

Abstract
The α9α10 nicotinic acetylcholine receptor (nAChR) has been characterized as an effective anti-pain target that functions through a non-opioid mechanism. However, as a pentameric ion channel comprised of two different subunits, the specific targeting of α9α10 nAChRs has proven challenging. Previously the 13-amino-acid peptide, RgIA, was shown to block α9α10 nAChRs with high potency and specificity. This peptide, characterized from the venom of the carnivorous marine snail, Conus regius, produced analgesia in several rodent models of chronic pain. Despite promising pre-clinical data in behavioral assays, the number of specific α9α10 nAChR antagonists remains small and the physiological mechanisms of analgesia remain cryptic. In this study, we implement amino-acid substitutions to definitively characterize the chemical properties of RgIA that contribute to its activity against α9α10 nAChRs. Using this mutational approach, we determined the vital role of biochemical side-chain properties and amino acids in the second loop that are amenable to substitutions to further engineer next-generation analogs for the blockade of α9α10 nAChRs.
Keywords: Structure-activity relationship, nAChR, Nicotinic, Conotoxin, Analgesia
1. INTRODUCTION
Mollusks from the genus Conus are predatory marine snails that hunt worms, snails, or fish. These 750+ cone snail species use a sophisticated hunting and envenomation strategy tailored to their prey. Conus venoms are complex, once estimated to contain 50–200 unique compounds per species. However, with the advancement of transcriptomics and proteomics, this estimate has expanded approximately 10-fold. The wide diversity of bioactive compounds in cone snail venoms enables the specific and potent blockade of a wide array of targets involved in neurotransmission in the prey species. Many of the characterized targets fall within the families of voltage- and ligand-gated ion channels and G-protein-coupled receptors (GPCRs) [1]. Due to their natural library of bioactive compounds, cone snails are regarded as a source of potential therapeutic molecules.
Peptides from Conus have been particularly useful in the characterization of nicotinic acetylcholine receptors (nAChRs). Neuronal nAChRs are comprised of five subunits, containing one or more α subunits (α2-α10), and typically one or more ß subunits (ß1–4). On an evolutionary basis, nAChR subunits can be divided into three distinct clades. The α-subunits, α7, α9, and α10, are considered to be the ancestral members and can form functional channels without the presence of β subunits. The second clade includes the rest of the α subunits and the third clade includes the β subunits, as well as the δ, γ, and ε subunits that contribute to the muscle subtype. [2, 3]. Competitive agonists and antagonists bind to a site located at the interface of adjacent α/α or α/β subunits.
The α9 subunit can form pentameric α9 homomers, but preferentially assembles with the α10 subunit to form α9α10 heteromers. α9α10 nAChRs are of particular interest in that antagonists of this subtype have been shown to alleviate trauma- and chemotherapy-induced nerve injury and the resulting chronic pain [4–12]. Selective action on α9α10 nAChRs is important given that agonists and positive allosteric modulators of the closely-related α7 nAChR subtype are also analgesic [5, 13, 14]. One of the few selective antagonists for α9α10 nAChRs is the α-conotoxin peptide, RgIA, which was originally characterized from the Atlantic worm-hunting snail, Conus regius (the royal cone snail). [15, 16]. This short cyclic peptide consists of thirteen amino acids, GCCSDPRCRYRCR, and has two disulfide bridges linking Cys2-Cys8 and Cys3-Cys12.
Although the native RgIA peptide has efficacy in pre-clinical rodent behavioral assays, it has nearly 300-fold lower potency on the human vs rat α9α10 nAChR. This difference in potency is sufficiently driven by a single amino acid substitution in the α9 subunit, where Thr86 in the rat receptor is changed to Ile86 in the human α9 subunit [17]. Additionally, the second loop of RgIA was implicated in its selectivity for α9α10 vs α7 nAChRs in a study that employed Ala substitution of RgIA residues. [Ala9]RgIA had increased affinity for α7 nAChRs and had approximately 1000-fold reduced affinity for α9α10 nAChRs [15]. Due to the high identity between RgIA and several other α-conotoxins, and the dramatic effects of a single amino acid mutation in the α9 subunit, we surmised that the second loop is not only responsible for the selectivity towards α9α10 nAChRs, but also amenable to substitution to enhance its selectivity and potency (Figure 1). Here we show that several properties of Arg7 in the first disulfide loop are critical for potency at both the rat and human receptor. In contrast we demonstrate that changes in the second disulfide loop can be fine-tuned to preferentially increase potency for the human α9α10 nAChR subtype. Structural analysis of these analogs indicate that these changes arise from differences in side chains, rather than alterations in peptide scaffold.
Figure 1:
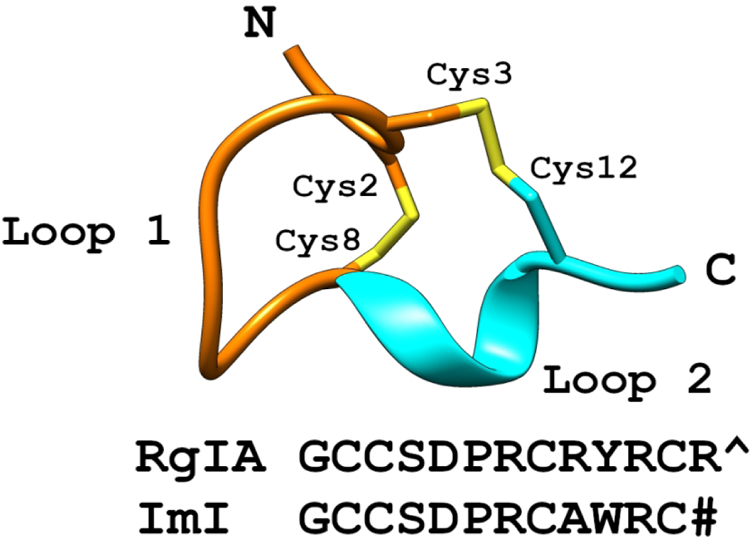
A cartoon of a prototypical α-conotoxin with the first loop (stick) colored orange, and the second loop (ribbon) colored cyan. The sequences of RgIA and the homologous ImI are shown, with a carboxylated C-terminus indicated by a caret (^) and an amidated C-terminus indicated by a pound-sign (#).
2. MATERIALS AND METHODS
Acetylcholine chloride, L-ascorbic acid, bovine serum albumin (BSA), 1,3-ethanediol, mineral oil, potassium chloride, penicillin/streptomycin, and thioanisole, were obtained from Sigma-Aldrich (St. Louis, MO). Acetonitrile, barium chloride dihydrate, calcium chloride dihydrate, HEPES, magnesium chloride hexahydrate, sodium pyruvate, trifluoroacetic acid (TFA), were obtained from Thermo-Fisher Scientific. Iodine, phenol, and potassium ferricyanide were obtained from Arcos Organics (Geel, Blegium). Clones of rat α9 and rat α10 cDNA in pGEMHe and pSGEM, respectively, were generously provided by A. Belen Elgoyhen (Universidad de Buenos Aires, Buenos Aires, Argentina).
2.1. Peptide Synthesis
RgIA and its analogs were synthesized using an Apex 396 automated peptide synthesizer (AAPPTec; Louisville, KY) applying standard solid-phase Fmoc (9-fluorenylmethyloxy-carbonyl) protocols. Peptides were constructed on preloaded Fmoc-Arg(Pbf)-Wang resin, Fmoc-Tyr(tBu)-Wang, Fmoc-Phe-Wang, Fmoc-Trp(Boc)-Wang, Fmoc-Lys(Boc)-Wang and RA-MBHA resin (Peptides International, Louisville, KY), Fmoc-Cit-Wang resin (Santa Cruze Biotechnology; Dallas, TX), Fmoc-1-Nal-Wang resin and Fmoc-2-Nal-Wang resin (Advanced Chemtech; Louisville, KY) with a substitution range 0.3–0.8 mmol/g. All standard amino acids were purchased from AAPPTec. Special amino acids were purchased as follows: Fmoc-L-Cit-OH and Fmoc-L-hArg(Pmc)-OH from Chem-Impex International, Inc. (Wood Dale, IL); Fmoc-Nar(Boc)2-OH from Iris-Biotech GMBH (Marktredwitz, Germany), Fmoc-4-COOH-Phe(OtBu)-OH from Bachem (Torrance, CA) and Fmoc-3-I-L-Tyr-OH from Peptides International.
Side-chain protective groups for the respective amino acids are as follows: Ser and Tyr, tert-butyl ether (tBu), Asp, 4-COOH-Phe, O-tert-butyl (OtBu), Arg, 2,2,4,6,7-pentamethyl-dihydrobenzofuran-5-sulfonyl (Pbf), Trp, Lys, Nar, tert-butyloxycarbonyl (Boc), hArg, 2,2,5,7,8-pentamethylchroman-6-sulfonyl (Pmc). Cys1 and Cys3 were trytl (Trt) protected; Cys2 and Cys4 were protected with acetamidomethyl (Acm) group.
For each coupling reaction ten-fold excess of standard amino acids was used and three- or five-fold excess of the unnatural amino acids. The coupling activation was achieved with one equivalent of 0.4 M benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP, Chem-Impex) and two equivalents of 2 M N,N-diisopropylethyl amine (DIPEA, Sigma-Aldrich, St. Louis, MO,) in N-methyl-2 pyrrolidone (NMP, Fisher Scientific, Fair Lawn, NJ) as the solvent. For standard amino acids, the coupling reaction was conducted for 60 min, and special amino acids were conducted for 90 min. Fmoc deprotection was carried out for 20 min with 20% piperidine (Alfa Aesar, Tewksbury, MA) in dimethylformamide (DMF, Fisher Scientific).
2.2. Peptide Oxidation
The linear RgIA (GCCSDPRCRYRCR) and its analogs were folded in two steps to facilitate the formation of intramolecular disulfide bonds between Cys2 and Cys8 and between Cys3 and Cys12, forming the 1–3, 2–4 disulfide arrangement that is characteristic of α-conotoxins. The linear peptide was cleaved from the Wang resin using reagent K (trifluoroacetic acid (TFA) (Fisher Bioreagents), phenol (Arcos Organics), 1,2-ethanediol (Sigma Aldrich), thioanisole (Sigma Aldrich), H2O / 9.0:0.75:0.25:0.5:0.5 ratio by volume). The first oxidation step to close Cys2-Cys8 bridge was performed under potassium ferricyanide (4.0 mM) (Arcos Organics) and tris HCl (20 mM) (Mallinckrodt Pharmaceuticals, Surrey, United Kingdom), pH 7.5 for 45 minutes and purified via reversed-phase high-performance liquid chromatography (RP-HPLC) using a C18 column. The second disulfide bridge between Cys3-Cys12 was formed in the solution consisting of iodine (5.0 mM) in H2O, acetonitrile (ACN) (Fisher Chemical), and TFA (72.0:25.0:3.0 ratio by volume) and again, purified through RP-HPLC.
2.3. cRNA Expression
Capped cRNA was prepared and Xenopus laevis oocytes were isolated as previously described [31, 32]. cRNA was injected into Xenopus oocytes using a Drummond 10 μL microdispenser (Drummond Scientific, Broomall, PA). Glass micropipettes pulled from thin-walled capillary tubes (ID 0.75 mm/OD 1.0 mm) (World Precision Instruments, Sarasota, FL) using a Narishige PN-3 micropipette puller (Narishige, Amityville, NY) were broken to an outer diameter of 22–25 μm. cRNA of α9 nAChR and α10 nAChR were mixed in a 1:1 ratio, diluted to 1000 ng/μL of each subunit, and 30–50 ng was injected. Once injected, the oocytes were incubated at 17°C in ND96 (96.0 mM NaCl, 2.0 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 5.0 mM HEPES, pH 7.1–7.5) supplemented with penicillin (100 U/mL) (Sigma-Aldrich), streptomycin (100 μg/mL) (Sigma-Aldrich), gentamicin (100 μg/mL) (Life Technologies), sulfamethoxazole (160 µg/mL) (Teva Pharmaceuticals, Parsippany, NJ), trimethoprim (32 µg/mL) (Teva Pharmaceuticals, Parsippany, NJ), amikacin (0.1 mg/mL) (Sagent Pharmaceuticals, Schaumburg, IL), and sodium pyruvate (2.5 mM). The incubation solution was exchanged daily. The oocytes were injected within three days of harvesting, and recordings were measured 2–5 days after injection.
2.4. Two Electrode Voltage Clamping
Injected Xenopus laevis oocytes were non-invasively immobilized in a cylindrical, 30 μL perfusion chamber casted in Sylgard and gravity perfused at approximately 2mL/minute. The ND96 solution used for perfusion was modified to substitute extracellular Ca2+ with Ba2+ to prevent the activation of calcium-activated chloride channels, and supplemented with 0.1 mg/mL bovine serum albumin (BSA) to reduce nonspecific binding. The BaND96 media (96.0 mM NaCl, 2.0 mM KCl, 1.8 mM BaCl2, 1.0 mM MgCl2, 9.0 μM CaCl2, 5.0 mM HEPES, pH 7.5) was degassed under house-vacuum for at least 30 minutes prior to the experiment to prevent the formation of air cavitation in the perfusion system. Glass electrodes filled with 3.0 M KCl, pulled to a resistance of 1–10 MΩ were used to record measurements. Oocytes were pulsed with a 1-second bolus of 100 μM ACh every 60 seconds for α9α10 nAChRs, recording current measurements for 30 seconds starting immediately with the ACh pulse. Once a stable baseline was reached, α-conopeptides were perfused until equilibrium was achieved. Dose-response concentrations were applied at half-log molar increments.
2.5. NMR Structural Analysis
Samples of RgIA and its analogs were prepared by dissolving peptide (∼1 mg) in 90% H2O, 10% D2O at a pH of ∼3. The spectra were recorded on a Bruker Avance III 600 MHz NMR spectrometer at 298 K and included total correlation spectroscopy (mixing time of 80 ms) and NOESY (mixing time of 200 ms). Chemical shifts were referenced to internal 2,2-dimethyl-2-silapentane-5-sulfonate at 0 ppm. The spectra were processed with Topspin (Bruker Biospin) and analyzed with CcpNMR analysis (version 2.4.1). Secondary αH chemical shifts were calculated as the difference between the observed αH chemical shifts and that of the corresponding residues in a random coil peptide [18].
2.6. Data Analysis
Baselines were fitted using the SSasymp Self-Starting Nls Asymptotic Regression Model in R: A language and environment for statistical computing [19]. The last 3 ACh responses obtained in a steady state were divided by the fitted baseline value to yield a % response in a given treatment condition. The dose-response data were fit to equation 1, where nH is the hill coefficient.
| (1) |
These data were fitted using non-linear regression analysis using GraphPad Prism Version 6.00 for Windows (GraphPad Software, La Jolla, California USA, http://www.graphpad.com). Mean IC50s and their respective standard error of the means were compared using an ordinary one-way analysis of variance (ANOVA) followed by a Dunnet’s multiple comparison test.
3. RESULTS
3.1. Arg7 side chain length and charge are essential for RgIA activity
RgIA is a highly positively charged peptide, with Arg constituting 4/13 residues. It potently blocked the rat α9α10 nAChR with an IC50 of 1.8 nM (Table 1). We investigated the role of Arg by substituting residues similar in charge and/or structure. Arginine has a side chain composed of a saturated aliphatic 3-carbon chain terminating in a guanidino group. To investigate the contribution of these functional groups, we substituted Arg7 with closely related homologs: citrulline (Cit), an uncharged arginine analog with similar steric properties but terminating in a carbamoylamino group; L-norarginine (Nar), a non-proteogenic arginine homolog with a sidechain that is one carbon-atom shorter than arginine, and L-homoarginine (hArg), an analog with a sidechain one carbon atom longer than Arg (Figure 2a–d). The neutralization of charge in the [Cit7]RgIA analog resulted in an effective loss of potency (IC50 >1000 nM) on the rat α9α10 nAChR. Likewise, although the side chains of Nar and hArg only differ from Arg by one carbon atom, both substitutions also effectively abolish activity at the rat α9α10 nAChR (IC50 > 1000 nM). Together, these substitutions indicate that the major biochemical properties of Arg7 are essential for RgIA activity, namely, that side chain length and charge are both critical. Finally, we examined the necessity of the 3-propylguanidine side chain of Arg by comparing it to a primary 4-aminobutyl side chain via [Lys7]RgIA. Again, the substitution resulted in an analog with an IC50 of >1000 nM.
Table 1:
Summary of the α-RgIA analog dose-response curves as obtained by two-electrode voltage clamp. Increases in potency >5-fold are indicated in bold and instances with no data are represented with dash (−) marks. The IC50 reflects the mean of at least 3 replicate samples. IC50 values are accompanied by their 95% confidence interval. Non-natural amino acids include L-citrulline (Cit), L-homoarginine (hArg), L-norarginine (Nar), and 3-iodo-L-tyrosine (Iyr).
| PEPTIDE | SEQUENCE | IC50 RAT | α9α10 (nM) | FOLD Δ RAT | IC50 HUMAN | α9α10 (nM) | FOLD Δ HUMAN |
|---|---|---|---|---|---|---|---|
| RgIA | GCCSDPRCRYRCR | 1.8 | (1.4 – 2.2) | 1.0 | 510 | (303 – 856) | 1.0 |
| [R7K]RgIA | GCCSDP( K )CRYRCR | >1000 | - | <0.002 | - | - | - |
| [R7Cit]RgIA | GCCSDP(Cit)CRYRCR | >1000 | - | <0.002 | - | - | - |
| [R7Hrg]RgIA | GCCSDP(Hrg)CRYRCR | >1000 | - | <0.002 | - | - | - |
| [R7Nrg]RgIA | GCCSDP(Nrg)CRYRCR | >1000 | - | <0.002 | - | - | - |
| [R9K]RgIA | GCCSDPRC( K )YRCR | >1000 | - | <0.002 | - | - | - |
| [R9Cit]RgIA | GCCSDPRC(Cit)YRCR | 7.1 | (6.8 – 8.1) | 0.25 | 182 | (134 – 248) | 2.8 |
| [R9Hrg]RgIA | GCCSDPRC(Hrg)YRCR | >1000 | - | <0.002 | - | - | - |
| [R9Nrg]RgIA | GCCSDPRC(Nrg)YRCR | >1000 | - | <0.002 | - | - | - |
| [Y10YI]RgIA | GCCSDPRCR(YI )RCR | 0.5 | (0.4 – 0.6) | 3.6 | 14.6 | (12.7 – 16.7) | 34.9 |
| [R11K]RgIA | GCCSDPRCRY( K )CR | 1.6 | (1.5 – 1.8) | 1.13 | 114 | (74.7 – 174) | 4.5 |
| [R11Q]RgIA | GCCSDPRCRY( Q )CR | 1.7 | (1.3 – 2.3) | 1.1 | 88 | (63.2 – 123) | 5.8 |
| [R11Cit]RgIA | GCCSDPRCRY(Cit)CR | 19.9 | (16.6 – 23.8) | 0.1 | >1000 | - | <0.5 |
| [R11Hrg]RgIA | GCCSDPRCRY(Hrg)CR | 1.5 | (1.3 – 1.9) | 1.2 | 40.3 | (31.1 – 52.1) | 12.7 |
| [R11Nrg]RgIA | GCCSDPRCRY(Nrg)CR | 11.4 | (9.4 – 13.9) | 0.16 | 213 | (174 – 261) | 2.4 |
| [R13K]RgIA | GCCSDPRCRYRC( K ) | 4.9 | (2.9 – 8.3) | 0.4 | >1000 | - | <0.5 |
| [R13Cit]RgIA | GCCSDPRCRYRC(Cit) | 3.3 | (2.2 – 5.1) | 0.5 | >1000 | - | <0.5 |
| [R13Y]RgIA | GCCSDPRCRYRC( Y ) | 0.3 | (0.2 – 0.3) | 6.9 | 2.1 | (1.7 – 2.6) | 242.9 |
Figure 2:
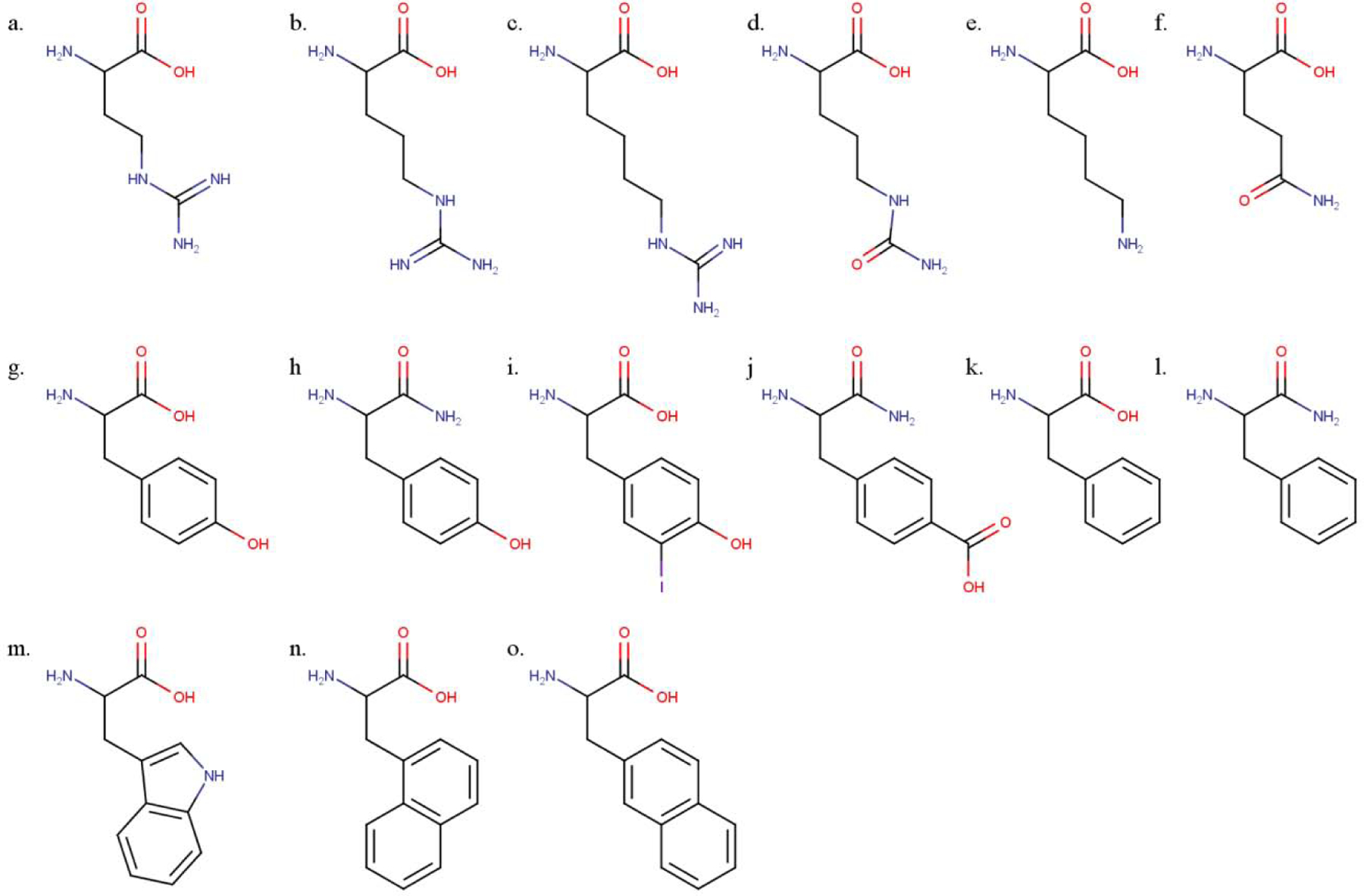
Structures of amino acids and analogs used in RgIA structure-activity relationship. (a) L-norarginine (Nar), (b) L-arginine (Arg), (c) L-homoarginine (hArg), (d) L-citrulline (Cit), (e) L-lysine (Lys), (f) L-glutamine (Gln), (g) L-tyrosine (Tyr), (h) L-tyrosine amide (Tyr#), (i) 3-iodo-L-tyrosine (Iyr), (j) L-4-carboxyphenylalanine amide (4-COOH-Phe#), (k) L-phenylalanine (Phe), (l) L-phenylalanine amide (Phe#), (m) L-tryptophan (Trp), (n) 1-naphthalenealanine (1-Nal), and (o) 2-naphthlenealanine (2-Nal).
3.2. Side chain length, but not charge is critical in position 9 of RgIA.
To investigate the importance of Arg9, analogs were synthesized that methodically altered only a side chain length or a charge. Shortening or extending the side chain of Arg9 by one carbon length via [Nar9]RgIA or [hArg9]RgIA mutations (Figure 2a–c), respectively, produced analogs with IC50 values >1000 nM on rat α9α10 nAChRs (Table 1). Thus, the change of a single carbon-length effectively abolished activity. A replacement of the Arg9 side chain with a 4-aminobutyl moiety via the [Lys9]RgIA (Figure 2e) analog also produced an IC50 value >1000 nM on the rat α9α10 nAChR. In contrast, neutralizing the charge via the [Cit9]RgIA (Figure 2d) mutation only decreased the affinity towards the rat α9α10 nAChR by approximately 4-fold (Table 1, Figure 3a).
Figure 3:
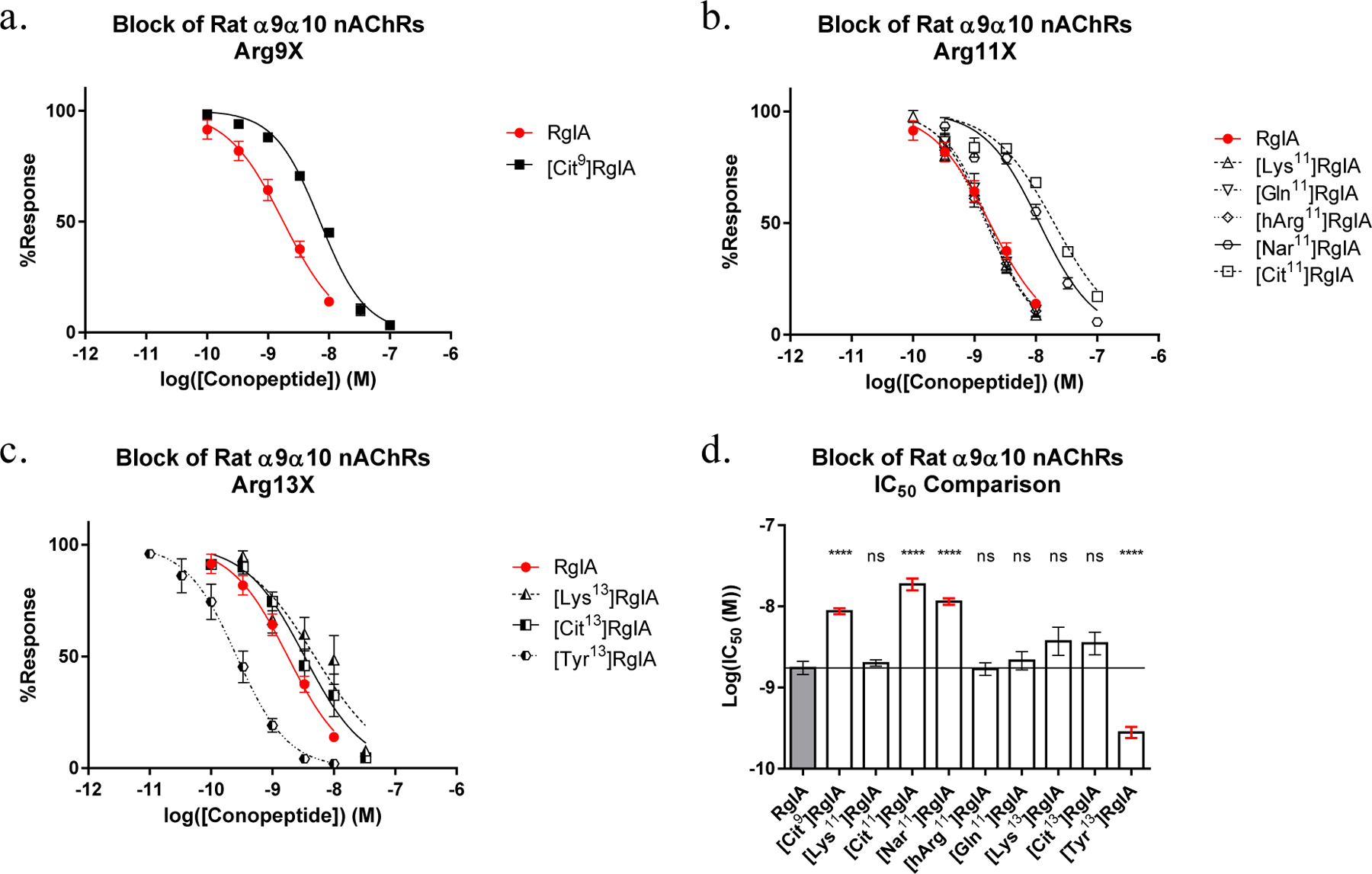
Dose response curves of (a) Arg9 substitutions. Not pictured [hArg9] and [Nar9]RgIA, both of which yielded IC50 values >1000 nM, (b) Arg11 substitutions, and (c) Arg13 substitutions, as obtained by two electrode voltage clamp. (d) The calculated IC50s were compared to RgIA using ordinary one-way ANOVA followed by a Dunnett’s multiple comparison test. Each data point represents at least three replicate samples accompanied with the standard error of the mean.
3.3. Substitution of Tyr10 or Arg11 selectively increases potency for the human α9α10 nAChR.
Contrary to the highly sensitive nature of Arg7 and Arg9, Tyr10 and Arg11 were more tolerant of amino acid substitutions (Table 1). In many cases, mutations in position 11 yielded minor increases in activity on the rat receptor accompanied with a major increase on the human α9α10 nAChR. Substitution of Tyr10 with a mono-iodinated 3-iodo-L-tyrosine (Iyr) yielded a 3.6-fold increase in activity on the rat receptor and a 34.9-fold increase on the human α9α10 nAChR. Substitution of Arg11 with Lys, Gln or hArg (Figure 2c,e,f) had little-to-no effect on potency for the rat α9α10 nAChR (Figure 3b). In contrast, replacement of Arg11 with Cit or Nar reduced activity by 11- and 6-fold, respectively, against the rat receptor. Despite decreased, or nearly unchanged, potency on the rat receptor, the [Nar11]RgIA analog yielded an approximately ~2.4-fold increase in potency on the human α9α10 nAChR and the [hArg11]RgIA analog showed an approximately 12.7-fold increase in potency (Figure 4).
Figure 4:
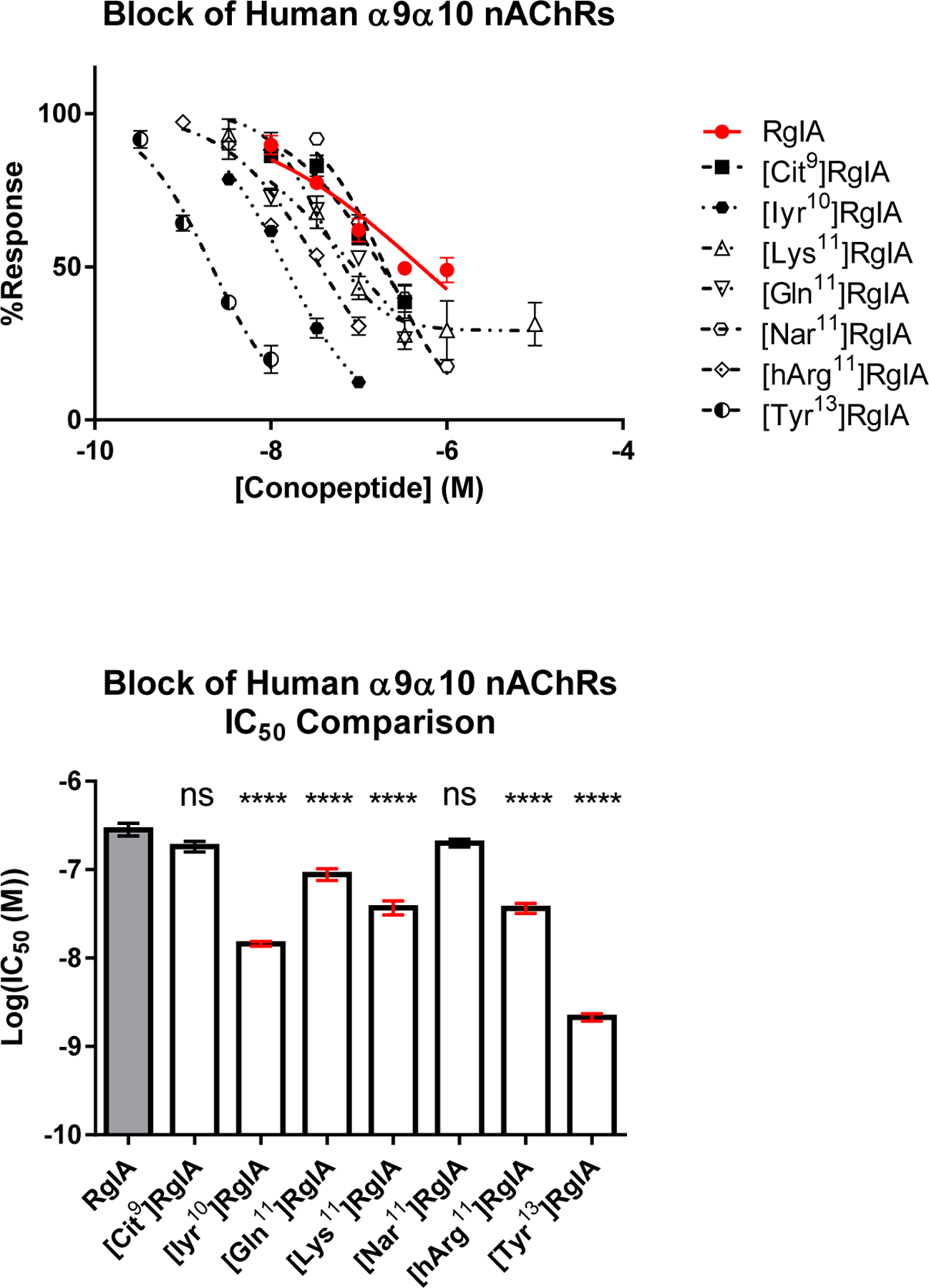
(a) Dose response curves of RgIA analogs tested against the human α9α10 nAChR, obtained by two-electrode voltage clamp. Each point represents at least three replicates displayed with the standard error of the mean (SEM). (b) The IC50 values of each RgIA analog compared to the native RgIA by ordinary one-way ANOVA followed by a Dunnett’s multiple comparison test, shown with the SEM. A designation of four asterisks (****) indicates a value of P <0.0001. Non-natural amino acids include L-citrulline (Cit), 3-iodo-L-tyrosine (Iyr) and L-homoarginine (hArg).
3.4. The C-terminal Arg13 is an engineerable hotspot
Based on the co-crystal structure of RgIA with the α9 nAChR extracellular domain (PDB 6HY7) and molecular modeling studies based on this structure, the C-terminal arginine (Arg13) appears to face outward from the acetylcholine binding site [20]. Due to the relatively unrestricted nature of this residue, we implemented a broader suite of substitutions in this position. Initial testing indicated that substitution of Arg13 with Lys or Cit did not significantly affect RgIA activity against the rat α9α10 nAChR (Figure 3c–d). Against the human receptor, however, both [Lys13]RgIA and [Cit13]RgIA decreased potency to an IC50 > 1000 nM.
Because the loss of the planar guanidino group via [Lys13]RgIA or the loss of a formal charge via [Cit13]RgIA disproportionately affected RgIA activity against the human receptor compared to the rat homolog, we further investigated the biochemical properties of the C-terminal residue and its effects on the human α9α10 nAChR.
We synthesized additional analogs using a suite of natural and non-natural amino acids. This panel included analogs featuring (i) π-orbital-rich aromatic groups and/or (ii) electronegative moieties capable of hydrogen-bond acceptance (Figure 2g–o). Analogs in this suite of substitutions were tested at 10 nM on the human α9α10 nAChR. This concentration was chosen to compare to the IC90 of [Tyr13]RgIA as a binary measure of whether the respective substitution increased antagonist activity. Notably, [Tyr13]RgIA and [1-Nal13] yielded an IC50 < 10 nM. Additionally, the amidation of the C-terminal carboxylic acid in [Tyr#13]RgIA works against the activity-benefit of [Tyr13]RgIA at 10 nM, resulting in a 52.8% response compared to a 19.8% response (p=0.0027 (**); unpaired t-test), respectively. The same trend is seen with the amidation of [Phe#13]RgIA compared to the free C-terminal carboxylic acid of [Phe13]RgIA, resulting in an average response of 31.6% compared to 12.1% (p=0.034 (*); unpaired t-test), respectively. Of all the substitutions, the [Tyr13]RgIA and [Phe13]RgIA analogs resulted in the greatest increase in potency against the human α9α10 nAChR, resulting in 19.8% and 12.11% response, respectively (Figure 5). Concentration-response analysis indicated that [Tyr13]RgIA was 6.9-fold more potent on the rat α9α10 nAChR (Figure 3d) and 243-fold more potent on the human α9α10 nAChR compared to native RgIA (Figure 4).
Figure 5:
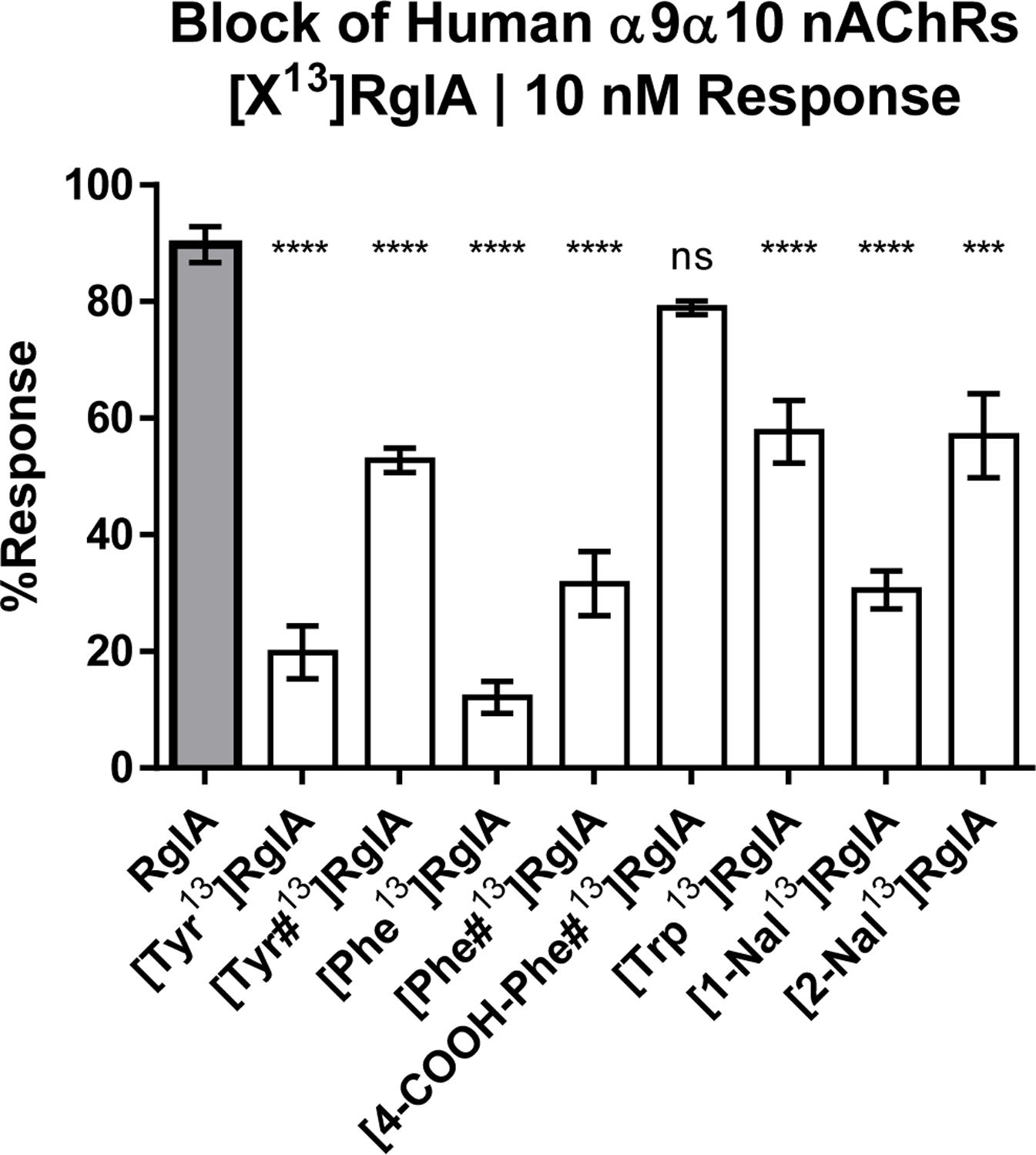
The comparison of Arg13 analogs tested against human α9α10 nAChRs, obtained by two-electrode voltage clamp. Each analog was flowed on at 10 nM and allowed to reach equilibrium. Each data plot represents the mean of at least 3 replicate samples accompanied with the standard error of the mean. The IC50 values of each RgIA analog compared to the native RgIA by ordinary one-way ANOVA followed by a Dunnett’s multiple comparison test, shown with the SEM. A designation of three asterisks (***) indicates a value of P<0.001, and four asterisks (****) indicates a value of P <0.0001. Non-natural amino acids were utilized to specifically test for electronegativity vs. aromaticity of the side chain moieties. Non-natural amino acids include L-tyrosine amide (Tyr#); L-phenylalanine amide (Phe#), L-4-carboxyl-phenylalanine amide (4-COOH-Phe#); 1-naphthalenealanine (1-Nal); 2-naphthalenealanine (2-Nal).
3.5. Structural Characterization of RgIA Analogs
The solution structures of several RgIA analogs were assessed by NMR. Representative analogs that had the greatest effects on activity were analyzed, including [hArg7], [Nar7], [Cit7], [hArg9], [Iyr10], [Gln11], and [Tyr13]RgIA. Comparison of secondary αH shifts with those of native RgIA revealed no significant changes, suggesting negligible effects of the sidechain substitutions upon backbone conformation of the peptides (Figure 6).
Figure 6:
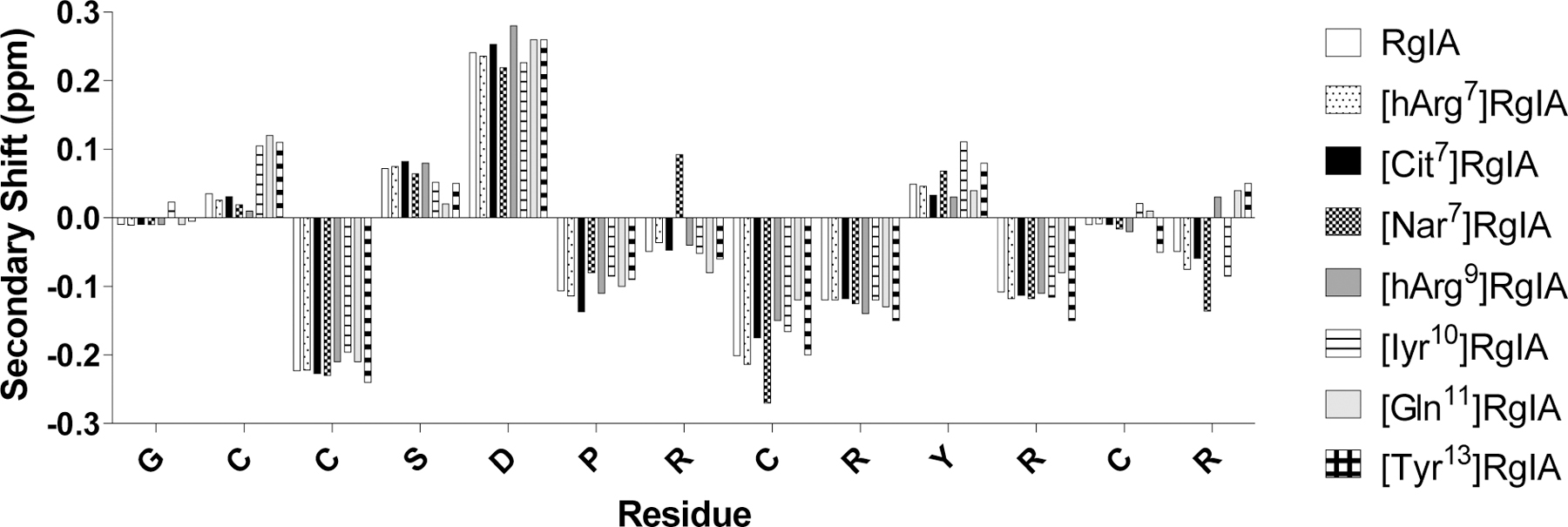
NMR readouts of secondary αH shifts of RgIA analogs in water. The shifts between the analogs are not considered significant. Non-natural amino acids include L-homoarginine (hArg), L-norarginine (Nar), and 3-Iodo-L-tyrosine (Iyr).
4. DISCUSSION
Alternatives to non-opioid analgesics remain limited. The 2019 Annual Surveillance Report of Drug-Related Risks and Outcomes, from the United States Centers for Disease Control and Prevention (CDC), estimated that of the 70,237 reported deaths due to drug overdose in 2017, 67.8% of these involved opioids, yet approximately 15% of the US population still filled at least one prescription for an opioid in 2018 [21]. The need for non-opioid drugs for the management of chronic pain is an ongoing and pressing matter.
Previously, we have shown that the α9α10 nAChR antagonist, RgIA, and its derivative analog, RgIA4 relieved pain in rodent models of physical nerve trauma and chemotherapy-induced neuropathic pain [5, 8–10]. Although efficacy in in vivo models was validated, the mechanism of analgesia stemming from the antagonism of α9α10 nAChRs has not yet been elucidated. There is some evidence that demonstrates immunomodulatory roles of α9α10 nAChRs that can be directly altered by RgIA [17, 22–25]. Currently, there are no commercially available probes specific to α9α10 nAChRs, that are amenable to functional studies, such as co-immunoprecipitation or flow cytometry to further characterize the mechanisms of RgIA-mediated analgesia. In this study, we identified biochemical properties within RgIA residues that may guide the development of α9α10 nAChR probes.
Although the blockade of α9α10 nAChRs has been implicated as a non-opioid target for pain relief, the list of specific antagonists remains short. Beyond RgIA, Vc1.1, GeXIVA, ZZ1–61c, ZZ-204G, and their derivatives, there are few characterized reagents that can specifically differentiate α9α10 nAChRs from other receptor subtypes in the acetylcholine-receptor family [4–12]. In part, this poor specificity is due to the high homology between the α9, α10, and α7 subunits. Until now, a thorough structure-activity-relationship study of RgIA and its interaction with the α9α10 nAChR has not been reported in detail. The unusually arginine-rich content of RgIA and GeXIVA has been of particular interest, spurring the development of oligoarginine peptides that can function as nAChR antagonists [26].
RgIA and Vc1.1 are both α-conotoxins from the A superfamily of Conus peptides, whereas GeXIVA is a structurally distinct O-superfamily peptide [12]. Nevertheless, each of these conotoxins are antagonists of α9α10 nAChRs and produce analgesia in animal models of neuropathic pain [4, 11, 27–33]. Vc1.1, and several other α-conotoxins, have also been shown to produce analgesic effects through the indirect inhibition of the neuronal N-type calcium channel, CaV2.2, via activation of GABAB receptors (GABABRs) [28–30]. Moreover, Vc1.1 and cyclic Vc1.1 (cVc1.1) were shown to inhibit the neuronal signaling of pain by reducing excitability of sensory neurons through GABABR-mediated mechanisms [31, 33]. In contrast, GeXIVA does not block N-type calcium channels [12]. While RgIA has also been proposed to produce analgesia through the activation of GABABRs, other studies show that the RgIA derivative, RgIA4, exhibits at least 1000-fold selectivity for α9α10 nAChRs vs. a panel of other molecular targets, including GABABRs [10, 29, 30]. RgIA4 has been shown to directly affect the release of inflammatory cytokines through α9α10 nAChRs on peripheral monocytes, as well as immune-cell accumulation around neuronal sites of damage, supporting a potential anti-neuroinflammatory relief pain mechanism [22, 32, 34].
In order to gain molecular insight into what makes an effective α9α10 nAChR antagonist, the biochemical characteristics of RgIA were investigated. The distal guanidino group of arginine can facilitate more hydrogen-bond and electrostatic interactions compared to the basic functional group of lysine, resulting in greater ionic-interaction stability [35]; these interactions may play a key role in RgIA function. The importance of the positioning and hydrogen-bonding properties in RgIA activity is most evident for Arg7 where changes of the length, charge, or functional group of the side chain effectively abolish RgIA activity on rat α9α10 nAChRs (IC50 > 1000 nM).
The highly-sensitive nature of the Arg9 side-chain length revealed a subtler but important characteristic; the position of this residue appears to be a primary contributor to RgIA activity. Although the co-crystal structure of RgIA and the α9 extracellular domain suggests that Arg9 and Tyr10 face inward into the binding site, our studies show that alterations to these positions can still benefit activity. The mono-iodination of the adjacent Tyr, [Iyr10]RgIA, for example, resulted in a 3.6-fold increased potency for the rat receptor and 34.9-fold increased potency for the human receptor (Table 1). In contrast, substitutions of Arg11 resulted in more nuanced changes, typically benefitting potency for both the rat and the human receptor; however, removal of the formal charge via [Cit11]RgIA decreases the potency >10-fold for the rat receptor and yields an IC50 > 1000 nM for the human α9α10 nAChR. Finally, Arg13 plays an important role in determining the potency of the ligands; substitution has the ability to selectively affect the potency towards the human receptor and the C-terminal carboxylic acid moiety likely contributes to the peptide activity. Replacement of Arg13 with Tyr significantly increased the potency of RgIA for human α9α10 nAChRs by 243-fold.
Previous studies showed that the single [Thr86Ile]α9 point mutation is sufficient to account for the lower potency of RgIA for the human vs. rat α9α10 nAChR [17]. In contrast, this current structure-activity relationship study highlights that changes in the second loop of RgIA can address this cross-species difference. α-Conotoxin ImI from Conus imperialis, shares an eight out of thirteen amino acid identity with RgIA, yet most potently blocks α7 and α3β2 nAChR subtypes [36, 37]. This sequence identity is entirely conserved through the first loop of RgIA and ImI, in which a three-amino-acid motif of Asp-Pro-Arg (DPR) was determined as essential for the potency of these peptides (Figure 1) [15]. Preliminary structure-activity studies of RgIA showed that several second loop mutations increased activity against the human α9α10 nAChR at 100 nM [10]. Of the analogs screened, the potency trends of analogs determined at 100 nM were consistent with those observed in the current study. In the present study we further demonstrated that analogs such as [Cit11]RgIA and [Lys13]RgIA, have an IC50 >1000 nM. Interestingly, despite the low activity for the human α9α10 nAChR, [Cit11]RgIA and [Lys13]RgIA had low nM activity for the rat α9α10 nAChR. Further, while several mutations in the second loop produced modest increases in potency against the rat α9α10 nAChR, the same mutations selectively enhanced potency against the human receptor. This is particularly pronounced with [Iyr10]RgIA and [Tyr13]RgIA analogs, where the substitutions disproportionately enhance activity on the human receptor. The development of RgIA analogs to target human α9α10 nAChRs, while maintaining activity against the rat receptor, is vital to ensure that preclinical data will translate into clinical settings. Understanding the biochemical properties responsible for RgIA activity can help inform the rational design of future α9α10 nAChR antagonists.
As peptide drugs, native RgIA and Vc1.1 are both susceptible to proteolytic degradation, reducing bioavailability. The cyclization of Vc1.1 has shown effective enhancement of bioavailability [38]. In an endeavor to understand the interactions of specific side chain moieties of RgIA and the α9α10 nAChR interface, we have also shown that the activity of the analogs can be maintained with the use of non-natural amino acids. This finding can be particularly useful in therapeutic development in that the incorporation of non-natural amino acids is often beneficial to the evasion of proteolytic cleavage by endogenous proteases. The incorporation of non-natural amino acids may further the opportunity to effectively improve on the pharmacokinetics of these peptide-based nAChR antagonists without compromising receptor potency.
Structurally, the lack of change in secondary αH shifts indicates that the primary contributors to activity are the side chain properties of RgIA residues, more so than backbone conformation. Therefore, understanding the nature of chemical interactions involved with the α9α10 nAChR binding interface and the preservation of backbone structure in the RgIA peptide may inform the design of small molecule drugs, especially those built off of modular scaffolds. By understanding key biochemical properties of RgIA, a broad structure-activity relationship dataset should help inform the rational design of peptides and small molecules, as well as facilitate in silico screening efforts to develop specific nAChR-targeted therapeutics.
Acknowledgements
Funding for this project was generously provided by the National Institutes of Health (R35-GM136430 & R01-GM103801), the United States Department of Defense (W81XWH1710413), and the Dale A. Stringfellow Fellowship granted through the School of Biological Sciences of the University of Utah. DJC is supported by an Australian Research Council Laureate Fellowship (FL150100146).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
The University of Utah holds patents on conopeptides including RgIA4 on which JMM is an inventor.
References
- [1].Lewis RJ, Dutertre S, Vetter I, Christie MJ, Conus venom peptide pharmacology, Pharmacol Rev 64(2) (2012) 259–98. [DOI] [PubMed] [Google Scholar]
- [2].Le Novere N, Changeux JP, Molecular evolution of the nicotinic acetylcholine receptor: an example of multigene family in excitable cells, J Mol Evol 40(2) (1995) 155–72. [DOI] [PubMed] [Google Scholar]
- [3].Tsunoyama K, Gojobori T, Evolution of nicotinic acetylcholine receptor subunits, Mol Biol Evol 15(5) (1998) 518–27. [DOI] [PubMed] [Google Scholar]
- [4].Satkunanathan N, Livett B, Gayler K, Sandall D, Down J, Khalil Z, Alpha-conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones, Brain Res 1059(2) (2005) 149–58. [DOI] [PubMed] [Google Scholar]
- [5].Vincler M, Wittenauer S, Parker R, Ellison M, Olivera BM, McIntosh JM, Molecular mechanism for analgesia involving specific antagonism of alpha9alpha10 nicotinic acetylcholine receptors, Proc Natl Acad Sci U S A 103(47) (2006) 17880–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Holtman JR, Dwoskin LP, Dowell C, Wala EP, Zhang Z, Crooks PA, McIntosh JM, The novel small molecule alpha9alpha10 nicotinic acetylcholine receptor antagonist ZZ-204G is analgesic, Eur J Pharmacol 670(2–3) (2011) 500–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li X, Hu Y, Wu Y, Huang Y, Yu S, Ding Q, Zhangsun D, Luo S, Anti-hypersensitive effect of intramuscular administration of alphaO-conotoxin GeXIVA[1,2] and GeXIVA[1,4] in rats of neuropathic pain, Prog Neuropsychopharmacol Biol Psychiatry 66 (2016) 112–9. [DOI] [PubMed] [Google Scholar]
- [8].Pacini A, Micheli L, Maresca M, Branca JJ, McIntosh JM, Ghelardini C, Di Cesare Mannelli L, The alpha9alpha10 nicotinic receptor antagonist alpha-conotoxin RgIA prevents neuropathic pain induced by oxaliplatin treatment, Exp Neurol 282 (2016) 37–48. [DOI] [PubMed] [Google Scholar]
- [9].Christensen SB, Hone AJ, Roux I, Kniazeff J, Pin JP, Upert G, Servent D, Glowatzki E, McIntosh JM, RgIA4 Potently Blocks Mouse alpha9alpha10 nAChRs and Provides Long Lasting Protection against Oxaliplatin-Induced Cold Allodynia, Front Cell Neurosci 11 (2017) 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Romero HK, Christensen SB, Di Cesare Mannelli L, Gajewiak J, Ramachandra R, Elmslie KS, Vetter DE, Ghelardini C, Iadonato SP, Mercado JL, Olivera BM, McIntosh JM, Inhibition of alpha9alpha10 nicotinic acetylcholine receptors prevents chemotherapy-induced neuropathic pain, Proc Natl Acad Sci U S A 114(10) (2017) E1825–E1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang H, Li X, Zhangsun D, Yu G, Su R, Luo S, The alpha9alpha10 Nicotinic Acetylcholine Receptor Antagonist alphaO-Conotoxin GeXIVA[1,2] Alleviates and Reverses Chemotherapy-Induced Neuropathic Pain, Mar Drugs 17(5) (2019) 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Luo S, Zhangsun D, Harvey PJ, Kaas Q, Wu Y, Zhu X, Hu Y, Li X, Tsetlin VI, Christensen S, Romero HK, McIntyre M, Dowell C, Baxter JC, Elmslie KS, Craik DJ, McIntosh JM, Cloning, synthesis, and characterization of alphaO-conotoxin GeXIVA, a potent alpha9alpha10 nicotinic acetylcholine receptor antagonist, Proc Natl Acad Sci U S A 112(30) (2015) E4026–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Di Cesare Mannelli L, Pacini A, Matera C, Zanardelli M, Mello T, De Amici M, Dallanoce C, Ghelardini C, Involvement of alpha7 nAChR subtype in rat oxaliplatin-induced neuropathy: effects of selective activation, Neuropharmacology 79 (2014) 37–48. [DOI] [PubMed] [Google Scholar]
- [14].Ulloa L, The vagus nerve and the nicotinic anti-inflammatory pathway, Nat Rev Drug Discov 4(8) (2005) 673–84. [DOI] [PubMed] [Google Scholar]
- [15].Ellison M, Feng ZP, Park AJ, Zhang X, Olivera BM, McIntosh JM, Norton RS, Alpha-RgIA, a novel conotoxin that blocks the alpha9alpha10 nAChR: structure and identification of key receptor-binding residues, J Mol Biol 377(4) (2008) 1216–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ellison M, Haberlandt C, Gomez-Casati ME, Watkins M, Elgoyhen AB, McIntosh JM, Olivera BM, Alpha-RgIA: a novel conotoxin that specifically and potently blocks the alpha9alpha10 nAChR, Biochemistry 45(5) (2006) 1511–7. [DOI] [PubMed] [Google Scholar]
- [17].Azam L, McIntosh JM, Molecular basis for the differential sensitivity of rat and human alpha9alpha10 nAChRs to alpha-conotoxin RgIA, J Neurochem 122(6) (2012) 1137–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wishart DS, Sykes BD, Richards FM, The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy, Biochemistry 31(6) (1992) 1647–51. [DOI] [PubMed] [Google Scholar]
- [19].Team RC, R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, 2016. [Google Scholar]
- [20].Zouridakis M, Papakyriakou A, Ivanov IA, Kasheverov IE, Tsetlin V, Tzartos S, Giastas P, Crystal Structure of the Monomeric Extracellular Domain of alpha9 Nicotinic Receptor Subunit in Complex With alpha-Conotoxin RgIA: Molecular Dynamics Insights Into RgIA Binding to alpha9alpha10 Nicotinic Receptors, Front Pharmacol 10 (2019) 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Centers for Disease Control and Prevention, 2019 Annual Surveillance Report of Drug-Related Risks and Outcomes — United States Surveillance Special Report, in: Centers for Disease Control and Prevention. U.S. Department of Health and Human Services; (Ed.) 2019, pp. 1–129. [Google Scholar]
- [22].Richter K, Mathes V, Fronius M, Althaus M, Hecker A, Krasteva-Christ G, Padberg W, Hone AJ, McIntosh JM, Zakrzewicz A, Grau V, Phosphocholine - an agonist of metabotropic but not of ionotropic functions of alpha9-containing nicotinic acetylcholine receptors, Sci Rep 6 (2016) 28660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hecker A, Kullmar M, Wilker S, Richter K, Zakrzewicz A, Atanasova S, Mathes V, Timm T, Lerner S, Klein J, Kaufmann A, Bauer S, Padberg W, Kummer W, Janciauskiene S, Fronius M, Schweda EK, Lochnit G, Grau V, Phosphocholine-Modified Macromolecules and Canonical Nicotinic Agonists Inhibit ATP-Induced IL-1beta Release, J Immunol 195(5) (2015) 2325–34. [DOI] [PubMed] [Google Scholar]
- [24].Zakrzewicz A, Richter K, Agne A, Wilker S, Siebers K, Fink B, Krasteva-Christ G, Althaus M, Padberg W, Hone AJ, McIntosh JM, Grau V, Canonical and Novel Non-Canonical Cholinergic Agonists Inhibit ATP-Induced Release of Monocytic Interleukin-1beta via Different Combinations of Nicotinic Acetylcholine Receptor Subunits alpha7, alpha9 and alpha10, Front Cell Neurosci 11 (2017) 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Peng H, Ferris RL, Matthews T, Hiel H, Lopez-Albaitero A, Lustig LR, Characterization of the human nicotinic acetylcholine receptor subunit alpha (alpha) 9 (CHRNA9) and alpha (alpha) 10 (CHRNA10) in lymphocytes, Life Sci 76(3) (2004) 263–80. [DOI] [PubMed] [Google Scholar]
- [26].Lebedev DS, Kryukova EV, Ivanov IA, Egorova NS, Timofeev ND, Spirova EN, Tufanova EY, Siniavin AE, Kudryavtsev DS, Kasheverov IE, Zouridakis M, Katsarava R, Zavradashvili N, Iagorshvili I, Tzartos SJ, Tsetlin VI, Oligoarginine Peptides, a New Family of Nicotinic Acetylcholine Receptor Inhibitors, Mol Pharmacol 96(5) (2019) 664–673. [DOI] [PubMed] [Google Scholar]
- [27].Hone AJ, Servent D, McIntosh JM, alpha9-containing nicotinic acetylcholine receptors and the modulation of pain, Br J Pharmacol 175(11) (2018) 1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Huynh TG, Cuny H, Slesinger PA, Adams DJ, Novel mechanism of voltage-gated N-type (Cav2.2) calcium channel inhibition revealed through alpha-conotoxin Vc1.1 activation of the GABA(B) receptor, Mol Pharmacol 87(2) (2015) 240–50. [DOI] [PubMed] [Google Scholar]
- [29].Callaghan B, Adams DJ, Analgesic alpha-conotoxins Vc1.1 and RgIA inhibit N-type calcium channels in sensory neurons of alpha9 nicotinic receptor knockout mice, Channels (Austin) 4(1) (2010) 51–4. [DOI] [PubMed] [Google Scholar]
- [30].Callaghan B, Haythornthwaite A, Berecki G, Clark RJ, Craik DJ, Adams DJ, Analgesic alpha-conotoxins Vc1.1 and Rg1A inhibit N-type calcium channels in rat sensory neurons via GABAB receptor activation, J Neurosci 28(43) (2008) 10943–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Castro J, Grundy L, Deiteren A, Harrington AM, O’Donnell T, Maddern J, Moore J, Garcia-Caraballo S, Rychkov GY, Yu R, Kaas Q, Craik DJ, Adams DJ, Brierley SM, Cyclic analogues of alpha-conotoxin Vc1.1 inhibit colonic nociceptors and provide analgesia in a mouse model of chronic abdominal pain, Br J Pharmacol 175(12) (2018) 2384–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Di Cesare Mannelli L, Cinci L, Micheli L, Zanardelli M, Pacini A, McIntosh JM, Ghelardini C, alpha-conotoxin RgIA protects against the development of nerve injury-induced chronic pain and prevents both neuronal and glial derangement, Pain 155(10) (2014) 1986–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sadeghi M, McArthur JR, Finol-Urdaneta RK, Adams DJ, Analgesic conopeptides targeting G protein-coupled receptors reduce excitability of sensory neurons, Neuropharmacology 127 (2017) 116–123. [DOI] [PubMed] [Google Scholar]
- [34].Richter K, Sagawe S, Hecker A, Kullmar M, Askevold I, Damm J, Heldmann S, Pohlmann M, Ruhrmann S, Sander M, Schluter KD, Wilker S, Konig IR, Kummer W, Padberg W, Hone AJ, McIntosh JM, Zakrzewicz AT, Koch C, Grau V, C-Reactive Protein Stimulates Nicotinic Acetylcholine Receptors to Control ATP-Mediated Monocytic Inflammasome Activation, Front Immunol 9 (2018) 1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sokalingam S, Raghunathan G, Soundrarajan N, Lee SG, A study on the effect of surface lysine to arginine mutagenesis on protein stability and structure using green fluorescent protein, PLoS One 7(7) (2012) e40410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ellison M, Gao F, Wang HL, Sine SM, McIntosh JM, Olivera BM, Alpha-conotoxins ImI and ImII target distinct regions of the human alpha7 nicotinic acetylcholine receptor and distinguish human nicotinic receptor subtypes, Biochemistry 43(51) (2004) 16019–26. [DOI] [PubMed] [Google Scholar]
- [37].Ellison M, McIntosh JM, Olivera BM, Alpha-conotoxins ImI and ImII. Similar alpha 7 nicotinic receptor antagonists act at different sites, J Biol Chem 278(2) (2003) 757–64. [DOI] [PubMed] [Google Scholar]
- [38].Clark RJ, Jensen J, Nevin ST, Callaghan BP, Adams DJ, Craik DJ, The engineering of an orally active conotoxin for the treatment of neuropathic pain, Angew Chem Int Ed Engl 49(37) (2010) 6545–8. [DOI] [PubMed] [Google Scholar]