

Abstract
Alcohol misuse and dependence is a widespread health problem. The central nucleus of the amygdala (CeA) plays important roles in both the anxiety associated with alcohol (ethanol) dependence and the increased alcohol intake that is observed during withdrawal in dependent animals. We and others have shown the essential involvement of the corticotrophin releasing factor (CRF) system in alcohol’s synaptic effects on the CeA and in the development of ethanol dependence. Another system that has been shown to be critically involved in the molecular underpinnings of alcohol dependence is the norepinephrine (NE) system originating in the locus coeruleus. Both the CRF and NE systems act in concert to facilitate a stress response: central amygdalar afferents release CRF in the locus coeruleus promoting widespread release of NE. In this study, we are the first to use fast-scan cyclic voltammetry to classify local electrically-evoked NE release in the CeA and to determine if acute alcohol and CRF modulate it. Evoked NE release is action potential dependent, is abolished after depletion of monoaminergic vesicles, differs pharmacologically from dopamine release, is insensitive to acute alcohol, and decreases in response to locally applied CRF. Taken together, these results indicate that NE release in the CeA is released canonically in a vesicular-dependent manner, and that while acute alcohol does not directly alter NE release, CRF decreases it. Our results suggest that CRF acts locally on NE terminals as negative feedback and potentially prevents hyperactivation of the CRF-norepinephrine stress pathway.
Keywords: stress, alcohol, CRF, norepinephrine, amygdala, voltammetry
1. Introduction
Alcohol use disorder (AUD) is one of the leading preventable causes of death both due to direct toxicity and exacerbation of comorbidities (Bauer et al., 2014). Alcoholism, or the neurophysiologic dependence on alcohol (ethanol), is behaviorally described as the compulsory seeking and drinking of alcohol, progressive loss of regulatory control, and finally, a negative emotion state in the absence of alcohol (Koob, 2013). This negative emotion state is characterized by the recruitment of brain stress circuitry, particularly in the central nucleus of the amygdala (CeA), the principle output nucleus of the amygdala (Koob, 2008; Koob and Le Moal, 2008; Roberto et al., 2010, 2012).
Two of the predominant stress systems in the brain are the norepinephrine (NE) and corticotropin releasing factor (CRF) signaling systems. Norepinephrine and CRF signaling within the extended amygdala are involved in stress-induced reinstatement of drug-seeking and are also necessary for the enhanced anxiety that accompanies protracted abstinence from chronic drug exposure (Smith and Aston-Jones, 2008). Norepinephrine neurons principally originate in the locus coeruleus (LC) of the brainstem and project diffusely to many different areas of the brain where NE is involved in a variety of neuropsychiatric states and disorders (Sara, 2009), and increased firing of LC NE neurons results in a state of increased anxiety and stress (McCall et al., 2015). The extrahypothalamic CRF system also is widely distributed throughout the brain including in the CeA (Sakanaka et al., 1986; Stenzelpoore et al., 1994; Curtis et al., 2002). Importantly, these two stress systems interact. There are NE LC neurons that directly project to the CeA (Campese et al., 2017; Finnell et al., 2019) and CRF+ neurons in the CeA that correspondingly project to the LC (Reyes et al., 2008, 2011; McCall et al., 2015), where CRF transmission in the LC increases anxiety (Lee et al., 2008; McCall et al., 2015). Functional connectivity between these two areas has given rise to a theory in which the CeA and the LC are involved in a “feed-forward loop” which escalates the brain stress response (Koob, 1999). According to this theory, the LC NE neurons projecting to the CeA are among the first ones to be recruited following a stressful event and they activate CeA CRF+ cells which project back to the LC, increasing tonic firing of LC NE neurons. Chronic alcohol dysregulates these systems (Koob, 2015) and increased levels of extracellular CRF in the CeA have been measured following acute withdrawal from alcohol (Roberto et al, 2010).
Despite how well characterized the relationship between the LC and the CeA is in stress response, little is known about how local NE release in the CeA is regulated and whether it is dysregulated by acute and/or chronic alcohol exposure. Thus, we sought to characterize the effects of alcohol and CRF on locally-evoked NE release in the CeA using fast-scan cyclic voltammetry (FSCV), an electrochemical technique that allows for sub-second monitoring of neurotransmitter release and kinetics. Interestingly, we found that while acute alcohol has no effect on NE release, CRF decreased electrically-evoked NE release in both naïve and dependent animals, indicating that CRF interactions in the CeA may provide local inhibition to the LC CeA feed-forward reciprocal loop.
2. Materials and Methods
2.1. Animals
Adult male Sprague Dawley rats (n = 45, 225 – 350 g) were obtained from Charles River (Raleigh, NC) and were given ad libitum access to food and water and maintained on a reverse light/dark cycle with lights on between 8 PM and 8 AM. All protocols and procedures were approved by The Scripps Research Institute and Brigham Young University Institutional Animal Care and Use Committee in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
2.2. Chronic Alcohol Treatment
Alcohol dependent rats (n = 6) were generated using the standard alcohol (ethanol) inhalation method of The Scripps Research Institute Alcohol Research Center in which rats were exposed to alcohol vapor for 5 – 7 weeks, 14 hours per day (Rogers et al., 1979; Roberto et al., 2004b, 2004a, 2010; Roberto and Siggins, 2006; Cruz et al., 2013; Kallupi et al., 2014). This procedure has been shown by us and others to generate alcohol dependence in rodents, as demonstrated by increased alcohol drinking behavior, anxiety-like behavior, and reward deficits (O’Dell et al., 2004; Gilpin et al., 2008; Roberto et al., 2010). On experimental days, alcohol dependent rats were maintained in the vapor chambers until slices were prepared. Slices were then maintained in alcohol-free solutions and all electrochemical recordings were performed in slices undergoing acute withdrawal (2–8 hours) as previously described (Kallupi et al., 2014; Roberto et al., 2004b, 2004a, Roberto et al., 2010, Varodayan et al., 2017b, 2017a). Blood alcohol levels (BALs) were measured 1–2 times per week from tail blood samples. The mean BAL was 151.70 ±10.46 mg/dL.
2.3. Brain slice preparation and drug application
Rats were briefly anesthetized with 3–5% isoflurane and decapitated. The brains were rapidly dissected and placed in oxygenated (95% O2, 5% CO2), ice-cold high sucrose cutting solution (pH 7.3–7.4) consisting of (in mM): 206.0 sucrose, 2.5 KCl, 0.5 CaCl2, 7.0 MgCl2, 1.2 NaH2PO4, 26.0 NaHCO3, 5.0 D-glucose, and 5.0 HEPES (Roberto et al., 2003, 2004b, 2004a; Varodayan et al., 2017a, 2017b). Coronal brain slices (400 μm) were obtained using a Leica VT1200 S vibratome. Slices were then incubated in oxygenated artificial cerebral spinal fluid (ACSF) which contained the following (in mM): 130 NaCl, 3.5 KCl, 2.0 CaCl2, 1.25 NaH2PO4, 1.5 MgSO4, 24.0 NAHCO3, and 10.0 D-glucose for 30 min at 37°C then 30 min at room temperature.
All drugs were obtained from Tocris Bioscience (Bristol, UK), unless otherwise notated. Drugs were bath-applied for slice voltammetry at the following concentrations (unless otherwise specified), which are based on Ki values (to attempt to prevent non-specific binding) when applicable: alcohol (44 mM; Pharmaco-Aaper, Brookfield, CT, USA), CRF (100 nM), yohimbine (25 μM), clonidine (10 uM), desipramine (50 μM), TTX (1 μM), and reserpine (10 μM).
2.4. Voltammetry recordings
For recording, slices were transferred to a recording chamber and continuously superfused with fresh, oxygenated ACSF (32–34 °C) at approximately 2 mL/min. Fast-scan cyclic voltammetry recordings were collected and analyzed using Demon Voltammetry and Analysis software (Yorgason et al., 2011a). Carbon fiber electrodes were custom made by aspirating a single carbon fiber into a borosilicate glass capillary tube (O.D. 1.2mm, I.D. 0.69mm; Sutter Instruments, Novato, CA) as previously described (Schilaty et al., 2014). The capillary tubes were then pulled using a model P-1000 horizontal electrode puller (Sutter Instruments). Under microscopic control, the carbon fibers were trimmed so that 100 – 150 μm of bare fiber protruded from the glass seal on the carbon fiber. During the recording, the electrode was linearly scanned (400 V/s) from −0.4 to 1.2 V and back to −0.4 V (Ag vs AgCl) and cyclic voltammograms were recorded at 10 Hz by means of a ChemClamp voltage clamp amplifier (Dagan Corporation, Minneapolis, MN). Electrodes were conditioned prior to insertion into tissue by applying the triangular waveform at 60 Hz for 5–10 min (Yorgason et al., 2017). Low noise electrodes were positioned ~75 μm below the surface of the tissue in the medial subdivision of the CeA. Tissue was electrically stimulated using a bipolar stimulator (30 Hz, 30 P, 1 msec pulsewidth for NE in CeA; 20 Hz, 10 P, 4 msec pulsewidth for dopamine (DA) in striatum) to evoke catecholamine release. Due to the unique challenges of recording the small signals, some recordings showed stimulation artifact during electrical stimulation (e.g. prominent negative signal in Fig 5 traces). When recording NE in the CeA, the tissue was stimulated every 4 minutes, and when recording dopamine in the striatum, the tissue was stimulated every 2 minutes.
Figure 5: CRF Decreases Evoked NE Release in the CeA.
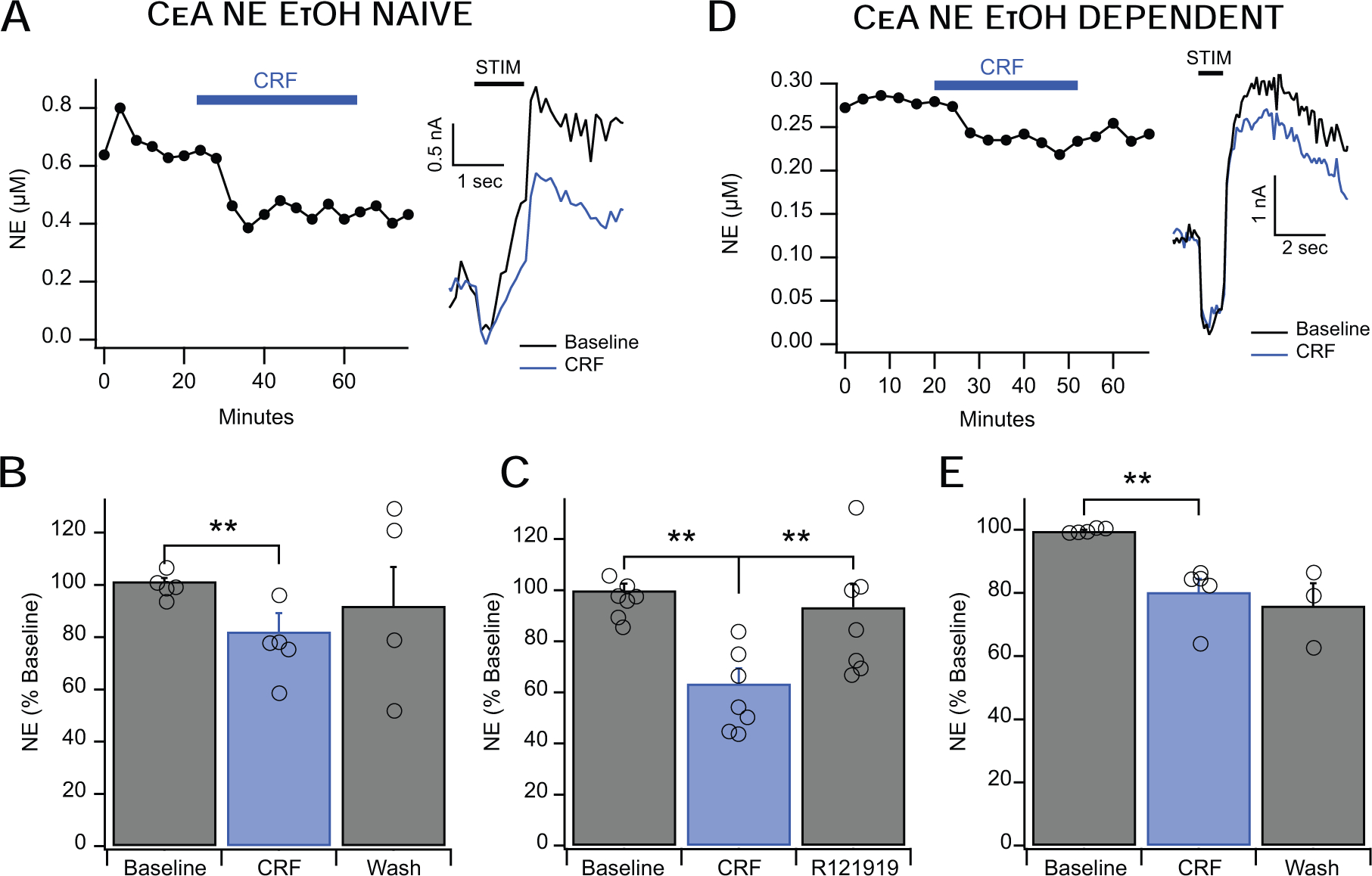
A&B. CRF (100 nM) decreased NE release in the CeA of alcohol-naïve animals to approximately 82% of baseline release levels with moderate (but statistically insignificant) washout. The left inset shows a representative time course of CRF’s action and the right inset shows representative current vs time traces. C. After CRF application, R121919 (1 μM) co-applied with CRF fully reversed the CRF-induced decrease in NE signal. D&E. CRF (100 nM) similarly decreased NE release in the CeA of alcohol-dependent animals to approximately 80% of baseline release levels, and this effect failed to wash out. The left inset shows a representative time course of CRF’s action and the right inset shows representative current vs time traces. It is important to note that while the representative insets show a difference in baseline levels of NE release between alcohol naïve and dependent animals, this effect is limited to the selected representative examples. This effect is averaged out when combined with aggregate data (see section 3.5). Error bars represent the SEM and *,** indicates a significance level of p < 0.05 and p < 0.01, respectively.
2.5. Data and Statistical Analysis
All catecholamine release was analyzed using Demon Voltammetry Analysis software. Evoked release was quantified at peak oxidation currents (peak heights/amplitude). Norepinephrine reuptake (clearance) was quantified using the time constant (τ) calculated by Demon Voltammetry using an single exponential fit function, and the goodness of fit was verified by calculating the Spearman correlation coefficient (Fig 1C) (Yorgason et al., 2011a). Norepinephrine concentrations were calculated by calibrating each electrode against 10 μM NE. Norepinephrine release was verified by comparing the color plot and voltammogram against the calibration voltammogram. Given the nearly identical oxidative potential of NE and DA, FSCV alone was not sufficient to discriminate between these two molecules: each experiment was verified to be either NE or DA pharmacologically as described previously (Park et al., 2011). Yohimbine, an α2 adrenergic receptor inhibitor, acts on the adrenergic autoreceptor and effectively disinhibits NE release – NE signals treated with yohimbine (25 μM) increased in amplitude whereas DA signals did not respond. Correspondingly, NE signals treated with desipramine (50 μM), a NET blocker, increased clearance time (τ), but desipramine had no effect on DA signals. While it is possible that α2 adrenergic receptors may be present on DA terminals in the CeA, potentially confounding this pharmacology-based discrimination method, a current literature search has not indicated that there is evidence for α2 adrenergic receptors on DA terminals in this region. When possible, the experimental pharmacology was applied first, and then washed out prior to species confirmation via either yohimbine or desipramine. This was done to prevent any potentiation or modulation of the experimental conditions. Exceptions to this are noted and explained below. To ensure biological variability and avoid potential bias, slices for each experimental condition came from at least 3 different animals.
Figure 1: Effects of Desipramine and Yohimbine on Evoked NE Release in the CeA.
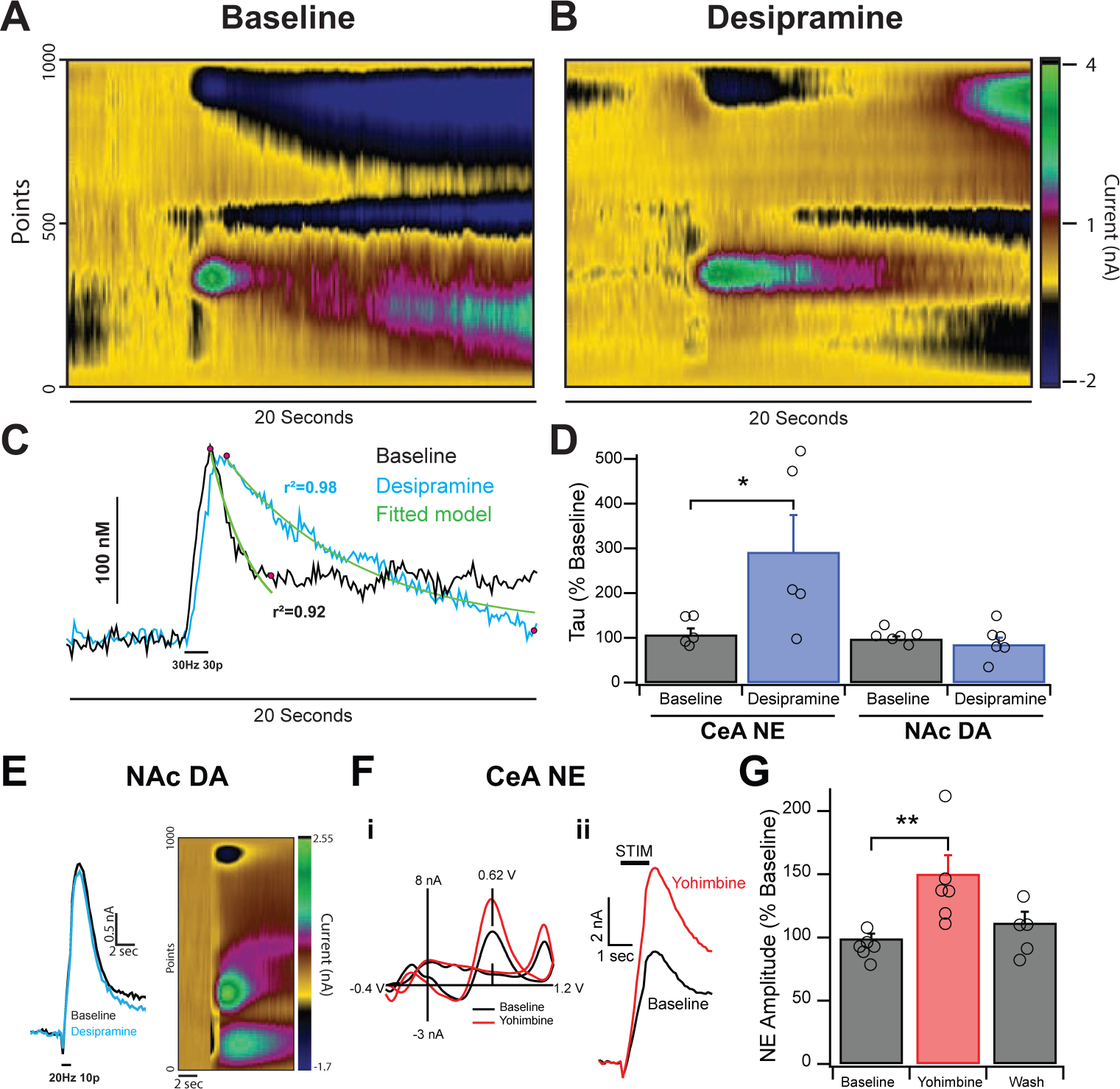
A&B. Colorplots depicting electrically evoked (30 Hz 30 pulse) NE release in the CeA before and after desipramine (50 μM). Stimulation triggers rapid release of neurotransmitter, which peaks and levels decay slowly to baseline. C. Current vs Time traces from colorplots in A&B at peak oxidation potentials. Traces were modeled using a single exponential fit before and after desipramine. Curve fits (shown in green) were based off a region of interest between the peak response and where the initial decay had stabilized (highlighted with pink dots). Reuptake times were calculated by averaging the half-life constant tau. D. Desipramine increased reuptake times of NE signals (left) to approximately 290% of baseline without affecting peak height. Desipramine failed to increase the half-life constant for exclusively DA signals (right) in the striatum. E. Stimulated DA release in the NAc showing no effect of desipramine on reuptake. F. Inset i shows the characteristic voltammogram of NE with a primary oxidative peak at 0.62 V and Yohimbine’s effects on NE release. Amplitude of this peak is directly correlated with volume of NE released from tissue. Inset ii shows current vs time plots of electrically-evoked NE release. G. Yohimbine (25 μM) increased the NE signal to approximately 150% of baseline levels, an effect that washed out after removal of drug. Error bars represent the SEM and * and ** indicate a significance level of p < 0.05 and 0.01 respectively.
Statistics were performed using JMP version 14 (JMP, SAS Institute Inc., Cary, NC) and data was visualized using Igor Pro 7 (Wavemetrics, Lake Oswego, OR). For groups of two variables, statistical significance was calculated using an unpaired, two-tailed Student’s t test. For groups of more than two variables but with only one factor, significance was determined using a one-way ANOVA, combined with either Tukey’s HSD test for post hoc analysis or Dunnett’s post hoc analysis in cases of multiple drugs all being compared back to the original baseline. For all figures, error bars represent the standard error of the mean (SEM), numbers in parentheses at the base of histograms are indicative of the total number of experiments for that condition, and *, **, *** denote significance levels of p < 0.05, p < 0.01, and p < 0.001, respectively.
3. Results
3.1. Norepinephrine signals increase in amplitude when treated with yohimbine and in clearance time when treated with desipramine.
Since NE and dopamine (DA) are identical when measured with FSCV, it was first necessary to reliably differentiate between these two monoamines. To address this, voltammetry was used in concert with desipramine (a NET blocker) and/or yohimbine (an α2 adrenergic receptor antagonist) to verify for the presence of NE (see section 2.5 for methodological details). Bath application of desipramine (50 μM) (Millan et al., 2001) increased NE signals in the CeA showed increased clearance time (τ) of 290 ± 82% compared to baseline clearance rates (Fig. 1A–D; F1,8=4.831, p < 0.05). Norepinephrine signal response to desipramine contrasts sharply with known DA signal response in the NAc, which failed to respond at all (Fig. 1D,E; F1,10=0.543, p = 0.49, ns). To investigate the potential of DAT being involved in this mechanism, we tested GBR 12909 (300 nM) on NE signals. GBR 12909 did not have an effect on the NE signal (F1,22=0.00516, p = 0.943 ns) in the CeA. Thus, uptake in the CeA appears to be mediated mostly via the NET. Predominantly NE signals responded to 25 μM yohimbine (Schwartz and Clark, 1998) with a signal amplitude increase to 150 ± 10% of baseline that reversed upon wash (Fig. 1F,G; F2,14=6.968, p < 0.01). Thus, blocking the adrenergic α2 receptor results in greater NE release in the CeA, likely by decreasing autoreceptor feedback inhibition. Signals that failed to respond to either yohimbine or desipramine were considered to be predominantly dopaminergic.
3.2. Clonidine decreases the NE signal, which can then be reversed by yohimbine
We then tested to see if clonidine (an α2 adrenergic receptor agonist) would inhibit NE expression due to the location of α2 adrenergic receptors on presynaptic adrenergic terminals and their role as autoreceptors (Starke et al., 1989). As expected, treatment of a NE signal with 10 μM clonidine (Jarrott et al., 1979) resulted in a decrease in signal amplitude to 66 ± 8% of baseline amplitude. Importantly, immediately after treatment with clonidine (10 μM), slices were treated with yohimbine (25 μM), which caused the NE signals to rebound to baseline levels (Fig. 2; F2,15= 6.46, p < 0.01).
Figure 2: Clonidine Suppresses Evoked NE Release, which can then be Restored by Yohimbine.
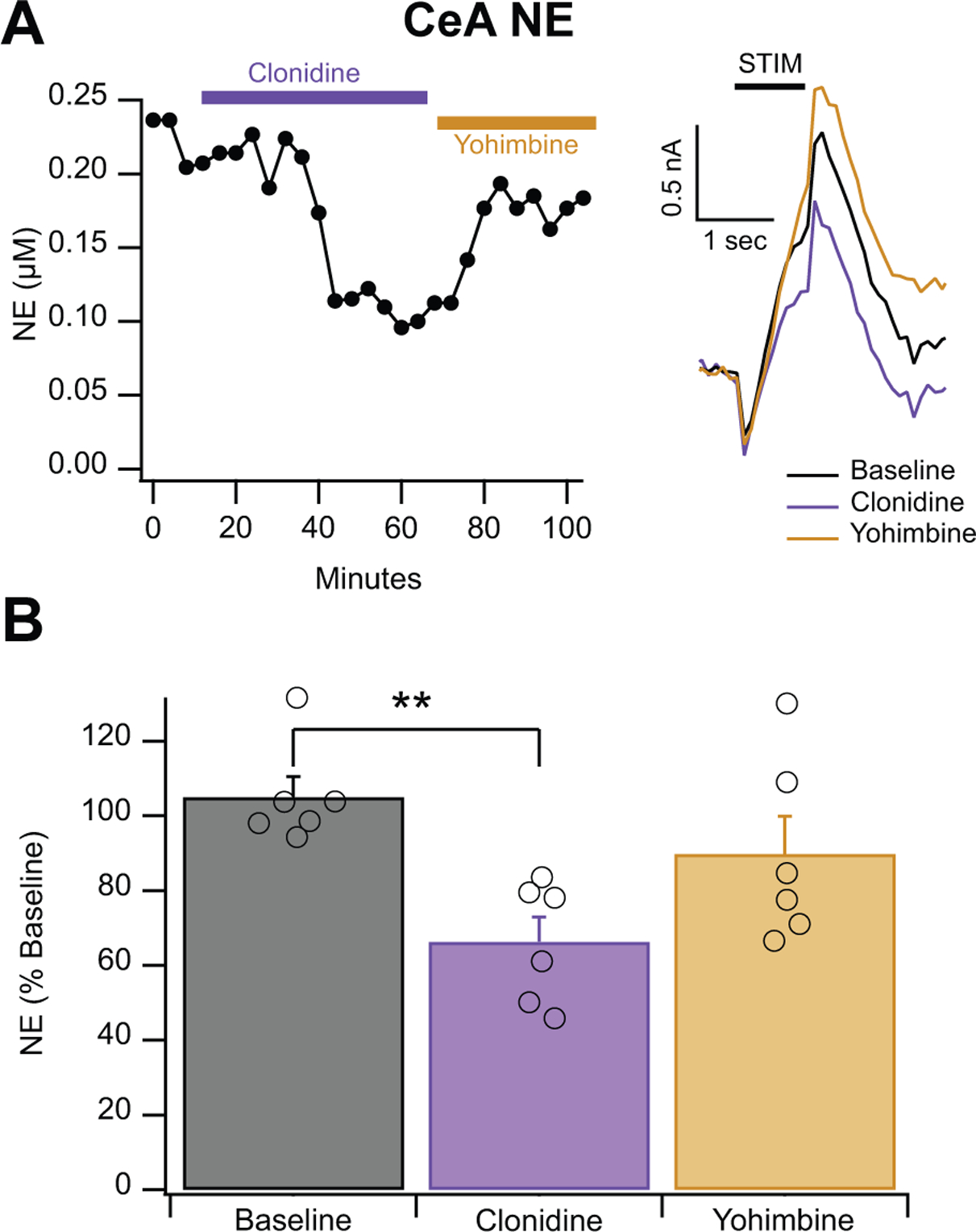
A. Representative recordings of clonidine (10 μM) followed by yohimbine (25 μM). Clonidine represses the NE signal, which is then restored by yohimbine. Inset shows representative traces. B. Cumulative histogram of showing clonidine (10 μM) reduces the NE signal to approximately 66% of baseline and the restoration of the signal by yohimbine (25 μM). Error bars represent the SEM and ** indicates a significance level of p < 0.01.
3.3. Norepinephrine release is dependent on both vesicles and action potentials
Reserpine (a VMAT blocker) is known to deplete monoaminergic vesicles (Beckstead et al., 2004). Norepinephrine signals treated with reserpine (10 μM) were decreased to 22 ± 11% of baseline (Fig. 3A,B; F1,3=113.5, p < 0.001), indicating that release is mainly dependent on functional vesicles. Similarly, 1.0 μM TTX (a voltage-gated Na+ channel inhibitor) decreased the NE signal in the CeA to 10 ± 8% of baseline amplitude (Fig. 3C,D; F1,3=17.64, p < 0.0001), demonstrating that NE release depends on action potentials. Since both reserpine and TTX are relatively irreversible antagonists, it was necessary to pharmacologically verify monoamine species prior to experimental condition. It is unlikely that this prior treatment affected the results as neither desipramine nor yohimbine are known to directly affect vesicular packaging or action potential generation and both drugs were washed out for at least 20 minutes prior to reserpine and TTX testing.
Figure 3: Reserpine and TTX Abolish Evoked NE Release in the CeA.
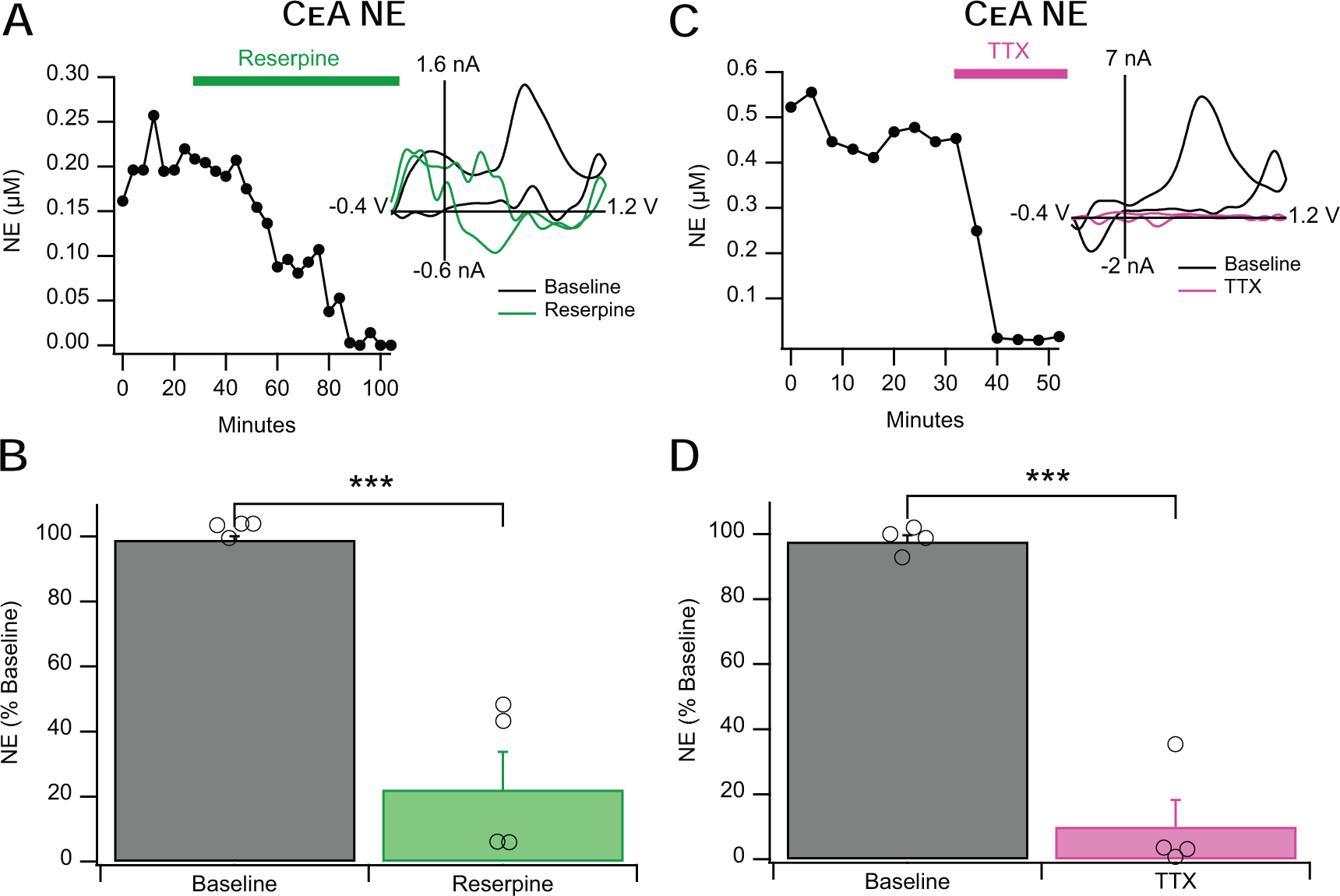
A&B. Reserpine (10 μM) attenuates evoked NE release in the CeA to approximately 22% of baseline levels. The left inset shows a representative time course of reserpine’s action and the right inset shows representative voltammograms. C&D. TTX (1.0 μM) similarly decreases evoked NE release to approximately 10% of baseline release levels. The left inset shows a representative time course of TTX’s action and the right inset shows representative voltammograms. Error bars represent the SEM and *** indicates a significance level of p < 0.001.
3.4. Acute alcohol does not alter NE release in CeA
Alcohol has been shown to have no effect on NE release in the basolateral amygdala (BLA) when NE is measured by microdialysis (Karkhanis et al., 2015), and is known to decrease DA release (measured by FSCV) in the NAc shell, which is part of the extended amygdala complex (Schilaty et al., 2014; Yorgason et al., 2015). However, to our knowledge, it is not known whether acute alcohol can impair NE release in the CeA and we are the first to investigate this. Here, we applied a dose that produces a maximal response on GABA release in the CeA (44 mM; Roberto et al., 2004b, 2003). Alcohol did not alter NE release (Fig. 4A,B; F2,13=0.0255, p =0.97 ns) in the CeA of alcohol naïve rats. We then tested a higher concentration of alcohol (66 mM) and found that it was also ineffective in altering NE release (F2,13=1.153, p =0.301 ns). Interestingly, when treated with acute 44 mM alcohol, signals insensitive to yohimbine (determined to be not NE) decreased to 52± 6% of baseline (Fig. 4C,D; F1,14=14.83, p < 0.01). Given that DA and NE have an identical electrochemical signature, we assume that this non-NE catecholamine is DA. This acute alcohol-induced decrease in signal in the CeA resembles the effect of EtOH on evoked DA release in the striatum (Schilaty et al., 2014; Yorgason et al., 2015), strengthening the conclusion that the signal is predominantly DA. Indeed, alcohol (66 mM) decreased DA signals in the NAc to 73% of the baseline signal (F2,10=11.11, p < 0.05). Together, this data shows that CeA NE release is relatively insensitive to alcohol’s inhibitory effects.
Figure 4: Alcohol does not Affect NE Release in the CeA.
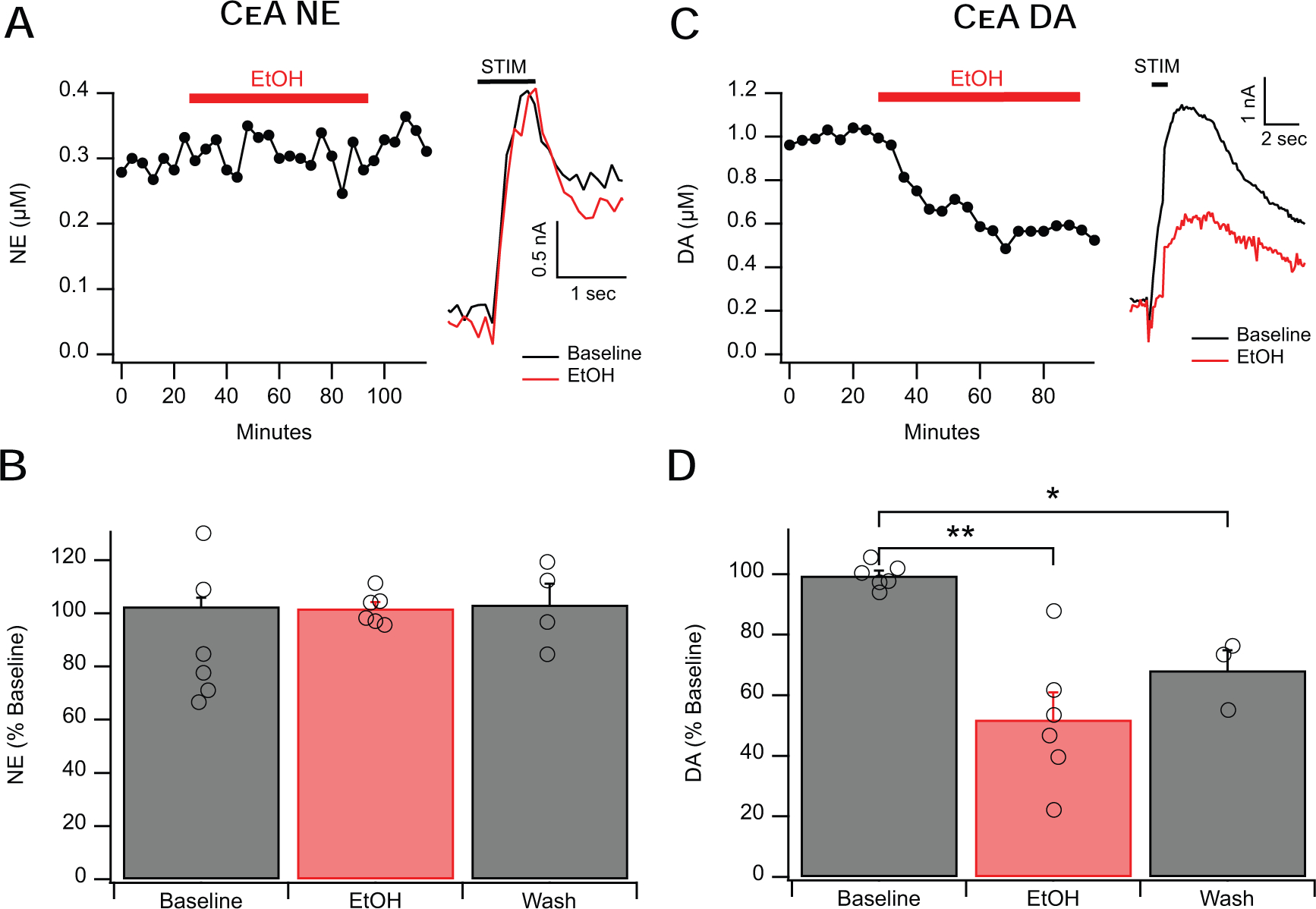
A&B. Alcohol (44 mM) failed to affect evoked NE release in the CeA. The left inset shows a representative time course of alcohol’s action and the right inset shows representative current vs time traces. C&D. Alcohol (44 mM) decreased evoked DA release in the CeA to approximately 52% of baseline release levels, an effect which did not fully wash out. The left inset shows a representative time course of alcohol’s action and the right inset shows representative current vs time traces. Error bars represent the SEM, numbers in histograms represent total number of experiments for the experimental condition. * and ** indicate a significance level of p < 0.05 and 0.01 respectively.
3.5. CRF decreases CeA NE release in both alcohol-naïve and alcohol-dependent rats
Given the complex interaction between the CRF and NE systems, we tested whether CRF would influence locally-evoked NE release. We hypothesized that acute application of CRF would increase NE release in the CeA and that this effect would be enhanced in alcohol-dependent animals. In contrast to our hypothesis, we found that CRF (100 nM; Roberto et al., 2010) decreased NE release in the CeA of alcohol-naïve rats to 82 ± 7% of baseline levels (Fig. 5A,B; F1,3= 9.1, p < 0.01). In a separate set of experiments, we applied CRF first followed by CRF co-applied by CRF1 receptor antagonist R121919 (1.0 μM). We found that the CRF induced-inhibition of NE release was reversed with R121919 (Fig. 5C; F2,12= 11.17, p < 0.05). Since CRF is known to play a role in alcohol dependence and drug-seeking behavior (Weiss et al., 2001), we also tested the effect of CRF on NE release in the CeA of alcohol-dependent rats. Similarly, in alcohol dependent rats, CRF (100 nM) also decreased NE release to 80 ± 5% of baseline (Fig. 5D,E; F1,4=158.6, p < 0.01). Interestingly, the magnitude of the CRF-induced decrease in the NE signaling was comparable in the two groups.
Additionally, comparable baseline NE peak amplitudes were observed in CeA of naïve and alcohol dependent animals, with average of 0.29 ± 0.09 μM NE and 0.27 ± 0.05 μM NE respectively (data not shown). There was no difference between baseline levels of release in alcohol-naïve and alcohol-dependent animals (F1,5=4.69, p = 0.84, ns, data not shown), suggesting that alcohol dependence does not affect baseline release of NE as measured by FSCV. The apparent difference in baseline NE release in the timecourses (Fig. 5 A,D) is limited to the specific representative traces.
4. Discussion
Alcoholism is a chronic relapsing disorder characterized by a compulsion to seek and consume alcohol and has been linked to the dysregulation of brain emotion systems, including both those that mediate reward and those that mediate stress. Alcohol dependence is characterized by an increased negative emotion state (Koob, 2008; Koob and Le Moal, 2008), and recruitment of the NE system of the LC and the CRF system of the CeA (Koob, 1999; Haass-Koffler et al., 2018). Interactions between these two stress systems have been studied and there is evidence for a feed-forward loop between the CeA and the LC, giving the potential for a rapid stress response (Koob, 1999; Lee et al., 2008; Reyes et al., 2008, 2011; McCall et al., 2015). While stress-induced NE release occurs in both the BLA and CeA (Galvez et al., 1996; Quirarte et al., 1998; Hatfield et al., 1999), little is known about the local regulatory control on NE release in the CeA and potential CRF modulation. Thus, in this study, we used FSCV to measure real-time, electrically-evoked release of NE and investigate the effects of acute CRF on NE transmission in the CeA of both alcohol naïve and dependent rats. To the best of our knowledge, we are the first to identify electrically-evoked NE release in the CeA of both alcohol-naïve and alcohol-dependent rats and characterize the physical nature of this NE release. Similar to what was observed in the BNST (Park et al., 2011), blocking NE reuptake and α2 autoreceptors resulted in prolonged signals with larger release. However, a direct comparison between the previous in vivo BNST study and the present CeA study is not possible since in vivo and in vitro slice preparations can produce very different results in release and uptake even in the brain region (for example, compare (Yorgason et al., 2011b, 2016).
In characterizing the NE signal, we used reserpine, a VMAT inhibitor which causes terminals to become depleted of monoamines, as vesicles require constant VMAT activity to maintain the pH gradient to hold monoamines in vesicles (Freyberg et al., 2016). Notably, reserpine reduced evoked NE signals in CeA to 22% of baseline levels, suggesting that this type of neurotransmission is vesicle dependent. Additionally, blockade of voltage-gated sodium channels using TTX reduced signals to 10% baseline levels, suggesting that these NE vesicles are dependent on canonical action potentials.
While we had originally hypothesized that CRF would increase NE transmission, we found that acute, bath-applied CRF decreases electrically-evoked NE transmission in the CeA of both alcohol-naïve and alcohol-dependent animals, and effect that was blocked in naïve rats with CRF1 receptor antagonism. This suggests that CRF locally inhibits NE release in the CeA. One speculation is that CRF acts on CRF1 and/or CRF2 receptors on presynaptic CeA NE terminals directly modulating NE transmission, but CRF could also act by modulating NE release via multiple indirect synaptic effects. We have previously showed that CRF increases locally evoked GABA transmission in the CeA via CRF1 receptors (Roberto et al., 2010; Varodayan et al., 2017b). In contrast, CRF decreases locally evoked glutamate release mainly via CRF1, and only partially via CRF2, receptors in CeA (Varodayan et al., 2017a). Notably, CRF-induced enhancement of CeA GABAergic transmission in alcohol dependent rats was greater than the effect observed in naïve animals (Roberto et al., 2010), whereas we did not observe differences in the CRF-induced evoked glutamatergic responses in the CeA of naïve versus alcohol-dependent rats (Varodayan et al., 2017a). Similar to the effects of CRF on evoked glutamate transmission, the effect of CRF on evoked NE response was not different between alcohol naïve and dependent animals. Thus, the present results indicate that the CRF system modulating NE terminals may undergo compensatory neuroadaptations that contribute to the maintenance of homeostasis during chronic ethanol exposure.
Our voltammetry data showing CRF-induced decrease of evoked NE in CeA contrasts with the findings of Su et al, who reported that CRF causes release of NE in the CeA (Su et al., 2015). To understand this discrepancy, it is necessary to recognize the difference in experimental conditions between these two studies. The current study was conducted solely in brain slices that largely eliminate macrocircuitry (afferent and efferent projections are sheared during the slice preparation, which includes loss of NE terminal regulation from the soma), whereas Su et al were using microdialysis in freely moving animals. Apart from the trauma induced by cannulation, the macrocircuitry of the feed-forward CeA-LC loop was intact. This limitation (lack of intact macrocircuitry) likely accounts for the difference between these two studies, but also affords valuable insights into the local, regulatory control on NE release in the CeA.
It is also important to note that acute alcohol had no effect on NE transmission in the CeA, when measured here using FSCV, similar to what has previously been shown in the BLA (Karkhanis et al., 2015). While alcohol clearly affects transmission of the biochemically-related DA in the striatum when measured with either FSCV or microdialysis, this effect appears to be indirect, likely through glycinergic or GABAergic intermediaries (Jonsson et al., 2014; Schilaty et al., 2014; Yorgason et al., 2015). While presumed DA signals in the CeA are affected by alcohol, it is important that there is not a similar mechanism affecting NE release, and that although alcohol is involved in a stress response, it does not appear to directly affect NE transmission in the CeA. Despite this lack of direct effects of alcohol on NE transmission, acute alcohol increases GABAergic transmission in the CeA via increased GABA release (Roberto et al., 2003, 2004a), likely diminishing CeA activity to the LC, and slowing CRF-containing driven LC NE activity. Additionally, while we speculate that CRF1 activation mediates this increase in GABA release, we can’t rule out a direct activation of CRF1 by alcohol. In our in vitro slice preparation without TTX, the local circuitry is maintained, thus, it is possible that other targets modulated by alcohol, including voltage-gated calcium channels () or other neuropeptidergic systems may blunt/counteract an alcohol effect on NE transmission. Finally, there could be other brainstem NE sources that may influence our measurements.
Both NE and CRF play crucial roles in behavioral aspects of addiction, especially the anxiogenic effects of drug withdrawal (Menzaghi et al., 1994; Heinrichs et al., 1995; Delfs et al., 2000). CRF release (measured using microdialysis) in the CeA is increased during withdrawal in alcohol-dependent animals (Merlo Pich et al., 1995), and appears to contribute to withdrawal-related anxiety. This withdrawal-related anxiety can be reduced by injection of CRF receptor antagonists into the CeA (Rassnick et al., 1993). Additionally, acute restraint stress induced rapid elevation in extracellular NE content in rat CeA (Galvez et al., 1996; Inglis and Moghaddam, 1999; Khoshbouei et al., 2002; Reznikov et al., 2007, 2009). Based on these studies, we hypothesize that NE and CRF may act in concert in the CeA to modulate stress-related components of alcohol addiction.
In conclusion, CRF decreases electrically-evoked NE release in the CeA by increasing inhibitory and/or decreasing excitatory control over the NE terminals in CeA. It is possible that CRF is released somatodendritically in the CeA as has been previously characterized (Dabrowska et al., 2013), providing a tight regulation of a CRF-evoked NE response from local CRF negative feedback in the CeA. In addition, local application of exogenous CRF modulates both GABAergic and glutamatergic signaling in the CeA (Roberto et al., 2010; Silberman and Winder, 2015; Varodayan et al., 2017a). Taken together, this indicates that increased CRF receptor activity in the CeA may increase anxiety-like behaviors without driving a global, NE-driven stress response through the LC. The CeA is a hub for negative emotional processing (reviewed in Gilpin, Herman & Roberto 2015) and serves as the major output nucleus of the entire amygdala complex, projecting to additional regions that regulate stress and anxiety-related behaviors. Therefore, CRF regulation of the NE system in the CeA may represent a novel mechanism within a key stress circuit that can lead to potential additional targets for alcohol-dependence treatment.
Highlights.
Evoked norepinephrine release in the CeA is vesicle and action potential dependent
CRF decreases norepinephrine release in the CeA
Norepinephrine release in the CeA is insensitive to alcohol
Acknowledgements
Support for this study was provided by National Institute on Alcohol Abuse and Alcoholism grants AA015566, AA013498, AA021491, AA017447, AA006420, AA027700, and AA020919 and by National Institute on Drug Abuse grant DA035958. The authors thank Dr. Florence Varodayan for the valuable comments on the manuscript. The authors declare no conflict of interest. This is TSRI manuscript number 29698.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bauer UE, Briss PA, Goodman RA, Bowman BA (2014) Prevention of chronic disease in the 21st century: Elimination of the leading preventable causes of premature death and disability in the USA. Lancet 384:45–52. [DOI] [PubMed] [Google Scholar]
- Beckstead MJ, Grandy DK, Wickman K, Williams JT (2004) Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron 42:939–946. [DOI] [PubMed] [Google Scholar]
- Campese VD, Soroeta JM, Vazey EM, Aston-Jones G, Ledoux JE, Sears RM (2017) Noradrenergic regulation of central amygdala in aversive pavlovian-to-instrumental transfer. eNeuro. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz MT, Herman MA, Cote DM, Ryabinin AE, Roberto M (2013) Ghrelin increases GABAergic transmission and interacts with ethanol actions in the rat central nucleus of the amygdala. Neuropsychopharmacology 38:364–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis AL, Bello NT, Connolly KR, Valentino RJ (2002) Corticotropin-releasing factor neurones of the central nucleus of the amygdala mediate locus coeruleus activation by cardiovascular stress. J Neuroendocrinol 14:667–682. [DOI] [PubMed] [Google Scholar]
- Dabrowska J, Hazra R, Guo JD, DeWitt S, Rainnie DG (2013) Central CRF neurons are not created equal: Phenotypic differences in CRF-containing neurons of the rat paraventricular hypothalamus and the bed nucleus of the stria terminalis. Front Neurosci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delfs JM, Zhu Y, Druhan JP, Aston-Jones G (2000) Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature 403:430–434. [DOI] [PubMed] [Google Scholar]
- Finnell JE, Moffitt CM, Hesser LA, Harrington E, Melson MN, Wood CS, Wood SK (2019) The contribution of the locus coeruleus-norepinephrine system in the emergence of defeatinduced inflammatory priming. Brain Behav Immun. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyberg Z et al. (2016) Mechanisms of amphetamine action illuminated through optical monitoring of dopamine synaptic vesicles in Drosophila brain. Nat Commun 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez R, Mesches MH, Mcgaugh JL (1996) Norepinephrine release in the amygdala in response to footshock stimulation. Neurobiol Learn Mem 66:253–257. [DOI] [PubMed] [Google Scholar]
- Gilpin NW, Herman MA, Roberto M (2015) The Central Amygdala as an Integrative Hub for Anxiety and Alcohol Use Disorders. Biol Psychiatry 77:859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpin NW, Richardson HN, Cole M, Koob GF (2008) Vapor inhalation of alcohol in rats. Curr Protoc Neurosci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass-Koffler CL, Swift RM, Leggio L (2018) Noradrenergic targets for the treatment of alcohol use disorder. Psychopharmacology (Berl) 235:1625–1634 Available at: http://link.springer.com/10.1007/s00213-018-4843-6 [Accessed May 30, 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfield T, Spanis C, McGaugh JL (1999) Response of amygdalar norepinephrine to footshock and GABAergic drugs using in vivo microdialysis and HPLC. Brain Res 835:340–345. [DOI] [PubMed] [Google Scholar]
- Heinrichs SC, Menzaghi F, Pich EM, Britton KT, Koob GF (1995) The Role of CRF in Behavioral Aspects of Stress. Ann N Y Acad Sci 771:92–104. [DOI] [PubMed] [Google Scholar]
- Inglis FM, Moghaddam B (1999) Dopaminergic innervation of the amygdala is highly responsive to stress. J Neurochem 72:1088–1094. [DOI] [PubMed] [Google Scholar]
- Jarrott B, Louis WJ, Summers1 RJ (1979) THE CHARACTERISTICS OF [3H]-CLONIDINE BINDING TO AN ca-ADRENOCEPTOR IN MEMBRANES FROM GUINEA-PIG KIDNEY. Br J Pharmac 65:663–670 Available at: http://onlinelibrary.wiley.com/store/10.1111/j.1476-5381.1979.tb07879.x/asset/j.1476-5381.1979.tb07879.x.pdf?v=1&t=jc2em82r&s=aeb7e12b6387b222b39959290c1a5a8ef49d03a1 [Accessed January 5, 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson S, Adermark L, Ericson M, Söderpalm B (2014) The involvement of accumbal glycine receptors in the dopamine-elevating effects of addictive drugs. Neuropharmacology 82:69–75. [DOI] [PubMed] [Google Scholar]
- Kallupi M, Varodayan FP, Oleata CS, Correia D, Luu G, Roberto M (2014) Nociceptin/Orphanin FQ Decreases Glutamate Transmission and Blocks Ethanol-Induced Effects in the Central Amygdala of Naive and Ethanol-Dependent Rats. Neuropsychopharmacology 39:1081–1092 Available at: http://www.nature.com/articles/npp2013308 [Accessed January 3, 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkhanis AN, Alexander NJ, Mccool BA, Weiner JL, Jones SR (2015) Chronic social isolation during adolescence augments catecholamine response to acute ethanol in the basolateral amygdala. Synapse 69:385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshbouei H, Cecchi M, Dove S, Javors M, Morilak DA (2002) Behavioral reactivity to stress: Amplification of stress-induced noradrenergic activation elicits a galanin-mediated anxiolytic effect in central amygdala. Pharmacol Biochem Behav 71:407–417. [DOI] [PubMed] [Google Scholar]
- Koob GF (1999) Corticotropin-releasing factor, norepinephrine, and stress. Biol Psychiatry 46:1167–1180. [DOI] [PubMed] [Google Scholar]
- Koob GF (2008) A role for brain stress systems in addiction. Neuron 59:11–34 Available at: http://linkinghub.elsevier.com/retrieve/pii/S0896-6273(08)00530-8%5Cnpapers://a16ed0bffd10-49f5-8692-d49b1396b516/Paper/p301%5Cnhttp://www.neuron.org/cgi/content/full/59/1/11/DC1/%5Cnpapers://a16ed0bf-fd10-49f5-8692-d49b1396b516/Paper/p18414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF (2013) Theoretical frameworks and mechanistic aspects of alcohol addiction: alcohol addiction as a reward deficit disorder. Curr Top Behav Neurosci 13:3–30 Available at: http://www.ncbi.nlm.nih.gov/pubmed/21744309 [Accessed December 28, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF (2015) The dark side of emotion: the addiction perspective. Eur J Pharmacol 753:73–87 Available at: http://www.ncbi.nlm.nih.gov/pubmed/25583178 [Accessed January 2, 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M (2008) Addiction and the Brain Antireward System. Annu Rev Psychol 59:29–53 Available at: http://www.annualreviews.org/doi/10.1146/annurev.psych.59.103006.093548. [DOI] [PubMed] [Google Scholar]
- Lee Y, Fitz S, Johnson PL, Shekhar A (2008) Repeated stimulation of CRF receptors in the BNST of rats selectively induces social but not panic-like anxiety. Neuropsychopharmacology 33:2586–2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall JG, Al-Hasani R, Siuda ER, Hong DY, Norris AJ, Ford CP, Bruchas MR (2015) CRH Engagement of the Locus Coeruleus Noradrenergic System Mediates Stress-Induced Anxiety. Neuron 87:606–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzaghi F, Rassnick S, Heinrichs S, Baldwin H, Pich EM, Weiss F, Koob GF (1994) The Role of Corticotropin‐Releasing Factor in the Anxiogenic Effects of Ethanol Withdrawal. Ann N Y Acad Sci 739:176–184. [DOI] [PubMed] [Google Scholar]
- Merlo Pich E, Lorang M, Yeganeh M, Rodriguez de Fonseca F, Raber J, Koob GF, Weiss F (1995) Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J Neurosci 15:5439–5447 Available at: http://www.ncbi.nlm.nih.gov/pubmed/7643193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ, Gobert A, Lejeune F, Newman-Tancredi A, Rivet JM, Auclair A, Peglion JL (2001) S33005, a novel ligand at both serotonin and norepinephrine transporters: I. Receptor binding, electrophysiological, and neurochemical profile in comparison with venlafaxine, reboxetine, citalopram, and clomipramine. J Pharmacol Exp Ther 298:565–580 Available at: http://www.ncbi.nlm.nih.gov/pubmed/11454918 [Accessed January 5, 2018]. [PubMed] [Google Scholar]
- O’Dell LE, Roberts AJ, Smith RT, Koob GF (2004) Enhanced alcohol self-administration after intermittent versus continuous alcohol vapor exposure. Alcohol Clin Exp Res 28:1676–1682. [DOI] [PubMed] [Google Scholar]
- Park J, Takmakov P, Wightman RM (2011) In vivo comparison of norepinephrine and dopamine release in rat brain by simultaneous measurements with fast-scan cyclic voltammetry. J Neurochem 119:932–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirarte GL, Galvez R, Roozendaal B, McGaugh JL (1998) Norepinephrine release in the amygdala in response to footshock and opioid peptidergic drugs. Brain Res 808:134–140. [DOI] [PubMed] [Google Scholar]
- Rassnick S, Heinrichs SC, Britton KT, Koob GF (1993) Microinjection of a corticotropin-releasing factor antagonist into the central nucleus of the amygdala reverses anxiogenic-like effects of ethanol withdrawal. Brain Res 605:25–32. [DOI] [PubMed] [Google Scholar]
- Reyes BAS, Carvalho AF, Vakharia K, Van Bockstaele EJ (2011) Amygdalar peptidergic circuits regulating noradrenergic locus coeruleus neurons: Linking limbic and arousal centers. Exp Neurol 230:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes BAS, Drolet G, Van Bockstaele EJ (2008) Dynorphin and stress-related peptides in rat locus coeruleus: Contribution of amygdalar efferents. J Comp Neurol 508:663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznikov LR, Grillo C a, Piroli GG, Pasumarthi RK, Reagan LP, Fadel J (2007) Acute stress-mediated increases in extracellular glutamate levels in the rat amygdala: differential effects of antidepressant treatment. Eur J Neurosci 25:3109–3114 Available at: http://www.ncbi.nlm.nih.gov/pubmed/17561824. [DOI] [PubMed] [Google Scholar]
- Reznikov LR, Reagan LP, Fadel JR (2009) Effects of acute and repeated restraint stress on gaba efflux in the rat basolateral and central amygdala. Brain Res 1256:61–68. [DOI] [PubMed] [Google Scholar]
- Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M, Cottone P, Madamba SG, Stouffer DG, Zorrilla EP, Koob GF, Siggins GR, Parsons LH (2010) Corticotropin Releasing Factor-Induced Amygdala Gamma-Aminobutyric Acid Release Plays a Key Role in Alcohol Dependence. Biol Psychiatry 67:831–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Gilpin NW, Siggins GR (2012) The central amygdala and alcohol: Role of γ-aminobutyric acid, glutamate, and neuropeptides. Cold Spring Harb Perspect Med 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Moore SD, Tallent MK, Siggins GR (2003) Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons. Proc Natl Acad Sci U S A 100:2053–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Stouffer DG, Parsons LH, Siggins GR (2004a) Increased GABA Release in the Central Amygdala of Ethanol- Dependent Rats. Alcohol Clin Exp Res 24:10159–10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Schweitzer P, Madamba SG, Stouffer DG, Parsons LH, Siggins GR (2004b) Acute and chronic ethanol alter glutamatergic transmission in rat central amygdala: an in vitro and in vivo analysis. J Neurosci 24:1594–1603 Available at: http://www.ncbi.nlm.nih.gov/pubmed/14973247%5Cnhttp://www.jneurosci.org/cgi/doi/10.1523/JNEUROSCI.5077-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Siggins GR (2006) Nociceptin/orphanin FQ presynaptically decreases GABAergic transmission and blocks the ethanol-induced increase of GABA release in central amygdala. Proc Natl Acad Sci U S A 103:9715–9720 Available at: http://www.ncbi.nlm.nih.gov/pubmed/16788074 [Accessed January 3, 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Wiener SG, Bloom FE (1979) Long-term ethanol administration methods for rats: advantages of inhalation over intubation or liquid diets. Behav Neural Biol 27:466–486. [DOI] [PubMed] [Google Scholar]
- Sakanaka M, Shibasaki T, Lederis K (1986) Distribution and efferent projections of corticotropin-releasing factor-like immunoreactivity in the rat amygdaloid complex. Brain Res 382:213–238. [DOI] [PubMed] [Google Scholar]
- Sara SJ (2009) The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci 10:211–223. [DOI] [PubMed] [Google Scholar]
- Schilaty ND, Hedges DM, Jang EY, Folsom RJ, Yorgason JT, McIntosh JM, Steffensen SC (2014) Acute ethanol inhibits dopamine release in the nucleus accumbens via α6 nicotinic acetylcholine receptors. J Pharmacol Exp Ther 349:559–567 Available at: http://www.ncbi.nlm.nih.gov/pubmed/24643637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz DD, Clark TP (1998) Selectivity of atipamezole, yohimbine and tolazoline for alpha-2 adrenergic receptor subtypes: Implications for clinical reversal of alpha-2 adrenergic receptor mediated sedation in sheep. J Vet Pharmacol Ther 21:342–347. [DOI] [PubMed] [Google Scholar]
- Silberman Y, Winder DG (2015) Ethanol and corticotropin releasing factor receptor modulation of central amygdala neurocircuitry: An update and future directions. Alcohol 49:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RJ, Aston-Jones G (2008) Noradrenergic transmission in the extended amygdala: Role in increased drug-seeking and relapse during protracted drug abstinence. Brain Struct Funct 213:43–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starke K, Gothert M, Kilbinger H (1989) Modulation of neurotransmitter release by presynaptic autoreceptors. Physiol Rev 69:864–989 Available at: http://www.ncbi.nlm.nih.gov/pubmed/2568648 [Accessed January 5, 2018]. [DOI] [PubMed] [Google Scholar]
- Stenzelpoore MP, Heinrichs SC, Rivest S, Koob GF, Vale WW (1994) Overproduction of Corticotropin-Releasing Factor in Transgenic Mice - a Genetic Model of Anxiogenic Behavior. J Neurosci 14:2579–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J, Tanaka Y, Muratsubaki T, Kano M, Kanazawa M, Fukudo S (2015) Injection of corticotropin-releasing hormone into the amygdala aggravates visceral nociception and induces noradrenaline release in rats. Neurogastroenterol Motil 27:30–39. [DOI] [PubMed] [Google Scholar]
- Varodayan FP, Correia D, Kirson D, Khom S, Oleata CS, Luu G, Schweitzer P, Roberto M (2017a) CRF modulates glutamate transmission in the central amygdala of naïve and ethanol-dependent rats. Neuropharmacology 125:418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varodayan FP, de Guglielmo G, Logrip ML, George O, Roberto M (2017b) Alcohol Dependence Disrupts Amygdalar L-Type Voltage-Gated Calcium Channel Mechanisms. J Neurosci 37:4593–4603 Available at: http://www.jneurosci.org/lookup/doi/10.1523/JNEUROSCI.3721-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss F, Ciccocioppo R, Parsons LH, Katner S, Liu X, Zorrilla EP, Valdez GR, Ben-Shahar O, Angeletti S, Richter RR (2001) Compulsive Drug-Seeking Behavior and Relapse. Ann N Y Acad Sci 937:1–26 Available at: http://doi.wiley.com/10.1111/j.1749-6632.2001.tb03556.x [Accessed January 9, 2018]. [DOI] [PubMed] [Google Scholar]
- Yorgason JT, Calipari ES, Ferris MJ, Karkhanis AN, Fordahl SC, Weiner JL, Jones SR (2016) Social isolation rearing increases dopamine uptake and psychostimulant potency in the striatum. Neuropharmacology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorgason JT, España RA, Jones SR (2011a) Demon Voltammetry and Analysis software: Analysis of cocaine-induced alterations in dopamine signaling using multiple kinetic measures. J Neurosci Methods 202:158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorgason JT, Jones SR, España RA (2011b) Low and high affinity dopamine transporter inhibitors block dopamine uptake within 5 sec of intravenous injection. Neuroscience. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorgason JT, Rose JH, McIntosh JM, Ferris MJ, Jones SR (2015) Greater ethanol inhibition of presynaptic dopamine release in C57BL/6J than DBA/2J mice: Role of nicotinic acetylcholine receptors. Neuroscience 284:854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorgason JT, Zeppenfeld DM, Williams JT (2017) Cholinergic Interneurons Underlie Spontaneous Dopamine Release in Nucleus Accumbens. J Neurosci 37:2086–2096 Available at: http://www.jneurosci.org/lookup/doi/10.1523/JNEUROSCI.3064-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]