. , (2016). De novo mutations in PDE10A cause childhood-onset chorea with bilateral striatal lesions. Am. J. Hum. Genet, 98, 763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Diggle CP, Sukoff Rizzo SJ, Popiolek M, Hinttala R, Schulke JP, Kurian MA, et al. , (2016). Biallelic mutations in PDE10A Lead to loss of striatal PDE10A and a hyperkinetic movement disorder with onset in infancy. Am. J. Hum. Genet, 98, 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Semple-Rowland SL, Green DA, (1994). Molecular characterization of the a′-subunit of cone photoreceptor cGMP phosphodiesterase in normal and rd chicken. Exp. Eye Res, 59, 365–372. [DOI] [PubMed] [Google Scholar]
- 56.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, et al. , (1985). Measurement of protein using bicinchoninic acid. Anal. Biochem, 150, 76–85. [DOI] [PubMed] [Google Scholar]
- 57.Cote RH, (2005). cGMP binding to the regulatory GAF domains of photoreceptor phosphodiesterase (PDE6). Methods Mol. Biol, 307, 141–154. [DOI] [PubMed] [Google Scholar]
- 58.Artemyev NO, Arshavsky VY, Cote RH, (1998). Photoreceptor phosphodiesterase: interaction of inhibitory g subunit and cyclic GMP with specific binding sites on catalytic subunits. Methods, 14, 93–104. [DOI] [PubMed] [Google Scholar]
- 59.Otwinowski Z, Minor W, (1997). Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 60.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, et al. , (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Emsley P, Lohkamp B, Scott WG, Cowtan K, (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Winn MD, Murshudov GN, Papiz MZ, (2003). Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol, 374, 300–321. [DOI] [PubMed] [Google Scholar]
- 63.Chu F, Baker PR, Burlingame AL, Chalkley RJ, (2010). Finding chimeras: a bioinformatics strategy for identification of cross-linked peptides. Mol. Cell. Proteomics, 9, 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trnka MJ, Baker PR, Robinson PJ, Burlingame AL, Chalkley RJ, (2014). Matching cross-linked peptide spectra: only as good as the worse identification. Mol. Cell. Proteomics, 13, 420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Webb B, Lasker K, Velazquez-Muriel J, Schneidman-Duhovny D, Pellarin R, Bonomi M, et al. , (2014). Modeling of proteins and their assemblies with the Integrative Modeling Platform. Methods Mol. Biol, 1091, 277–295. [DOI] [PubMed] [Google Scholar]
- 66.Bitencourt-Ferreira G, de Azevedo WF Jr., (2019). Molecular docking simulations with ArgusLab. Methods Mol. Biol, 2053, 203–220. [DOI] [PubMed] [Google Scholar]
- 67.Humphrey W, Dalke A, Schulten K, (1996). VMD: visual molecular dynamics. J. Mol. Graph, 14, 33–38 (27–8). [DOI] [PubMed] [Google Scholar]
- 68.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. , (2005). Scalable molecular dynamics with NAMD. J. Comput. Chem, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, et al. , (1998). All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- 70.Mackerell AD Jr., Feig M, Brooks CL 3rd., (2004). Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem, 25, 1400–1415. [DOI] [PubMed] [Google Scholar]
- 71.Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, et al. , (2017). CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brown DK, Penkler DL, Sheik Amamuddy O, Ross C, Atilgan AR, Atilgan C, et al. , (2017). MD-TASK: a software suite for analyzing molecular dynamics trajectories. Bioinformatics, 33, 2768–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A, (1995). NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR, 6, 277–293. [DOI] [PubMed] [Google Scholar]
- 74.Lee W, Tonelli M, Markley JL, (2015). NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics, 31, 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bahrami A, Assadi AH, Markley JL, Eghbalnia HR, (2009). Probabilistic interaction network of evidence algorithm and its application to complete labeling of peak lists from protein NMR spectroscopy. PLoS Comput. Biol, 5, e1000307,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee W, Westler WM, Bahrami A, Eghbalnia HR, Markley JL, (2009). PINE-SPARKY: graphical interface for evaluating automated probabilistic peak assignments in protein NMR spectroscopy. Bioinformatics, 25, 2085–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wishart DS, Sykes BD, (1994). The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR, 4, 171–180. [DOI] [PubMed] [Google Scholar]
- 78.Li Y, Jain N, Limpanawat S, To J, Quistgaard EM, Nordlund P, et al. , (2015). Interaction between human BAP31 and respiratory syncytial virus small hydrophobic (SH) protein. Virology, 482, 105–110. [DOI] [PubMed] [Google Scholar]
- 79.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, et al. , (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res, 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.