

Abstract
Vitamin C (ascorbic acid) is a water-soluble antioxidant and a cofactor for a large number of enzymes. It is present in all tissues and especially abundant in corneal epithelium, stem cells, and neurons. Although similar to thiols in its ability to react with many reactive oxygen species (ROS), ascorbate is much better (>100× faster) than glutathione at scavenging of primary ROS (superoxide radical and singlet oxygen). Ascorbate appears to be especially important for elimination of O2•– in the nucleus which contains little or no SOD activity. Cofactor functions of ascorbate involve the maintenance of activity of Fe(II)/2-oxoglutarate-dependent dioxygenases via reduction of Fe(III). The most prominent activity of ascorbate-dependent dioxygenases in the cytoplasm is hydroxylation of prolines in proteins involved in the formation of extracellular matrix and regulation of metabolism and hypoxia responses. In the nucleus, ascorbate is important for oxidative demethylation of 5-methylcytosine in DNA (by TET proteins) and removal of methyl groups from histone lysines (by JmjC demethylases). Differentiation and other cellular reprograming processes involving DNA demethylation are especially sensitive to ascorbate insufficiency. High doses of vitamin C alone or in combinations with drugs produced cancer-suppressive effects which involved redox, immune, and epigenetic mechanisms. Solutions to vitamin C deficiency in cultured cells are discussed to improve the physiological relevance of in vitro models. An abundance of vitamin C in rodents limits their ability to fully recapitulate human sensitivity to adverse health effects of malnutrition and xenobiotics, including neurotoxicity, lung injury, and intergenerational and other epigenetic effects.
Vitamin C is an essential micronutrient to humans and other primates caused by loss-of-function mutations in l-gulono-1.4-lactone oxidase (encoded by GULO gene) which catalyzes the final step in ascorbic acid biosynthesis from glucose. The majority of eukaryotes synthesize ascorbic acid and yeast, producing a related compound erythro-ascorbic acid. Vitamin C is found in the diet and in vivo in its reduced (ascorbic acid) or oxidized (dehydroascorbic acid) forms (Figure 1A). Due to its low pKa = 4.2 at the C3 hydroxyl, ascorbic acid is present almost completely (>99%) as ascorbate anion at physiological pH. Severe vitamin C deficiency in humans leads to scurvy, which is a fatal disease that can be cured only by a restored intake of vitamin C.1 Several overt clinical manifestations of scurvy (reopening of old wounds, bleeding, loss of teeth, gums, and skin abnormalities) are clearly linked to the long-established role of ascorbate in the production of mature collagen. Other symptoms and high-fatality rates were more difficult to explain by defects in collagen formation, pointing to a broader set of essential activities of ascorbate. Although the occurrence of scurvy is rare in the modern world, the incidence of a significant vitamin C deficiency is still common even in the wealthy countries.2−4 A recent popularity of animal product-based (ketogenic and similar) diets can potentially increase the incidence or extent of vitamin C insufficiency. Vitamin C deficiency is at least partially responsible for intergenerational epigenetic effects produced by a nutritional deprivation during pregnancy.5 The main physiological functions of ascorbate include its activities as an antioxidant and a critical cofactor for a growing number of enzymes that are directly or indirectly involved in multiple cellular activities. Resurgence of research interest in vitamin C is further fueled by recent discoveries of its importance in the main biochemical processes regulating epigenetics and its emerging promise in cancer treatment. These new findings indicate that the role of ascorbate in modulation of cellular responses to toxicants, drugs, and other stressors extends beyond its antioxidant activities.
Figure 1.

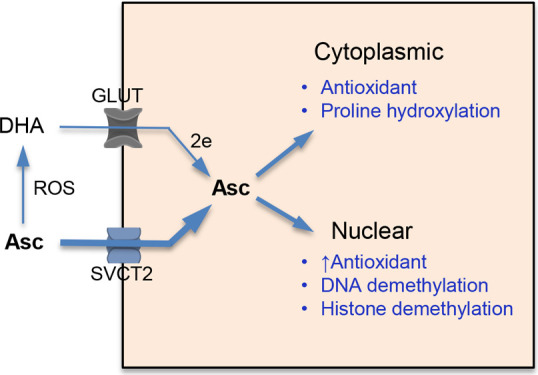
Chemical forms and cellular metabolism of vitamin C. (A) Structures of the main forms of vitamin C. At physiological pH, reduced vitamin C predominantly exists as ascorbate anion due to its low pKa = 4.2. (B) Cellular uptake of reduced (ascorbate, Asc) and oxidized (dehydroascorbic acid, DHA) forms of vitamin C. Extracellular concentrations of dehydroascorbic acid and, consequently, the importance of its uptake are higher for cells that release large amounts of oxidants during the inflammatory responses (neutrophils, for example). Extracellular reduction of Fe(III) by ascorbate is another source of oxidized vitamin C which enters cells through GLUT glucose transporters. Fe(II) is taken up by cells via the divalent metal transporter DMT1.
Vitamin C Uptake and Concentrations In Vivo
Ascorbate enters cells through sodium-dependent vitamin C transporters (SVCT) 1 and 2.6−8 SVCT1 (SLC23A1) is important for ascorbate absorption in the intestine and, especially, for reabsorption in the kidney. A more ubiquitously expressed SVCT2 (SLC23A2) is responsible for ascorbate uptake by cells in a majority of tissues. Consistent with its major physiological importance, SVCT2 knockout mice die almost immediately after birth.9 Dehydroascorbic acid structurally resembles glucose and enters cells via GLUT transporters, which is followed by its rapid reduction to ascorbate (Figure 1B). Human plasma contains on average approximately 50 μM vitamin C, which predominantly comprises ascorbate. Cellular concentrations of ascorbate are much higher and range from 1 to 5 mM for most tissues, but they reach 10 mM and higher in neurons, adrenal glands, and corneal epithelium.10,11 Ascorbate is the most dramatically elevated metabolite in human and mouse hematopoietic stem cells (up to 18×) in comparison to their more differentiated progeny.12 Based on the established role of ascorbate in stem cells (discussed below), it is possible that many other if not all stem cells in vivo contain much higher ascorbate levels than the bulk of any tissue containing differentiated cells. Although plasma concentrations of dehydroascorbic are low (1–2 μM), human erythrocytes (cells lacking SVCT1/2 proteins) are very efficient at its uptake through the GLUT1 transporter and are able to accumulate plasma-level concentrations of vitamin C, which are proposed to constitute its physiological reservoir in humans.13 Ascorbate export mechanisms of from cells are currently unknown. High levels of ascorbate in tissues (20–200× over plasma concentration) are also frequently viewed as a systemic reservoir of vitamin C in humans. The postulated reservoir role is probably not the primary biological reason for high ascorbate in human tissues, as equally high tissue concentrations of ascorbate are also present in mice and rats that continuously produce copious amounts of vitamin C (∼100× over recommended intake in humans). There is now clear experimental evidence that very high ascorbate concentrations are in fact necessary for the normal functions of some cells (stem cells, for example). Although a scurvy-level vitamin C deficiency is currently rare, significantly lower ascorbate levels are frequently found among cancer patients, which in experimental models promoted cancer aggressiveness and limited responses to several chemotherapeutics.
Antioxidant Activities in Cells
Ascorbate is generally known as a water-soluble antioxidant. However, the importance of its specific protective activities and the overall role as the radical scavenger and antioxidant are not well covered or even completely ignored in some otherwise comprehensive reviews of cellular antioxidant defenses. Among small antioxidants, glutathione typically receives a lot of attention, although, as discussed below, it is clearly an inferior direct scavenger of the primary cellular oxidants in comparison to ascorbate. Underappreciation of ascorbate could be related to its (near) absence in cultured cells, which are the most commonly used biological models in studies of oxidative stress. In ascorbate-devoid cells, glutathione certainly takes on the role of the key small antioxidant in the direct detoxification of reactive oxygen species (ROS). A large portion of cellular damage by ROS frequently results from the reactive products of lipid oxidation, such as acrolein, crotonaldehyde, and 4-hydroxy-2-nonenal. Glutathione and other small thiols protect against toxicity of aldehydes, especially the most toxic group of α,β-unsaturated aldehydes, through the formation of inactive conjugates.14–16 This property is not shared by ascorbate which acts as a reducer (electron donor) and does not form covalent bonds with reactive carbonyls. However, as discussed below, ascorbate is much more effective than glutathione in the elimination of primary ROS.
Ascorbate is a broad spectrum antioxidant as it can react with a range of organic radicals and ROS. One-electron reduction of ROS and other radicals by ascorbate results in the formation of the resonance-stabilized ascorbate radical, which is completely unreactive with other biomolecules and is primarily lost via disproportionation reaction with another ascorbate radical, yielding ascorbate and dehydroascorbic acid.17 A broad radical/ROS reactivity is not unique to ascorbate, as it is also found among thiols including glutathione which is present in cells at comparable concentrations to those of ascorbate. For example, both glutathione and ascorbate are very effective at removal of a highly toxic hydroxyl radical (•OH). The reactivity of ascorbate and glutathione with the most abundant cellular ROS, H2O2, is very limited (rate constants 2 × 10 M–1 s–1 and 0.9 M–1 s–1, respectively)18 and unlikely to be physiologically significant. The importance of glutathione in suppression of H2O2 toxicity stems from its role as a cofactor for glutathione peroxidases (GPXs), especially a very effective cytoplasmic enzyme GPX1 which is present in cells at an ∼1 μM concentration and has the rate constant of 2 × 107 M–1 s–1 for H2O2.123 Multiplication of rate constants by cellular concentrations of GPX1 and ascorbate in vivo (5 mM) gives rates of H2O2 elimination equal to 20 s–1 and 0.01 s–1, respectively. Thus, the enzymatic activity of GPX1 is expected to be 2000× more effective at detoxification of H2O2 in comparison to ascorbate and even higher relative to the non-enzymatic removal with glutathione. Catalase and peroxiredoxin-1 (PRDX1) represent other abundant and very effective enzymatic systems for elimination of H2O2 by converting it into H2O, further indicating that H2O2 detoxification in cells is principally based on the biological mechanisms and likely includes minimal contributions from direct chemical reactions with small antioxidants. Although ascorbate is a highly hydrophilic molecule, it also suppresses oxidation of lipids by reducing the tocopheroxyl radical and thereby maintaining the levels of the main lipid-soluble antioxidant, vitamin E.19,20 Consistent with this role, addition of physiological levels of ascorbate inhibited lipid peroxide-dependent cell death (ferroptosis) in cultured cells.21 Glutathione is probably even more important for prevention of ferroptosis due to its role as a cofactor for the main lipid peroxide-eliminating enzyme GPX4.22 Thus, both ascorbate and glutathione suppress lipid peroxidation and ferroptosis but through mechanistically distinct mechanisms.
The main difference in the direct scavenging of the main ROS between ascorbate and glutathione (small thiols in general) is found in their reactivity with the superoxide anion radical (O2•–), which is >100× higher for ascorbate than for glutathione. Rate constant for ascorbate is 2.9 × 105 M–1 s–1 (mean of two studies),23,24 and the highest reported rate constant for glutathione is 1.1 × 103 M–1 s–1.25 Other estimates of rate constant for glutathione are much lower.15,18 O2•– is the primary cellular ROS arising during the respiratory metabolism in mitochondria, physiological enzymatic activities (NADPH oxidases, xanthine oxidase) and redox cycling of various drugs and toxicants.15,18 Dismutation of O2•– by the cellular enzymes SOD1/2 produces hydrogen peroxide (H2O2), whereas the reaction of O2•– with a weak oxidant nitric oxide (•NO) yields a secondary oxidant peroxynitrite (ONOO–). H2O2 and ONOO– can subsequently serve as sources of tertiary oxidants, including a very reactive OH• radical (Figure 2). Considering its abundance (10 μM or more)26 and a very high activity of cytoplasmic SOD1 in the removal of O2•– (k = 2 × 109 M–1 s–1),27 it is important to ask whether ascorbate can make a meaningful contribution to the elimination of this primary ROS. Using cellular concentrations and rate constants for calculations of their reaction rates (SOD1: 10 μM × 2 × 109 M–1 s–1 = 2 × 104 s–1; ascorbate: 5 mM × 2.9 × 105 M–1 s–1 = 1.5 × 103 s–1), SOD1 appears to be 13.3× more effective in the elimination of O2•– than ascorbate. This difference translates into a 7% contribution of ascorbate to the removal of cytoplasmic superoxide. In cells with its particularly high concentrations (stem cells, neurons, corneal epithelium), ascorbate should make a larger impact on scavenging O2•– and more significantly complement SOD1 activity. Although its superoxide-scavenging activity diminishes the formation of peroxynitrite, ascorbate is known to accelerate a release of •NO from S-nitrosoglutathione, which is a major physiological carrier of nitric oxide. Use of specific metal chelators in cell culture found a dependence of this activity on the availability of redox-active Cu.28 Since cellular concentrations of free Cu ions are tightly controlled, ascorbate-promoted decomposition of S-nitrosoglutathione appears to occur more likely outside the cells.
Figure 2.

Main cellular ROS and antioxidant mechanisms. Superoxide anion radical (O2•–) is the primary ROS produced by various physiological and pathophysiological processes. Reduction of O2•– by SODs or ascorbate (Asc) produces a less reactive but more diffusible H2O2 which is detoxified by catalase, peroxiredoxin 1 (PRDX1), and glutathione peroxidase 1 (GPX1). Reaction of H2O2 with free Fe(II) and less frequently Cu(I) yields a highly damaging •OH radical. Hydroxyl and other reactive radicals are also formed during the decomposition of peroxynitrite generated in the reaction of nitric oxide radical (•NO) with O2•–. In comparison with the biological processes, ascorbate makes no significant contribution to the detoxification of H2O2 and plays a minor role in the removal of cytoplasmic O2•– in the majority of cells. Secondary radicals (tertiary ROS) arising from H2O2 and ONOO– lack specialized enzymatic defenses and are primarily detoxified by small antioxidants such as ascorbate and small thiols (glutathione). Cells also lack enzymatic processes for removal of singlet oxygen (1O2) which is effectively eliminated by ascorbate via a two-electron reduction to H2O2. In contrast to the cytoplasm, the nucleus has a minimal or no SOD activity, which elevates the importance of ascorbate in scavenging superoxide in this cellular compartment.
In contrast to H2O2 and O2•–, tertiary ROS (•OH, •CO3–, and •NO2 radicals) (Figure 2) lack enzymatic defenses and are primarily eliminated in direct reactions with ascorbate and small thiols. Ascorbate is expected to be more important than glutathione in detoxification of nitrogen dioxide and carbonyl radicals. Both •CO3– and •NO2 preferentially react with the thiolate anion (RS–),29 which constitutes only a small fraction (5%) of the total glutathione at physiological pH. The predominance of the RSH form is advantageous for the reaction of glutathione with •OH, which proceeds through hydrogen abstraction.15 The rate constants for the reactions of •OH with glutathione (1.6 × 1010 M–1 s–1) and ascorbate (4.5 × 109 M–1 s–1)30 are very high and only moderately different, indicating that the relative contributions of these antioxidants to the detoxification of hydroxyl radical are largely a function of their cellular concentrations.
Another primary cellular oxidant that is effectively removed by ascorbate is singlet oxygen (1O2), which is an electronically excited state of O2. Although it is not a radical, 1O2 is a strong oxidant and mutagen. Singlet oxygen is produced in photoreactions of UV and visible light with biomolecules and released by certain enzymatic reactions (lipid peroxidases, for example).31 Cells lack specialized enzymes for detoxification of singlet oxygen and rely on small antioxidants for protection against this oxidant. The rate constant for reaction of ascorbate with 1O2 is very high (k = 3 × 108 M–1 s–1)32 and comparable to those of specialized enzymes detoxifying superoxide or hydrogen peroxide. Unlike reactions with other ROS, ascorbate acts as a two-electron donor in the elimination of singlet oxygen:
Scavenging of 1O2 by glutathione is also relatively fast (k = 2.4 × 106 M–1 s–1)33 but still approximately 100× slower than that by ascorbate. The singlet oxygen-reactive form of glutathione is the thiolate anion which is present as a minor fraction at physiological pH (pKa = 8.7 for glutathione). Thus, ascorbate is clearly a dominant small molecule scavenger of 1O2. A biological support for its importance in detoxification of 1O2 is the presence of high concentrations of ascorbate in human corneal epithelial cells (∼8 mM)34 which are continuously exposed to UV and visible light and, therefore, experience an extensive formation of singlet oxygen. Consistent with this interpretation, nocturnal animals have significantly lower ascorbate levels in their corneal epithelium in comparison to diurnal species. Protective effects of corneal ascorbate against UV-induced DNA damage have been confirmed in vivo.35,36 Another example where ascorbate reacts with a potent toxicant via two-electron transfer is reduction of carcinogenic chromium(VI).37 Reaction of Cr(VI) with ascorbate yields non-oxidizing Cr(IV). If Cr(VI) reacts with thiols, reduction proceeds through an one-electron transfer, and the resulting product, Cr(V), is a strong oxidant causing DNA breakage38 and phosphorylation of the transcription factor p53 by the oxidant-sensitive kinase ATM.39
Radical-scavenging activities of antioxidants are recognized as being important for all physiological functions of cells, including preservation of genome stability. The main ROS-detoxifying enzymes exhibit a clear intracellular compartmentalization. SOD1, PRDX1, and GPX1 are primarily cytoplasmic, SOD2 is a mitochondrial enzyme, and catalase resides in peroxisomes. The nucleus is the second largest compartment in the majority of cells, and it does not appear to contain significant levels of these antioxidant enzymes under normal conditions. A severe oxidative stress can cause a nuclear translocation of SOD1, which is linked to the regulation of transcription of the antioxidant genes.40 The paucity of enzymatic defenses makes the nucleus more dependent on the anti-ROS activity of small antioxidants. Nuclear concentrations of ascorbate have not been specifically established yet, but they are expected to be similar to those in cytoplasm since nuclear pores allow a free diffusion of molecules with molecular weight <1000 Da. The molecular weight of the ascorbate anion (175.1) is much smaller than this threshold, which should promote a near equilibrium between nuclear and cytoplasmic pools. Immunomicroscopy-based measurements in human cultured cells found that nuclear ascorbate levels were higher than those in the cytoplasm.41 However, it is unclear whether the employed aldehyde fixation for immunomicroscopy can be effective for retention and detection of the small molecule ascorbate. In the (near) absence of SODs, ascorbate is expected to play a dominant role in the removal of superoxide inside the nucleus. The apparent exclusion of SOD1 from the nucleus could be linked to the avoidance of the deleterious genotoxic effects of this enzyme which causes strand breaks when it comes in contact with DNA.42,43 Similar to cytoplasm, ascorbate should also act as a main reducer of mutagenic singlet oxygen. H2O2, the product of 1O2 and O2•– reduction by ascorbate, is less reactive and more diffusible and can be enzymatically eliminated upon its entry into the cytoplasm. The effectiveness of ascorbate in the removal of nuclear O2•– can be judged from the extent of the nuclear oxidant-sensitive signaling. The redox-active drug bleomycin causes DNA breaks and releases superoxide, causing activation of the nuclear kinase ATM via DNA damage-dependent and direct oxidation-mediated mechanisms, respectively. Physiological concentrations of ascorbate produced a strong suppression of the direct oxidation-mediated activation of ATM and phosphorylation of its nucleoplasmic target CHK2.44 These observations experimentally support the notion that ascorbate plays a major role in detoxification of O2•– in the nucleus. Stem cells contain much higher concentrations of ascorbate in comparison to their differentiated progeny,12 which was linked to the need for the cofactor activity of ascorbate in DNA demethylation activity and epigenetic reprogramming of gene expression in stem cells. It is unclear why reduction of Fe(III) to Fe(II) in DNA demethylases (TET proteins) would require >10× higher ascorbate concentrations than in other members of the Fe(II)/2-oxoglutarate-dependent dioxygenases. Another possibility is that high ascorbate concentrations are not needed as much for the Fe(III) reduction in TET proteins as for the elimination of O2•– produced during aborted catalytic cycles, which otherwise can inactivate the enzyme. The active intermediate in TET enzymes is Fe(II)-O2 which, if not used catalytically, will form Fe(III)-O2•– with a potential to produce damage locally or upon the release of O2•–. A similar mechanism is responsible for the release of superoxide and self-inactivation of bleomycin by its Fe(II)-bound O2, which occurs in the absence of the external substrate.44,45 The uncoupling between oxygen activation and substrate oxidation occurs with a relatively high frequency in Fe(II)-dependent microsomal CYP450 monooxygenases. These enzymes release superoxide as a result of the decay of the initial Fe(II)-O2 oxycomplex.46 For many substrates, the coupling efficiencies are below 10%, indicating that a large fraction of Fe-activated oxygen in CYP450s is released as ROS.15 The uncoupling frequency in oxidative demethylation of DNA by TET proteins is currently unknown.
Cofactor for Cytoplasmic Enzymes
Ascorbate is a soluble cofactor for more than 60 cellular enzymes, which is indicative of its broad impact on physiological processes and consistent with multiple clinical symptoms in scurvy and developmental abnormalities arising as a result of maternal vitamin C deficiency during pregnancy. The main class of enzymes engaging ascorbate as the unbound cofactor are Fe(II)/2-oxoglutarate-dependent dioxygenases. 2-Oxoglutarate acts a cosubstrate and Fe(II) as the O2-binding cofactor. The catalytic reaction of these enzymes involves a transfer of one oxygen atom from Fe(II)-bound O2 onto the substrate and production of a hydroxylation product. The other oxygen atom is transferred to 2-oxoglutarate causing its decarboxylation and release of succinate and CO2 (Figure 3A). At the end of the catalytic cycle, Fe(II) is oxidized to Fe(III) which is unable to bind O2.47 Collagen-modifying prolyl-4-hydroxylases are a classic example of dioxygenases that use ascorbate to restore their enzymatic activity by reducing Fe(III) to Fe(II). Hydroxylation of prolyl residues is essential for the correct folding of collagen fibers and the resulting formation of stable extracellular matrices.48 In scurvy, the loss of mature collagen fibers is responsible for opening of old wounds, skin deformities, bleedings, and loss of teeth. Although scurvy is uncommon in modern societies, significantly depleted levels of vitamin C are frequently found in cancer patients.16 This insufficiency can weaken extracellular matrix and cell–cell attachment and promote tumor invasion and metastasis.
Figure 3.

Role of ascorbate in the activity of Fe(II)/2-oxoglutarate-dependent dioxygenases. (A) General mechanism of oxidative hydroxylation by Fe(II)/2-oxoglutarate-dependent dioxygenases. Fe(II)-bound O2 serves as a source of oxygen atoms in oxidative hydroxylation of a substrate and the cosubstrate 2-oxoglutarate. Ascorbate (Asc) acts as a reducer of Fe(III) to Fe(II), which restores activity of dioxygenases after each catalytic cycle. (B) Constitutive degradation of hypoxia-inducible transcription factors HIF-1α and HIF-2α in normoxic cells. HIF-1α/2α are continuously produced and then immediately targeted for degradation in normoxia via O2-dependent hydroxylation of specific Pro residues (Pro402 and Pro564 in HIF-1α). Hydroxylated prolines are recognized by VHL, which triggers polyubiquitination (Ubn) of both HIFs and their subsequent proteolysis by proteasomes. Ascorbate acts a preferred reducer of Fe(III) to Fe(II) to maintain activity of HIF-targeting prolyl hydroxylases PHD1–3. Another hydroxylation site in HIF-1α in normoxic conditions is Asn803, which regulates binding of the transcription coactivators p300/CBP (not shown). Asn803 hydroxylation is mediated by the Fe(II)/2-oxoglutarate-dependent dioxygenase FIH, whose activity is promoted by ascorbate through reduction of Fe(III) to Fe(II). (C) Removal of genomic 5-methylcytosine (5-mC) by a sequential oxidation of the methyl group by TET enzymes. Ascorbate is required for reduction of Fe(III) to Fe(II) to restore TET activity after each catalytic cycle. BER, base excision repair; 5-hmC, 5-hydroxymethylcytosine; 5-fC, 5-formylcytosine; and 5-caC, 5-carboxycytosine. (D) Role of ascorbate in demethylation of histone lysines. Jumanji C (JmjC) domain-containing histone demethylases catalyzes the removal of a methyl group from tri-, di-, and monomethylated ε-amino groups of lysine. Hydroxymethyl group, the product of demethylase activity, is unstable and spontaneously released in the form of formaldehyde. As with other dioxygenases, ascorbate is involved in reduction of Fe(III) to Fe(II) to restore activity of JmjC demethylases after each reaction cycle.
Ascorbate is also involved in the control of stability of the hypoxia-inducible factors HIF-1α and HIF-2α via stimulation of their proline hydroxylation by PHD1–3.49−52 Proline-hydroxylated HIFs are recognized by the VHL-containing E3 ubiquitin ligase which induces their polyubiquitination and a subsequent degradation by 26S proteasomes (Figure 3B). Stabilization of HIF-1α/HIF-2α proteins occurs rapidly upon a loss of O2, but it can also be induced by divalent metal ions (Co2+, Ni2+, Mn2+) competing with Fe(II) for PHD binding, by 2-oxoglutarate analogues and by ROS. Oxidants such as H2O2 can directly damage functionally important Cys-SH groups in PHD1–3,53,54 but the inhibition of HIF1/2-Pro hydroxylation activity, especially in cell culture, can be further exacerbated by oxidant-induced depletion of ascorbate.50,55 The absence of Pro hydroxylation leads to a rapid stabilization of HIF-1α/HIF-2α which then bind their stable subunit HIF-1β leading to the nuclear translocation of the heterodimers. A full activation of HIF-1α also requires the loss of the FIH-mediated hydroxylation of Asn803, which permits the recruitment of the transcription coactivators p300/CBP.56 FIH is another Fe(II)/2-oxoglutarate-dependent dioxygenase whose hydroxylation activity is promoted by ascorbate.52 Activation of the transcriptional factor HIF-1 in solid tumors is known to enhance their invasiveness and metastasis.57 This aggressive phenotype is at least in part dependent on the ascorbate deficiency in the tumors. For example, an investigation of human endometrial cancers found that high-grade tumors had the highest HIF-1α protein levels and were ascorbate-deficient.58 In the Gulo–/– mouse model of ascorbate deficiency, restoration of physiological levels of vitamin C inhibited tumor growth and HIF-1 pathway activity.59Gulo–/– mice that were kept on the ascorbate-deficient diet also showed a slower formation and growth of tumors formed by the hypoxia-mimicking carcinogen nickel.60 A vitamin C-deficient state in human malignancies stems from multiple causes such as a poor accumulation of ascorbate by some cancers, a limited blood circulation in solid tumors, and low systemic ascorbate levels found among many cancer patients.
HIF1/2 are the best known, but not the only, targets of prolyl hydroxylases PHD1–3. A recently discovered novel substrate of PHD2 is a serine-threonine protein kinase AKT which regulates a metabolic branch in mitogenic signaling from growth factor receptors. In response to growth factor stimulation, AKT is activated by phosphorylation at Thr308 which is reversed by protein phosphatase 2A. The recruitment of this phosphatase occurs in response to Pro hydroxylation of AKT by PHD2.61 Another biochemical process in the cytoplasm that has long been considered as dependent on ascorbate is carnitine production. The first step in carnitine biosynthesis is catalyzed by 6-N-trimethyllysine hydroxylase which is also a member of the Fe(II)/2-oxoglutarate-dependent family of dioxygenases. Carnitine is necessary for the transport of long-chain fatty acids into the mitochondria and ATP production via β-oxidation. Surprisingly, mice with a severe deficiency in tissue vitamin C (<2% relative to normal animals) did not show any significant changes in carnitine levels in serum and multiple organs.62 It remains unclear whether 6-N-trimethyllysine hydroxylase has a very low Km for ascorbate or is more promiscuous in the choice of the reducers for restoration of catalytic Fe(II) from Fe(III).
Ascorbate in Epigenetic Regulation
Reprogramming of gene expression is the most recently discovered fundamental biological process that is strongly affected by cellular vitamin C. The lateness of this discovery for ascorbate is related to the fact that biochemical mechanisms for how two critical steps in the control of gene expression, demethylation of 5-methylC in DNA and lysine demethylation of histones, have been defined only relatively recently. Cell culture and in vivo studies have clearly shown that ascorbate was important for the erasure of the epigenetic memory in embryonic stem cells or differentiated cells by acting as a cofactor for the family of ten−eleven translocation (TET1–3) proteins that remove cytosine methylation in DNA.63 Cytosine methylation occurs at its C5 position in CpG dinucleotides and usually acts as a transcription-repressive mark. TET enzymes are Fe(II)-dependent dioxygenases that catalyze a series of consecutive oxidations of 5-methylcytosine by first hydroxylating it to 5-hydroxymethylcytosine, which is further oxidized to 5-formylcytosine and 5-carboxycytosine. The last two oxidation products are then removed during base excision repair by thymine DNA glycosylase and replaced with the unmethylated cytosine (Figure 3C). In addition to in vitro assays with recombinant TET proteins, there is clear evidence for the importance of ascorbate in demethylation of 5-methylcytosine in various cultured cells and in vivo.64−66 Addition of ascorbate to human embryonic stem cells produced a dramatic impact on their epigenome, involving a widespread and specific DNA demethylation of 1847 genes.67 The ascorbate-demethylated group of genes included the gene sets that experienced a loss of 5-methylcytosine during differentiation of human embryonic stem cells and contained bivalent marks. A high ascorbate content of hematopoietic stem cells was necessary for their DNA demethylation, TET2-dependent gene expression signature and stimulation of differentiation and suppression of leukemia development.12,68 Erasure of epigenetic memory to create induced pluripotent stem cells was dependent on active DNA demethylation by TET proteins which were shown to be activated by ascorbate through reduction of Fe(III) to Fe(II).69 In addition to its differentiation effects in the developing brain,70 TET-dependent regional demethylation of DNA continuously occurs in neurons in the adult brain, which is important for neuronal activity and memory formation.71−73 In a mouse model of neurodegeneration, vitamin C deficiency impaired brain cognition and increased amyloid accumulation and deposition.74 Efficient DNA demethylation in hematopoietic stem cells required higher ascorbate concentrations than other ascorbate-promoted enzymatic processes.12,68 This property of TET-mediated DNA demethylation could be a reason for the unusually high ascorbate concentrations maintained in hematopoietic stem cells and neurons in vivo. These cells are also particularly sensitive to oxidative stress, as evidenced by the residence of hematopoietic stem cells in the hypoxic niche of the bone marrow and numerous neurodegenerative conditions associated with a compromised ability of the brain cells to deal with oxidants or their damage to proteins or DNA. Neurons appear to be continuously producing large amounts of superoxide that in addition to SOD activity requires normal levels of ascorbate for its efficient elimination.75
Post-translational histone modifications represent a second and more dynamic level of epigenetic regulation of gene expression in chromatin. Methylation of lysines located in the N-terminal tails is one of the well-characterized modifications that occurs in chromatin-deposited histones. The most frequent sites of lysine methylation are K4, K9, K27, K36, and K79 in histone H3 and K20 in histone H4. Lysine can be mono-, di-, and trimethylated at its N-6 position. Trimethylation of histone H3 at K4, K36, and K79 is associated with actively transcribed genes, whereas the presence of trimethylated K9 and K27 is associated with gene repression.76,77 Methylation status of histone lysines is dynamically regulated in response to transcriptional needs of cells. Although methylation of histone lysines was known for a long time, lysine demethylases have been discovered relatively recently. There are two classes of histone lysine demethylases that differ in their catalytic mechanisms and substrate specificity. The majority of lysine demethylases belong to the class of JmjC domain-containing Fe(II)/2-oxoglutarate-dependent dioxygenases (>20 enzymes) that collectively can demethylate lysines at any position in histones and are capable of demethylating mono-, di-, or trimethylated N-6. A pair of flavin adenine dinucleotide-dependent amine oxidases LSD1 (KDM1A) and LSD2 (KDM1B) represent a second class of histone lysine demethylases.78,79 LSD1 and LSD2 can remove a methyl group only from mono- or dimethylated lysines and act specifically on methylated lysine-4 in histone H3. Amine oxidation in the methylated lysine by LSD1/2 generates an unstable imine which is spontaneously hydrolyzed, releasing formaldehyde and yielding unmethylated lysine.
JmjC histone demethylases catalyze hydroxylation of a methyl group in lysines, which results in its dissociation in the form of formaldehyde (Figure 3D).80 Similar to other Fe(II)/2-oxoglutarate-dependent dioxygenases, JmjC histone demethylases require ascorbate for their optimal catalytic activity. In vitro histone lysine demethylation by JmjC hydroxylases was inhibited when ascorbate was omitted from the assay.81,82 In cellular models, ascorbate was identified as a critical factor for histone demethylation and epigenetic changes during reprogramming of somatic cells into induced pluripotent stem cells by acting as the activator of H3K36 and H3K9 demethylases.83,84 Ascorbate-induced demethylation of H3K9me2 in stem cells resulted from a specific activation of histone demethylases Kdm3a and Kdm3b. Importantly, vitamin C-stimulated Kdm3a/b-mediated H3K9me2 demethylation and TET-mediated DNA demethylation were independent processes.85 It is expected that nuclear ascorbate is a major stimulative factor for activity of all JmjC histone demethylases, although the degree of their sensitivity to its levels relative to other reducers of Fe(III) may vary.
Vitamin C in Cancer Therapy
Two open-label studies in the 1970s led by Nobel prize-winning biochemist Linus Pauling and conducted in terminal cancer patients using intravenous administration of large doses of vitamin C have found significant beneficial effects on survival.86,87 However, follow-up double-blind, placebo-controlled clinical trials did not show any survival benefits of vitamin C in cancer patients,88,89 which effectively shut down for a long time the whole notion that vitamin C could work as an anticancer treatment. What was not appreciated until recently is the difference in the delivery of pharmacological doses of vitamin C between early positive studies and the later negative trials. In the follow-up trials, vitamin C was given orally, which is now known to provide only a limited increase in systemic ascorbate which is tightly regulated via the rates of absorption and urinary excretion.90,91 Therapeutic effects of large doses of vitamin C have been linked to a preferential accumulation of dehydroascorbic acid by cancer cells due to their common overexpression of GLUT transporters. In colorectal cancers, the overexpression of GLUT1 glucose transporter and hyperaccumulation of dehydroascorbic acid were specifically associated with mutations in KRAS and BRAF oncogenes.92 GLUT1 is also strongly upregulated by hypoxic signaling, and human clear cell renal carcinomas lacking the HIF1α/2α-targeting ubiquitin ligase VHL were more sensitive to killing by high doses of vitamin C through its hyperaccumulation.93 The cause of death in cancer cells that hyperaccumulated dehydroascorbic acid was linked to the exhaustion of energy resources and glutathione which were consumed by the reduction of dehydroascorbic acid to ascorbate.92 Overexpression of GLUT1 is associated with a metabolic reprogramming of cancer cells toward aerobic glycolysis (Warburg effect), which makes them more dependent on the inefficient ATP generation in glycolysis and, therefore, more vulnerable to energy depletion by dehydroascorbate reduction. Several studies have also linked a hyperaccumulation of ascorbate in cancer cells with the increased production of oxidants as a result of elevated levels of labile iron which, unlike its protein-bound form, readily catalyzes the Fenton reaction with the resulting generation of a highly toxic •OH radical.94−96 Oxidative and energy stresses by high vitamin C doses were associated with the induction of DNA damage and PARP hyperactivation-dependent cytotoxicity.93,97,98 A mechanistically distinct process based on the increased differentiation of stem cells operates in the suppression of leukemia by vitamin C. Patients with myelodysplastic syndrome and acute myeloid leukemia frequently carry inactivating mutations in TET2, which leads to a DNA hypomethylation phenotype and impaired differentiation of hematopoietic stem cells. Treatment with vitamin C mimicked TET restoration by enhancing 5-methylcytosine oxidation and promoting differentiation of stem cells and suppression of leukemia in mouse models and in primary patient-derived xenografts.12,68 A combination of vitamin C with clinically used drugs or ionizing radiation enhanced the killing of cancer cells of different histological origins.16,99,100 For many combined regiments, the increased killing of cancer cells can be attributed to their stressed energy metabolism and elevated ROS, diminishing their ability to survive chemotherapeutic or radiation treatments. In the case of bleomycin, a drug using Fe(II)-activated oxygen to oxidize and break DNA, ascorbate enhanced its DNA-damaging activity by acting as a very effective reducer of Fe(III) to Fe(II).44 The administration of high doses of vitamin C or possibly just restoration of its levels in ascorbate-deficient patients can also promote epigenetic and transcriptomic changes, making cancer cells more susceptible to specific cancer treatments, as it was found for ascorbate-induced sensitization of melanoma to BET inhibitors.101 Vitamin C has also diminished the aggressiveness of many cancers by inhibiting their growth, invasiveness, and metastasis.16,102 These beneficial responses were promoted by a decreased hypoxic signaling produced by a more efficient proline hydroxylation of HIF-1/HIF-2 and, consequently, their more rapid degradation in ascorbate-enriched cells. These transcription factors are known to promote motility, invasiveness, and metastasis of cancer cells and are markers of aggressive tumors.57 In ascorbate-deficient tumors, vitamin C supplementation could also boost the production of mature collagen48 and consequently, strengthen cell–cell adherence and the extracellular matrix, which would exert suppressive effects on the ability of cancer cells to migrate and invade surrounding tissue and reach distant sites. Another broad mechanism which has been overlooked so far in practically all preclinical models is the impact of vitamin C on anticancer immune responses. Humor tumor xenografts are grown in immunodeficient mice in order to avoid their immune rejection. Thus, this commonly used in vivo model excludes any contribution of the immune cells to antitumor responses. A recent study of lymphoma growth in the immunocompetent mice showed that vitamin C administration was synergistic with a clinically used anti-PD1 immune checkpoint treatment via increased DNA demethylation in tumor and immune cells.103 The combination therapy strongly increased intratumor infiltration of CD8+ T lymphocytes, granzyme B production by cytotoxic T cells and natural killer cells, and interleukin-12 biosynthesis by antigen-presenting cells compared with anti-PD1 alone. These findings raise a possibility that vitamin C may act a general promoter of immune responses against many cancers that are responsive to anti-PD1/PD-L1 therapy.
Vitamin C Deficiency in Cultured Cells and Its Remediation
Various tissue culture media have been historically formulated to provide robust growth of cells by abundantly supplying them with energy-generating and macromolecule-building nutrients. Much less attention has been given to micronutrients, most of which are provided via the addition of fetal bovine serum to growth media. Vitamin C is a such micronutrient that is absent in the majority of cell culture media. The typical addition of 10% fetal bovine serum to culture media theoretically should provide the 1/10th of the physiological concentration of vitamin C, which in practice is much lower due to its loss during storage of serum and serum-supplemented media. Overnight fed cells typically contain low micromolar concentrations of vitamin C (5–20 μM),104,105 which is approximately 1% of its levels in cells in vivo. Vitamin C usually becomes undetectable in cultured cells at 2 days postfeeding. Primary and other cells growing in growth factor-supplemented synthetic media without addition of serum are completely devoid of vitamin C. Although rodents produce their own ascorbic acid, its synthesis occurs in the liver,106−108 and all nonhepatic rodent cells in culture are also vitamin C deficient. It is unclear how much ascorbic acid biosynthesis remains in primary rodent hepatocytes which exhibit major changes in their metabolic activities within a few days in culture. A monolayer of rodent hepatocytes would also need to continuously generate massive amounts of ascorbic acid to establish and maintain its physiological concentration in a very large volume of media relative to the number of cells in the dishes. In the absence of extracellular vitamin C in culture, primary and other cells rapidly lose their cellular ascorbate. Thus, it is likely that even rodent hepatocytes in culture are vitamin C deficient.
A full restoration of vitamin C levels in cells can be achieved by the addition of dehydroascorbic acid that readily enters cells via ubiquitously expressed GLUT transporters. Glucose competitively inhibits the uptake of dehydroascorbic acid, which hinders restoration of cellular ascorbate when high-glucose growth media are used. A switch to a low-glucose growth medium or a balanced salt solution (Krebs buffer) supplemented with low glucose (0.5–1 mM) and a regular serum percentage can result in the full restoration of physiological levels of ascorbate in cells after only 1–2 h of incubation.44,104 Expression of GLUT transporters is impacted by many factors, and the optimal concentrations of dehydroascorbic acid should be determined for each cell line. A more gradual delivery of vitamin C, but usually only to subphysiological levels, can be accomplished by the supplementation of culture media with ascorbate (daily additions of 50–200 μM). Even when ascorbate is used as a source of vitamin C supplementation, its easy oxidation in media can lead to a significant formation and uptake of dehydroascorbic acid. This is especially common for cells grown in serum-free media which contain large amounts of iron to compensate for the absence of transferrin-mediated iron uptake from serum. The addition of ascorbate-2-phosphate, a more stable derivative of ascorbate, typically leads to higher levels of cellular vitamin C, and it has a lower risk of the formation of ROS in the growth media. Ascorbate-2-phosphate is not redox-active, and the release of ascorbate occurs after its dephosphorylation in cells.
Methodological challenges associated with vitamin C supplementation in culture include its relatively rapid loss from cells.109,110 Consequently, cells should be used either shortly after restoration of ascorbate concentrations or periodic additions of a freshly prepared stock of ascorbic acid in cold H2O are employed for a prolonged maintenance of vitamin C levels in cells. Even though oxidation of ascorbate to dehydroascorbic acid still yields a biologically active form of vitamin C, it is relatively unstable and undergoes irreversible hydrolysis and further rearrangements. Incubations with excessive amounts of vitamin C can cause cytotoxicity,122,111 which can result either from the overloading of cells with dehydroascorbic acid when its reduction to ascorbate consumes important cellular reducers or from excessive amounts of ascorbate in O2-rich media where it reacts with iron and catalyzes the production of toxic ROS. Cancer cells are characterized by a strongly elevated uptake of glucose and overexpression of GLUT transporters, which makes them more sensitive to killing by high doses of dehydroascorbic acid.92,93 Vitamin C restoration in cells can be assessed by its direct measurements and/or by a functional test such as a suppression of ATM-dependent phosphorylation of its targets KAP1 and/or CHK2 by chromate.113
Assays for Vitamin C Forms
Irrespective of the use of HPLC or microplate readers, the majority of the currently used assays for vitamin C in biological samples can be divided into two groups based on whether they measure its reduced (ascorbic acid) or oxidized (dehydroascorbic acid) forms. Ascorbic acid is measured by electrochemical detectors coupled with HPLC or by spectrophotometric methods detecting Fe(II) formation from Fe(III) (known as the FRASC assay - ferric reducing/antioxidant and ascorbic acid). Fluorescence-based assays (HPLC or microplate reader versions) detect the formation of dehydroascorbic acid conjugates with dyes such as o-phenylenediamine114 or 1,2-diamino-4,5-dimethoxybenzene.110 For measurements of total vitamin C by electrochemical detection, samples are treated with a reducer to convert dehydroascorbic acid into ascorbic acid.115 In the FRASC assay, dehydroascorbic acid is not detectable, which may result in a significant underestimation of total vitamin C due to its ready autoxidation in samples ex vivo. Assays based on the conjugation of dyes with dehydroascorbic acid use pretreatments of samples with ascorbate oxidase or TEMPOL to convert all vitamin C into its oxidized form. A detection of both oxidized and reduced forms of vitamin C has been challenging due to a very small percentage of the dehydroascorbic acid that is typically present in biological samples (tissues, cells or plasma/serum). Dehydroascorbic acid values were typically calculated from the differences in HPLC-electrochemical measurements between samples treated with reducer (total ascorbate) and untreated samples (ascorbate only).115 These estimates are not very precise or accurate as they derive from subtraction of a large number from only a slightly larger number. In assays utilizing dehydroascorbate conjugation with dyes, omission of ascorbate oxidase or TEMPOL still results in a very significant reaction of probes with ascorbate through catalysis of its oxidation. LC-MS is currently the only method for the direct determination of both abundant ascorbate and scarce dehydroascorbic acid.92
Concluding Remarks: Vitamin C in Mechanistic Toxicology/Pharmacology
The multitude of antioxidant and biochemical functions of ascorbate makes it important that biologically relevant concentrations of vitamin C are included in studies of toxic and other stress responses in cultured cells. Vitamin C addition is routinely used in the field of stem cell biology, but this practice has not yet been commonly accepted in toxicology/pharmacology or stress biology in general even when epigenetic changes or ROS effects are specifically examined. For oxidants, cellular ascorbate was found to exert major effects on the type and magnitude of genotoxic events44,104 and epigenetic alterations.116 In addition to the changes in the magnitude of cytotoxicity or other general responses, ascorbate can dramatically alter the relative importance of specific signaling mechanisms activated in cells by carcinogens and DNA-damaging cancer drugs.16,44,110,113 The importance of extracellular ascorbate should also be considered when a chemical is known to react with ascorbate directly or with Fe(II) that is abundantly produced in ascorbate-supplemented media.124 Histone lysine demethylation is a dynamic process that occurs continuously during transcription cycles and in response to promoter binding by transcription factors. The important function of ascorbate as a cofactor for histone demethylases raises a possibility that transcriptomic and, consequently, downstream biological responses to many redox-inactive xenobiotics and stressors can also be affected to some extent by nuclear ascorbate.
The importance of ascorbate in the regulation of cellular reprogramming and gene expression via its cofactor functions for the DNA-demethylating TET enzymes and histone lysine demethylases raises a possibility that populations with vitamin C insufficiency could be particularly susceptible to adverse health effects of epigenetic toxicants. Mice and rats continuously produce large amounts of vitamin C, which may limit their sensitivity in capturing intergenerational and other epigenetic consequences of human malnutrition and its combination with other stressors or drugs. Clearly established roles of ascorbate in neurodevelopment, memory formation, and the development of neurodegenerative processes71−74 raise a possibility that individuals with low systemic levels of vitamin C could be especially vulnerable to neurotoxic effects of chemicals and/or other stressors. This vulnerability would be difficult to detect in standard rodent models, although the use of Gulo–/– mice or similarly deficient rats can be readily adapted to assess the impact of vitamin C insufficiency.
Vitamin C levels in rodent and human tissues are generally similar, but concentrations of ascorbate in the lung lining fluids of laboratory rodents are more than 10× higher than those in humans.118−120 This difference accounts for a rapid extracellular inactivation of soluble chromate in rodent lungs, making them resistant to carcinogenesis by this Cr(VI) form in contrast to its carcinogenicity in human lungs. Slowly solubilizing particles of chromium(VI) escape this detoxification by extracellular ascorbate and induce genotoxicity upon their intracellular dissolution.117 Oxidative stress in the lung is a common adverse effect for many inhaled toxicants. The use of standard laboratory rodents containing very high levels of ascorbate in the lung lining fluid represents an additional uncertainty in their use for the mechanistic assessment of human health risks by airborne contaminants.
Biography
Anatoly Zhitkovich is Professor of Medical Science in the Department of Pathology and Laboratory Medicine at Brown University. He received B.Sc. with distinction in biochemistry from Belarussian State University and a Ph.D. in biochemistry from Belarussian Academy of Sciences. Prof. Zhitkovich has long-standing research interests in mechanisms of DNA damage by carcinogens and anticancer drugs and activation of stress signaling networks engaging DNA repair, cell cycle checkpoints, and cell death programs. One of the research directions in his lab has been the elucidation of the role of ascorbate in redox metabolism and the biological activity of carcinogenic metals and metal-containing drugs.
Research is supported by grants ES008786, ES028072, and ES031002 from the National Institute of Environmental Health Sciences.
The author declares no competing financial interest.
References
- Magiorkinis E.; Beloukas A.; Diamantis A. (2011) Scurvy: past, present and future. Eur. J. Intern. Med. 22 (2), 147–152. 10.1016/j.ejim.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Rowe S.; Carr A. C. (2020) Global Vitamin C Status and Prevalence of Deficiency: A Cause for Concern?. Nutrients 12 (7), 2008. 10.3390/nu12072008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marik P. E.; Liggett A. (2019) Adding an orange to the banana bag: vitamin C deficiency is common in alcohol use disorders. Crit Care. 23 (1), 165. 10.1186/s13054-019-2435-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindran P.; Wiltshire S.; Das K.; Wilson R. B. (2018) Vitamin C deficiency in an Australian cohort of metropolitan surgical patients. Pathology 50 (6), 654–658. 10.1016/j.pathol.2018.07.004. [DOI] [PubMed] [Google Scholar]
- DiTroia S. P.; Percharde M.; Guerquin M. J.; Wall E.; Collignon E.; Ebata K. T.; Mesh K.; Mahesula S.; Agathocleous M.; Laird D. J.; Livera G.; Ramalho-Santos M. (2019) Maternal vitamin C regulates reprogramming of DNA methylation and germline development. Nature 573 (7773), 271–275. 10.1038/s41586-019-1536-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukaguchi H.; Tokui T.; Mackenzie B.; Berger U. V.; Chen X. Z.; Wang Y.; Brubaker R. F.; Hediger M. A. (1999) A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature 399 (6731), 70–75. 10.1038/19986. [DOI] [PubMed] [Google Scholar]
- Wang H.; Dutta B.; Huang W.; Devoe L. D.; Leibach F. H.; Ganapathy V.; Prasad P. D. (1999) Human Na(+)-dependent vitamin C transporter 1 (hSVCT1): primary structure, functional characteristics and evidence for a non-functional splice variant. Biochim. Biophys. Acta, Biomembr. 1461 (1), 1–9. 10.1016/S0005-2736(99)00182-0. [DOI] [PubMed] [Google Scholar]
- Bürzle M.; Hediger M. A. (2012) Functional and physiological role of vitamin C transporters. Curr. Top. Membr. 70, 357–375. 10.1016/B978-0-12-394316-3.00011-9. [DOI] [PubMed] [Google Scholar]
- Sotiriou S.; Gispert S.; Cheng J.; Wang Y.; Chen A.; Hoogstraten-Miller S.; Miller G. F.; Kwon O.; Levine M.; Guttentag S. H.; Nussbaum R. L. (2002) Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nat. Med. 8 (5), 514–517. 10.1038/0502-514. [DOI] [PubMed] [Google Scholar]
- Kojo S. (2004) Vitamin C: basic metabolism and its function as an index of oxidative stress. Curr. Med. Chem. 11 (8), 1041–1064. 10.2174/0929867043455567. [DOI] [PubMed] [Google Scholar]
- Padayatty S. J.; Levine M. (2016) Vitamin C: the known and the unknown and Goldilocks. Oral Dis. 22 (6), 463–493. 10.1111/odi.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agathocleous M.; Meacham C. E.; Burgess R. J.; Piskounova E.; Zhao Z.; Crane G. M.; Cowin B. L.; Bruner E.; Murphy M. M.; Chen W.; Spangrude G. J.; Hu Z.; DeBerardinis R. J.; Morrison S. J. (2017) Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549 (7673), 476–481. 10.1038/nature23876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montel-Hagen A.; Kinet S.; Manel N.; Mongellaz C.; Prohaska R.; Battini J. L.; Delaunay J.; Sitbon M.; Taylor N. (2008) Erythrocyte Glut1 triggers dehydroascorbic acid uptake in mammals unable to synthesize vitamin C. Cell 132 (6), 1039–1048. 10.1016/j.cell.2008.01.042. [DOI] [PubMed] [Google Scholar]
- Zhitkovich A. (2019) N-acetylcysteine: antioxidant, aldehyde scavenger, and more. Chem. Res. Toxicol. 32, 1318–1319. 10.1021/acs.chemrestox.9b00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvez S.; Long M. J. C.; Poganik J. R.; Aye Y. (2018) Redox signaling by reactive electrophiles and oxidants. Chem. Rev. 118, 8798–8888. 10.1021/acs.chemrev.7b00698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaszczak W.; Barczak W.; Masternak J.; Kopczynski P.; Zhitkovich A.; Rubis B. (2019) Vitamin C as a modulator of the response to cancer therapy. Molecules 24 (3), 453. 10.3390/molecules24030453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielski B. H. J.; Allen A. O.; Schwarz H. A. (1981) Mechanism of disproportionation of ascorbate radicals. J. Am. Chem. Soc. 103, 3516–3518. 10.1021/ja00402a042. [DOI] [Google Scholar]
- Winterbourn C. C. (2008) Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 4 (5), 278–286. 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- Ng C. F.; Schafer F. Q.; Buettner G. R.; Rodgers V. G. (2007) The rate of cellular hydrogen peroxide removal shows dependency on GSH: mathematical insight into in vivo H2O2 and GPx concentrations. Free Radical Res. 41 (11), 1201–1211. 10.1080/10715760701625075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer J. E.; Slater T. F.; Willson R. L. (1979) Direct observation of a free radical interaction between vitamin E and vitamin C. Nature 278 (5706), 737–738. 10.1038/278737a0. [DOI] [PubMed] [Google Scholar]
- Buettner G. R. (1993) The pecking order of free radicals and antioxidants: lipid peroxidation, alpha-tocopherol, and ascorbate. Arch. Biochem. Biophys. 300 (2), 535–543. 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- Lőrincz T.; Holczer M.; Kapuy O.; Szarka A. (2019) The Interrelationship of Pharmacologic Ascorbate Induced Cell Death and Ferroptosis. Pathol. Oncol. Res. 25 (2), 669–679. 10.1007/s12253-018-0539-9. [DOI] [PubMed] [Google Scholar]
- Yang W. S.; Kim K. J.; Gaschler M. M.; Patel M.; Shchepinov M. S.; Stockwell B. R. (2016) Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. U. S. A. 113 (34), E4966–4975. 10.1073/pnas.1603244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikimi M. (1975) Oxidation of ascorbic acid with superoxide anion generated by the xanthine-xanthine oxidase system. Biochem. Biophys. Res. Commun. 63 (2), 463–468. 10.1016/0006-291X(75)90710-X. [DOI] [PubMed] [Google Scholar]
- Cabelli D. E.; Bielski B. H. J. (1983) Kinetics and mechanism for the oxidation of ascorbic acid.ascorbate by HO2/O2– radicals. A pulse radiolysis and stopped flow photolysis study. J. Phys. Chem. 87, 1809–1812. 10.1021/j100233a031. [DOI] [Google Scholar]
- Sueishi Y.; Hori M.; Ishikawa M.; Matsu-Ura K.; Kamogawa E.; Honda Y.; Kita M.; Ohara K. (2014) Scavenging rate constants of hydrophilic antioxidants against multiple reactive oxygen species. J. Clin. Biochem. Nutr. 54 (2), 67–74. 10.3164/jcbn.13-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurobe N.; Suzuki F.; Okajima K.; Kato K. (1990) Sensitive enzyme immunoassay for human Cu/Zn superoxide dismutase. Clin. Chim. Acta 187 (1), 11–20. 10.1016/0009-8981(90)90257-S. [DOI] [PubMed] [Google Scholar]
- Rakhit R.; Chakrabartty A. (2006) Structure, folding, and misfolding of Cu,Zn superoxide dismutase in amyotrophic lateral sclerosis. Biochim. Biophys. Acta, Mol. Basis Dis. 1762 (11–12), 1025–1037. 10.1016/j.bbadis.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Xu A.; Vita J. A.; Keaney J. F. Jr. (2000) Ascorbic acid and glutathione modulate the biological activity of S-nitrosoglutathione. Hypertension 36 (2), 291–295. 10.1161/01.HYP.36.2.291. [DOI] [PubMed] [Google Scholar]
- Samuni Y.; Goldstein S.; Dean O. M.; Berk M. (2013) The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta, Gen. Subj. 1830 (8), 4117–4129. 10.1016/j.bbagen.2013.04.016. [DOI] [PubMed] [Google Scholar]
- Hata K.; Urushibara A.; Yamashita S.; Lin M.; Muroya Y.; Shikazono N.; Yokoya A.; Fu H.; Katsumura Y. (2015) Chemical repair activity of free radical scavenger edaravone: reduction reactions with dGMP hydroxyl radical adducts and suppression of base lesions and AP sites on irradiated plasmid DNA. J. Radiat. Res. 56 (1), 59–66. 10.1093/jrr/rru079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanofsky J. R. (1989) Singlet Oxygen Production by Biological Systems. Chem.-Biol. Interact. 70, 1–28. 10.1016/0009-2797(89)90059-8. [DOI] [PubMed] [Google Scholar]
- Kramarenko G. G.; Hummel S. G.; Martin S. M.; Buettner G. R. (2006) Ascorbate reacts with singlet oxygen to produce hydrogen peroxide. Photochem. Photobiol. 82 (6), 1634–1637. 10.1562/2006-01-12-RN-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devasagayam T. P.; Sundquist A. R.; Di Mascio P.; Kaiser S.; Sies H. (1991) Activity of thiols as singlet molecular oxygen quenchers. J. Photochem. Photobiol., B 9 (1), 105–116. 10.1016/1011-1344(91)80008-6. [DOI] [PubMed] [Google Scholar]
- Brubaker R. F.; Bourne W. M.; Bachman L. A.; McLaren J. W. (2000) Ascorbic acid content of human corneal epithelium. Invest. Ophthalmol. Vis. Sci. 41 (7), 1681–1683. [PubMed] [Google Scholar]
- Reddy V. N.; Giblin F. J.; Lin L. R.; Chakrapani B. (1998) The effect of aqueous humor ascorbate on ultraviolet-B-induced DNA damage in lens epithelium. Invest. Ophthalmol. Vis. Sci. 39 (2), 344–350. [PubMed] [Google Scholar]
- Ringvold A.; Anderssen E.; Kjønniksen I. (1998) Ascorbate in the corneal epithelium of diurnal and nocturnal species. Invest. Ophthalmol. Vis. Sci. 39 (13), 2774–2777. [PubMed] [Google Scholar]
- DeLoughery Z.; Luczak M. W.; Zhitkovich A. (2014) Monitoring Cr intermediates and reactive oxygen species with fluorescent probes during chromate reduction. Chem. Res. Toxicol. 27, 843–851. 10.1021/tx500028x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer J.; Reynolds M.; Stoddard L.; Zhitkovich A. (2006) Causes of DNA single-strand breaks during reduction of chromate by glutathione in vitro and in cells. Free Radical Biol. Med. 40, 1981–1992. 10.1016/j.freeradbiomed.2006.01.028. [DOI] [PubMed] [Google Scholar]
- Luczak M. W.; Krawic C.; Zhitkovich A. (2019) p53 activation by Cr(VI): a transcriptionally limited response induced by ATR kinase in S-phase. Toxicol. Sci. 172, 11–22. 10.1093/toxsci/kfz178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang C. K.; Liu Y.; Thomas J.; Zhang Y.; Zheng X. F. (2014) Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat. Commun. 5, 3446. 10.1038/ncomms4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Németh C. E.; Nemoda Z.; Lőw P.; Szabó P.; Horváth E. Z.; Willaert A.; Boel A.; Callewaert B. L.; Coucke P. J.; Colombi M.; Bánhegyi G.; Margittai É. (2019) Decreased Nuclear Ascorbate Accumulation Accompanied with Altered Genomic Methylation Pattern in Fibroblasts from Arterial Tortuosity Syndrome Patients. Oxid. Med. Cell. Longevity 2019, 8156592. 10.1155/2019/8156592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowjat W. K.; Kharatishvili M.; Costa M. (1996) DNA and RNA strand scission by copper, zinc and manganese superoxide dismutases. BioMetals 9 (4), 327–335. 10.1007/BF00140601. [DOI] [PubMed] [Google Scholar]
- Han Y.; Shen T.; Jiang W.; Xia Q.; Liu C. (2007) DNA cleavage mediated by copper superoxide dismutase via two pathways. J. Inorg. Biochem. 101 (2), 214–224. 10.1016/j.jinorgbio.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Rubis B.; Luczak M. W.; Krawic C.; Zhitkovich A. (2019) Vitamin C increases DNA breaks and suppresses DNA damage-independent activation of ATM by bleomycin. Free Radical Biol. Med. 136, 12–21. 10.1016/j.freeradbiomed.2019.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger R. M.; Peisach J.; Horwitz S. B. (1981) Activated bleomycin. A transient complex of drug, iron, and oxygen that degrades DNA. J. Biol. Chem. 256 (22), 11636–11644. [PubMed] [Google Scholar]
- Grinkova Y. V.; Denisov I. G.; McLean M. A.; Sligar S. G. (2013) Oxidase uncoupling in heme monooxygenases: human cytochrome P450 CYP3A4 in Nanodiscs. Biochem. Biophys. Res. Commun. 430, 1223–1227. 10.1016/j.bbrc.2012.12.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam M. S.; Leissing T. M.; Chowdhury R.; Hopkinson R. J.; Schofield C. J. (2018) 2-Oxoglutarate-Dependent Oxygenases. Annu. Rev. Biochem. 87, 585–620. 10.1146/annurev-biochem-061516-044724. [DOI] [PubMed] [Google Scholar]
- Myllyharju J. (2008) Prolyl 4-hydroxylases, key enzymes in the synthesis of collagens and regulation of the response to hypoxia, and their roles as treatment targets. Ann. Med. 40 (6), 402–417. 10.1080/07853890801986594. [DOI] [PubMed] [Google Scholar]
- Knowles H. J.; Raval R. R.; Harris A. L.; Ratcliffe P. J. (2003) Effect of ascorbate on the activity of hypoxia-inducible factor in cancer cells. Cancer Res. 63 (8), 1764–1768. [PubMed] [Google Scholar]
- Salnikow K.; Donald S. P.; Bruick R. K.; Zhitkovich A.; Phang J. M.; Kasprzak K. S. (2004) Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J. Biol. Chem. 279, 40337–40344. 10.1074/jbc.M403057200. [DOI] [PubMed] [Google Scholar]
- Vissers M. C.; Gunningham S. P.; Morrison M. J.; Dachs G. U.; Currie M. J. (2007) Modulation of hypoxia-inducible factor-1 alpha in cultured primary cells by intracellular ascorbate. Free Radical Biol. Med. 42 (6), 765–772. 10.1016/j.freeradbiomed.2006.11.023. [DOI] [PubMed] [Google Scholar]
- Flashman E.; Davies S. L.; Yeoh K. K.; Schofield C. J. (2010) Investigating the dependence of the hypoxia-inducible factor hydroxylases (factor inhibiting HIF and prolyl hydroxylase domain 2) on ascorbate and other reducing agents. Biochem. J. 427 (1), 135–142. 10.1042/BJ20091609. [DOI] [PubMed] [Google Scholar]
- Briggs K. J.; Koivunen P.; Cao S.; Backus K. M.; Olenchock B. A.; Patel H.; Zhang Q.; Signoretti S.; Gerfen G. J.; Richardson A. L.; Witkiewicz A. K.; Cravatt B. F.; Clardy J.; Kaelin W. G. Jr. (2016) Paracrine induction of HIF by glutamate in breast cancer: EglN1 senses cysteine. Cell 166, 126–39. 10.1016/j.cell.2016.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G.; Won H. S.; Lee Y. M.; Choi J. W.; Oh T. I.; Jang J. H.; Choi D. K.; Lim B. O.; Kim Y. J.; Park J. W.; Puigserver P.; Lim J. H. (2016) Oxidative Dimerization of PHD2 is Responsible for its Inactivation and Contributes to Metabolic Reprogramming via HIF-1α Activation. Sci. Rep. 6, 18928. 10.1038/srep18928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagé E. L.; Chan D. A.; Giaccia A. J.; Levine M.; Richard D. E. (2008) Hypoxia-inducible factor-1alpha stabilization in nonhypoxic conditions: role of oxidation and intracellular ascorbate depletion. Mol. Biol. Cell 19 (1), 86–94. 10.1091/mbc.e07-06-0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lando D.; Peet D. J.; Gorman J. J.; Whelan D. A.; Whitelaw M. L.; Bruick R. K. (2002) FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 16, 1466–1471. 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schito L.; Semenza G. L. (2016) Hypoxia-inducible factors: master regulators of cancer progression. Trends Cancer. 2, 758–770. 10.1016/j.trecan.2016.10.016. [DOI] [PubMed] [Google Scholar]
- Kuiper C.; Molenaar I. G.; Dachs G. U.; Currie M. J.; Sykes P. H.; Vissers M. C. (2010) Low ascorbate levels are associated with increased hypoxia-inducible factor-1 activity and an aggressive tumor phenotype in endometrial cancer. Cancer Res. 70 (14), 5749–5758. 10.1158/0008-5472.CAN-10-0263. [DOI] [PubMed] [Google Scholar]
- Campbell E. J.; Vissers M. C.; Bozonet S.; Dyer A.; Robinson B. A.; Dachs G. U. (2015) Restoring physiological levels of ascorbate slows tumor growth and moderates HIF-1 pathway activity in Gulo(−/−) mice. Cancer Med. 4 (2), 303–314. 10.1002/cam4.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasprzak K. S.; Diwan B. A.; Kaczmarek M. Z.; Logsdon D. L.; Fivash M. J.; Salnikow K. (2011) Effects of ascorbic acid on carcinogenicity and acute toxicity of nickel subsulfide, and on tumor transplants growth in gulonolactone oxidase knock-out mice and wild-type C57BL mice. Toxicol. Appl. Pharmacol. 257 (1), 32–37. 10.1016/j.taap.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J.; Chakraborty A. A.; Liu P.; et al. (2016) pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science 353 (6302), 929–932. 10.1126/science.aad5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa H.; Sato Y.; Tanaka Y.; Inai Y.; Amano A.; Iwama M.; Kondo Y.; Handa S.; Murata A.; Nishikimi M.; Goto S.; Maruyama N.; Takahashi R.; Ishigami A. (2008) Vitamin C is not essential for carnitine biosynthesis in vivo: verification in vitamin C-depleted senescence marker protein-30/gluconolactonase knockout mice. Biol. Pharm. Bull. 31 (9), 1673–1679. 10.1248/bpb.31.1673. [DOI] [PubMed] [Google Scholar]
- Wu X.; Zhang Y. (2017) TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet. 18 (9), 517–534. 10.1038/nrg.2017.33. [DOI] [PubMed] [Google Scholar]
- Blaschke K.; Ebata K. T.; Karimi M. M.; et al. (2013) Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 500 (7461), 222–226. 10.1038/nature12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor E. A.; Court B. L.; Young J. I.; Wang G. (2013) Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J. Biol. Chem. 288 (19), 13669–13674. 10.1074/jbc.C113.464800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin R.; Mao S. Q.; Zhao B.; et al. (2013) Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 135 (28), 10396–10403. 10.1021/ja4028346. [DOI] [PubMed] [Google Scholar]
- Chung T. L.; Brena R. M.; Kolle G.; et al. (2010) Vitamin C promotes widespread yet specific DNA demethylation of the epigenome in human embryonic stem cells. Stem Cells 28 (10), 1848–1855. 10.1002/stem.493. [DOI] [PubMed] [Google Scholar]
- Cimmino L.; Dolgalev I.; Wang Y.; et al. (2017) Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 170 (6), 1079–1095. 10.1016/j.cell.2017.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hore T. A.; von Meyenn F.; Ravichandran M.; et al. (2016) Retinol and ascorbate drive erasure of epigenetic memory and enhance reprogramming to naïve pluripotency by complementary mechanisms. Proc. Natl. Acad. Sci. U. S. A. 113 (43), 12202–12207. 10.1073/pnas.1608679113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X. B.; Kim M.; Kim S. Y.; Yi S. H.; Rhee Y. H.; Kim T.; Lee E. H.; Park C. H.; Dixit S.; Harrison F. E.; Lee S. H. (2015) Vitamin C facilitates dopamine neuron differentiation in fetal midbrain through TET1- and JMJD3-dependent epigenetic control manner. Stem Cells 33 (4), 1320–1332. 10.1002/stem.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. U.; Su Y.; Zhong C.; Ming G. L.; Song H. (2011) Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145 (3), 423–434. 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaas G. A.; Zhong C.; Eason D. E.; et al. (2013) TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron 79 (6), 1086–1093. 10.1016/j.neuron.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes C.; Sousa N.; Pinto L.; Marques C. J. (2019) TET enzymes in neurophysiology and brain function. Neurosci. Biobehav. Rev. 102, 337–344. 10.1016/j.neubiorev.2019.05.006. [DOI] [PubMed] [Google Scholar]
- Dixit S.; Bernardo A.; Walker J. M.; Kennard J. A.; Kim G. Y.; Kessler E. S.; Harrison F. E. (2015) Vitamin C deficiency in the brain impairs cognition, increases amyloid accumulation and deposition, and oxidative stress in APP/PSEN1 and normally aging mice. ACS Chem. Neurosci. 6 (4), 570–581. 10.1021/cn500308h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y.; Sasaki T.; Sato Y.; Amano A.; Aizawa S.; Iwama M.; Handa S.; Shimada N.; Fukuda M.; Akita M.; Lee J.; Jeong K. S.; Maruyama N.; Ishigami A. (2008) Vitamin C depletion increases superoxide generation in brains of SMP30/GNL knockout mice. Biochem. Biophys. Res. Commun. 377 (1), 291–296. 10.1016/j.bbrc.2008.09.132. [DOI] [PubMed] [Google Scholar]
- Black J. C.; Van Rechem C.; Whetstine J. R. (2012) Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol. Cell 48, 491–507. 10.1016/j.molcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun K.; Jeon J.; Park K.; Kim J. (2017) Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 49 (4), e324. 10.1038/emm.2017.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y.; Lan F.; Matson C.; et al. (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119 (7), 941–953. 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Karytinos A.; Forneris F.; Profumo A.; et al. (2009) A novel mammalian flavin-dependent histone demethylase. J. Biol. Chem. 284 (26), 17775–17782. 10.1074/jbc.M109.003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose R. J.; Kallin E. M.; Zhang Y. (2006) JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 7 (9), 715–727. 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- Tsukada Y.; Fang J.; Erdjument-Bromage H.; et al. (2006) Histone demethylation by a family of JmjC domain-containing proteins. Nature 439 (7078), 811–816. 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- Whetstine J. R.; Nottke A.; Lan F.; et al. (2006) Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 125 (3), 467–481. 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- Wang T.; Chen K.; Zeng X.; et al. (2011) The histone demethylases Jhdm1a/1b enhance somatic cell reprogramming in a vitamin-C-dependent manner. Cell Stem Cell. 9 (6), 575–587. 10.1016/j.stem.2011.10.005. [DOI] [PubMed] [Google Scholar]
- Chen J.; Liu H.; Liu J.; et al. (2013) H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat. Genet. 45 (1), 34–42. 10.1038/ng.2491. [DOI] [PubMed] [Google Scholar]
- Ebata K. T.; Mesh K.; Liu S.; et al. (2017) Vitamin C induces specific demethylation of H3K9me2 in mouse embryonic stem cells via Kdm3a/b. Epigenet. Chromatin 10, 36. 10.1186/s13072-017-0143-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron E.; Pauling L. (1976) Supplemental ascorbate in the supportive treatment of cancer: prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. U. S. A. 73 (10), 3685–3689. 10.1073/pnas.73.10.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron E.; Pauling L. (1978) Supplemental ascorbate in the supportive treatment of cancer: reevaluation of prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. U. S. A. 75 (9), 4538–4542. 10.1073/pnas.75.9.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creagan E. T.; Moertel C. G.; O’Fallon J. R.; et al. (1979) Failure of high-dose vitamin C (ascorbic acid) therapy to benefit patients with advanced cancer. A controlled trial. N. Engl. J. Med. 301 (13), 687–690. 10.1056/NEJM197909273011303. [DOI] [PubMed] [Google Scholar]
- Moertel C. G.; Fleming T. R.; Creagan E. T.; Rubin J.; O’Connell M. J.; Ames M. M. (1985) High-dose vitamin C versus placebo in the treatment of patients with advanced cancer who have had no prior chemotherapy. A randomized double-blind comparison. N. Engl. J. Med. 312 (3), 137–141. 10.1056/NEJM198501173120301. [DOI] [PubMed] [Google Scholar]
- Padayatty S. J.; Sun H.; Wang Y.; et al. (2004) Vitamin C pharmacokinetics: implications for oral and intravenous use. Ann. Intern. Med. 140 (7), 533–537. 10.7326/0003-4819-140-7-200404060-00010. [DOI] [PubMed] [Google Scholar]
- Parrow N. L.; Leshin J. A.; Levine M. (2013) Parenteral ascorbate as a cancer therapeutic: a reassessment based on pharmacokinetics. Antioxid. Redox Signaling 19 (17), 2141–2156. 10.1089/ars.2013.5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J.; Mullarky E.; Lu C.; Bosch K. N.; Kavalier A.; Rivera K.; Roper J.; Chio I. I.; Giannopoulou E. G.; Rago C.; Muley A.; Asara J. M.; Paik J.; Elemento O.; Chen Z.; Pappin D. J.; Dow L. E.; Papadopoulos N.; Gross S. S.; Cantley L. C. (2015) Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 350 (6266), 1391–1396. 10.1126/science.aaa5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W.; Wang Y.; Xu Y.; et al. (2014) The hypoxia-inducible factor renders cancer cells more sensitive to vitamin C-induced toxicity. J. Biol. Chem. 289 (6), 3339–3351. 10.1074/jbc.M113.538157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfeld J. D.; Sibenaller Z. A.; Mapuskar K. A.; et al. (2017) O2·- and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell 31 (4), 487–500. 10.1016/j.ccell.2017.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt K. E.; Falls K. C.; Schoenfeld J. D.; et al. (2018) Augmentation of intracellular iron using iron sucrose enhances the toxicity of pharmacological ascorbate in colon cancer cells. Redox Biol. 14, 82–87. 10.1016/j.redox.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L.; Zhang L.; Wang S.; Zhao B.; Lv H.; Shang P. (2020) Labile iron affects pharmacological ascorbate-induced toxicity in osteosarcoma cell lines. Free Radical Res. 54, 385. 10.1080/10715762.2020.1744577. [DOI] [PubMed] [Google Scholar]
- Herst P. M.; Broadley K. W.; Harper J. L.; McConnell M. J. (2012) Pharmacological concentrations of ascorbate radiosensitize glioblastoma multiforme primary cells by increasing oxidative DNA damage and inhibiting G2/M arrest. Free Radical Biol. Med. 52 (8), 1486–1493. 10.1016/j.freeradbiomed.2012.01.021. [DOI] [PubMed] [Google Scholar]
- Buranasudja V.; Doskey C. M.; Gibson A. R.; et al. (2019) Pharmacologic Ascorbate Primes Pancreatic Cancer Cells for Death by Rewiring Cellular Energetics and Inducing DNA Damage. Mol. Cancer Res. 17 (10), 2102–2114. 10.1158/1541-7786.MCR-19-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y.; Chapman J.; Levine M.; Polireddy K.; Drisko J.; Chen Q. (2014) High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci. Transl. Med. 6 (222), 222ra18. 10.1126/scitranslmed.3007154. [DOI] [PubMed] [Google Scholar]
- Alexander M. S.; Wilkes J. G.; Schroeder S. R.; et al. (2018) Pharmacologic Ascorbate Reduces Radiation-Induced Normal Tissue Toxicity and Enhances Tumor Radiosensitization in Pancreatic Cancer. Cancer Res. 78 (24), 6838–6851. 10.1158/0008-5472.CAN-18-1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafi S.; Camarena V.; Volmar C. H.; et al. (2018) Vitamin C Sensitizes Melanoma to BET Inhibitors. Cancer Res. 78 (2), 572–583. 10.1158/0008-5472.CAN-17-2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha J.; Roomi M. W.; Ivanov V.; Kalinovsky T.; Niedzwiecki A.; Rath M. (2013) Ascorbate supplementation inhibits growth and metastasis of B16FO melanoma and 4T1 breast cancer cells in vitamin C-deficient mice. Int. J. Oncol. 42 (1), 55–64. 10.3892/ijo.2012.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchtel R. A.; Bhagat T.; Pradhan K.; et al. (2020) High-dose ascorbic acid synergizes with anti-PD1 in a lymphoma mouse model. Proc. Natl. Acad. Sci. U. S. A. 117 (3), 1666–1677. 10.1073/pnas.1908158117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds M.; Stoddard L.; Bespalov I.; Zhitkovich A. (2007) Ascorbate acts as a highly potent inducer of chromate mutagenesis and clastogenesis: linkage to DNA breaks in G2 phase by mismatch repair. Nucleic Acids Res. 35, 465–476. 10.1093/nar/gkl1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLoughery Z.; Luczak M. W.; Ortega-Atienza S.; Zhitkovich A. (2015) DNA double-strand breaks by Cr(VI) are targeted to euchromatin and cause ATR-dependent phosphorylation of histone H2AX and its ubiquitination. Toxicol. Sci. 143 (1), 54–63. 10.1093/toxsci/kfu207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mårtensson J.; Meister A. (1992) Glutathione deficiency increases hepatic ascorbic acid synthesis in adult mice. Proc. Natl. Acad. Sci. U. S. A. 89 (23), 11566–11568. 10.1073/pnas.89.23.11566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horio F.; Shibata T.; Naito Y.; Nishikimi M.; Yagi K.; Yoshida A. (1993) L-gulono-gamma-lactone oxidase is not induced in rats by xenobiotics stimulating L-ascorbic acid biosynthesis. J. Nutr. Sci. Vitaminol. 39 (1), 1–9. 10.3177/jnsv.39.1. [DOI] [PubMed] [Google Scholar]
- Smirnoff N. (2018) Ascorbic acid metabolism and functions: A comparison of plants and mammals. Free Radical Biol. Med. 122, 116–129. 10.1016/j.freeradbiomed.2018.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsten P.; Amitai G.; Kehrl J.; Dhariwal K. R.; Klein H. G.; Levine M. (1990) Millimolar concentrations of ascorbic acid in purified human mononuclear leukocytes. Depletion and reaccumulation. J. Biol. Chem. 265 (5), 2584–2587. [PubMed] [Google Scholar]
- Reynolds M.; Zhitkovich A. (2007) Cellular vitamin C increases chromate toxicity via a death program requiring mismatch repair but not p53. Carcinogenesis 28, 1613–1620. 10.1093/carcin/bgm031. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Negoro T.; Satoh K.; Jiang Y.; Hashimoto K.; Kikuchi H.; Nishikawa H.; Miyata T.; Yamamoto Y.; Nakano K.; Yasumoto E.; Nakayachi T.; Mineno K.; Satoh T.; Sakagami H. (2001) Synergistic cytotoxic action of vitamin C and vitamin K3. Anticancer Res. 21 (5), 3439–3444. [PubMed] [Google Scholar]
- Jamison J. M.; Gilloteaux J.; Nassiri M. R.; Venugopal M.; Neal D. R.; Summers J. L. (2004) Cell cycle arrest and autoschizis in a human bladder carcinoma cell line following Vitamin C and Vitamin K3 treatment. Biochem. Pharmacol. 67 (2), 337–351. 10.1016/j.bcp.2003.08.040. [DOI] [PubMed] [Google Scholar]
- Luczak M. W.; Green S. E.; Zhitkovich A. (2016) Different ATM signaling in response to chromium(VI) metabolism via ascorbate and nonascorbate reduction: implications for in vitro models and toxicogenomics. Environ. Health Perspect. 124, 61–66. 10.1289/ehp.1409434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vislisel J. M.; Schafer F. Q.; Buettner G. R. (2007) A simple and sensitive assay for ascorbate using a plate reader. Anal. Biochem. 365 (1), 31–39. 10.1016/j.ab.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis S. A.; Paule R. C.; Ziegler R. G. (1990) Ascorbic and dehydroascorbic acids measured in plasma preserved with dithiothreitol or metaphosphoric acid. Clin. Chem. 36 (10), 1750–1755. 10.1093/clinchem/36.10.1750. [DOI] [PubMed] [Google Scholar]
- Niu Y.; DesMarais T. L.; Tong Z.; Yao Y.; Costa M. (2015) Oxidative stress alters global histone modification and DNA methylation. Free Radical Biol. Med. 82, 22–28. 10.1016/j.freeradbiomed.2015.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawic C.; Zhitkovich A. (2018) Toxicological Antagonism among Welding Fume Metals: Inactivation of Soluble Cr(VI) by Iron. Chem. Res. Toxicol. 31 (11), 1172–1184. 10.1021/acs.chemrestox.8b00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vliet A.; O’Neill C. A.; Cross C. E.; Koostra J. M.; Volz W. G.; Halliwell B.; Louie S. (1999) Determination of low-molecular-mass antioxidant concentrations in human respiratory tract lining fluids. Am. J. Physiol. 276, L289–296. 10.1152/ajplung.1999.276.2.L289. [DOI] [PubMed] [Google Scholar]
- Slade R.; Stead A. G.; Graham J. A.; Hatch G. E. (1985) Comparison of lung antioxidant levels in humans and laboratory animals. Am. Rev. Respir. Dis. 131, 742–746. 10.1164/arrd.1985.131.5.742. [DOI] [PubMed] [Google Scholar]
- Suzuki Y.; Fukuda K. (1990) Reduction of hexavalent chromium by ascorbic acid and glutathione with special reference to the rat lung. Arch. Toxicol. 64, 169–176. 10.1007/BF02010721. [DOI] [PubMed] [Google Scholar]
- Krawic C.; Luczak M. W.; Zhitkovich A. (2017) Variation in extracellular detoxification is a link to different carcinogenicity among chromates in rodent and human lungs. Chem. Res. Toxicol. 30, 1720–1729. 10.1021/acs.chemrestox.7b00172. [DOI] [PMC free article] [PubMed] [Google Scholar]