

Abstract
The cholinergic anti-inflammatory reflex (CAIR) represents an important homeostatic regulatory mechanism for sensing and controlling the body’s response to inflammatory stimuli. Vagovagal reflexes are an integral component of CAIR whose anti-inflammatory effects are mediated by acetylcholine (ACh) acting at α7 nicotinic acetylcholine receptors (α7nAChR) located on cells of the immune system. Recently, it is appreciated that CAIR and α7nAChR also participate in the control of metabolic homeostasis. This has led to the understanding that defective vagovagal reflex circuitry underlying CAIR might explain the coexistence of obesity, diabetes, and inflammation in the metabolic syndrome. Thus, there is renewed interest in the α7nAChR that mediates CAIR, particularly from the standpoint of therapeutics. Of special note is the recent finding that α7nAChR agonist GTS-21 acts at L-cells of the distal intestine to stimulate the release of two glucoregulatory and anorexigenic hormones: glucagon-like peptide-1 (GLP-1) and peptide YY (PYY). Furthermore, α7nAChR agonist PNU 282987 exerts trophic factor-like actions to support pancreatic β-cell survival under conditions of stress resembling diabetes. This review provides an overview of α7nAChR function as it pertains to CAIR, vagovagal reflexes, and metabolic homeostasis. We also consider the possible usefulness of α7nAChR agonists for treatment of obesity, diabetes, and inflammation.
Keywords: α7nAChR, cholinergic anti-inflammatory reflex, obesity, diabetes, GLP-1, PYY
1. Introduction
The pathogenesis of obesity and type 2 diabetes (T2D) are intertwined in that these two disorders participate in a metabolic syndrome in which abnormalities of appetite control, body weight, energy expenditure, glycemia, and lipid metabolism exist in combination with elevated blood pressure and low-grade systemic inflammation [1]. Here, we review the potential for a new α7 nicotinic acetylcholine receptor (α7nAChR) agonist therapy to treat the metabolic syndrome, a concept based on published data (see Table 1). The approach exploits the cholinergic anti-inflammatory reflex (CAIR) that is mediated by the vagus nerve (VN; Cranial Nerve X). In this circuit (Figure 1), pro-inflammatory cytokines stimulate VN sensory afferents to initiate a vagovagal reflex conveyed through the central nervous system (CNS), ultimately leading to acetylcholine (ACh) release from parasympathetic VN efferents of the autonomic nervous system (ANS) [2]. As established for CAIR, this ACh then activates the α7nAChR on immune system cells to suppress cytokine production and to produce an anti-inflammatory effect [3]. Here we propose that defects of the vagovagal reflex underlying CAIR explain, at least in part, why obesity, T2D, and inflammation coexist in the metabolic syndrome [4, 5]. This concept is consistent with the established ability of vagovagal reflexes to control appetite, body weight, energy expenditure, and glycemia [6, 7]. Thus, there is renewed interest in the α7nAChR that mediates CAIR, particularly from the standpoint of therapeutics and drug discovery. This review provides an overview of α7nAChR expression and function as it pertains to CAIR, vagovagal reflexes, and metabolic homeostasis. We also consider the possible usefulness of α7nAChR agonists for treatment of obesity, diabetes, and inflammation.
Table 1.
In vivo and in vitro tissue-specific effects of α7nAChR agonists
| Tissue/Cell/Organism | Agonist | Effect | Reference |
|---|---|---|---|
| Adipocyte (3T3-L1) | GTS-21 | Anti-Inflammatory Inhibits NF-κB | [162] |
| Adipocyte (human) | PNU 282987 | Anti-Inflammatory | [37] |
| Adipocyte (mouse) | PNU 282987 | STAT3 activation | [39] |
| Adipocyte (mouse, human) | ICH3 | Anti-Inflammatory Enhanced viability | [96] |
| Muscle (mouse, C2C12) | PNU 282987 | STAT3 activation Glucose uptake | [39] |
| Skeletal Muscle (DIO mouse) | ICH3 | Improved insulin sensitivity | [96] |
| Vascular smooth muscle (Rat) | Nicotine GTS-21 | Insulin signaling p44/42 MAPK α7nAChR expression | [163] |
| Liver (Kupffer cells) | Nicotine | Protects against ConA-induced hepatitis | [22] |
| Liver (Kupffer cells) | Nicotine PNU 282987 | Protects against Fas-induced hepatocyte apoptosis | [21] |
| CNS (mouse hypothalamus) | PNU 282987 | Anorexic JAK2/STAT3 activation | [164] |
| Islets of Langerhans (mouse) | PNU 282987 | Anti-Inflammatory Anti-apoptosis | [152] |
| Enteroendocrine (L-cells) | GTS-21 | Enhanced viability GLP-1 secretion | [130] |
| Keratinocyte (human) | AR-R17779 Tropisetron | Anti-Inflammatory Suppress TNFα | [165] |
| db/db obese mouse | TC-7020 | Decreased weight, food intake. Reduced blood glucose, triglycerides, TNFα. | [98] |
| db/db, DIO mouse | Nicotine | Improved glucose homeostasis, insulin sensitivity. | [38] |
| Zucker fatty rat | Nicotine | Improved glucose homeostasis, insulin sensitivity. | [111] |
Fig. 1.
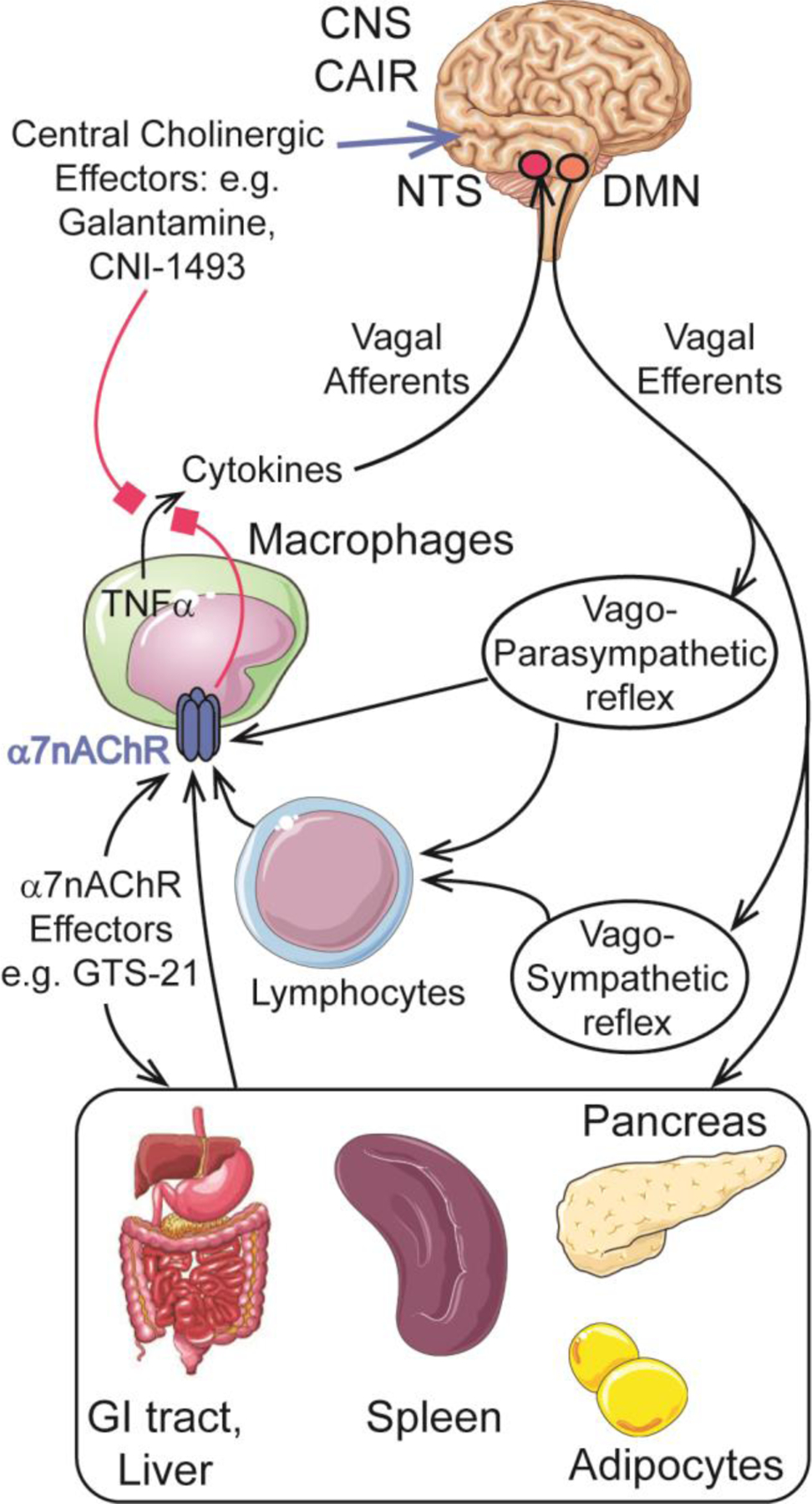
Neurocircuitry of vagovagal reflexes underlying CAIR and mediated by α7nAChR with consequent engagement of multiple organ systems including the central nervous system (CNS) brainstem nucleus tractus solitarius (NTS) and the dorsal motor nucleus (DMN) of the vagus nerve so that TNFα proinflammatory cytokine production is inhibited in cells of the immune system. Black arrows indicate stimulatory effects, whereas red boxes indicate inhibitory effects
2. Role of CAIR and α7nAChR in adaptation to inflammation
2.1. Overview
Infection, tissue trauma, and other disease states such as arthritis and inflammatory bowel disease cause local and/or systemic inflammation. When inflammation is uncontrolled or excessive, tissue damage results from uncontrolled release of pro-inflammatory cytokines (TNF, IL-1), histones, high-mobility group box 1 protein, mitochondrial DNA, and lipopolysaccharide (LPS) [8]. Under these conditions, the activation and release of inflammatory substances from white blood cells and other tissues triggers homeostatic self-protective processes that are mediated by CAIR so as to prevent further tissue damage [2, 3] (Figure 1). CAIR initiates ACh release from VN efferents, thereby stimulating the α7nAChR on immune system cells to suppress pro-inflammatory cytokine production [9]. Simultaneously, CAIR, acting through the α7nAChR, stimulates production and release of anti-inflammatory substances (IL-4, IL-10, TGFβ) [9].
Initiation of CAIR requires a vagovagal reflex in which afferent input to the brainstem nucleus tractus solitarius (NTS) stimulates efferent output originating from the dorsal motor nucleus of the vagus (DMV) [2, 3] (Figure 1). CAIR targets lymphocytes, macrophages, dendritic cells, adipocytes, keratinocytes, endothelial cells, and epithelial cells of the intestine and lung to suppress inflammation [10–12]. Importantly, the neural circuitry of CAIR may not be exclusively engaged by pro-inflammatory cytokines since vagovagal reflexes are also stimulated by metabolites, nutrients, and intestinal hormones [13]. Since vagovagal reflexes play an important role in the control of metabolic homeostasis [4, 6, 7], the neural circuitry of CAIR is well situated to participate in the dual control of inflammation and whole-body metabolism in the healthy and disease states.
2.2. Physiological and clinical relevance of CAIR
The VN afferents that mediate CAIR express receptors for multiple inflammatory molecules including IL-1, TNF, IgG FC, LPS, and prostaglandins [14]. For example, stimulation of these afferents after intraperitoneal administration of IL-1 or LPS activates CAIR to counteract the febrile and acute phase responses of inflammation [15]. Subdiaphragmatic vagotomy abrogates the anti-inflammatory effects of CAIR [14], whereas electrical stimulation of the VN produces an anti-inflammatory effect analogous to CAIR. Vagal nerve stimulation (VNS) ameliorates the inflammatory action of intravenously administered LPS, while also reducing hepatic TNF synthesis, lowering serum levels of TNF, and suppressing shock [16]. Thus, CAIR provides a rapid anti-inflammatory reflex to counteract uncontrolled release of pro-inflammatory cytokines.
Multiple neural networks of CAIR exist, not all of which require VN release of ACh acting directly at α7nAChR on immune system cells. For example, in the spleen, a vagosympathetic reflex exists in which ACh released from VN efferents stimulates the splenic nerve to initiate norepinephrine release, which in turn stimulates ACh release from a subset of T cells that express the ACh biosynthetic enzyme choline acetyltransferase [17] (Figure 1). Resultant stimulation of α7nAChR on adjacent splenic macrophages suppresses TNF production to reduce inflammation [17]. On the other hand, in the gastrointestinal tract, VN efferents innervate nNOS-VIP-ACh positive enteric neurons to stimulate ACh release, which then acts at intestinal resident macrophages to exert an α7nAChR-mediated effect to inhibit TNF production and inflammation [18, 19]. VN efferents participating in CAIR also innervate the liver, and their stimulation attenuates inflammation under conditions of LPS-induced sepsis, hepatitis, and ischemia-reperfusion [20]. These hepatic VN efferents activate α7nAChR to reduce Fas-induced apoptosis of hepatocytes, most likely through an effect on Kupffer cells to reduce reactive oxygen species production [21]. Nicotine activation of α7nAChR in Kupffer cells is also reported to reduce concanavalin A-induced hepatitis through the inhibition of NF-κB [22].
When considering the anti-inflammatory effects of CAIR summarized above, it is important to note that in vivo studies using α7nAChR knockout (KO) mice demonstrate that this receptor mediates the anti-inflammatory effects of VNS under conditions of LPS administration [11]. This finding is consistent with the identification of α7nAChR as the target of nicotine in assays monitoring suppression of cytokine release in LPS-treated macrophages [23]. VNS that emulates CAIR also influences food intake, body weight, fat mass, glycemia, and insulin sensitivity [13, 24–34]. For example, VNS in Zucker diabetic fatty (ZDF) rats improves glycemic control, lowers levels of glycated hemoglobin (HbA1c), and improves insulin sensitivity in muscle and liver [30]. In obese mini-pigs, VNS decreases body weight and subcutaneous fat mass, and is coupled to improved insulin sensitivity, and improved brain and skeletal muscle glucose uptake [31]. Furthermore, rats with diet-induced obesity (DIO) also demonstrate reductions in food intake and body weight in response to VNS [28]. What relevance such findings have to humans is not yet certain, and in fact a block of VN conduction is reported to lower body weight in humans [35, 36]. Still, these findings obtained in multiple models do demonstrate that VNS leads to significant metabolic alterations consistent with a major role for CAIR in metabolic homeostasis.
From a medical systems physiology standpoint, it is noteworthy that subcutaneous mature adipocytes from obese human subjects display reduced α7nAChR expression, and that this anomaly is reversible after dieting to achieve significant weight loss [37]. It is further demonstrated that α7nAChR activation induces an anti-inflammatory effect in human adipocytes that is potentially beneficial so as to ameliorate this component of the metabolic syndrome [37]. These human studies compliment findings obtained using leptin receptor-deficient obese mice (db/db) or DIO mice where CAIR activation improves insulin sensitivity and insulin-sensitive glucose uptake in adipose tissue [38], and also in liver and muscle [39]. Thus, complex inter-organ communication is under the control of vagovagal reflexes that mediate CAIR, and that are also of major relevance to the control of metabolic homeostasis [40]. Dysfunctions of this inter-organ communication are likely to be important contributing factors to the metabolic syndrome.
Finally, is especially interesting that VNS increases circulating levels of glucagon-like peptide-1 (GLP-1), an intestinal “incretin” hormone that lowers levels of blood glucose, suppresses appetite, delays gastric emptying, improves cardiovascular function, and counteracts inflammation [41–43]. This action of VNS is mediated by VN efferents that innervate the intestinal wall where enteroendocrine L-cells that release GLP-1 are located [43]. These L-cells also release Peptide YY (PYY), a hormone that suppresses appetite [43]. Thus, an attractive hypothesis is that the afferent and efferent components of vagovagal reflexes mediating CAIR overlap functionally and anatomically with vagovagal reflex circuitry controlling whole-body metabolism and endocrine system function.
2.3. Therapeutic approaches targeting vagovagal reflex circuitry
It is increasingly recognized that inflammation and metabolic decompensation in the disease state result from a loss of function in afferent and/or efferent limbs of vagovagal reflex circuitry [7]. For example, consumption of a high fat diet (HFD) results in VN dysfunction accompanied by obesity, hyperglycemia, and adipose tissue inflammation in rodents and humans [44, 45]. Mechanistically, chronic ingestion of the HFD results in a decreased sensitivity of VN afferents to metabolites, nutrients, and hormones that modulate vagovagal reflex function through their direct effects at the peripheral terminations of these sensory neurons [46, 47]. For example, the stimulation of VN afferent activity by intestinally absorbed glucose is diminished in the UCD-T2D rat model of human T2D in which there is adult-onset obesity, hyperglycemia, and adipose tissue inflammation [48].
Collectively, these findings suggest that new therapies designed to reverse or compensate for a loss of vagovagal reflex function might constitute novel therapeutic options when considering how to combat obesity, diabetes, and inflammation. In this regard, VNS might serve as one method by which to override defective vagovagal reflex function. However, a pharmacological approach based on the use of medications that emulate CAIR is also conceivable. Investigational agents include the anti-cytokine CNI-1493 and the acetylcholinesterase (AChE) galantamine that acts within the CNS at muscarinic ACh receptors to stimulate VN efferent activity emulating CAIR [49] (Figure 1). They also include α7nAChR agonists that were previously investigated for use in the treatment of inflammatory, neurodegenerative, and psychiatric disorders [50–53]. In the following sections we discuss α7nAChR agonists that serve as lead compounds for treatment of obesity, diabetes, and inflammation.
3. Molecular, pharmacological, and signaling properties of α7nAChR
When reviewing the potential usefulness of α7nAChR agonists as therapeutic agents, it is first necessary to briefly summarize available information concerning their molecular target of action, the α7nAChR [54–57]. It is one member of the nAChR family, the first member of which was cloned from the electric organ of the Torpedo ray [58]. All such nACh receptors are comprised of pentameric assemblies derived from a gene pool of 17 available α, β, γ, δ, and ε protein subunits that co-assemble to form ligand-gated cation channels [59]. Besides their classic role in nicotinic cholinergic transmission at the motor neuron end plate on skeletal muscle, multiple isoforms of nAChR are expressed in numerous non-neuronal cell types throughout the body [59]. Unlike most other isoforms of nAChR that are solely heteropentameric in structure, the α7nAChR is formed as either a homopentameric channel comprised solely of α subunits, or as a heteropentamer of α7β2 subunits in which two α7 subunits must be present for functionality [60, 61].
The tissue-specific subunit composition of each isoform of nAChR determines its functional and pharmacological properties [62]. One feature of α7nAChR is that it has high selectivity and permeability for Ca2+ in comparison to other nAChRs [63]. Thus, α7nAChR agonists exert Ca2+-mediated actions to regulate signaling pathways and gene expression networks [64, 65] (Figure 2). Ca2+ entry through α7nAChR can also initiate Ca2+-induced Ca2+ release from intracellular Ca2+ stores, thereby generating an additional increase of cytosolic [Ca2+] that amplifies this effect [66]. Such ionotropic properties of α7nAChR are complemented by a metabotropic signaling function in which it interacts with Gq heterotrimeric G proteins to stimulate phospholipase C, generate IP3, and further mobilize intracellular Ca2+ [67].
Fig. 2.
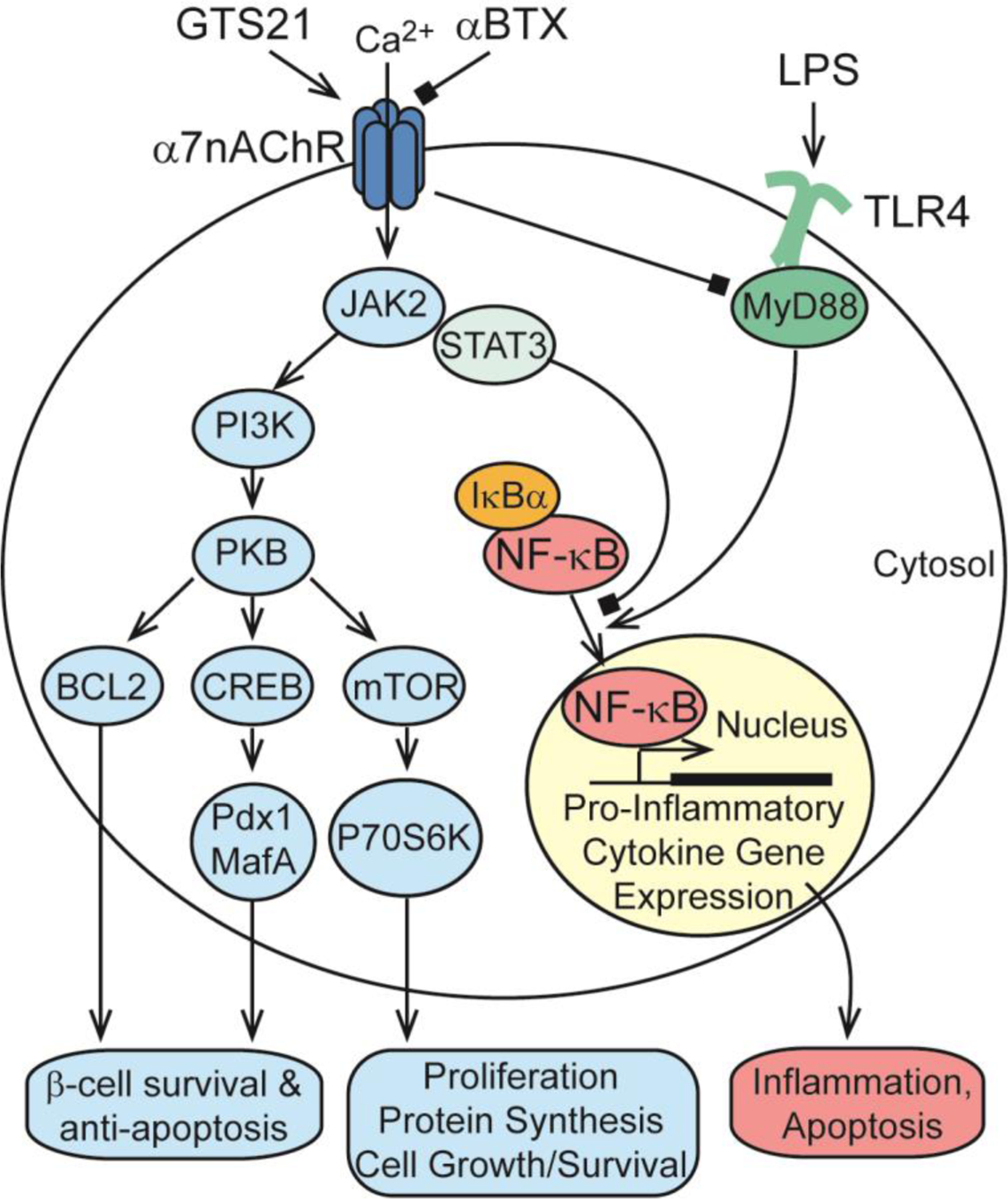
Growth factor-like signal transduction pathways under the control of α7nAChR and relevant to CAIR, proinflammatory cytokine production, cell growth/survival, inflammation, apoptosis, and pancreatic β-cell survival. For additional details concerning α7nAChR and β-cell survival, see Gupta et al. [152].
α7nAChR opens in response to low concentrations of nicotine, but then rapidly desensitizes [57]. These properties contrast with the α3β4 nAChR in autonomic ganglia that is insensitive to low concentrations of nicotine, and that is slow to desensitize [68]. Since α7nAChR is not expressed at high levels in autonomic ganglia [69, 70], α7nAChR agonists are less likely to exert undesirable ANS side effects related to direct ganglionic stimulation. Investigations of α7nAChR function are facilitated by the availability of mice with a KO of CHRNA7, the gene that codes for α7nAChR [71]. Furthermore, an armamentarium of α7nAChR agonists and antagonists is available [72–75]. Selective partial or full agonists for stimulation of α7nAChR include choline, GTS-21, PNU 282987, PHA 568487 and ICH3 (see Table 2). Compliments to conventional agonists are the “silent” agonists, such as m-bromo PEP and NS 6740, that induce receptor desensitization with little or no channel opening, but that still play a role in regulating inflammation [76, 77]. Positive allosteric modulators (PAMs) that enhance agonist action at α7nAChR include 5-hydroxyindole (5HI), PNU 120596, A 867744, and B 973B. PAMs fall into two categories. Type-1 PAMs such as 5HI simply increase agonist-evoked current amplitude, whereas type II PAMs such as PNU 120596 increase current amplitude and also reactivate desensitized receptors [78]. Available α7nAChR antagonists include α-bungarotoxin and methyllcaconitine. The selectivities of these compounds are not absolute, and in fact GTS-21 exhibits off-target antagonist actions at serotonin type-3 receptors (5-HT3R) when tested at high concentrations [79]. Interestingly, the CHRNA7 gene underwent a partial duplication to produce CHRFAM7A [80, 81]. Its gene product dupα7 acts as a dominant negative regulator of human α7nAChR [82]. Thus, dupα7 has the potential to modulate CAIR [83], but it is not discussed in detail here.
Table 2.
Representative α7nAChR agonists and EC50 values for α7nAChR stimulation
α7nAChR agonists also regulate JAK2, STAT3, PI3K/PKB, BCL2, CREB, mTOR, p70S6K, and NF-κB signaling pathways [12, 84–86] (Figure 2). In the absence of inflammatory stimuli, NF-κB subunits RelA and p50 are located in the cytoplasm in association with the inhibitory protein IκBα. Inflammatory mediator such as LPS activate Toll-like receptors (TLR4) in association with their MyD88 adaptor proteins, which triggers the phosphorylation and degradation of IκBα. This releases free NF-κB subunits so that they can translocate to the nucleus, bind to specific DNA regulatory elements, and stimulate pro-inflammatory gene expression (Figure 2). α7nAChR agonists inhibit this inflammatory response by suppressing NF-κB nuclear translocation [12, 84–86].
4. α7nAChR regulation of inflammation in obesity
Obesity is a key risk factor for metabolic abnormalities including insulin resistance and T2D, as wells as atherosclerosis and nonalcoholic fatty liver disease (NAFLD) [87]. Furthermore, obesity and inflammation are linked with respect to their mutual contributions to the metabolic syndrome [1]. Thus, it is significant that α7nAChR agonists act in animal models of obesity to suppress inflammation and to improve insulin sensitivity in liver, muscle and adipose tissue [88–93]. When considering α7nAChR agonist action in the context of obesity, it is useful to summarize relevant features of adipocyte biology. In this regard, adipose tissue was at one time considered to be a metabolically inactive reservoir of stored excess calories. However, it is now recognized that adipocytes secrete metabolites (free fatty acids, glycerol), hormones (leptin, adiponectin, resistin), and inflammatory mediators (TNF and IL-6) [94]. A balance between pro- and anti-inflammatory actions of these secretory products determines the progression or regression of adipose tissue inflammation.
α7nAChR agonists exert their beneficial effects in adipocytes by down regulating the activities of pro-inflammatory mediators, while simultaneously upregulating the activities of anti-inflammatory mediators [2, 3]. Importantly, α7nAChR agonists also counteract inflammatory monocyte infiltration in adipose tissue, a phenomenon under the control of the chemokine monocyte chemoattractant protein-1 (MCP-1) [95]. Similarly, α7nAChR agonists counteract the accumulation and activation of adipose tissue macrophages (ATMs) within fat [38]. This is significant because activated ATMs with the (M1) phenotype secrete inflammatory cytokines (TNF, IL-1, IL-6) that trigger inflammation via the IκB kinase β and c-Jun N-terminal kinase (JNK) pathways [87–90].
Some interesting observations concerning human adipocyte heterogeneity are revealed in studies using the α7nAChR agonist ICH3 [96]. When human adipocytes from obese donors are studied in vitro, ICH3 differentially affects IL6 and adiponectin gene expression in visceral adipose tissue (VAT) as compared to subcutaneous adipose tissue (SAT) [96]. This is a gene-specific effect because VAT and SAT show no change in TNFα or leptin expression in response to ICH3 [96]. Such differential actions of ICH3 in VAT and SAT are understandable if there exists fat depot-specific actions of α7nAChR agonists. VAT and SAT have different developmental origins, and it is VAT that is associated with metabolic dysfunction, whereas SAT tends to be protective [97]. Thus, α7nAChR agonist action in VAT might be especially relevant when considering how α7nAChR stimulation reduces excess adiposity. A similar fat depot-specific action of ICH3 is also observed for DIO mice where epididymal fat mass is reduced by ICH3, whereas perirenal and subcutaneous fat mass are not affected [96]. In these same mice, ICH3 improves insulin sensitivity and glucose tolerance [96].
5. α7nAchR regulation of inflammation in diabetes
We now consider what role α7nAChR plays in the control of inflammation in the context of diabetes, as inferred from studies of α7nAChR agonist action in animal models of hyperglycemia. Leptin receptor-deficient db/db mice are one such model in which hyperglycemia, obesity, and low-grade systemic inflammation coexist. Administration of α7nAChR agonist TC-7020 to these db/db mice leads to reduced levels of circulating pro-inflammatory TNFα, and also reduced levels of blood glucose and triglycerides [98]. Such effects of TC-7020 are blocked by α7nAChR antagonist methyllycaconitine or by JAK2 inhibitor AG-490, as expected if the α7nAChR-mediated anti-inflammatory action of TC-7020 is linked to improved glucoregulation [98]. Consistent with these findings, treatment with the broad-spectrum nAChR agonist nicotine improves insulin sensitivity and glucoregulation in db/db mice [38]. This is also the case for a mouse model of DIO and hyperglycemia resulting from a HFD [39, 99]. Conversely, mice with a KO of α7nAChR exhibit insulin resistance, impaired glucose tolerance, and a reduced ability of nicotine to suppress pro-inflammatory cytokine production [38]. Findings are also reported for Sprague-Dawley rats in which nicotine treatment improves insulin sensitivity and glucoregulation [39]. This ability of nicotine to improve insulin sensitivity is reproduced by α7nAChR agonist PNU 282987, and is lost in α7nAChR KO mice [39]. Such findings are in general agreement with one report that the appearance of hepatic insulin resistance, steatosis, and inflammatory macrophage infiltration is accelerated in α7nAChR KO mice fed the HFD [100]. In contrast, wild-type mice fed the HFD exhibit reduced steatosis and inflammatory infiltration when treated with α7nAChR agonist PNU 282987 [100].
It is interesting that when maternal pregnant mice are fed the HFD, the male offspring of these mice exhibit reduced hepatic α7nAChR expression that is accompanied by insulin resistance [101]. These abnormalities correlate with elevated levels of TNFα and a reduced ability of insulin to stimulate phosphorylation of PKB in hepatocytes of the male offspring [101]. Such findings are understandable if HFD-induced fetal metabolic “programming” during gestation leads to downregulation of hepatic α7nAChR expression, with consequent insulin resistance leading to metabolic decompensation in progeny mice [101, 102].
Another interesting aspect of this story concerns galantamine, an AChE inhibitor that has CNS penetrance and that has been studied in patients with metabolic syndrome [103, 104]. Galantamine slows metabolic inactivation of ACh, thereby increasing the availability of ACh for synaptic transmission mediated by all isoforms of ACh receptors [105]. Surprisingly, galantamine also binds to the nAChR to allosterically enhance its activation by ACh both pre- and post- synaptically [106, 107]. In mice fed the HFD, galantamine decreases plasma levels of IL-6, leptin, MCP-1, and resistin, while increasing levels of adiponectin [108]. Furthermore, galantamine lowers plasma glucose, insulin, homeostatic model assessment of insulin resistance score, and hepatic steatosis [108]. One clinical trial testing galantamine in patients with metabolic syndrome demonstrates reductions in plasma TNFα and leptin, and increased levels of adiponectin and IL-10 [104]. Galantamine also decreases plasma glucose, insulin resistance, and heart rate variability in patients with metabolic syndrome [104]. In summary, these studies demonstrate the ability of α7nAChR agonists and galantamine to improve glycemic control and insulin sensitivity, in part through modulation of adipose tissue inflammation in experimental models of diabetes.
6. α7nAChR regulation of appetite and body weight
Our understanding of α7nAChR agonist action that is relevant to appetite and body weight control is aided by prior studies concerning actions of nicotine in adipose tissue or the CNS. From a systems physiology standpoint, nicotine reduces appetite and food intake while also increasing energy expenditure so that body weight is reduced [109]. When considering the peripheral action of nicotine, it is established that in rodents, a nicotine-stimulated reduction of body weight is proportional to reduced fat mass and an anti-inflammatory effect [110]. For example, in the fa/fa leptin receptor-deficient Zucker rat model of obesity, nicotine reduces body weight, lowers levels of TNFα in visceral fat tissues, and reduces fasting levels of blood glucose, while also improving glucose tolerance [111]. To what extent α7nAChR participates in such effects of nicotine is uncertain, especially from the standpoint of its peripheral expression in human adipose tissue. For example, in studies of fat depot SAT isolated from obese human donors not administered nicotine, levels of α7nAChR mRNA and protein are decreased [37]. This finding raises questions concerning whether or not sufficient numbers of α7nAChR exist in SAT of obese subjects in order for nicotine to reduce fat mass. Still, the α7nAChR agonist PNU 282987 produces an anti-inflammatory effect in these same samples of SAT [37]. Furthermore, in SAT samples obtained from obese human donors after a 3-month weight loss intervention, increased levels of α7nAChR levels are detectable [37]. Potentially, pharmacological administration of an α7nAChR agonist targeting both SAT and VAT (as discussed above) might be an effective strategy to reduce adipose inflammation, increase adipocyte energy expenditure, and reduce fat mass in patients with obesity.
Nicotine also exerts direct stimulatory actions within the CNS to suppress appetite, reduce food intake, increase energy expenditure, and reduce body weight [112]. α7nAChR is expressed at high levels in the hypothalamus [112], and its stimulation by nicotine influences proopiomelanocortin (POMC), neuropeptide Y (NPY), melanin-concentrating hormone, GABA, glutamate, dopamine, and serotonin neurotransmitter circuitry relevant to the control of appetite, food intake, energy expenditure, and body weight [112]. Still, formal proof that α7nAChR does in fact mediate these multiple actions of nicotine within the CNS will require cell type-specific knockouts of α7nAChR gene expression in discreet populations of neurons.
It is remarkable that nicotine activates GLP-1 positive neurons in the NTS, an effect associated with reduced nicotine self-administration in mice [113]. This constitutes a negative feedback “satiety” sensor for nicotine, and it involves GLP-1 acting as a neurotransmitter to excite medial habenular projections to the interpeduncular nucleus so that nicotine reward is decreased [113]. An intriguing possibility is that α7nAChR mediates stimulatory effects of nicotine at GLP-1 positive NTS neurons. If so, VN afferent input to the NTS might activate brainstem neural circuits that stimulate ACh release which then signals through α7nAChR to stimulate GLP-1 release from these same GLP-1 positive neurons. As summarized below, efferent VN input to the intestinal wall may also drive GLP-1 release from L-cells in a α7nAChR-mediated manner. Thus, a situation may exist in which VN efferents release ACh to stimulate L-cell GLP-1 release, thereby allowing GLP-1 to excite VN afferents that project to the NTS, and that in turn stimulate GLP-1 release from NTS neurons. Potentially, coordinate gene expression underlies this neural circuitry in which α7nAChR biosynthesis is linked to GLP-1 biosynthesis in the NTS and L-cells.
Finally, α7nAChR agonists reduce food intake and weight gain in mouse models of DIO [96, 99]. Thus, clinical testing in humans seems warranted in order to evaluate whether α7nAChR agonists have sufficient efficacy to achieve body weight reduction. Potentially, α7nAChR agonists might serve as an add-on therapy in combination with newer approaches to pharmacological weight reduction. For example, liraglutide is a synthetic GLP-1 receptor agonist that emulates the appetite suppressing and blood glucose-lowering actions of incretin hormone GLP-1 [114–116]. Just as impressive, combined administration of naltrexone and bupropion is an effective therapy for weight reduction in obese and diabetic subjects [114, 117, 118]. Still, the adverse side effect of nausea is a frequent complicating factor when administering appetite-suppressant drugs that have strong CNS penetrance [119, 120]. Whether this is also the case for α7nAChR agonists administered to obese subjects is not yet reported.
7. α7nAChR regulation of enteroendocrine L-cell function and viability
Enteroendocrine cells (EEC) are specialized epithelial cells that line the wall of the intestine and that release hormones into the systemic circulation in response to nutrients (e.g., glucose, lipids, amino acids) present within the intestinal lumen after a meal [121]. Specialized microvilli on the apical (i.e., luminal) membrane of the EEC act as nutrient sensors by virtue of their expression of multiple subtypes of nutrient transporters (e.g., Na+-dependent glucose or amino acid transporters) [122]. These microvilli also express nutrient responsive G protein-coupled receptors (GPCRs) that include the T1R2 amino acid taste receptor, the GPR120 fatty acid receptor, and the TGR5 bile acid receptor [123]. Nutrients acting at the apical membrane of the EEC cell raise intracellular levels of cAMP and Ca2+, thereby stimulating hormone release from the basolateral membrane [124].
In addition to such nutrient-stimulated hormone release, there is also neural control mediated by the ANS and the enteric nervous system (ENS) [125] (Figure 3). EEC hormone release is under the control of vagovagal reflexes, and ACh released from VN efferents acts not only at EEC muscarinic ACh receptors (mAChR), but also at the α7nAChR. For example, L-cells of the distal intestine synthesize GLP-1 and Peptide YY (PYY) [126], and α7nAChR stimulation results in the co-secretion of both peptides (Figure 3). Whereas attention previously focused on nutrient control of GLP-1 and PYY release [121], L-cells are innervated by VN efferents [125]. Thus, neural regulation of EEC hormone secretion is of major importance to glucoregulation and appetite control [127], and in fact VN stimulation releases GLP-1 from L-cells [128, 129].
Fig. 3.
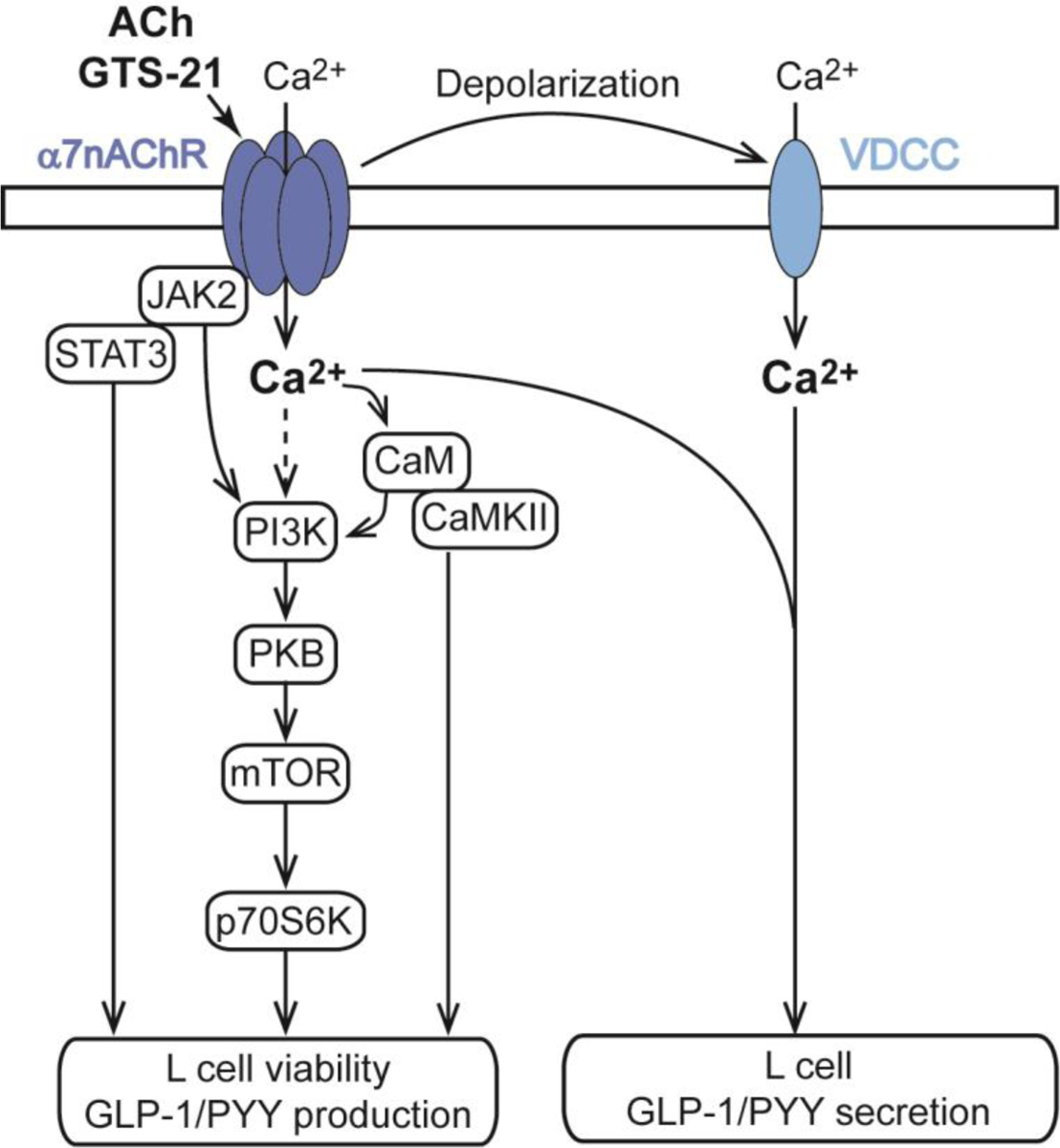
Dual role of α7nAChR to promote enteroendocrine L-cell GLP-1 and PYY secretion, and to also enhance L-cell viability under conditions of hyperglycemic stress. For additional details see Wang et al. [130].
As reported by Wang et al. (2018), studies using RT-PCR, Western blot, or immunofluorescence analysis reveal that α7nAChR is detectable in murine intestine, in the mouse L-cell line GLUTag, the mouse EEC line STC-1, and in the human EEC line NCI-H716 [130]. Double immunofluorescence labeling also demonstrates co-expression of α7nAChR and GLP-1 in primary mouse intestinal cell cultures enriched with L-cells, and in sections of mouse ileum [130]. Moreover, the α7nAChR agonist GTS-21 stimulates GLP-1 release from primary cultures of mouse intestinal cells that are enriched with L-cells [130]. Studies of GLUTag cells demonstrate that this action of GTS-21 results from its ability to promote membrane depolarization, Ca2+ influx, and Ca2+-dependent exocytosis of GLP-1 [130] (Figure 3). As expected, the GLP-1 secretagogue action of GTS-21 is abrogated by pretreatment with the α7nAChR antagonist α-bungarotoxin, or after siRNA-mediated knockdown of α7nAChR expression [130].
The above-summarized in vitro secretagogue actions of GTS-21 correlate with its in vivo ability to raise levels of circulating GLP-1 in C57BL/6 mice [130]. More recently, we find that GTS-21 improves oral glucose tolerance in wild-type mice but not α7nAChR KO mice (Q. Meng and R.N. Cooney, unpublished findings). Since GLP-1 is an established blood glucose-lowering hormone [43], this glycemic action of GTS-21 might be indirect (i.e., mediated by GLP-1 released from L-cells). However, an action of GTS-21 independent of L-cell GLP-1 release has not been ruled out. This possibility could be tested in studies using the specific GLP-1 receptor antagonist Ex (9–39) [43]. Defining the exact locus of GTS-21 action relevant to glucoregulation will require a more refined mouse genetics approach using cell type-specific knockouts of α7nAChR in L-cells or other cells types (e.g., ANS or CNS neurons) that express α7nAChR and that might participate in glucoregulation.
GTS-21 also stimulates growth factor signaling pathways in L-cells (Figure 3). Western blot analysis demonstrates that GTS-21 promotes PI3K, PKB, and mTOR/p70S6K mediated phosphorylation [130]. These effects correlate with the action of GTS-21 to counteract glucotoxicity in an in vitro assay of L-cell viability using GLUTag cells [130]. GTS-21 slows apoptosis under conditions of hyperglycemia, and all such actions of GTS-21 are blocked by inhibitors for each of these kinases [130]. Remarkably, buffering of intracellular Ca2+ to low levels blocks PI3K, PKB, and mTOR/p70S6K mediated phosphorylation in response to GTS-21 [130]. Conversely, inhibitors of PI3K, PKB, and mTOR/p70S6K suppress Ca2+-dependent GLP-1 secretion in response to GTS-21 [130]. Thus, Ca2+ entry through α7nAChR serves as an ionic coupling factor in support of protein kinase-mediate pro-survival and secretagogue actions GTS-21 (Figure 3). Furthermore, GLP-1 release is conditional on a novel permissive action of PI3K, PKB, and mTOR/p70S6K to support exocytosis.
Interestingly, attempts to ameliorate T2D in clinical studies have not yet been fruitful when testing GLP-1 secretagogues that are GPCR agonists, and that stimulate free fatty acid receptors (TAK-875), or bile acid receptors (SB-756050), or fatty acid amide receptors (APD597) located on L-cells. Potentially, a new therapeutic strategy that is instead based on the use of α7nAChR agonists to restore L-cell number and function will provide a means with which to treat T2D. In fact, the intestinal distribution and numbers of EEC are altered in T2D [131]. Thus, strategies to increase L-cell turnover, number, and differentiated state might be useful as means to treat T2D [131]. In this regard, α7nAChR agonists might be applicable to this purpose, although this has yet to be tested.
The GLP-1 secretagogue action of GTS-21 summarized above is accompanied by its ability to stimulate the release of PYY (1–36) from L-cells [130]. This is significant since circulating PYY (1–36) is converted to PYY (3–36) that transits across the blood brain barrier whereupon it binds to the type 2 isoform of neuropeptide Y receptor (NPY2R) located in the hypothalamus [126]. In this manner, PYY (3–36) exerts an anorexigenic appetite suppressing effect mediated by NPY2R [126]. Simultaneously, GLP-1 excites VN afferents that innervate the NTS, thereby exerting an additional anorexigenic effect [132, 133]. Based on these findings, α7nAChR agonists that simultaneously stimulate GLP-1 and PYY release might find a usefulness for treatment of the metabolic syndrome. If so, they would be predicted to suppress appetite, reduce body weight, enhance liver, fat, and skeletal muscle insulin sensitivity, and improved glycemia in T2D.
There is also a potential role for α7nAChR as a determinant of GLP-1 release from α-cells located in the islets of Langerhans. Normally, these α-cells release glucagon, a hormone that stimulates hepatic glucose production to counteract hypoglycemia. However, under conditions of stress resembling T2D, a phenotypic conversion occurs in which α-cells attain the ability to synthesize and release GLP-1 [134–136]. This intra-islet GLP-1 then acts as a paracrine hormone to stimulate GLP-1 receptors located on β-cells within the islets [134]. Resultant GLP-1 receptor activation not only protects β-cells from apoptosis induced by glucolipotoxicity [137, 138], but it also enhances insulin biosynthesis and secretion [139–142]. Recently, we established an ability of GTS-21 to stimulate GLP-1 release from the mouse α-cell line αTC1.6. Furthermore, we find that GTS-21 upregulates prohormone convertase (PC1/3) expression to stimulate conversion of proglucagon to GLP-1 (O.G. Chepurny and Q. Meng, unpublished findings). Thus, α7nAChR agonists might serve as intra-islet GLP-1 secretagogues for treatment of T2D.
8. α7nAChR regulation of β-cell adaptation to stress
Preganglionic VN efferents in rats and mice innervate pancreatic ganglia where they synapse on postganglionic neurons that project to the islets of Langerhans [143]. ACh released from these VN efferents stimulates insulin secretion from islet β-cells [144]. Furthermore, this ACh exerts a proliferative effect to stimulate β-cell replication in rats and mice so that β-cell “mass” is increased [145]. In humans there is VN control of insulin secretion [146, 147], but cholinergic VN innervation of human islets is sparse [148]. Instead, ACh is released from human islet α-cells so that it acts as a paracrine transmitter to stimulate ACh receptors on β-cells [149]. ACh action at β-cells is mediated by assemblies of nAChR isoforms comprised of α2, α3, α4, α5, α7, β2 and β4 subunits, of which α7 and β2 are most abundantly expressed in mouse islets [150–153]. ACh also stimulates multiple isoforms of β-cell mAChR, of which the M3 mAChR is of high significance [154] (Figure 4).
Fig. 4.

The nicotinic α7nAChR mediates actions of ACh to counteract endoplasmic reticulum stress and to protect against apoptosis in pancreatic β-cells. However, Ca2+ entry through α7nAChR fails to stimulate β-cell insulin secretion. Instead, ACh action at muscarinic M3 receptors promotes insulin secretion through the mobilization of intracellular Ca2+, while also upregulating protein kinase C (PKC) activity. Abbreviations: A-UPR, adaptive unfolded protein response; T-UPR, terminal unfolded protein response; IRE1α, inositol-requiring enzyme 1α.
Paradoxically, nicotine inhibits insulin release rather than stimulating it [150, 153]. This indicates that nAChR activation fails to sufficiently raise levels of [Ca2+] within “active zones” of exocytosis where insulin secretion occurs [155]. However, “priming” of islets with nicotine enhances the ability of mAChR agonist oxotremorine to stimulate insulin secretion [154]. Such findings indicate that nAChR function in β-cells is not conventional with respect to standard secretagogue actions of nicotine and ACh observed in other endocrine cell types (e.g., adrenal chromaffin cells that secrete epinephrine). Thus, the question arises as to what role α7nAChR plays in β-cell biology? As summarized below, the β-cell α7nAChR mediates a trophic factor-like action of α7nAChR agonists to counteract apoptosis and to enhance β-cell survival in mouse models of diabetes (Figure 4).
Gupta et al. provide the most definitive study validating an ability of an α7nAChR agonist (PNU 282987) to counteract hyperglycemia, suppress β-cell apoptosis, and to preserve β-cell mass (BCM) through a β-cell pro-survival effect that is monitored in the multiple low-dose streptozotocin (MLDS) model of diabetes using mice [152]. In this model, steptozotocin (STZ) induces β-cell apoptosis and reduces BCM, while also inducing an inflammatory response (insulitis) in which immune cells release cytokines that further accelerate β-cell death [156]. This MLDS model resembles, but does not fully recapitulate, some features of autoimmune type 1 diabetes (T1D) in which immune destruction of β-cells leads to insulin insufficiency and sustained hyperglycemia [156].
When PNU 282987 is administered for 2 weeks on a daily basis to MLDS mice, it exerts a significant long-term blood glucose-lowering effect measured in an intra-peritoneal glucose tolerance test [152]. This action of PNU 282987 is attributable to its ability to protect against the decrease of BCM that streptozotocin induces. Thus, PNU 282987 partially reverses insulin insufficiency in the MLDS model [152]. TUNEL assays with MLDS pancreas slices reveal that PNU 282987 preserves BCM by counteracting STZ-induced apoptosis [152]. Mechanistically, PNU 282987 stimulates the JAK2-STAT3 anti-inflammatory signaling pathway, and it also upregulates PKB, BCL2, and CREB pro-survival signaling pathways [152]. Importantly, these effects are measured in islets of wild-type mice but not α7nAChR KO mice [152]. PNU 282987 also counteracts streptozotocin-induced depletion of β-cell transcription factors Pdx1 and MafA that are important to establishment of β-cell identity in the fully differentiated state [152] (refer to Figure 2). Thus, PNU 282987 preserves gene expression important to β-cell survival, while also preserving the ability of β-cells to synthesize and release insulin [152].
Additional β-cell anti-apoptosis actions of PNU 282987 and nicotine are reported by Ishibashi et al. for studies of the rat β-cell line INS-1 and the human β-cell line EndoC-βH1[157]. These studies focus on how α7nAChR stimulation counteracts the “terminal” unfolded protein response (T-UPR) that induces apoptosis, and that results from excessive endoplasmic reticulum (ER) stress [158] (Figure 4). In healthy β-cells there normally exists an “adaptive” UPR (A-UPR) that does not induce apoptosis, and that is instead cytoprotective in that it monitors secretory protein folding and assembly in the ER. When secretory protein demand increases (e.g., upregulation of ER proinsulin content), the A-UPR is activated to enhance ER protein folding capacity, to degrade ER unfolded proteins, and to reduce the “load” of secretory proteins transported to the plasma membrane for exocytosis [158]. However, under conditions of excessive β-cell ER stress (e.g., failure of ER proinsulin to fold properly), the T-UPR is initiated with consequent β-cell apoptosis [158]. A key event underlying the T-UPR is hyperactivation of the UPR sensor IRE1α (inositol-requiring enzyme 1α) [158]. This leads to hyperactivation of endoribonuclease (RNase), cleavage of ER-localized mRNAs, ribosomal RNAs, and micro RNAs, so that apoptosis is initiated [158]. Ishibashi et al. find that nicotine inhibits IRE1α activation in response to ER stress [157]. Specifically, induction of T-UPR by treatment with tunicamycin (an inhibitor of ER protein folding) leads to an autophosphorylation of IRE1α that signals its activation, an effect inhibited by treatment with nicotine [157]. This effect of nicotine correlates with its ability to suppress tunicamycin-induced upregulation of the T-UPR markers XBP1s (spliced X-box binding protein) and TXNIP (thioredoxin-interacting protein). PNU 282987 mimics these cytoprotective actions of nicotine, and these effects are absent after siRNA knockdown of α7nAChR [157].
As summarized above, α7 and β2 are the predominant isoforms nAChR subunits found in mice islets. Thus, it is significant that Somm et al. report that glucose-stimulated insulin secretion is not impaired in islets of α7β2nAChR mice in which there is a double KO of α7 and β2 [151]. This finding reinforces the view that α7β2nAChR action in mouse islets is unrelated to short-term control of insulin secretion. Importantly, Klee et al. further substantiate the long-term trophic role α7 plays by demonstrating an ability of nicotine to exert a pro-survival effect to counteract cytokine-induced apoptosis in islets of mice with a KO of β2 but not α7 [159]. These investigators also report a major species difference in which α5 mRNA is expressed at higher levels than α7 in human islets [159]. However, this analysis is open to interpretation since whole islets contain multiple endocrine cell types. Ideally, such studies will be repeated using pure preparations of β-cells [159].
Studies of Klee et al. also shed light on the physiological basis of α7nAChR agonist action in β-cells. In mouse islets, α7nAChR agonist choline reduces cytokine-induced activation of caspase3, a mediator of apoptosis [159]. Furthermore, choline counteracts cytokine-induced elevation of [Ca2+]i, endoplasmic reticulum stress, and mitochondrial outer membrane permeabilization (MOMP), all of which are key events leading to apoptosis. Collectively, these findings are of potential medical significance relevant to T1D since choline also counteracts apoptosis induced by treatment of human islets with pro-inflammatory cytokines (IL-1β, TNFα, IFNγ) [159]. Still, the direct involvement of β-cell α7nAChR as the principal molecular target of choline action in human β-cells remains not fully established in the absence of a targeted KO or knockdown of the various α subunits expressed within human β-cells.
9. Emerging clinical strategies relevant to anti-obesity therapeutics
An increasingly popular clinical procedure for treatment of obesity is based on the use of implantable electrical devices that allow chronic stimulation of the vagus nerve [7]. These implantable VNS devices are safe and effective so that a 12-month stimulation regimen provides excess weight loss of ca. 25%, an effect accompanied by rapid improvements in glycemic control and blood pressure that are long lasting [160]. The efficacy of VNS for weight reduction is understandable in view of the above summarized roles of CAIR, vagovagal reflexes, and α7nAChR to control metabolic homeostasis. However, disadvantages of VNS include the need for implant surgery and the high cost of the implantable device. A small percentage of patients complain of pain at the neuroregulatory site, and mild to moderate symptoms of heartburn, dysphagia and/or nausea [36]. Replacement of device batteries and electrodes is also a complicating factor. Still, this approach is substantially less intrusive than bariatric surgery options such as gastric bypass surgery or sleeve gastrectomy for treatment of obesity. What is presently unknown is whether the efficacy of VNS for weight reduction might be enhanced by co-administration of an α7nAChR agonist. This combinatorial approach is plausible in view of prior clinical testing of investigational α7nAChR agonists that were demonstrated to be generally well tolerated. For example, there was suitable patient compliance when evaluating α7nAChR agonist efficacy for treatment of schizophrenia and cognitive dysfunction [50]. Such studies established that GTS-21 is generally well tolerated, with a low frequency of adverse side effects (eczema, dermatitis, dizziness, headache, and orthostatic hypotension) when it is administered at doses of up to 450 mg/day [161].
Finally, it is of note that an alternative approach targeting CNS cholinergic neurotransmission is based on the use of the AChE inhibitor galantamine. It slows hydrolytic degradation of ACh so that it exerts a generalized effect to stimulate multiple AChR subtypes including muscarinic and nicotinic ACh receptors [104]. In a 12-week clinical trial, galantamine attenuates inflammation, reduces insulin resistance, and improves glycemia [104]. Galantamine is FDA-approved to treat dementia, and it is well tolerated, with only three adverse events in six study patients [104]. However, nausea, vomiting, diarrhea, dizziness, and weight loss are infrequently reported. Intriguingly, anti-inflammatory effects of galantamine appear to be mediated by its CNS site of action at ACh receptors in which it upregulates efferent VN outflow that emulates CAIR [3]. Therefore, it will be interesting to evaluate to what extent α7nAChR participates in the beneficial metabolic actions of galantamine reported to date.
10. Conclusion
A principal conclusion to be drawn from this review of the literature is that vagovagal reflexes are mediated by shared assemblies of VN afferent and efferent circuits that are of importance to homeostatic control of inflammation, appetite, body weight, energy expenditure, and glycemia (Figure 5). VN sensory input that contributes to this control is relayed through the CNS and ANS, and it is initiated by direct actions of inflammatory cytokines, nutrients, metabolites, and hormones binding to their receptors located on the peripheral terminations of VN afferents in multiple organ systems. α7nAChR plays an important role as a determinant of VN efferent control of immune cell function, while it also participates in the ANS regulation of EEC hormone secretion (GLP-1, PYY), intestinal L-cell viability, and pancreatic β-cell survival. Here, we propose that defects of the vagal reflex circuitry underlying CAIR explain, at least in part, why inflammation, obesity, and diabetes coexist in the metabolic syndrome. Potentially, such defects may be overridden by direct electrical stimulation of the VN in order to achieve a therapeutic effect. However, a pharmacological approach using α7nAChR agonists might achieve a similar outcome. To date, α7nAChR is primarily considered to be a target for drug discovery relevant to treatment of neurodegenerative and psychiatric disorders. Still, absent their potential deleterious side effects owing to CNS, ANS, or ENS α7nAChR activation, available evidence indicates a likely role for investigational α7nAChR agonists as a new experimental treatment for the metabolic syndrome.
Fig. 5.
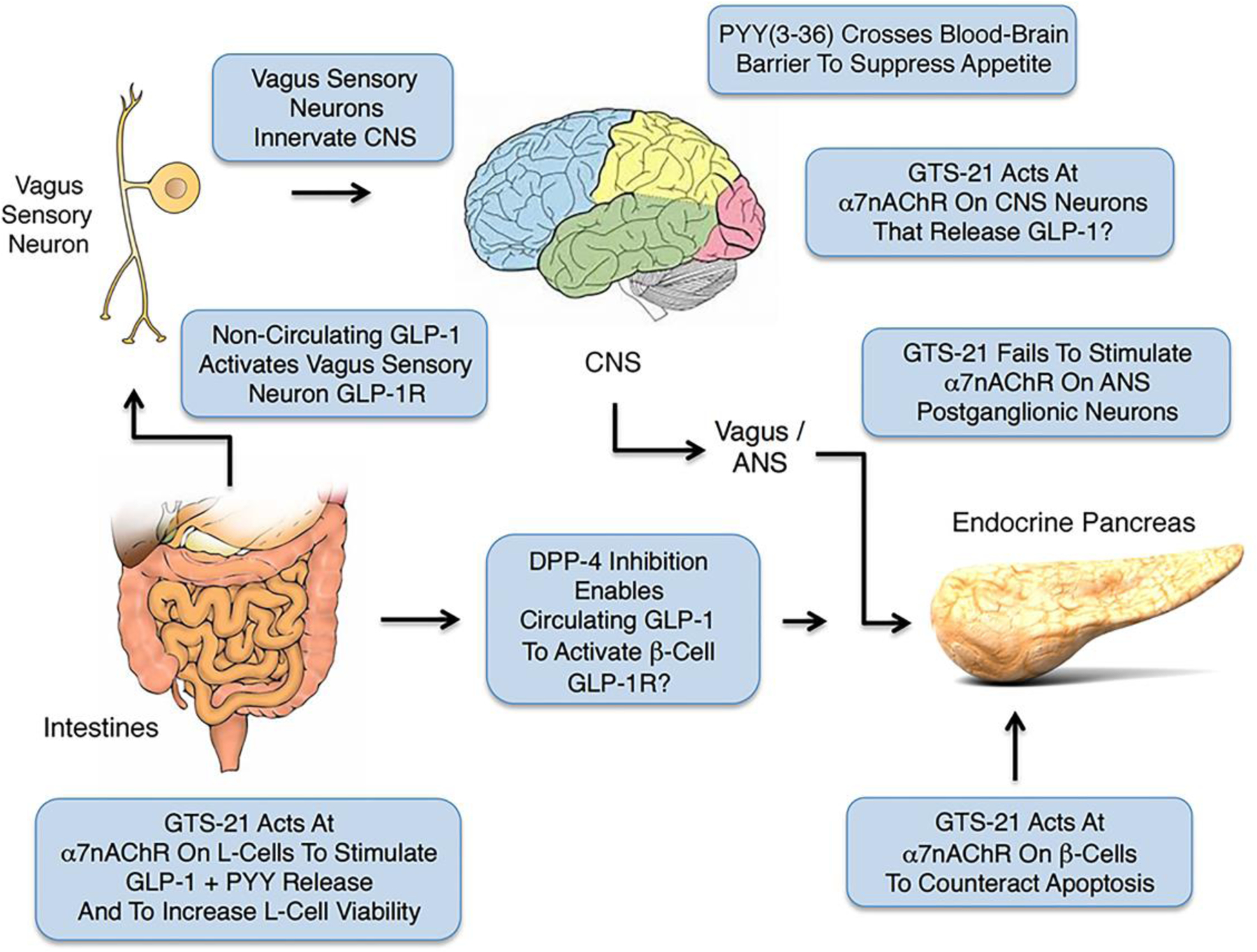
α7nAChR agonist GTS-21 participates in the control of GLP-1 and PYY release from L-cells, while also protecting β-cells from apoptosis. Note that GLP-1 released from L-cells initiates vagovagal reflexes that transmit through CNS and ANS to influence whole-body metabolism, whereas PYY crosses the blood brain barrier to suppress appetite. Dipeptidylpeptidase-4 (DPP-4) inhibitors enhance the glycemic action of GLP-1 by slowing its normally fast metabolic inactivation. It is presently uncertain whether levels of circulating GLP-1 are sufficiently high to stimulate the β-cell GLP-1 receptor even when T2D patients are administered DPP-4 inhibitors. Instead, DPP-4 inhibitors may primarily enhance GLP-1 action at the GLP-1R located on VN afferents. For additional details see Burcelin et al. [133].
Acknowledgments
Funding: Supported in part by 1R01DK122332 (GGH, RNC) and 5R01DK069575 (GGH)
Abbreviations
- α7nAChR
α7 nicotinic acetylcholine receptor
- ACh
acetylcholine
- AChE
acetylcholinesterase
- ANS
autonomic nervous system
- ATM
adipose tissue macrophage
- BCL2
B-cell lymphoma 2
- BCM
beta-cell mass
- CAIR
cholinergic anti-inflammatory reflex
- CNS
central nervous system
- CREB
cAMP response element-binding protein
- DIO
diet-induced obesity
- DMV
dorsal motor nucleus of the vagus
- EEC
enteroendocrine cell
- ENS
enteric nervous system
- ER
endoplasmic reticulum
- GABA
gama aminobutyric acid
- GPCR
G protein-coupled receptor
- GLP-1
glucagon-like peptide-1
- GTS-21
3-(2,4-dimethoxy-benzylidene)anabaseine
- HbA1c
glycated hemoglobin 1c
- HFD
high fat diet
- IFN
interferon
- IgG Fc
immunoglobulin G fragment crystallizable
- IκBα
inhibitor of nuclear factor kappa B alpha
- IL
interleukin
- IRE1α
inositol-requiring enzyme 1α
- KO
knockout
- JAK2
Janus kinase 2
- JNK
c-Jun N-terminal kinase
- LPS
lipopolysaccharide
- MCP-1
chemokine monocyte chemoattractant protein-1
- MLDS
multiple low-dose streptozotocin
- MOMP
mitochondrial outer membrane permeabilization
- MyD88
myeloid differentiation factor 88
- mTOR
mammalian target of rapamycin
- NAFLD
non-alcoholic fatty liver disease
- NF-κB
nuclear factor kappa B
- NOS
nictric oxide synthase
- NPY
neuropeptide Y
- NPY2R
neuropeptide Y2 receptor
- NTS
nucleus tractus solitarius
- PAM
positive allosteric modulator
- p70S6K
ribosomal protein S6 kinase beta-1
- PC1/3
prohormone convertase 1/3
- PI3K
phosphatidylinositol 3-kinase
- PKA
protein kinase A
- PKB
protein kinase B
- PNU 282987
N-(3R)-1-Azabicyclo[2.2.2]oct-3-yl-4-chlorobenzamide
- POMC
proopiomelanocortin
- PYY
peptide YY
- RNase
endoribonuclease
- SAT
subcutaneous adipose tissue
- siRNA
small interfering RNA
- STAT3
signal transducer and activator of transcription 3
- STZ
streptozotocin
- XBP1s
spliced X-box binding protein
- T1D
type 1 diabetes
- T2D
type 2 diabetes
- TGF
transforming growth factor
- TXNIP
thioredoxin-interacting protein
- TLR4
Toll-like receptor-4
- TNF
tumor necrosis factor
- UCD-T2D
UC Davis type 2 diabetes model rat
- UPR
unfolded protein response
- VAT
visceral adipose tissue
- VIP
vasoactive intestinal polypeptide
- VN
vagus nerve
- ZDF
Zucker diabetic fatty rat
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflicts of Interest: The authors have no financial interests to disclose.
Ethics Approval: N/A
Consent to participate: N/A
Consent for publication: N/A
Availability of data: N/A
Code Availability: N/A
References
- 1.Cornier MA, Dabelea D, Hernandez TL, Lindstrom RC, Steig AJ, Stob NR et al. The metabolic syndrome. Endocr Rev. 2008;29(7):777–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pavlov VA, Tracey KJ. The vagus nerve and the inflammatory reflex-linking immunity and metabolism. Nat Rev Endocrinol. 2012;8(12):743–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang EH, Chavan SS, Pavlov VA. Cholinergic control of inflammation, metabolic dysfunction, and cognitive impairment in obesity-associated disorders: mechanisms and novel therapeutic opportunities. Front Neurosci. 2019;13:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berthoud HR, Neuhuber WL. Vagal mechanisms as neuromodulatory targets for the treatment of metabolic disease. Ann N Y Acad Sci. 2019;1454(1):42–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonaz B, Sinniger V, Pellissier S. The vagus nerve in the neuro-immune axis: implications in the pathology of the gastrointestinal tract. Front Immunol. 2017;8:1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Browning KN, Verheijden S, Boeckxstaens GE. The vagus nerve in appetite regulation, mood, and intestinal inflammation. Gastroenterology. 2017;152(4):730–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Lartigue G. Role of the vagus nerve in the development and treatment of diet-induced obesity. The Journal of physiology. 2016;594(20):5791–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kany S, Vollrath JT, Relja B. Cytokines in inflammatory disease. Int J Mol Sci. 2019;20(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnston GR, Webster NR. Cytokines and the immunomodulatory function of the vagus nerve. Br J Anaesth. 2009;102(4):453–62. [DOI] [PubMed] [Google Scholar]
- 10.Rosas-Ballina M, Goldstein RS, Gallowitsch-Puerta M, Yang L, Valdés-Ferrer SI, Patel NB et al. The selective alpha7 agonist GTS-21 attenuates cytokine production in human whole blood and human monocytes activated by ligands for TLR2, TLR3, TLR4, TLR9, and RAGE. Mol Med. 2009;15(7–8):195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421(6921):384–8. [DOI] [PubMed] [Google Scholar]
- 12.Yoshikawa H, Kurokawa M, Ozaki N, Nara K, Atou K, Takada E et al. Nicotine inhibits the production of proinflammatory mediators in human monocytes by suppression of I-kappaB phosphorylation and nuclear factor-kappaB transcriptional activity through nicotinic acetylcholine receptor alpha7. Clin Exp Immunol. 2006;146(1):116–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Masi EB, Valdes-Ferrer SI, Steinberg BE. The vagus neurometabolic interface and clinical disease. Int J Obes (Lond). 2018;42(6):1101–11. [DOI] [PubMed] [Google Scholar]
- 14.Chavan SS, Pavlov VA, Tracey KJ. Mechanisms and therapeutic relevance of neuro-immune communication. Immunity. 2017;46(6):927–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goehler LE, Gaykema RP, Nguyen KT, Lee JE, Tilders FJ, Maier SF et al. Interleukin-1beta in immune cells of the abdominal vagus nerve: a link between the immune and nervous systems? J Neurosci. 1999;19(7):2799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405(6785):458–62. [DOI] [PubMed] [Google Scholar]
- 17.Rosas-Ballina M, Olofsson PS, Ochani M, Valdes-Ferrer SI, Levine YA, Reardon C et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 2011;334(6052):98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abot A, Cani PD, Knauf C. Impact of intestinal peptides on the enteric nervous system: novel approaches to control glucose metabolism and food intake. Frontiers in endocrinology. 2018;9:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nezami BG, Srinivasan S. Enteric nervous system in the small intestine: pathophysiology and clinical implications. Current gastroenterology reports. 2010;12(5):358–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Metz CN, Pavlov VA. Vagus nerve cholinergic circuitry to the liver and the gastrointestinal tract in the neuroimmune communicatome. American journal of physiology Gastrointestinal and liver physiology. 2018;315(5):G651–g8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hiramoto T, Chida Y, Sonoda J, Yoshihara K, Sudo N, Kubo C. The hepatic vagus nerve attenuates Fas-induced apoptosis in the mouse liver via alpha7 nicotinic acetylcholine receptor. Gastroenterology. 2008;134(7):2122–31. [DOI] [PubMed] [Google Scholar]
- 22.Zhao J, Park S, Kim JW, Qi J, Zhou Z, Lim CW et al. Nicotine attenuates concanavalin A-induced liver injury in mice by regulating the alpha7-nicotinic acetylcholine receptor in Kupffer cells. International immunopharmacology. 2020;78:106071. [DOI] [PubMed] [Google Scholar]
- 23.Jamal Uddin M, Joe Y, Zheng M, Blackshear PJ, Ryter SW, Park JW et al. A functional link between heme oxygenase-1 and tristetraprolin in the anti-inflammatory effects of nicotine. Free radical biology & medicine. 2013;65:1331–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahren B, Taborsky GJ Jr. The mechanism of vagal nerve stimulation of glucagon and insulin secretion in the dog. Endocrinology. 1986;118(4):1551–7. [DOI] [PubMed] [Google Scholar]
- 25.Berthoud HR. The vagus nerve, food intake and obesity. Regul Pept. 2008;149(1–3):15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bugajski AJ, Gil K, Ziomber A, Zurowski D, Zaraska W, Thor PJ. Effect of long-term vagal stimulation on food intake and body weight during diet induced obesity in rats. J Physiol Pharmacol. 2007;58 Suppl 1:5–12. [PubMed] [Google Scholar]
- 27.Burneo JG, Faught E, Knowlton R, Morawetz R, Kuzniecky R. Weight loss associated with vagus nerve stimulation. Neurology. 2002;59(3):463–4. [DOI] [PubMed] [Google Scholar]
- 28.Dai F, Yin J, Chen JDZ. Effects and mechanisms of vagal nerve stimulation on body weight in diet-induced obese rats. Obesity surgery. 2020;30(3):948–56. [DOI] [PubMed] [Google Scholar]
- 29.de Lartigue G, Diepenbroek C. Novel developments in vagal afferent nutrient sensing and its role in energy homeostasis. Curr Opin Pharmacol. 2016;31:38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li S, Zhai X, Rong P, McCabe MF, Wang X, Zhao J et al. Therapeutic effect of vagus nerve stimulation on depressive-like behavior, hyperglycemia and insulin receptor expression in Zucker fatty rats. PloS one. 2014;9(11):e112066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malbert CH, Picq C, Divoux JL, Henry C, Horowitz M. Obesity-associated alterations in glucose metabolism are reversed by chronic bilateral stimulation of the abdominal vagus nerve. Diabetes. 2017;66(4):848–57. [DOI] [PubMed] [Google Scholar]
- 32.Pardo JV, Sheikh SA, Kuskowski MA, Surerus-Johnson C, Hagen MC, Lee JT et al. Weight loss during chronic, cervical vagus nerve stimulation in depressed patients with obesity: an observation. Int J Obes (Lond). 2007;31(11):1756–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sobocki J, Fourtanier G, Estany J, Otal P. Does vagal nerve stimulation affect body composition and metabolism? Experimental study of a new potential technique in bariatric surgery. Surgery. 2006;139(2):209–16. [DOI] [PubMed] [Google Scholar]
- 34.Val-Laillet D, Biraben A, Randuineau G, Malbert CH. Chronic vagus nerve stimulation decreased weight gain, food consumption and sweet craving in adult obese minipigs. Appetite. 2010;55(2):245–52. [DOI] [PubMed] [Google Scholar]
- 35.Arterburn DE, Fisher DP. The current state of the evidence for bariatric surgery. Jama. 2014;312(9):898–9. [DOI] [PubMed] [Google Scholar]
- 36.Ikramuddin S, Blackstone RP, Brancatisano A, Toouli J, Shah SN, Wolfe BM et al. Effect of reversible intermittent intra-abdominal vagal nerve blockade on morbid obesity: the ReCharge randomized clinical trial. Jama. 2014;312(9):915–22. [DOI] [PubMed] [Google Scholar]
- 37.Cancello R, Zulian A, Maestrini S, Mencarelli M, Della Barba A, Invitti C et al. The nicotinic acetylcholine receptor alpha7 in subcutaneous mature adipocytes: downregulation in human obesity and modulation by diet-induced weight loss. Int J Obes (Lond). 2012;36(12):1552–7. [DOI] [PubMed] [Google Scholar]
- 38.Wang X, Yang Z, Xue B, Shi H. Activation of the cholinergic antiinflammatory pathway ameliorates obesity-induced inflammation and insulin resistance. Endocrinology. 2011;152(3):836–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu TY, Guo LL, Wang P, Song J, Le YY, Viollet B et al. Chronic exposure to nicotine enhances insulin sensitivity through alpha7 nicotinic acetylcholine receptor-STAT3 pathway. PloS one. 2012;7(12):e51217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Priest C, Tontonoz P. Inter-organ cross-talk in metabolic syndrome. Nat Metab. 2019;1(12):1177–88. [DOI] [PubMed] [Google Scholar]
- 41.Burcelin R, Gourdy P. Harnessing glucagon-like peptide-1 receptor agonists for the pharmacological treatment of overweight and obesity. Obes Rev. 2017;18(1):86–98. [DOI] [PubMed] [Google Scholar]
- 42.Müller TD, Finan B, Bloom SR, D’Alessio D, Drucker DJ, Flatt PR et al. Glucagon-like peptide 1 (GLP-1). Mol Metab. 2019;30:72–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nadkarni P, Chepurny OG, Holz GG. Regulation of glucose homeostasis by GLP-1. Prog Mol Biol Transl Sci. 2014;121:23–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kentish SJ, Vincent AD, Kennaway DJ, Wittert GA, Page AJ. High-Fat Diet-induced obesity ablates gastric vagal afferent circadian rhythms. J Neurosci. 2016;36(11):3199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kentish S, Li H, Philp LK, O’Donnell TA, Isaacs NJ, Young RL et al. Diet-induced adaptation of vagal afferent function. The Journal of physiology. 2012;590(1):209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daly DM, Park SJ, Valinsky WC, Beyak MJ. Impaired intestinal afferent nerve satiety signalling and vagal afferent excitability in diet induced obesity in the mouse. The Journal of physiology. 2011;589(Pt 11):2857–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kentish SJ, O’Donnell TA, Isaacs NJ, Young RL, Li H, Harrington AM et al. Gastric vagal afferent modulation by leptin is influenced by food intake status. The Journal of physiology. 2013;591(7):1921–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee J, Cummings BP, Martin E, Sharp JW, Graham JL, Stanhope KL et al. Glucose sensing by gut endocrine cells and activation of the vagal afferent pathway is impaired in a rodent model of type 2 diabetes mellitus. Am J Physiol Regul Integr Comp Physiol. 2012;302(6):R657–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bernik TR, Friedman SG, Ochani M, DiRaimo R, Ulloa L, Yang H et al. Pharmacological stimulation of the cholinergic antiinflammatory pathway. The Journal of experimental medicine. 2002;195(6):781–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beinat C, Banister SD, Herrera M, Law V, Kassiou M. The therapeutic potential of alpha7 nicotinic acetylcholine receptor (alpha7 nAChR) agonists for the treatment of the cognitive deficits associated with schizophrenia. CNS Drugs. 2015;29(7):529–42. [DOI] [PubMed] [Google Scholar]
- 51.Ishikawa M, Hashimoto K. α7 nicotinic acetylcholine receptor as a potential therapeutic target for schizophrenia. Curr Pharm Des. 2011;17(2):121–9. [DOI] [PubMed] [Google Scholar]
- 52.Pohanka M. Alpha7 nicotinic acetylcholine receptor is a target in pharmacology and toxicology. Int J Mol Sci. 2012;13(2):2219–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Terry AV Jr., Callahan PM. α7 nicotinic acetylcholine receptors as therapeutic targets in schizophrenia: Update on animal and clinical studies and strategies for the future. Neuropharmacology. 2020;170:108053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chini B, Raimond E, Elgoyhen AB, Moralli D, Balzaretti M, Heinemann S. Molecular cloning and chromosomal localization of the human alpha 7-nicotinic receptor subunit gene (CHRNA7). Genomics. 1994;19(2):379–81. [DOI] [PubMed] [Google Scholar]
- 55.Orr-Urtreger A, Seldin MF, Baldini A, Beaudet AL. Cloning and mapping of the mouse alpha 7-neuronal nicotinic acetylcholine receptor. Genomics. 1995;26(2):399–402. [DOI] [PubMed] [Google Scholar]
- 56.Peng X, Katz M, Gerzanich V, Anand R, Lindstrom J. Human alpha 7 acetylcholine receptor: cloning of the alpha 7 subunit from the SH-SY5Y cell line and determination of pharmacological properties of native receptors and functional alpha 7 homomers expressed in Xenopus oocytes. Mol Pharmacol. 1994;45(3):546–54. [PubMed] [Google Scholar]
- 57.Séguéla P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13(2):596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Changeux JP. The nicotinic acetylcholine receptor: the founding father of the pentameric ligand-gated ion channel superfamily. J Biol Chem. 2012;287(48):40207–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kalamida D, Poulas K, Avramopoulou V, Fostieri E, Lagoumintzis G, Lazaridis K et al. Muscle and neuronal nicotinic acetylcholine receptors. Structure, function and pathogenicity. The FEBS journal. 2007;274(15):3799–845. [DOI] [PubMed] [Google Scholar]
- 60.Andersen N, Corradi J, Sine SM, Bouzat C. Stoichiometry for activation of neuronal alpha7 nicotinic receptors. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(51):20819–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nielsen BE, Minguez T, Bermudez I, Bouzat C. Molecular function of the novel alpha7beta2 nicotinic receptor. Cellular and molecular life sciences : CMLS. 2018;75(13):2457–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu J, Lukas RJ. Naturally-expressed nicotinic acetylcholine receptor subtypes. Biochem Pharmacol. 2011;82(8):800–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uteshev VV. alpha7 nicotinic ACh receptors as a ligand-gated source of Ca2+ ions: the search for a Ca2+ optimum. Adv Exp Med Biol. 2012;740:603–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Corradi J, Bouzat C. Understanding the bases of function and modulation of α7 nicotinic receptors: implications for drug discovery. Mol Pharmacol. 2016;90(3):288–99. [DOI] [PubMed] [Google Scholar]
- 65.King JR, Ullah A, Bak E, Jafri MS, Kabbani N. Ionotropic and metabotropic mechanisms of allosteric modulation of α7 nicotinic receptor intracellular calcium. Mol Pharmacol. 2018;93(6):601–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu Q, Berg DK. Actin filaments and the opposing actions of CaM kinase II and calcineurin in regulating alpha7-containing nicotinic receptors on chick ciliary ganglion neurons. J Neurosci. 1999;19(23):10280–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.King JR, Nordman JC, Bridges SP, Lin MK, Kabbani N. Identification and characterization of a G protein-binding cluster in α7 nicotinic acetylcholine receptors. J Biol Chem. 2015;290(33):20060–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grady SR, Wageman CR, Patzlaff NE, Marks MJ. Low concentrations of nicotine differentially desensitize nicotinic acetylcholine receptors that include alpha5 or alpha6 subunits and that mediate synaptosomal neurotransmitter release. Neuropharmacology. 2012;62(5–6):1935–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mao D, Yasuda RP, Fan H, Wolfe BB, Kellar KJ. Heterogeneity of nicotinic cholinergic receptors in rat superior cervical and nodose ganglia. Mol Pharmacol. 2006;70(5):1693–9. [DOI] [PubMed] [Google Scholar]
- 70.Simeone X, Karch R, Ciuraszkiewicz A, Orr-Urtreger A, Lemmens-Gruber R, Scholze P et al. The role of the nAChR subunits α5, β2, and β4 on synaptic transmission in the mouse superior cervical ganglion. Physiol Rep. 2019;7(6):e14023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Orr-Urtreger A, Göldner FM, Saeki M, Lorenzo I, Goldberg L, De Biasi M et al. Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J Neurosci. 1997;17(23):9165–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gulsevin A, Papke RL, Horenstein N. In silico modeling of the α7 nicotinic acetylcholine receptor: new pharmacological challenges associated with multiple modes of signaling. Mini Rev Med Chem. 2020;20(10):841–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kem WR. The brain alpha7 nicotinic receptor may be an important therapeutic target for the treatment of Alzheimer’s disease: studies with DMXBA (GTS-21). Behav Brain Res. 2000;113(1–2):169–81. [DOI] [PubMed] [Google Scholar]
- 74.Papke RL, Lindstrom JM. Nicotinic acetylcholine receptors: Conventional and unconventional ligands and signaling. Neuropharmacology. 2020;168:108021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Williams DK, Wang J, Papke RL. Positive allosteric modulators as an approach to nicotinic acetylcholine receptor-targeted therapeutics: advantages and limitations. Biochem Pharmacol. 2011;82(8):915–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Briggs CA, Gronlien JH, Curzon P, Timmermann DB, Ween H, Thorin-Hagene K et al. Role of channel activation in cognitive enhancement mediated by alpha7 nicotinic acetylcholine receptors. Br J Pharmacol. 2009;158(6):1486–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Godin JR, Roy P, Quadri M, Bagdas D, Toma W, Narendrula-Kotha R et al. A silent agonist of alpha7 nicotinic acetylcholine receptors modulates inflammation ex vivo and attenuates EAE. Brain, behavior, and immunity. 2020;87:286–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gronlien JH, Hakerud M, Ween H, Thorin-Hagene K, Briggs CA, Gopalakrishnan M et al. Distinct profiles of alpha7 nAChR positive allosteric modulation revealed by structurally diverse chemotypes. Mol Pharmacol. 2007;72(3):715–24. [DOI] [PubMed] [Google Scholar]
- 79.Gurley DA, Lanthorn TH. Nicotinic agonists competitively antagonize serotonin at mouse 5-HT3 receptors expressed in Xenopus oocytes. Neurosci Lett. 1998;247(2–3):107–10. [DOI] [PubMed] [Google Scholar]
- 80.Gault J, Robinson M, Berger R, Drebing C, Logel J, Hopkins J et al. Genomic organization and partial duplication of the human alpha7 neuronal nicotinic acetylcholine receptor gene (CHRNA7). Genomics. 1998;52(2):173–85. [DOI] [PubMed] [Google Scholar]
- 81.Riley B, Williamson M, Collier D, Wilkie H, Makoff A. A 3-Mb map of a large segmental duplication overlapping the alpha7-nicotinic acetylcholine receptor gene (CHRNA7) at human 15q13-q14. Genomics. 2002;79(2):197–209. [DOI] [PubMed] [Google Scholar]
- 82.Araud T, Graw S, Berger R, Lee M, Neveu E, Bertrand D et al. The chimeric gene CHRFAM7A, a partial duplication of the CHRNA7 gene, is a dominant negative regulator of alpha7*nAChR function. Biochem Pharmacol. 2011;82(8):904–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.de Lucas-Cerrillo AM, Maldifassi MC, Arnalich F, Renart J, Atienza G, Serantes R et al. Function of partially duplicated human α77 nicotinic receptor subunit CHRFAM7A gene: potential implications for the cholinergic anti-inflammatory response. J Biol Chem. 2011;286(1):594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Baez-Pagan CA, Delgado-Velez M, Lasalde-Dominicci JA. Activation of the macrophage alpha7 nicotinic acetylcholine receptor and control of inflammation. J Neuroimmune Pharmacol. 2015;10(3):468–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol. 2005;6(8):844–51. [DOI] [PubMed] [Google Scholar]
- 86.Marrero MB, Bencherif M. Convergence of alpha 7 nicotinic acetylcholine receptor-activated pathways for anti-apoptosis and anti-inflammation: central role for JAK2 activation of STAT3 and NF-kappaB. Brain Res. 2009;1256:1–7. [DOI] [PubMed] [Google Scholar]
- 87.Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology. 2007;132(6):2169–80. [DOI] [PubMed] [Google Scholar]
- 88.Han JM, Levings MK. Immune regulation in obesity-associated adipose inflammation. J Immunol. 2013;191(2):527–32. [DOI] [PubMed] [Google Scholar]
- 89.Sutherland JP, McKinley B, Eckel RH. The metabolic syndrome and inflammation. Metab Syndr Relat Disord. 2004;2(2):82–104. [DOI] [PubMed] [Google Scholar]
- 90.Phosat C, Panprathip P, Chumpathat N, Prangthip P, Chantratita N, Soonthornworasiri N et al. Elevated C-reactive protein, interleukin 6, tumor necrosis factor alpha and glycemic load associated with type 2 diabetes mellitus in rural Thais: a cross-sectional study. BMC Endocr Disord. 2017;17(1):44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett. 2008;582(1):97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rehman K, Akash MS. Mechanisms of inflammatory responses and development of insulin resistance: how are they interlinked? J Biomed Sci. 2016;23(1):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tzanavari T, Giannogonas P, Karalis KP. TNF-alpha and obesity. Curr Dir Autoimmun. 2010;11:145–56. [DOI] [PubMed] [Google Scholar]
- 94.Coelho M, Oliveira T, Fernandes R. Biochemistry of adipose tissue: an endocrine organ. Archives of medical science : AMS. 2013;9(2):191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jiao ZY, Wu J, Liu C, Wen B, Zhao WZ, Du XL. Nicotinic α7 receptor inhibits the acylation stimulating protein-induced production of monocyte chemoattractant protein-1 and keratinocyte-derived chemokine in adipocytes by modulating the p38 kinase and nuclear factor-κB signaling pathways. Molecular medicine reports. 2016;14(4):2959–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Scabia G, Cancello R, Dallanoce C, Berger S, Matera C, Dattilo A et al. ICH3, a selective alpha7 nicotinic acetylcholine receptor agonist, modulates adipocyte inflammation associated with obesity. J Endocrinol Invest. 2020;43(7):983–93. [DOI] [PubMed] [Google Scholar]
- 97.Chau YY, Bandiera R, Serrels A, Martínez-Estrada OM, Qing W, Lee M et al. Visceral and subcutaneous fat have different origins and evidence supports a mesothelial source. Nat Cell Biol. 2014;16(4):367–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marrero MB, Lucas R, Salet C, Hauser TA, Mazurov A, Lippiello PM et al. An alpha7 nicotinic acetylcholine receptor-selective agonist reduces weight gain and metabolic changes in a mouse model of diabetes. J Pharmacol Exp Ther. 2010;332(1):173–80. [DOI] [PubMed] [Google Scholar]
- 99.Hasan MK, Friedman TC, Sims C, Lee DL, Espinoza-Derout J, Ume A et al. alpha7-nicotinic acetylcholine receptor agonist ameliorates nicotine plus high-fat diet-induced hepatic steatosis in male mice by inhibiting oxidative stress and stimulating AMPK signaling. Endocrinology. 2018;159(2):931–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li DJ, Zhao T, Xin RJ, Wang YY, Fei YB, Shen FM. Activation of alpha7 nicotinic acetylcholine receptor protects against oxidant stress damage through reducing vascular peroxidase-1 in a JNK signaling-dependent manner in endothelial cells. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2014;33(2):468–78. [DOI] [PubMed] [Google Scholar]
- 101.Costa SO, Souza CM, Lanza PG, Sartori JO, Ignacio-Souza LM, Candreva T et al. Maternal high fat diet consumption reduces liver alpha7 nicotinic cholinergic receptor expression and impairs insulin signalling in the offspring. Sci Rep. 2020;10(1):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhu Z, Cao F, Li X. Epigenetic Programming and Fetal Metabolic Programming. Frontiers in endocrinology. 2019;10:764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lilienfeld S. Galantamine--a novel cholinergic drug with a unique dual mode of action for the treatment of patients with Alzheimer’s disease. CNS drug reviews. 2002;8(2):159–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Consolim-Colombo FM, Sangaleti CT, Costa FO, Morais TL, Lopes HF, Motta JM et al. Galantamine alleviates inflammation and insulin resistance in patients with metabolic syndrome in a randomized trial. JCI insight. 2017;2(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mucke HA. The case of galantamine: repurposing and late blooming of a cholinergic drug. Future Sci OA. 2015;1(4):Fso73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Maelicke A, Samochocki M, Jostock R, Fehrenbacher A, Ludwig J, Albuquerque EX et al. Allosteric sensitization of nicotinic receptors by galantamine, a new treatment strategy for Alzheimer’s disease. Biol Psychiatry. 2001;49(3):279–88. [DOI] [PubMed] [Google Scholar]
- 107.Texidó L, Ros E, Martín-Satué M, López S, Aleu J, Marsal J et al. Effect of galantamine on the human alpha7 neuronal nicotinic acetylcholine receptor, the Torpedo nicotinic acetylcholine receptor and spontaneous cholinergic synaptic activity. Br J Pharmacol. 2005;145(5):672–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Satapathy SK, Ochani M, Dancho M, Hudson LK, Rosas-Ballina M, Valdes-Ferrer SI et al. Galantamine alleviates inflammation and other obesity-associated complications in high-fat diet-fed mice. Mol Med. 2011;17(7–8):599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jo YH, Talmage DA, Role LW. Nicotinic receptor-mediated effects on appetite and food intake. J Neurobiol. 2002;53(4):618–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Winders SE, Grunberg NE. Effects of nicotine on body weight, food consumption and body composition in male rats. Life Sci. 1990;46(21):1523–30. [DOI] [PubMed] [Google Scholar]
- 111.Liu RH, Kurose T, Matsukura S. Oral nicotine administration decreases tumor necrosis factor-alpha expression in fat tissues in obese rats. Metabolism. 2001;50(1):79–85. [DOI] [PubMed] [Google Scholar]
- 112.McFadden KL, Cornier MA, Tregellas JR. The role of alpha-7 nicotinic receptors in food intake behaviors. Front Psychol. 2014;5:553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tuesta LM, Chen Z, Duncan A, Fowler CD, Ishikawa M, Lee BR et al. GLP-1 acts on habenular avoidance circuits to control nicotine intake. Nat Neurosci. 2017;20(5):708–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Barrea L, Pugliese G, Muscogiuri G, Laudisio D, Colao A, Savastano S. New-generation anti-obesity drugs: naltrexone/bupropion and liraglutide. An update for endocrinologists and nutritionists. Minerva Endocrinol. 2020;45(2):127–37. [DOI] [PubMed] [Google Scholar]
- 115.Khalil H, Ellwood L, Lord H, Fernandez R. Pharmacological Treatment for Obesity in Adults: An Umbrella Review. Ann Pharmacother. 2020;54(7):691–705. [DOI] [PubMed] [Google Scholar]
- 116.Montan PD, Sourlas A, Olivero J, Silverio D, Guzman E, Kosmas CE. Pharmacologic therapy of obesity: mechanisms of action and cardiometabolic effects. Ann Transl Med. 2019;7(16):393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Greenway FL, Fujioka K, Plodkowski RA, Mudaliar S, Guttadauria M, Erickson J et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376(9741):595–605. [DOI] [PubMed] [Google Scholar]
- 118.Hollander P, Gupta AK, Plodkowski R, Greenway F, Bays H, Burns C et al. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care. 2013;36(12):4022–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Borner T, Workinger JL, Tinsley IC, Fortin SM, Stein LM, Chepurny OG et al. Corrination of a GLP-1 receptor agonist for glycemic control without emesis. Cell Rep. 2020;31(11):107768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mietlicki-Baase EG, Liberini CG, Workinger JL, Bonaccorso RL, Borner T, Reiner DJ et al. A vitamin B12 conjugate of exendin-4 improves glucose tolerance without associated nausea or hypophagia in rodents. Diabetes Obes Metab. 2018;20(5):1223–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gribble FM, Reimann F. Enteroendocrine Cells: Chemosensors in the intestinal epithelium. Annu Rev Physiol. 2016;78:277–99. [DOI] [PubMed] [Google Scholar]
- 122.Gribble FM, Reimann F. Function and mechanisms of enteroendocrine cells and gut hormones in metabolism. Nat Rev Endocrinol. 2019;15(4):226–37. [DOI] [PubMed] [Google Scholar]
- 123.Mace OJ, Tehan B, Marshall F. Pharmacology and physiology of gastrointestinal enteroendocrine cells. Pharmacol Res Perspect. 2015;3(4):e00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Reimann F, Habib AM, Tolhurst G, Parker HE, Rogers GJ, Gribble FM. Glucose sensing in L cells: a primary cell study. Cell metabolism. 2008;8(6):532–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ye L, Liddle RA. Gastrointestinal hormones and the gut connectome. Curr Opin Endocrinol Diabetes Obes. 2017;24(1):9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.De Silva A, Bloom SR. Gut Hormones and Appetite Control: a focus on PYY and GLP-1 as therapeutic targets in obesity. Gut Liver. 2012;6(1):10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sandoval D, Dunki-Jacobs A, Sorrell J, Seeley RJ, D’Alessio DD. Impact of intestinal electrical stimulation on nutrient-induced GLP-1 secretion in vivo. Neurogastroenterol Motil. 2013;25(8):700–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yin J, Ji F, Gharibani P, Chen JD. Vagal nerve stimulation for glycemic control in a rodent model of type 2 diabetes. Obesity surgery. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rocca AS, Brubaker PL. Role of the vagus nerve in mediating proximal nutrient-induced glucagon-like peptide-1 secretion. Endocrinology. 1999;140(4):1687–94. [DOI] [PubMed] [Google Scholar]
- 130.Wang D, Meng Q, Leech CA, Yepuri N, Zhang L, Holz GG et al. alpha7 nicotinic acetylcholine receptor regulates the function and viability of L cells. Endocrinology. 2018;159(9):3132–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jorsal T, Rhee NA, Pedersen J, Wahlgren CD, Mortensen B, Jepsen SL et al. Enteroendocrine K and L cells in healthy and type 2 diabetic individuals. Diabetologia. 2018;61(2):284–94. [DOI] [PubMed] [Google Scholar]
- 132.Cabou C, Burcelin R. GLP-1, the gut-brain, and brain-periphery axes. Rev Diabet Stud. 2011;8(3):418–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Burcelin R, Gourdy P, Dalle S. GLP-1-based strategies: a physiological analysis of differential mode of action. Physiology (Bethesda). 2014;29(2):108–21. [DOI] [PubMed] [Google Scholar]
- 134.Smith EP, An Z, Wagner C, Lewis AG, Cohen EB, Li B et al. The role of β cell glucagon-like peptide-1 signaling in glucose regulation and response to diabetes drugs. Cell metabolism. 2014;19(6):1050–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.O’Malley TJ, Fava GE, Zhang Y, Fonseca VA, Wu H. Progressive change of intra-islet GLP-1 production during diabetes development. Diabetes Metab Res Rev. 2014;30(8):661–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Donath MY, Burcelin R. GLP-1 effects on islets: hormonal, neuronal, or paracrine? Diabetes Care. 2013;36 Suppl 2(Suppl 2):S145–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Urusova IA, Farilla L, Hui H, D’Amico E, Perfetti R. GLP-1 inhibition of pancreatic islet cell apoptosis. Trends Endocrinol Metab. 2004;15(1):27–33. [DOI] [PubMed] [Google Scholar]
- 138.Farilla L, Bulotta A, Hirshberg B, Li Calzi S, Khoury N, Noushmehr H et al. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology. 2003;144(12):5149–58. [DOI] [PubMed] [Google Scholar]
- 139.Leech CA, Dzhura I, Chepurny OG, Kang G, Schwede F, Genieser HG et al. Molecular physiology of glucagon-like peptide-1 insulin secretagogue action in pancreatic β cells. Prog Biophys Mol Biol. 2011;107(2):236–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Holz GG IV, Kühtreiber WM, Habener JF. Pancreatic beta-cells are rendered glucose-competent by the insulinotropic hormone glucagon-like peptide-1(7–37). Nature. 1993;361(6410):362–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Holz GG, Chepurny OG. Diabetes outfoxed by GLP-1? Sci STKE. 2005;2005(268):pe2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chepurny OG, Hussain MA, Holz GG. Exendin-4 as a stimulator of rat insulin I gene promoter activity via bZIP/CRE interactions sensitive to serine/threonine protein kinase inhibitor Ro 31–8220. Endocrinology. 2002;143(6):2303–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Li W, Yu G, Liu Y, Sha L. Intrapancreatic ganglia and neural regulation of pancreatic endocrine secretion. Front Neurosci. 2019;13:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gilon P, Henquin JC. Mechanisms and physiological significance of the cholinergic control of pancreatic beta-cell function. Endocr Rev. 2001;22(5):565–604. [DOI] [PubMed] [Google Scholar]
- 145.Moullé VS, Tremblay C, Castell AL, Vivot K, Ethier M, Fergusson G et al. The autonomic nervous system regulates pancreatic β-cell proliferation in adult male rats. Am J Physiol Endocrinol Metab. 2019;317(2):E234–e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ahrén B, Holst JJ. The cephalic insulin response to meal ingestion in humans is dependent on both cholinergic and noncholinergic mechanisms and is important for postprandial glycemia. Diabetes. 2001;50(5):1030–8. [DOI] [PubMed] [Google Scholar]
- 147.Ahrén B. Autonomic regulation of islet hormone secretion--implications for health and disease. Diabetologia. 2000;43(4):393–410. [DOI] [PubMed] [Google Scholar]
- 148.Rodriguez-Diaz R, Abdulreda MH, Formoso AL, Gans I, Ricordi C, Berggren PO et al. Innervation patterns of autonomic axons in the human endocrine pancreas. Cell metabolism. 2011;14(1):45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Rodriguez-Diaz R, Dando R, Jacques-Silva MC, Fachado A, Molina J, Abdulreda MH et al. Alpha cells secrete acetylcholine as a non-neuronal paracrine signal priming beta cell function in humans. Nature Medicine. 2011;17(7):888–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yoshikawa H, Hellström-Lindahl E, Grill V. Evidence for functional nicotinic receptors on pancreatic beta cells. Metabolism. 2005;54(2):247–54. [DOI] [PubMed] [Google Scholar]
- 151.Somm E, Guérardel A, Maouche K, Toulotte A, Veyrat-Durebex C, Rohner-Jeanrenaud F et al. Concomitant alpha7 and beta2 nicotinic AChR subunit deficiency leads to impaired energy homeostasis and increased physical activity in mice. Mol Genet Metab. 2014;112(1):64–72. [DOI] [PubMed] [Google Scholar]
- 152.Gupta D, Lacayo AA, Greene SM, Leahy JL, Jetton TL. Beta-cell mass restoration by alpha7 nicotinic acetylcholine receptor activation. J Biol Chem. 2018;293(52):20295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ganic E, Singh T, Luan C, Fadista J, Johansson JK, Cyphert HA et al. MafA-controlled nicotinic receptor expression is essential for insulin secretion and is impaired in patients with type 2 diabetes. Cell Rep. 2016;14(8):1991–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Duttaroy A, Zimliki CL, Gautam D, Cui Y, Mears D, Wess J. Muscarinic stimulation of pancreatic insulin and glucagon release is abolished in m3 muscarinic acetylcholine receptor-deficient mice. Diabetes. 2004;53(7):1714–20. [DOI] [PubMed] [Google Scholar]
- 155.Bokvist K, Eliasson L, Ammälä C, Renström E, Rorsman P. Co-localization of L-type Ca2+ channels and insulin-containing secretory granules and its significance for the initiation of exocytosis in mouse pancreatic β-cells. Embo j. 1995;14(1):50–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.King AJ. The use of animal models in diabetes research. Br J Pharmacol. 2012;166(3):877–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ishibashi T, Morita S, Kishimoto S, Uraki S, Takeshima K, Furukawa Y et al. Nicotinic acetylcholine receptor signaling regulates inositol-requiring enzyme 1α activation to protect β-cells against terminal unfolded protein response under irremediable endoplasmic reticulum stress. J Diabetes Investig. 2020;11(4):801–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–6. [DOI] [PubMed] [Google Scholar]
- 159.Klee P, Bosco D, Guerardel A, Somm E, Toulotte A, Maechler P et al. Activation of nicotinic acetylcholine receptors decreases apoptosis in human and female murine pancreatic islets. Endocrinology. 2016;157(10):3800–8. [DOI] [PubMed] [Google Scholar]
- 160.Shikora S, Toouli J, Herrera MF, Kulseng B, Zulewski H, Brancatisano R et al. Vagal blocking improves glycemic control and elevated blood pressure in obese subjects with type 2 diabetes mellitus. J Obes. 2013;2013:245683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kitagawa H, Takenouchi T, Azuma R, Wesnes KA, Kramer WG, Clody DE et al. Safety, pharmacokinetics, and effects on cognitive function of multiple doses of GTS-21 in healthy, male volunteers. Neuropsychopharmacology. 2003;28(3):542–51. [DOI] [PubMed] [Google Scholar]
- 162.Wu J, Jiao ZY, Zhang Z, Tang ZH, Zhang HH, Lu HL et al. Cross-talk between alpha7 nAChR-mediated cholinergic pathway and acylation stimulating protein signaling in 3T3-L1 adipocytes: role of NFkappaB and STAT3. Biochem Cell Biol. 2015;93(4):335–42. [DOI] [PubMed] [Google Scholar]
- 163.Wada T, Naito M, Kenmochi H, Tsuneki H, Sasaoka T. Chronic nicotine exposure enhances insulin-induced mitogenic signaling via up-regulation of alpha7 nicotinic receptors in isolated rat aortic smooth muscle cells. Endocrinology. 2007;148(2):790–9. [DOI] [PubMed] [Google Scholar]
- 164.Souza CM, do Amaral CL, Souza SC, de Souza ACP, de Cássia Alves Martins I, Contieri LS et al. JAK2/STAT3 pathway is required for α7nAChR-dependent expression of POMC and AGRP neuropeptides in male mice. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2019;53(4):701–12. [DOI] [PubMed] [Google Scholar]
- 165.Stegemann A, Bohm M. Tropisetron via alpha7 nicotinic acetylcholine receptor suppresses tumor necrosis factor-alpha-mediated cell responses of human keratinocytes. Exp Dermatol. 2019;28(3):276–82. [DOI] [PubMed] [Google Scholar]
- 166.Meyer EM, Kuryatov A, Gerzanich V, Lindstrom J, Papke RL. Analysis of 3-(4-hydroxy, 2-Methoxybenzylidene)anabaseine selectivity and activity at human and rat alpha-7 nicotinic receptors. J Pharmacol Exp Ther. 1998;287(3):918–25. [PubMed] [Google Scholar]
- 167.Bodnar AL, Cortes-Burgos LA, Cook KK, Dinh DM, Groppi VE, Hajos M et al. Discovery and structure-activity relationship of quinuclidine benzamides as agonists of alpha7 nicotinic acetylcholine receptors. J Med Chem. 2005;48(4):905–8. [DOI] [PubMed] [Google Scholar]
- 168.Wishka DG, Walker DP, Yates KM, Reitz SC, Jia S, Myers JK et al. Discovery of N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]furo[2,3-c]pyridine-5-carboxamide, an agonist of the alpha7 nicotinic acetylcholine receptor, for the potential treatment of cognitive deficits in schizophrenia: synthesis and structure--activity relationship. J Med Chem. 2006;49(14):4425–36. [DOI] [PubMed] [Google Scholar]
- 169.Acker BA, Jacobsen EJ, Rogers BN, Wishka DG, Reitz SC, Piotrowski DW et al. Discovery of N-[(3R,5R)-1-azabicyclo[3.2.1]oct-3-yl]furo[2,3-c]pyridine-5-carboxamide as an agonist of the alpha7 nicotinic acetylcholine receptor: in vitro and in vivo activity. Bioorg Med Chem Lett. 2008;18(12):3611–5. [DOI] [PubMed] [Google Scholar]
- 170.Dallanoce C, Magrone P, Matera C, Frigerio F, Grazioso G, De Amici M et al. Design, synthesis, and pharmacological characterization of novel spirocyclic quinuclidinyl-Delta2-isoxazoline derivatives as potent and selective agonists of alpha7 nicotinic acetylcholine receptors. ChemMedChem. 2011;6(5):889–903. [DOI] [PubMed] [Google Scholar]
- 171.Matera C, Dondio G, Braida D, Ponzoni L, De Amici M, Sala M, Dallanoce C. In vivo and in vitro ADMET profiling and in vivo pharmacodynamic investigations of a selective alpha7 nicotinic acetylcholine receptor agonist with a spirocyclic Delta(2)-isoxazoline molecular skeleton. Eur J Pharmacol. 2018;820:265–273. [DOI] [PubMed] [Google Scholar]
- 172.Di Cesare ML, Pacini A, Matera C, Zanardelli M, Mello T, De Amici M, Dallanoce C, Ghelardini C. Involvement of alpha7 nAChR subtype in rat oxaliplatin-induced neuropathy: effects of selective activation. Neuropharmacology 2014;79:37–48. [DOI] [PubMed] [Google Scholar]
- 173.Bristow LJ, Easton AE, Li YW, Sivarao DV, Lidge R, Jones KM et al. The novel, nicotinic alpha7 receptor partial agonist, BMS-933043, improves cognition and sensory processing in preclinical models of schizophrenia. PloS one. 2016;11(7):e0159996. [DOI] [PMC free article] [PubMed] [Google Scholar]