

Abstract
Pancreatic cancer is among the most well characterized cancer types, yet a large proportion of the heritability of pancreatic cancer risk remains unclear. Here we performed a large transcriptome-wide association study (TWAS) to systematically investigate associations between genetically predicted gene expression in normal pancreas tissue and pancreatic cancer risk. Using data from 305 subjects of mostly European descent in the Genotype-Tissue Expression Project, we built comprehensive genetic models to predict normal pancreas tissue gene expression, modifying the UTMOST (unified test for molecular signatures). These prediction models were applied to the genetic data of 8,275 pancreatic cancer cases and 6,723 controls of European ancestry. Thirteen genes showed an association of genetically predicted expression with pancreatic cancer risk at a false discovery rate (FDR) ≤ 0.05, including seven previously reported genes (INHBA, SMC2, ABO, PDX1, RCCD1, CFDP1, and PGAP3) and six novel genes not yet reported for pancreatic cancer risk (6q27: SFT2D1 (odds ratio (OR) (95% confidence interval (CI)): 1.54 (1.25–1.89)); 13q12.13: MTMR6 (OR (95% CI): 0.78 (0.70–0.88)); 14q24.3: ACOT2 (OR (95% CI): 1.35 (1.17–1.56)); 17q12: STARD3 (OR (95% CI): 6.49 (2.96–14.27)); 17q21.1: GSDMB (OR (95% CI): 1.94 (1.45–2.58)); and 20p13: ADAM33 (OR (95% CI): 1.41 (1.20–1.66))). The associations for ten of these genes (SFT2D1, MTMR6, ACOT2, STARD3, GSDMB, ADAM33, SMC2, RCCD1, CFDP1, and PGAP3) remained statistically significant even after adjusting for risk SNPs identified in previous GWAS. Collectively, this analysis identified novel candidate susceptibility genes for pancreatic cancer that warrant further investigation.
Keywords: transcriptome-wide association study, genetic factors, pancreatic cancer, gene expression
Introduction
Pancreatic cancer is the third leading cause of cancer death in the United States, and its incidence has continued to increase in recent years (1). There are several established risk factors for pancreatic cancer, including tobacco smoking, heavy alcohol consumption, obesity, chronic pancreatitis, type 2 diabetes, and family history of pancreatic cancer (2). Inherited rare mutations in hereditary pancreatic cancer explain only a small fraction of genetic heritability (3). Due to the nonspecific symptoms in earlier stages, this malignancy is usually detected at a late stage, resulting in a 5-year survival rate of only 9% (1). Currently, there is no effective screening test available for pancreatic cancer. Therefore, there is an urgent need to better characterize the etiology of pancreatic cancer and develop effective early detection and/or screening strategies.
Since 2009, several GWAS have been performed to identify common susceptibility variants associated with pancreatic cancer risk, including studies conducted by the Pancreatic Cancer Cohort Consortium (PanScan I, II, III) and the Pancreatic Cancer Case Control Consortium (PanC4) primarily focusing on Europeans, as well as studies conducted in East Asians. To date, nearly two dozen common risk variants have been identified for pancreatic cancer risk (3–9). Many pancreatic cancer risk variants identified by GWAS, however, are not located in coding regions, but in gene regulatory elements (10). It has been hypothesized that a large proportion of GWAS reported association signals may be due to regulatory effects of susceptibility variants on the gene expression of disease target genes (11,12). For pancreatic cancer, the genes responsible for the reported associations remain unknown for the large majority of the GWAS-identified risk loci.
Recently, gene-based approaches, such as Transcriptome-wide association study (TWAS) design, have been developed to uncover novel candidate disease susceptibility genes by assessing associations between genetically predicted gene expression with disease risk (13,14). Unlike GWAS that tests individual genetic variants, TWAS aggregates the effect of multiple SNPs into a single biologically meaningful testing unit, thus significantly improving the power for gene identification. Such a design also confers an advantage for disease gene discovery since direct profiling of the transcriptome in relevant (normal) human tissues in a sufficient sample size is expensive and often difficult to carry out. Such an approach using genetic instruments could potentially reduce the influence of several biases commonly encountered in typical epidemiological studies, including selection bias, residual confounding, and reverse causation. Besides discovering novel genetic loci, TWAS design can also potentially identify candidate target genes of GWAS identified risk variants. To date, such a TWAS design has been applied to uncover candidate susceptibility genes for multiple cancer types, including breast cancer, ovarian cancer, prostate cancer, and melanoma (11,12,15–17). A recent TWAS study for pancreatic cancer risk has been conducted, in which 25 significant gene-level associations, including 14 at 11 novel loci, were identified (18). In this study, authors assessed gene expression genetic imputation in 48 tissue types beyond the pancreas tissue. Focusing on the pancreas, the most relevant tissue for pancreatic cancer, the authors evaluated both tumor adjacent normal pancreas tissue (the Laboratory of Translational Genomics dataset, n=95) and normal pancreas tissue (the Genotype-Tissue Expression dataset (GTEx), n=174). It is known that tumor growth can influence gene expression levels in surrounding tissues, and some gene expression might be substantially altered in tumor-adjacent normal tissue compared with that in normal tissue from subjects without cancer. Therefore, ideally, to study pancreatic cancer susceptibility genes, normal pancreas tissue from healthy subjects should be used. Recently, data from the final version (v8) of the GTEx project have been released. In this dataset, 305 subjects, primarily of European ancestries, have both genotyping and normal pancreas tissue transcriptome data available. Leveraging this largest available reference dataset for normal pancreas tissue, we applied the state-of-the-art modeling strategy of UTMOST (unified test for molecular signatures), to generate comprehensive normal pancreatic tissue gene expression genetic models. We conducted a large pancreatic cancer TWAS (19) to identify additional candidate pancreatic cancer susceptibility genes.
Methods
Transcriptome and genome data from the GTEx project
We used transcriptome and genome data from the GTEx v8 (The database of Genotypes and Phenotypes (dbGaP) accession: phs000424.v8.p2) to develop genetic imputation models for genes expressed in normal pancreatic tissue. Details of the GTEx v8 dataset have been described elsewhere (https://gtexportal.org/home/documentationPage). In brief, genomic information of 838 subjects was collected using whole genome sequencing (WGS), as performed by the Broad Institute’s Genomics Platform. Details of RNA sequencing experiments, quality control (QC) of the gene expression data, and genomic data have been described elsewhere (20,21).
Building pancreatic tissue gene expression prediction models
The cross-tissue UTMOST framework was used to build pancreas tissue gene expression genetic models (19). Here we modified the model training approach to obtain a reliable estimate of the imputation performance. SNPs within 1 Mb upstream and downstream of the gene body were considered as predictor variables in the model. To reduce the computational burden, LD-pruning (r2=0.9) was performed before model training. It has been shown that there is no significant difference in prediction quality from applying LD pruning (13). The residual of the normalized TPM was used for model building after adjusting for covariates of age, sex, sequencing platform, the first five principal components (PCs), and probabilistic estimation of expression residuals (PEER) factors. In the joint-tissue prediction model, the effect sizes were estimated by minimizing the loss function with a LASSO penalty on the columns (within-tissue effects) and a group-LASSO penalty on the rows (cross-tissue effects). The group penalty term implemented sharing of the information from feature (SNP) selection across all the tissues. The optimization problem uses two hyperparameters, λ1 and λ2, for the within-tissue and cross-tissue penalization, respectively. Five-fold cross-validation was performed for hyperparameter tuning.
Here, we modified the original model training by unifying the hyperparameter pairs to avoid the overestimation of the prediction performance. Briefly, λ1 and λ2 were initialized using the range of pre-trained lambdas from single-tissue elastic net models. For each gene, 25 lambda pairs (five for each lambda) were generated. In our modified version, the 25 lambda pairs were consistent across the five-fold cross-validation, while the original UTMOST assigned different lambdas for each fold. The unified hyperparameter pairs made the different folds comparable, thus avoiding the performance overestimation in a retrained model. The optimization of the joint model was initialized by single-tissue weights generated in each fold and the optimization stopped if the training error in each training set or the related tuning error was higher than the previous step. After the five-fold training, one of the 25 lambda pairs was selected as the best lambda pair according to the average tuning error across the five folds. The prediction performance was evaluated by the correlation between the predicted and observed expression levels in the combined tuning set. Models with Pearson’s correlation r > 0.1 and P < 0.05 were used in subsequent analysis.
Associations between genetically predicted gene expression and pancreatic cancer risk
For our association analysis, we leveraged GWAS conducted in PanScan I, PanScan II, PanScan III, and PanC4, downloaded from dbGaP (Study Accession: phs000206.v5.p3 and phs000648.v1.p1). The detailed information for these GWAS has been described elsewhere (4–7,22,23). Briefly, genotyping was performed on the Illumina HumanHap550, 610-Quad, OmniExpress, and OmiExpressExome arrays, respectively. We performed standard QC according to the guidelines recommended by the consortia (3). We excluded study subjects who were related to each other, with missing information on age or sex, had gender discordance, were non-European ancestry based on genetic estimation, and with a low call rate (less than 94% and 98% in PanScan and PanC4, respectively). We also removed duplicated SNPs and those with a high missing call rate (of at least 6% and 2% in PanScan and PanC4, respectively), or with violations of Hardy-Weinberg equilibrium (HWE) (of P < 1×10−7 and P < 1×10−4 in PanScan and PanC4, respectively). In PanC4 dataset, we additionally excluded variants with minor allele frequency (MAF) < 0.005, with more than one mendelian error in HapMap control trios, with more than two discordant calls in study duplicates, and those with sex difference in allele frequency > 0.2 or in heterozygosity > 0.3 for autosomes/XY. The genotype imputation was conducted with a reference panel of the Haplotype Reference Consortium (r1.1 2016), using Minimac4 after phasing with Eagle v2.4 (24).
Imputed SNPs with an imputation quality of at least 0.3 were retained. We then evaluated the associations between individual SNPs and pancreatic cancer risk after adjusting for age, sex and top principal components (25).
We investigated the associations of genetically predicted gene expression in pancreas tissue with pancreatic cancer risk using the summary statistics generated from 8,275 cases and 6,723 controls of European ancestry. Using S-PrediXcan (26), the associations of genetically predicted gene expression were estimated based on prediction weights, GWAS summary statistics, and an SNP-correlation (LD) matrix (14,15). In brief, the formula:
was used to estimate the Z-score of the association between predicted gene expression and pancreatic cancer risk. Here wlg represents the weight of SNP l for predicting the expression of gene g, and represent the GWAS association regression coefficient and its standard error for SNP l, and and represent the estimated variances of SNP l and the predicted expression of gene g respectively. For a majority of the tested genes, most of the corresponding predicting SNPs were available in GWAS datasets and used for the association analyses (e.g., ≥80% predicting SNPs used for 90.2% of the tested genes). A Benjamini-Hochberg false discovery rate (FDR) corrected P-value threshold of ≤ 0.05 was used to determine significant associations. The FDR analysis was performed using ‘p.adjust’ function in R (27). We further conducted conditional analysis with adjustments of previously identified pancreatic cancer risk variants to assess whether the associations between genetically predicted gene expression and pancreatic cancer risk in the main analyses were independent of the risk variants identified in GWAS. Previously reported pancreatic cancer risk SNPs that are available in the current dataset (rs10094872, rs11655237, rs1486134, rs1517037, rs1561927, rs16986825, rs17688601, rs2736098, rs2816938, rs2941471, rs35226131, rs3790844, rs401681, rs4795218, rs505922, rs6971499, rs7190458, rs78417682, rs9543325, rs9581943, and rs9854771) were adjusted for in the conditional analysis using individual level data.
To evaluate whether the TWAS identified genes can improve the risk prediction of pancreatic cancer, we compared the baseline model (PRS1) including age, sex, top principal components, and GWAS identified risk variants with another model (PRS2) in which predicted expression of TWAS identified genes were also included. In PRS2 model, we did not include the three genes with associations of genetic predicted expression were shown to be influenced by risk variants, as well as exclude one other gene (PGAP3) correlated with STARD3. We compared the Area Under the ROC curve (AUC) of both models. Analyses were conducted using R version 4.0.1 (2020-06-06).
Results
Gene expression prediction model building
The overall study flow was presented in Figure 1. The flowchart of quality control and prediction model training in the reference dataset was shown in Supplementary Figure 1. Using the modified UTMOST framework, we generated prediction models for 8,479 genes with performance r > 0.1 and P < 0.05. Detailed information regarding the number of models built according to different performance thresholds and gene types is shown in Supplementary Table 1.
Figure 1.

Study design flow chart
Associations of predicted gene expression in pancreas tissue with pancreatic cancer risk
Of the 8,433 genes tested, we identified 13 genes whose genetically predicted expression was associated with pancreatic cancer risk at P ≤ 8.00×10−5, a false discovery rate (FDR)-corrected significance level (Tables 1 and 2; Figure 2). Of these, six were novel genes which have not been reported in previous studies (6q27: SFT2D1; 13q12.13: MTMR6; 14q24.3: ACOT2; 17q12: STARD3; 17q21.1: GSDMB; 20p13: ADAM33) (Table 1) and seven genes were previously reported (7p14.1: INHBA; 9q31.1: SMC2; 9q34.2: ABO; 13q12.2: PDX1; 15q26.1: RCCD1; 16q23.1: CFDP1; 17q12: PGAP3) (Table 2). The 25 lambda pairs and the corresponding prediction performance in the tuning set for prediction models of the 13 associated genes are shown in Supplementary Table 2.
Table 1.
Pancreatic cancer associations for six novel genes that have not been previously reported
| Region | Gene name* | Classificationa | R2b | No. of SNPs in prediction models | Associations based on S-PrediXcan analyses | Associations in conditional analyses adjusting for known risk variantsb | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| OR | 95% CI | P valuec | FDR P value | OR | 95% CI | P value after adjusting for known risk variantsd | |||||
| 6q27 | SFT2D1 | protein | 0.07 | 3 | 1.54 | 1.25 – 1.89 | 5.40 × 10−5 | 0.04 | 1.50 | 1.18–1.90 | 9.29 × 10−4 |
| 13q12.13 | MTMR6 | protein | 0.25 | 41 | 0.78 | 0.70 – 0.88 | 4.94 × 10−5 | 0.04 | 0.79 | 0.70–0.90 | 4.33 × 10−4 |
| 14q24.3 | ACOT2 | protein | 0.09 | 80 | 1.35 | 1.17 – 1.56 | 6.06 × 10−5 | 0.04 | 1.40 | 1.20–1.63 | 2.22 × 10−5 |
| 17q12 | STARD3 | protein | 0.01 | 11 | 6.49 | 2.96 – 14.27 | 3.59 × 10−6 | 0.01 | 5.11 | 2.10–12.48 | 3.59 × 10−4 |
| 17q21.1 | GSDMB | protein | 0.03 | 44 | 1.94 | 1.45 – 2.58 | 7.03 × 10−6 | 0.01 | 2.09 | 1.51–2.87 | 7.64 × 10−6 |
| 20p13 | ADAM33 | protein | 0.11 | 35 | 1.41 | 1.20 – 1.66 | 4.43 × 10−5 | 0.04 | 1.44 | 1.20–1.72 | 1.10 × 10−4 |
Protein: protein coding genes;
R2: prediction performance (R2) derived using GTEx data;
P value: derived from association analyses of 8,275 cases and 6,723 controls; associations with FDR-corrected P value ≤ 0.05 considered significant;
Adjusted risk SNPs include known pancreatic cancer risk SNPs that are available in current datasets: rs3790844, rs2816938, rs1486134, rs9854771, rs401681, rs2736098, rs35226131, rs6971499, rs17688601, rs78417682, rs1561927, rs10094872, rs2941471, rs505922, rs9543325, rs9581943, rs7190458, rs11655237, rs4795218, rs1517037, and rs16986825.
Bold ones represent associations with P < 5.93 × 10−6, based on Bonferroni correction of 8,433 tests (0.05/8,433).
Table 2.
Pancreatic cancer associations for seven genes that have been reported in a previous TWAS study
| Region | Gene name | Classification | R2a | No. of SNPs in prediction models | Associations based on S-PrediXcan analysesa | Associations in conditional analyses adjusting for known risk variantsb | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| OR | 95% CI | P value | FDR P value | OR | 95% CI | P valueb | |||||
| 7p14.1 | INHBA | protein | 0.05 | 14 | 0.54 | 0.43 – 0.68 | 1.32 × 10−7 | 2.22 × 10−4 | 0.75 | 0.55–1.01 | 0.06 |
| 9q31.1 | SMC2 | protein | 0.02 | 20 | 2.88 | 1.98 – 4.18 | 3.84 × 10−8 | 1.08 × 10−4 | 2.78 | 1.90–4.07 | 1.58 × 10−7 |
| 9q34.2 | ABO | transcript | 0.57 | 32 | 1.24 | 1.15 – 1.34 | 1.59 × 10−8 | 6.69 × 10−5 | 0.95 | 0.86–1.05 | 0.33 |
| 13q12.2 | PDX1 | protein | 0.06 | 6 | 0.49 | 0.40 – 0.61 | 5.10 × 10−11 | 4.30 × 10−7 | 0.66 | 0.33–1.30 | 0.23 |
| 15q26.1 | RCCD1 | protein | 0.32 | 18 | 0.84 | 0.77 – 0.91 | 7.17 × 10−5 | 0.05 | 0.85 | 0.77–0.93 | 3.92 × 10−4 |
| 16q23.1 | CFDP1 | protein | 0.07 | 57 | 1.52 | 1.31 – 1.77 | 6.17 × 10−8 | 1.30 × 10−4 | 1.48 | 1.26–1.74 | 1.75 × 10−6 |
| 17q12 | PGAP3 | Protein | 0.29 | 50 | 1.20 | 1.11–1.30 | 1.38 × 10−5 | 0.02 | 1.19 | 1.08–1.31 | 2.90 × 10−4 |
R2: prediction performance (R2) derived using GTEx data; P value: derived from association analyses of 8,275 cases and 6,723 controls; associations with FDR-corrected P value ≤ 0.05 considered significant;
Adjusted risk SNPs include known pancreatic cancer risk SNPs that are available in current datasets: rs3790844, rs2816938, rs1486134, rs9854771, rs401681, rs2736098, rs35226131, rs6971499, rs17688601, rs78417682, rs1561927, rs10094872, rs2941471, rs505922, rs9543325, rs9581943, rs7190458, rs11655237, rs4795218, rs1517037, and rs16986825.
Bold ones represent associations with P < 5.93 × 10−6, based on Bonferroni correction of 8,433 tests (0.05/8,433).
Figure 2.
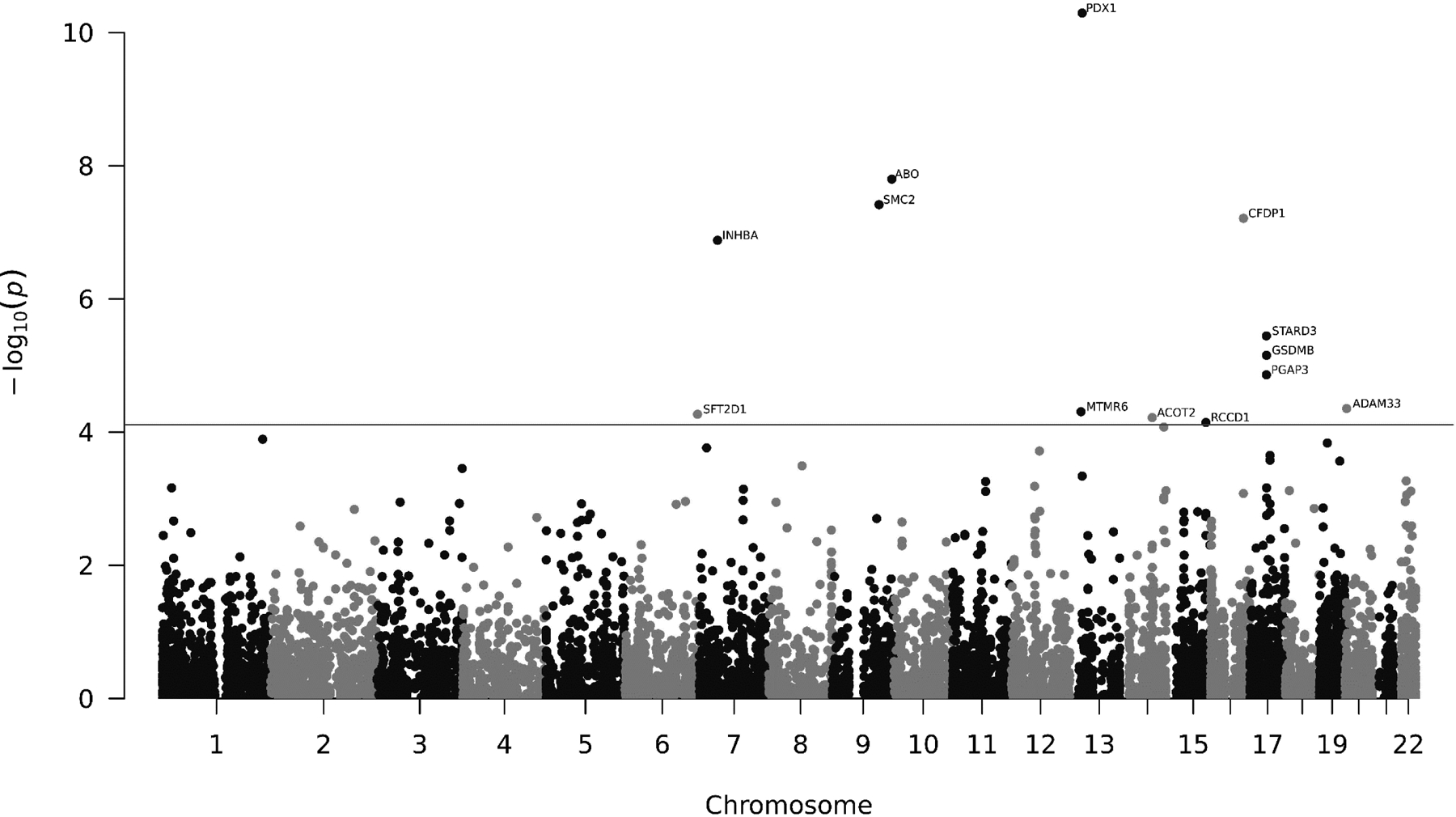
Manhattan plot of association results from the pancreatic cancer transcriptome-wide association study. The horizon line represents P = 8.00 × 10−5 (FDR-corrected P value ≤ 0.05). Each dot represents the genetically predicted expression of one specific gene by pancreatic tissue prediction models. The x axis represents the genomic position of the corresponding gene, and the y axis represents the negative logarithm of the association P value.
Except for PDX1, ABO, and CFDP1, other ten genes are at least 500kb away from any risk variant reported in previous GWAS of pancreatic cancer (Table 3). An association between lower genetically predicted expression and increased pancreatic cancer risk was observed for INHBA (7p14.1), PDX1 (13q12.2), MTMR6 (13q12.13), and RCCD1 (15q26.1). Conversely, an association between higher genetically predicted expression and increased pancreatic cancer risk was identified for SFT2D1 (6q27), SMC2 (9q31.1), ABO (9q34.2), ACOT2 (14q24.3), CFDP1 (16q23.1), PGAP3 (17q12), STARD3 (17q12), GSDMB (17q21.1), and ADAM33 (20p13). Based on stratified analysis according to age (< 70 years old or ≥70 years old), association estimates of these 13 genes were largely consistent between the two groups (Table 4).
Table 3.
Distances between TWAS identified associated genes and closest risk SNPs
| TWAS identified genes | Closest risk SNP on the same Chr | Position (build37) | Distance to the risk SNP (kb) | ||
|---|---|---|---|---|---|
| Gene name | Chr | Position (build37) | |||
| SFT2D1 | 6 | 166733516–166755991 | – | – | – |
| INHBA | 7 | 41728601–41742706 | rs78417682 | 47488903 | 5,746 |
| ABO | 9 | 136130563–136150630 | rs505922 | 136149229 | 1 |
| SMC2 | 9 | 106856213–106903700 | rs505922 | 136149229 | 29,246 |
| MTMR6 | 13 | 25820339–25861704 | rs9581943 | 28493997 | 2,632 |
| PDX1 | 13 | 28494168–28500451 | rs9581943 | 28493997 | 0.17 |
| ACOT2 | 14 | 74034324–74042362 | rs9543325 | 73916628 | 118 |
| RCCD1 | 15 | 91498106–91506355 | – | – | – |
| CFDP1 | 16 | 75327608–75467387 | rs7190458 | 75263661 | 70 |
| STARD3 | 17 | 37793333–37820454 | rs4795218 | 36078510 | 1,715 |
| GSDMB | 17 | 38060848–38074903 | rs4795218 | 36078510 | 1,982 |
| PGAP3 | 17 | 37827375–37844310 | rs4795218 | 36078510 | 1,749 |
| ADAM33 | 20 | 3648620–3662755 | – | – | – |
Table 4.
Associations of TWAS identified genes and pancreatic cancer risk stratified by age
| Gene name | <70 years old a | ≥70 years old a | P value for interaction | ||||
|---|---|---|---|---|---|---|---|
| OR | 95% CI | P value | OR | 95% CI | P value | ||
| SFT2D1 | 1.38 | 1.03–1.85 | 0.032 | 1.90 | 1.29–2.79 | 1.21 × 10−3 | 0.16 |
| MTMR6 | 0.82 | 0.70–0.96 | 0.013 | 0.73 | 0.59–0.90 | 3.61 × 10−3 | 0.35 |
| ACOT2 | 1.39 | 1.15–1.68 | 5.71 × 10−4 | 1.35 | 1.05–1.74 | 0.019 | 0.73 |
| STARD3 | 6.59 | 2.18–19.87 | 8.67 × 10−4 | 3.09 | 0.73–13.04 | 0.13 | 0.39 |
| GSDMB | 1.90 | 1.28–2.81 | 1.45 × 10−3 | 2.22 | 1.31–3.76 | 3.05 × 10−3 | 0.63 |
| ADAM33 | 1.52 | 1.21–1.91 | 2.92 × 10−4 | 1.41 | 1.05–1.89 | 0.024 | 0.64 |
| INHBA | 0.54 | 0.40–0.71 | 2.10 × 10−5 | 0.62 | 0.42–0.90 | 0.013 | 0.66 |
| SMC2 | 2.61 | 1.63–4.18 | 7.03 × 10−5 | 3.18 | 1.72–5.89 | 2.43 × 10−4 | 0.69 |
| ABO | 1.20 | 1.09–1.32 | 2.29 × 10−4 | 1.35 | 1.19–1.53 | 4.74 × 10−6 | 0.14 |
| PDX1 | 0.46 | 0.35–0.60 | 2.14 × 10−8 | 0.59 | 0.41–0.84 | 3.46 × 10−3 | 0.25 |
| RCCD1 | 0.87 | 0.78–0.97 | 0.015 | 0.81 | 0.70–0.94 | 5.16 × 10−3 | 0.42 |
| CFDP1 | 1.46 | 1.21–1.77 | 9.74 × 10−5 | 1.62 | 1.25–2.09 | 2.73 × 10−4 | 0.60 |
| PGAP3 | 1.23 | 1.09–1.38 | 5.64 × 10−4 | 1.11 | 0.95–1.29 | 0.19 | 0.30 |
To determine whether the observed associations between genetically predicted gene expression and pancreatic cancer risk were independent of GWAS identified association signals, we performed individual level data analyses adjusting for GWAS-identified risk SNPs (4–7,22,23). For all six novel genes (SFT2D1, MTMR6, ACOT2, STARD3, GSDMB, and ADAM33) and four previously reported genes (SMC2, RCCD1, CFDP1 and PGAP3), the association remained significant (Tables 1 and 2). This suggests that the predicted expression of these genes may be associated with pancreatic cancer risk at least partially independent of the GWAS-identified risk variants. For three known genes (INHBA, ABO, and PDX1), their associations became insignificant after adjusting for known risk SNPs (Table 2), suggesting that their gene expression associations may be influenced by the known risk SNPs. The associations of age, sex, top principle components, and known risk variants with pancreatic cancer risk in conditional analyses were shown in Supplementary Table 3.
We compared performance of the two models, PRS1 including age, sex, top principal components, and GWAS identified risk variants, and PRS2 which also included TWAS identified genes, for risk prediction of pancreatic cancer. Compared with PRS1 (AUC=0.621), PRS2 (AUC=0.633) has a better performance, with AUC increased by 1.2% (Figure 3).
Figure 3.
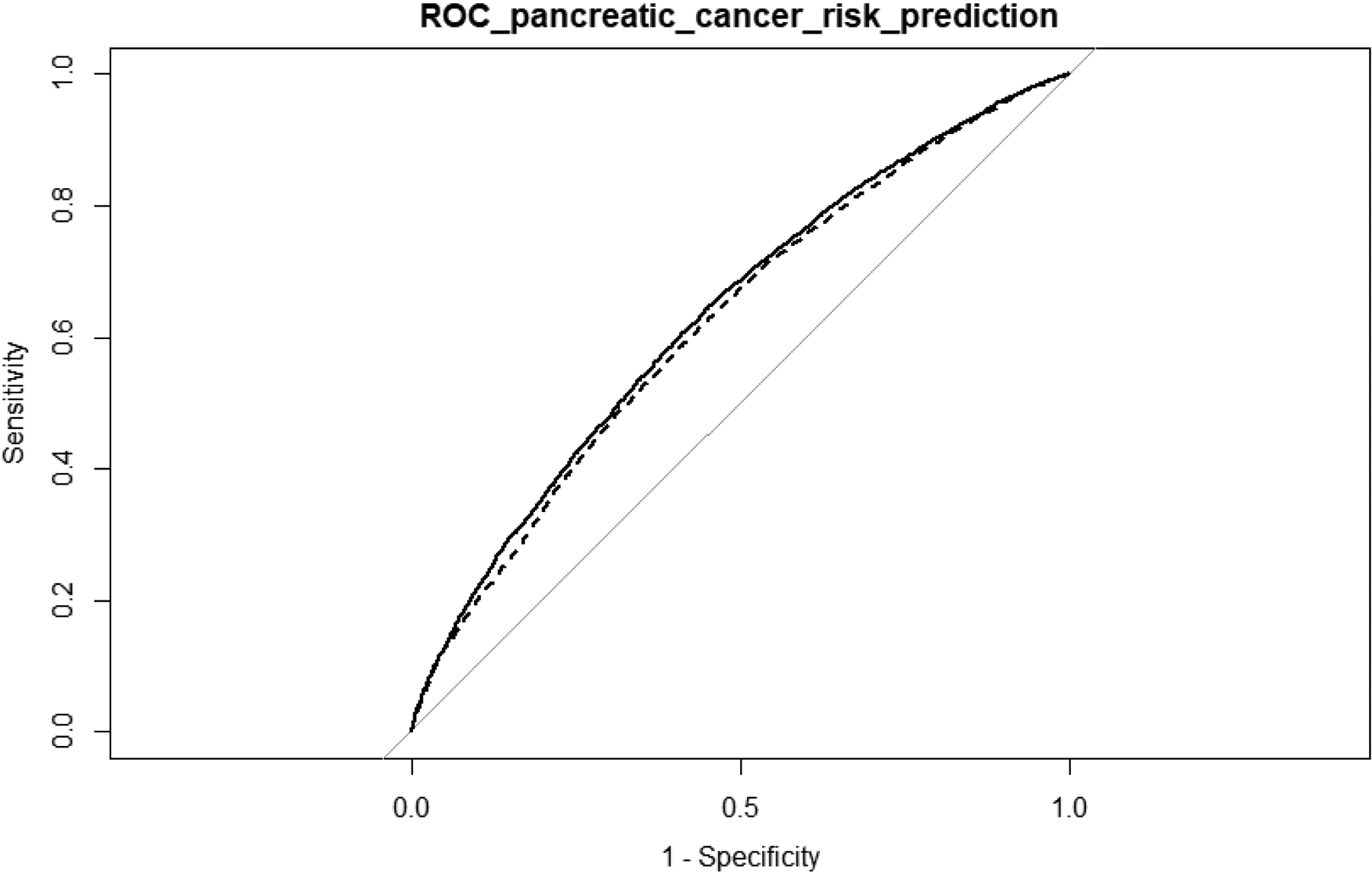
Performance of prediction models with and without incorporating TWAS identified genes in pancreatic cancer risk prediction. The dash line represents the PRS1 including age, sex, top principal components, and GWAS identified risk variants. The solid line represents PRS2 which also included TWAS identified genes. The AUC for PRS1 and PRS2 are 0.621 and 0.633, respectively.
Discussion
Leveraging the largest available reference dataset for normal pancreas tissue transcriptome and a joint-tissue genetic modeling strategy for gene expression, we performed a comprehensive TWAS study to evaluate the relationship between genetically predicted gene expression in pancreas tissue and pancreatic cancer risk. We identified 13 genes whose genetically predicted expression was associated with pancreatic cancer risk (FDR ≤ 0.05), including six novel genes. Even after adjusting for risk SNPs identified in previous GWAS studies, the associations for ten genes (six novel genes and four reported genes) remained statistically significant. Our study provides substantial new information to improve the understanding of genetics and etiology for pancreatic cancer.
Several novel genes that we identified in this study have been shown to play potential roles in regulating lipid trafficking (StAR-related lipid transfer domain containing 3, STARD3), cancer progression (ADAM metallopeptidase domain 33, ADAM33), apoptosis (Gasdermin-B, GSDMB; Myotubularin-related protein 6, MTMR6), and vesicle fusion (SFT2 domain containing 3, SFT2D3). StAR (steroidogenic acute regulatory protein) is a member of a subfamily of lipid trafficking protein which localizes to the membranes of late endosomes and is involved in cholesterol transport (28,29). STARD3 has been shown to be co-amplified with HER2/neu over-expression (30,31) and associated with shorter overall and disease-free survival in breast cancer patients (32). Vassilev B et al. suggested that STARD3 over-expression resulted in increased cholesterol biosynthesis and Src kinase activity in breast cancer cells (33). Moreover, STARD3 is also over-expressed in the development of gastric cancer (34) and prostate cancer (35). The present study shows that increased genetically predicted expression of STARD3 was associated with increased risk of pancreatic cancer. This direction of effect with pancreatic cancer is consistent with the patterns for breast, gastric and prostate cancer. ADAM33 encodes a protein that is a type I transmembrane glycoprotein. Members of ADAM family are membrane-immobilized proteins that are related to snake venom double integrin structurally. The protein is involved in cell adhesion and plays an important role in cancer progression (36). The over-expression of ADAM33 was found to contribute to the pathogenesis of sinonasal inverted papillomas (37), laryngeal carcinoma (38), and gastric cancer (39). Interestingly, in other work, reduced ADAM33 gene expression was associated with increased risk of breast cancer (40), triple-negative breast cancer and basal-like markers, as well as shorter metastasis-free survival and overall survival of breast cancer (41). GSDMB (17q21.1) encodes a member of the gasdermin-domain containing protein family that is potentially involved in the regulation of apoptosis in cancer (42,43). Human GSDMB is transcribed in proliferating normal epithelial cells. Over-expression of GSDMB is associated with reduced survival and increased metastasis in breast cancer patients (44,45), and correlated with carcinogenesis and progression of uterine cervix cancer (46). It was also identified as a potential oncogene for esophageal squamous cell carcinoma and gastric cancer (47). MTMR6 (13q12.13) encodes Myotubularin-related protein 6 which is a catalytically active member of the myotubularin (MTM) family. The formation of the MTMR6-MTMR9 complex could regulate DNA damage-induced apoptosis (48). The expression of MTMR6 was higher in ovarian tumor tissues compared with tumor-adjacent normal tissues (48). SFT2D1 (6q27) encodes SFT2 domain-containing protein 1. SFT2 is a non-essential membrane protein and localized to late-Golgi compartment. SFT2 plays an important role in the process of vesicle fusion with the Golgi complex. Low SFT2D1 gene expression predicted poor outcome in high-risk neuroblastoma patients (49).
Seven genes showing a significant association in our study have been reported in an earlier TWAS for pancreatic cancer risk (18). The directions of their associations of genetically predicted expression were consistent. Some earlier studies have suggested that five of them are potentially associated with pancreatic cancer. PDX1 (pancreatic and duodenal homeobox 1) is a “master regulator” of pancreas development. PDX1 is a crucial product of the developing pancreas, and plays a crucial role in preventing pancreatic intraepithelial neoplasia that precedes pancreatic ductal adenocarcinoma (50,51). Previous research has identified rs3818626 in SMC2 (structural maintenance of chromosomes 2) to be associated with pancreatic cancer risk (52). ABO (alpha 1–3-N-acetylgalactosaminyltransferase and alpha 1–3-galactosyltransferase) located at the 9q34 region and its encoding protein was the basis of the ABO blood group system which was biosynthesized by A and B-transferases (53). Multiple studies have suggested ABO to be associated with risk of pancreatic cancer (5,54). The gene PGAP3 (post-GPI attachment to proteins phospholipase 3) encodes a glycosylphosphatidylinositol (GPI)-specific phospholipase that primarily localizes to the Golgi apparatus. The tethering of proteins to plasma membranes via posttranslational GPI-anchoring plays a key role in protein sorting and trafficking (55). Walsh N et al. identified three PGAP3 polymorphisms to be potentially relevant to risk of pancreatic ductal adenocarcinoma (6). INHBA (inhibin subunit beta A) encodes a member of the TGF-beta (transforming growth factor-beta) superfamily of proteins, and the encoded protein is processed to generate a subunit of the dimeric activin and inhibin protein complexes proteolytically (56). It was identified that INHBA was over-expressed in pancreatic tumors and associated with reduced patient survival (57). Although based on literature search we did not identify studies reporting link between two other genes (INHBA and RCC1 domain-containing protein 1(RCCD1) and pancreatic cancer, they have been reported to be potentially related to several other tumors (Supplementary Table 4).
Of the other 18 genes identified in Zhong et al., for six of them (CELA3B, SMUG1, BTBD6, SUPT4H1, PGPEP1 and ZDHHC11B), we were able to build their corresponding genetic prediction models (18). Of these, SMUG1, BTBD6, and PGPEP1 were also nominally significant at P<0.05 with the direction consistent with that reported in Zhong et al., and the P=0.10 for SUPT4H1 with the same direction (Supplementary Table 5). For the 12 remaining genes, we were not able to build genetic prediction models with R2≥ 0.01 using the UTMOST method. It is worth noting that, for six of these 12 genes, namely, TERT, CLPTM1L, SMC2-AS1, RP11–80H5.9, BCAR1, and TMEM170A, their associations identified in Zhong et al. were based on prediction models in non-pancreas tissues. For several others, the associations were identified based on prediction models built using data from tumor-adjacent normal pancreas tissue from cancer patients instead of normal pancreas tissue from healthy subjects. As we noted in the Introduction, some gene expression traits might be substantially altered in tumor-adjacent normal tissues due to the somatic changes. Further research is warranted to better characterize the associations of these genes with pancreatic cancer.
Compared with many other existing methods, the joint-tissue strategy of UTMOST confers significant advantages. Many other methods, such as PrediXcan and TWAS/FUSION, do not take into consideration the similarity of genetic regulation for many genes across different human tissues (58,59), thereby posing a challenge when the effective number of the corresponding tissue samples is low (60). UTMOST is a powerful method to jointly analyze data from multiple genetically-correlated tissues, thus significantly improving the accuracy of expression imputation in available tissues to enhance power for gene discovery. Based on assessments in internal cross validation and external validation, the gene expression imputation accuracy can be significantly improved for the UTMOST strategy compared with PrediXcan method, as well as the Bayesian Sparse Linear Mixed-effects Model (BSLMM), a method used in TWAS/FUSION (14). In the previous pancreatic cancer TWAS, PrediXcan and TWAS/FUSION methods were used to develop gene expression prediction models for pancreas and individual non-pancreas tissues (18).
The sample size for association analysis in this study was large, which could provide high statistical power to detect associations for genes with a relatively high cis-heritability (h2). For example, our study has 80% statistical power to detect an association with pancreatic cancer risk at P < 8.00×10−5 (similar to FDR < 0.05) with an OR of 1.28 or higher per one standard deviation increase (or decrease) in the expression level of genes with an h2 of 0.1 or higher. The design of using genetic instruments reduces selection bias and potential influence due to reverse causation. On the other hand, several potential limitations need to be acknowledged. First, the associations identified in this study do not necessarily imply causality. Aligned with other reports, although TWAS is useful for prioritizing causal genes, false positive findings could exist for some of the identified associations (61). Several reasons can potentially induce these, including correlated expression across individuals, correlated predicted expression, as well as shared variants (61). In our study, two identified genes, STARD3 and PGAP3, are both located in region 17q12. Future functional investigation will better characterize whether the identified genes play a causal role in pancreatic tumorigenesis. Second, in TWAS design the estimated genetically regulated component of gene expression but not the overall expression is evaluated, thus the relationship between overall gene expression and diseases cannot be directly inferred from TWAS and need to be assessed in different studies. Third, in the current study for the identified associated genes, we are not able to evaluate whether their associations with pancreatic cancer risk differ according to family history of pancreatic cancer and tumor stage/grade due to a lack of relevant information. Future work investigating this is needed to better understand the associations.
Besides improving understanding of genetics and etiology of pancreatic cancer, the identification of candidate susceptibility genes may also improve risk prediction of this deadly malignancy. In our evaluation, the prediction model incorporating TWAS identified genes confers an improved performance compared with a model without such TWAS identified genes (Figure 3). On the other hand, the current prediction model only serves for illustration purpose and additional work is needed to better evaluate performance of the model incorporating TWAS identified genes for predicting pancreatic cancer risk. For example, additional risk factors for pancreatic cancer, such as smoking, heavy alcohol consumption, obesity, chronic pancreatitis, type 2 diabetes, and family history of pancreatic cancer can be further included in such a model. Secondly, in the current study we only applied the intuitive logistic regression model. More sophisticated models can be explored to evaluate whether new models can be developed with improved performance. Thirdly, we used the PanScan/PanC4 data, based on which the associated genes and risk variants were identified, for evaluating model performance. Ideally, the performance of such a prediction model could be assessed in independent datasets that have not been used for the identification of these genes and variants to provide an unbiased assessment.
In conclusion, in this large-scale TWAS study of pancreatic cancer, we identified 13 genes whose genetically predicted expression was associated with pancreatic cancer risk, including six novel genes. Ten of these genes remained statistically significant after adjusting for risk SNPs identified in previous GWAS studies. Further investigation of these genes will provide new insights into the biology and genetics of pancreatic cancer.
Supplementary Material
Statement of significance:
A Transcriptome-wide association analysis identifies seven previously reported and six novel candidate susceptibility genes for pancreatic cancer risk.
Acknowledgements
This study is supported by the University of Hawaii Cancer Center. Lang Wu is supported by NCI R00 CA218892. Nancy J Cox is supported by U01HG009086. Eric Gamazon is supported by the National Human Genome Research Institute of the National Institutes of Health under Award Number R35HG010718 and R01HG011138. Qizhi Yao is supported by VA Merit Award 1 I01 CX001822-01A2 (PI: Yao). Duo Liu is partially supported by the Harbin Medical University Cancer Hospital. Yanfa Sun is partially supported by the Department of Education of Fujian Province, P R China. The datasets used for the analyses described in this manuscript were obtained from dbGaP at http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap through dbGaP accession phs000206.v5.p3 and phs000648.v1.p1. The authors would like to thank all of the individuals for their participation in the parent PanScan/PanC4 studies and all the researchers, clinicians, technicians and administrative staff for their contribution to the studies. The PanScan study was funded in whole or in part with federal funds from the National Cancer Institute (NCI), US National Institutes of Health (NIH) under contract number HHSN261200800001E. Additional support was received from NIH/NCI K07 CA140790, the American Society of Clinical Oncology Conquer Cancer Foundation, the Howard Hughes Medical Institute, the Lustgarten Foundation, the Robert T. and Judith B. Hale Fund for Pancreatic Cancer Research and Promises for Purple. A full list of acknowledgments for each participating study is provided in the Supplementary Note of the manuscript with PubMed ID: 25086665. For the PanC4 GWAS study, the patients and controls were derived from the following PANC4 studies: Johns Hopkins National Familial Pancreas Tumor Registry, Mayo Clinic Biospecimen Resource for Pancreas Research, Ontario Pancreas Cancer Study (OPCS), Yale University, MD Anderson Case Control Study, Queensland Pancreatic Cancer Study, University of California San Francisco Molecular Epidemiology of Pancreatic Cancer Study, International Agency of Cancer Research and Memorial Sloan Kettering Cancer Center. This work is supported by NCI R01CA154823 Genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number HHSN2682011000111. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing financial interests
Eric Gamazon receives an honorarium from the journal Circulation Research of the American Heart Association as a member of the Editorial Board. There is no potential conflict of interests for other authors.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA: A Cancer Journal for Clinicians; 2020;70:7–30 [DOI] [PubMed] [Google Scholar]
- 2.Moore A, Donahue T. Pancreatic Cancer. Jama 2019;322:1426- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petersen GM. Familial pancreatic cancer. 2016. Elsevier; p 548–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klein AP, Wolpin BM, Risch HA, Stolzenberg-Solomon RZ, Mocci E, Zhang M, et al. Genome-wide meta-analysis identifies five new susceptibility loci for pancreatic cancer. Nature communications 2018;9:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Childs EJ, Mocci E, Campa D, Bracci PM, Gallinger S, Goggins M, et al. Common variation at 2p13. 3, 3q29, 7p13 and 17q25. 1 associated with susceptibility to pancreatic cancer. Nature genetics 2015;47:911–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolpin BM, Rizzato C, Kraft P, Kooperberg C, Petersen GM, Wang Z, et al. Genome-wide association study identifies multiple susceptibility loci for pancreatic cancer. Nature genetics 2014;46:994–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petersen GM, Amundadottir L, Fuchs CS, Kraft P, Stolzenberg-Solomon RZ, Jacobs KB, et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22. 1, 1q32. 1 and 5p15. 33. Nature genetics 2010;42:224–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Low S-K, Kuchiba A, Zembutsu H, Saito A, Takahashi A, Kubo M, et al. Genome-wide association study of pancreatic cancer in Japanese population. PloS one 2010;5:e11824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu C, Miao X, Huang L, Che X, Jiang G, Yu D, et al. Genome-wide association study identifies five loci associated with susceptibility to pancreatic cancer in Chinese populations. Nature genetics 2012;44:62–6 [DOI] [PubMed] [Google Scholar]
- 10.Zhang M, Lykke-Andersen S, Zhu B, Xiao W, Hoskins JW, Zhang X, et al. Characterising cis-regulatory variation in the transcriptome of histologically normal and tumour-derived pancreatic tissues. Gut 2018;67:521–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu L, Wang J, Cai Q, Cavazos TB, Emami NC, Long J, et al. Identification of novel susceptibility loci and genes for prostate cancer risk: a transcriptome-wide association study in over 140,000 European descendants. Cancer research 2019;79:3192–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu L, Shi W, Long J, Guo X, Michailidou K, Beesley J, et al. A transcriptome-wide association study of 229,000 women identifies new candidate susceptibility genes for breast cancer. Nature genetics 2018;50:968–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, et al. A gene-based association method for mapping traits using reference transcriptome data. Nature genetics 2015;47:1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nature genetics 2016;48:245–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu Y, Beeghly-Fadiel A, Wu L, Guo X, Li B, Schildkraut JM, et al. A transcriptome-wide association study among 97,898 women to identify candidate susceptibility genes for epithelial ovarian cancer risk. Cancer research 2018;78:5419–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mancuso N, Gayther S, Gusev A, Zheng W, Penney KL, Kote-Jarai Z, et al. Large-scale transcriptome-wide association study identifies new prostate cancer risk regions. Nature communications 2018;9:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang T, Choi J, Kovacs MA, Shi J, Xu M, Goldstein AM, et al. Cell-type–specific eQTL of primary melanocytes facilitates identification of melanoma susceptibility genes. Genome research 2018;28:1621–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhong J, Jermusyk A, Wu L, Hoskins JW, Collins I, Mocci E, et al. A Transcriptome-Wide Association Study Identifies Novel Candidate Susceptibility Genes for Pancreatic Cancer. JNCI: Journal of the National Cancer Institute 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu Y, Li M, Lu Q, Weng H, Wang J, Zekavat SM, et al. A statistical framework for cross-tissue transcriptome-wide association analysis. Nature genetics 2019;51:568–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Consortium G The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015;348:648–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Consortium G Genetic effects on gene expression across human tissues. Nature 2017;550:204–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amundadottir L, Kraft P, Stolzenberg-Solomon RZ, Fuchs CS, Petersen GM, Arslan AA, et al. Genome-wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nature genetics 2009;41:986–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang M, Wang Z, Obazee O, Jia J, Childs EJ, Hoskins J, et al. Three new pancreatic cancer susceptibility signals identified on chromosomes 1q32. 1, 5p15. 33 and 8q24. 21. Oncotarget 2016;7:66328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nature genetics 2016;48:1279–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu J, Shu X, Guo X, Liu D, Bao J, Milne RL, et al. Associations between genetically predicted blood protein biomarkers and pancreatic cancer risk. Cancer Epidemiology and Prevention Biomarkers 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barbeira AN, Dickinson SP, Bonazzola R, Zheng J, Wheeler HE, Torres JM, et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nature communications 2018;9:1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Team RC. R: A language and environment for statistical computing. Vienna, Austria; 2013. [Google Scholar]
- 28.Wilhelm LP, Wendling C, Védie B, Kobayashi T, Chenard MP, Tomasetto C, et al. STARD 3 mediates endoplasmic reticulum-to-endosome cholesterol transport at membrane contact sites. The EMBO journal 2017;36:1412–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lapillo M, Salis B, Palazzolo S, Poli G, Granchi C, Minutolo F, et al. First-of-its-kind STARD3 Inhibitor: In Silico Identification and Biological Evaluation as Anticancer Agent. ACS medicinal chemistry letters 2019;10:475–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kauraniemi P, Bärlund M, Monni O, Kallioniemi A. New amplified and highly expressed genes discovered in the ERBB2 amplicon in breast cancer by cDNA microarrays. Cancer research 2001;61:8235–40 [PubMed] [Google Scholar]
- 31.Dressman MA, Baras A, Malinowski R, Alvis LB, Kwon I, Walz TM, et al. Gene expression profiling detects gene amplification and differentiates tumor types in breast cancer. Cancer research 2003;63:2194–9 [PubMed] [Google Scholar]
- 32.Vinatzer U, Dampier B, Streubel B, Pacher M, Seewald MJ, Stratowa C, et al. Expression of HER2 and the coamplified genes GRB7 and MLN64 in human breast cancer: quantitative real-time reverse transcription-PCR as a diagnostic alternative to immunohistochemistry and fluorescence in situ hybridization. Clinical cancer research 2005;11:8348–57 [DOI] [PubMed] [Google Scholar]
- 33.Vassilev B, Sihto H, Li S, Hölttä-Vuori M, Ilola J, Lundin J, et al. Elevated levels of StAR-related lipid transfer protein 3 alter cholesterol balance and adhesiveness of breast cancer cells: potential mechanisms contributing to progression of HER2-positive breast cancers. The American journal of pathology 2015;185:987–1000 [DOI] [PubMed] [Google Scholar]
- 34.Yun S, Yoon K, Lee S, Kim E, Kong S, Choe J, et al. PPP1R1B-STARD3 chimeric fusion transcript in human gastric cancer promotes tumorigenesis through activation of PI3K/AKT signaling. Oncogene 2014;33:5341–7 [DOI] [PubMed] [Google Scholar]
- 35.Stigliano A, Gandini O, Cerquetti L, Gazzaniga P, Misiti S, Monti S, et al. Increased metastatic lymph node 64 and CYP17 expression are associated with high stage prostate cancer. Journal of endocrinology 2007;194:55–61 [DOI] [PubMed] [Google Scholar]
- 36.White JM. ADAMs: modulators of cell–cell and cell–matrix interactions. Current opinion in cell biology 2003;15:598–606 [DOI] [PubMed] [Google Scholar]
- 37.Stasikowska-Kanicka O, Wągrowska-Danilewicz M, Danilewicz M. Immunohistochemical study on ADAM33 in sinonasal inverted papillomas and squamous cell carcinomas of the larynx. Archives of Medical Science: AMS 2016;12:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Topal O, Erinanc H, Ozer C, Canpolat E, Celik S, Erbek S. Expression of “a disintegrin and metalloproteinase-33”(ADAM-33) protein in laryngeal squamous cell carcinoma. The Journal of Laryngology & Otology 2012;126:511–5 [DOI] [PubMed] [Google Scholar]
- 39.Kim K-E, Song H, Hahm C, Yoon SY, Park S, Lee H-r, et al. Expression of ADAM33 is a novel regulatory mechanism in IL-18-secreted process in gastric cancer. The Journal of Immunology 2009;182:3548–55 [DOI] [PubMed] [Google Scholar]
- 40.Yang PJ, Hou MF, Tsai EM, Liang SS, Chiu CC, Ou-Yang F, et al. Breast cancer is associated with methylation and expression of the a disintegrin and metalloproteinase domain 33 (ADAM33) gene affected by endocrine-disrupting chemicals. Oncology reports 2018;40:2766–77 [DOI] [PubMed] [Google Scholar]
- 41.Manica GC, Ribeiro CF, De Oliveira MA, Pereira IT, Chequin A, Ramos EA, et al. Down regulation of ADAM33 as a predictive biomarker of aggressive breast cancer. Scientific reports 2017;7:44414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Croteau-Chonka DC, Qiu W, Martinez FD, Strunk RC, Lemanske RF Jr, Liu AH, et al. Gene expression profiling in blood provides reproducible molecular insights into asthma control. American journal of respiratory and critical care medicine 2017;195:179–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016;535:111–6 [DOI] [PubMed] [Google Scholar]
- 44.Molina-Crespo Á, Cadete A, Sarrio D, Gámez-Chiachio M, Martinez L, Chao K, et al. Intracellular delivery of an antibody targeting Gasdermin-B reduces HER2 breast cancer aggressiveness. Clinical Cancer Research 2019;25:4846–58 [DOI] [PubMed] [Google Scholar]
- 45.Hergueta-Redondo M, Sarrio D, Molina-Crespo Á, Vicario R, Bernadó-Morales C, Martínez L, et al. Gasdermin B expression predicts poor clinical outcome in HER2-positive breast cancer. Oncotarget 2016;7:56295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun Q, Yang J, Xing G, Sun Q, Zhang L, He F. Expression of GSDML associates with tumor progression in uterine cervix cancer. Translational oncology 2008;1:73-IN1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saeki N, Usui T, Aoyagi K, Kim DH, Sato M, Mabuchi T, et al. Distinctive expression and function of four GSDM family genes (GSDMA-D) in normal and malignant upper gastrointestinal epithelium. Genes, Chromosomes and Cancer 2009;48:261–71 [DOI] [PubMed] [Google Scholar]
- 48.Zou J, Chang S-C, Marjanovic J, Majerus PW. MTMR9 increases MTMR6 enzyme activity, stability, and role in apoptosis. Journal of Biological Chemistry 2009;284:2064–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ognibene M, Morini M, Garaventa A, Podestà M, Pezzolo A. Identification of a minimal region of loss on chromosome 6q27 associated with poor survival of high-risk neuroblastoma patients. Cancer Biology & Therapy 2020;21:391–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shih HP, Seymour PA, Patel NA, Xie R, Wang A, Liu PP, et al. A gene regulatory network cooperatively controlled by Pdx1 and Sox9 governs lineage allocation of foregut progenitor cells. Cell reports 2015;13:326–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roy N, Hebrok M. Regulation of cellular identity in cancer. Developmental cell 2015;35:674–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feng Y, Liu H, Duan B, Liu Z, Abbruzzese J, Walsh KM, et al. Potential functional variants in SMC2 and TP53 in the AURORA pathway genes and risk of pancreatic cancer. Carcinogenesis 2019;40:521–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rummel SK, Ellsworth RE. The role of the histoblood ABO group in cancer. Future Science OA 2016;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Y, Liu L, Huang Y, Zheng H, Li L. Association of ABO polymorphisms and pancreatic Cancer/Cardiocerebrovascular disease: a meta-analysis. BMC medical genetics 2020;21:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fujita M, Umemura M, Yoko-o T, Jigami Y. PER1 is required for GPI-phospholipase A2 activity and involved in lipid remodeling of GPI-anchored proteins. Molecular biology of the cell 2006;17:5253–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brown CW, Houston-Hawkins DE, Woodruff TK, Matzuk MM. Insertion of Inhbb into the Inhba locus rescues the Inhba-null phenotype and reveals new activin functions. Nature genetics 2000;25:453–7 [DOI] [PubMed] [Google Scholar]
- 57.Zhong X, Pons M, Poirier C, Jiang Y, Liu J, Sandusky GE, et al. The systemic activin response to pancreatic cancer: Implications for effective cancer cachexia therapy. Journal of cachexia, sarcopenia and muscle 2019;10:1083–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Consortium G, Laboratory D. Genetic effects on gene expression across human tissues. Nature 2017;550:204–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu X, Finucane HK, Gusev A, Bhatia G, Gazal S, O’Connor L, et al. Functional architectures of local and distal regulation of gene expression in multiple human tissues. The American Journal of Human Genetics 2017;100:605–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huan T, Joehanes R, Song C, Peng F, Guo Y, Mendelson M, et al. Genome-wide identification of DNA methylation QTLs in whole blood highlights pathways for cardiovascular disease. Nat Commun 2019;10:4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wainberg M, Sinnott-Armstrong N, Mancuso N, Barbeira AN, Knowles DA, Golan D, et al. Opportunities and challenges for transcriptome-wide association studies. Nature genetics 2019;51:592–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.