

Abstract
The BTBR T+Itpr3tf/J (BTBR) mouse has been used as a complex genetic model of Autism Spectrum Disorders (ASD). While the specific mechanisms underlying BTBR behavioral phenotypes are poorly understood, prior studies have implicated profound differences in innate immune system control of pro-inflammatory cytokines. Innate immune activation and elevated pro-inflammatory cytokines are also detected in blood of children with ASD. In this study, we examined how underlying BTBR genetic variants correspond to strain-specific changes in chromatin accessibility, resulting in a pro-inflammatory response specifically in BTBR bone marrow derived macrophages (BMDM). In response to repeated lipopolysaccharide (LPS) treatments, C57BL/6J (C57) BMDM exhibited intact endotoxin tolerance. In contrast, BTBR BMDM exhibited hyper-responsive expression of genes that were normally tolerized in C57. This failure in formation of endotoxin tolerance in BTBR was mirrored at the level of chromatin accessibility. Using ATAC-seq, we specifically identified promoter and enhancer regions with strain-specific differential chromatin accessibility both at baseline and in response to LPS. Regions with strain-specific differences in chromatin accessibility were significantly enriched for BTBR genetic variants, such that an average of 22% of the differential chromatin regions had at least one variant. Together, these results demonstrate that BTBR genetic variants contribute to altered chromatin responsiveness to endotoxin challenge resulting in hyper-responsive innate immunity in BTBR. These findings provide evidence for an interaction between complex genetic variants and differential epigenetic regulation of innate immune responses.
Introduction
The ability of peripheral innate immune cells to control pro-inflammatory cytokines during periods of repeated or prolonged exposure to immune activating stimuli is critical for preventing tissue damage(Liu et al., 2019). Cells that compose the innate immune system such as monocytes, macrophages, natural killer cells and microglia were traditionally thought to respond rapidly and nonspecifically to immune stimuli and lack immune memory(Netea et al., 2016, 2011; Wendeln et al., 2018). However, recent evidence has demonstrated a form of innate immune memory in which immune activation can alter a secondary response to either the same or different pathogen exposure. In macrophages, repeated exposure to lipopolysaccharide (LPS) induces a refractory period of immune activation termed endotoxin tolerance. This form of innate immune memory results from epigenetic suppression of genes associated with inflammation (tolerance) and continued expression of genes coding for anti-microbial molecules (non-tolerance)(Foster et al., 2007; Novakovic et al., 2016; Saeed et al., 2014). Mechanistic studies have demonstrated that innate immune memory is critically dependent on epigenetic reprograming that produces transcriptional changes in immune signaling, metabolism and ultimately enhances the immune cell’s capacity to appropriately respond to stimulation(Novakovic et al., 2016; Saeed et al., 2014). How these epigenetic regulatory mechanisms become dysfunctional in cases of chronic inflammation is of great interest considering the large number of disease states characterized by increased inflammation.
Numerous studies have shown increased innate immune activation in individuals with autism spectrum disorders (ASD)(Ashwood et al., 2011; Enstrom et al., 2010; Goines and Ashwood, 2013; Krakowiak et al., 2017; Zerbo et al., 2014). These include increased activated monocyte populations circulating in the periphery or increased cytokines related to monocyte/macrophage activation(Hughes et al., 2018). For example, increased IL-β was found in newborn blood spots from infants that later went on to develop ASD compared to typically developing children(Krakowiak et al., 2017). Monocytes isolated from children with ASD compared to typically developing children show elevated baseline cytokine levels, as well as a hyper-response in cytokine levels that were associated with increased behavioral impairments in the children(Enstrom et al., 2010). Several additional lines of evidence point to increased microglia activation in ASD brain, including activated microglial morphology(Morgan et al., 2014, 2012, 2010), enhanced pro-inflammatory cytokines(Smith et al., 2012; Tarkowski et al., 2003), increased immune gene expression(Gandal et al., 2018a, 2018b; Gupta et al., 2014; Parikshak et al., 2016), and real-time PET scan analyses(Suzuki et al., 2013).
In animal models relevant to ASD, elevated innate immune responses have also been shown(Onore et al., 2013; Schwartzer et al., 2015, 2013). The BTBR T+Itpr3tf/J (BTBR) mouse strain shows a natural lack of sociability, increased repetitive behaviors, and impaired cognition(McFarlane et al., 2008; Moy et al., 2007; Roullet and Crawley, 2011; Scattoni et al., 2011). Similar to monocytes isolated from children with ASD, macrophages from the BTBR mouse strain have a skewed pro-inflammatory phenotype and show a larger cytokine response to LPS challenge than macrophages from C57BL/6J (C57) mice(Onore et al., 2013). The immune alterations in BTBR have been correlated with the presentation of ASD relevant behaviors(Onore et al., 2013), suggesting a potential link between immune and behavioral dysfunction. Consistent with this hypothesis, replacement of the immune system with wildtype bone marrow rescued the social impairments but not the repetitive behaviors in BTBR(Schwartzer et al., 2017). BTBR mice were originally derived in the 1950s from an inbred strain carrying the at (nonagouti; black and tan) and wildtype T (brachyury) mutations and then subsequently crossed with mice with the tufted (Itpr3tf) allele (http://jaxmice.jax.org/strain/002282.html). The BTBR genome is divergent from that of C57, including over 6 million SNPs and 1.6 million insertions and deletions (InDels)(Jones-Davis et al., 2013; Pstkov et al., 2005), similar to differences between any two human individuals(Auton et al., 2015). In combination with the observed immune abnormalities, the BTBR mouse strain is an interesting model for ASD that captures aspects of complex genetic, environmental, and immune components of the disorder.
To uncover mechanisms that may explain BTBR genetic differences in regulation of cytokine responses, we examined gene expression and chromatin dynamics in response to immune challenge and tolerance using bone marrow derived macrophages (BMDM) cultures. We specifically assessed endotoxin responses in BMDM cultures(Foster et al., 2007) from BTBR and C57 strains to test the hypothesis that the chronic hyper-inflammatory state observed in BTBR mice may be a result of altered chromatin dynamics and immune gene regulation. Using a combination of gene expression, epigenetic, genetic, and bioinformatic approaches, we identified key regions of differential chromatin regulation between the strains that related to both altered gene expression patterns and underlying strain genetic variants. Together our findings indicate that genetic variation between strains significantly contributes to altered chromatin accessibility and LPS-induced gene expression in BMDM cultures, culminating in higher baseline and LPS responsivity in the BTBR strain.
Results
BTBR BMDM hyper-respond to LPS immune challenge
To test the hypothesis that BTBR strain differences in inflammation can be observed at a level of gene responsivity to LPS challenge and tolerance, we compared transcript levels in BMDM cultures from juvenile postnatal day 28 (P28) male BTBR (n=10) and C57BL6/J (C57) (n=9) mice. After seven days in culture the differentiated macrophages were re-plated (3 wells per mouse) and then treated with either media, one or two doses of LPS (100 ng/ml) (Figure 1) for a within animal treatment design. This protocol has been utilized previously to examine endotoxin tolerance in cultured BMDM(Foster et al., 2007). We confirmed the expression patterns of gene targets involved in forming endotoxin tolerance using a custom Fluidigm Biomark HD Delta gene assay containing gene targets previously identified(Foster et al., 2007) as either tolerized (reduced expression upon the second LPS exposure) or non-tolerized (fail to suppress expression upon the second LPS exposure) (Figure 2 and Table S1). Of the 47 genes on the array, 44 genes showed significant regulation in at least one condition in response to LPS and were then compared across strains and LPS treatment conditions (Table S1).
Figure 1.
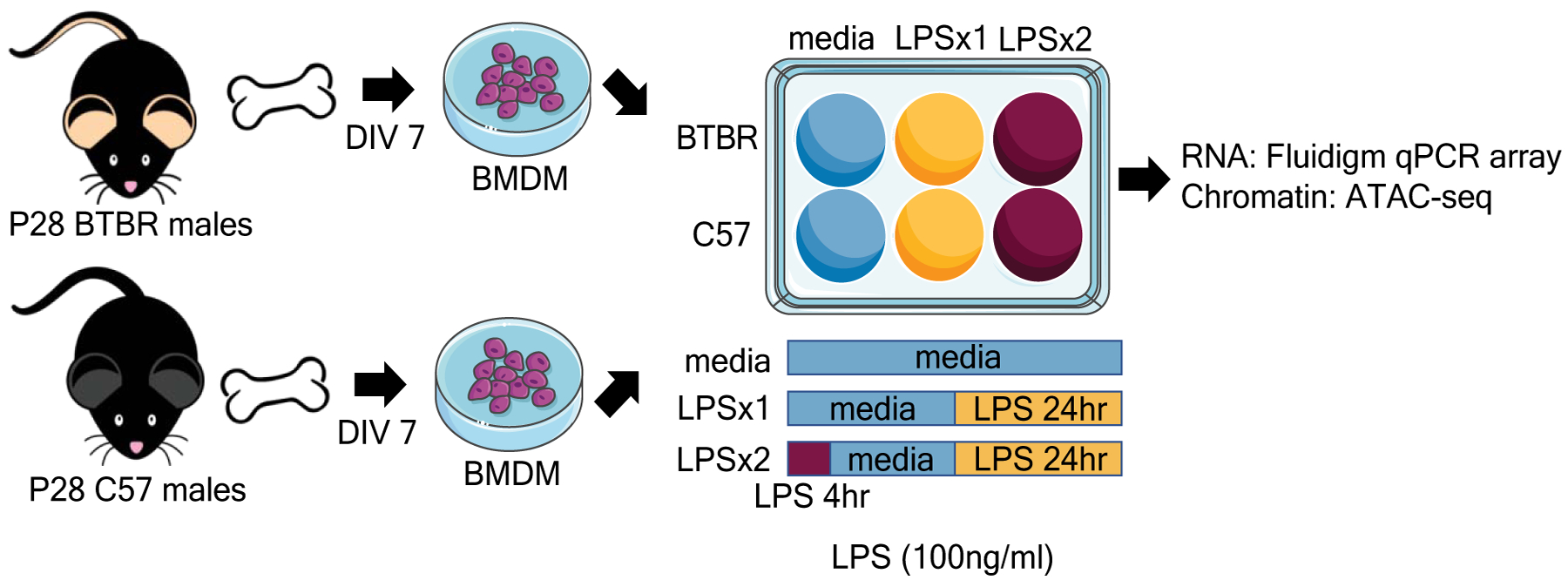
Experimental Design. Bone marrow was extracted from postnatal day 28 (P28) male BTBR or C57 mice and differentiated in culture for seven days (DIV 7) to form bone marrow derived macrophages (BMDM). BMDM from each mouse were re-seeded into three wells and treated with either media, one dose of LPS for 24hrs (LPSx1) or given a 4hrs LPS pretreatment followed by a 24hrs LPS treatment (LPSx2). All LPS treatments were at 100ng/ml of culture media. Following treatment, cells were collected for RNA expression analysis (Fluidigm array) or chromatin accessibility (ATAC-sequencing).
Figure 2.
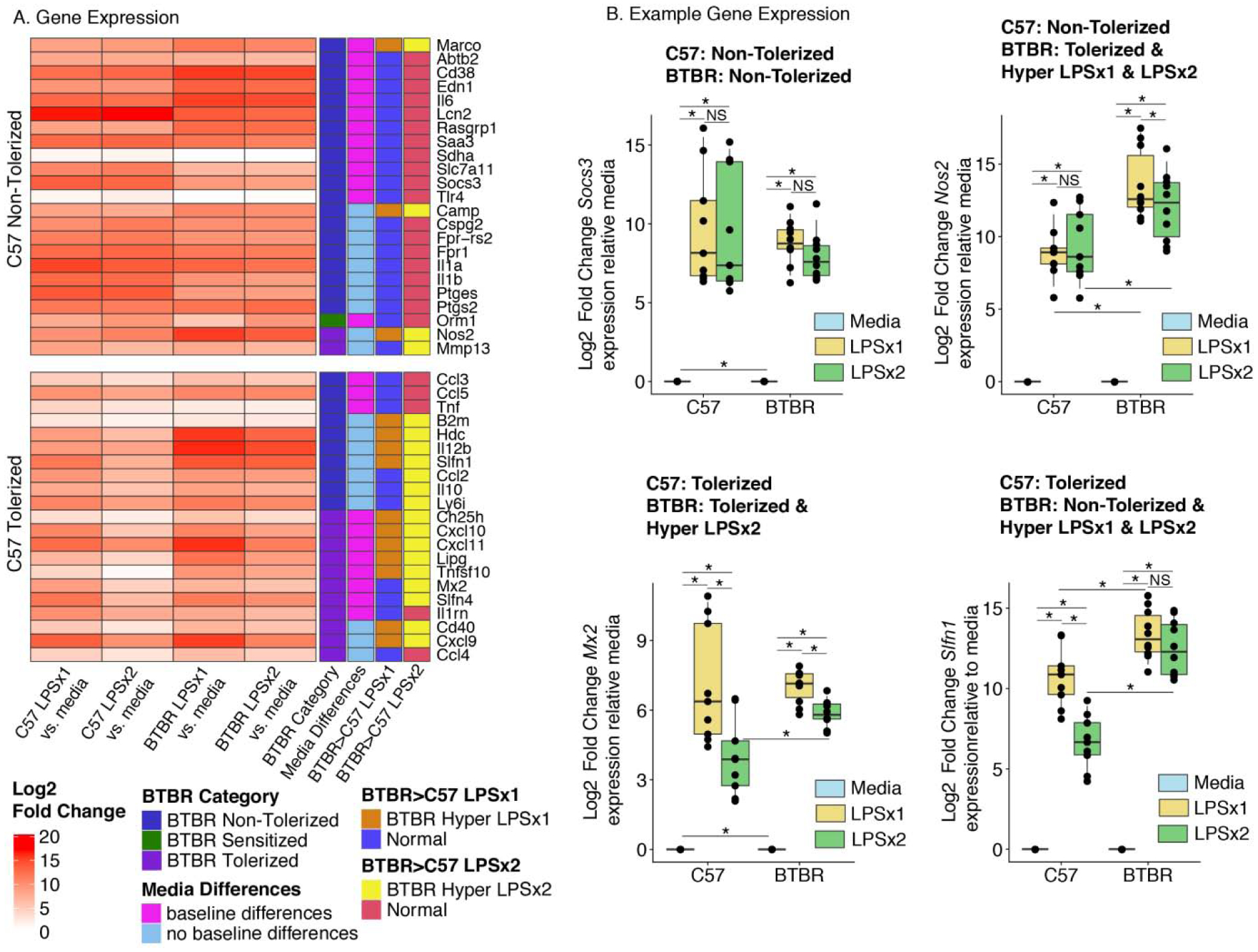
BTBR BMDM Hyper-Respond to LPS Stimulation A. Log2 Fold Change relative to media values (within each animal) for genes on the Fluidigm qPCR expression array that show either non-tolerized or tolerized patterns of expression in C57 BMDM. Gene expression patterns were classified based on Benjamini-Hochberg (BH) corrected posthoc t-test comparisons following a linear regression analysis (see methods) (Table S1) and are displayed as categories to the right of the expression heatmap for each gene. B. Boxplots of individual example genes for Log2 Fold Change gene expression. Boxes are defined by the 25th and 75th quartiles for each condition with median values shown as the horizontal line. Whiskers are defined by the 5th and 95th percentile values. Individual values are shown as overlaid points. * BH p < 0.05. Categorizations given above each plot. See Table S1 for statistics. n=9 C57 mice and n=10 BTBR mice, per animal each were re-plated into three treatment conditions (within animal design).
In C57 BMDM, 21 out of the 44 examined genes showed an expression pattern consistent with tolerization, defined as a significant (ANOVA and Benjamini-Hochberg (BH) corrected posthoc comparison p<0.05) increase from baseline (media) with a single dose of LPS (LPSx1) and an attenuated response when the cultures had been pre-treated with LPS (LPSx2) (Figure 2 and Table 1) (LPSx2<LPSx1 BH p<0.05). Of the 44 genes examined, 23 showed a non-tolerized pattern in which the pre-treatment with LPS failed to produce an attenuation of the second response (LPSx1 ~ LPSx2, non-significant BH p>0.05). In contrast, for the BTBR BMDMs, only 13/44 genes showed a tolerized pattern of expression (LPSx2<LPSx1 BH p<0.05), 30/47 showed a non-tolerized pattern (LPSx1 ~ LPSx2, non-significant BH p>0.05), and one showed sensitization (LPSx2>LPSx1 BH p<0.05). Many of the examined genes (24/44) showed significant but small magnitude strain-specific differences in media condition (C57 media > BTBR media or C57 media < BTBR media BH p<0.05) (Figure 2, Table 1), suggesting that strain alone influences baseline regulation of some immune genes similar to what has been observed previously for other strains(Verena M. Link et al., 2018).
Table 1.
Gene Expression Categories By Strain
| Category | #genes | % of total genes (44) |
|---|---|---|
| Baseline Differences | 24 | 55 |
| C57 Tolerized | 21 | 48 |
| BTBR Tolerized | 13 | 30 |
| C57 Non-Tolerized | 23 | 52 |
| BTBR Non-Tolerized | 30 | 68 |
| BTBR > C57LPSx1 | 14 | 32 |
| BTBR > C57LPSx2 | 20 | 45 |
Shown are the number of genes and the percentage of genes out of the total 44 with significant LPS regulation for each category.
When compared between strains, the majority of genes with a non-tolerized pattern in C57 also showed a non-tolerized pattern in BTBR (20/23 or 87%). For genes with a tolerized pattern in C57, only about half (11/21 or 52%) showed a similar tolerized pattern in the BTBR BMDM. Consistent with previous observations at the protein level in the BTBR mice, many of the examined genes showed a hyper-response to LPS treatment (LPSx1 BTBR>C57 BH p<0.05 and/or LPSx2 BTBR>C57 BH p<0.05) (Table 1). For the C57 tolerized genes, 76% (16/21) showed a significant hyper-response in BTBR to either one or both LPS treatments. Together, these results suggest that BTBR BMDM show increased responsivity to LPS, that is particularly true of genes that normally become tolerized to LPS in C57.
Chromatin accessibility across strains in response to LPS
To test the hypothesis that strain-specific differences in chromatin accessibility may dictate the transcriptional response profile observed in the BTBR BMDM, we performed Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq)(Buenrostro et al., 2013) on C57 and BTBR BMDMs stimulated with media, LPSx1 or LPSx2 treatments. As shown in Figure 1, BMDM cultures were prepared from male juvenile C57 mice (n=4) and BTBR mice (n=4). After differentiation, BMDM from each mouse were re-plated into three wells (within animal design) and subsequently treated with either media, LPSx1 or LPSx2 (Figure 1) and prepared for ATAC-seq. There were no differences in alignment, any of the quality control (QC) metrics or in the number of filtered or unfiltered reads between any of the sample conditions (Table S2 and Figure S1). Two treatment samples (C57 media sample 3 and C57 LPSx2 sample 3) failed final QC assessment and were excluded from subsequent analysis (Figure S2 and S3). Differential regions of accessibility for ATAC-seq were identified within nucleosome free regions (NFR, reads <147bp) for each biological replicate. Consensus peaks that were found in at least 3 out of 4 biological replicates for each condition were then used for comparisons (106,176 total) (Figure S4 and Table S2). A comparison of the two strains for each treatment condition using quartile normalized read counts demonstrated that the majority of promoter containing peaks (within 3kb of the TSS) and putative enhancer regions (>3kb from TSS) were consistent between the two strains across treatment conditions (Figure S5).
To statistically test for regions with differential accessibility between strains and LPS treatments, we compared normalized read counts within the consensus peaks. Using a general linear model with effects for strain and LPS treatment and a correction for the within subject’s correlation from the repeated measures design, we identified 62,146 significant Differentially Accessible Regions (DARs) (BH corrected p < 0.05) (Table S2 and Figure S6). Chromatin accessibility levels (normalized read counts within significant peaks) across samples clustered most strongly by LPS condition (Figure 3A). A single LPS treatment resulted in a significant increase (BH corrected p < 0.05) in chromatin accessibility in 4,764 peaks in C57 and 9,972 peaks in BTBR (LPSx1-DARs). A second LPS dose resulted in 6,499 peaks in C57 and 8,415 peaks in BTBR (LPSx2-DARs). Similarly, chromatin accessibility decreases were also observed in response to LPSx1 and LPSx2 (Figure 3B and C, Table S2) (BH corrected p < 0.05).
Figure 3.
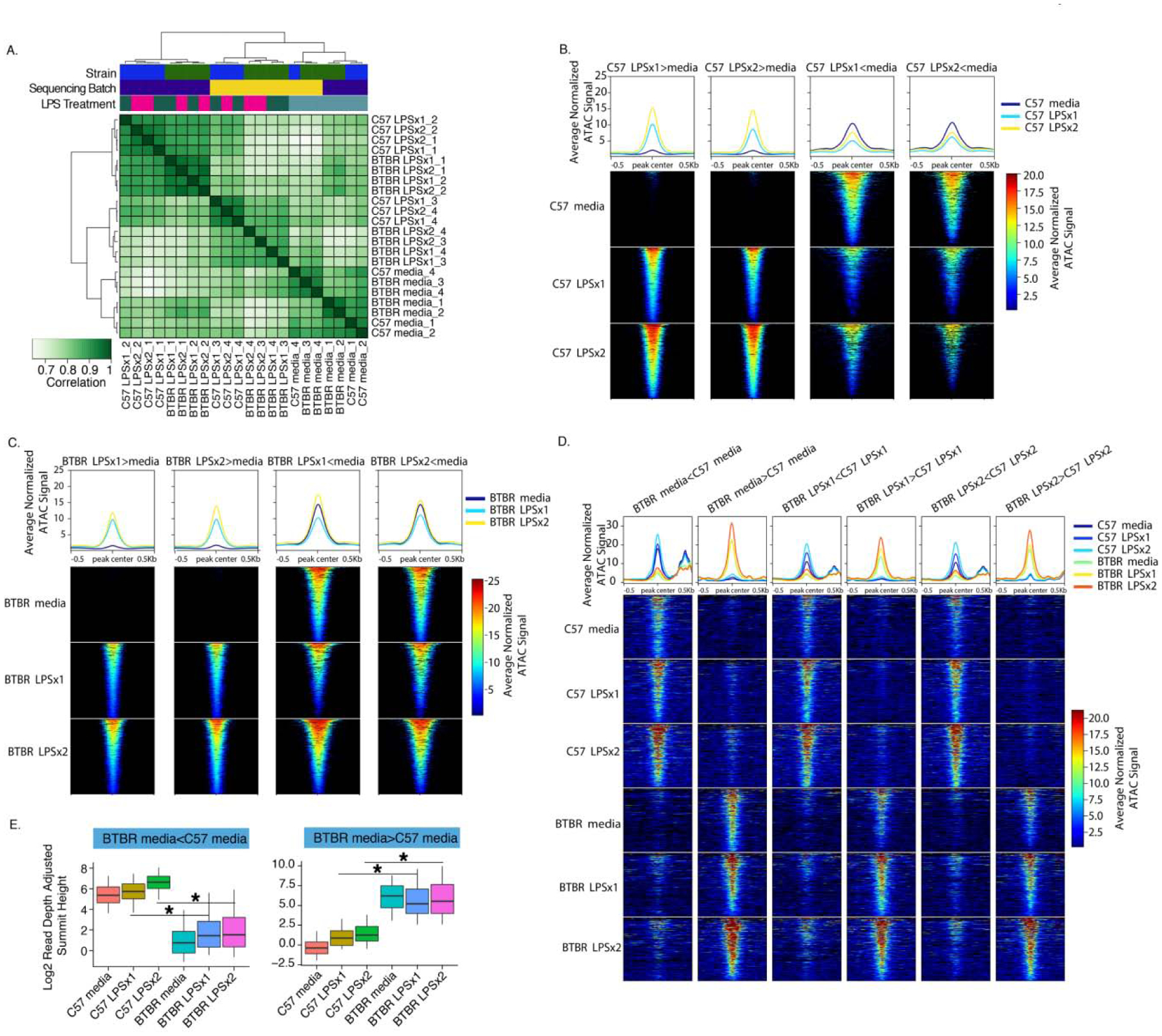
Strain-independent and Strain-specific Chromatin Responses to LPS. A. Clustered heatmap of ATAC-seq signal (maximum read pileup value normalized to relative library size) for all significant peaks identified across all conditions. B. Average ATAC-seq signal (reads normalized to sequencing depth and averaged across replicates) for DARs identified in C57 in response to LPS. Line plots show means across DARs for each comparison. Each row of the heatmap shows signal averaged across individual DARs. C. Same as B but for BTBR. D. Same as B but for regions with differential accessibility between strains for each LPS treatment condition. E. Boxplots for the average ATAC-seq signal (reads normalized to sequencing depth) for DARs identified in the baseline conditions. Boxes are defined by the 25th and 75th quartiles for each condition with median values shown as the horizontal line. Whiskers are defined by the 5th and 95th percentile values. ANOVA with Benjamini-Hochberg corrected posthoc comparisons *p-value < 0.05. All statistics and comparisons are shown in Table S5.
To compare the strains, we first examined the impact of strain on the baseline media condition, identifying 731 DARs (Figure 3D and Table S2). Peaks with lower accessibility in BTBR (367 peaks with BTBR media signal < C57 media signal) were significantly enriched within distal intergenic regions (Table S3) and showed enrichment (Fisher’s exact test BH corrected p<0.05) for previously published lists of enhancers sensitive to immune stimulation in BMDM cultures(Barish et al., 2010; Ghisletti et al., 2010; Verena M. Link et al., 2018) (Figures S7 and Table S4). Peaks with higher accessibility in BTBR at baseline (364 BTBR media signal > C57 media signal) were also significantly enriched in distal intergenic regions (Table S3) but not in immune enhancers(Barish et al., 2010; Ghisletti et al., 2010; Verena M. Link et al., 2018) (Figure S7), suggesting that a small subset of immune-responsive enhancer elements are suppressed at baseline in the BTBR BMDM cultures.
Regions with strain-specific accessibility differences at baseline were also differential upon treatment with LPS (Figure 3D). Specifically, regions with lower accessibility in the BTBR media condition were less likely to be open upon LPS treatment compared to C57 and vice versa (Figure 3D). To directly test this observation, we compared the average read-depth normalized ATAC signal within the 731 DARs with strain differences at baseline (Figure 3E and Table S5). As predicted, regions showing lower accessibility in BTBR versus C57 at baseline also showed lower accessibility in response to LPS treatment (ANOVA, C57 LPSx1>BTBR LPSx1 and C57 LPSx2>BTBR LPSx2 BH corrected posthoc p < 0.05). In comparison, regions with higher accessibility in BTBR than C57 at baseline also showed higher levels of LPS response (ANOVA, C57 LPSx1<BTBR LPSx1 and C57 LPSx2<BTBR LPSx2 BH corrected posthoc p < 0.05) (Figure 3E, Table S5). Together, these results indicate that the baseline chromatin accessibility differences between the strains can influence subsequent LPS response. Interestingly, the regions with lower accessibility in BTBR compared to C57 at baseline are also enriched for published immune regulatory enhancers(Barish et al., 2010; Ghisletti et al., 2010; Verena M. Link et al., 2018) (Figure S7), suggesting a difference in enhancer accessibility in the BTBR.
We next compared the response to LPS between the two strains. LPS-DARs identified in C57 were significantly shared with LPS-DARs identified in BTBR (Permutation overlap analysis FDR < 0.05) (Table S5). However, there were a small number of regions that showed differential accessibility in response to LPSx1 or LPSx2 between the strains (Figure 3D and Table S2). There were 633 peaks with BTBR>C57 in response to LPSx1 and 682 in response to LPSx2. There were 816 peaks with lower accessibility in BTBR (BTBR<C57) in response to LPSx1 and 826 in response to LPSx2. Regions with lower accessibility in BTBR were enriched for numerous immune regulatory enhancers(Barish et al., 2010; Ghisletti et al., 2010; Verena M. Link et al., 2018) (Figure S7), further supporting differential enhancer accessibility in response to LPS in BTBR.
BTBR genetic variants are enriched within regions of differential chromatin accessibility between strains
To test the hypothesis that genetic differences between the two strains may influence their differential chromatin responsiveness, we utilized previously published BTBR genetic variants compared to the C57 reference genome(Jones-Davis et al., 2013) (Figure 4). In BTBR, both SNPs and InDels (Insertions and Deletions) are distributed over numerous genomic features including splice sites, introns, UTRs, coding regions, promoters and intergenic regions (Figure 4A). To test the impact of BTBR genetic variants on chromatin accessibility, DARs were examined for enrichment of BTBR SNPs and InDels. Permutation testing examined the enrichment of BTBR variants within each set of DARs against the background of all consensus peaks. Strain-specific DARs at media baseline and in response to LPS were significantly enriched for both BTBR SNPs and InDels (Figure 4B and Table S6), while strain-independent LPS-DARs were not enriched for these genetic variants. These results support the hypothesis that natural genetic variation between strains may contribute to some of the altered chromatin state at baseline and in response to LPS.
Figure 4.
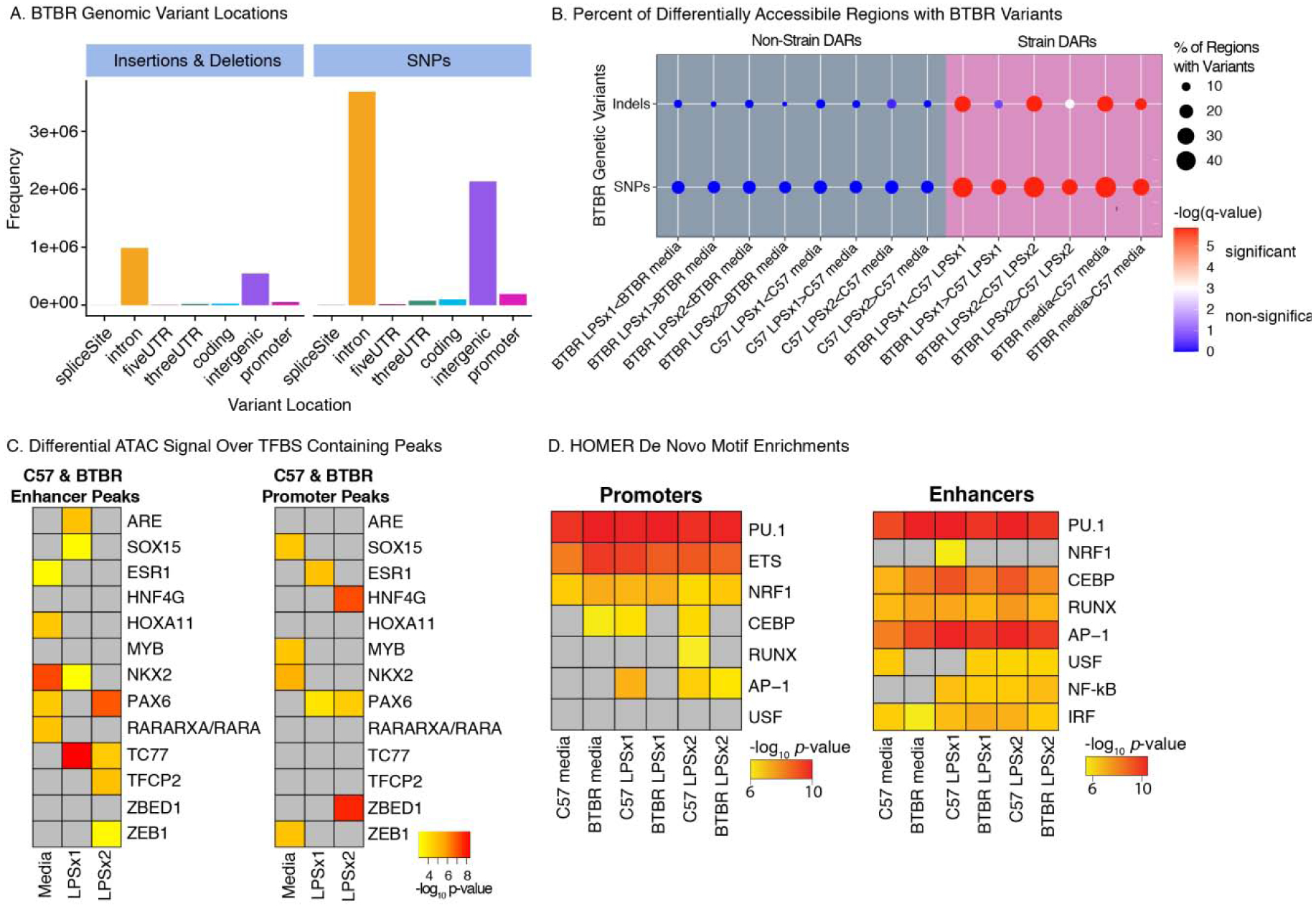
BTBR Genetic Variants Impact Chromatin Accessibility and Transcription Factor Binding Sites A. BTBR Insertions and Deletions (InDels) and SNPs from the Mouse Genomes Project (https://www.sanger.ac.uk/science/data/mouse-genomes-project) occur in numerous genomic elements including introns and intergenic regions. B. Percent of DARs containing BTBR SNPs or InDels. Dots are colored by -log(FDR corrected q-value) for permutation enrichment between BTBR variant and chromatin accessibility regions (see Table S6 for statistics). Significant enrichments (q<0.05) are colored red and non-significant (q>0.05) are colored blue. Dots are sized by the percentage of regions with variants. C. MMARGE analysis results showing chromatin accessibility between strains significantly differs for peaks containing these TF binding motifs with genetic variants. Peaks were defined for each treatment condition (consensus peaks) and subdivided into promoters (< 3 kb from TSS) and enhancers (non-promoters). D. De novo motif enrichment analysis was performed with MMARGE for ATAC-seq consensus peaks of each strain and treatment condition. For C and D, significant enrichments are shown as -log10 p- values. Non-significant enrichments are in grey.
To further test the hypothesis that genetic variation between strains may alter accessibility to specific transcription factor binding motifs, variant analysis was performed for each peak set using MMARGE (Motif Mutation Analysis for Regulatory Genomic Elements)(Verena M Link et al., 2018). MMARGE determines the significance of transcription factor (TF) binding motifs by evaluating correlation between the genetic variations interrupting the motif and the level of chromatin accessibility. MMARGE calculates the distribution of chromatin accessibility between two strains for all loci with the motif of interest in the first strain and compares it with the distribution of chromatin accessibility at all loci with the motif in the other strain. If the presence of the motif is important for chromatin accessibility, the distributions will not overlap and therefore be significant. We separated the loci into those within promoters (< 3 kb from TSS) compared to putative enhancers (non-promoters). Out of the 287 TF binding motifs tested, genetic variation leading to differences in TF binding motifs between the strains were significant for 13 motifs (Figure 4C), suggesting that genetic variation between the strains may contribute to differential accessibility at these loci. For example, strain dependent chromatin accessibility was impacted by genetic variants at LPSx1 and LPSx2 responsive enhancers containing the motif of TCF7, a repressive transcription factor that plays important roles in the regulation of Runx splicing and has been implied in macrophage functions in various other tissues (Link et al., 2018a). In a complementary approach, we applied MMARGE’s de novo motif analysis to the same set of consensus peaks divided into promoters and enhancers (Figure 4D). In enhancers and promoters across treatment conditions and strains, we found significant motif enrichments for the canonical macrophage transcription factors PU.1, CEBP, AP-1, but did not find evidence for genetic stain differences differentially impacting their binding motifs across conditions.
Differential chromatin accessibility mirrors non-tolerized differential gene expression in response to LPS
To test the hypothesis that strain-specific differences in chromatin accessibility impact expression of tolerized and non-tolerized genes in response to LPS treatment, we further divided LPS-DARs into regions that significantly changed in response relative to the media condition, only to the first LPS treatment (LPSx1only), only the second treatment (LPSx2only) or showed similar responses to both treatments (LPSx1&2) (Table S6). We compared DARs within promoters (< 3 kb from TSS) to DEGs from the array (Figure 5 and Table S6). We hypothesized that genes that respond to LPS with non-tolerized patterns of expression would open chromatin at their promoters in response to LPS and maintain that open state across multiple LPS treatments (media < LPSx1&2). As predicted, DEGs showing a non-tolerized pattern of expression were significantly enriched for genes with promoter DARs with increased accessibility upon LPSx1 and LPSx2 relative to media for both C57 and BTBR (Figure 5A). For example, the promoter region of Socs3 is open and expression is maintained across LPS treatment conditions (Figure 5B) in both strains. Non-tolerized genes for both strains were also significantly enriched for regions with increased accessibility only in response to the second LPS dose (media < LPSx2only), indicating changes in chromatin structure may occur around promoters of non-tolerized genes that facilitates their continued expression. Non-tolerized genes from both strains were also enriched for regions of accessibility that were higher in C57 compared to BTBR in response to LPS, suggesting less dynamic chromatin responses to LPS in the BTBR. Overall, promoters of non-tolerized genes in both strains appear to maintain a permissive transcriptional state for on-going gene expression in response to repeated LPS.
Figure 5.
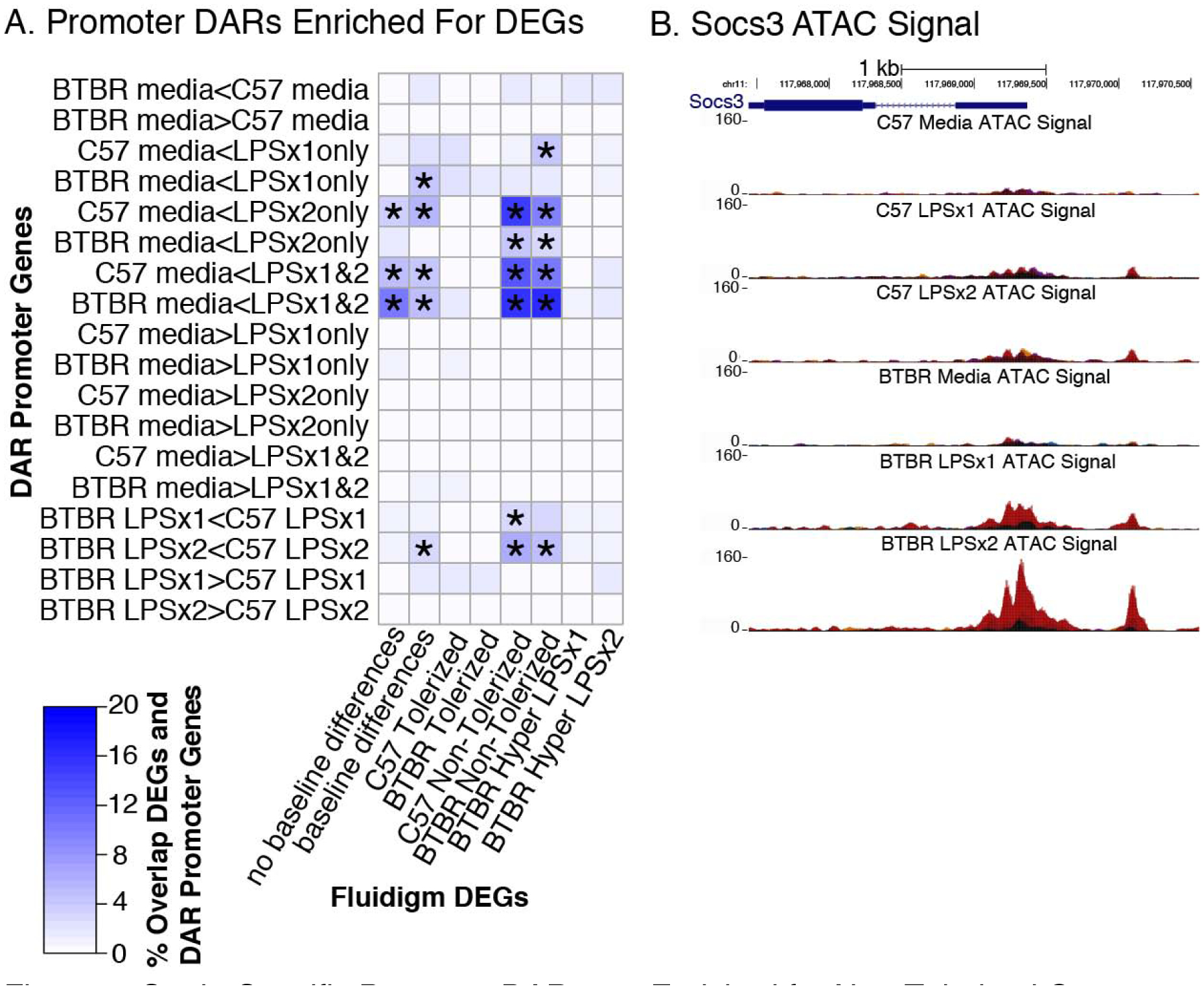
Strain-Specific Promoter DARs are Enriched for Non-Tolerized Genes A. Differentially expressed genes (DEGs) from Figure 2 were examined for enrichment of DARs within promoter regions of genes. The heatmap is colored by the percent of overlap between each row/column comparison. Significant enrichments (Fisher’s exact test) are denoted with * for all FDR corrected p-value < 0.05. B. Normalized ATAC-seq signal over the promoter region of the Socs3 gene. Socs3 shows a non-tolerized pattern of gene expression (Figure 2B) and shows increased accessibility at its promoter across treatments. Replicates are shown in different colors for each UCSC genome browser track.
Similarly, we predicted that genes that tolerize expression in response to repeated LPS treatment would be enriched for peaks only present in response to LPSx1 (media < LPSx1only). However, we found no significant enrichments between tolerized genes and chromatin accessibility regions in response to any LPS condition in either strain, suggesting that regulation of tolerization of these genes may occur at an epigenomic level other than chromatin accessibility. We also identified positive enrichments between genes with differences at baseline between the strains and regions that respond to LPS, supporting the previous conclusions that a subset of LPS responsive genes may be impacted by changes in chromatin structure at baseline. However, the same regions were also enriched for genes without media differences in expression, indicating that the baseline strain impacts are likely gene specific and only apply to a subset of LPS responsive genes. We did not find any significant enrichments between the identified DEGs from the array and BTBR genetic variants found within either the gene coding region or within promoters (Table S7). This suggests that the relationship between chromatin accessibility at the promoter and gene expression is independent of the direct impact of promoter genetic variants and instead may be due to altered long-range interactions between distal genomic regulatory elements and promoters.
Discussion
This study relates naturally occurring genetic variation to changes in chromatin accessibility and gene expression in innate immune cells from a mouse model of ASD. Similar to previous findings in human monocytes(Enstrom et al., 2010), BTBR BMDM show a hyper-response to immune stimulation. At the level of gene expression, approximately half of the genes examined that tolerize expression to repeated LPS in C57 failed to tolerize in BTBR. Instead in the BTBR many of these genes showed larger and sustained expression levels in response to repeated LPS. To examine potential changes in chromatin regulation as underlying immune differences, we identified DARs responsive to both LPS and strain effects. Overall chromatin dynamics in response to LPS were similar between the strains, indicating global chromatin regulation mechanisms were not responsible for the observed immune differences. We did identify regions with altered chromatin accessibility in response to LPS and between strains, largely within distal enhancers. Furthermore, regions with differential accessibility between strains were enriched for BTBR genetic variants, demonstrating an association between genetic variation and chromatin regulation of inflammatory response. We further explored this relationship in terms of altered transcription factor interactions and identified several candidates where strain dependent chromatin accessibility was impacted by genetic variants. Chromatin accessibility over promoter regions was related to gene expression of non-tolerized genes but not tolerized genes. Together, this is the first work to directly examine how epigenetic differences regulate gene expression in BTBR immune cells and highlight the interaction between natural genetic variation, chromatin, and immune responsiveness.
Understanding how genetic variants impact chromatin regulation, gene expression and cellular function is key for understanding how genetic risk alleles contribute to human disease. Genome wide association studies (GWAS) have identified human genetic variants associated with complex traits and increased risk for human disease. The majority of these SNPs occur outside of gene coding regions and within regions of accessible chromatin(Maurano et al., 2012) that can be cell-type specific enhancers(Nott et al., 2019). In a comprehensive comparison of BMDM from 5 mouse strains (not including BTBR), strain-specific gene expression resulted primarily from genetic variation in distal regulatory elements(Verena M. Link et al., 2018). Under baseline conditions there was a large variation in gene expression with 10% of expressed transcripts varying by at least 4-fold between strains. In response to treatment with a Toll Like Receptor 4 agonist Kdo2- lipid A (KLA), distinctions between the strains emerged but the general response to KLA was conserved between strains, similar to our findings with LPS treatment. The majority of the strain differences in gene expression were not explained by genetic differences in promoter sequences but instead by cis-variation in distal putative enhancers. Histone marks and long-range chromatin interactions revealed strain-dependent impacts of cis-regulatory domains on transcription factor binding and gene expression(Verena M. Link et al., 2018). Our findings of BTBR versus C57 strain-specific differences in BMDM responsiveness are consistent with these findings. Strain and LPS-DARs were significantly enriched for distal intergenic regions (but not promoters) and for previously identified immune-sensitive enhancers in BMDM. BTBR genetic variants were not enriched within non-strain DARs or the coding region or promoters of DEGs, supporting a role for distal regulation. Recent work has also identified long-range enhancer-promoter interactions as mediating the impact of human risk allele SNPs on gene expression regulation(Nott et al., 2019). Using similar long-range chromatin interaction techniques (Fang et al., 2016), future work could directly examine how BTBR and other strain genetic variants within enhancers alter long-range promoter interactions.
These findings also suggest that naturally occurring genetic variation in humans may influence mechanisms for regulating innate immune responses and specifically responses to repeated pathogen exposure. Previous work in human monocytes has identified numerous genetic variants that contribute to regulation in response to immune stimulation(Fairfax et al., 2014) as well as in mediating the immune response during sepsis(Davenport et al., 2016). Understanding how individual genetic variation may impact chromatin regulation and gene expression in immune cells is critical for developing screening strategies for predicting individual patient outcomes and new treatment development(Davenport et al., 2016). Future work is needed to investigate natural human genetic variation in endotoxin tolerance responsiveness in human macrophages from patient populations such as ASD.
The relationship between chromatin accessibility and gene expression is complex, as chromatin state does not always directly predict expression(Verena M. Link et al., 2018). Prior work in BMDM in response to endotoxin tolerance identified loss in chromatin accessibility over the promoters of several exemplar tolerized genes(Foster et al., 2007). While this is consistent with our findings at some promoters, we did not observe enrichment for tolerized genes and changes in promoter chromatin accessibility. This suggests that tolerized genes are suppressed by additional mechanisms. Previous work has suggested critical roles for histone modifications(Foster et al., 2007; Novakovic et al., 2016; Saeed et al., 2014) as well as differential transcription factor binding in response to immune stimulation(Verena M. Link et al., 2018). The majority of enriched transcription factor binding motifs within DARs were similar between strains, however we did identify several candidate transcription factors potentially impacted by changes in chromatin accessibility, both within promoters and putative enhancers. Additional work will be needed to directly test the impact of BTBR genetic variants on epigenomic mechanisms in regulating gene expression required for the formation of endotoxin tolerance in innate immune cells.
Genetic variants may only be one factor contributing to the chromatin accessibility differences observed in the BTBR in response to repeated LPS treatment. Previous work in T cell populations from BTBR mice has demonstrated altered protein levels of several important immune-mediating transcription factors. In CD4+ T-cells, BTBR showed increased levels of RORγt, T-bet, and GATA-3(Bakheet et al., 2017), suggesting altered transcription factor signaling. Similarly, in BTBR BMDM disruption of upstream signaling cascades may alter transcription factor function and recruitment, potentially impacting chromatin remodeling complexes and contributing to altered chromatin accessibility and immune gene expression. The alterations in BTBR immune function have been suggested to contribute to the development and manifestation of ASD relevant behaviors in BTBR(Heo et al., 2011; Onore et al., 2013). Several lines of evidence point to ongoing and prominent activation of innate myeloid immune cells in ASD(Ashwood et al., 2011; Enstrom et al., 2010; Hu et al., 2018; Inga Jácome et al., 2016; Molloy et al., 2006). Furthermore, we and others have shown increased activation of circulating myeloid cells in the periphery following ex vivo stimulation, including changes in gene expression and the release of pro-inflammatory cytokines(Ashwood et al., 2011; Enstrom et al., 2010; Mendizabal et al., 2016). The degree of myeloid cell activation was associated with more impaired behaviors and increased burden of co-morbidities in children with ASD(Ashwood et al., 2011). Our finding suggests that BTBR is a relevant model for studying the complex genetic architecture of immune alterations in ASD.
This work focused on immune gene regulatory mechanisms in bone marrow derived macrophages. BMDM cultures are a commonly used system for examining molecular mechanisms of immune function but may not fully capture all of the in vivo aspects of immune cell regulation. For example, the epigenome of tissue resident macrophages adapt to their tissue of residence providing a unique chromatin regulatory state dictating gene expression(Lavin et al., 2014). This may be extremely important in future investigation of tissue resident macrophages such as brain microglia that are exquisitely tuned to their local environments(Gosselin et al., 2017). Future work will be needed to characterize immune tolerance and chromatin regulation in additional tissue resident immune cell populations. Understanding the molecular mechanisms underlying the altered immune cell function in ASD models and human immune cell populations will lead to novel insights into immune dysfunction in ASD and identification of new immune-based therapeutic targets.
Methods
BMDM Cultures and LPS Treatment
All experiments were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. All procedures were approved by the Institutional Animal Care and Use Committee of the University of California, Davis. Bone marrow cells were isolated from postnatal day 28 BTBR T+ Itpr3tf/J (Jax 002282) and C57BL6/J (Jax 000664) male mice. Isolated cells from each mouse were cultured for seven days to generate bone marrow derived macrophages (BMDM) in DMEM/F12 media supplemented with 10% fetal bovine serum and 1% Penicillin-Streptomycin (base media). BMDM from each mouse were then reseeded at a density of 5×105 cells per well of a 6 well plate and incubated overnight. Cultures from each mouse were then treated with either media, one dose LPS (LPSx1) or two doses of LPS (LPSx2). BMDM were treated with media (media or LPSx1) or 100ng LPS/ml for four hours (LPSx2). Cultures were then washed twice with HBSS and incubated overnight in base media. Twenty hours later cultures were treated with media (media) or 100ng LPS/ml for 24 hours (LPSx1 and LPSx2). Following treatment cells were collected for RNA or ATAC-seq.
Fluidigm Biomark HD Delta Gene Expression Assay
Gene expression for selected tolerized and non-tolerized gene targets identified from (Foster et al., 2007) were examined in two independent experiments of LPS treatment of BMDM. Gene expression analysis was performed using the Fluidigm Biomark HD Delta gene assay on a 48.48 Integrated Fluidic Circuit. Delta gene assay design was performed by Fluidigm for equivalent amplification between the two genotypes and primer sequences are listed in Supplemental Table S2. An amount of 2.5ng of total RNA was used as input for cDNA synthesis (Fluidigm, 100–6472 B1) followed by 10 cycles of pre-amplification (Fluidigm, 100–5875 C1) using a mix of all forward and reverse primers. Samples were then diluted five-fold and analyzed on a 48.48 IFC and Biomark HD machine in duplicate or triplicate (Fluidigm, 100–9791 B1). Cycle threshold (Ct) values below detection were replaced with the max Ct value of 30. Ct values were averaged across technical replicates and then normalized to Hprt expression using the delta Ct method (Ct gene – Ct Hprt). For each gene, the delta Ct values were then used for group comparisons by ANOVA with strain, LPS treatment and the interaction with experimental batch considered as covariates in the analysis. Pairwise comparisons between conditions were then made using t-tests with posthoc Benjamini-Hochberg corrections for multiple comparisons. For display purposes, the delta delta Ct values for each gene were then calculated relative to the Ct value for the media treated control for each animal and relative fold change expression levels were calculated by 2^(-delta delta Ct).
Gene expression levels were defined as non-tolerized if expression increased significantly with one dose of LPS compared to media (BH corrected posthoc media < LPSx1 p-value < 0.05) and there was not a significant difference between one and two doses of LPS (BH corrected posthoc LPSx1 vs LPSx2 p-value > 0.05). Genes were defined as tolerized if expression increased significantly with one dose of LPS compared to media (BH corrected posthoc media < LPSx1 p-value < 0.05) and expression was lower with two doses of LPS compared to one dose of LPS (LPSx1 > LPSx2 BH corrected posthoc p-value < 0.05). Genes were defined as sensitized if expression increased significantly with one dose of LPS compared to media (BH corrected posthoc media < LPSx1 p-value < 0.05) and expression was further increased with two doses of LPS compared to one dose of LPS (LPSx1 < LPSx2 BH corrected posthoc p-value < 0.05). Genes with a hyper induction in BTBR compared to C57 were categorized for Hyper LPSx1 (BH corrected posthoc C57 LPSx1 < BTBR LPSx1 p-value < 0.05) and Hyper LPSx2 (BH corrected posthoc C57 LPSx2 < BTBR LPSx2 p-value < 0.05). Media Baseline differences were defined by a significant BH corrected posthoc comparison between C57 and BTBR media conditions with a p-value < 0.05.
ATAC-seq Library Preparation
ATAC-seq was performed as described in(Buenrostro et al., 2015). Briefly, cultures of 50,000 BMDM were washed once in ice cold 1xPBS and then resuspended gently in 100μl ATAC-seq lysis buffer (1 mM TrisCl, pH 7.4, 1 mM NaCl, 0.3 mM MgCl2, and 0.01% Triton-X 100) to release nuclei. Nuclei were then pelleted (1,000xg, 10min, 4°C) and transposed in 2.5μl Tn5 (Nextera Tn5 transposase, Illumina), 12.5μl TD reaction buffer and 10.5μl water for 40min at 37°C with rotation. Following transposition, DNA was purified (MinElute PCR Purification Kit, Qiagen) and stored at −80°C. Samples were then thawed on ice and unique barcodes(Buenrostro et al., 2015) were added to each sample using 5 cycles of PCR(Buenrostro et al., 2015). A side PCR reaction was taken and used as input for qPCR for an additional 20 cycles to determine the optimal number of additional PCR cycles for each library (the cycle number that corresponds to one-third of the maximum fluorescent intensity). Each sample was then amplified by the determined number of PCR cycles and then purified (MinElute PCR Purification Kit, Qiagen). Final libraries were then size selected for fragments between 100–1000bp using AMPure XP beds (Beckman Coulter) and quality was checked by DNA High Sensitivity bioanalyzer. Libraries were pooled (6 per pool) and sequenced across four lanes of the HiSeq2500 to generate 50bp paired end reads.
BTBR genome analysis
BTBR SNPs and Indels were downloaded from the Mouse Genomes Project (https://www.sanger.ac.uk/science/data/mouse-genomes-project) and transferred from mm9 to mm10 using CrossMap (version 0.2.8)(Zhao et al., 2014). The program MMARGE (Motif Mutation Analysis for Genomic Elements, version 1)(Verena M Link et al., 2018) was used to create a modified version of the mm10 genome that incorporated BTBR SNPs and Indels (MMARGE.pl prepare_files). The BTBR genome was used to generate bowtie2 index files for subsequent alignments of BTBR sequencing data. SNPs and InDels were examined for variant location using the locateVariants function in the R package VariantAnnotation(Obenchain et al., 2014) and the mouse organism database (org.Mm.eg.db).
ATAC-seq Alignment, Quality Control, and peak calling
All fastq files were trimmed using trimmomatic (version 0.38) to remove paired end adapters, minimum MAPQ score of 30, leading and trailing 3bp, and to a minimum length of 15bp. Trimmed fastq files were then aligned to either the mm10 genome or a modified mm10 that incorporated BTBR genetic changes using bowtie2(Langmead and Salzberg, 2012) with MMARGE(Verena M Link et al., 2018) with default settings for paired end data. BTBR aligned data were then shifted back to mm10 standard genome coordinates using MMARGE.pl shift(Verena M Link et al., 2018). For all samples aligned reads were then filtered using samtools (version 1.8) to remove PCR duplicates, unpaired reads, unmapped reads, and secondary alignments. To assess transposition efficiency and the proportion of nucleosome free reads compared to mono and di- nucleosomes, histograms of the distribution of insert sizes was created using picard-tools (version 2.18.4) CollectInsertSizeMetrics. The filtered reads were then shifted to account for the Tn5 insertions site (+4bp to positive strand and −5bp from negative strand) using the perl script ATAC_BAM_shifter_gappedAlign.pl (https://github.com/TheJacksonLaboratory/ATAC-seq/blob/master/auyar/). Shifted reads were filtered for nucleosome free regions (read length < 147bp) and mitochondrial reads were removed using samtools. Biological replicates for each strain and treatment condition were compared for consistency using deeptools(Ramírez et al., 2016). For each sample, bamCoverage was used to make bigwig files with read coverage normalized to 1x sequencing depth using an effective genome size for mm10 of 2,652,783,500 with chrM, chrX and chrY excluded from normalization. The bin size was set to 10bp and the mm10 blacklist regions were excluded. Biological replicates were then compared within each condition using multiBigwigSummary followed by plotCorrelation with --corMethod spearman –skipZeros.
Filtered reads were then used for peak calling with HOMER(Heinz et al., 2010) (- style factor -center -F 0 -L 0 -C 0 -fdr 0.01 -minDist 200 -size 200). The resulting peak files were then filtered to remove mm10 blacklist regions using bedtools. Biological replicates were compared using a Spearman’s correlation with Deeptools(Ramírez et al., 2016). Two C57 samples failed initial QC and were removed from subsequent analysis (see results). Alignments, read counts, filtering and peak counts were examined for differences between experimental conditions using a repeated measures ANOVA with factors for LPS treatment, strain and the interaction with a repeated measures factor for subject.
ATAC-seq Differential Accessibility Analysis
Differential accessibility regions were identified from HOMER peaks called within NFRs using the R packages DiffBind(Stark and Brown, 2011), edgeR(Robinson et al., 2010), limma and voom(Ritchie et al., 2015). Individual peak files for each sample were combined into a dba object and the configuration was set for paired end sequencing. Consensus peaks between biological replicates for each condition were defined using dba.peakset as peaks occurring within 3 out of 4 biological replicates with peak overlap of at least 1bp. The fraction of reads in peaks (FRIP) was then calculated for each sample for all consensus peaks as an additional QC measure. Raw reads were then counted within all consensus peaks for each sample with summits set to True, and scaling to control samples set to false using Diffbind dba.count. The resulting counts were normalized for the total sequencing library size (after filtering described above) using edgeR with calcNormFactors and method set to Relative Log Expression (RLE), where the median library is calculated from the geometric mean of all columns and the median ratio of each sample to the median library is taken as the scale factor(Robinson et al., 2010). The RLE normalized counts were then further normalized for common, trended and tagwise dispersion using a design matrix with factors for strain and LPS treatment and a zero intercept. To account for the repeated measures aspect of the experimental design, the dgList object was then passed to voom and sample weights were calculated using the duplicateCorrelation function with block set to sample replicate. The final call to lmFit included the blocking factor and the correlation, resulting in a model that accounts for the correlation within subjects. Individual planned comparisons were then conducted using makeContrasts, contrast.fit, eBayes, and topTable. All significant regions (Benjamini-Hochberg corrected p < 0.05) were then further separated by the direction of accessibility change between conditions.
To examine the overlap between region conditions, region files were compared using the resampleRegions function for the number of overlaps in the R package regioneR(Gel et al., 2016). Region lists were tested for increased overlap compared to overlap expected by chance using 1000 permutation tests of random resampling from the universe of all consensus peaks identified in the experiment. The resulting permuted p- values were then FDR corrected. The same approach was used to overlap regions with BTBR SNPs and InDels. Pairwise overlap heatmaps were made using the R package Intervene(Khan and Mathelier, 2017).
Promoter DAR-DEG Enrichment Analysis
Peak lists for regions with increased accessibility upon LPS stimulation were further parsed into regions that responded only to one LPS dose (LPSx1only), only two LPS doses (LPSx2only) or in response to both LPS doses (LPSx1&x2) using the R package valr(A. Riemondy et al., 2017). Regions were annotated using the R package ChIPseeker with the overlap function set to “all”, which sets the gene overlap with the peak to be reported as nearest gene independent of whether it is a TSS region or not. DARs in promoters (TSS to 3kb upstream) were assigned to the corresponding ensemble gene ID. The DAR-promoter genes were then compared to DEGs using a one-tailed Fisher’s exact test with an FDR correction to p<0.05, against a background set of all consensus peaks found within promoters.
Visualization of Average ATAC-seq Signal Over Peak Regions
Visualization of average ATAC-seq signal was done using Deeptools(Ramírez et al., 2016). Average bigwig files for each condition were made using (http://wresch.github.io/2014/01/31/merge-bigwig-files.html) from the read depth normalized bigwig files described above. The resulting bigwig files were then compared to specified peak regions using Deeptools(Ramírez et al., 2016) computeMatrix reference-point --referencePoint center -b 500 -a 500 –skipZeros followed by plotHeatmap --refPointLabel “peak center” --averageTypeSummaryPlot mean -- perGroup --colorMap RdBu --yMax 40.
Region Enrichments
Enrichment of ATAC-seq regions within previously published regulatory regions was performed using a the R package LOLA(Sheffield and Bock, 2015) with a one-tailed Fisher’s exact test with FDR correction to p<0.05 across all comparisons. Region lists include regions identified in ENCODE ChIP-seq for mouse BMDM (www.encodeproject.org), and ChIP-seq target regions from several transcription factors in mouse BMDM with and without immune stimulation(Barish et al., 2010; Ghisletti et al., 2010; Verena M. Link et al., 2018).
MMARGE’s mutation analysis
Consensus bigwig file from each condition were merged between the different strains with HOMER’s mergePeaks and split into promoter and enhancer regions based on their distance to the closest TSS (peaks less than 3kb from the nearest TSS were defined as promoters, the rest as enhancers). These files were annotated with raw read counts using HOMER and then quantile normalized using R’s normalization. MMARGE’s mutation_analysis(Verena M Link et al., 2018) was performed on each peak file using natural genetic variation from C57 and BTBR, as well as -no_correction. The results of this analysis were summarized with MMARGE’s summary_heatmap and a significance threshold of 0.05.
Modified Homer Motif Enrichment Analysis
To analyze enriched de novo motifs, MMARGE’s modified de novo motif analysis from HOMER was used. The analysis was performed on each peak file from each treatment in each strain for promoters and enhancers. The original sequence per peak was extracted and HOMER’s de novo motif finding algorithm was used for these sequences.
Scatterplots
To visualize the ATAC-seq data in scatter plots, peak files generated for MMARGE’s mutation analysis were used and plotted in R as log2 values.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (R01ES021707, R01NS081913 to J.M.L., RO1HD090214, RO1 MH118209, R21MH116383 to P.A.), Johnson Foundation. Brain & Behavior Research Foundation (NARSAD Young Investigator Award to A.V.C.), National Institutes of Mental Health (1K01MH116389-01A1 K01 Mentored Research Scientist Development award to A.V.C). This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley (NIH S10 OD018174 Instrumentation Grant) and the University of California, Davis Intellectual and Developmental Disabilities Research Center (IDDRC) (NIH U54 HD079125)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
The author(s) declare no competing interests.
Data Availability
Raw and processed sequencing data is available at NCBI GEO accession number GSE131446. All analysis code is available on the first author’s github (https://github.com/aciernia/BTBR-BMDM-Endotoxin-Tolerance) or in public repositories.
References
- A. Riemondy K, M. Sheridan R, Gillen A, Yu Y, G. Bennett C, R. Hesselberth J., 2017. valr: Reproducible genome interval analysis in R. F1000Research 6, 1025 10.12688/f1000research.11997.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van de Water J, 2011. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun 25, 40–45. 10.1016/j.bbi.2010.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR, 2015. A global reference for human genetic variation. Nature 526, 68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakheet SA, Alzahrani MZ, Ansari MA, Nadeem A, Zoheir KMA, Attia SM, Al-Ayadhi LY, Ahmad SF, 2017. Resveratrol Ameliorates Dysregulation of Th1, Th2, Th17, and T Regulatory Cell-Related Transcription Factor Signaling in a BTBR T + tf/J Mouse Model of Autism. Mol. Neurobiol 10.1007/s12035-016-0066-1 [DOI] [PubMed] [Google Scholar]
- Barish GD, Yu RT, Karunasiri M, Ocampo CB, Dixon J, Benner C, Dent AL, Tangirala RK, Evans RM, 2010. Bcl-6 and NF-κB cistromes mediate opposing regulation of the innate immune response. Genes Dev. 24, 2760–2765. 10.1101/gad.1998010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ, 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, Greenleaf WJ, 2015. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 1–9. 10.1002/0471142727.mb2129s109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport EE, Burnham KL, Radhakrishnan J, Humburg P, Hutton P, Mills TC, Rautanen A, Gordon AC, Garrard C, Hill AVS, Hinds CJ, Knight JC, 2016. Genomic landscape of the individual host response and outcomes in sepsis: A prospective cohort study. Lancet Respir. Med 4, 259–271. 10.1016/S2213-2600(16)00046-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enstrom AM, Onore CE, Van de Water JA, Ashwood P, 2010. Differential monocyte responses to TLR ligands in children with autism spectrum disorders. Brain. Behav. Immun 24, 64–71. 10.1016/j.bbi.2009.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairfax BP, Humburg P, Makino S, Naranbhai V, Wong D, Lau E, Jostins L, Plant K, Andrews R, McGee C, Knight JC, 2014. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science (80-. ). 343, 1–30. 10.1126/science.1246949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Yu M, Li G, Chee S, Liu T, Schmitt AD, Ren B, 2016. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res. 10.1038/cr.2016.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster SL, Hargreaves DC, Medzhitov R, 2007. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447, 972–978. 10.1038/nature05836 [DOI] [PubMed] [Google Scholar]
- Gandal MJ, Haney J, Parikshak N, Leppa V, Horvath S, Geschwind DH, 2018a. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science (80-. ). 693, 693–697. 10.1101/040022 [DOI] [Google Scholar]
- Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, Won H, van Bakel H, Varghese M, Wang Y, Shieh AW, Haney J, Parhami S, Belmont J, Kim M, Moran Losada P, Khan Z, Mleczko J, Xia Y, Dai R, Wang D, Yang YT, Xu M, Fish K, Hof PR, Warrell J, Fitzgerald D, White K, Jaffe AE, PsychENCODE Consortium P, Peters MA, Gerstein M, Liu C, Iakoucheva LM, Pinto D, Geschwind DH, 2018b. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, eaat8127 10.1126/science.aat8127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gel B, Díez-Villanueva A, Serra E, Buschbeck M, Peinado MA, Malinverni R, 2016. RegioneR: An R/Bioconductor package for the association analysis of genomic regions based on permutation tests. Bioinformatics 32, 289–291. 10.1093/bioinformatics/btv562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisletti S, Barozzi I, Mietton F, Polletti S, De Santa F, Venturini E, Gregory L, Lonie L, Chew A, Wei C-L, Ragoussis J, Natoli G, 2010. Identification and Characterization of Enhancers Controlling the Inflammatory Gene Expression Program in Macrophages. Immunity 32, 317–328. 10.1016/j.immuni.2010.02.008 [DOI] [PubMed] [Google Scholar]
- Goines PE, Ashwood P, 2013. Cytokine dysregulation in autism spectrum disorders (ASD): possible role of the environment. Neurotoxicol Teratol 36, 67–81. 10.1016/j.ntt.2012.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, Jaeger BN, O’Connor C, Fitzpatrick C, Pasillas MP, Pena M, Adair A, Gonda DG, Levy ML, Ransohoff RM, Gage FH, Glass CK, 2017. An environment-dependent transcriptional network specifies human microglia identity. Science (80-. ). June 23, 6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J, West AB, Arking DE, 2014. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat. Commun 5, 5748 10.1038/ncomms6748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK, al., et, 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–89. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo Y, Zhang Y, Gao D, Miller VM, Lawrence DA, 2011. Aberrant immune responses in a mouse with behavioral disorders. PLoS One 6, e20912 10.1371/journal.pone.0020912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu CC, Xu X, Xiong GL, Xu Q, Zhou BR, Li CY, Qin Q, Liu CX, Li HP, Sun YJ, Yu X, 2018. Alterations in plasma cytokine levels in chinese children with autism spectrum disorder. Autism Res. 11, 989–999. 10.1002/aur.1940 [DOI] [PubMed] [Google Scholar]
- Hughes HK, Mills Ko E, Rose D, Ashwood P, 2018. Immune Dysfunction and Autoimmunity as Pathological Mechanisms in Autism Spectrum Disorders. Front. Cell. Neurosci 10.3389/fncel.2018.00405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inga Jácome M, Morales Chacòn L, Vera Cuesta H, Maragoto Rizo C, Whilby Santiesteban M, Ramos Hernandez L, Noris García E, González Fraguela M, Fernandez Verdecia C, Vegas Hurtado Y, Siniscalco D, Gonçalves C, Robinson-Agramonte M, 2016. Peripheral Inflammatory Markers Contributing to Comorbidities in Autism. Behav. Sci. (Basel). 6, 29 10.3390/bs6040029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones-Davis DM, Yang M, Rider E, Osbun NC, da Gente GJ, Li J, Katz AM, Weber MD, Sen S, Crawley J, Sherr EH, 2013. Quantitative Trait Loci for Interhemispheric Commissure Development and Social Behaviors in the BTBR T+ tf/J Mouse Model of Autism. PLoS One 8, e61829 10.1371/journal.pone.0061829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Mathelier A, 2017. Intervene: A tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinformatics 18, 287 10.1186/s12859-017-1708-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakowiak P, Goines PE, Tancredi DJ, Ashwood P, Hansen RL, Hertz-Picciotto I, Van de Water J, 2017. Neonatal Cytokine Profiles Associated With Autism Spectrum Disorder. Biol. Psychiatry 81, 442–451. 10.1016/j.biopsych.2015.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL, 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I, 2014. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326. 10.1016/j.cell.2014.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link Verena M., Duttke SH, Chun HB, Holtman IR, Westin E, Hoeksema MA, Abe Y, Skola D, Romanoski CE, Tao J, Fonseca GJ, Troutman TD, Spann NJ, Strid T, Sakai M, Yu M, Hu R, Fang R, Metzler D, Ren B, Glass CK, 2018. Analysis of Genetically Diverse Macrophages Reveals Local and Domain-wide Mechanisms that Control Transcription Factor Binding and Function. Cell 173, 1796–1809.e17. 10.1016/j.cell.2018.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link, Verena M, Romanoski CE, Metzler D, Glass CK, 2018. MMARGE: Motif Mutation Analysis for Regulatory Genomic Elements. Nucleic Acids Res. 46, 7006– 7021. 10.1093/nar/gky491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Cao S, Zhou Y, Xiong Y, 2019. Recent advances in endotoxin tolerance. J. Cell. Biochem 120, 56–70. 10.1002/jcb.27547 [DOI] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, Shafer A, Neri F, Lee K, Kutyavin T, Stehling-Sun S, Johnson AK, Canfield TK, Giste E, Diegel M, Bates D, Hansen RS, Neph S, Sabo PJ, Heimfeld S, Raubitschek A, Ziegler S, Cotsapas C, Sotoodehnia N, Glass I, Sunyaev SR, Kaul R, Stamatoyannopoulos JA, 2012. Systematic localization of common disease- associated variation in regulatory DNA. Science (80-. ). 10.1126/science.1222794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane HG, Kusek GK, Yang M, Phoenix JL, Bolivar VJ, Crawley JN, 2008. Autism-like behavioral phenotypes in BTBR T+tf/J mice. Genes, Brain Behav. 7, 152–163. 10.1111/j.1601-183X.2007.00330.x [DOI] [PubMed] [Google Scholar]
- Mendizabal I, Shi L, Keller TE, Konopka G, Preuss TM, Hsieh TF, Hu E, Zhang Z, Su B, Yi SV, 2016. Comparative Methylome Analyses Identify Epigenetic Regulatory Loci of Human Brain Evolution. Mol. Biol. Evol 33, 2947– 2959. 10.1093/molbev/msw176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy CA, Morrow AL, Meinzen-Derr J, Schleifer K, Dienger K, Manning-Courtney P, Altaye M, Wills-Karp M, 2006. Elevated cytokine levels in children with autism spectrum disorder. J. Neuroimmunol 172, 198–205. 10.1016/j.jneuroim.2005.11.007 [DOI] [PubMed] [Google Scholar]
- Morgan JT, Barger N, Amaral DG, Schumann CM, 2014. Stereological Study of Amygdala Glial Populations in Adolescents and Adults with Autism Spectrum Disorder. PLoS One 9, e110356 10.1371/journal.pone.0110356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JT, Chana G, Abramson I, Semendeferi K, Courchesne E, Everall IP, 2012. Abnormal microglial-neuronal spatial organization in the dorsolateral prefrontal cortex in autism. Brain Res 1456, 72–81. 10.1016/j.brainres.2012.03.036 [DOI] [PubMed] [Google Scholar]
- Morgan JT, Chana G, Pardo CA, Achim C, Semendeferi K, Buckwalter J, Courchesne E, Everall IP, 2010. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol Psychiatry 68, 368–376. 10.1016/j.biopsych.2010.05.024 [DOI] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Young NB, Perez A, Holloway LP, Barbaro RP, Barbaro JR, WILSON LM, Threadgill DW, Lauder JM, Magnuson TR, Crawley JN, 2007. Mouse behavioral tasks relevant to autism: Phenotypes of 10 inbred strains. Behav. Brain Res 176, 4–20. 10.1016/j.bbr.2006.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Joosten LAB, Latz E, Mills KHG, Natoli G, Stunnenberg HG, O’Neill LAJ, Xavier RJ, 2016. Trained immunity: A program of innate immune memory in health and disease. Science 352, aaf1098. 10.1126/science.aaf1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Quintin J, Van Der Meer JWM, 2011. Trained immunity: A memory for innate host defense. Cell Host Microbe. 10.1016/j.chom.2011.04.006 [DOI] [PubMed] [Google Scholar]
- Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y, Keulen Z, Pasillas MP, O’Connor C, Nickl CK, Schafer ST, Shen Z, Rissman RA, Brewer JB, Gosselin D, Gonda DD, Levy ML, Rosenfeld MG, McVicker G, Gage FH, Ren B, Glass CK, 2019. Brain cell type–specific enhancer–promoter interactome maps and disease-risk association. Science (80-. ). 10.1126/science.aay0793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novakovic B, Habibi E, Wang SY, Arts RJW, Davar R, Megchelenbrink W, Kim B, Kuznetsova T, Kox M, Zwaag J, Matarese F, van Heeringen SJ, Janssen-Megens EM, Sharifi N, Wang C, Keramati F, Schoonenberg V, Flicek P, Clarke L, Pickkers P, Heath S, Gut I, Netea MG, Martens JHA, Logie C, Stunnenberg HG, 2016. β-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 167, 1354–1368.e14. 10.1016/j.cell.2016.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenchain V, Lawrence M, Carey V, Gogarten S, Shannon P, Morgan M, 2014. VariantAnnotation: A Bioconductor package for exploration and annotation of genetic variants. Bioinformatics 30, 2076–2078. 10.1093/bioinformatics/btu168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onore CE, Careaga M, Babineau BA, Schwartzer JJ, Berman RF, Ashwood P, 2013. Inflammatory macrophage phenotype in BTBR T+tf/J mice. Front Neurosci 7, 158 10.3389/fnins.2013.00158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, Hartl C, Leppa V, Ubieta L.T. de la T, Huang J, Lowe JK, Blencowe BJ, Horvath S, Geschwind DH, 2016. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540, 423–427. 10.1038/nature20612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pstkov PM, Graber JH, Churchill GA, Dipetrillo K, King BL, Paigen K, 2005. Evidence of a large-scale functional organization of mammalian chromosomes. PLoS Genet. 1, 312–322. 10.1371/journal.pgen.0010033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T, 2016. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. 10.1093/nar/gkw257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK, 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–40. 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roullet FI, Crawley JN, 2011. Reduced scent marking and ultrasonic vocalizations in the BTBR T+tf/J mouse model of autism. 10, 35–43. 10.1111/j.1601-183X.2010.00582.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed S, Quintin J, Kerstens HHD, Rao NA, Matarese F, Cheng S, Ratter J, Ent MA Van Der, Sharifi N, Janssen-megens EM, Ter M, 2014. Epigenetic programming during monocyte to macrophage differentiation and trained innate immunity. Science (80-. ). 345, 1–26. 10.1126/science.1251086.Epigenetic [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scattoni ML, Ricceri L, Crawley JN, Elena VR, Superiore I, 2011. Unusual repertoire of vocalizations in adult BTBR T+tf/J mice during three types of social encounters. 10, 44–56. 10.1111/j.1601-183X.2010.00623.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzer JJ, Careaga M, Chang C, Onore CE, Ashwood P, 2015. Allergic fetal priming leads to developmental, behavioral and neurobiological changes in mice. Transl Psychiatry 5, e543 10.1038/tp.2015.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzer JJ, Careaga M, Onore CE, Rushakoff JA, Berman RF, Ashwood P, Schwartzer JJ, Careaga M, Berman RF, Ashwood P, 2013. Maternal immune activation and strain specific interactions in the development of autism-like behaviors in mice. Transl Psychiatry 3, 220–226. 10.1038/tp.2013.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzer JJ, Onore CE, Rose D, Ashwood P, 2017. C57BL/6J bone marrow transplant increases sociability in BTBR T+ Itpr3tf/J mice. Brain. Behav. Immun 59, 55–61. 10.1016/j.bbi.2016.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield NCN, Bock C, 2015. LOLA: Enrichment analysis for genomic region sets and regulatory elements in R and Bioconductor. Bioinformatics. 10.1093/bioinformatics/btv612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA, Das A, Ray SK, Banik NL, 2012. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull 10.1016/j.brainresbull.2011.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark R, Brown G, 2011. DiffBind : differential binding analysis of ChIP-Seq peak data. Bioconductor 1–27. [Google Scholar]
- Suzuki K, Sugihara G, Ouchi Y, Nakamura K, Futatsubashi M, Takebayashi K, Yoshihara Y, Omata K, Matsumoto K, Tsuchiya KJ, Iwata Y, Tsujii M, Sugiyama T, Mori N, 2013. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 70, 49–58. 10.1001/jamapsychiatry.2013.272 [DOI] [PubMed] [Google Scholar]
- Tarkowski E, Liljeroth AM, Minthon L, Tarkowski A, Wallin A, Blennow K, 2003. Cerebral pattern of pro-and anti-inflammatory cytokines in dementias, in: Brain Research Bulletin: Elsevier Inc., pp. 255–260. 10.1016/S0361-9230(03)00088-1 [DOI] [PubMed] [Google Scholar]
- Wendeln A-C, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, Wagner J, Häsler LM, Wild K, Skodras A, Blank T, Staszewski O, Datta M, Centeno TP, Capece V, Islam MR, Kerimoglu C, Staufenbiel M, Schultze JL, Beyer M, Prinz M, Jucker M, Fischer A, Neher JJ, 2018. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556, 332–338. 10.1038/s41586-018-0023-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbo O, Yoshida Cathleen, Grether JK, Van de Water Judy, Ashwood P, Delorenze GN, Hansen RL, Kharrazi M, Croen LA, Baio J, Blumberg S, Bramlett M, Kogan M, Schieve L, Jones J, Lu M, Kim YS, Leventhal B, Koh Y, Fombonne E, Laska E, Lim E, Cheon K, Kim S, Kim YK, Lee H, Song D, Grinker R, Kanner L, Ashwood P, Wills S, Water, J Van de, Croen L, Yoshida CK, Odouli R, Newman T, Goines PE, Ashwood P, Goines PE, Croen L, Braunschweig D, Yoshida CK, Grether J, Hansen R, Kharrazi M, Ashwood P, Water, J Van de, Jyonouchi H, Sun S, Le H, Li X, Chauhan A, Sheikh A, Patil S, Chauhan V, Li XM, Ji L, Brown T, Malik M, Pardo C, Vargas D, Zimmerman A, Vargas D, Nascimbene C, Krishnan C, Zimmerman A, Pardo C, Enstrom A, Krakowiak P, Onore C, Pessah IN, Hertz-Picciotto I, Hansen RL, Water, JA Van de, Ashwood P, Grether JK, Croen L, Anderson M, Nelson K, Yolken RH, Heuer L, Ashwood P, Schauer J, Goines P, Krakowiak P, Hertz-Picciotto I, Hansen R, Croen L, Pessah IN, Water, J Van de, Heuer LS, Rose M, Ashwood P, Water, J Van de, Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Water, J Van de, Suzuki K, Matsuzaki H, Iwata K, Kameno Y, Shimmura C, Kawai S, Yoshihara Y, Wakuda T, Takebayashi K, Takagai S, Matsumoto K., Tsuchiya K, Iwata Y, Nakamura K, Tsujii M, Sugiyama T, Mori N, Onore C, Careaga M, Ashwood P, Dinarello C, Bauer S, Kerr B, Patterson P, Rostene W, Kitabgi P, Parsadaniantz S, Ponzio N, Servatius R, Beck K, Marzouk A, Kreider T, Smith S, Li J, Garbett K, Mirnics K, Patterson P, Atladottir H, Henriksen T, Schendel D, Parner E, Atladottir H, Thorsen P, Ostergaard L, Schendel D, Lemcke S, Abdallah M, Parner E, Brown A, Sourander A, Hinkka-Yli-Salomaki S, McKeague I, Sundvall J, Surcel H, Chess S, Deykin E, MacMahon B, Zerbo O, Iosif A, Walker C, Ozonoff S, Hansen RL, Hertz-Picciotto I, Zerbo O, Qian Y, Yoshida Cathleen, Grether JK, Water, J Van de, Croen L, Abdallah MW, Larsen N, Grove J, Bonefeld-Jorgensen E, Norgaard-Pedersen B, Hougaard D, Mortensen E, Abdallah MW, Larsen N, Grove J, Norgaard-Pedersen B, Thorsen P, Mortensen E, Hougaard D, Abdallah MW, Larsen N, Grove J, Norgaard-Pedersen B, Thorsen P, Mortensen E, Hougaard D, Abdallah MW, Larsen N, Mortensen E, Atladottir H, Norgaard-Pedersen B, Bonefeld-Jorgensen E, Grove J, Hougaard D, Al-Ayadhi L, Mostafa G, Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah IN, Water, J Van de, Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah IN, Water, J Van de, Manzardo A, Henkhaus R, Dhillon S, Butler M, Nelson P, Kuddo T, Song E, Dambrosia J, Kohler S, Satyanarayana G, Vandunk C, Grether JK, Nelson K, Jager, W De, Bourcier K, Rijkers G, Prakken B, Seyfert-Margolis V, Croen L, Goines P, Braunschweig D, Yolken R, Yoshida CK, Grether JK, Fireman B, Kharrazi M, Hansen RL, Water, J Van de, Yeargin-Allsopp M, Rice C, Karapurkar T, Doernberg N, Boyle C, Murphy C, Dammann O, O’Shea T, Rostene W, Guyon A, Kular L, Godefroy D, Barbieri F, Bajetto A, Banisadr G, Callewaere C, Conductier G, Rovere C, Melik-Parsadaniantz S, Florio T, 2014. Neonatal cytokines and chemokines and risk of Autism Spectrum Disorder: the Early Markers for Autism (EMA) study: a case-control study. J. Neuroinflammation 11, 113 10.1186/1742-2094-11-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Sun Z, Wang J, Huang H, Kocher JP, Wang L, 2014. CrossMap: A versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30, 1006–1007. 10.1093/bioinformatics/btt730 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.